
The Effect of Ultrasound on the Crystallisation of Paracetamol in the Presence of Structurally Similar Impurities

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methodology
2.1. Materials
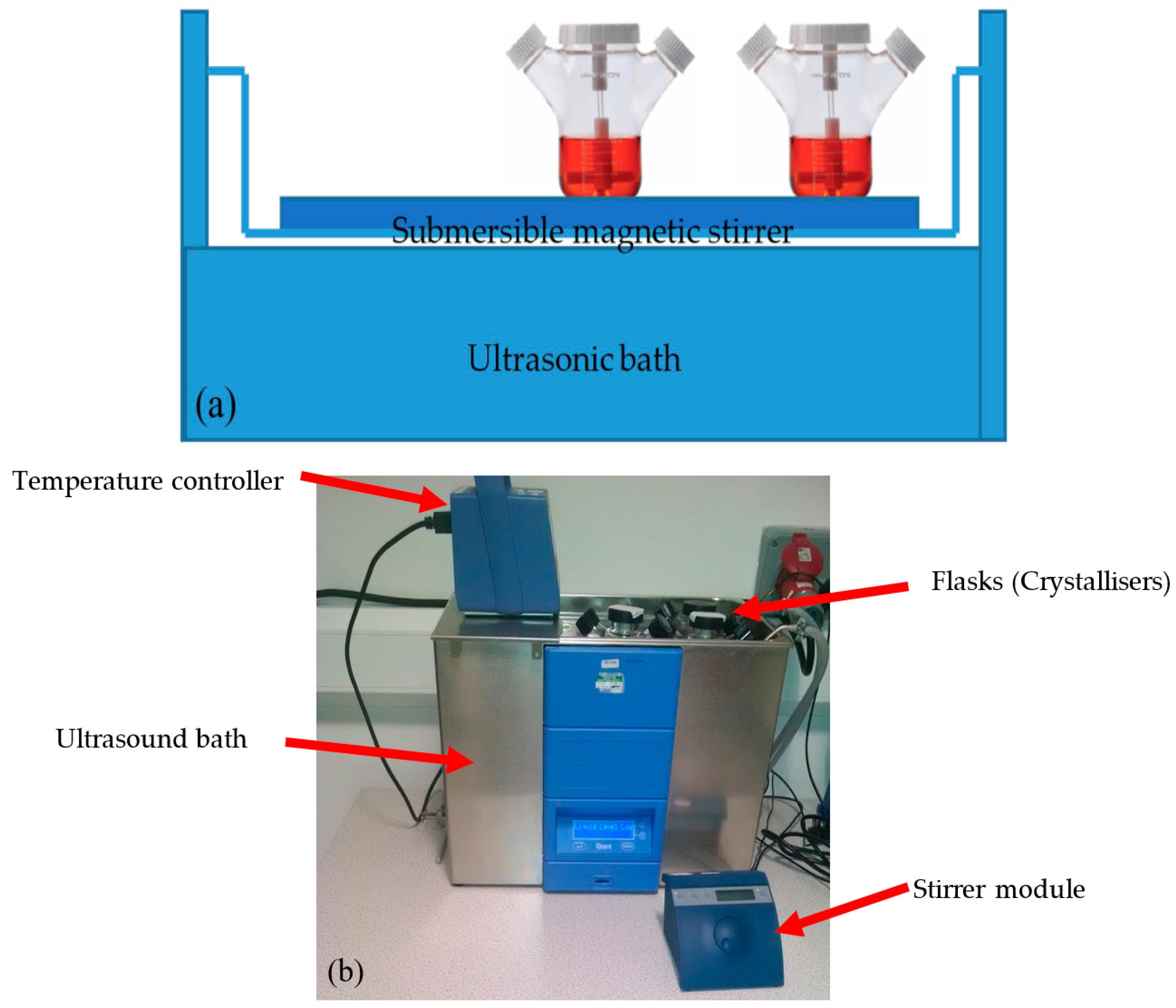
2.2. Equipment Apparatus
2.2.1. Solubility Measurements
Gravimetric Method
Detection of the Dissolution Temperature Method Using Crystalline-Technobis Crystallisation Systems
2.2.2. Sono-Crystallisation Studies
2.3. Experimental Method
2.3.1. Solubility
Gravimetric Method
Dissolution Temperature Detection Method
2.3.2. Ultrasonic Intensity Measurements
2.3.3. Sono-Crystallisation
2.3.4. Impurity Analysis Using HPLC
3. Results
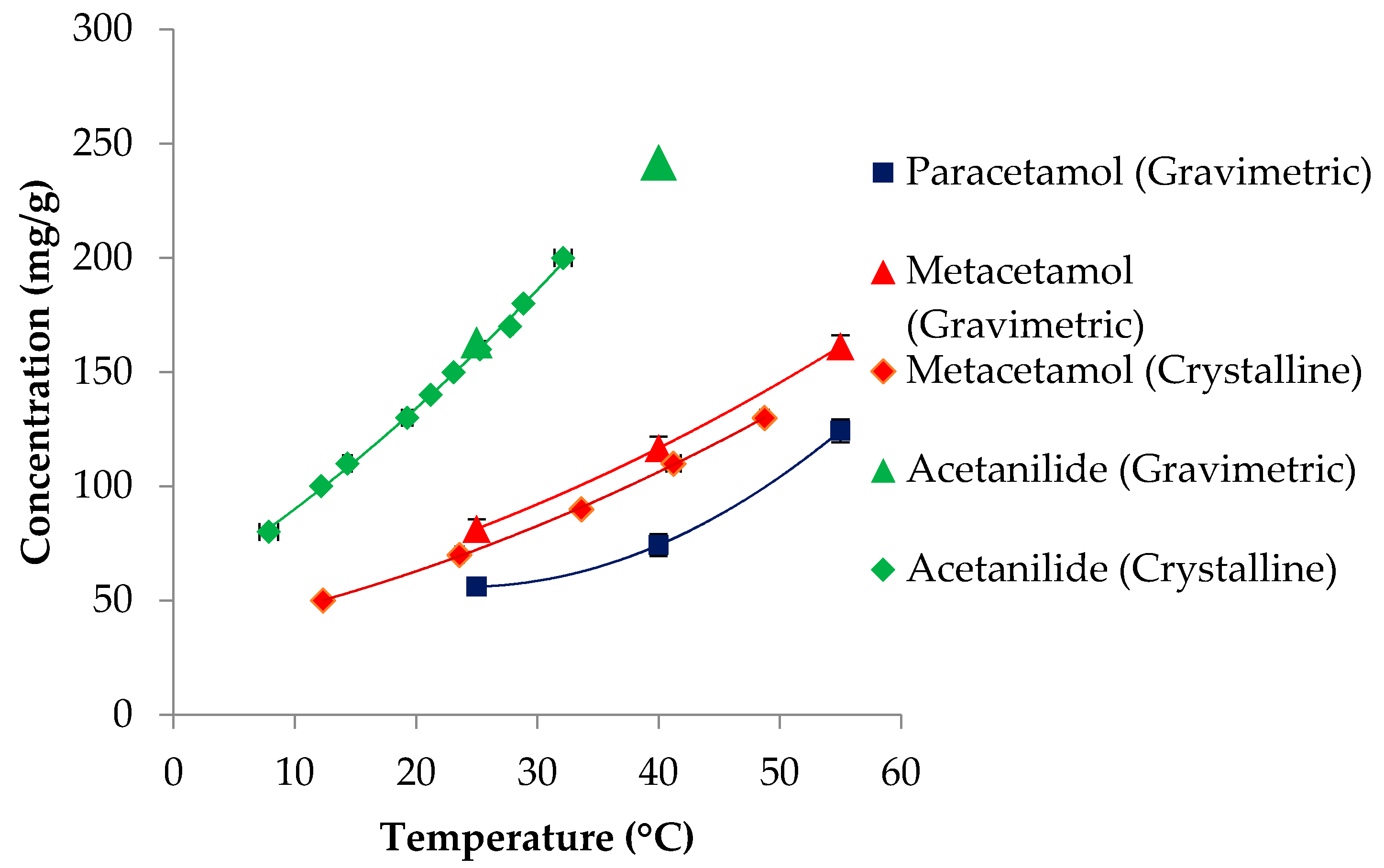
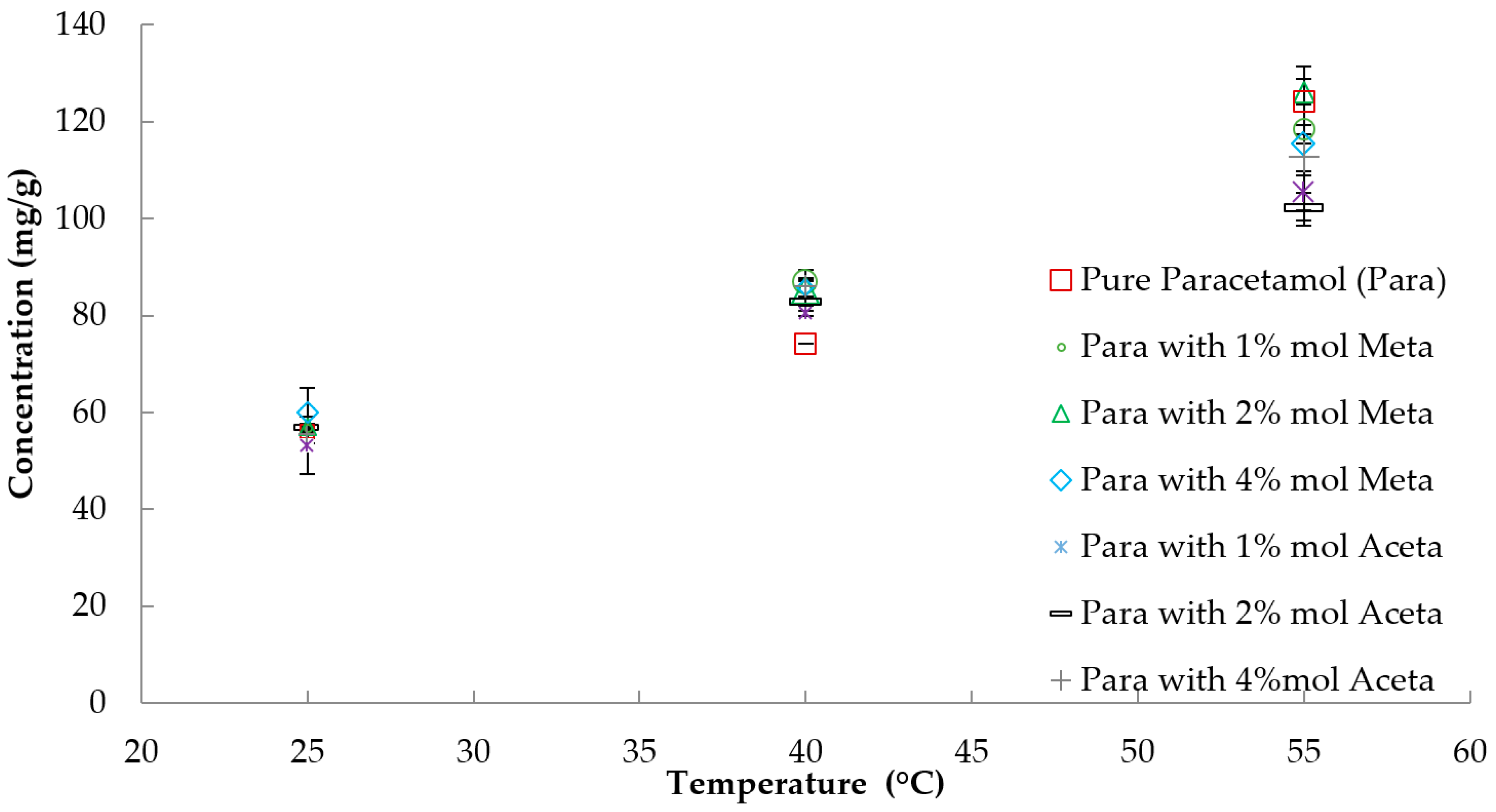
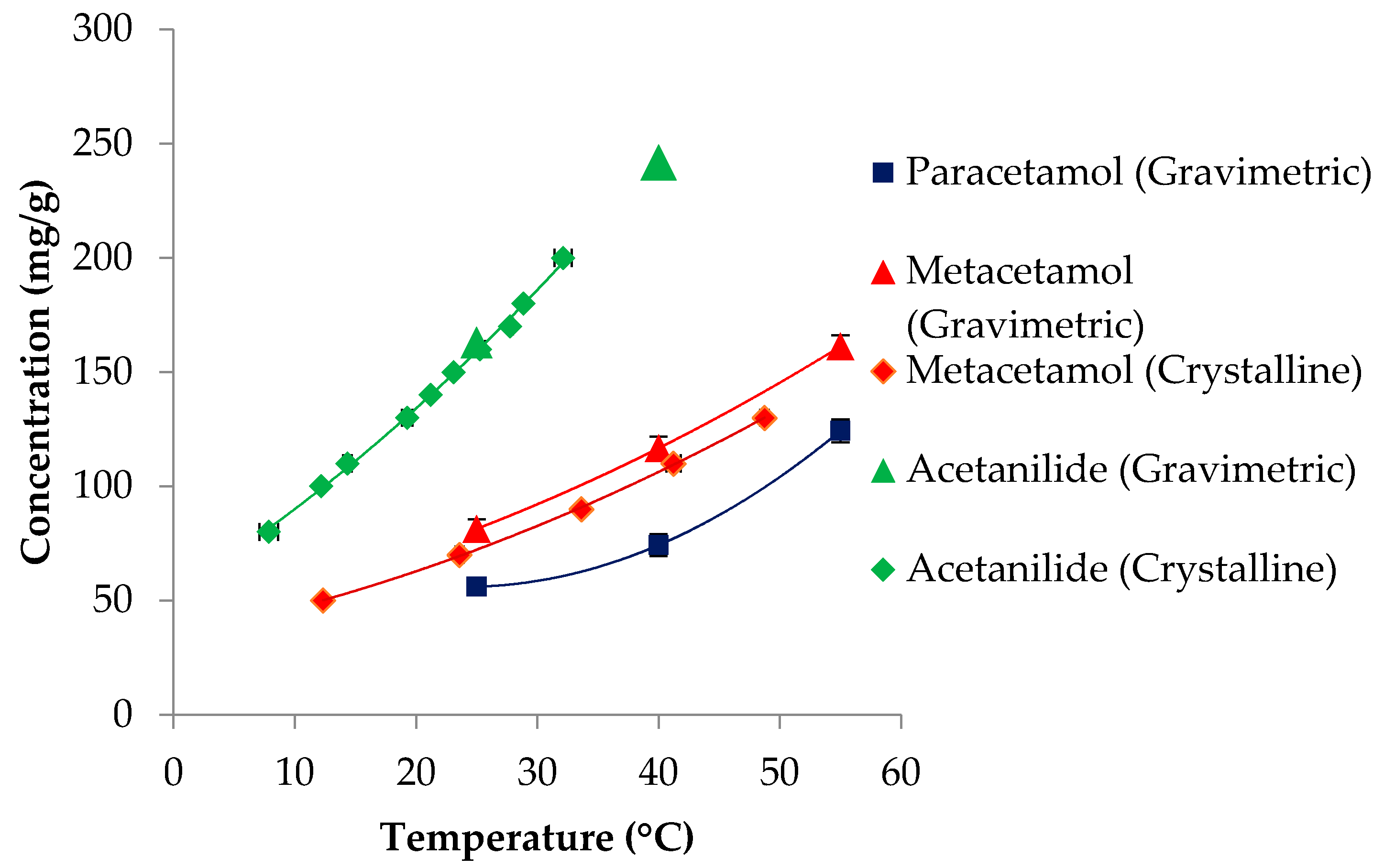
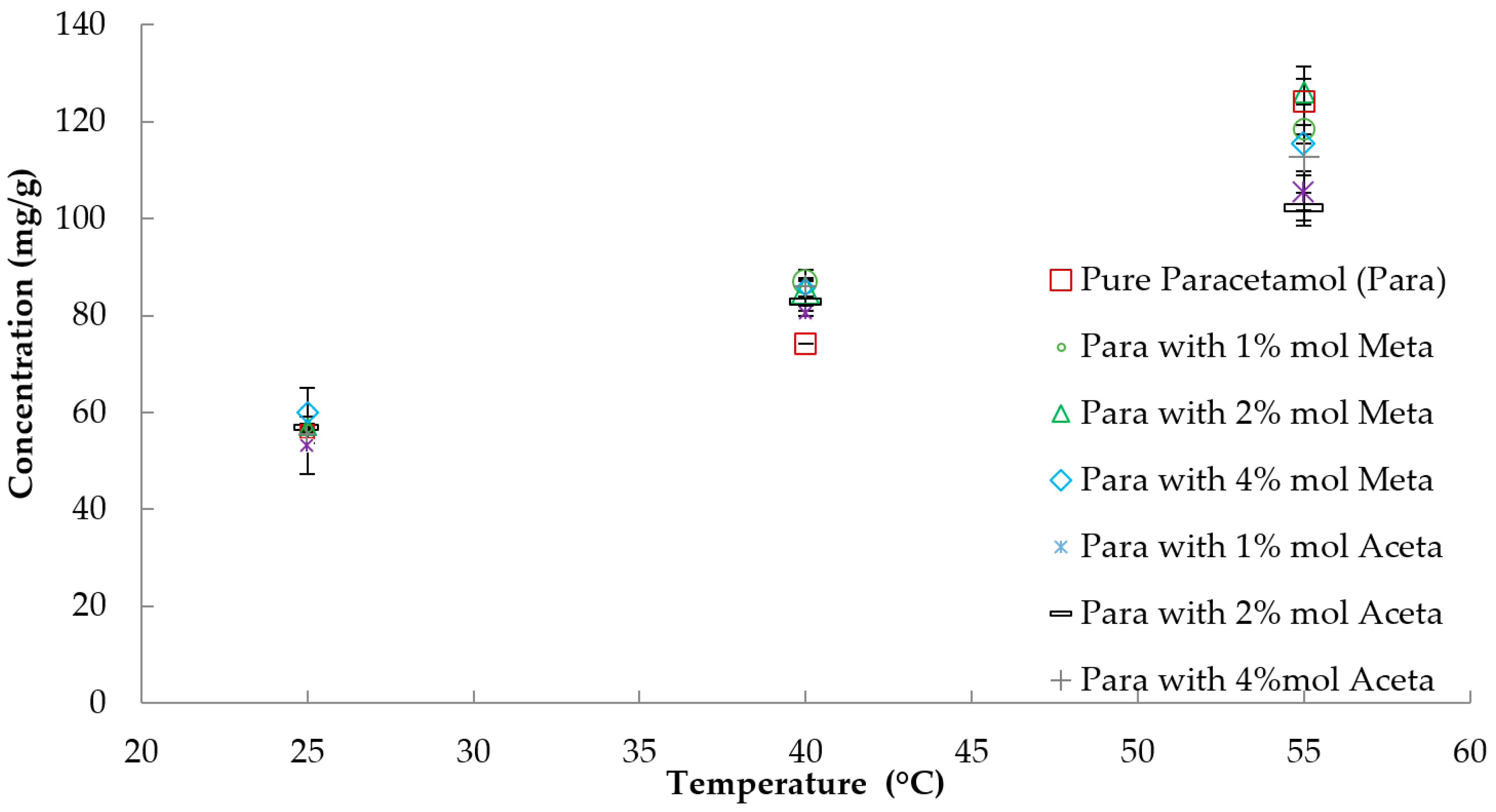
3.1. Solubility
3.2. Ultrasonic Intensity Measurements
3.3. Sonocrystallisation
3.3.1. Crystallisation Temperature
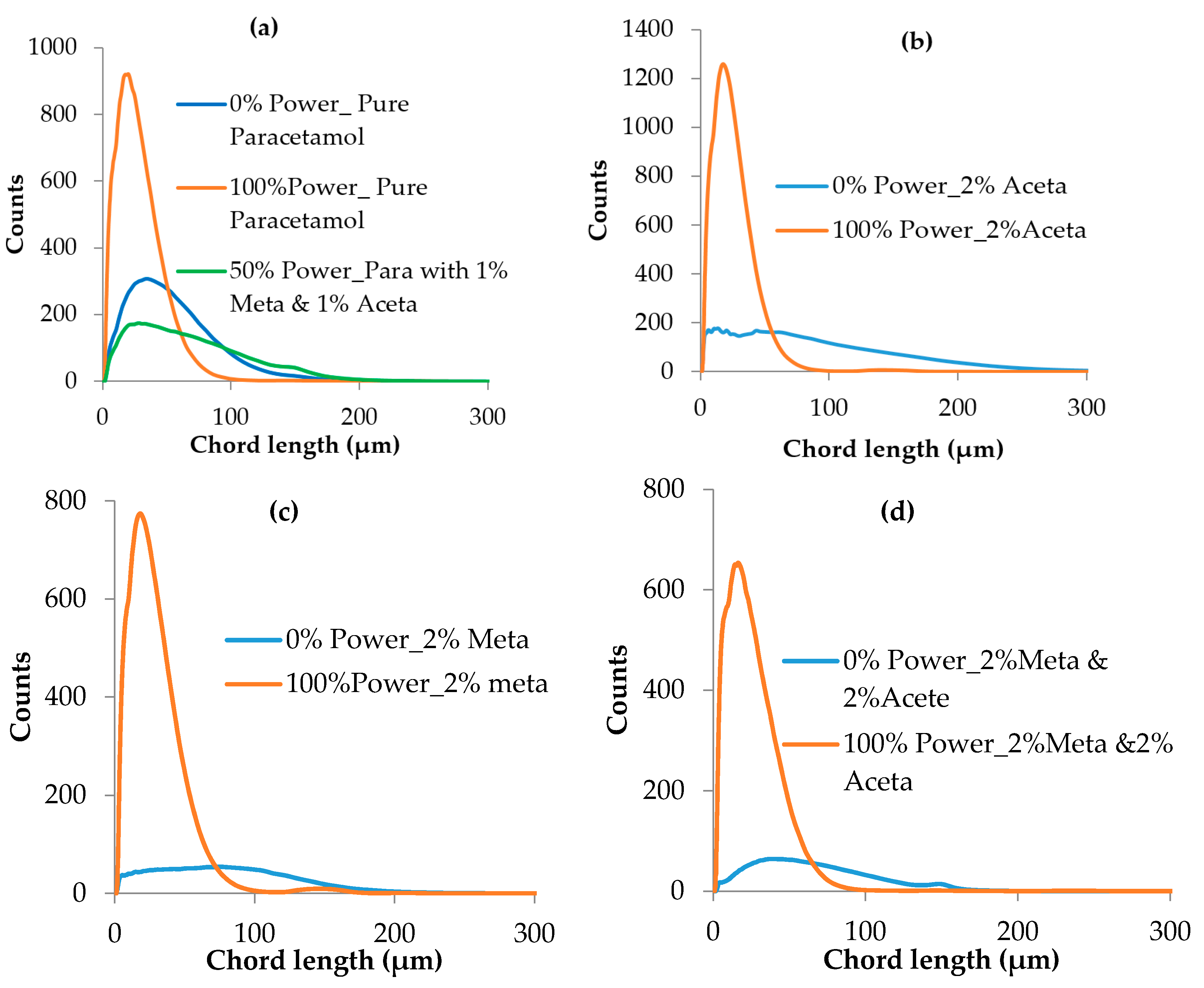
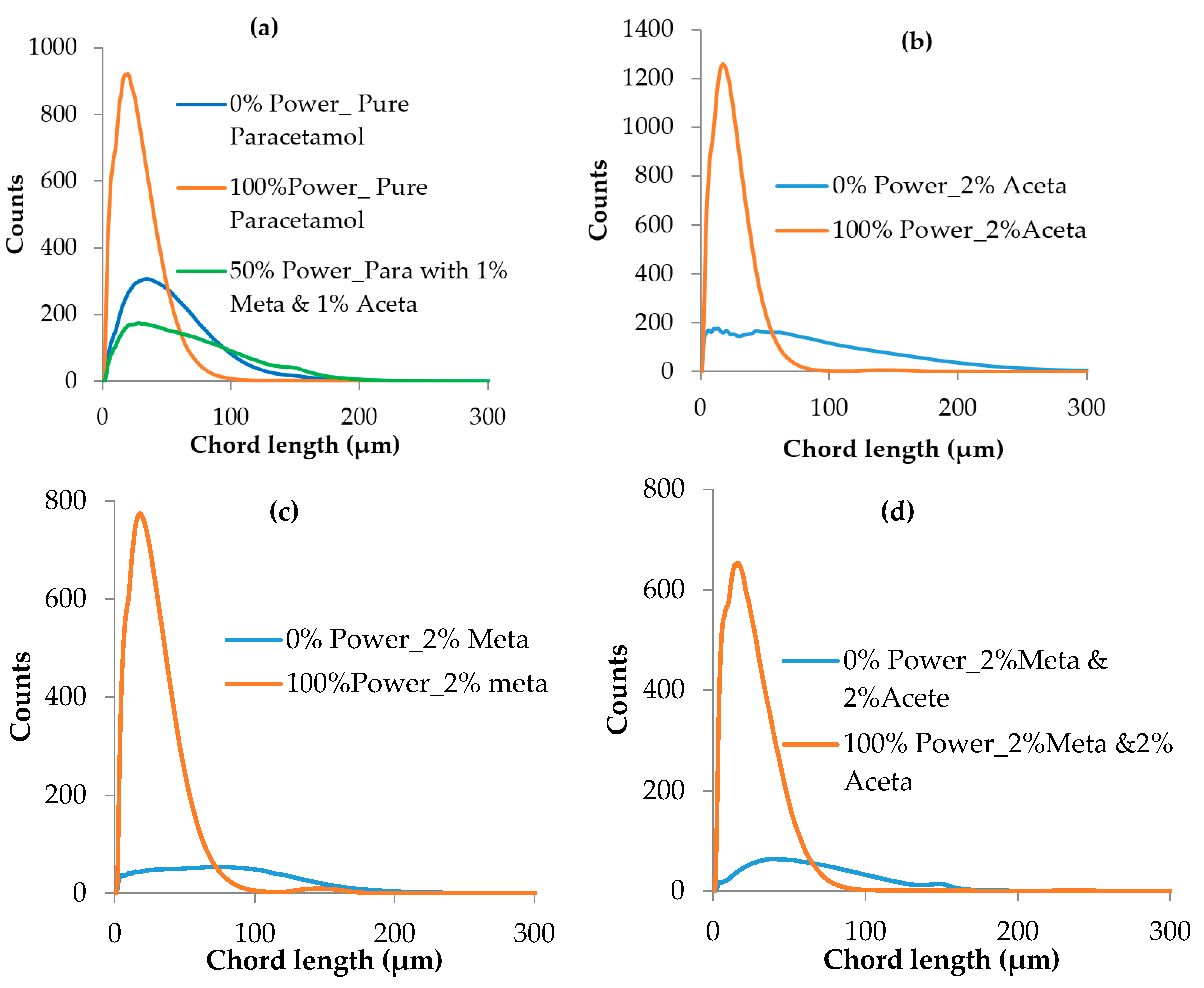
3.3.2. Crystal Size Distribution
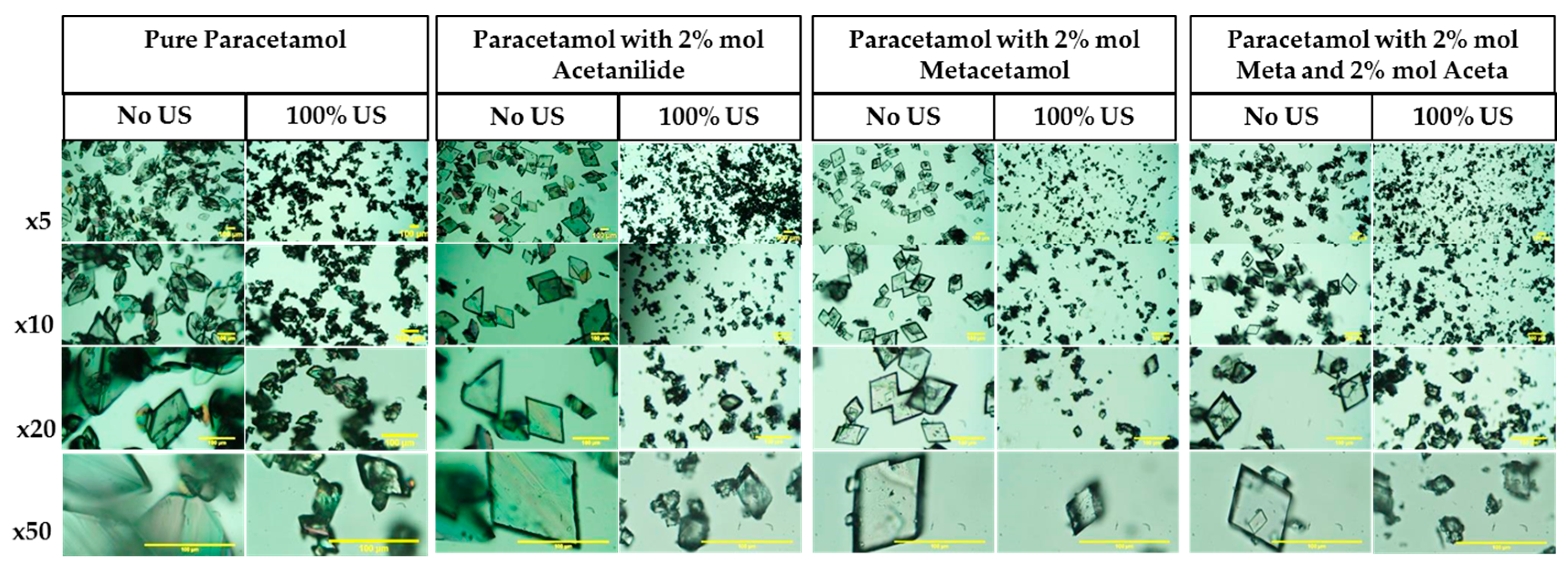
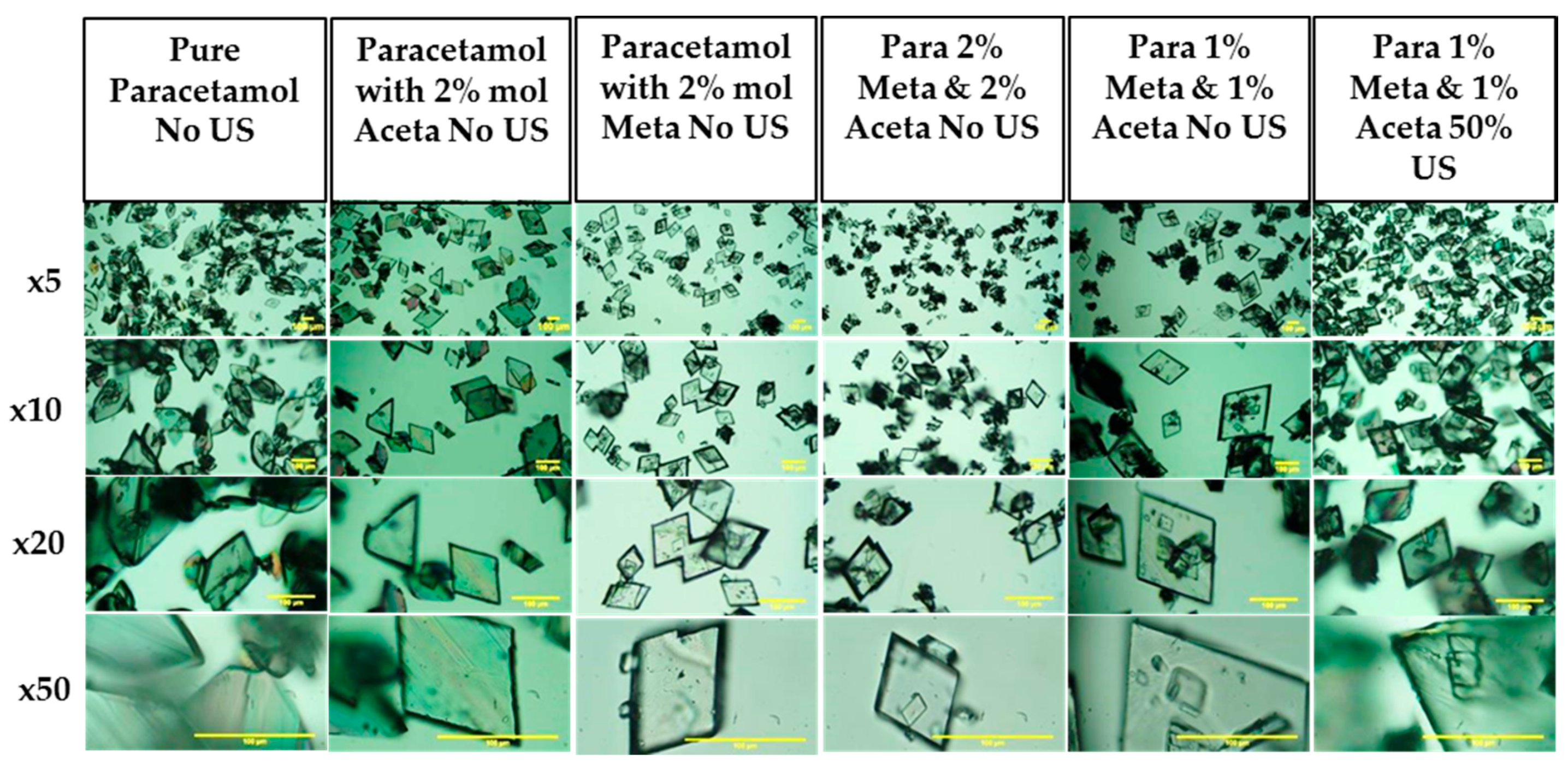
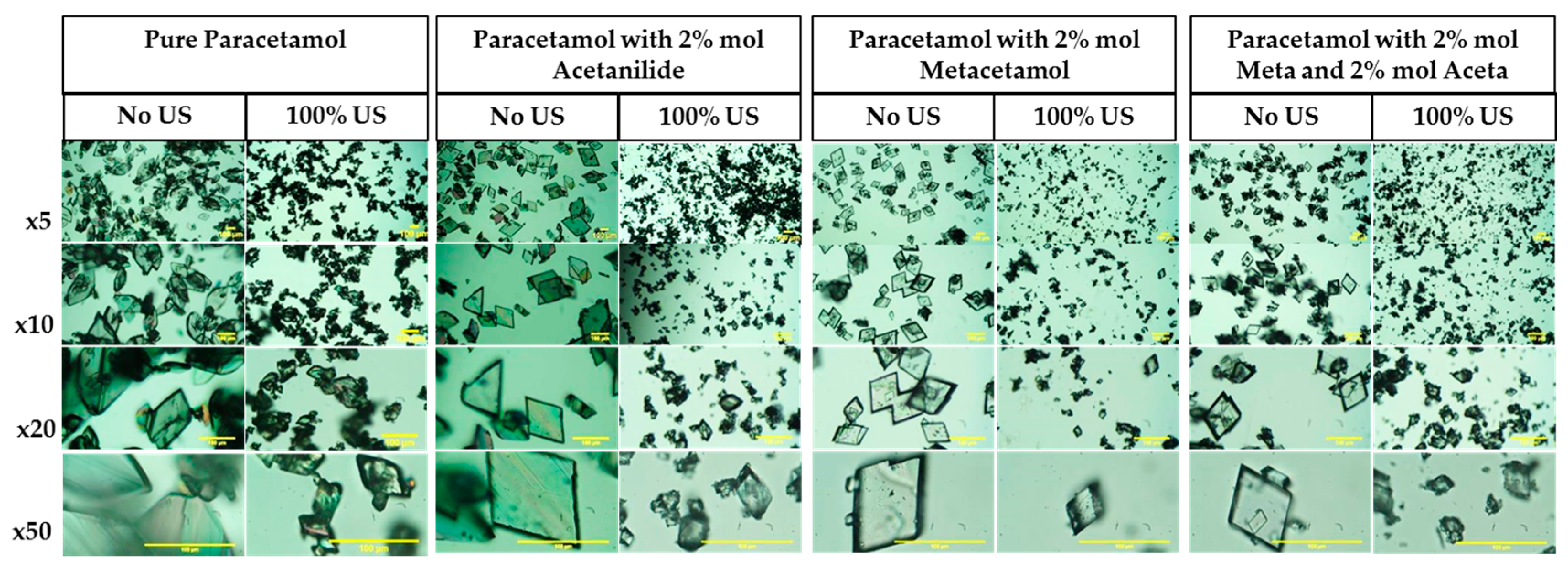
3.3.3. Crystal Habit
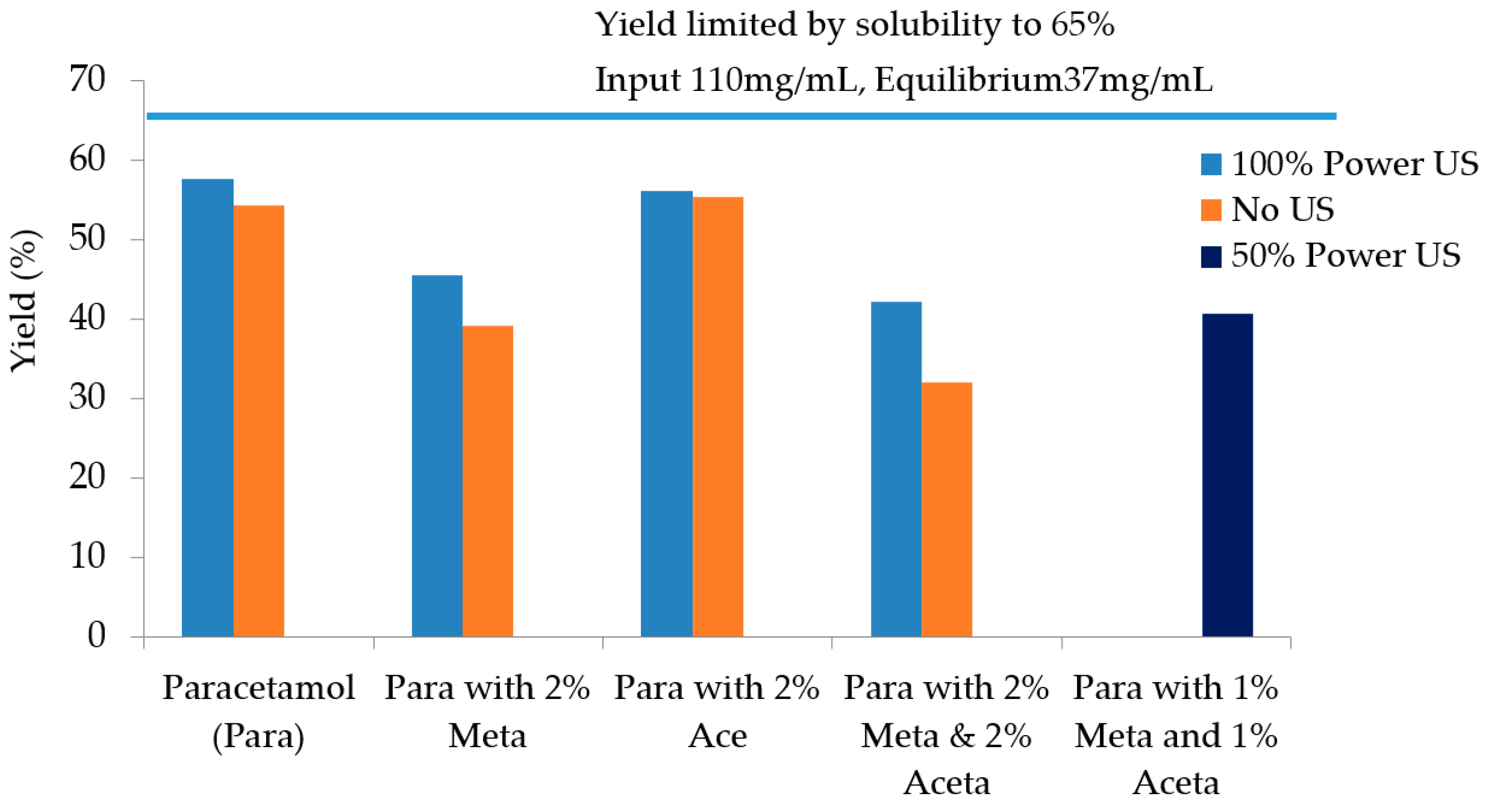
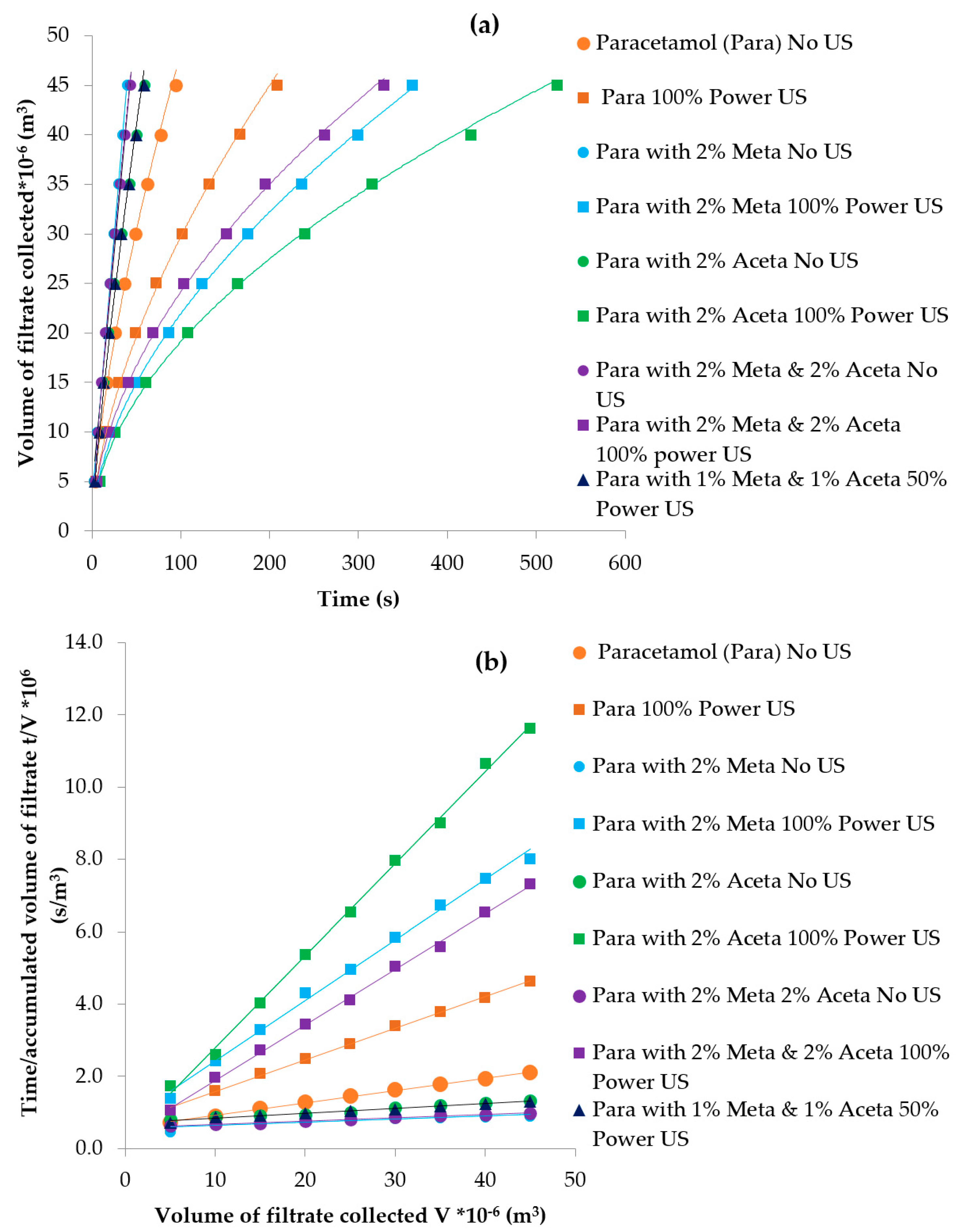
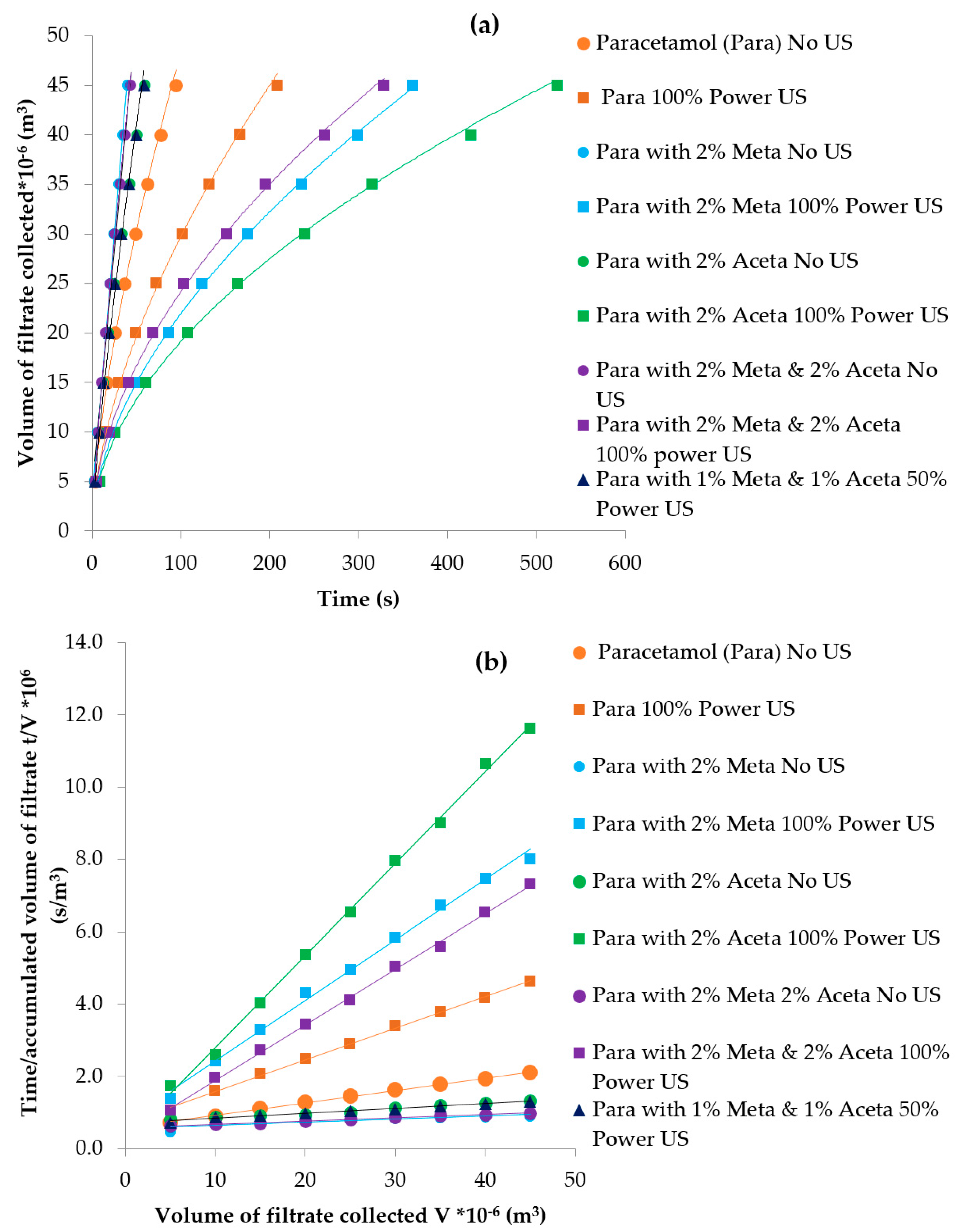
3.3.4. Particle Isolation by Filtration
3.3.5. Crystal Purity
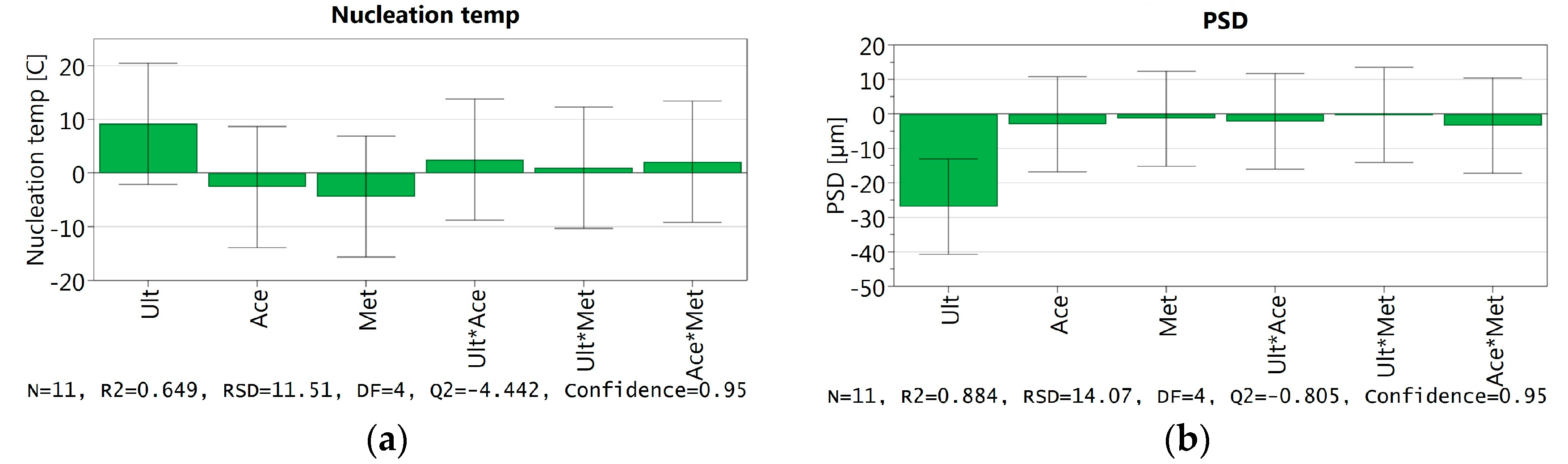
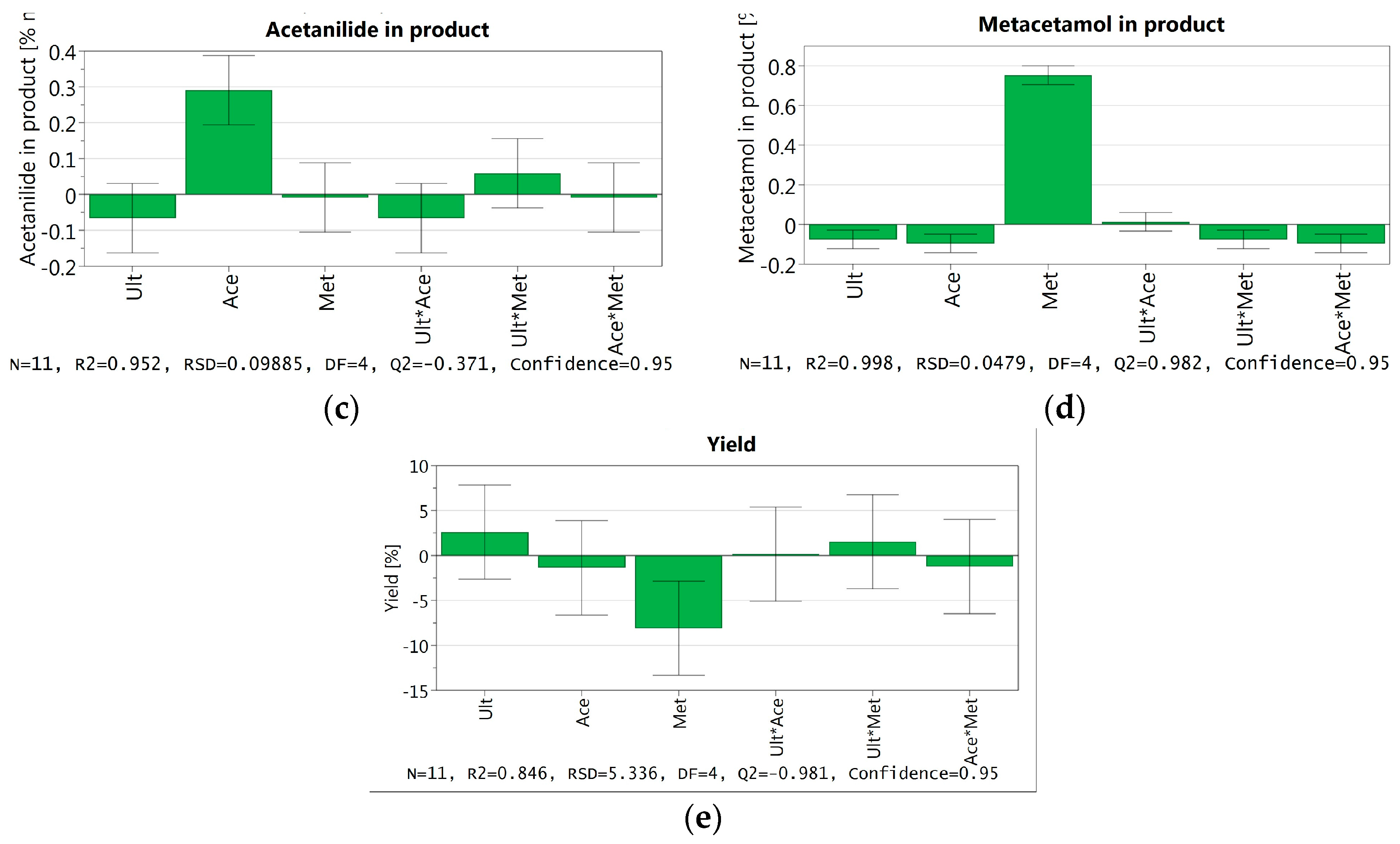
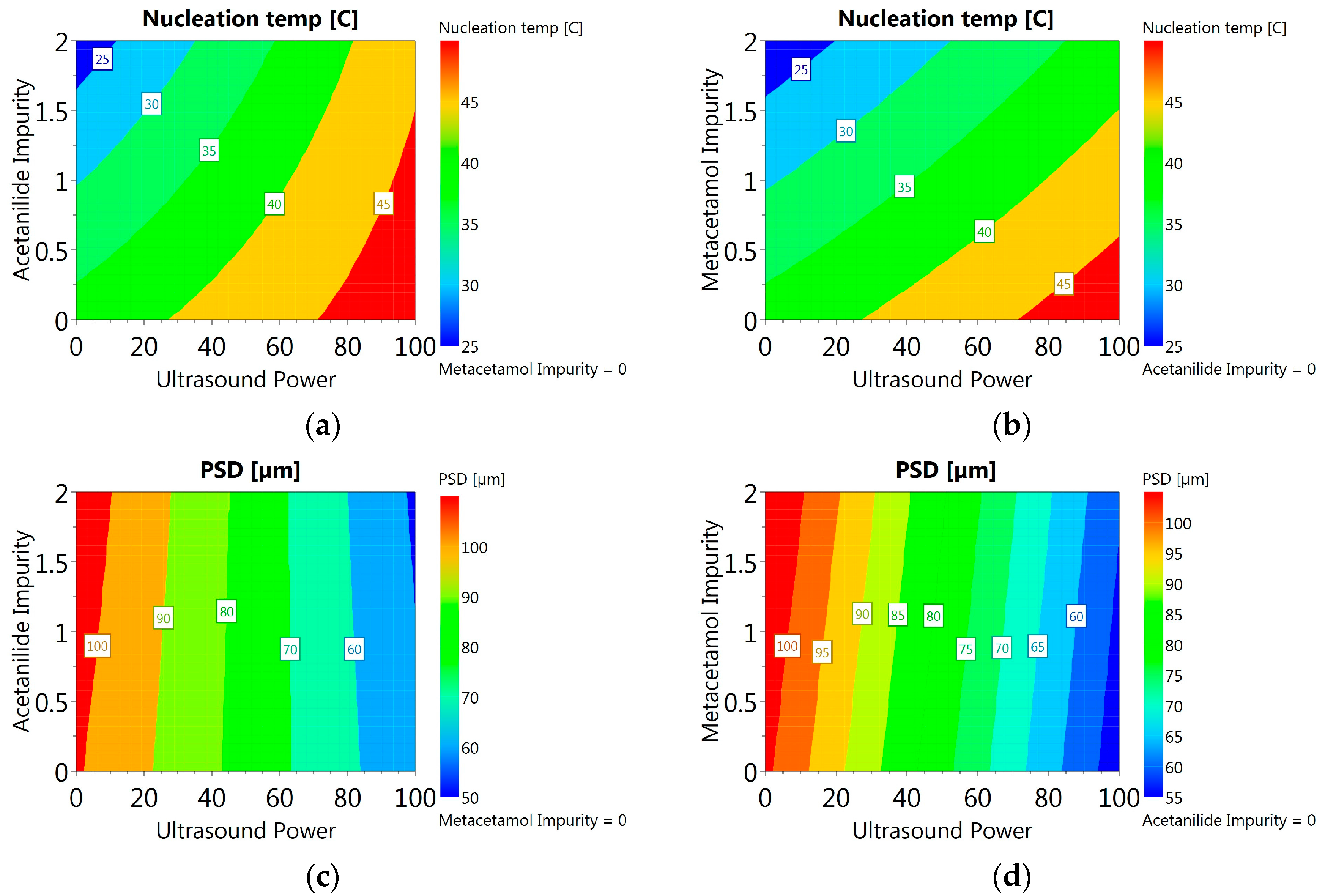
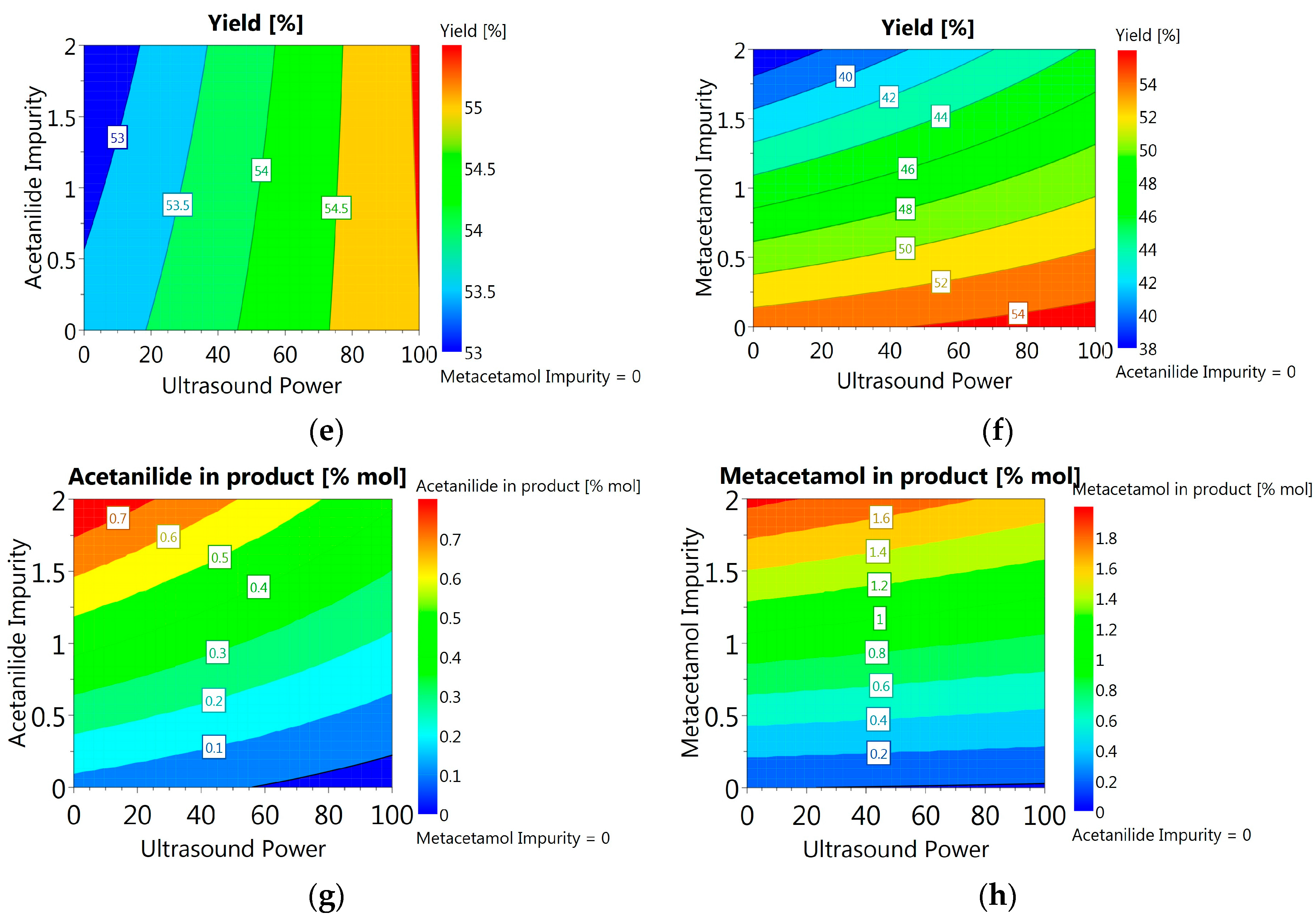
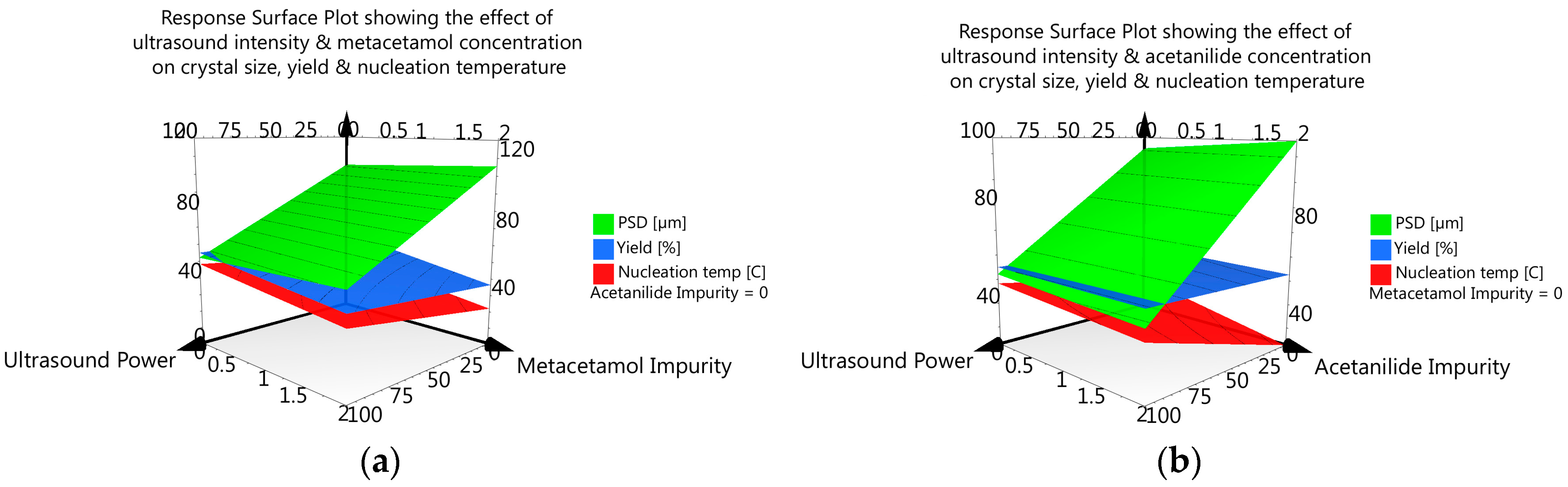
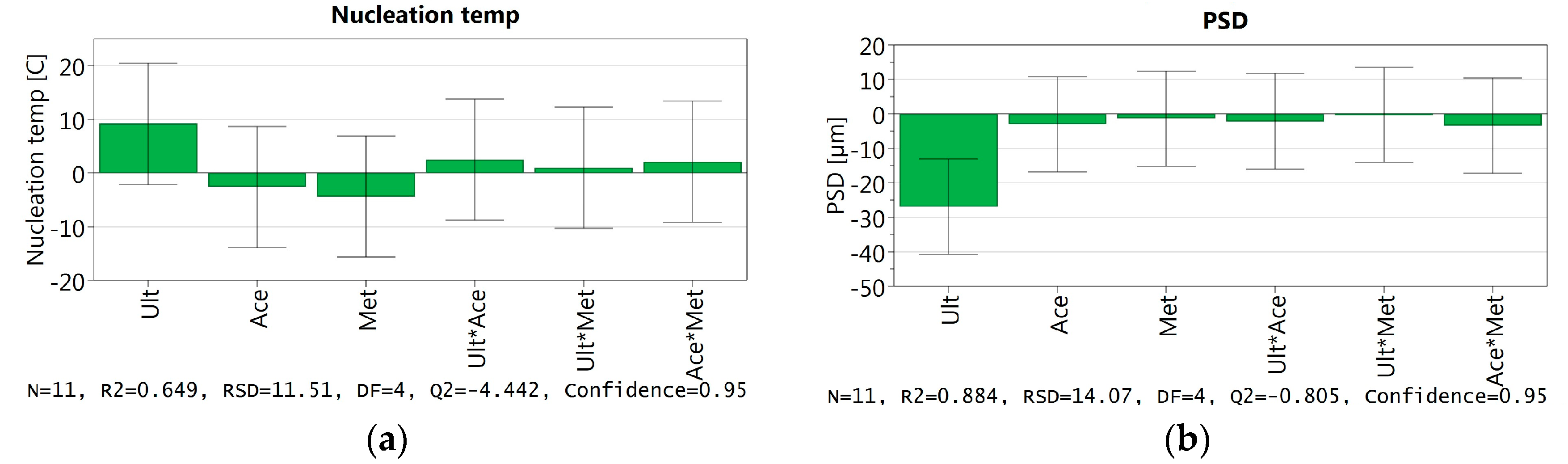
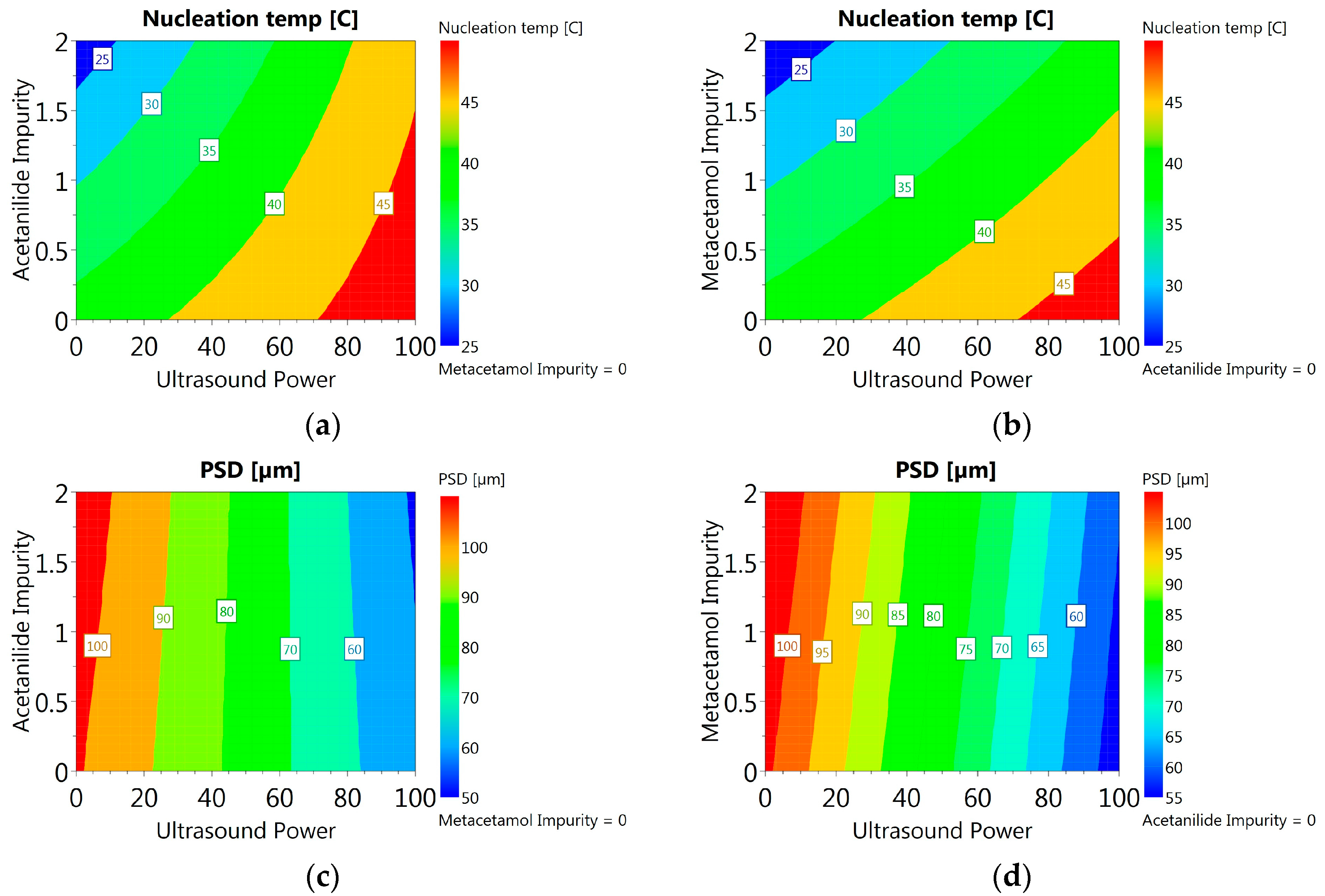
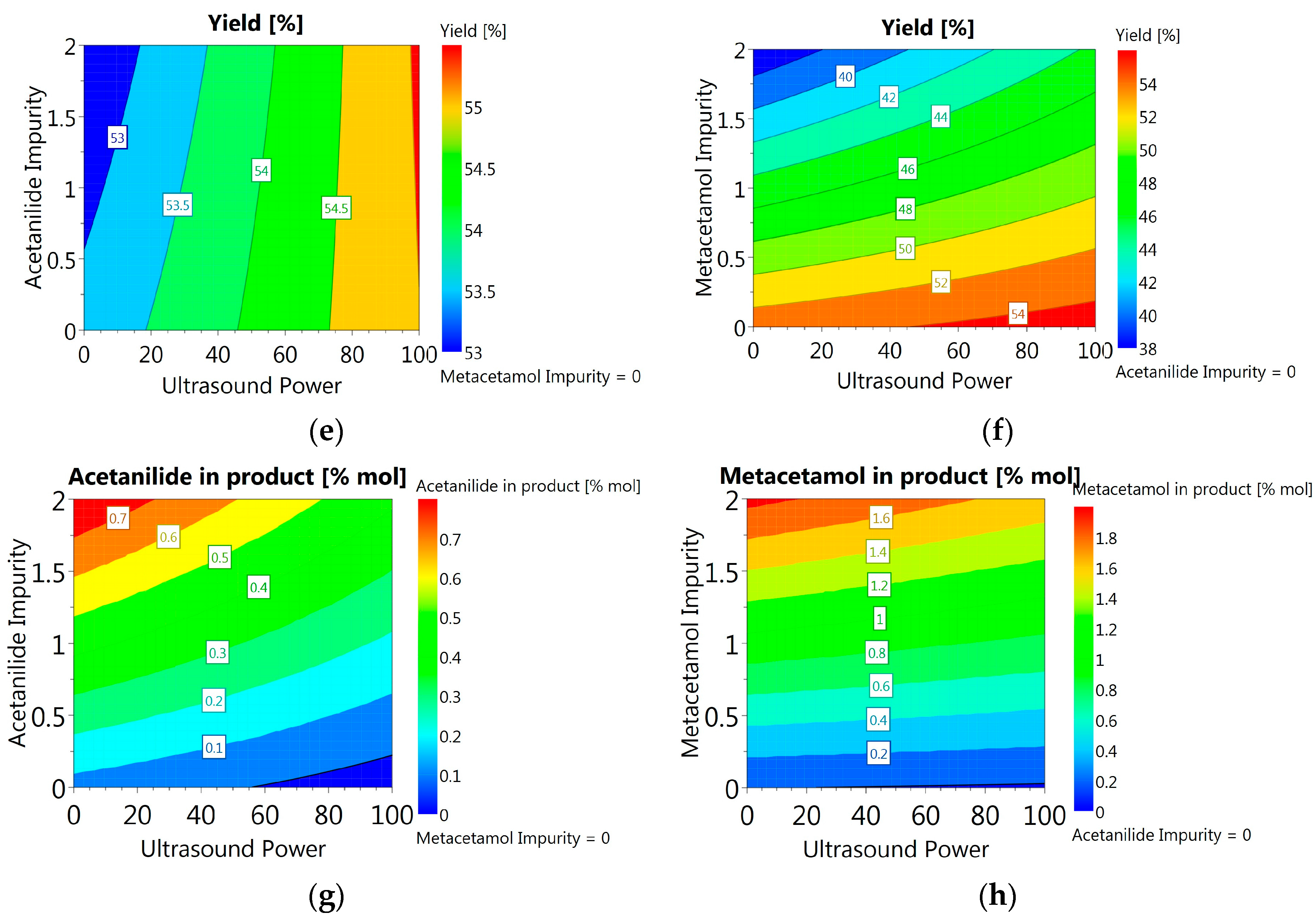
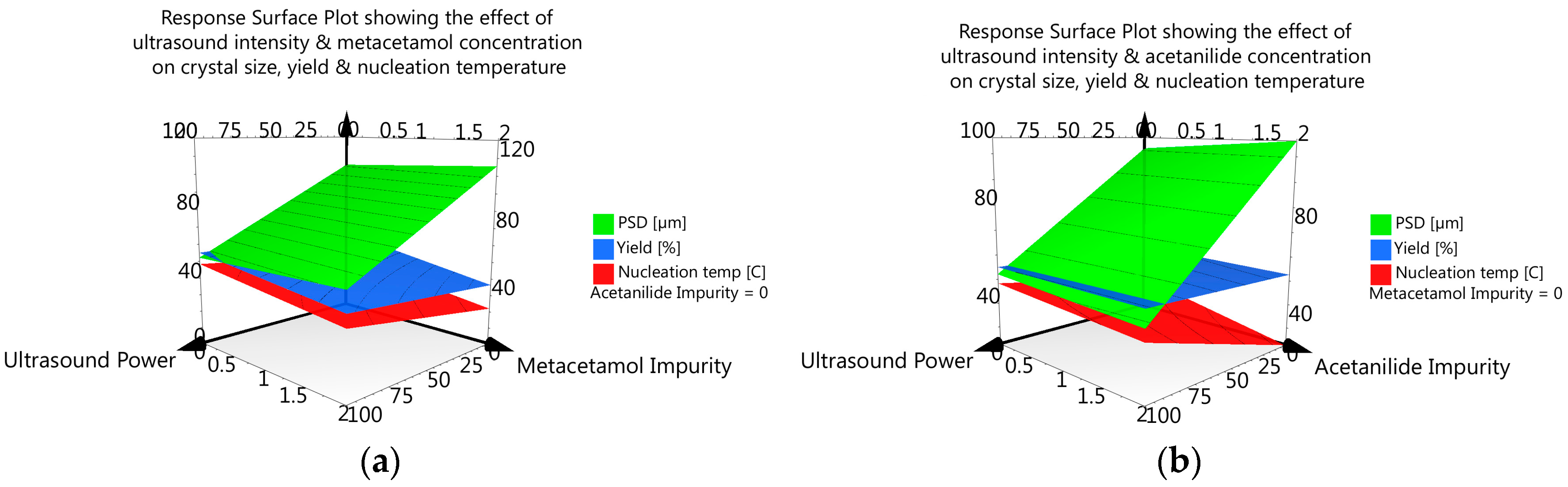
3.3.6. DoE and Multivariate Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Beckmann, W. Crystallization: Basic Concepts and Industrial Applications; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Hendriksen, B.A.; Grant, D.J.; Meenan, P.; Green, D.A. Crystallisation of paracetamol (acetaminophen) in the presence of structurally related substances. J. Cryst. Growth 1998, 183, 629–640. [Google Scholar] [CrossRef]
- Prasad, K.V.; Ristic, R.I.; Sheen, D.B.; Sherwood, J.N. Crystallization of paracetamol from solution in the presence and absence of impurity. Int. J. Pharm. 2001, 215, 29–44. [Google Scholar] [CrossRef]
- Thompson, C.; Davies, M.C.; Roberts, C.J.; Tendler, S.J.B.; Wilkinson, M.J. The effects of additives on the growth and morphology of paracetamol (acetaminophen) crystals. Int. J. Pharm. 2004, 280, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Saleemi, A.; Onyemelukwe, I.I.; Nagy, Z. Effects of a structurally related substance on the crystallization of paracetamol. Front. Chem. Sci. Eng. 2013, 7, 79–87. [Google Scholar] [CrossRef]
- Hendriksen, B.A.; Grant, D.J. The effect of structurally related substances on the nucleation kinetics of paracetamol (acetaminophen). J. Cryst. Growth 1995, 156, 252–260. [Google Scholar] [CrossRef]
- Rauls, M.; Bartosch, K.; Kind, M.; Kuch, S.; Lacmann, R.; Mersmann, A. The influence of impurities on crystallization kinetics–A case study on ammonium sulfate. J. Cryst. Growth 2000, 213, 116–128. [Google Scholar] [CrossRef]
- Sangwal, K.; Mielniczek-Brzoska, E. Effect of impurities on metastable zone width for the growth of ammonium oxalate monohydrate crystals from aqueous solutions. J. Cryst. Growth 2004, 267, 662–675. [Google Scholar] [CrossRef]
- Podder, J. The study of impurities effect on the growth and nucleation kinetics of potassium dihydrogen phosphate. J. Cryst. Growth 2002, 237, 70–75. [Google Scholar] [CrossRef]
- Poornachary, S.K.; Lau, G.; Chow, P.S.; Tan, R.B.H.; George, N. The Effect and Counter-Effect of Impurities on Crystallization of an Agrochemical Active Ingredient: Stereochemical Rationalization and Nanoscale Crystal Growth Visualization. Cryst. Growth Des. 2011, 11, 492–500. [Google Scholar] [CrossRef]
- Mukuta, T.; Lee, A.Y.; Kawakami, T.; Myerson, A.S. Influence of impurities on the solution-mediated phase transformation of an active pharmaceutical ingredient. Cryst. Growth Des. 2005, 5, 1429–1436. [Google Scholar] [CrossRef]
- Févotte, F.; Févotte, G. A method of characteristics for solving population balance equations (PBE) describing the adsorption of impurities during crystallization processes. Chem. Eng. Sci. 2010, 65, 3191–3198. [Google Scholar] [CrossRef] [Green Version]
- Judge, R.A.; Forsythe, E.L.; Pusey, M.L. The effect of protein impurities on lysozyme crystal growth. Biotechnol. Bioeng. 1998, 59, 776–785. [Google Scholar] [CrossRef]
- Sangwal, K. Effects of impurities on crystal growth processes. Prog. Cryst. Growth Charact. Mater. 1996, 32, 3–43. [Google Scholar] [CrossRef]
- Scott, C.; Black, S. In-line analysis of impurity effects on crystallisation. Org. Process Res. Dev. 2005, 9, 890–893. [Google Scholar] [CrossRef]
- Poornachary, S.K.; Chow, P.S.; Tan, R.B. Effect of solution speciation of impurities on α-glycine crystal habit: A molecular modeling study. J. Cryst. Growth 2008, 310, 3034–3041. [Google Scholar] [CrossRef]
- Repic, O. Principles of Process Research and Chemical Development in the Pharmaceutical Industry; John Wiley & Sons: Hoboken, NJ, USA, 1998. [Google Scholar]
- Chow, R.; Blindt, R.; Chivers, R.; Povey, M. A study on the primary and secondary nucleation of ice by power ultrasound. Ultrasonics 2005, 43, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Crespo, R.; Martins, P.M.; Gales, L.; Rocha, F.; Damas, A.M. Potential use of ultrasound to promote protein crystallization. J. Appl. Crystallogr. 2010, 43, 1419–1425. [Google Scholar] [CrossRef]
- Devarakonda, S.; Evans, J.M.B.; Myerson, A.S. Impact of Ultrasonic Energy on the Crystallization of Dextrose Monohydrate. Cryst. Growth Des. 2003, 3, 741–746. [Google Scholar] [CrossRef]
- Dodds, J.; Espitalier, F.; Louisnard, O.; Grossier, R.; David, R.; Hassoun, M.; Baillon, F.; Gatumel, C.; Lyczko, N. The Effect of Ultrasound on Crystallisation-Precipitation Processes: Some Examples and a New Segregation Model. Part. Part. Syst. Charact. 2007, 24, 18–28. [Google Scholar] [CrossRef]
- Guo, Z.; Zhang, M.; Li, H.; Wang, J.; Kougoulos, E. Effect of ultrasound on anti-solvent crystallization process. J. Cryst. Growth 2005, 273, 555–563. [Google Scholar] [CrossRef]
- Higaki, K.; Ueno, S.; Koyano, T.; Sato, K. Effects of ultrasonic irradiation on crystallization behavior of tripalmitoylglycerol and cocoa butter. J. Am. Oil Chem. Soc. 2001, 78, 513–518. [Google Scholar] [CrossRef]
- Kordylla, A.; Koch, S.; Tumakaka, F.; Schembecker, G. Towards an optimized crystallization with ultrasound: Effect of solvent properties and ultrasonic process parameters. J. Cryst. Growth 2008, 310, 4177–4184. [Google Scholar] [CrossRef]
- De Castro, M.D.L.; Priego-Capote, F. Ultrasound-assisted crystallization (sonocrystallization). Ultrason. Sonochem. 2007, 14, 717–724. [Google Scholar]
- Lyczko, N.; Espitalier, F.; Louisnard, O.; Schwartzentruber, J. Effect of ultrasound on the induction time and the metastable zone widths of potassium sulphate. Chem. Eng. J. 2002, 86, 233–241. [Google Scholar] [CrossRef]
- Miyasaka, E.; Kato, Y.; Hagisawa, M.; Hirasawa, I. Effect of ultrasonic irradiation on the number of acetylsalicylic acid crystals produced under the supersaturated condition and the ability of controlling the final crystal size via primary nucleation. J. Cryst. Growth 2006, 289, 324–330. [Google Scholar] [CrossRef]
- Narducci, O.; Jones, A.G.; Kougoulos, E. Continuous crystallization of adipic acid with ultrasound. Chem. Eng. Sci. 2011, 66, 1069–1076. [Google Scholar] [CrossRef]
- Nývlt, J.; Žáček, S. Effect of Ultrasonics on Agglomeration. Cryst. Res. Technol. 1995, 30, 1055–1063. [Google Scholar] [CrossRef]
- Patrick, M.; Blindt, R.; Janssen, J. The effect of ultrasonic intensity on the crystal structure of palm oil. Ultrason. Sonochem. 2004, 11, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Ramisetty, K.A.; Pandit, A.B.; Gogate, P.R. Ultrasound-Assisted Antisolvent Crystallization of Benzoic Acid: Effect of Process Variables Supported by Theoretical Simulations. Ind. Eng. Chem. Res. 2013, 52, 17573–17582. [Google Scholar] [CrossRef]
- Sayan, P.; Sargut, S.T.; Kiran, B. Effect of ultrasonic irradiation on crystallization kinetics of potassium dihydrogen phosphate. Ultrason. Sonochem. 2011, 18, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Virone, C.; Kramer, H.J.M.; van Rosmalen, G.M.; Stoop, A.H.; Bakker, T.W. Primary nucleation induced by ultrasonic cavitation. J. Cryst. Growth 2006, 294, 9–15. [Google Scholar] [CrossRef]
- Boels, L.; Wagterveld, R.M.; Mayer, M.J.; Witkamp, G.J. Seeded calcite sonocrystallization. J. Cryst. Growth 2010, 312, 961–966. [Google Scholar] [CrossRef]
- Dhumal, R.; Biradar, S.; Paradkar, A.; York, P. Ultrasound Assisted Engineering of Lactose Crystals. Pharm. Res. 2008, 25, 2835–2844. [Google Scholar] [CrossRef] [PubMed]
- Eder, R.J.P.; Schrank, S.; Besenhard, M.O.; Roblegg, E.; Gruber-Woelfler, H.; Khinast, J.G. Continuous Sonocrystallization of Acetylsalicylic Acid (ASA): Control of Crystal Size. Cryst. Growth Des. 2012, 12, 4733–4738. [Google Scholar] [CrossRef]
- Guo, Z.; Jones, A.G.; Li, N.; Germana, S. High-speed observation of the effects of ultrasound on liquid mixing and agglomerated crystal breakage processes. Powder Technol. 2007, 171, 146–153. [Google Scholar] [CrossRef]
- Kim, S.; Wei, C.; Kiang, S. Crystallization process development of an active pharmaceutical ingredient and particle engineering via the use of ultrasonics and temperature cycling. Org. Process Res. Dev. 2003, 7, 997–1001. [Google Scholar] [CrossRef]
- Kougoulos, E.; Marziano, I.; Miller, P.R. Lactose particle engineering: Influence of ultrasound and anti-solvent on crystal habit and particle size. J. Cryst. Growth 2010, 312, 3509–3520. [Google Scholar] [CrossRef]
- Li, H.; Wang, J.; Bao, Y.; Guo, Z.; Zhang, M. Rapid sonocrystallization in the salting-out process. J. Cryst. Growth 2003, 247, 192–198. [Google Scholar] [CrossRef]
- Nalajala, V.S.; Moholkar, V.S. Investigations in the physical mechanism of sonocrystallization. Ultrason. Sonochem. 2011, 18, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.R.; Murthy, Z.V.P. Effect of process parameters on crystal size and morphology of lactose in ultrasound-assisted crystallization. Cryst. Res. Technol. 2011, 46, 243–248. [Google Scholar] [CrossRef]
- Wohlgemuth, K.; Kordylla, A.; Ruether, F.; Schembecker, G. Experimental study of the effect of bubbles on nucleation during batch cooling crystallization. Chem. Eng. Sci. 2009, 64, 4155–4163. [Google Scholar] [CrossRef]
- Amara, N.; Ratsimba, B.; Wilhelm, A.-M.; Delmas, H. Crystallization of potash alum: Effect of power ultrasound. Ultrason. Sonochem. 2001, 8, 265–270. [Google Scholar] [CrossRef]
- Li, H.; Li, H.; Guo, Z.; Liu, Y. The application of power ultrasound to reaction crystallization. Ultrason. Sonochem. 2006, 13, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Ruecroft, G.; Hipkiss, D.; Ly, T.; Maxted, N.; Cains, P.W. Sonocrystallization: The Use of Ultrasound for Improved Industrial Crystallization. Org. Process Res. Dev. 2005, 9, 923–932. [Google Scholar] [CrossRef]
- Gracin, S.; Uusi-Penttilä, M.; Rasmuson, Å.C. Influence of Ultrasound on the Nucleation of Polymorphs of p-Aminobenzoic Acid. Cryst. Growth Des. 2005, 5, 1787–1794. [Google Scholar] [CrossRef]
- Louhi-Kultanen, M.; Karjalainen, M.; Rantanen, J.; Huhtanen, M.; Kallas, J. Crystallization of glycine with ultrasound. Int. J. Pharm. 2006, 320, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Shirgaonkar, I.Z.; Pandit, A.B. Effect of Sonication on Crystal Properties. Sep. Sci. Technol. 1995, 30, 2239–2243. [Google Scholar] [CrossRef]
- Suslick, K.S. The chemical effects of ultrasound. Sci. Am. 1989, 260, 80–86. [Google Scholar] [CrossRef]
- Apfel, R.E. Sonic effervescence: A tutorial on acoustic cavitation. J. Acoust. Soc. Am. 1997, 101, 1227–1237. [Google Scholar] [CrossRef]
- Lauterborn, W.; Ohl, C.-D. Cavitation bubble dynamics. Ultrason. Sonochem. 1997, 4, 65–75. [Google Scholar] [CrossRef]
- Lauterborn, W.; Kurz, T.; Mettin, R.; Ohl, C. Experimental and theoretical bubble dynamics. Adv. Chem. Phys. 1999, 110, 295–380. [Google Scholar]
- Price, C.J. Take some solid steps to improve crystallization. Chem. Eng. Prog. 1997, 93, 34–43. [Google Scholar]
- Zamanipoor, M.H.; Mancera, R.L. The emerging application of ultrasound in lactose crystallisation. Trends Food Sci. Technol. 2014, 38, 47–59. [Google Scholar] [CrossRef]
- Knorr, D.; Zenker, M.; Heinz, V.; Lee, D.-U. Applications and potential of ultrasonics in food processing. Trends Food Sci. Technol. 2004, 15, 261–266. [Google Scholar] [CrossRef]
- Mason, T.; Paniwnyk, L.; Lorimer, J. The uses of ultrasound in food technology. Ultrason. Sonochem. 1996, 3, S253–S260. [Google Scholar] [CrossRef]
- Jiang, M.; Papageorgiou, C.D.; Waetzig, J.; Hardy, A.; Langston, M.; Braatz, R.D. Indirect ultrasonication in continuous slug-flow crystallization. Cryst. Growth Des. 2015, 15, 2486–2492. [Google Scholar] [CrossRef]
- Rossi, D.; Jamshidi, R.; Saffari, N.; Kuhn, S.; Gavriilidis, A.; Mazzei, L. Continuous-flow sonocrystallization in droplet-based microfluidics. Cryst. Growth Des. 2015, 15, 5519–5529. [Google Scholar] [CrossRef]
- Siddique, H.; Brown, C.J.; Houson, I.; Florence, A.J. Establishment of a Continuous Sonocrystallization Process for Lactose in an Oscillatory Baffled Crystallizer. Org. Process Res. Dev. 2015, 19, 1871–1881. [Google Scholar] [CrossRef] [Green Version]
- Haisa, M.; Kashino, S.; Kawai, R.; Maeda, H. The Monoclinic Form of p-Hydroxyacetanilide. Acta Crystallogr. B 1976, 32, 1283–1285. [Google Scholar] [CrossRef]
- Haisa, M.; Kashino, S.; Maeda, H. The orthorhombic form of p-hydroxyacetanilide. Acta Crystallogr. B 1974, 30, 2510–2512. [Google Scholar] [CrossRef]
- Perrin, M.-A.; Neumann, M.A.; Elmaleh, H.; Zaske, L. Crystal structure determination of the elusive paracetamol Form III. Chem. Commun. 2009, 3181–3183. [Google Scholar] [CrossRef] [PubMed]
- Ristic, R.; Finnie, S.; Sheen, D.; Sherwood, J. Macro-and micromorphology of monoclinic paracetamol grown from pure aqueous solution. J. Phys. Chem. B 2001, 105, 9057–9066. [Google Scholar] [CrossRef]
- Ripperger, S.; Gösele, W.; Alt, C.; Loewe, T. Filtration, 1. fundamentals. In Ullmann’s Encyclopedia of Industrial Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2009; Volume 14, pp. 677–709. [Google Scholar]
- Behera, S.; Ghanty, S.; Ahmad, F.; Santra, S.; Banerjee, S. UV-visible spectrophotometric method development and validation of assay of paracetamol tablet formulation. Int. J. Chem. Anal. Sci. 2012, 3, 2–6. [Google Scholar] [CrossRef]
- Ungnade, H.E. Ultraviolet Absorption Spectra of Acetanilides. J. Am. Chem. Soc. 1954, 76, 5133–5135. [Google Scholar] [CrossRef]
- Granberg, R.A.; Rasmuson, Å.C. Solubility of Paracetamol in Pure Solvents. J. Chem. Eng. Data 1999, 44, 1391–1395. [Google Scholar] [CrossRef]
- Baena, Y.; Pinzón, J.A.; Barbosa, H.J.; Martínez, F. Temperature-dependence of the solubility of some acetanilide derivatives in several organic and aqueous solvents. Phys. Chem. Liq. 2004, 42, 603–613. [Google Scholar] [CrossRef]
- Barrio, M.; Huguet, J.; Rietveld, I.B.; Robert, B.; Céolin, R.; Tamarit, J.-L. The Pressure-Temperature Phase Diagram of Metacetamol and Its Comparison to the Phase Diagram of Paracetamol. J. Pharm. Sci. 2017, 106, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Drebushchak, V.A.; McGregor, L.; Rychkov, D.A. Cooling rate “window” in the crystallization of metacetamol form II. J. Therm. Anal. Calorim. 2017, 127, 1807–1814. [Google Scholar] [CrossRef]
- Chow, R.; Blindt, R.; Kamp, A.; Grocutt, P.; Chivers, R. The microscopic visualisation of the sonocrystallisation of ice using a novel ultrasonic cold stage. Ultrason. Sonochem. 2004, 11, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Chow, R.; Blindt, R.; Chivers, R.; Povey, M. The sonocrystallisation of ice in sucrose solutions: Primary and secondary nucleation. Ultrasonics 2003, 41, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Kurotani, M.; Miyasaka, E.; Ebihara, S.; Hirasawa, I. Effect of ultrasonic irradiation on the behavior of primary nucleation of amino acids in supersaturated solutions. J. Cryst. Growth 2009, 311, 2714–2721. [Google Scholar] [CrossRef]
- Dhumal, R.S.; Biradar, S.V.; Paradkar, A.R.; York, P. Particle engineering using sonocrystallization: Salbutamol sulphate for pulmonary delivery. Int. J. Pharm. 2009, 368, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Raman, V.; Abbas, A. Experimental investigations on ultrasound mediated particle breakage. Ultrason. Sonochem. 2008, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Wagterveld, R.; Boels, L.; Mayer, M.; Witkamp, G. Visualization of acoustic cavitation effects on suspended calcite crystals. Ultrason. Sonochem. 2011, 18, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.R.; Zeiger, B.W.; Suslick, K.S. Sonocrystallization and sonofragmentation. Ultrason. Sonochem. 2014, 21, 1908–1915. [Google Scholar] [CrossRef] [PubMed]
- Jordens, J. Application of Acoustic Energy in Crystallization Processes. Ph.D. Thesis, KU Leuven, Leuven, Belgium, 2016. [Google Scholar]
- Gielen, B.; Kusters, P.; Jordens, J.; Thomassen, L.C.J.; Van Gerven, T.; Braeken, L. Energy efficient crystallization of paracetamol using pulsed ultrasound. Chem. Eng. Process. Process Intensif. 2017, 114, 55–66. [Google Scholar] [CrossRef]
- Kaur Bhangu, S.; Ashokkumar, M.; Lee, J. Ultrasound Assisted Crystallization of Paracetamol: Crystal Size Distribution and Polymorph Control. Cryst. Growth Des. 2016, 16, 1934–1941. [Google Scholar] [CrossRef]
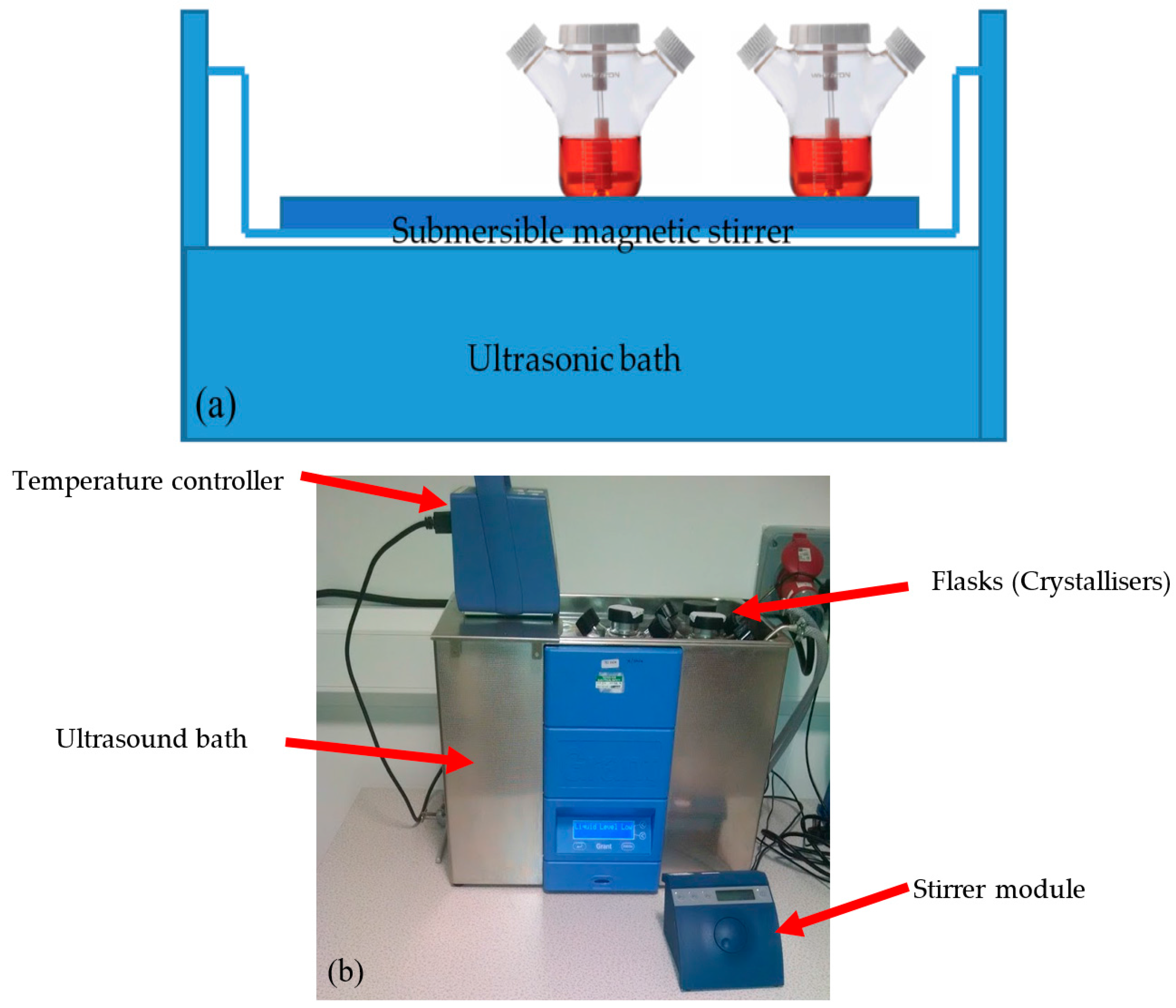



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Molecular Weight (g/mol) | Melting Point (K) | ΔHfusion (kJ mol-1) | Measured Solubility in 3-methyl-1-butanol (mg/g) | ||
|---|---|---|---|---|---|---|
| 25 °C | 40 °C | 55 °C | ||||
| Paracetamol | 151.16 | 443.6 [68]; 442.9 [69] | 27.1 [68]; 30.72 [69] | 54.8 | 74.3 | 124.3 |
| Metacetamol | 151.16 | 420 [70,71] | 26.0 [71]; 28.8 [70] | 81.4 | 116.7 | 162.6 |
| Acetanilide | 135.17 | 387.5 [69] | 20.30 [69] | 162.8 | 241.8 | - |
| Positions on the Stirrer plate at the Bottom of the Ultrasonic Bath | Ultrasonic Intensity (mW/cm2) | |
|---|---|---|
| 50% Ultrasonic Power | 100% Ultrasonic Power | |
| 1 | 0.13 ± 0.02 | 1.25 ± 0.13 |
| 2 | 0.25 ± 0.03 | 1.68 ± 0.25 |
| 3 | 0.24 ± 0.02 | 1.32 ± 0.09 |
| 4 | 0.12 ± 0.04 | 0.9 ± 0.07 |
| Average values | 0.19 ± 0.07 | 1.29 ± 0.32 |
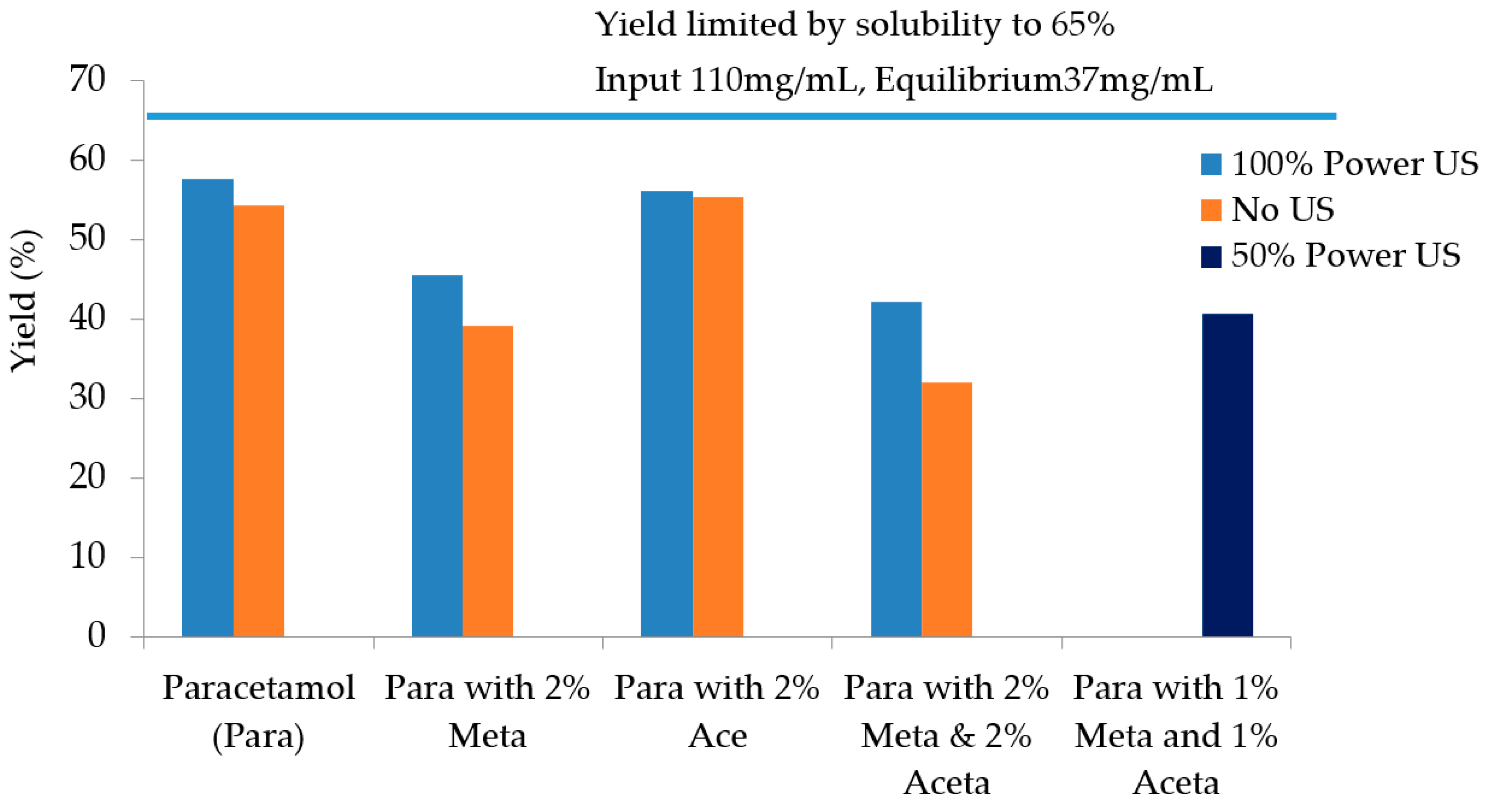
| Experiments | Tcrys (°C) | PSD Mean Chord Length (µm) | Filtration Time (s) | Filtration Rate (m3/s) | Cake Resistance (m/kg) | Yield |
|---|---|---|---|---|---|---|
| Pure paracetamol (100% power US) | 41.3 | 47–55 | 209 | 1.90 × 10−7 | 7.03 × 1011 | 57.7% |
| Pure paracetamol (no US) | 36.0 | 85–100 | 95 | 4.29 ×1 0−7 | 2.62 × 109 | 54.3% |
| 2 mol% Metacetamol (100% power US) | 36.4 | 47 | 361 | 1.07 × 10−7 | 1.52 × 1010 | 45.5% |
| 2 mol% Metacetamol (no US) | After maintaining at 15 °C for approx. 1.5 h | 100–109 | 45 | 1.06 × 10−7 | 9.02 × 108 | 39.1% |
| 2 mol% Acetanilide (US 100%P) | 43 | 40 | 561 | 7.32 × 10−7 | 1.88 × 1010 | 56.2% |
| 2 mol% Acetanilide (no US) | 15.5 °C | 105 | 54 | 7.01 × 10−7 | 1.02 × 109 | 55.3% |
| 2 mol% Metacetamol & Acetanilide (US 100% power) | 34.4 | 37.3 | 346 | 1.19 × 10−7 | 1.51 × 1010 | 42.2% |
| 2 mol% Metacetamol & Acetanilide (no US) | After maintaining at 15 °C for approx. 2.5 h | 87–90 | 48 | 9.61 × 10−7 | 1.14 × 109 | 32.0% |
| 1 mol% Metacetamol & 1 mol% Acetanilide (50% US power) | 44 | 87 | 61 | 7.19 × 10−7 | 1.38 × 109 | 40.6% |
| Experiment Conditions | Compounds | Percentage of Compounds in Crystal Product (%) | Percentage of Compounds in Mother Liquor (%) | ||
|---|---|---|---|---|---|
| With US | No US | With US | No US | ||
| Pure Paracetamol | Paracetamol | 100 | 100 | 100 | 100 |
| Metacetamol | 0 | 0 | 0 | 0 | |
| Acetanilide | 0 | 0 | 0 | 0 | |
| Paracetamol with 2% Acetanilide | Paracetamol | 99.62 | 99.15 | 94.65 | 96.02 |
| Acetanilide | 0.35 | 0.85 | 5.35 | 3.98 | |
| Paracetamol with 2% Metacetamol | Paracetamol | 98.47 | 98.12 | 96.05 | 96.43 |
| Metacetamol | 1.52 | 1.88 | 3.95 | 3.57 | |
| Paracetamol with 2% Metacetamol and 2% Acetanilide | Paracetamol | 98.1 | 97.98 | 91.37 | 91.49 |
| Metacetamol | 1.19 | 1.44 | 3.91 | 3.92 | |
| Acetanilide | 0.55 | 0.58 | 4.72 | 4.59 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.T.H.; Khan, A.; Bruce, L.M.; Forbes, C.; O’Leary, R.L.; Price, C.J. The Effect of Ultrasound on the Crystallisation of Paracetamol in the Presence of Structurally Similar Impurities. Crystals 2017, 7, 294. https://doi.org/10.3390/cryst7100294
Nguyen TTH, Khan A, Bruce LM, Forbes C, O’Leary RL, Price CJ. The Effect of Ultrasound on the Crystallisation of Paracetamol in the Presence of Structurally Similar Impurities. Crystals. 2017; 7(10):294. https://doi.org/10.3390/cryst7100294
Chicago/Turabian StyleNguyen, Thai T. H., Azeem Khan, Layla M. Bruce, Clarissa Forbes, Richard L. O’Leary, and Chris J. Price. 2017. "The Effect of Ultrasound on the Crystallisation of Paracetamol in the Presence of Structurally Similar Impurities" Crystals 7, no. 10: 294. https://doi.org/10.3390/cryst7100294