Modulating Nucleation by Kosmotropes and Chaotropes: Testing the Waters



1
Department of Chemistry, Universitätsstr. 10, University of Konstanz, Konstanz 78464, Germany
2
Freiburg Institute for Advanced Studies, Albert-Ludwigs-Universität Freiburg, Freiburg 79104, Germany
3
Centre for Biosystem Analysis, Albert-Ludwigs-Universität Freiburg, Freiburg 79104, Germany
*
Authors to whom correspondence should be addressed.
Crystals 2017, 7(10), 302; https://doi.org/10.3390/cryst7100302
Submission received: 24 August 2017
/
Accepted: 4 October 2017
/
Published: 6 October 2017
(This article belongs to the Special Issue Biological and Biogenic Crystallization)
Abstract
:Water is a fundamental solvent sustaining life, key to the conformations and equilibria associated with solute species. Emerging studies on nucleation and crystallization phenomena reveal that the dynamics of hydration associated with mineral precursors are critical in determining material formation and growth. With certain small molecules affecting the hydration and conformational stability of co-solutes, this study systematically explores the effects of these chaotropes and kosmotropes as well as certain sugar enantiomers on the early stages of calcium carbonate formation. These small molecules appear to modulate mineral nucleation in a class-dependent manner. The observed effects are finite in comparison to the established, strong interactions between charged polymers and intermediate mineral forms. Thus, perturbations to hydration dynamics of ion clusters by co-solute species can affect nucleation phenomena in a discernable manner.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Water is one of the most abundant molecules in the universe [1]. It affects global processes such as erosion and climate as well as phenomena at atomic length scales such as molecular configurations and interactions. As a ubiquitous cellular constituent, water molecules influence biomolecular conformations and interactions [2,3]. However, the influences of hydration and bulk water on chemical processes are not completely understood. Historically, certain salts have been linked with an ability to influence water structure, and thereby the conformational states of co-solute macromolecules. This gradually led to the nomenclature of solute additives as either structure-breakers or structure-makers, corresponding to their respective stabilizing or disruptive effect on the short-range order i.e., hydrogen bonding networks in the liquid [2,4]. However, convincing evidence for a significant perturbation of bulk water structure by solutes still remains lacking.
Alternate explanations are provided for the observed effects of ionic species on macromolecules [3]. For instance, direct interactions of ions with macromolecules and their first shell of hydration play key roles in the hydrophobic collapse and solubility of poly(N-isopropylacrylamide) [5]. Simulation studies also show that ions alter hydrogen bonding, salt bridges, and hydrophobic interactions that underlie macromolecular conformations [6,7]. These studies suggest that the Hofmeister series emerges from interactions of the ions with macromolecules and the associated hydration, and not due to structural perturbations to the bulk solvent. Therefore the configurational and chemical properties of solute macromolecules are important factors that determine the consequences of ionic interactions [3,8,9].
Certain small organic molecules are also implicated in affecting the hydration of co-solutes. For example, organisms thriving in extreme habitats modify solvent effects by sequestering high solute contents [10]. Members of this solute family (compensatory kosmotropes or osmolytes [11]) are generally compatible with biochemical processes and provide a mechanism for the evasion of osmotic stress without extensive covalent modifications of biomacromolecules [10]. This is essential for the survival of organisms including bacterial and fungal spores, rotifers, and tardigrades [12,13]. The mechanistic functions of certain osmolytes (e.g., glycerol and betaine) may be due to their exclusion from the immediate vicinity of co-solutes [14,15]. These kosmotropes may also decrease the solubility of hydrophobic, potentially toxic molecules by enhancing their aggregation [16]. Being polar molecules with a negligible net charge, kosmotropes prefer forming hydrogen bonds with water molecules and therefore are excluded from the hydration shell of hydrophobic patches on macromolecules. On these lines, a ‘solvophobic thermodynamic force’ is described for the unfavorable interactions between the osmolytes and peptide backbone that increase the free energy for macromolecular denaturation [17]. Another hypothesis for the activity of kosmotropes is the occurrence of microdomains of high and low density states of water in which osmolytes (kosmotropes) and chaotropes preferentially partition. In this scenario, the thermodynamic cost for altering the equilibrium between the density states is related to the conformational stability of macromolecular co-solutes [18].
In comparison to kosmotropes, in aqueous solutions of chaotropes, the solubility of non-polar solutes is enhanced. For instance, urea and guanidine significantly increase the solubility of biomolecules; however at the cost of denaturation, i.e., a loss of native macromolecular structure [19,20]. These small molecules interact with co-solutes and weaken interactions with solvent molecules. This leads to destabilized native conformations and an increased water-accessible surface area of macromolecules [16,21]. A preferential accumulation of chaotropic metabolites is also shown to support the growth of certain microbes at low temperatures [22]. Thus the effects of chaotropes and kosmotropes on the conformation and hydration of co-solutes arises from their preferential partitioning to either the bulk solvent phase or the water-solute interfaces and therefore is determined by the physicochemical nature of hydrated co-solutes [23]. Given the fundamental roles of these interactions in biomolecular structure and function, their impacts on nucleation and crystallization behavior in nature also require elucidation.
Of the natural minerals, calcium carbonate (CaCO3) is produced at a rate of 5 billion tons per year in the oceans and is a key industrial material on account of its role as a filler and scalant material [24,25,26]. It is a crucial raw material for the production of Portland cement (about 4 Gt manufactured per year). With advances in the resolution of analytical techniques, several key aspects of mineral nucleation and growth including pre-nucleation clusters (PNCs) and oriented attachment have been identified [27,28,29,30,31,32]. In these processes, the role of water as a bulk solvent and hydration molecule is fundamentally important. The formation of ion-clusters from free species requires a certain release of ion-associated hydration [28,29]. During the phase separation of hydrated PNCs, the dynamics of the water hydrogen network are affected and allow localizing a binodal limit for liquid-liquid demixing. This process yields dense calcium carbonate droplets, which are precursors to solid amorphous calcium carbonate (ACC) [33,34,35]. Recently, it was shown that different background ions influence PNC stabilities as well as ACC solubilities, which was rationalized by effects of the spectator ions on the hydration shells [36]. Considering the lower density of PNCs in comparison to that of liquid/gel-like and solid ACC, a significant ion cluster-associated hydration endures in the nucleated phase [37]. These intermediate mineral forms are characterized by distinct water contents. Stable forms of biogenic and synthetic ACC also contain structural water [38,39,40,41]. During the process of crystallization, the ACC undergoes steps of hydration loss and follows an energetically downhill sequence from a short-lived anhydrous form to crystalline phases [42,43]. This transformation process is significantly affected by the water content of the bulk solvent, affecting the polymorph selectivity of CaCO3 [44]. In the absence of additives stabilizing crystalline surfaces, the solvent conditions also determine the reconstruction of high energy faces [45]. These studies reflect a dynamic equilibrium between free (bulk) and ion-associated (hydration/structural) water during mineral formation, the importance of which is reflected by the distinct nature of the H-bond network of hydration shells in terms of density, structure, and dynamics [46,47,48]. Thus the bulk and mineral-associated solvent molecules play critical roles from the primary to final stages of mineralization, encompassing PNCs, liquid, and amorphous intermediates and crystals.
The roles of solute-associated and bulk solvent as “additives” are also demonstrated for several material systems. Considering calcium sulphate, solvent polarity, i.e., its water-withdrawing ability, has a direct bearing on the polymorph selection of bassanite and gypsum [49]. For organic crystals, the solvent composition also determines the selective nucleation of polymorphic forms [50]. For oriented attachment processes, the solvent modulates interactions with organic ligands and also provides a certain degree of particle movement that enables co-orientation and coalescence [31,51,52,53,54]. Thus the solvent phase commonly affects nucleation and crystallization processes as well as the subsequent material properties including crystallography, morphology, and polymorph.
In view of certain co-solutes modulating molecular hydration and also the significant contributions of solvent parameters towards nucleation and crystal growth, the aim of this study is to understand, “Do chaotropes and kosmotropes affect mineral nucleation?” For this purpose, small molecules typically classified as chaotropes and kosmotropes are quantitatively evaluated as additives during the nucleation of CaCO3. The additives tested are betaine, ectoine, trehalose, sorbitol, mannitol, glycine, urea, thiourea, and guanidium. With reports of the stereoisomers of mono- and oligo-saccharides in chaotropic and kosmotropic behavior [55], D- and L-forms of sugars are also investigated. The analytical approach involves potentiometric titrations performed by dosing calcium chloride into a solution of an additive dissolved in carbonate buffer. During the course of the experiment, the pH is kept constant by the counter-titration of a base. Similar experiments have been previously performed to assess the effects of diverse additives including synthetic polymers [26], biomolecules such as amino acids [56], carbohydrates, polysaccharides [57,58], proteins [59], and peptides [60] as well as inorganic species such as silica and magnesium [61,62,63,64]. By employing ion-selective electrodes and precise reaction conditions, the diverse roles of additives on different aspects of mineral nucleation including the stability of PNCs, inhibition or promotion of nucleation, and the nature of the nucleated phase are demonstrated.
2. Results
2.1. On Kosmotropes and Chaotropes
Dataset analyses for the time developments of free Ca2+ ion and pH are performed as described in previous literature [28,56,57]. For titrations performed in carbonate buffer, the detected free Ca2+ ion concentration increases linearly with time at a rate significantly lower than that of Ca2+ ion addition to water. This is due to the association of Ca2+ ions and carbonate species that result in diminished detected free Ca2+ ion contents [28]. The linear slope corresponding to the pre-nucleation regime describes the formation and stability of PNCs. A lower slope indicates an equilibrium shift towards cluster-associated Ca2+ ions i.e., bound Ca2+ and vice versa. For example, the evolution of free Ca2+ ions as well as the added base contents to maintain constant pH in absence or presence of different additives are represented in Figure 1. The constant pH level is required to quantitatively compare the effects of the chao-/kosmotropic additives during the early stages of calcium carbonate formation because it ensures a constant carbonate/bicarbonate ratio and removes corresponding, otherwise disturbing effects on supersaturation and ionic speciation.
As examples of kosmotropes and chaotropes in the field of cellular stress biology [65], the effects of trehalose and urea are compared to reference experiments at pH 9.75. These additives do not influence the time required for mineral nucleation but do significantly affect the equilibrium between free and carbonate-associated Ca2+ ions, as evident from the slope in the prenucleation regime before the sudden Ca2+ concentration drop upon nucleation. From Figure 1A, it is evident that trehalose leads to free Ca2+ ion contents higher than those in the reference experiments. On the other hand, urea stabilizes ion-clusters, presenting significant lower amounts of free Ca2+ ions. These observations are also reflected by the amount of base required for maintaining constant pH conditions (Figure 1B). This amount indicates the carbonate ions bound in the different stages of the experiment [28]. For instance, urea, as an effective stabilizer of ion-clusters, necessitates larger quantities of base solutions to maintain constant pH conditions. The stronger and weaker binding in presence of urea and trehalose, respectively, is seen in two independent measurements (calcium ion selective electrode and pH titration), and thus, the effect of the additives cannot be an effect of specific electrode artifacts. Therefore, given its sensitivity and quantitative nature, this methodology is suitable for addressing the effects of small organic molecules on mineral nucleation.
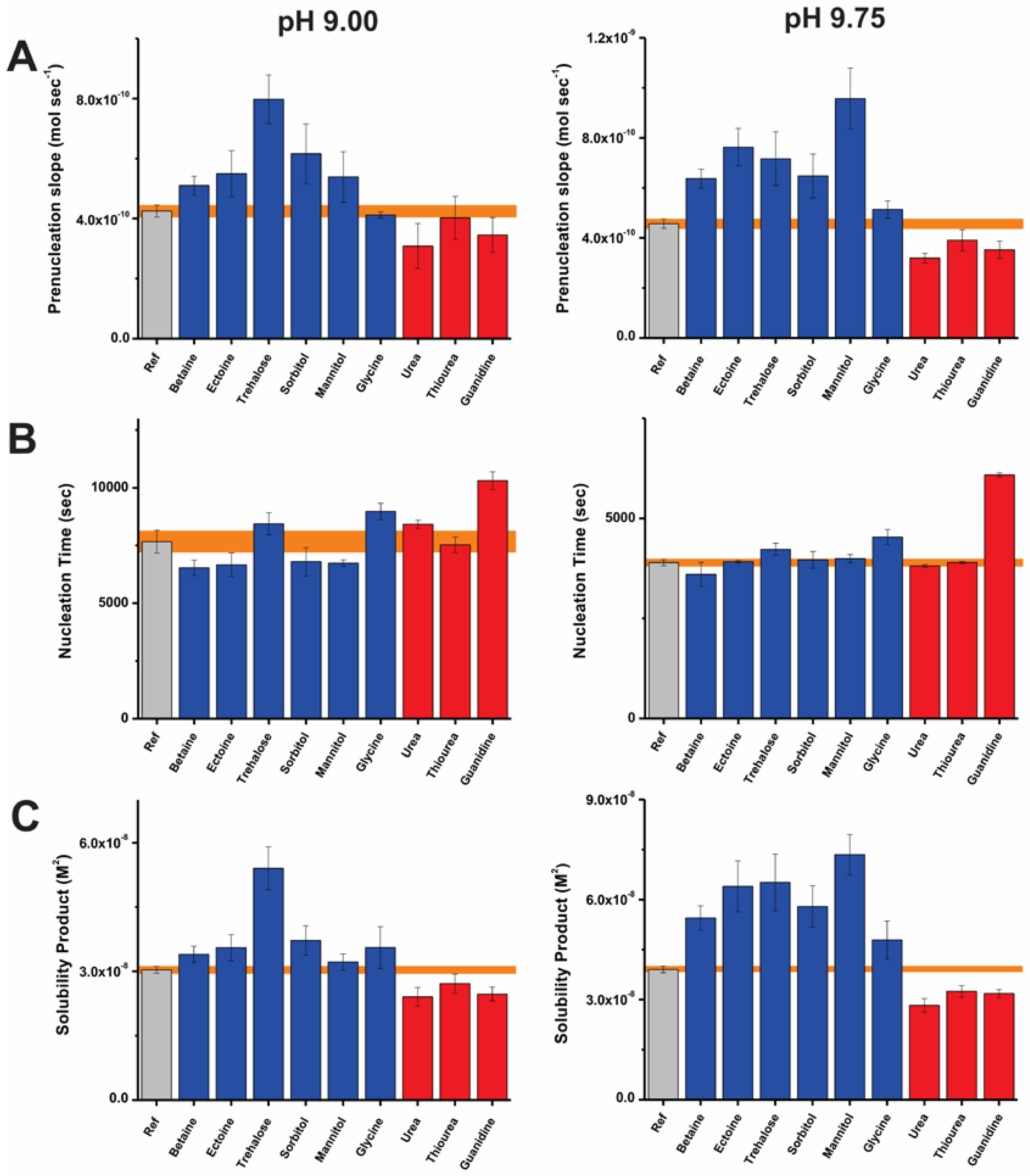
Figure 2A represents the slopes of the pre-nucleation regime in the absence and presence of additives at pH 9.0 and 9.75. Trehalose, sorbitol, ectoine, mannitol, and betaine induce increases in the pre-nucleation slope in comparison to the relative reference experiments at both pH values. At pH 9.0, this corresponds to percentage increases of about 87, 44, 29, 26, and 20%, respectively. At higher pH (9.75), these effects are more pronounced, leading to corresponding increases of about 41, 56, 66, 109, and 39% in the pre-nucleation slope. In this regard, trehalose and mannitol suppress ion-association and are effective pH-dependent destabilizers of PNCs. In view of the pre-nucleation regime, opposite outcomes are elicited by chaotropes. This is evident from the decreased slope of the pre-nucleation regimes corresponding to 30, 22, and 14% in presence of urea, guanidium and thiourea, respectively at pH 9.75. These effects are diminished at pH 9.0, however, urea retains its effect as a PNC stabilizer at pH 9.0 and 9.75. In view of the observed equilibrium shifts between free and bound ion species, the distinct bicarbonate and carbonate ratio appear to impart a pH-dependent physicochemistry to the PNCs, thus consequently determining additive-ion and –ion cluster interactions.
The effects of the small molecules on the time required for mineral nucleation are represented in Figure 2B. As introduced in previous studies [56,57], we apply a scaling factor (F) defined as the quotient of the average nucleation time in presence of an additive and that of the corresponding reference. At pH 9.75, several additives exhibit F values close to unity viz. no observed effect on nucleation time. As exceptions, glycine and guanidine inhibit nucleation, with corresponding F values of 1.03 and 1.1 at pH 9.0 and 1.2 and 1.6 at pH 9.75, all respectively. Previously, the inhibition of nucleation was attributed to the colloidal stabilization of nanoscopic precursors against aggregation [61], but it might also be related to an influence of the additives on the size of pre-nucleation clusters [66]. In either case, the increasing extent of inhibition at higher pH might suggest the involvement of electrostatic interactions driven by pH-related additive speciation. In the case of glycine, this is supported by the predominance of its base form above pH 9.5 (Figure S1). The inhibitory effects introduced by these molecules are small, in comparison to those in the presence of polymeric additives such as poly(acrylic acid), poly(aspartic acid), carboxymethyl cellulose, and heparin [25,26,56,57]. Certain small molecules promote mineral nucleation at pH 9.0. The corresponding F values presented by kosmotropes such as betaine, ectoine, and mannitol are 0.76, 0.77, and 0.78. It is intriguing that although minor, a nucleation promoting effect is induced by these small molecules. A similar effect is noted in the presence of D-arabinose, D-galactose, and sucrose [57]. In the scope of this study, the exact reasons underlying the weak nucleation promoting effect cannot be identified. However, we speculate that the promotion of mineral nucleation is related to the liquid–liquid phase separation, during which PNCs become nanodroplets that can subsequently yield either ACC or crystalline particles.
Potentiometric titrations also elucidate the nature of mineral products [26,28]. The estimated solubility products of the nucleated CaCO3 phase are depicted in Figure 2C. Overall, the kosmotropes and chaotropes lead to the formation of more and less stable phases after nucleation in relation to reference experiments, respectively. The reference experiments present solubility product values of 3.0 × 10−8 and 3.8 × 10−8 M2 after nucleation at pH 9.0 and 9.75, in agreement with previous reports within experimental accuracy [28,56,57]. As a general trend, mineral products nucleated in the presence of kosmotropes viz. betaine, ectoine, trehalose, sorbitol and mannitol exhibit solubility products higher than those of the reference experiments. The most soluble mineral products are formed in presence of trehalose and mannitol corresponding to mean values of 5.4 × 10−8 and 7.4 × 10−8 M2 at pH 9.0 and 9.75, respectively. On the other hand, urea, thiourea, and guanidine induce less soluble mineral products. At pH 9.0, the corresponding average values of the solubility product are between 2.4 × 10−8 and 2.7 × 10−8 M2 indicating the presence of a crystalline polymorph (possibly vaterite). At pH 9.75, these values range between 2.8 × 10−8 and 3.2 × 10−8 M2, which are lower than the solubility product of mineral particles nucleated in reference experiments (3.8 × 10−8 M2). This reflects the presence of either a relatively short-lived amorphous product or a phase with lower solubility (such as proto-calcitic ACC or vaterite), which are distinct from the proto-vaterite ACC phase produced in the reference experiment at pH 9.75 [38]. Thus, kosmotropes and chaotropes have distinct influences on the solubility product of the mineral phase nucleated.
In order to explore possible relations between the observed effects of additives on mineral nucleation and their activity as promotors or disruptors of macromolecular conformation, a scatter plot for the pre-nucleation slopes, which indicate the amount of formed PNCs (the lower the slope the more PNCs are formed) and post-nucleation solubility products is presented (Figure 3). Additives that induce equilibrium shifts towards ion-association, also stimulate the formation of nucleated phase with lower solubility values. This relation has been previously identified with respect to conditions of pH and also in the presence of certain additives [28,67,68]. The applied color scheme represents a chao/kosmotropicity scale based on a previous systematic study wherein (i) chaotropes are defined as solutes that induce conformational disorder in macromolecules by either weakening water-macromolecule interactions or associating non-covalently with macromolecules and (ii) kosmotropes are solutes that preferentially hydrogen bond with water molecules and promote intermolecular interactions [69]. Within the scope of this definition, the effects of the chaotropes on mineral nucleation might be due to direct interactions with ions and ion-cluster species. On the other hand, the kosmotropes suppress ion-association and promote nucleation products with higher solubilities on account of the changes in the hydration state of the inorganic species. In validation of the findings that additive-controlled equilibrium shifts towards ion-association also stimulate the nucleation of mineral phases with lower solubility values (Figure 3), gas-diffusion mineralization experiments are performed (Figure S2). Ectoine and betaine lead to the formation of a mixture of crystalline polymorphs, including calcite and vaterite. On the other hand, guanidine and urea predominantly produce calcite. It is important to consider that the experimental observations are consistent with the original use of terms “chaotrope” and “kosmotrope,” as previously discussed [3,69], and also that the chao-/kosmotorpic activities of salts and organic molecules can considerably deviate in solution mixtures. This discrepancy can originate from the non-additive effects of salts and organic molecules in inducing a net kosmotropic or chaotropic effect. For instance, simulated martian brine solutions are predominantly kosmotropic despite being constituted with chaotropic salts such as chlorides of iron and magnesium [70].
2.2. On Sugar Stereoisomers
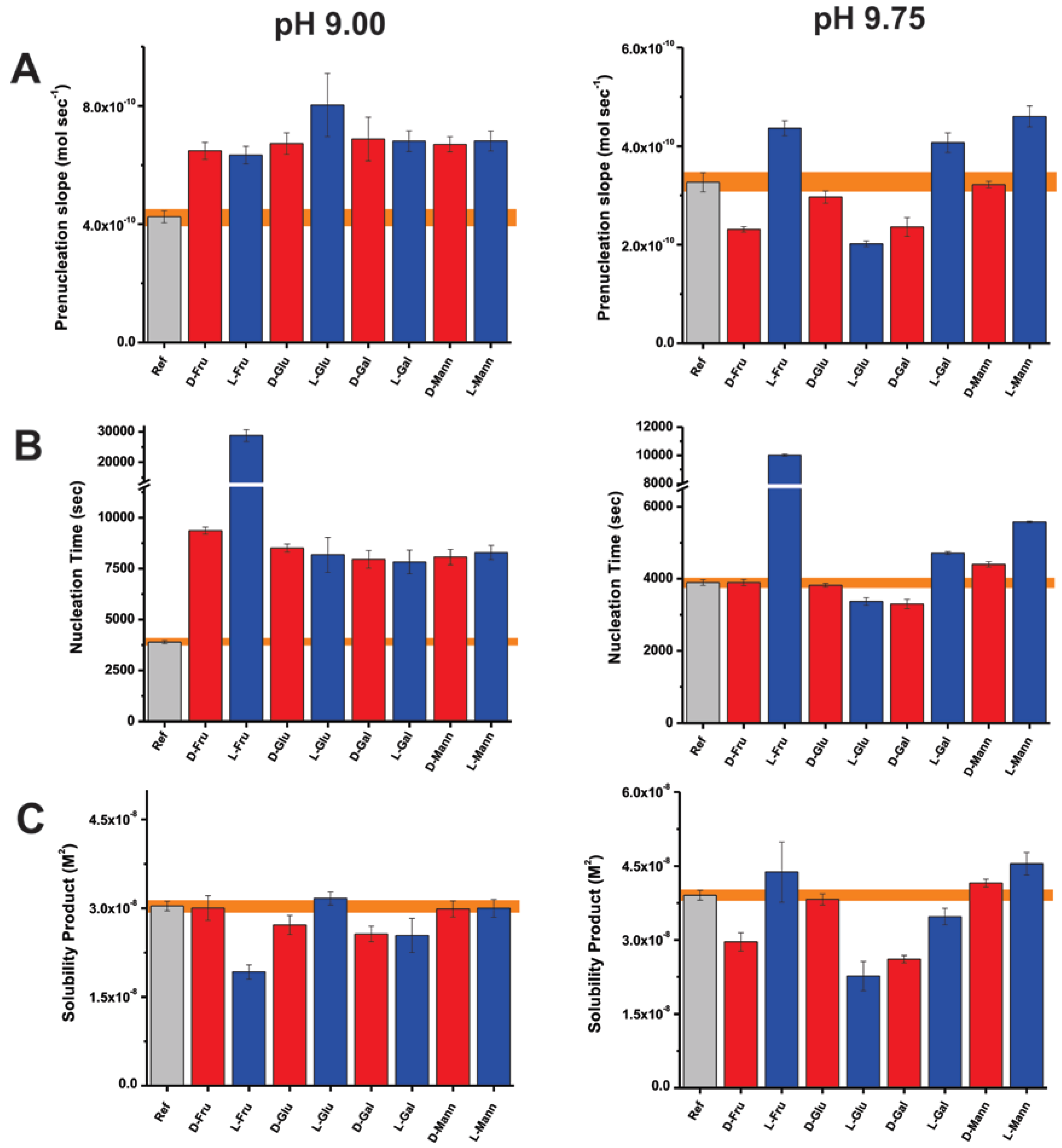
In order to examine possible contributions from a model that proposes solute-induced perturbations of the bulk structure of water, we investigate the effects of four sugar enantiomer pairs. The model suggests that liquid water consists of rapidly exchanging high and low density micro-domains, and the equilibrium between these domains can be altered by dissolved solutes [18,55]. This is supported by distinct elution profiles for glucose enantiomers, suggested to involve micro-domain equilibrium shifts, wherein the bioactive enantiomer prefers a less dense aqueous environment and L-glucose favors a more dense water state [55]. Based on this hypothesis, the individual enantiomers are expected to have distinct effects on nucleation independent of solution pH. However, at pH 9.75, L-glucose stabilizes PNCs and leads to thermodynamically more stable nucleation products (Figure 4). In relation, D-glucose induces no significant effects on the nucleation of CaCO3 particles. At lower pH, the effects of L-glucose are minor relative to those at pH 9.75. Under kinetically controlled mineralization, both enantiomers produce calcite as the predominant mineral polymorph (Figure S2). In light of these results, a pH-independent perturbation to the bulk solvent structure by the enantiomeric additives is not validated or is too weak and eclipsed by direct interactions to have observable effects on mineral nucleation.
Similar trends are noted for three sugar enantiomers, namely fructose, galactose and mannose. For these additives, the distinct effects of the D and L isomers on mineralization at pH 9.75 are relatively nullified at pH 9.0 (Figure 4). For instance, D-galactose and D-fructose stabilize PNCs and induce nucleation products with lower solubilities relative to the reference experiments, specifically at pH 9.75. Therefore, the effects of sugars on mineralization cannot be absolutely associated with either the D or L enantiomeric forms but are also determined by pH-dependent parameters such as the CO32−:HCO3− ratio and the additive conformation. Conditions of pH can alter the chemical nature of ion-clusters and the subsequent interactions between additives and mineral precursors [68,71]. Therefore, the selective trends of mineral nucleation are likely due to direct interactions with mineral species and associated hydration rather than perturbations to the bulk water phase.
3. Discussion
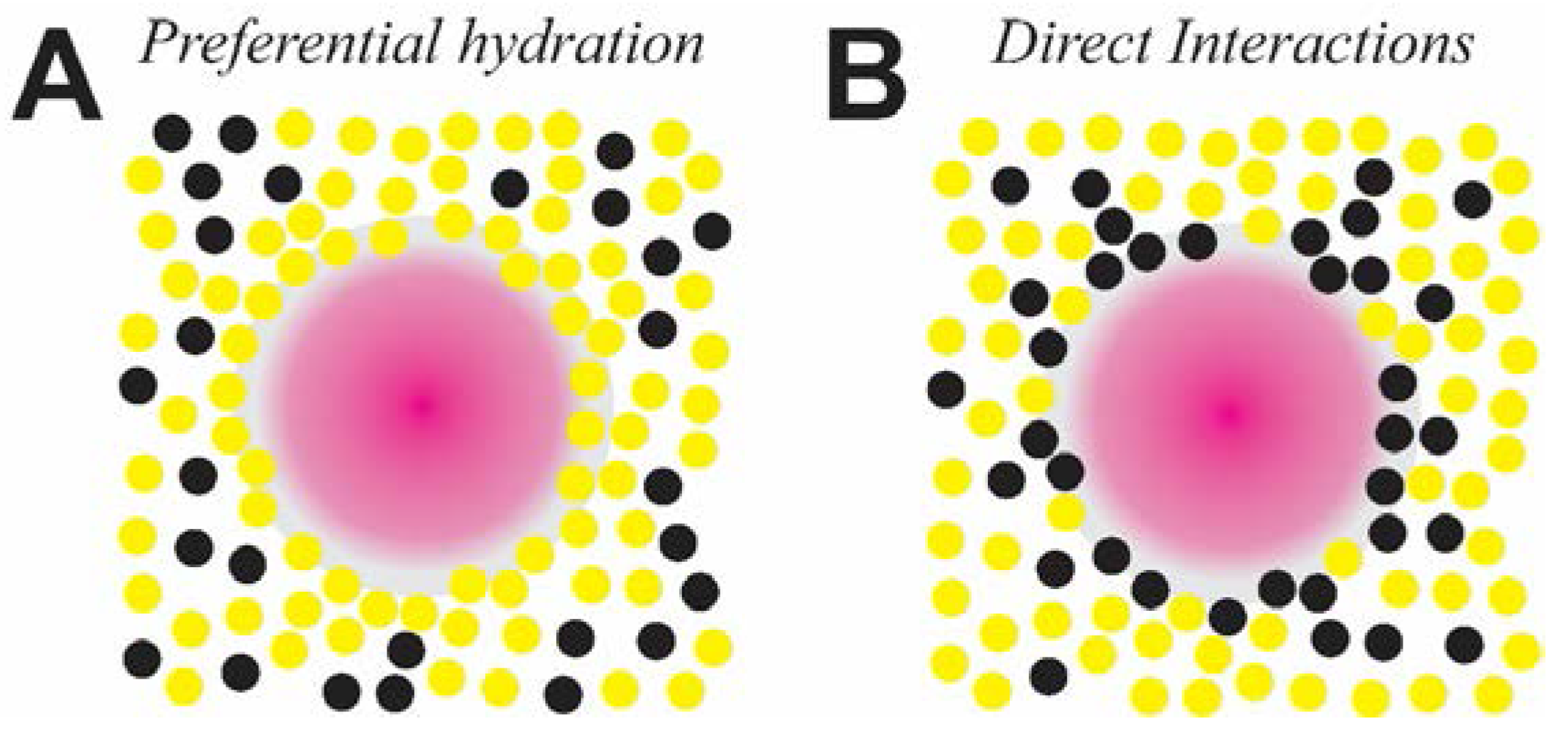
This study identifies the distinct trends of the early stages of precipitation of calcium carbonate in the presence of small molecules, classified on their chao/kosmotropic nature and stereochemistry. Bearing in mind the similar size regimes but distinct physicochemistry of ion-associates and macromolecules, the following mechanisms are proposed for the additive-controlled nucleation process. First, the destabilizers of ion-association, such as betaine and ectoine, are possibly excluded from the hydration shells of ion-clusters. Since ion association of CaCO3 PNCs is driven by the release of hydration waters [29], stabilized hydration shells would have adverse effects. Indeed, molecular dynamics simulations reveal that ectoine is preferentially excluded from macromolecular surfaces, leading to a dense hydration layer [72]. During the early stages of mineralization, similar interactions might suppress dehydration processes related to ion-association and their aggregation/coalescence toward mineral precursors with a larger degree of structural order (Figure 5A). Certain polyalcohols such as mannitol and sorbitol also destabilize PNCs and lead to more soluble nucleation products, relative to the reference experiments. The polyalcohols lead to pronounced destabilization of PNCs in comparison to the sugars, possibly due to the conformational flexibility originating from the lack of a constraining ring structure. Substantial increases in the surface tension of water by polyalcohols also lead to a preferential hydration of biomolecular co-solutes [73,74]. Similar interactions might apply to the formation of PNCs and other transient mineral phases in the presence of polyalcohols, during which dehydration reactions underlying the maturation of mineral precursors are inhibited to a certain extent. Among the kosmotropes tested, trehalose is the most potent in suppressing ion-association. A possible explanation is the property of trehalose molecules in effectively forming hydrogen bonds with water molecules distributed homogenously in the solvent [75,76]. Therefore, in addition to increased surface tension of the solvent, the ion species weakly associated with trehalose are possibly also broadly distributed in the solvent and inhibited against ion-association.
An interesting observation is the promotion of nucleation (F < 1.0) in the presence of certain kosmotropes and sugars. The observed effects might be due to an interference with the liquid-liquid demixing and dehydration processes involved in particle nucleation and affecting its kinetics [33,77,78,79]. The free energy change of solvation involves contributions from the formation of the cavity, which the solute occupies, and solute-solvent interactions (van der Waals and electrostatic) [80], expressed as follows:
This equation shows that kosmotropic additives that generally lower the solubility of solutes (salting out) suppress the formation of solvent cavities (first term in the above equation), and/or unfavorably affect the solute-solvent interactions (second and third term). Either effect could render liquid-like mineral precursors relatively more transient in presence of these additives by promoting the dehydration of inorganic liquid-like intermediate, thereby accelerating the nucleation of solid particles in a minor but occasionally detectable manner (this observation is not general for kosmotropes).
Considering the stabilizing effects of chaotropes toward PNCs, their influence is likely due to direct interactions with the inorganic species (Figure 5B). Previous investigations show that urea binds to the hydrophilic surfaces of proteins and stabilizes non-native conformations by usurping hydrogen bonds with hydrophilic surfaces [81]. The basis for this surfactant-like action of chaotropes is enthalpy driven and related to changes in interfacial energies and hydrophobic interactions [81,82,83,84]. On similar lines, as polar nonelectrolytes, urea, thiourea and guanidine possibly capture the PNCs and serve as a “molecular glue” between the ion-clusters. This proposed mechanism is supported by the crystal structure of an urea derivative, wherein hydrogen-bond interactions with carbonate species involve two NH···O bonds [85]. Such interactions can potentially promote ion-association and shift the equilibrium towards stable clusters, subsequently yielding nucleation products that are thermodynamically more stable (Figure 2).
In view of these nucleation and mineralization studies (Figure 3, Figure S2), the accumulation of the compensatory kosmotropes such as trehalose and ectoine by halophilic microbes might serve as a means to suppress intracellular ion-association and mineral nucleation processes detrimental to survival of the organisms. Such effects may be enhanced by molecular crowding i.e., high cytosolic accumulation of these additives [86]. In this regard, the interactions between macromolecules and ions or ion-clusters are relevant toward defining the geochemical boundary conditions for life and predicting planetary habitability [69,87]. Such investigations indicate the non-additive nature of net chaotropic and kosmotrophic effects of solution mixtures of diverse salts and organic molecules. These might have crucial contributions during additive-controlled mineralization processes.
We also show that the role of chirality is not limited to crystal growth but also encompasses mineral nucleation. Previous studies show that calcite crystals grown in the presence of either L or D aspartic acid present asymmetry associated with the distribution of crystal steps and terraces [88,89], and even the CaCO3 polymorph could be controlled by the chirality of an interacting amino acid [90]. The amplification of these interactions at macroscopic length scales is demonstrated by the distinct chirality of vaterite toroids and the helicity of potassium dichromate crystals tuned by the enantiospecificity of amino acids [91,92]. In view of mineral nucleation, we show that the sugar enantiomers present distinct interactions with mineral precursors and as a consequence affect the nucleation process in a pH-dependent manner. Thus with the distinct effects of amino acid and sugar enantiomers on mineral formation and growth, Lewis Carrol’s allusion to the consequences of chirality “Perhaps looking-glass milk isn’t good to drink” stands affirmed [93,94].
In summary, this systematic study explores the effects of small organic molecules on mineral nucleation. Additive-controlled mineralization is often represented as a two-component system involving the inorganic phase and the additive species, the solvent properties often being underrepresented. However, the contributions of solvophobic forces and weak interactions between the solvent molecules, additives, and material building units also appear crucial. It should be noted that PNCs are chemically distinct with highly dynamic configurations in comparison to the relative structural organization and rigidity of macromolecules. Therefore, future studies addressing the hydration dynamics during ion-association and phase transformation will certainly further elucidate the solvent phase as an active participant in nucleation and crystallization phenomena.
4. Materials and Methods
For the mineralization studies, the chemicals used are calcium chloride (1 M solution, Fluka, Happague, NY, USA), hydrochloric acid (1 M solution, Merck, Darmstadt, Germany,), sodium hydroxide (0.01 M, Alfa Aesar, Karlsruhe, Germany; 1 M, Merck, Darmstadt, Germany), sodium bicarbonate (Riedel de-Haën, Seelze, Germany), sodium carbonate (anhydrous, Sigma-Aldrich, Taufkirchen, Germany), sodium chloride (VWR Prolabo, Darmstadt, Germany), and ammonium carbonate (Acros Organics, Morris Plains, NJ, USA). The additives investigated include betaine (BioUltra, >99%, Sigma, Deisenhofen, Germany), ectoine (>99%, Sigma-Aldrich), D-(+)-trehalose from Saccharomyces cerevisiae (>99%, Sigma-Aldrich), D-sorbitol (>99%, Aldrich), D-mannitol (>98%, Sigma-Aldrich), glycine (>99%, Merck), urea (>99%, Merck), thiourea (>99%, Sigma-Aldrich), and guanidine (>99%, Sigma-Aldrich). The sugar additives used are D-(-)-fructose (Merck), D-galactose (≥ 97%, Merck), α-D-(+)-glucose (Sigma-Aldrich, ACS reagent), D-mannose (≥ 98%, Merck), L-(+)-fructose (>95%, Carbosynth, Compton, Berkshire, UK), L-(-)-galactose (≥ 99%, Carbosynth), L-(-)-glucose (≥ 99%, Sigma-Aldrich) and L-mannose (≥ 98%, Merck).
Potentiometric titrations are performed with a commercial titration system from Metrohm (Filderstadt, Germany), in which an apparatus (Titrando 905) controls two dosing units (Dosino 800), operated by a customized software (Tiamo v2.2). A CaCl2 (10 mM) solution is added at a fixed rate of 0.01 mL/min into 20 mL carbonate buffer (10 mM) containing additive (10 mM) [26,28,95]. Concurrently, the pH is maintained constant by the automatic counter-titration of NaOH (10 mM). The pH and free calcium contents are monitored by a glass electrode (Metrohm, No. 6.0256.100) and a Ca2+ ion-selective electrode (ISE, Metrohm, No. 6.0508.110). The ISE calibration is done by titrating CaCl2 into 20 mL water at constant pH using the aforementioned methodology. All titrations were performed with stirring at 900 rpm and at room temperature with a minimum of three repetitions. The pH development and detected Ca2+ contents enable the quantitative evaluations of Ca2+ and CO32− ions that occur in free and in bound states at a given time. Gas diffusion is performed by exposing solution mixtures containing CaCl2 (10 mM) and additive (10 mM) to ammonium carbonate vapors in a previously described format [56].
Supplementary Materials
The following are available online at www.mdpi.com/2073-4352/7/10/302/s1, Figure S1: Simulated speciation of glycine in relation to pH conditions, Figure S2: Representative SEM images of CaCO3 particles formed via gas diffusion experiments.
Acknowledgments
A.R. acknowledges fellowships from the Konstanz Research School Chemical Biology and Freiburg Institute for Advanced Studies. D.G. is a Research Fellow of the Zukunftskolleg of the University of Konstanz.
Author Contributions
Experimental tasks and preparation of the preliminary manuscript draft were done by A. R. Project supervision as well as detailed editing of manuscript were performed by D.G. and H.C.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Encrenaza, T.; Spohnb, T. Water in the solar system. In Encyclopedia of Astrobiology; Springer Berlin Heidelberg: Heidelberg, Germany, 2014. [Google Scholar]
- Ball, P. Water as an active constituent in cell biology. Chem. Rev. 2008, 108, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Ball, P.; Hallsworth, J.E. Water structure and chaotropicity: Their uses, abuses and biological implications. Phys. Chem. Chem. Phys. 2015, 17, 8297–8305. [Google Scholar] [CrossRef] [PubMed]
- Kunz, W.; Henle, J.; Ninham, B.W. ‘Zur lehre von der wirkung der salze’ (about the science of the effect of salts): Franz hofmeister's historical papers. Curr. Opin. Colloid Inter. 2004, 9, 19–37. [Google Scholar] [CrossRef]
- Zhang, Y.; Furyk, S.; Bergbreiter, D.E.; Cremer, P.S. Specific ion effects on the water solubility of macromolecules: Pnipam and the hofmeister series. J. Am. Chem. Soc. 2005, 127, 14505–14510. [Google Scholar] [CrossRef] [PubMed]
- Mason, P.E.; Dempsey, C.E.; Vrbka, L.; Heyda, J.; Brady, J.W.; Jungwirth, P. Specificity of ion-protein interactions: Complementary and competitive effects of tetrapropylammonium, guanidinium, sulfate, and chloride ions. J. Phys. Chem. B 2009, 113, 3227–3234. [Google Scholar] [CrossRef] [PubMed]
- McCammick, E.M.; Gomase, V.S.; McGenity, T.J.; Timson, D.J.; Hallsworth, J.E. Water-hydrophobic compound interactions with the microbial cell. In Handbook of Hydrocarbon and Lipid Microbiology; Springer Berlin Heidelberg: Heidelberg, Germany, 2010. [Google Scholar]
- Kunz, W. Specific ion effects in colloidal and biological systems. Curr. Opin. Colloid Inter. 2010, 15, 34–39. [Google Scholar] [CrossRef]
- Vrbka, L.; Jungwirth, P.; Bauduin, P.; Touraud, D.; Kunz, W. Specific ion effects at protein surfaces: A molecular dynamics study of bovine pancreatic trypsin inhibitor and horseradish peroxidase in selected salt solutions. J. Phys. Chem. B 2006, 110, 7036–7043. [Google Scholar] [CrossRef] [PubMed]
- Yancey, P.H.; Clark, M.E.; Hand, S.C.; Bowlus, R.D.; Somero, G.N. Living with water stress: Evolution of osmolyte systems. Science 1982, 217, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Lever, M.; Blunt, J.W.; Maclagan, R.G.A.R. Some ways of looking at compensatory kosmotropes and different water environments. Comp. Biochem. Physiol. A 2001, 130, 471–486. [Google Scholar] [CrossRef]
- Crowe, J.; Crowe, L. Membrane integrity in anhydrobiotic organisms: Toward a mechanism for stabilizing dry cells. In Water and Life; Springer: Berlin, Germany, 1992; pp. 87–103. [Google Scholar]
- Albertyn, J.; Hohmann, S.; Thevelein, J.M.; Prior, B.A. GPD1, which encodes glycerol-3-phosphate dehydrogenase, is essential for growth under osmotic stress in saccharomyces cerevisiae, and its expression is regulated by the high-osmolarity glycerol response pathway. Mol. Cell. Biol. 1994, 14, 4135–4144. [Google Scholar] [CrossRef] [PubMed]
- Gekko, K.; Timasheff, S.N. Mechanism of protein stabilization by glycerol: Preferential hydration in glycerol-water mixtures. Biochemistry 1981, 20, 4667–4676. [Google Scholar] [CrossRef] [PubMed]
- Courtenay, E.S.; Capp, M.W.; Anderson, C.F.; Record, M.T. Vapor pressure osmometry studies of osmolyte-protein interactions: Implications for the action of osmoprotectants in vivo and for the interpretation of “osmotic stress” experiments in vitro. Biochemistry 2000, 39, 4455–4471. [Google Scholar] [CrossRef] [PubMed]
- Moelbert, S.; Normand, B.; De Los Rios, P. Kosmotropes and chaotropes: Modelling preferential exclusion, binding and aggregate stability. Biophy. Chem. 2004, 112, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Bolen, D.W.; Baskakov, I.V. The osmophobic effect: Natural selection of a thermodynamic force in protein folding. J. Mol. Biol. 2001, 310, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Wiggins, P.M. High and low density intracellular water. Cell. Mol. Biol. 2001, 47, 735–744. [Google Scholar] [PubMed]
- Nozaki, Y.; Tanford, C. The solubility of amino acids, diglycine, and triglycine in aqueous guanidine hydrochloride solutions. J. Biol. Chem. 1970, 245, 1648–1652. [Google Scholar] [PubMed]
- De Xammar Oro, J. Role of co-solute in biomolecular stability: Glucose, urea and the water structure. J. Biol. Phys. 2001, 27, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Timasheff, S.N. Protein-solvent preferential interactions, protein hydration, and the modulation of biochemical reactions by solvent components. Proc. Natl. Acad. Sci. USA 2002, 99, 9721–9726. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.P.; Megaw, J.; Magill, C.L.; Nowotarski, K.; Williams, J.P.; Bhaganna, P.; Linton, K.; Patterson, M.F.; Underwood, G.J.C.; Mswaka, A.Y.; et al. Solutes determine the temperature windows for microbial survival and growth. Proc. Natl. Acad. Sci. USA 2010, 107, 7835–7840. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, J.D.; Olteanu, A.; Tripathy, A.; Pielak, G.J. Impact of protein denaturants and stabilizers on water structure. J. Am. Chem. Soc. 2004, 126, 1958–1961. [Google Scholar] [CrossRef] [PubMed]
- Rothon, R.; Paynter, C. Calcium carbonate fillers. In Encyclopedia of Polymers and Composites; Palsule, S., Ed.; Springer Berlin Heidelberg: Heidelberg, Germany, 2015; pp. 1–9. [Google Scholar]
- Verch, A.; Gebauer, D.; Antonietti, M.; Cölfen, H. How to control the scaling of CaCO3: A “fingerprinting technique” to classify additives. Phys. Chem. Chem. Phys. 2011, 13, 16811–16820. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, D.; Cölfen, H.; Verch, A.; Antonietti, M. The multiple roles of additives in CaCO3 crystallization: A quantitative case study. Adv. Mat. 2009, 21, 435–439. [Google Scholar] [CrossRef]
- Demichelis, R.; Raiteri, P.; Gale, J.D.; Quigley, D.; Gebauer, D. Stable prenucleation mineral clusters are liquid-like ionic polymers. Nat. Comm. 2011, 2, 590. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, D.; Völkel, A.; Cölfen, H. Stable prenucleation calcium carbonate clusters. Science 2008, 322, 1819–1822. [Google Scholar] [CrossRef] [PubMed]
- Kellermeier, M.; Raiteri, P.; Berg, J.K.; Kempter, A.; Gale, J.D.; Gebauer, D. Entropy drives calcium carbonate ion association. Chemphyschem 2015, 17, 3535–3541. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, N.; Cölfen, H.; Pinna, N.; Antonietti, M.; Nassif, N. Superstructures of calcium carbonate crystals by oriented attachment. Cryst. Growth Des. 2005, 5, 1317–1319. [Google Scholar] [CrossRef]
- Penn, R.L.; Banfield, J.F. Imperfect oriented attachment: Dislocation generation in defect-free nanocrystals. Science 1998, 281, 969–971. [Google Scholar] [CrossRef] [PubMed]
- De Yoreo, J.J.; Gilbert, P.U.; Sommerdijk, N.A.; Penn, R.L.; Whitelam, S.; Joester, D.; Zhang, H.; Rimer, J.D.; Navrotsky, A.; Banfield, J.F. Crystallization by particle attachment in synthetic, biogenic, and geologic environments. Science 2015, 349, aaa6760. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, F.; Wolf, S.L.P.; Born, B.; Luong, T.Q.; Cölfen, H.; Gebauer, D.; Havenith, M. Water dynamics from THz Spectroscopy reveal the locus of a liquid-liquid binodal limit in aqueous CaCO3 solutions. Angew. Chem. Int. Ed. 2017, 56, 490–495. [Google Scholar]
- Gower, L.B. Biomimetic model systems for investigating the amorphous precursor pathway and its role in biomineralization. Chem. Rev. 2008, 108, 4551–4627. [Google Scholar] [CrossRef]
- Wallace, A.F.; Hedges, L.O.; Fernandez-Martinez, A.; Raiteri, P.; Gale, J.D.; Waychunas, G.A.; Whitelam, S.; Banfield, J.F.; De Yoreo, J.J. Microscopic evidence for liquid-liquid separation in supersaturated CaCO3 solutions. Science 2013, 341, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Cara, A.; Putnis, C.V.; Rodriguez-Navarro, C.; Ruiz-Agudo, E. Hydration effects on the stability of calcium carbonate pre-nucleation species. Minerals 2017, 7, 126. [Google Scholar] [CrossRef]
- Gebauer, D.; Cölfen, H. Prenucleation clusters and non-classical nucleation. Nano. Today 2011, 6, 564–584. [Google Scholar] [CrossRef]
- Gebauer, D.; Gunawidjaja, P.N.; Ko, J.; Bacsik, Z.; Aziz, B.; Liu, L.; Hu, Y.; Bergström, L.; Tai, C.W.; Sham, T.K. Proto-calcite and proto-vaterite in amorphous calcium carbonates. Angew. Chem. 2010, 49, 8889–8891. [Google Scholar] [CrossRef] [PubMed]
- Addadi, L.; Raz, S.; Weiner, S. Taking advantage of disorder: Amorphous calcium carbonate and its roles in biomineralization. Adv. Mat. 2003, 15, 959–970. [Google Scholar] [CrossRef]
- Politi, Y.; Arad, T.; Klein, E.; Weiner, S.; Addadi, L. Sea urchin spine calcite forms via a transient amorphous calcium carbonate phase. Science 2004, 306, 1161–1164. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, J.H.; Checa, A.G.; Gale, J.D.; Gebauer, D.; Sainz-Díaz, C.I. Calcium carbonate polyamorphism and its role in biomineralization: How many amorphous calcium carbonates are there? Angew. Chem. Inter. Ed. 2012, 51, 11960–11970. [Google Scholar] [CrossRef] [PubMed]
- Radha, A.; Forbes, T.Z.; Killian, C.E.; Gilbert, P.; Navrotsky, A. Transformation and crystallization energetics of synthetic and biogenic amorphous calcium carbonate. Proc. Natl. Acad. Sci. USA 2010, 107, 16438–16443. [Google Scholar] [CrossRef] [PubMed]
- Ihli, J.; Wong, W.C.; Noel, E.H.; Kim, Y.Y.; Kulak, A.N.; Christenson, H.K.; Duer, M.J.; Meldrum, F.C. Dehydration and crystallization of amorphous calcium carbonate in solution and in air. Nat. Comm. 2014, 5, 3169. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-R.; Cai, A.-H.; Liu, R.; Pan, H.-H.; Tang, R.-K.; Cho, K. The roles of water and polyelectrolytes in the phase transformation of amorphous calcium carbonate. J. Cryst. Growth 2008, 310, 3779–3787. [Google Scholar] [CrossRef]
- Luo, Y.; Sonnenberg, L.; Cölfen, H. Novel method for generation of additive free high-energy crystal faces and their reconstruction in solution. Cryst. Growth Des. 2008, 8, 2049–2051. [Google Scholar] [CrossRef]
- Merzel, F.; Smith, J.C. Is the first hydration shell of lysozyme of higher density than bulk water? Proc. Natl. Acad. Sci. USA 2002, 99, 5378–5383. [Google Scholar] [CrossRef] [PubMed]
- Pizzitutti, F.; Marchi, M.; Sterpone, F.; Rossky, P.J. How protein surfaces induce anomalous dynamics of hydration water. J. Phys. Chem. B 2007, 111, 7584–7590. [Google Scholar] [CrossRef] [PubMed]
- Raiteri, P.; Gale, J.D. Water is the key to nonclassical nucleation of amorphous calcium carbonate. J. Am. Chem. Soc. 2010, 132, 17623–17634. [Google Scholar] [CrossRef] [PubMed]
- Tritschler, U.; Kellermeier, M.; Debus, C.; Kempter, A.; Cölfen, H. A simple strategy for the synthesis of well-defined bassanite nanorods. CrystEngComm 2015, 17, 3772–3776. [Google Scholar] [CrossRef]
- Khoshkhoo, S.; Anwar, J. Crystallization of polymorphs: The effect of solvent. J. Phys. D 1993, 26, B90. [Google Scholar] [CrossRef]
- Niederberger, M.; Cölfen, H. Oriented attachment and mesocrystals: Non-classical crystallization mechanisms based on nanoparticle assembly. Phys. Chem. Chem. Phys. 2006, 8, 3271–3287. [Google Scholar] [CrossRef] [PubMed]
- Polleux, J.; Pinna, N.; Antonietti, M.; Hess, C.; Wild, U.; Schlögl, R.; Niederberger, M. Ligand functionality as a versatile tool to control the assembly behavior of preformed titania nanocrystals. Chem. Eur. J. 2005, 11, 3541–3551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Banfield, J.F. Interatomic coulombic interactions as the driving force for oriented attachment. CrystEngComm 2014, 16, 1568–1578. [Google Scholar] [CrossRef]
- Fichthorn, K.A. Atomic-scale aspects of oriented attachment. Chem. Eng. Sci. 2015, 121, 10–15. [Google Scholar] [CrossRef]
- Wiggins, P. Life depends upon two kinds of water. PLoS ONE 2008, 3, e1406. [Google Scholar] [CrossRef] [PubMed]
- Picker, A.; Kellermeier, M.; Seto, J.; Gebauer, D.; Cölfen, H. The multiple effects of amino acids on the early stages of calcium carbonate crystallization. Z. Krist. Cryst. Mat. 2012, 227, 744–757. [Google Scholar] [CrossRef]
- Rao, A.; Berg, J.K.; Kellermeier, M.; Gebauer, D. Sweet on biomineralization: Effects of carbohydrates on the early stages of calcium carbonate crystallization. Eur. J. Min. 2014, 26, 537–552. [Google Scholar] [CrossRef]
- Rao, A.; Fernández, M.S.; Cölfen, H.; Arias, J.L. Distinct effects of avian egg derived anionic proteoglycans on the early stages of calcium carbonate mineralization. Cryst. Growth Des. 2015, 15, 2052–2056. [Google Scholar] [CrossRef]
- Rao, A.; Seto, J.; Berg, J.K.; Kreft, S.G.; Scheffner, M.; Cölfen, H. Roles of larval sea urchin spicule SM50 domains in organic matrix self-assembly and calcium carbonate mineralization. J. Struc. Biol. 2013, 183, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, D.; Verch, A.; Borner, H.G.; Cölfen, H. Influence of selected artificial peptides on calcium carbonate precipitation—A quantitative study. Cryst. Growth Des. 2009, 9, 2398–2403. [Google Scholar] [CrossRef]
- Kellermeier, M.; Gebauer, D.; Melero-García, E.; Drechsler, M.; Talmon, Y.; Kienle, L.; Cölfen, H.; García-Ruiz, J.M.; Kunz, W. Colloidal stabilization of calcium carbonate prenucleation clusters with silica. Adv. Func. Mat. 2012, 22, 4301–4311. [Google Scholar] [CrossRef]
- Kellermeier, M.; Picker, A.; Kempter, A.; Cölfen, H.; Gebauer, D. A straightforward treatment of activity in aqueous CaCO3 solutions and the consequences for nucleation theory. Adv. Mat. 2014, 26, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.L.; Jähme, K.; Gebauer, D. Synergy of mg 2+ and poly (aspartic acid) in additive-controlled calcium carbonate precipitation. CrystEngComm 2015, 17, 6857–6862. [Google Scholar] [CrossRef]
- Verch, A.; Antonietti, M.; Cölfen, H. Mixed calcium-magnesium pre-nucleation clusters enrich calcium. Z. Krist. Cryst. Mat. 2012, 227, 718–722. [Google Scholar] [CrossRef]
- De Lima Alves, F.; Stevenson, A.; Baxter, E.; Gillion, J.L.; Hejazi, F.; Hayes, S.; Morrison, I.E.; Prior, B.A.; McGenity, T.J.; Rangel, D.E.; et al. Concomitant osmotic and chaotropicity-induced stresses in Aspergillus wentii: Compatible solutes determine the biotic window. Curr. Genetics 2015, 61, 457–477. [Google Scholar] [CrossRef] [PubMed]
- Zahn, D. Thermodynamics and kinetics of prenucleation clusters, classical and non-classical nucleation. Chemphyschem 2015, 16, 2069–2075. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, D.; Kellermeier, M.; Gale, J.D.; Bergstrom, L.; Cölfen, H. Pre-nucleation clusters as solute precursors in crystallisation. Chem. Soc. Rev. 2014, 43, 2348–2371. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Vásquez-Quitral, P.; Fernández, M.S.; Berg, J.K.; Sánchez, M.; Drechsler, M.; Neira-Carrillo, A.; Arias, J.L.; Gebauer, D.; Cölfen, H. Ph-dependent schemes of calcium carbonate formation in the presence of alginates. Cryst. Growth Des. 2016, 16, 1349–1359. [Google Scholar] [CrossRef]
- Cray, J.A.; Russell, J.T.; Timson, D.J.; Singhal, R.S.; Hallsworth, J.E. A universal measure of chaotropicity and kosmotropicity. Env. Microbiol. 2013, 15, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Fox-Powell, M.G.; Hallsworth, J.E.; Cousins, C.R.; Cockell, C.S. Ionic strength is a barrier to the habitability of Mars. Astrobiology 2016, 16, 427–442. [Google Scholar] [CrossRef]
- Rao, A.; Huang, Y.C.; Cölfen, H. Additive speciation and phase behavior modulate mineralization. J. Phys. Chem. C 2017. [Google Scholar] [CrossRef]
- Yu, I.; Jindo, Y.; Nagaoka, M. Microscopic understanding of preferential exclusion of compatible solute ectoine: Direct interaction and hydration alteration. J. Phys. Chem. B 2007, 111, 10231–10238. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, J.K.; Bhat, R. Thermal stability of proteins in aqueous polyol solutions: Role of the surface tension of water in the stabilizing effect of polyols. J. Phys. Chem. B 1998, 102, 7058–7066. [Google Scholar] [CrossRef]
- Kaushik, J.K.; Bhat, R. Why is trehalose an exceptional protein stabilizer? An analysis of the thermal stability of proteins in the presence of the compatible osmolyte trehalose. J. Biol. Chem. 2003, 278, 26458–26465. [Google Scholar] [CrossRef] [PubMed]
- Lerbret, A.; Bordat, P.; Affouard, F.; Descamps, M.; Migliardo, F. How homogeneous are the trehalose, maltose, and sucrose water solutions? An insight from molecular dynamics simulations. J. Phys. Chem. B 2005, 109, 11046–11057. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.K.; Roy, I. Effect of trehalose on protein structure. Prot. Sci. 2009, 18, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Gutowski, K.E.; Broker, G.A.; Willauer, H.D.; Huddleston, J.G.; Swatloski, R.P.; Holbrey, J.D.; Rogers, R.D. Controlling the aqueous miscibility of ionic liquids: Aqueous biphasic systems of water-miscible ionic liquids and water-structuring salts for recycle, metathesis, and separations. J. Am. Chem. Soc. 2003, 125, 6632–6633. [Google Scholar] [CrossRef] [PubMed]
- Abdolrahimi, S.; Nasernejad, B.; Pazuki, G. Influence of process variables on extraction of cefalexin in a novel biocompatible ionic liquid based-aqueous two phase system. Phys. Chem. Chem. Phys. 2015, 17, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Bewernitz, M.A.; Gebauer, D.; Long, J.; Cölfen, H.; Gower, L.B. A metastable liquid precursor phase of calcium carbonate and its interactions with polyaspartate. Faraday Discuss. 2012, 159, 291–312. [Google Scholar] [CrossRef]
- Colominas, C.; Luque, F.J.; Teixidó, J.; Orozco, M. Cavitation contribution to the free energy of solvation: Comparison of different formalisms in the context of MST calculations. Chem. Phys. 1999, 240, 253–264. [Google Scholar] [CrossRef]
- Bennion, B.J.; Daggett, V. The molecular basis for the chemical denaturation of proteins by urea. Proc. Natl. Acad. Sci. USA 2003, 100, 5142–5147. [Google Scholar] [CrossRef] [PubMed]
- Wallqvist, A.; Covell, D.; Thirumalai, D. Hydrophobic interactions in aqueous urea solutions with implications for the mechanism of protein denaturation. J. Am. Chem. Soc. 1998, 120, 427–428. [Google Scholar] [CrossRef]
- Zangi, R.; Zhou, R.; Berne, B.J. Urea’s action on hydrophobic interactions. J. Am. Chem. Soc. 2009, 131, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- England, J.L.; Pande, V.S.; Haran, G. Chemical denaturants inhibit the onset of dewetting. J. Am. Chem. Soc. 2008, 130, 11854–11855. [Google Scholar] [CrossRef] [PubMed]
- Boiocchi, M.; Del Boca, L.; Gómez, D.E.; Fabbrizzi, L.; Licchelli, M.; Monzani, E. Nature of urea-fluoride interaction: Incipient and definitive proton transfer. J. Am. Chem. Soc. 2004, 126, 16507–16514. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Cölfen, H. On the biophysical regulation of mineral growth: Standing out from the crowd. J. Struc. Biol. 2016, 196, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Yakimov, M.M.; La Cono, V.; Spada, G.L.; Bortoluzzi, G.; Messina, E.; Smedile, F.; Arcadi, E.; Borghini, M.; Ferrer, M.; Schmitt-Kopplin, P.; et al. Microbial community of the deep-sea brine Lake Kryos seawater–brine interface is active below the chaotropicity limit of life as revealed by recovery of mRNA. Env. Microbiol. 2015, 17, 364–382. [Google Scholar] [CrossRef] [Green Version]
- Addadi, L.; Weiner, S. Biomineralization: Crystals, asymmetry and life. Nature 2001, 411, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Orme, C.; Noy, A.; Wierzbicki, A.; McBride, M.; Grantham, M.; Teng, H.; Dove, P.; DeYoreo, J. Formation of chiral morphologies through selective binding of amino acids to calcite surface steps. Nature 2001, 411, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.E.; Loges, N.; Mathiasch, B.; Panthöfer, M.; Mey, I.; Janshoff, A.; Tremel, W. Phase selection of calcium carbonate through the chirality of adsorbed amino acids. Angew. Chem. Int. Ed. 2007, 46, 5618–5623. [Google Scholar] [CrossRef] [PubMed]
- Oaki, Y.; Imai, H. Amplification of chirality from molecules into morphology of crystals through molecular recognition. J. Am. Chem. Soc. 2004, 126, 9271–9275. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Pacella, M.S.; Athanasiadou, D.; Nelea, V.; Vali, H.; Hazen, R.M.; Gray, J.J.; McKee, M.D. Chiral acidic amino acids induce chiral hierarchical structure in calcium carbonate. Nat. Comm. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Barron, L. From cosmic chirality to protein structure and function: Lord Kelvin’s legacy. QJM 1997, 90, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Heilbronner, E.; Dunitz, J.D. Reflections on Symmetry in Chemistry and Elsewhere; John Wiley & Sons: Hoboken, NJ, USA, 1993. [Google Scholar]
- Kellermeier, M.; Cölfen, H.; Gebauer, D. Investigating the early stages of mineral precipitation by potentiometric titration and analytical ultracentrifugation. Methods Enzymol. 2013, 532, 45–69. [Google Scholar] [PubMed]
Figure 1.
Developments of (A) free Ca2+ ions and (B) added NaOH contents for titrations performed at pH 9.75 in absence of additives (black) and the presence of either 10 mM urea (red) or trehalose (blue) in relation to the total Ca2+ ion contents. Zones shaded grey represent ±1 standard deviation for triplicate experiments. (A) The dotted line represents the development of total amount of Ca2+ ions dosed into the buffer.
Figure 1.
Developments of (A) free Ca2+ ions and (B) added NaOH contents for titrations performed at pH 9.75 in absence of additives (black) and the presence of either 10 mM urea (red) or trehalose (blue) in relation to the total Ca2+ ion contents. Zones shaded grey represent ±1 standard deviation for triplicate experiments. (A) The dotted line represents the development of total amount of Ca2+ ions dosed into the buffer.
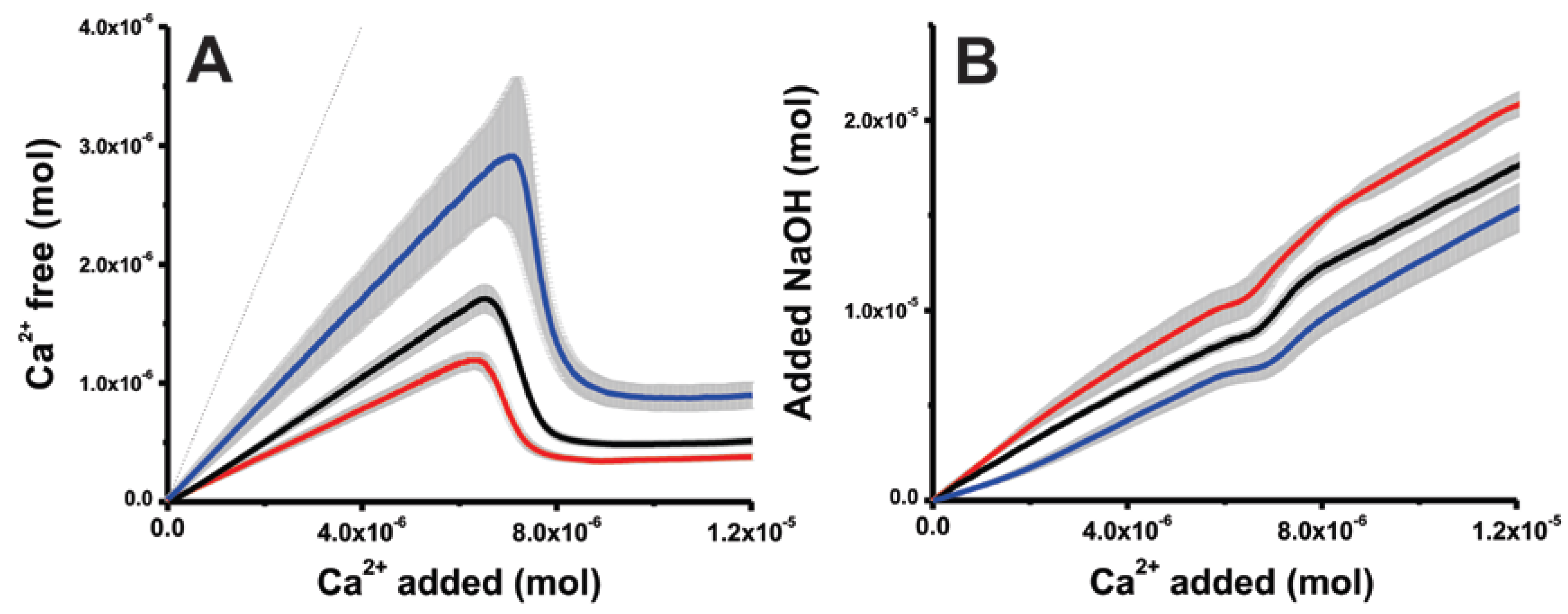
Figure 2.
Bar plots representing the early stages of CaCO3 nucleation in terms of the (A) pre-nucleation slope, (B) time required for nucleation and (C) solubility of initially nucleated phase at pH 9.0 (left) and 9.75 (right) in the presence of kosmotropic (blue) and chaotropic (red) additives and also in reference (Ref, gray) experiments. Error bars depict ± one standard deviation and corresponding values for reference experiments are shaded (orange).
Figure 2.
Bar plots representing the early stages of CaCO3 nucleation in terms of the (A) pre-nucleation slope, (B) time required for nucleation and (C) solubility of initially nucleated phase at pH 9.0 (left) and 9.75 (right) in the presence of kosmotropic (blue) and chaotropic (red) additives and also in reference (Ref, gray) experiments. Error bars depict ± one standard deviation and corresponding values for reference experiments are shaded (orange).
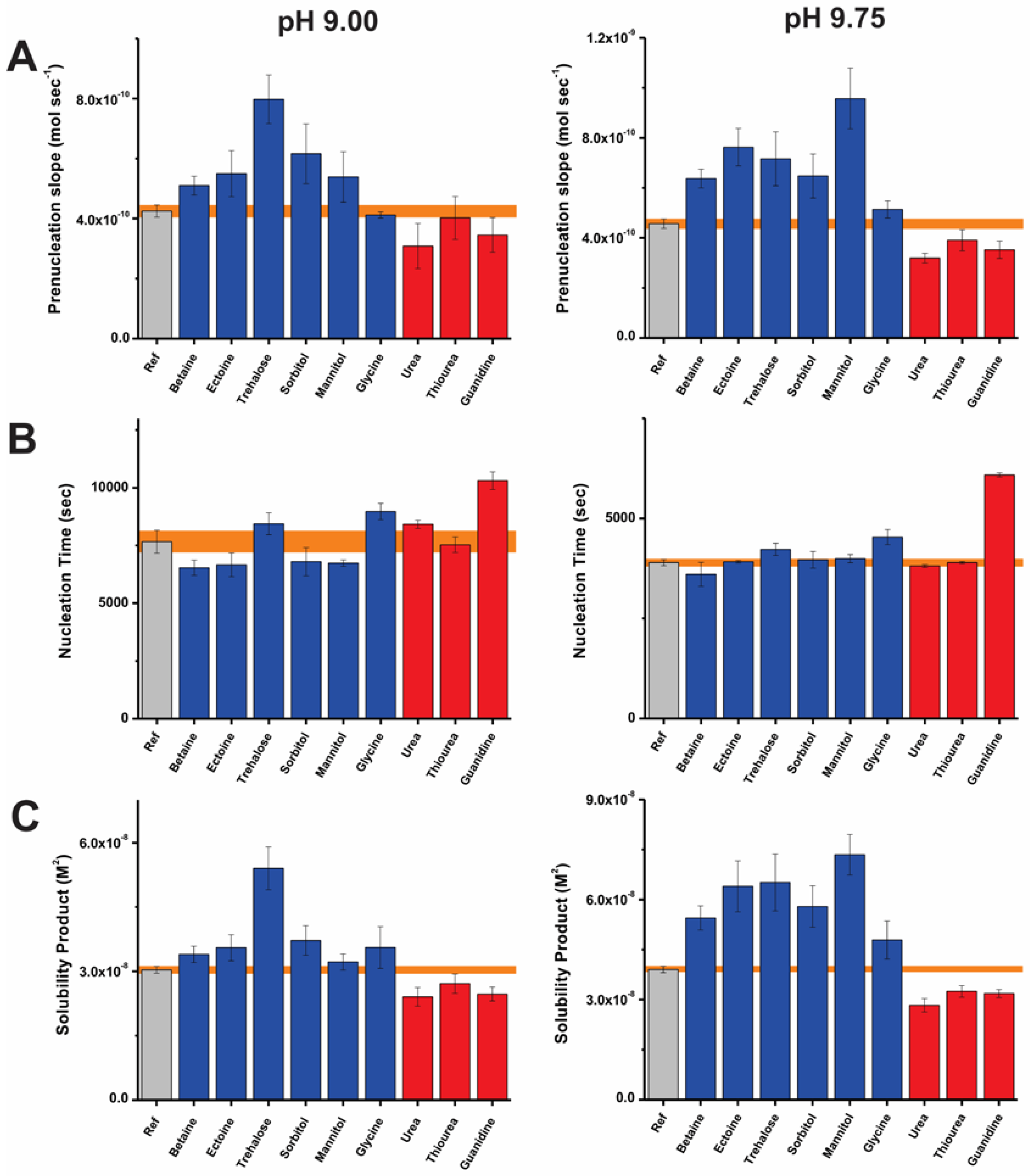
Figure 3.
Scatter plots representing slopes of the pre-nucleation regime and corresponding solubility products of phases formed after nucleation at (A) pH 9.0 and (B) 9.75 in reference (black) and additive-containing (colored) titrations. The color scale represents the relative chao(C)-/kosmotropic(K) activities of guanidine, urea, mannitol, sorbitol, trehalose, glycine, betaine, and ectoine as determined by systematic studies on agar gelation [69]. Error bars depict ± one standard deviation.
Figure 3.
Scatter plots representing slopes of the pre-nucleation regime and corresponding solubility products of phases formed after nucleation at (A) pH 9.0 and (B) 9.75 in reference (black) and additive-containing (colored) titrations. The color scale represents the relative chao(C)-/kosmotropic(K) activities of guanidine, urea, mannitol, sorbitol, trehalose, glycine, betaine, and ectoine as determined by systematic studies on agar gelation [69]. Error bars depict ± one standard deviation.
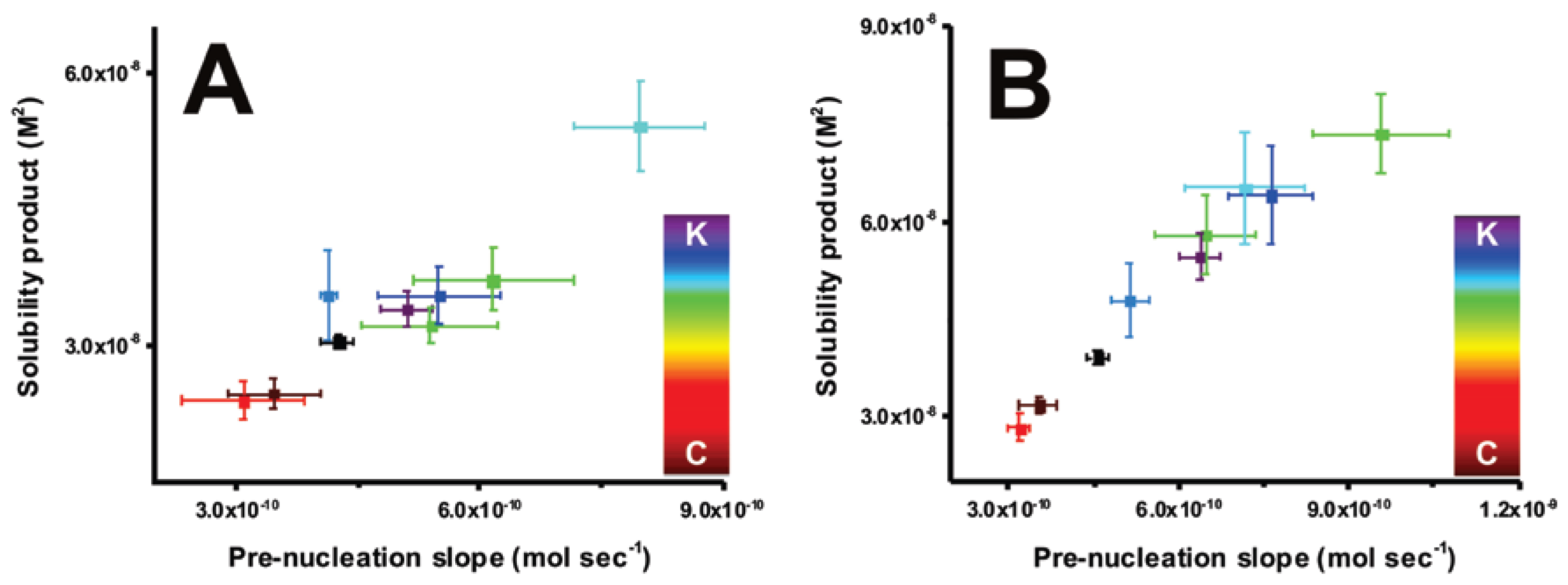
Figure 4.
Bar plots representing the early stages of CaCO3 nucleation in terms of the (A) pre-nucleation slope, (B) time required for nucleation, and (C) solubility of initially nucleated phase at pH 9.0 (left) and 9.75 (right) for additive-containing and reference (Ref) experiments. The acronyms indicate fructose (Fru), glucose (Glu), galactose (Gal), and mannose (Mann) in D (red) and L (blue) forms. Error bars depict ± one standard deviation. Error values for reference experiments are shaded (orange).
Figure 4.
Bar plots representing the early stages of CaCO3 nucleation in terms of the (A) pre-nucleation slope, (B) time required for nucleation, and (C) solubility of initially nucleated phase at pH 9.0 (left) and 9.75 (right) for additive-containing and reference (Ref) experiments. The acronyms indicate fructose (Fru), glucose (Glu), galactose (Gal), and mannose (Mann) in D (red) and L (blue) forms. Error bars depict ± one standard deviation. Error values for reference experiments are shaded (orange).

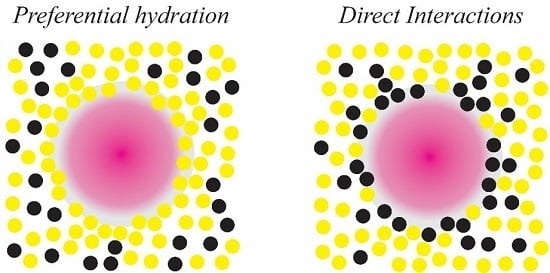
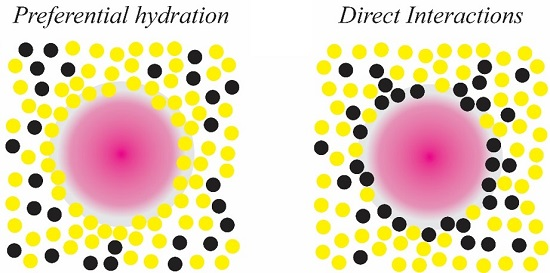
Figure 5.
Schematic representations of the effects of the small organic molecules (black) and solvent molecules (yellow) on mineral precursors (pink), indicating (A) a preferential hydration of mineral co-solutes and (B) direct organic-inorganic interactions.
Figure 5.
Schematic representations of the effects of the small organic molecules (black) and solvent molecules (yellow) on mineral precursors (pink), indicating (A) a preferential hydration of mineral co-solutes and (B) direct organic-inorganic interactions.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rao, A.; Gebauer, D.; Cölfen, H. Modulating Nucleation by Kosmotropes and Chaotropes: Testing the Waters. Crystals 2017, 7, 302. https://doi.org/10.3390/cryst7100302
AMA Style
Rao A, Gebauer D, Cölfen H. Modulating Nucleation by Kosmotropes and Chaotropes: Testing the Waters. Crystals. 2017; 7(10):302. https://doi.org/10.3390/cryst7100302
Chicago/Turabian StyleRao, Ashit, Denis Gebauer, and Helmut Cölfen. 2017. "Modulating Nucleation by Kosmotropes and Chaotropes: Testing the Waters" Crystals 7, no. 10: 302. https://doi.org/10.3390/cryst7100302
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.