
Lewis Acid Properties of Tetrel Tetrafluorides—The Coincidence of the σ-Hole Concept with the QTAIM Approach


Abstract
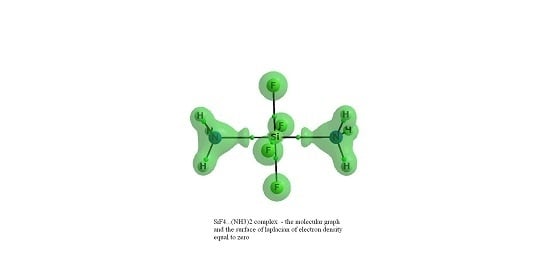
:
1. Introduction
2. Computational Details
3. Results and Discussion
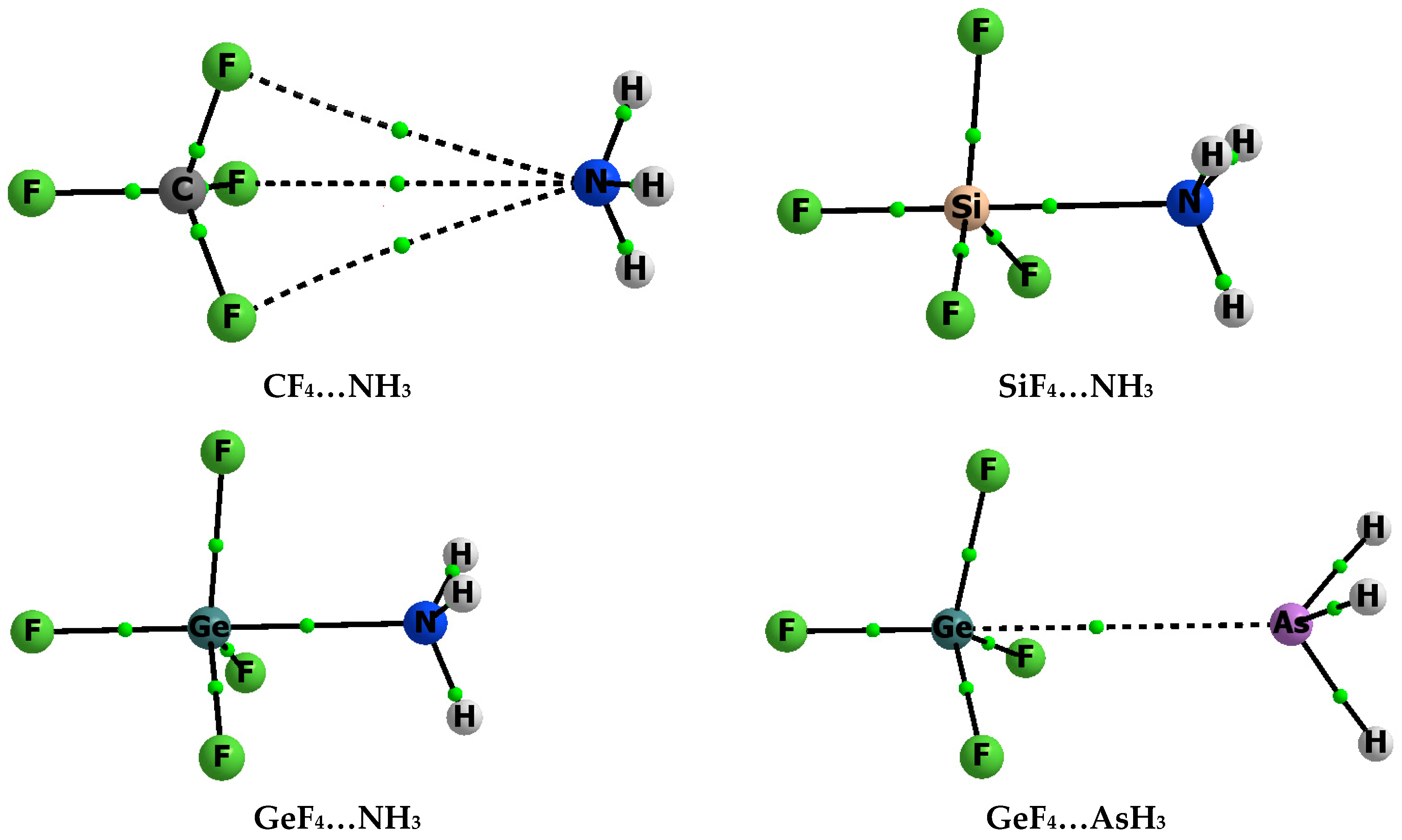
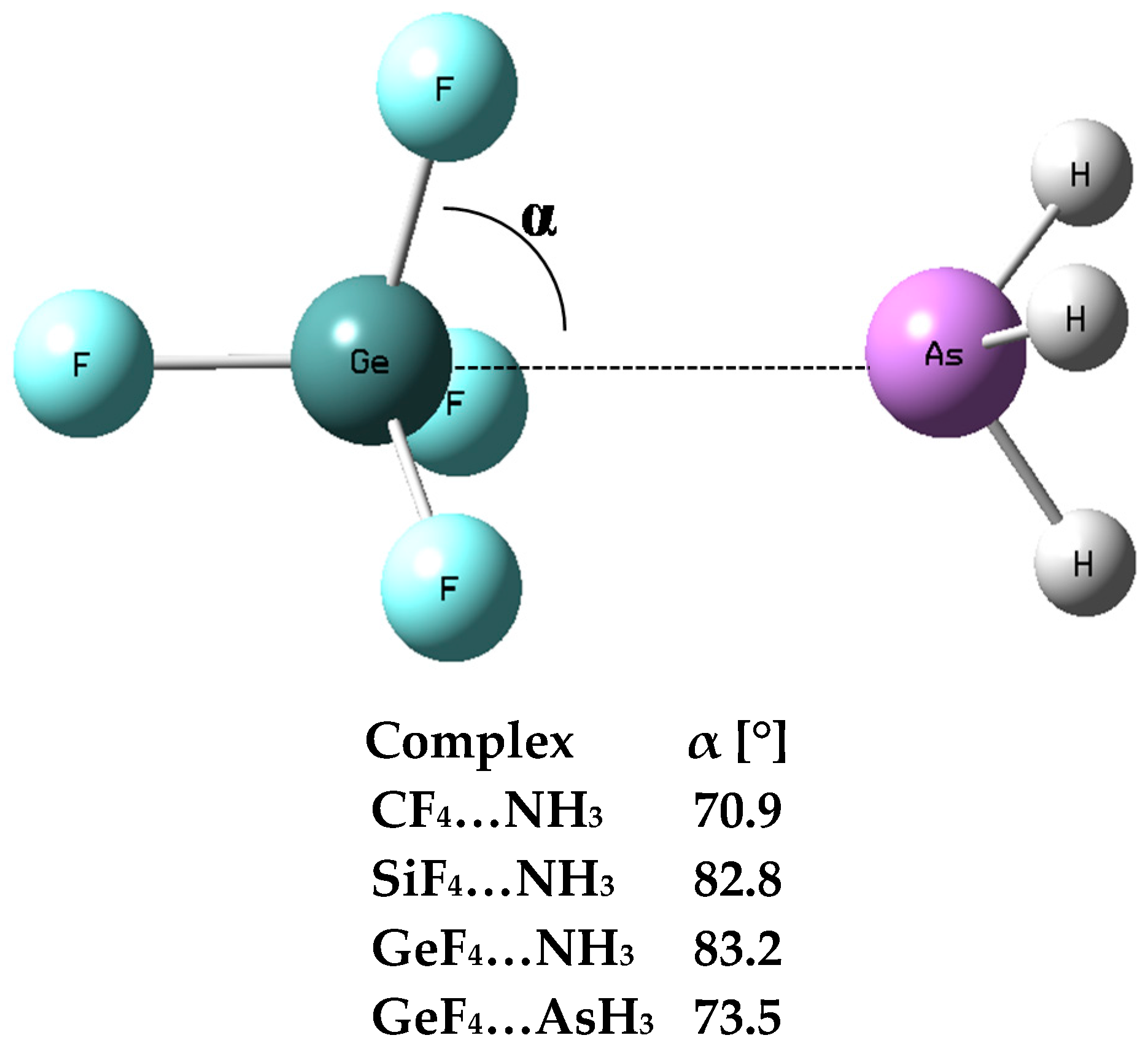
3.1. Interaction and Binding Energies
3.2. Electron Charge Shifts Resulting from Complexation—QTAIM Approach
3.3. Electron Charge Shifts Resulting from Complexation—NBO Approach
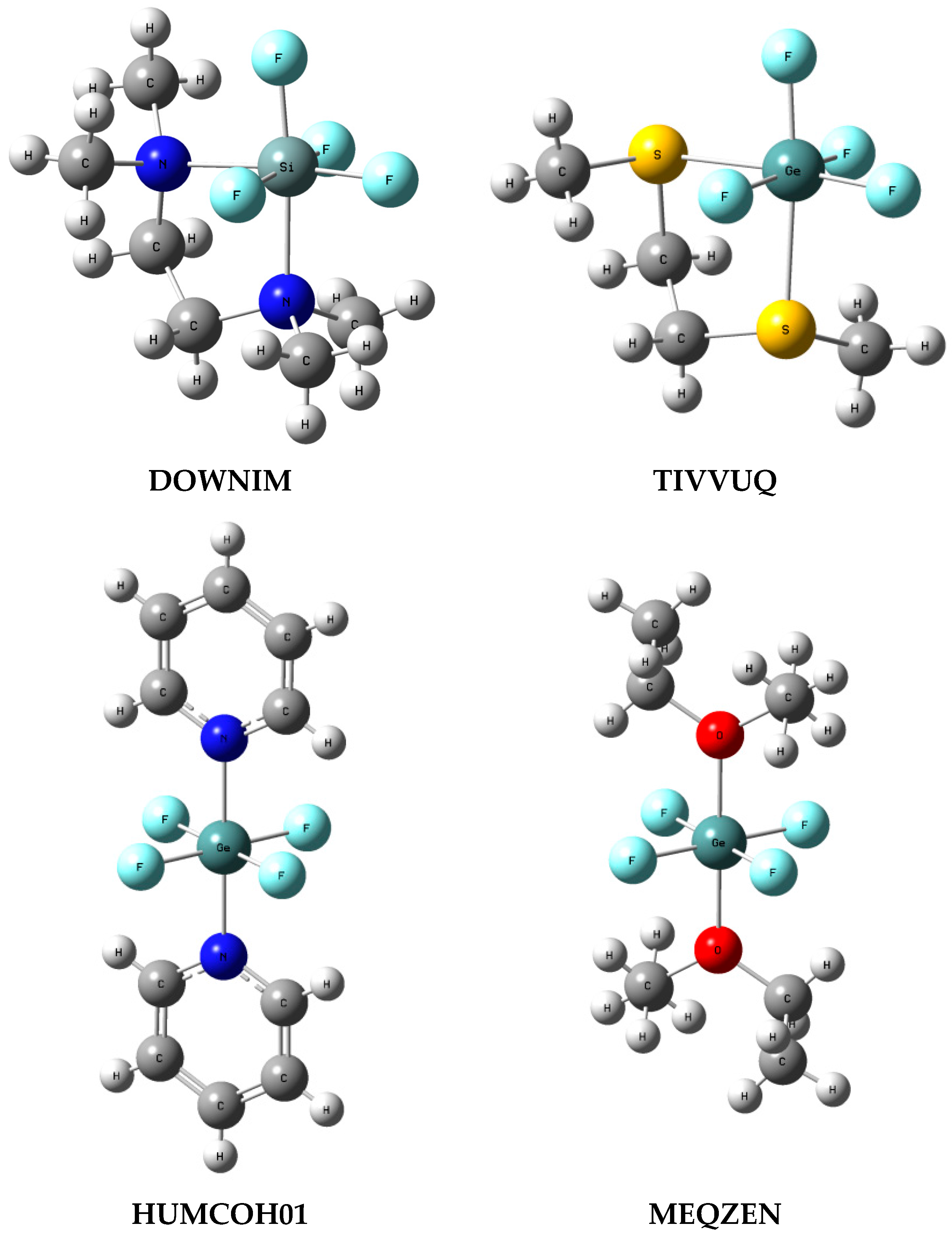
3.4. ZF4 Moiety in Crystal Structures
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Scheiner, S. (Ed.) Molecular Interactions. From van der Waals to Strongly Bound Complexes; John Wiley & Sons: Chichester, UK, 1997.
- Schneider, H.-J. Binding Mechanisms in Supramolecular Chemistry. Angew. Chem. Int. Ed. 2009, 48, 3924–3977. [Google Scholar]
- Hobza, P.; Müller-Dethlefs, K. Non-Covalent Interactions, Theory and Experiment. Royal Society of Chemistry; Thomas Graham House, Science Park, Milton Road: Cambridge, UK, 2010. [Google Scholar]
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer: Berlin, Germany, 1991. [Google Scholar]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Grabowski, S.J.; Leszczynski, J. (Eds.) Hydrogen Bonding—New Insights; Volume 3 of the Series: Challenges and Advances in Computational Chemistry and Physics; Springer: Berlin, Germany, 2006.
- Grabowski, S.J. (Ed.) Analysis of Hydrogen Bonds in Crystals (Printed Edition of the Special Issue Published in Crystals); MDPI: Basel, Switzerland; Beijing/Wuhan, China; Barcelona, Spain, 2016.
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the σ-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Lane, P.; Concha, M.C. Electrostatically driven complexes of SiF4 with amines. Int. J. Quantum Chem. 2009, 109, 3773–3780. [Google Scholar] [CrossRef]
- Bundhun, A.; Ramasami, P.; Murray, J.S.; Politzer, P. Trends in σ-hole strengths and interactions of F3MX molecules (M = C, Si, Ge and X = F, Cl, Br, I). J. Mol. Model. 2012, 19, 2739–2746. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding Interaction Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Tetrel bond—σ-hole bond as a preliminary stage of the SN2 reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7758. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. Halogen Bonding: An Interim Discussion. ChemPhysChem 2013, 14, 2145–2151. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Janjić, G.V.; Zarić, S.D. σ-Hole Interactions of Covalently-Bonded Nitrogen, Phosphorus and Arsenic: A Survey of Crystal Structures. Crystals 2014, 4, 12–31. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. Aerogen Bonding Interaction: A New Supramolecular Force? Angew. Chem. Int. Ed. 2015, 54, 7340–7343. [Google Scholar] [CrossRef] [PubMed]
- Chehayber, J.M.; Nagy, S.T.; Lin, C.S. Ab initio studies of complexes between SF4 and ammonia. Can. J. Chem. 1984, 62, 27–31. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, I.; Elguero, J. Molecular Complexes between Silicon Derivatives and Electron-Rich Groups. J. Phys. Chem. A 2001, 105, 743–749. [Google Scholar] [CrossRef]
- Helminiak, H.M.; Knauf, R.R.; Danforth, S.J.; Phillips, J.A. Structural and Energetic Properties of Acetonitrile–Group IV (A & B) Halide Complexes. J. Phys. Chem. A 2014, 118, 4266–4277. [Google Scholar] [PubMed]
- Sedlak, R.; Stasyuk, O.A.; Fonseca Guerra, C.; Rézač, J.; Růžička, A.; Hobza, P. New Insight into the Nature of Bonding in the Dimers of Lappert’s Stannylene and Its Ge Analogs: A Quantum Mechanical Study. J. Chem. Theory Comput. 2016, 12, 1696–1704. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Tetrel bonds, penta- and hexa-coordinated tin and lead centres. Appl. Organometal. Chem. 2017, e3727. [Google Scholar] [CrossRef]
- Yamashita, M.; Yamamoto, Y.; Akiba, K.-Y.; Hashizume, D.; Iwasaki, F.; Takagi, N.; Nagase, S. Syntheses and Structures of Hypervalent Pentacoordinate Carbon and Boron Compounds Bearing an Anthracene Skeleton—Elucidation of Hypervalent Interaction Based on X-ray Analysis and DFT Calculation. J. Am. Chem. Soc. 2005, 127, 4354–4371. [Google Scholar] [CrossRef] [PubMed]
- Akiba, K.-Y.; Moriyama, Y.; Mizozoe, M.; Inohara, H.; Nishii, T.; Yamamoto, Y.; Minoura, M.; Hashizume, D.; Iwasaki, F.; Takagi, N.; et al. Synthesis and Characterization of Stable Hypervalent Carbon Compounds (10-C-5) Bearing a 2,6-Bis(p-substituted phenyloxymethyl)benzene Ligand. J. Am. Chem. Soc. 2005, 127, 5893–5901. [Google Scholar] [CrossRef] [PubMed]
- Akiba, K. Hypervalent Carbon Compounds: Can Hexavalent Carbon Exist? In Organo Main Group Chemistry; Akiba, K., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; Chapter 12; pp. 251–268. [Google Scholar]
- Sohail, M.; Panisch, R.; Bowden, A.; Bassindale, A.R.; Taylor, P.G.; Korlyukov, A.A.; Arkhipov, D.E.; Male, L.; Callear, S.; Coles, S.J.; et al. Pentacoordinate silicon complexes with dynamic motion resembling a pendulum on the SN2 reaction pathway. Dalton Trans. 2013, 42, 10971–10981. [Google Scholar] [CrossRef] [PubMed]
- Pelzer, S.; Neumann, B.; Stammler, H.-G.; Ignat’ev, N.; Hoge, B. Synthesis of bis(pentafluoroethyl)germanes. Chem. Eur. J. 2016, 22, 4758–4763. [Google Scholar] [CrossRef] [PubMed]
- Pelzer, S.; Neumann, B.; Stammler, H.-G.; Ignat’ev, N.; Hoge, B. Hypervalent Pentafluoroethylgermanium Compounds, [(C2F5)nGeX5-n]− and [(C2F5)3GeF3]2− (X = F, Cl; n = 2–5). Chem. Eur. J. 2016, 22, 16460–16466. [Google Scholar] [CrossRef] [PubMed]
- Bouška, M.; Dostal, L.; Růžička, A.; Jambor, R. Stabilization of Three-Coordinated Germanium(II) and Tin(II) Cations by a Neutral Chelating Ligand. Organometallics 2013, 32, 1995–1999. [Google Scholar] [CrossRef]
- Miller, D.B.; Sisler, H.H. Observations on the Addition Compound of Silicon Tetrafluoride and Ammonia. J. Am. Chem. Soc. 1955, 77, 4998–5000. [Google Scholar] [CrossRef]
- Ault, B.S. Matrix-isolation studies of Lewis acid/base interactions: Infrared spectra of the 1:1 adduct SiF4.NH3. Inorg. Chem. 1981, 20, 2817–2822. [Google Scholar] [CrossRef]
- Urban, R.-D.; Rouillé, G.; Takami, M. Free jet IR spectroscopy of SiF4-N2 and SiF4-CO complexes in the 10 μm region. J. Mol. Struct. 1997, 413–414, 511–519. [Google Scholar] [CrossRef]
- Walther, A.M.; Ault, B.S. Infrared matrix isolation study of intermediate molecular complexes: Complexes of tetrafluorogermane with oxygen-containing bases. Inorg. Chem. 1984, 23, 3892–3897. [Google Scholar] [CrossRef]
- Davis, M.F.; Levason, W.; Reid, G.; Webster, M.; Zhang, W. The first examples of germanium tetrafluoride and tin tetrafluoride complexes with soft thioether coordination—Synthesis, properties and crystal structures. Dalton Trans. 2008, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.F.; Levason, W.; Reid, G.; Webster, M. Complexes of germanium(IV) fluoride with phosphane ligands: Structural and spectroscopic authentication of germanium(IV) phosphane complexes. Dalton Trans. 2008, 17, 2261–2269. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1, Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations. III. The second row atoms, Al-Ar. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar]
- Metz, B.; Stoll, H.; Dolg, M. Small-core multiconfiguration-Dirac–Hartree–Fock-adjusted pseudopotentials for post-d main group elements: Application to PbH and PbO. J. Chem. Phys. 2000, 113, 2563–2569. [Google Scholar] [CrossRef]
- Peterson, K.A. Systematically convergent basis sets with relativistic pseudopotentials. I. Correlation consistent basis sets for the post-d group 13–15 elements. J. Chem. Phys. 2003, 119, 11099–11112. [Google Scholar] [CrossRef]
- Piela, L. Ideas of Quantum Chemistry; Elsevier Science Publishers: Amsterdam, The Netherlands, 2007; pp. 684–691. [Google Scholar]
- Grabowski, S.J.; Sokalski, W.A. Different types of hydrogen bonds: Correlation analysis of interaction energy components. J. Phys. Org. Chem. 2005, 18, 779–784. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–561. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules, A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Wiley-VCH. Quantum Theory of Atoms in Molecules: Recent Progress in Theory and Application; Matta, C., Boyd, R.J., Eds.; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Keith, T.A. AIMAll (Version 11.08.23), TK Gristmill Software: Overland Park, KS, USA, 2011.
- Weinhold, F.; Landis, C. Valency and Bonding, A Natural Bond Orbital Donor—Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Weinhold, F. NBO 5.0, Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2001.
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.J.; et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Bader, R.F.W. Bond Paths Are Not Chemical Bonds. J. Phys. Chem. A 2009, 113, 10391–10396. [Google Scholar] [CrossRef] [PubMed]
- Maris, A.; Favero, L.B.; Velino, B.; Caminati, W. Pyridine-CF4: A Molecule with a Rotating Cap. J. Phys. Chem. A 2013, 117, 11289–11292. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. What is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 11, 2597–2625. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, S.; Morrison, I. The chemical character of the intermolecular bonds of seven phases of ice as revealed by AB initio calculation of electron densities. Chem. Phys. Lett. 2000, 317, 97–102. [Google Scholar] [CrossRef]
- Arnold, W.D.; Oldfield, E. The Chemical Nature of Hydrogen Bonding in Proteins via NMR: J-Couplings, Chemical Shifts, and AIM Theory. J. Am. Chem. Soc. 2000, 122, 12835–12841. [Google Scholar] [CrossRef]
- Rozas, I.; Alkorta, I.; Elguero, J. Behavior of ylides containing N, O, and C atoms as hydrogen bond acceptors. J. Am. Chem. Soc. 2000, 122, 1154–11161. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Manoharan, M.; Peabody, S.; Weinhold, F. Electronic Basis of Improper Hydrogen Bonding: A Subtle Balance of Hyperconjugation and Rehybridization. J. Am. Chem. Soc. 2003, 125, 5973–5987. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.; Allen, F.H.; Willett, P. The scientific impact of the Cambridge Structural Database: A citation-based study. J. Appl. Cryst. 2010, 43, 811–824. [Google Scholar] [CrossRef]
- Gillespie, R.J. Fifty years of the VSEPR model. Coord. Chem. Rev. 2008, 252, 1315–1327. [Google Scholar] [CrossRef]
- Gillespie, R.J.; Popelier, P.L.A. Chemical Bonding and Molecular Geometry; Oxford University Press: Oxford, UK, 2001. [Google Scholar]



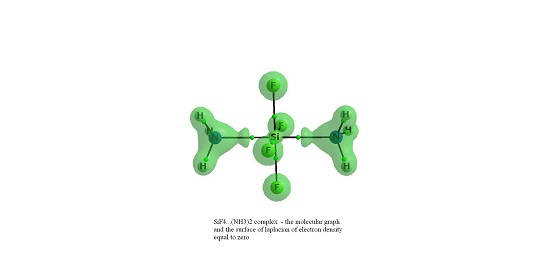{kind=link}
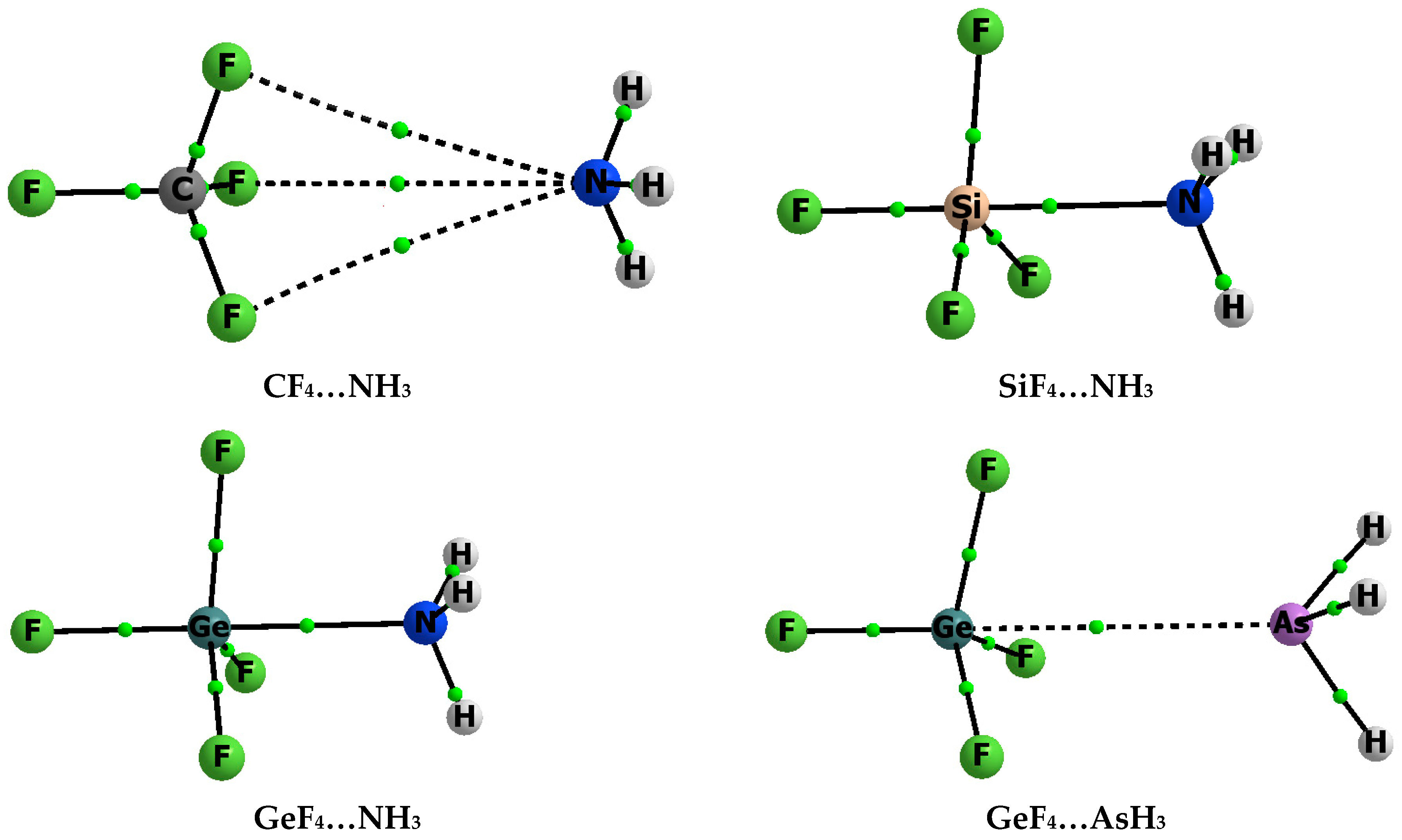{kind=link}
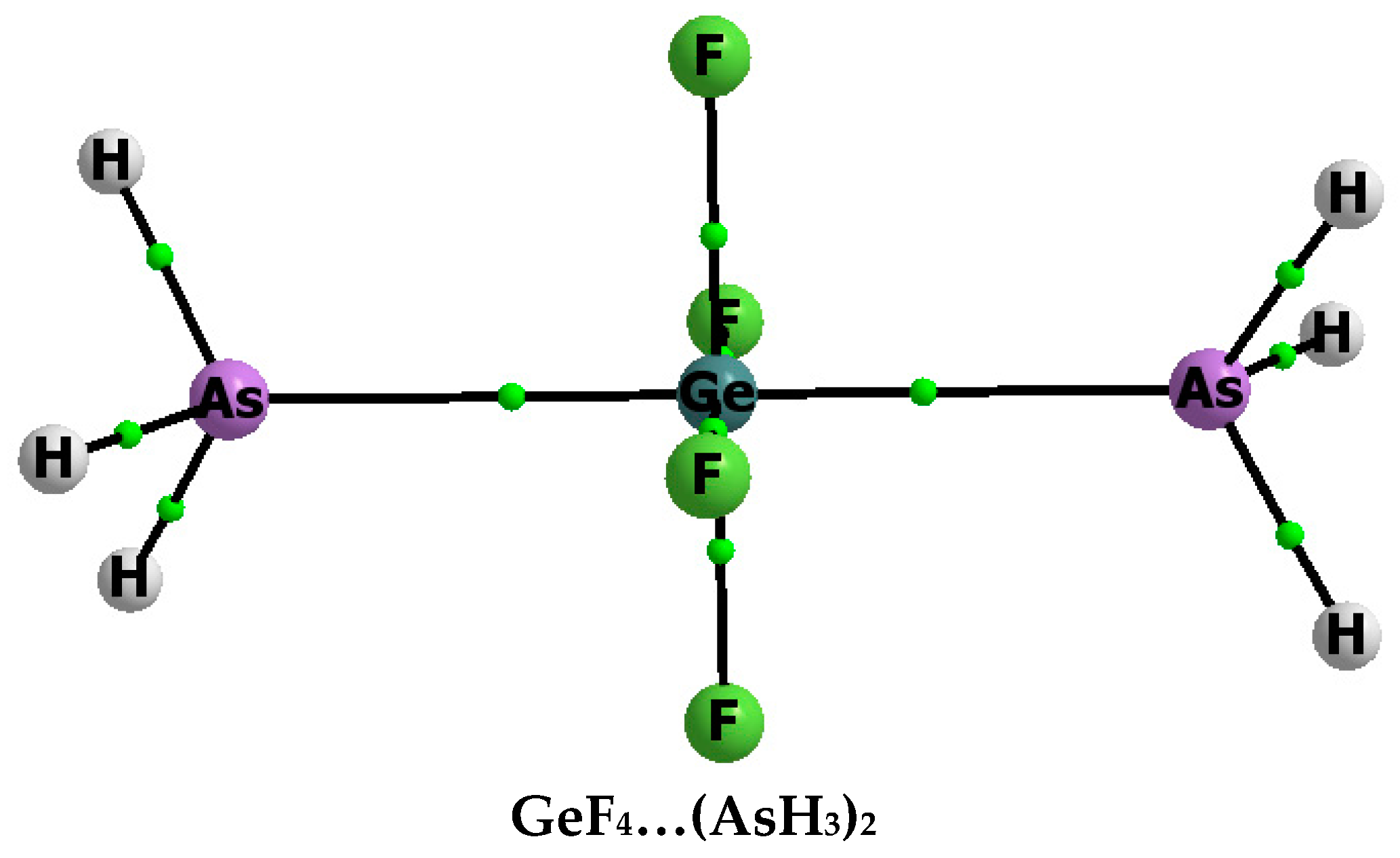{kind=link}
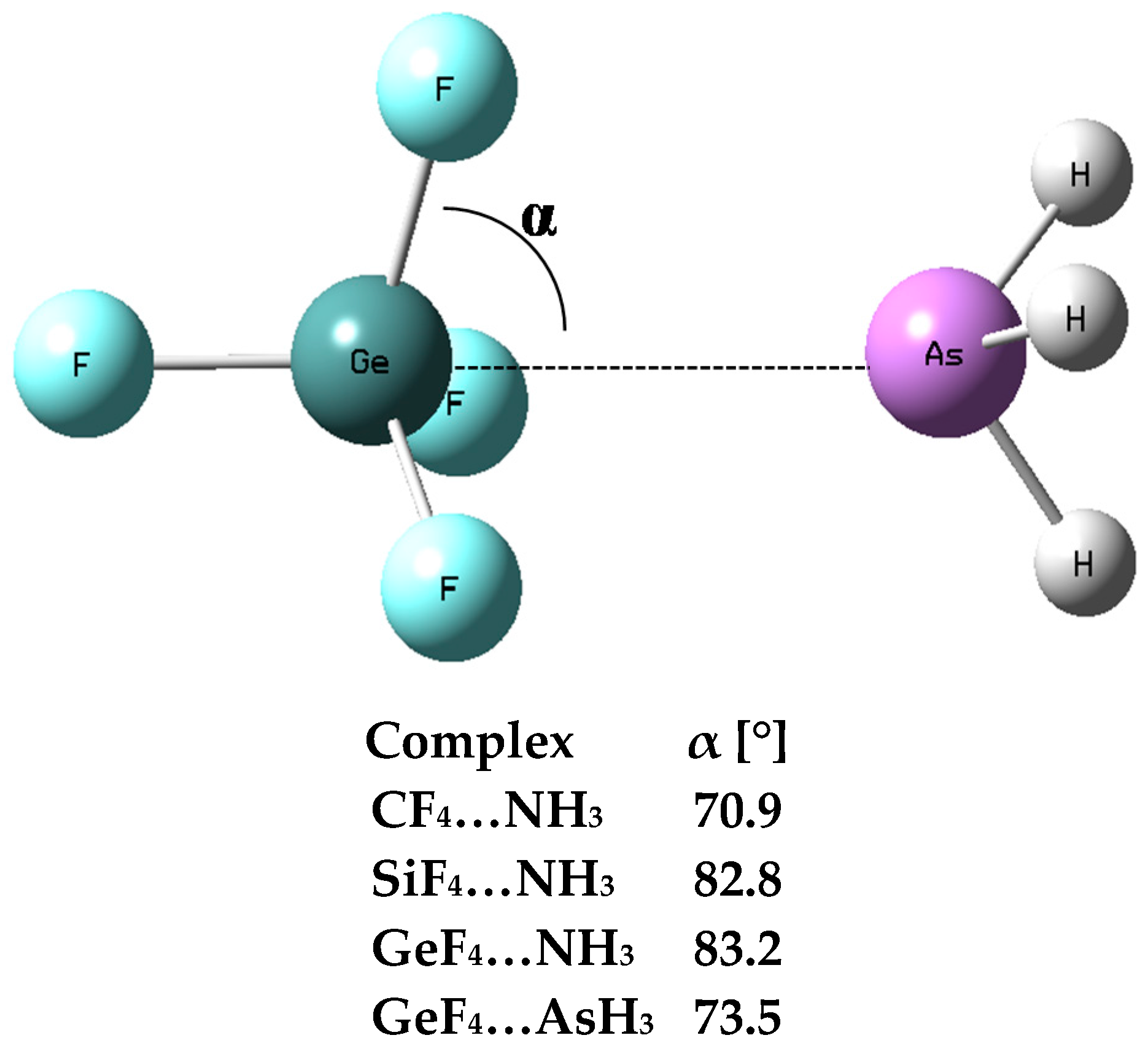{kind=link}
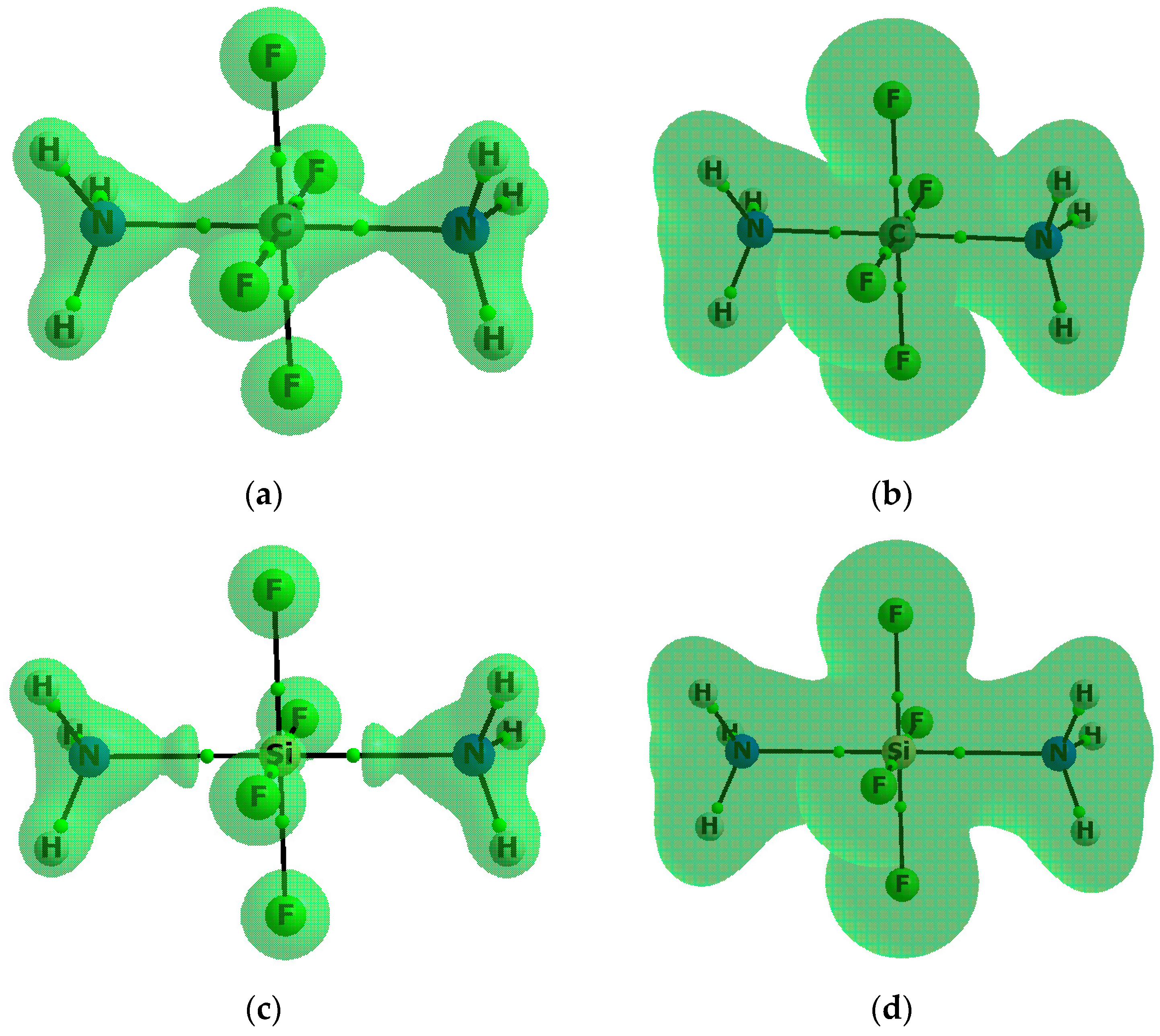{kind=link}
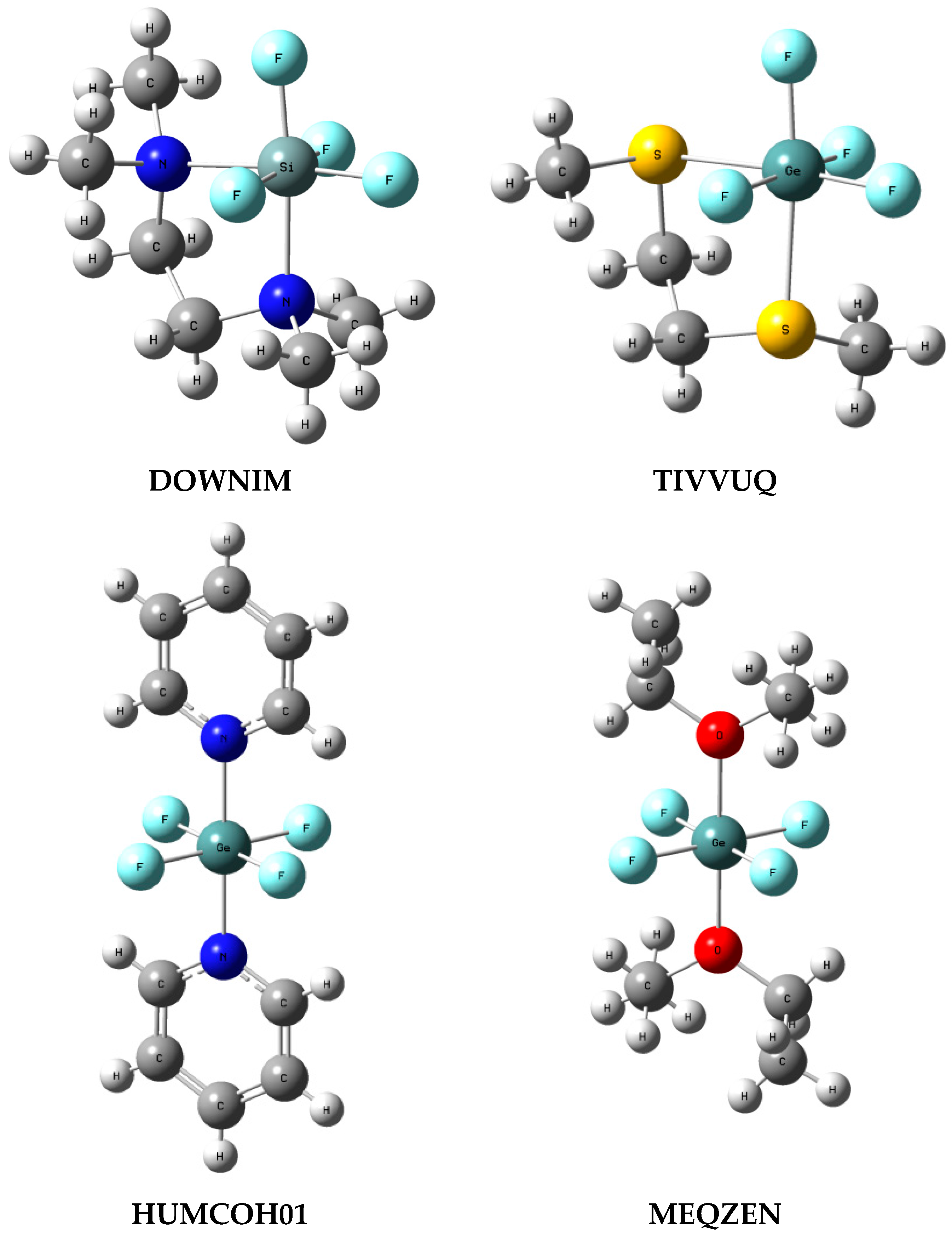{kind=link}
| Complex | Z–B | Eint | Ebin | BSSE | Edef |
|---|---|---|---|---|---|
| CF4–NH3 | 3.658 | −1.1 | −1.1 | 0.3 | 0.0 |
| CF4–(NH3)2 | 1.658 | −29.1 | 90.1 | 3.0 | 119.2 |
| CF4–(NH3)2 * | 1.658 | −76.8 | 91.9 | 6.5 | 168.7 |
| SiF4–NH3 | 2.072 | −29.6 | −8.5 | 2.5 | 21.1 |
| SiF4–(NH3)2 | 1.940 | −44.4 | −14.7 | 2.4 | 29.7 |
| SiF4–(NH3)2 * | 1.940 | −93.9 | −23.7 | 4.4 | 70.2 |
| GeF4–NH3 | 2.080 | −33.8 | −16.2 | 4.7 | 17.6 |
| GeF4–(NH3)2 | 1.997 | −42.4 | −19.2 | 4.9 | 23.2 |
| GeF4–(NH3)2 * | 1.997 | −90.5 | −34.2 | 10.9 | 56.3 |
| GeF4–AsH3 | 3.399 | −2.8 | −1.8 | 1.9 | 1.0 |
| GeF4–(AsH3)2 | 2.543 | −22.3 | 10.3 | 5.1 | 32.6 |
| GeF4–(AsH3)2 * | 2.543 | −48.1 | 11.5 | 10.1 | 59.6 |
| Complex | ρBCP | VBCP | GBCP | HBCP | ∇2ρBCP |
|---|---|---|---|---|---|
| CF4–NH3 * | 0.003 | −0.002 | 0.002 | 0.001 | 0.012 |
| CF4–(NH3)2 | 0.183 | −0.211 | 0.069 | −0.142 | −0.292 |
| SiF4–NH3 | 0.062 | −0.098 | 0.074 | −0.024 | 0.200 |
| SiF4–(NH3)2 | 0.081 | −0.147 | 0.113 | −0.033 | 0.319 |
| GeF4–NH3 | 0.083 | −0.117 | 0.081 | −0.036 | 0.184 |
| GeF4–(NH3)2 | 0.098 | −0.153 | 0.106 | −0.047 | 0.235 |
| GeF4–AsH3 | 0.012 | −0.006 | 0.006 | 0.000 | 0.026 |
| GeF4–(AsH3)2 | 0.062 | −0.052 | 0.028 | −0.025 | 0.012 |
| Moiety | QZF4 | QF * | QZ | PLZF * | Z-F * | PLZB |
|---|---|---|---|---|---|---|
| CF4 | 0 | −0.316 | 1.264 | 28.7 | 1.321 | not applied |
| CF4–NH3 | 0 | −0.349 (−0.358) | 1.404 | 28.7 (28.5) | 1.319 (1.327) | no occurrence |
| CF4–(NH3)2 | −1.111 | −0.522 | 0.977 | 17.7 | 1.559 | 22.8 |
| SiF4 | 0 | −0.634 | 2.537 | 12.4 | 1.574 | not applied |
| SiF4–NH3 | −0.163 | -0.692 (−0.696) | 2.608 | 11.1(10.4) | 1.610 (1.612) | 6.8 |
| SiF4–(NH3)2 | −0.467 | −0.717 | 2.399 | 9.7 | 1.675 | 9.7 |
| GeF4 | 0 | −0.628 | 2.511 | 13.5 | 1.687 | not applied |
| GeF4–NH3 | −0.183 | −0.706 (−0.704) | 2.640 | 10.9 (10.2) | 1.724 (1.719) | 7.0 |
| GeF4–(NH3)2 | −0.465 | −0.735 | 2.476 | 8.5 | 1.776 | 8.8 |
| GeF4–AsH3 | −0.017 | −0.684 (−0.691) | 2.727 | 12.7 (12.4) | 1.691 (1.694) | no occurrence |
| GeF4–(AsH3)2 | −0.738 | −0.733 | 2.195 | 8.5 | 1.769 | 14.9 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabowski, S.J. Lewis Acid Properties of Tetrel Tetrafluorides—The Coincidence of the σ-Hole Concept with the QTAIM Approach. Crystals 2017, 7, 43. https://doi.org/10.3390/cryst7020043
Grabowski SJ. Lewis Acid Properties of Tetrel Tetrafluorides—The Coincidence of the σ-Hole Concept with the QTAIM Approach. Crystals. 2017; 7(2):43. https://doi.org/10.3390/cryst7020043
Chicago/Turabian StyleGrabowski, Sławomir J. 2017. "Lewis Acid Properties of Tetrel Tetrafluorides—The Coincidence of the σ-Hole Concept with the QTAIM Approach" Crystals 7, no. 2: 43. https://doi.org/10.3390/cryst7020043
APA StyleGrabowski, S. J. (2017). Lewis Acid Properties of Tetrel Tetrafluorides—The Coincidence of the σ-Hole Concept with the QTAIM Approach. Crystals, 7(2), 43. https://doi.org/10.3390/cryst7020043