
Atomistic Modelling of Si Nanoparticles Synthesis




Abstract
:1. Introduction
2. Model and Computational Details
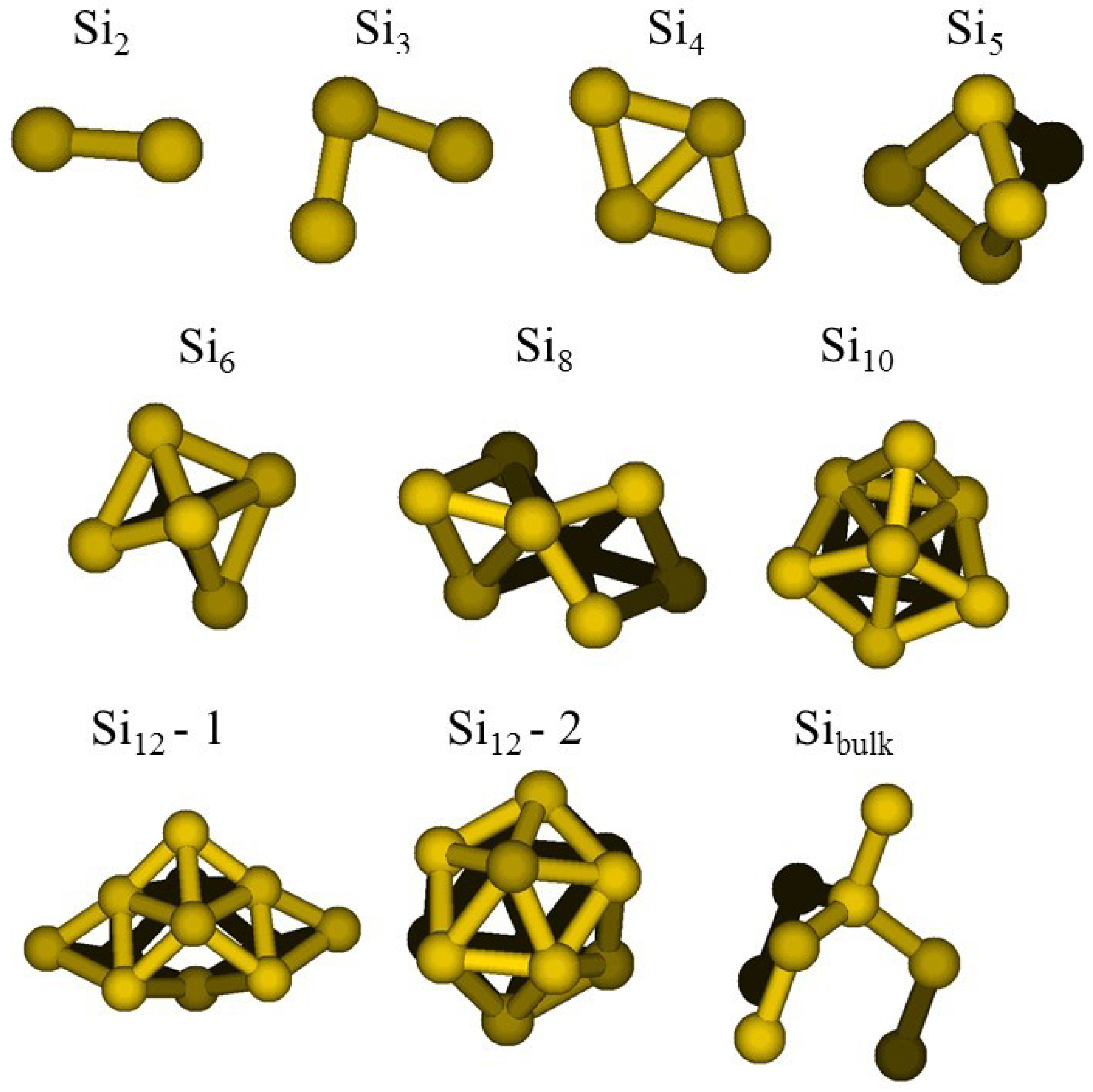
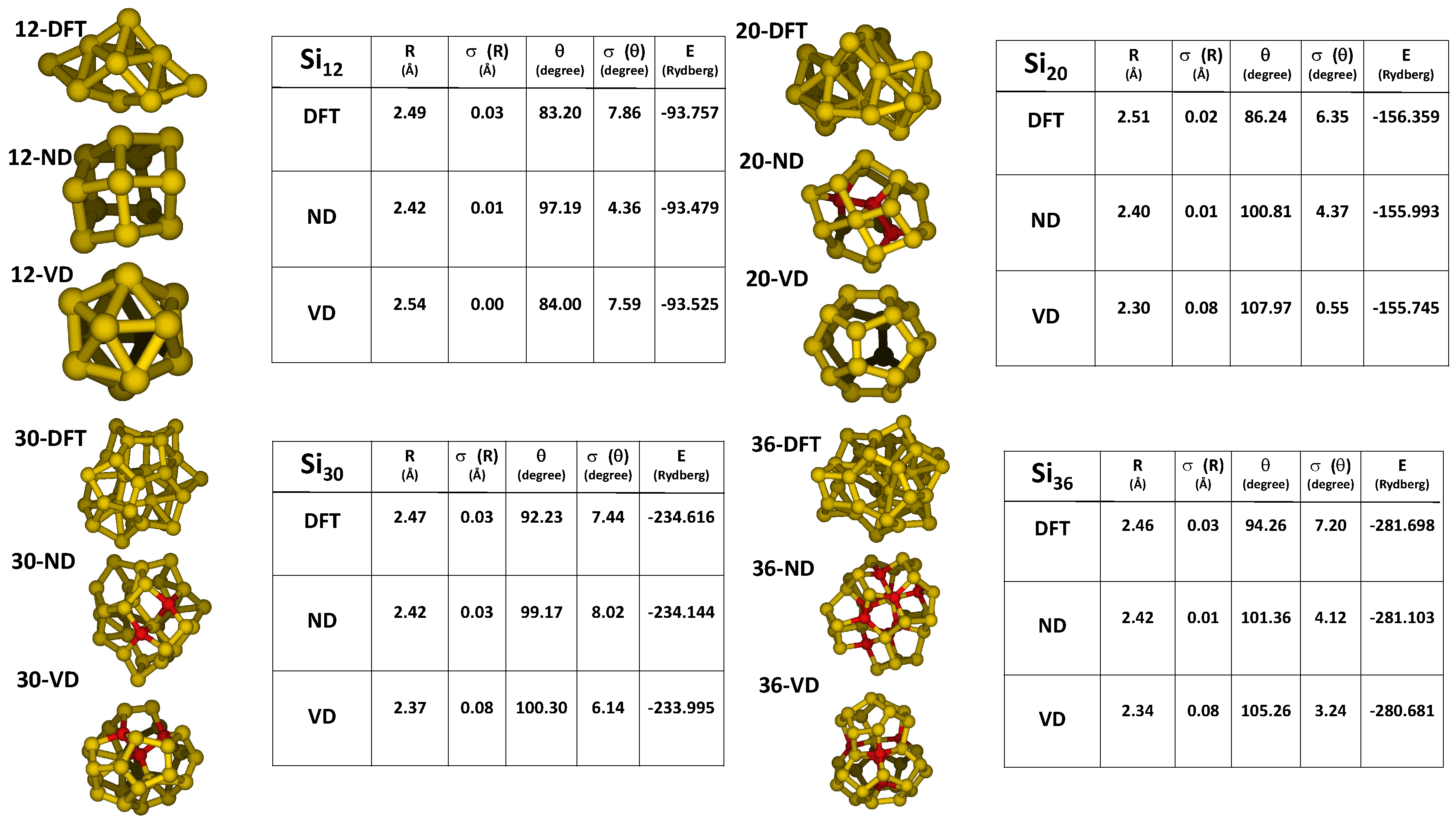
2.1. Optimization of the ReaxFF Parameters
2.2. Reactive Molecular Dynamics
- Simulation box: an appropriate value of the length of the cubic simulation box has been chosen in order to reduce the effect of the periodic boundary conditions. The number of events that occur on the border becomes less and less relevant than the number of total events when the size of the box is increased. In the limit of a box with an infinite size, the biasing constraint of the translational periodicity disappears. According to a series of simulations in which the volume of a cubic unit cell has been progressively increased (from a minimum of nm to a maximum of nm), the volume chosen was nm.
- Temperature: each simulation has been carried out at fixed average temperature. The temperature range between 1800 and 2400 K has been explored by increasing the temperature by steps of 100 K in each simulation.
- Atom density: the number of atoms within the simulation box has been chosen on the basis of particular experimental conditions: total gas pressure within the plasma reactor around 1 atmosphere (standard ambient pressure). On the basis of this latter value, the density of the simulation gas (which is composed of Si and Ar atoms) is about 3.6 × 10 atoms/m, which corresponds—in the chosen cubic box—to an ensemble of about 12,000 atoms.
- Composition of the mixture: the content of the (Si, Ar) mixture has been varied according to the following percentages: (i) 25% Si and 75% Ar; (ii) 50% Si and 50% Ar; and (iii) 75% Si and 25% Ar.
- Time step: The time step of the MD simulations has been set to 2 fs, having verified that a smaller time step of 0.5 fs reproduces the same results at both the qualitative and quantitative levels.
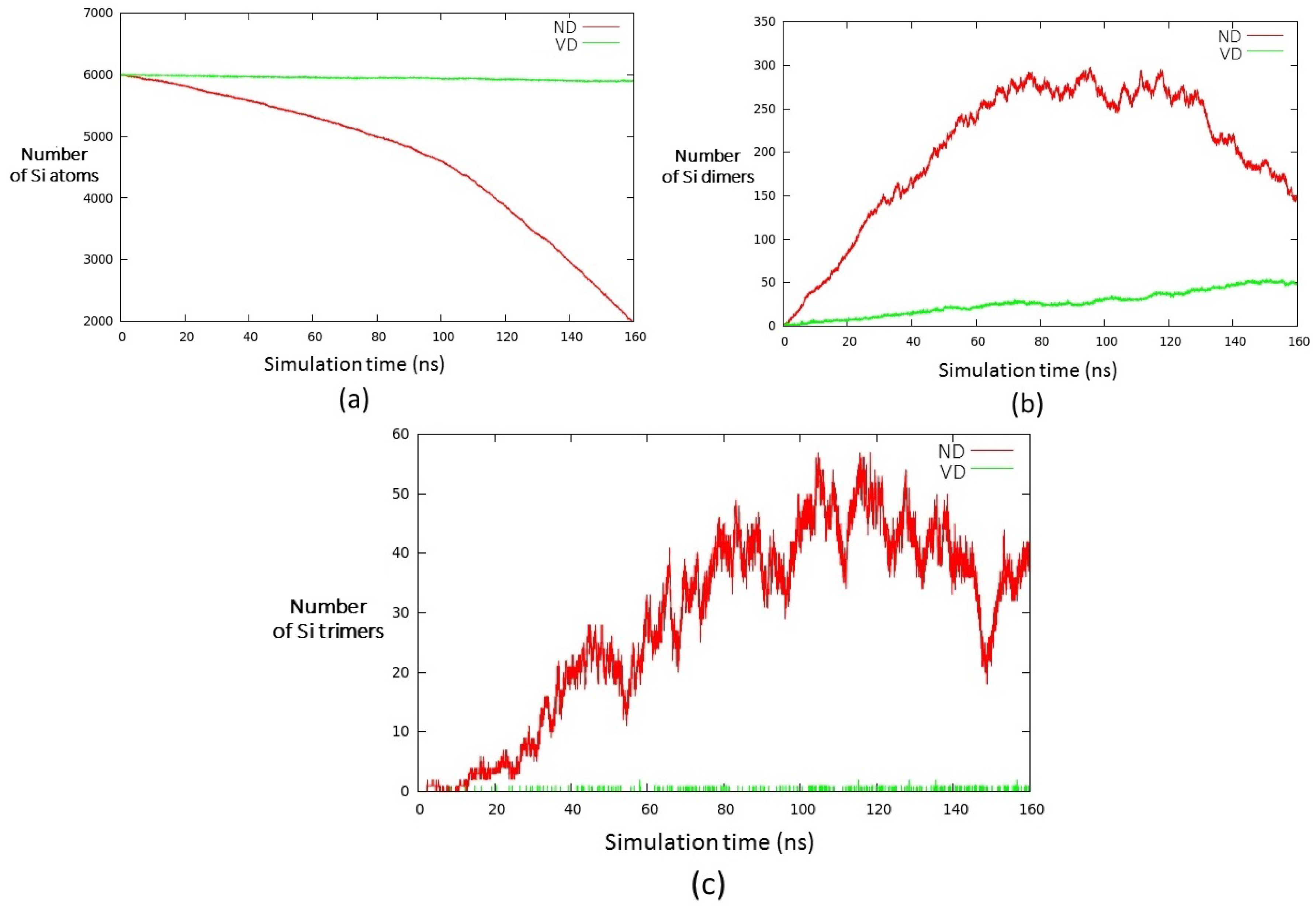
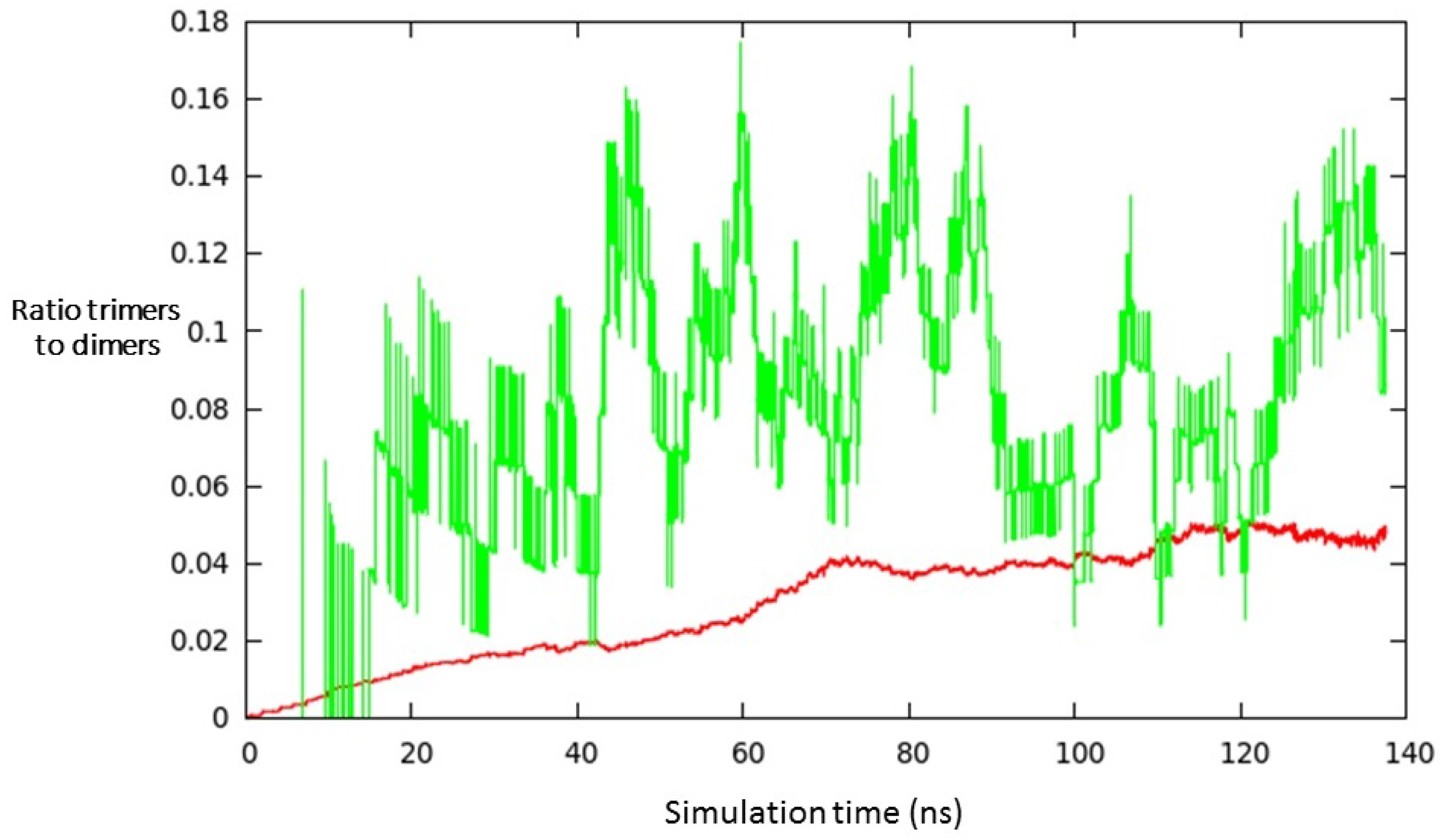
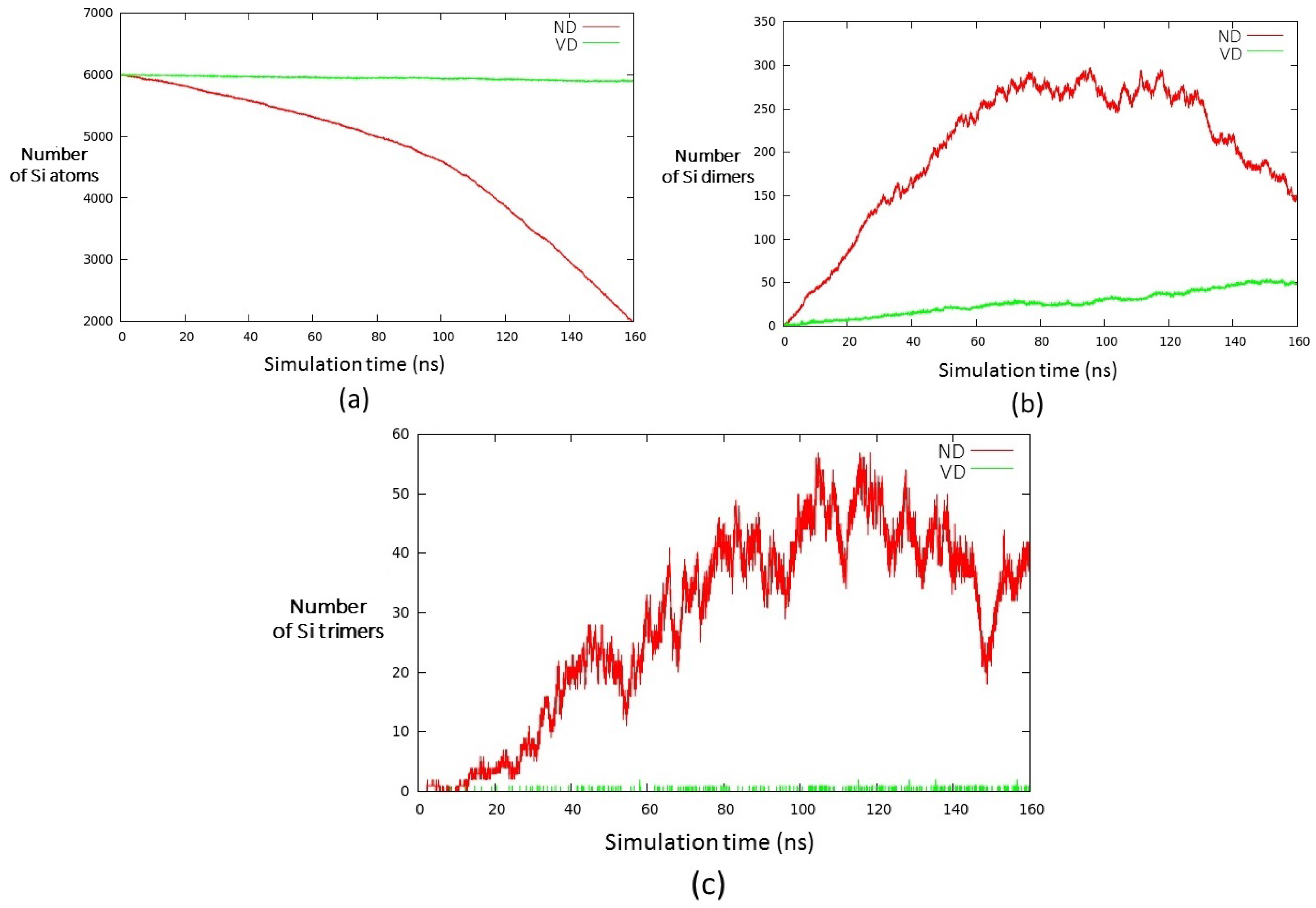
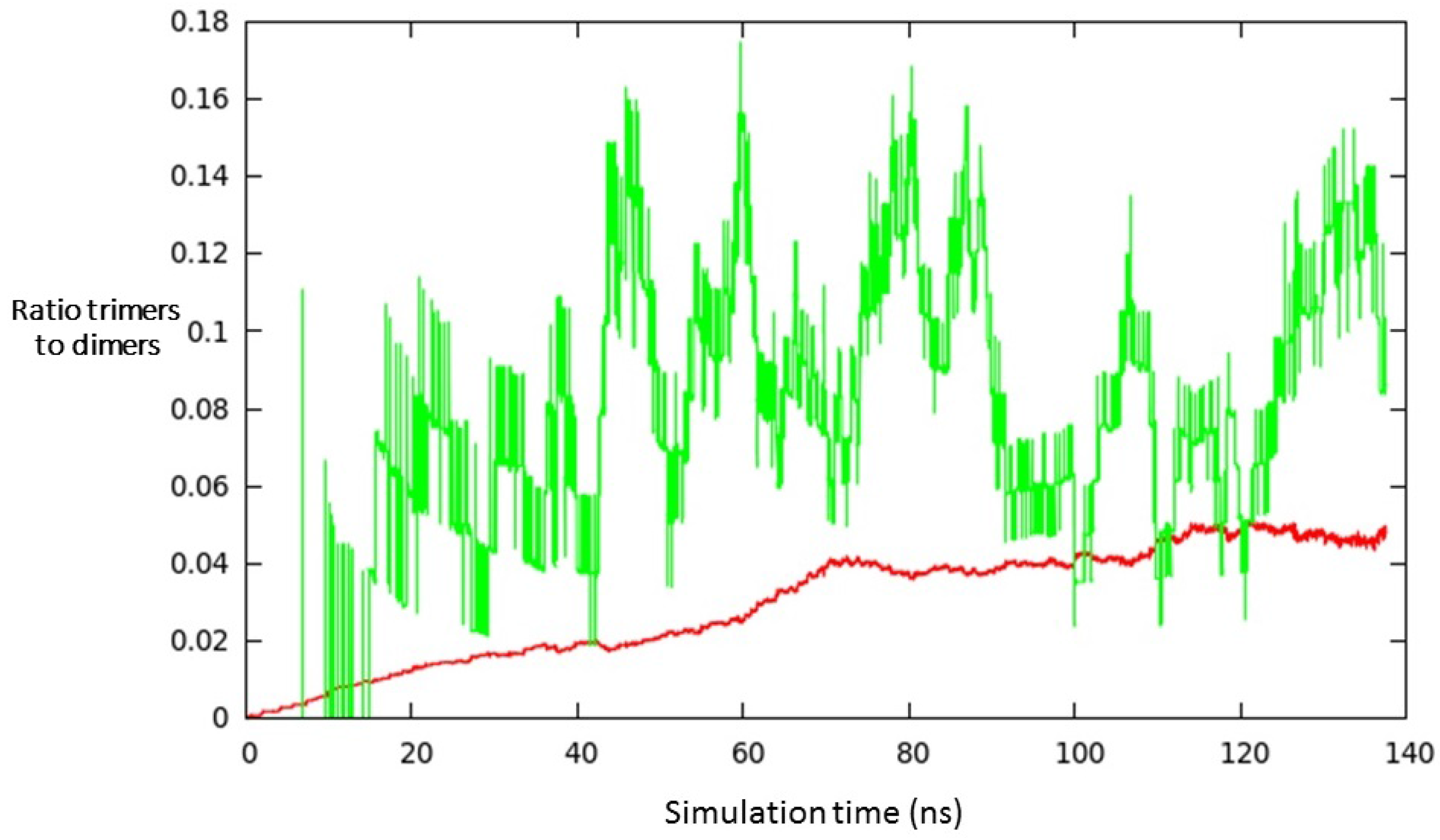
3. Results and Discussion
- (i)
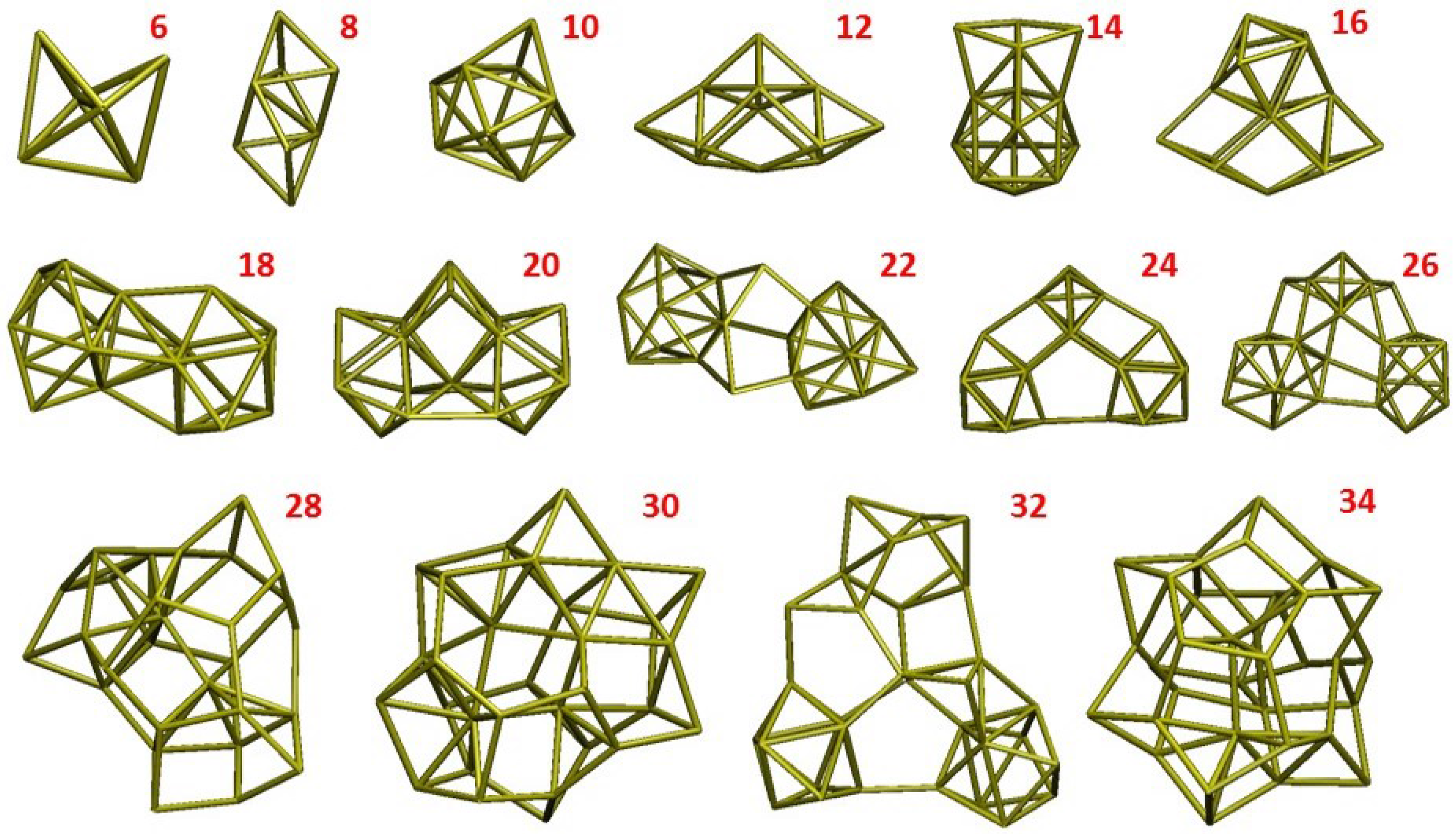
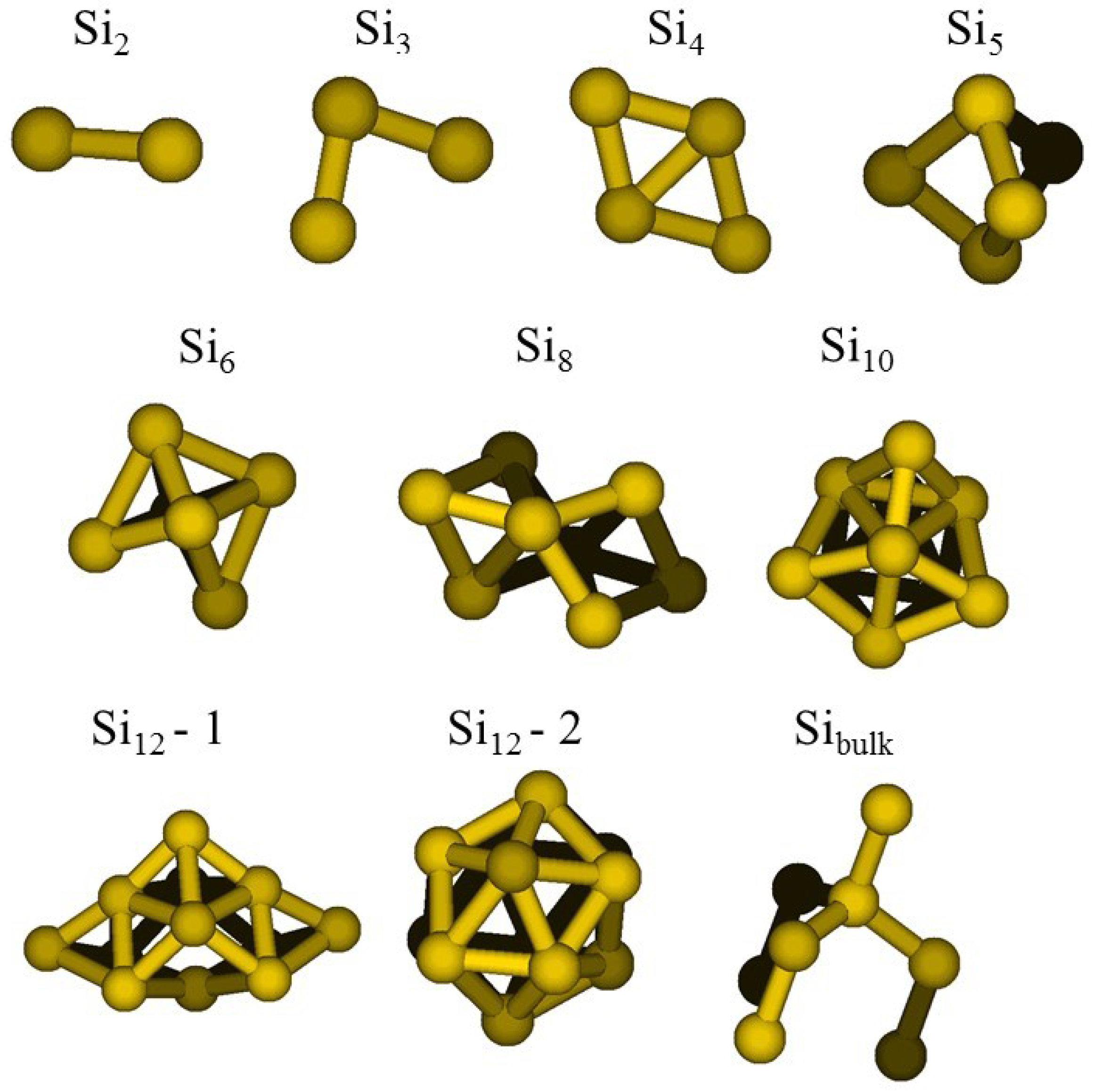
- a compact arrangement of atoms with coordination numbers and coordination angles much different from bulk silicon, where the coordination number is strictly four and the angles are perfectly tetrahedral; in the DFT-BH optimized structures, instead, the number of nearest neighbours can be as large as 6 and even 7, and the bond angles are remarkably different from tetrahedral;
- (ii)
- cluster surfaces are not smooth, but sharp with the presence of convex re-entrances, leaving the surface atoms with a very low coordination of one, or maximum two, neighbours.
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shigeta, M.; Murphys, A.B. Thermal plasmas for nanofabrication. J. Phys. D Appl. Phys. 2011, 44, 174025–174041. [Google Scholar] [CrossRef]
- Koirala, R.; Pratsinisa, S.E.; Baiker, A. Synthesis of catalytic materials in flames: Opportunities and challenges. Chem. Soc. Rev. 2016, 45, 3053–3068. [Google Scholar] [CrossRef] [PubMed]
- Schwade, B.; Roth, P. Simulation of nano-particle formation in a wall-heated aerosol reactor including coalescence. J. Aerosol Sci. 2003, 34, 339–357. [Google Scholar] [CrossRef]
- Wales, D.; Doye, P.K. Global Optimization by Basin-Hopping and the Lowest Energy Structures of Lennard-Jones Clusters Containing up to 110 Atoms. J. Phys. Chem. A 1997, 101, 5111–5116. [Google Scholar] [CrossRef]
- Van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: A Reactive Force Field for Hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- Kashchiev, D. Nucleation: Basic Theory with Applications; Butterworth-Heinemann: Oxford, UK, 2000. [Google Scholar]
- Newsome, D.A.; Sengupta, D.; Foroutan, H.; Russo, M.F.; van Duin, A.C. Oxidation of Silicon Carbide by O2 and H2O: A ReaxFF Reactive Molecular Dynamics Study, Part I. J. Phys. Chem. C 2012, 116, 16111–16121. [Google Scholar] [CrossRef]
- Han, Y.; Jiang, D.; Zhang, J.; Li, W.; Gan, Z.; Gu, J. Development, applications and challenges of ReaxFF reactive force field in molecular simulations. Front. Chem. Sci. Eng. 2016, 10, 16–38. [Google Scholar] [CrossRef]
- Li, C.; Monti, S.; Carravetta, V. Journey toward the Surface: How Glycine Adsorbs on Titania in Water Solution. J. Phys. Chem. C 2012, 116, 18318–18326. [Google Scholar] [CrossRef]
- Monti, S.; Li, C.; Carravetta, V. Reactive dynamics simulation of monolayer and multilayer adsorption of glycine on Cu(110). J. Phys. Chem. C 2013, 117, 5221–5228. [Google Scholar] [CrossRef]
- Carravetta, V.; Monti, S.; Li, C.; Ågren, H. Theoretical simulations of structure and X-ray photoelectron spectra of glycine and diglycine adsorbed on Cu(110). Langmuir 2013, 29, 10194–10204. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Monti, S.; Ågren, H.; Carravetta, V. Cysteine on TiO2(110): A theoretical study by reactive dynamics and photoemission spectra simulation. Langmuir 2014, 30, 8819–8828. [Google Scholar] [CrossRef] [PubMed]
- Monti, S.; Li, C.; Ågren, H.; Carravetta, V. Dropping a Droplet of Cysteine Molecules on a Rutile (110) Interface: Reactive versus Nonreactive Classical Molecular Dynamics Simulations. J. Phys. Chem. C 2015, 119, 6703–6712. [Google Scholar] [CrossRef]
- Monti, S.; Carravetta, V.; Ågren, H. Simulation of Gold Functionalization with Cysteine by Reactive Molecular Dynamics. J. Phys. Chem. Lett. 2016, 7, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, Á.M.; Mocellin, A.; Monti, S.; Li, C.; Marinho, R.R.T.; Medina, A.; Agren, H.; Carravetta, V.; de Brito, A.N. Surface-Altered Protonation Studied by Photoelectron Spectroscopy and Reactive Dynamics Simulations. J. Phys. Chem. Lett. 2015, 6, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Singh, V.; Grammatikopoulos, P.; Cassidy, C.; Kengo, A.; Sowwan, M.; Nordlund, K.; Djurabekova, F. Crystallization of silicon nanoclusters with inert gas temperature control. Phys. Rev. B 2015, 91, 35419–35431. [Google Scholar] [CrossRef]
- Zhao, J.; Baibuz, E.; Vernieres, J.; Grammatikopoulos, P.; Jansson, V.; Nagel, M.; Steinhauer, S.; Sowwan, M.; Kuronen, A.; Nordlund, K.; et al. Formation Mechanism of Fe Nanocubes by Magnetron Sputtering Inert Gas Condensation. ACS Nano 2016, 10, 4684–4694. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Cassidy, C.; Grammatikopoulos, P.; Djurabekova, F.; Nordlund, K.; Sowwan, M. Heterogeneous Gas-Phase Synthesis and Molecular Dynamics Modeling of Janus and Core-Satellite Si-Ag Nanoparticles. J. Phys. Chem. C 2014, 118, 13869–13875. [Google Scholar] [CrossRef]
- Janssens, G.O.A.; Baekelandt, B.G.; Toufar, H.; Mortier, W.J.; Schoonheydt, R.A. Comparison of Cluster and Infinite Crystal Calculations on Zeolites with the Electronegativity Equalization Method (EEM). J. Phys. Chem. 1995, 99, 3251–3258. [Google Scholar] [CrossRef]
- Mortier, W.J.; Ghosh, S.K.; Shankar, S. Electronegativity-equalization method for the calculation of atomic charges in molecules. J. Am. Chem. Soc. 1986, 108, 4315–4320. [Google Scholar] [CrossRef]
- Liu, B.; Lu, Z.Y.; Pan, B.; Wang, C.Z.; Ho, K.M.; Shvartsburg, A.A.; Jarrold, M.F. Ionization of medium-sized silicon clusters and the geometries of the cations. J. Chem. Phys. 1998, 109, 9401–9409. [Google Scholar] [CrossRef]
- Hudgins, R.R.; Imai, M.; Jarrold, M.F.; Dugourd, P. High-resolution ion mobility measurements for silicon cluster anions and cations. J. Chem. Phys. 1999, 111, 7865. [Google Scholar] [CrossRef]
- Bachels, T.; Schäfer, R. Binding energies of neutral silicon clusters. Chem. Phys. Lett. 2000, 324, 365–372. [Google Scholar] [CrossRef]
- Muller, J.; Liu, B.; Shvartsburg, A.A.; Ogut, S.; Chelikowsky, J.R.; Siu, K.W.M.; Ho, K.M.; Gantefor, G. Spectroscopic evidence for the tricapped trigonal prism structure of semiconductor clusters. Phys. Rev. Lett. 2000, 85, 1666–1669. [Google Scholar] [CrossRef] [PubMed]
- Kaxiras, E.; Jackson, K. Shape of Small Silicon Clusters. Phys. Rev. Lett. 1993, 71, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.M.; Shvartsburg, A.A.; Pan, B.; Lu, Z.Y.; Wang, C.Z.; Wacker, J.G.; Fye, J.L.; Jarrold, M.F. Structures of medium-sized silicon clusters. Nature 1998, 392, 582–585. [Google Scholar]
- Mitas, L.; Grossman, J.; Stich, I.; Tobik, J. Silicon Clusters of Intermediate Size: Energetics, Dynamics, and Thermal Effects. Phys. Rev. Lett. 2000, 84, 1479–1482. [Google Scholar] [CrossRef] [PubMed]
- Rata, I.; Shvartsburg, A.A.; Horoi, M.; Frauenheim, T.; Siu, K.W.M.; Jackson, K.A. Single-parent evolution algorithm and the optimization of Si clusters. Phys. Rev. Lett. 2000, 85, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, G.; Ding, F.; Lee, H.; Shen, W.; Zhao, J. Structural transition of Si clusters and their thermodynamics. Chem. Phys. Lett. 2001, 341, 529–534. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, Q.; Jena, P.; Rao, B.K.; Kawazoe, Y. Stabilization of Si60 Cage Structure. Phys. Rev. Lett. 2003, 90, 135503. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Zeng, X.C. Global geometry optimization of silicon clusters described by three empirical potentials. J. Chem. Phys. 2003, 119, 1442–1450. [Google Scholar] [CrossRef]
- Jackson, K.A.; Horoi, M.; Chaudhuri, I.; Frauenheim, T.; Shvartsburg, A.A. Unraveling the shape transformation in silicon clusters. Phys. Rev. Lett. 2004, 93, 013401. [Google Scholar] [CrossRef]
- Li, B.X. Stability of medium-sized neutral and charged silicon clusters. Phys. Rev. B Condens. Matter Mater. Phys. 2005, 71, 235311. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, X.; Wang, G.; Zhao, J. Optimally stuffed fullerene structures of silicon nanoclusters. Phys. Rev. B Condens. Matter Mater. Phys. 2005, 71, 113412. [Google Scholar] [CrossRef]
- Ona, O.; Bazterra, V.E.; Caputo, M.C.; Facelli, J.C.; Fuentealba, P.; Ferraro, M.B. Modified genetic algorithms to model cluster structures in medium-sized silicon clusters: Si18–Si60. Phys. Rev. A 2006, 73, 1–11. [Google Scholar] [CrossRef]
- Tereshchuk, P.L.; Khakimov, Z.M.; Umarova, F.T.; Swihart, M.T. Energetically competitive growth patterns of silicon clusters: Quasi-one-dimensional clusters versus diamond-like clusters. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 76, 125418. [Google Scholar] [CrossRef]
- Zhou, R.L.; Pan, B.C. Low-lying isomers of Si and Si (n = 31–50) clusters. J. Chem. Phys. 2008, 128, 234302. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Shao, N.; Zeng, X.C. Structures and relative stability of medium- and large-sized silicon clusters. VI. Fullerene cage motifs for low-lying clusters Si39, Si40, Si50, Si60, Si70, and Si80. J. Chem. Phys. 2008, 128, 104316. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density Functional Theory of Electronic Structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Metropolis, N.; Ulam, S. The Monte Carlo method. J. Am. Stat. Assoc. 1949, 44, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Barcaro, G.; Carravetta, V.; Monti, S.; Sementa, L. Force Field Parameters for Reactive Molecular Dynamics Simulation of Si Nanoparticles. J. Chem. Theory Comput. 2017. in preparation. [Google Scholar]
- LAMMPS Molecular Dynamics Simulator. Available online: http://lammps.sandia.gov (accessed on 12 February 2017).
- Sevast’yanov, V.G.; Nosatenko, P.Y.; Gorskii, V.V.; Ezhov, Y.S.; Sevast’yanov, D.V.; Simonenko, E.P.; Kuznetsov, N.T. Experimental and theoretical determination of the saturation vapor pressure of silicon in a wide range of temperatures. Russ. J. Inorg. Chem. 2010, 55, 2073–2088. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Si 25% | Si 25% | Si 25% | Si 50% | Si 50% | Si 50% | Si 75% | Si 75% | Si 75% | |
|---|---|---|---|---|---|---|---|---|---|
| System | N = 20 | N = 30 | N = 40 | N = 20 | N = 30 | N = 40 | N = 20 | N = 30 | N = 40 |
| T = 1800 K | 165 | 176 | 229 | 89 | 91 | 103 | 47 | 61 | 71 |
| T = 1900 K | 156 | 219 | 266 | 70 | 106 | 132 | 54 | 77 | 84 |
| T = 2000 K | 205 | 249 | 273 | 90 | 123 | 142 | 58 | 77 | 89 |
| T = 2100 K | 219 | 271 | 305 | 89 | 115 | 156 | 67 | 78 | 83 |
| T = 2200 K | 199 | 314 | 332 | 121 | 137 | 169 | 78 | 96 | 112 |
| T = 2300 K | 247 | 340 | 366 | 125 | 143 | 208 | 67 | 95 | 114 |
| T = 2400 K | 270 | 391 | 126 | 198 | 86 | 119 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barcaro, G.; Monti, S.; Sementa, L.; Carravetta, V. Atomistic Modelling of Si Nanoparticles Synthesis. Crystals 2017, 7, 54. https://doi.org/10.3390/cryst7020054
Barcaro G, Monti S, Sementa L, Carravetta V. Atomistic Modelling of Si Nanoparticles Synthesis. Crystals. 2017; 7(2):54. https://doi.org/10.3390/cryst7020054
Chicago/Turabian StyleBarcaro, Giovanni, Susanna Monti, Luca Sementa, and Vincenzo Carravetta. 2017. "Atomistic Modelling of Si Nanoparticles Synthesis" Crystals 7, no. 2: 54. https://doi.org/10.3390/cryst7020054
APA StyleBarcaro, G., Monti, S., Sementa, L., & Carravetta, V. (2017). Atomistic Modelling of Si Nanoparticles Synthesis. Crystals, 7(2), 54. https://doi.org/10.3390/cryst7020054