
On the Importance of Halogen–Halogen Interactions in the Solid State of Fullerene Halides: A Combined Theoretical and Crystallographic Study



Abstract
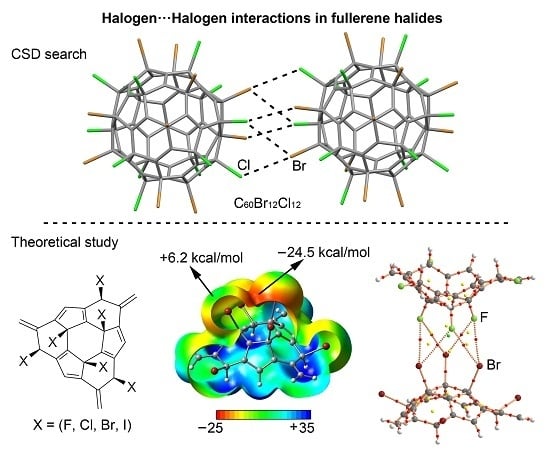
:
1. Introduction
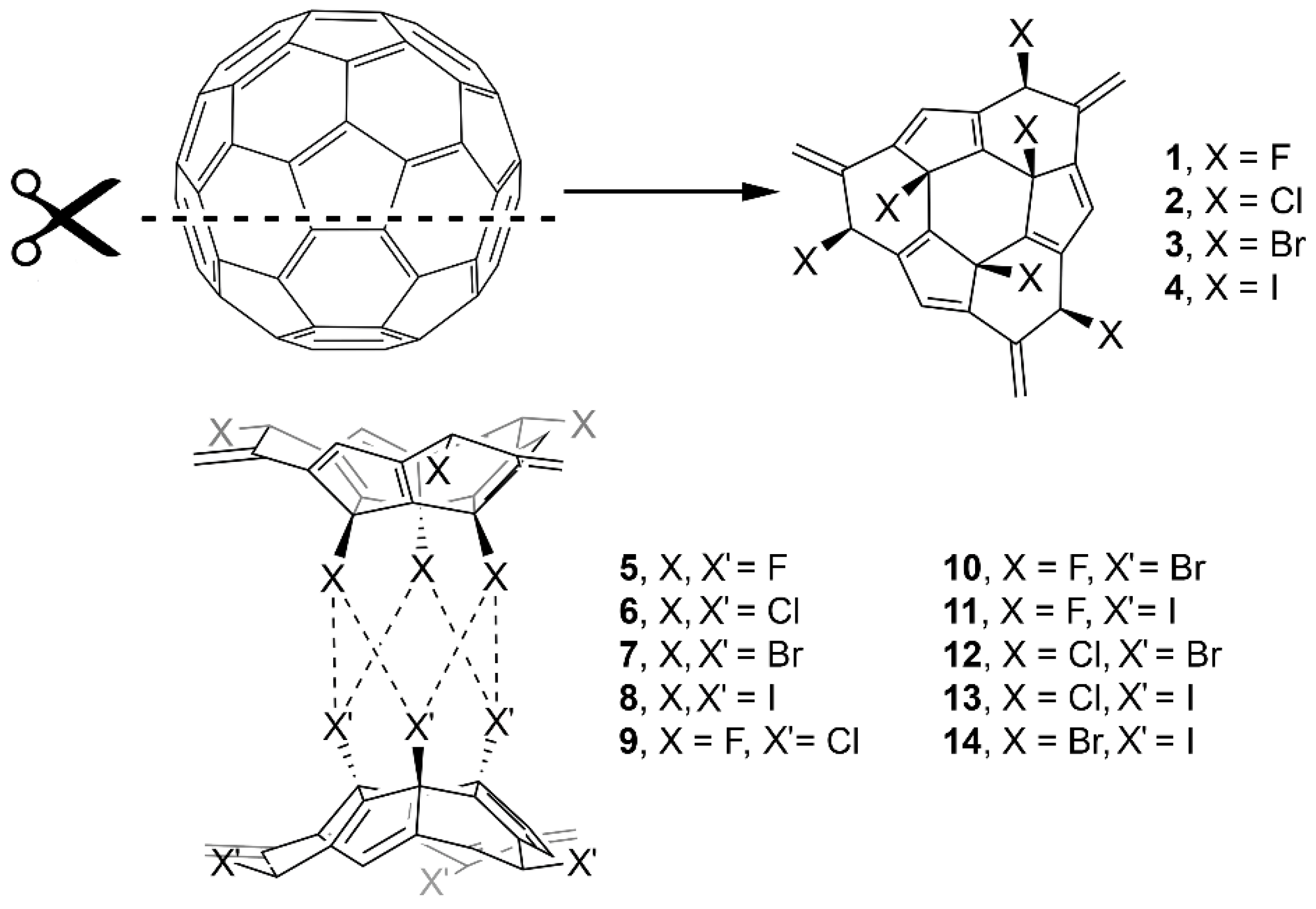
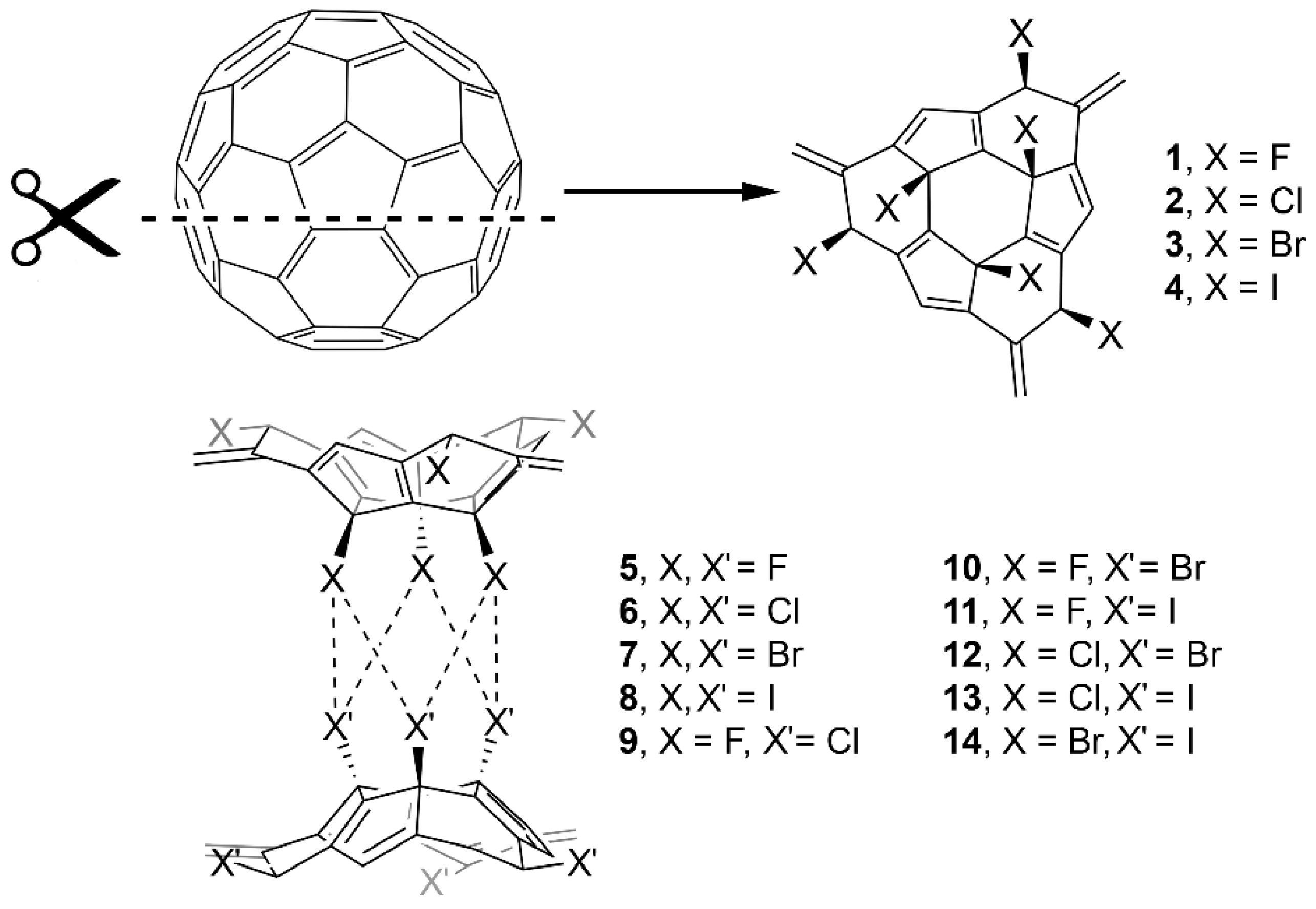
2. Results and Discussion
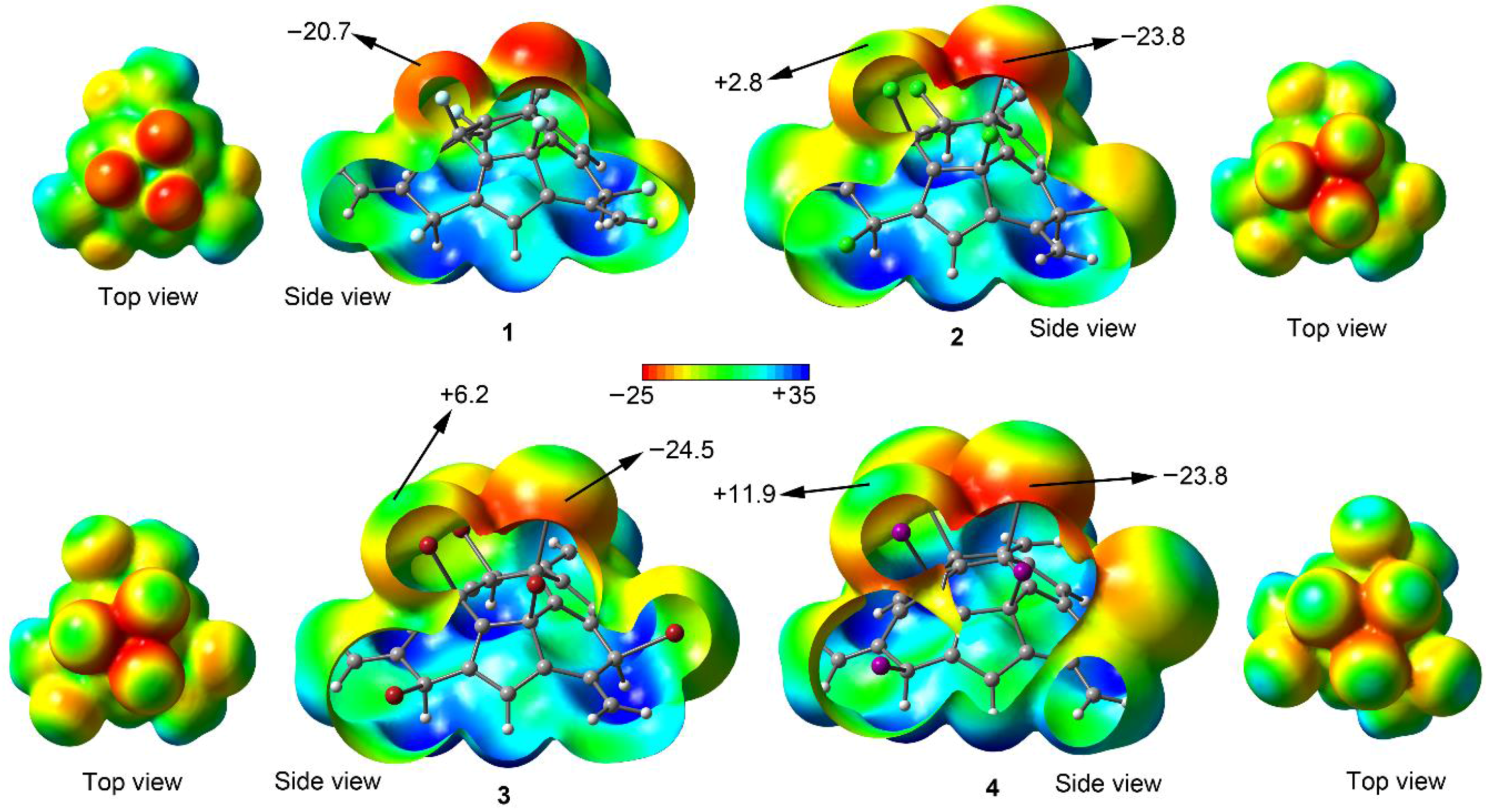
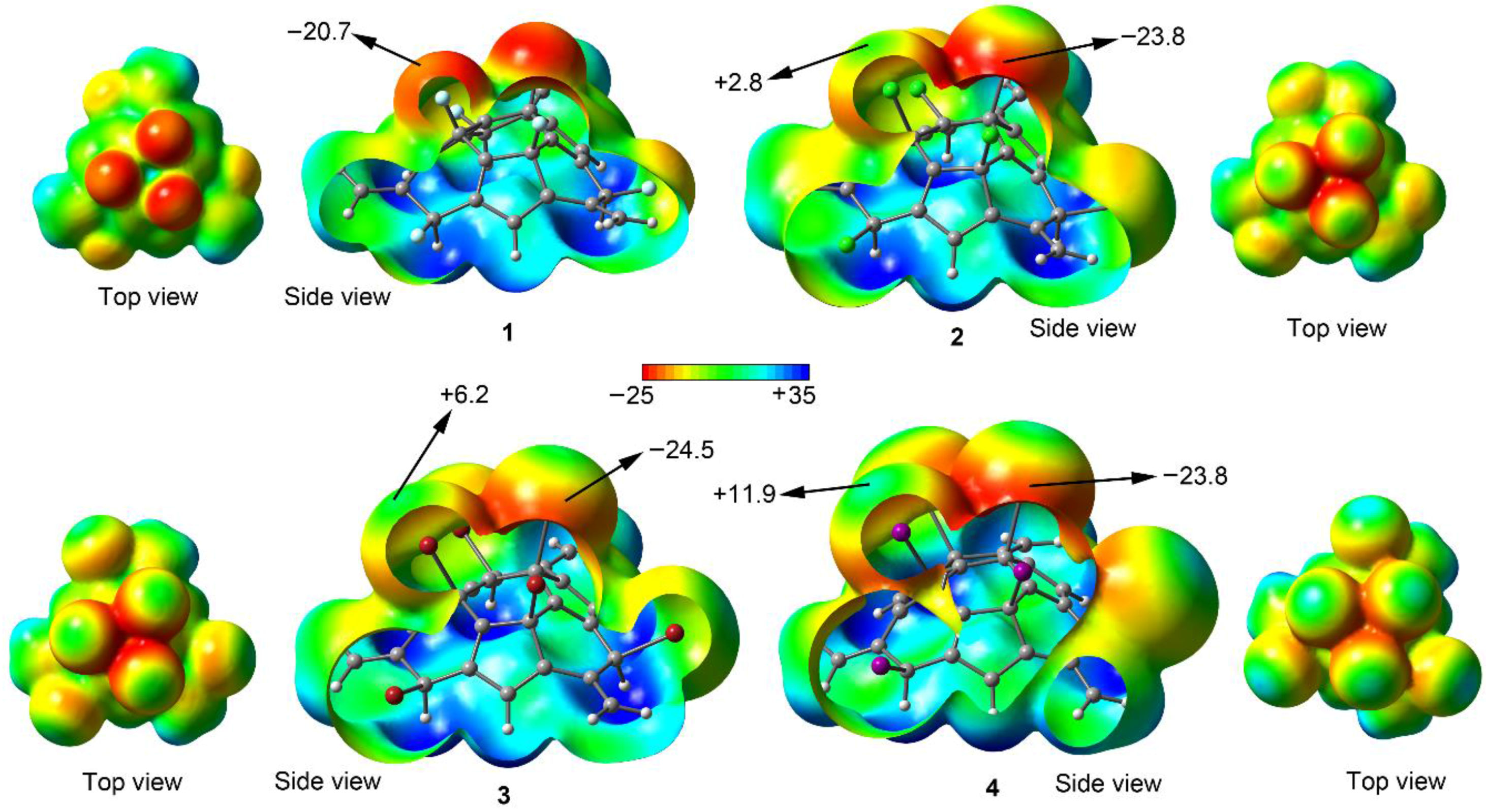
2.1. Preliminary MEP Analysis
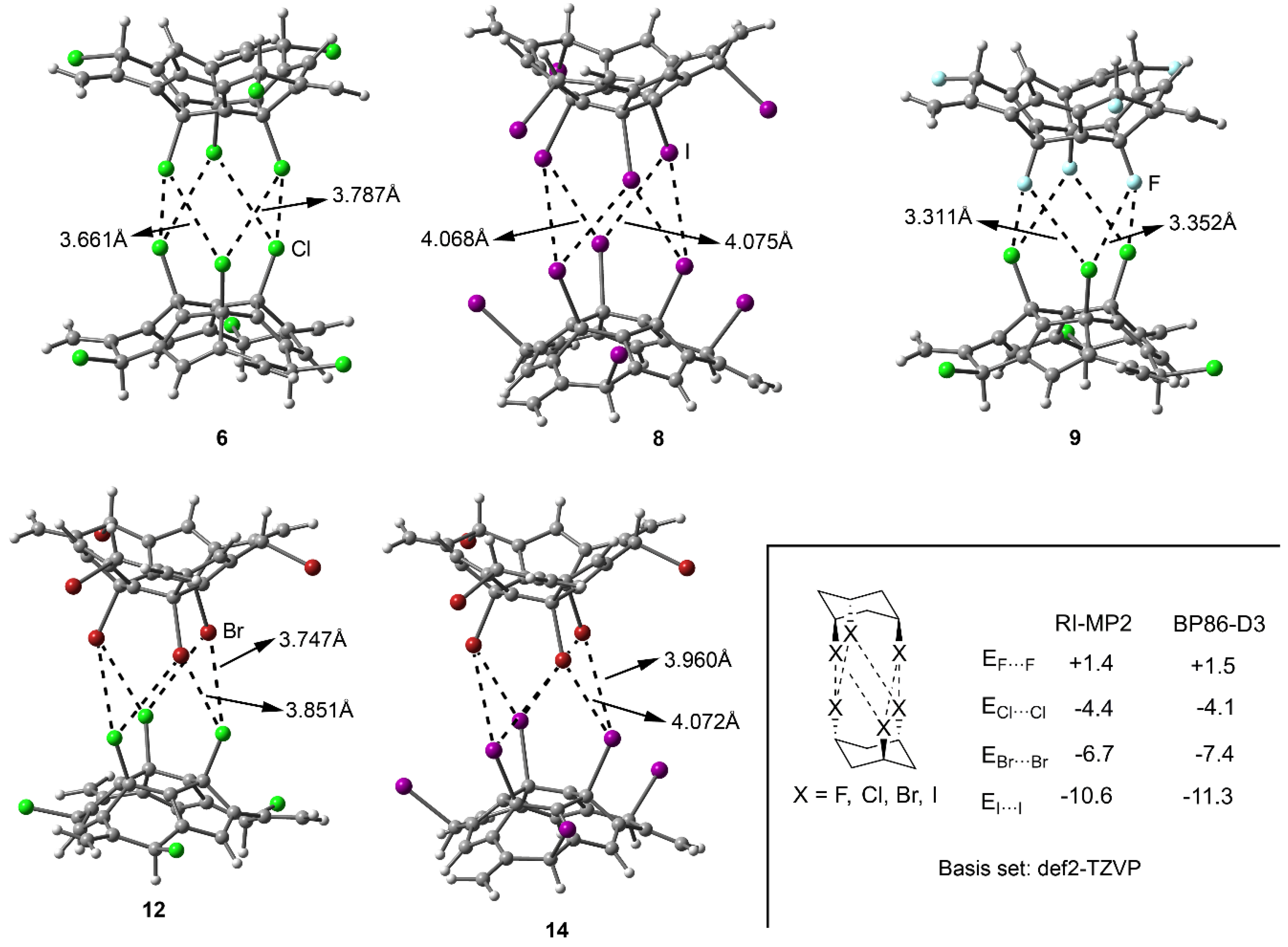
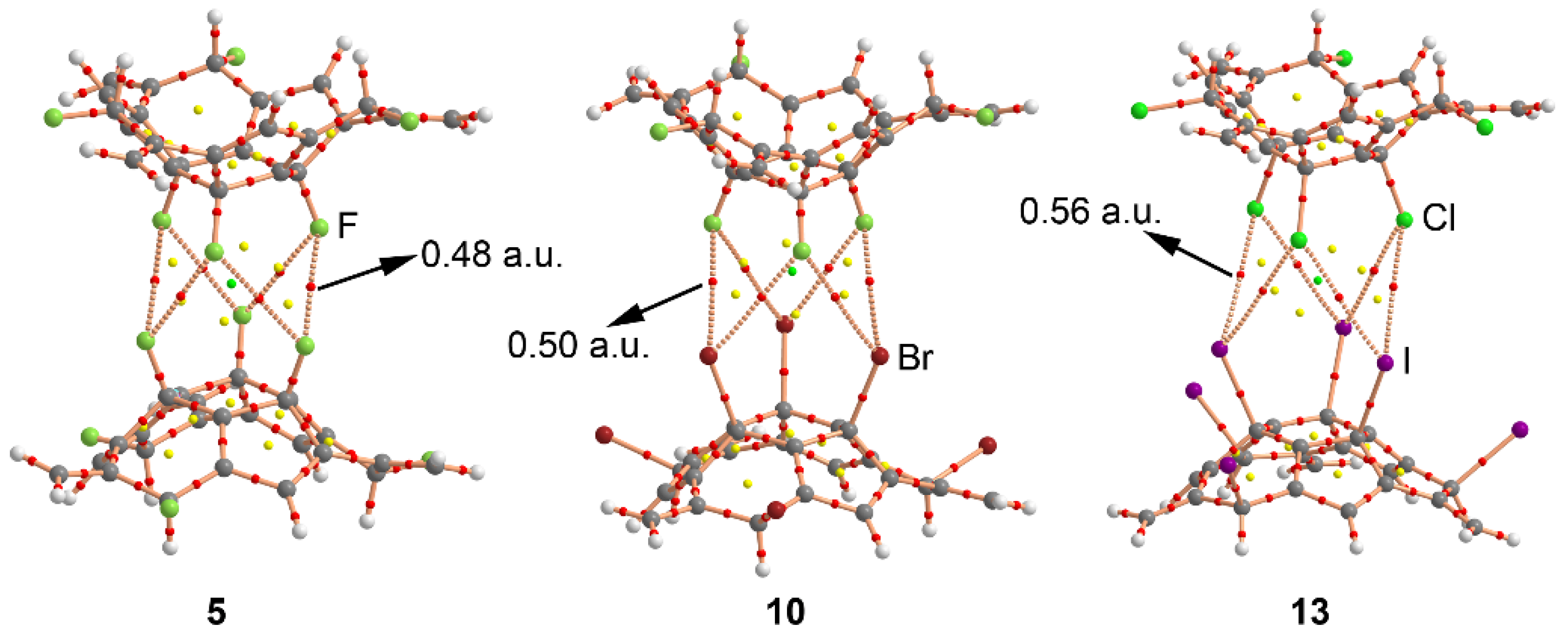
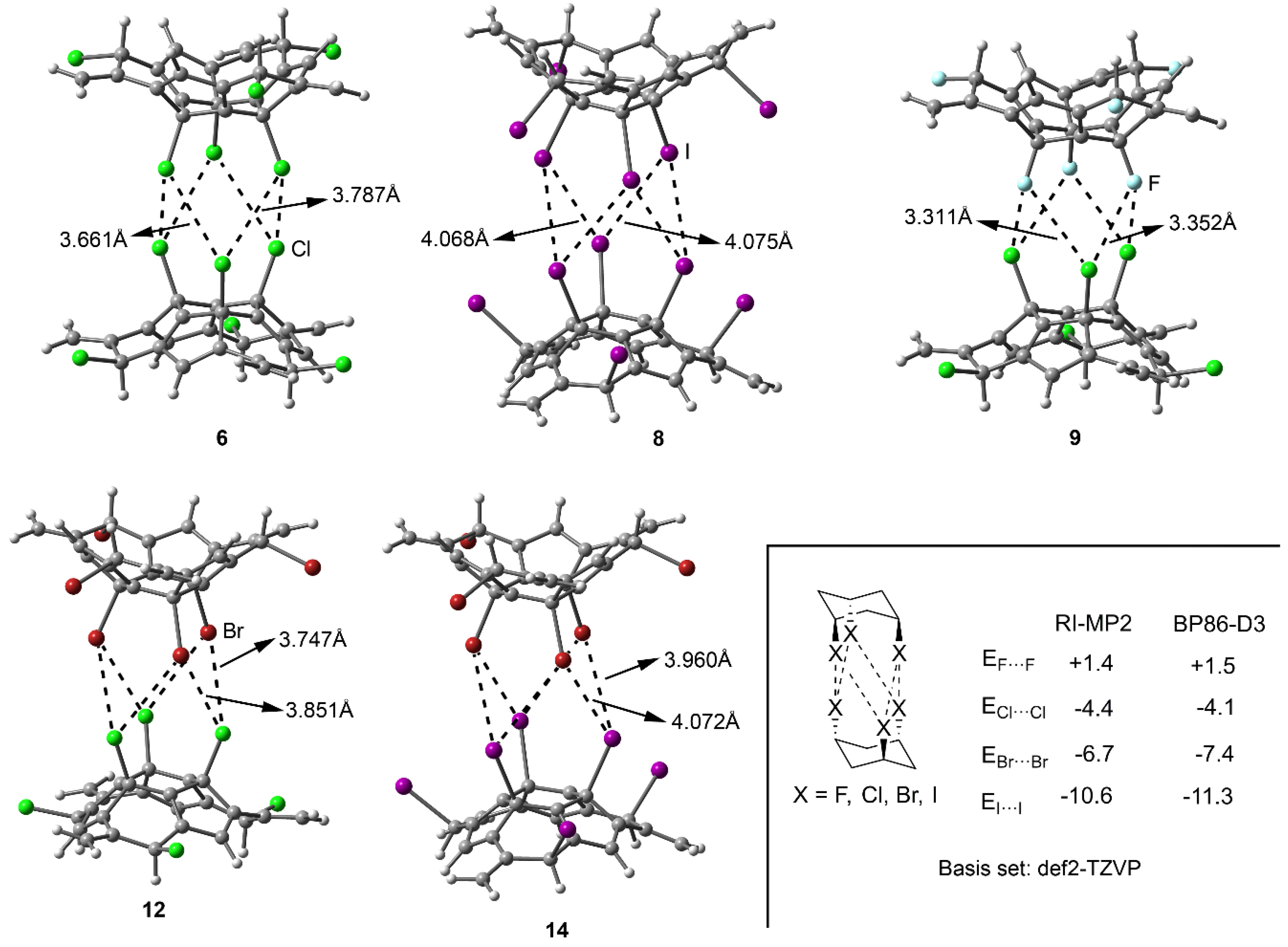
2.2. Energetic and Geometric Results
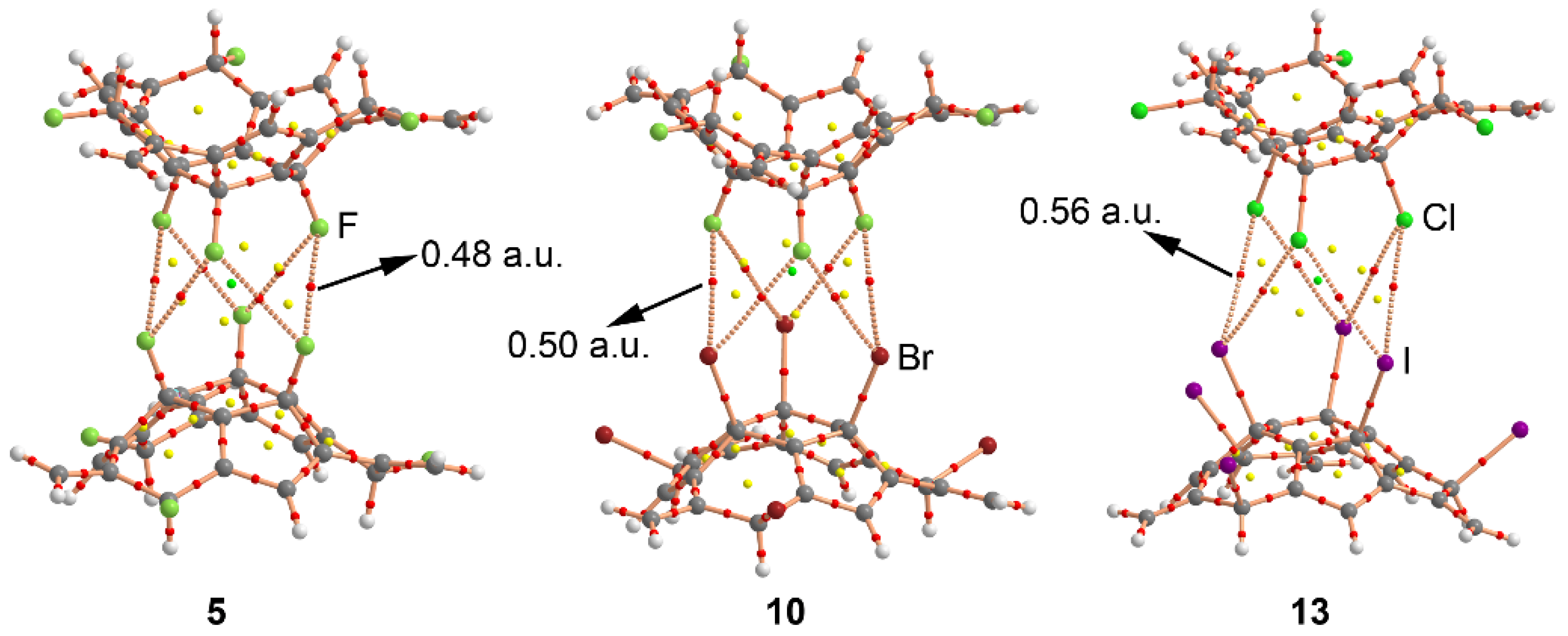
2.3. AIM and NBO Analyses
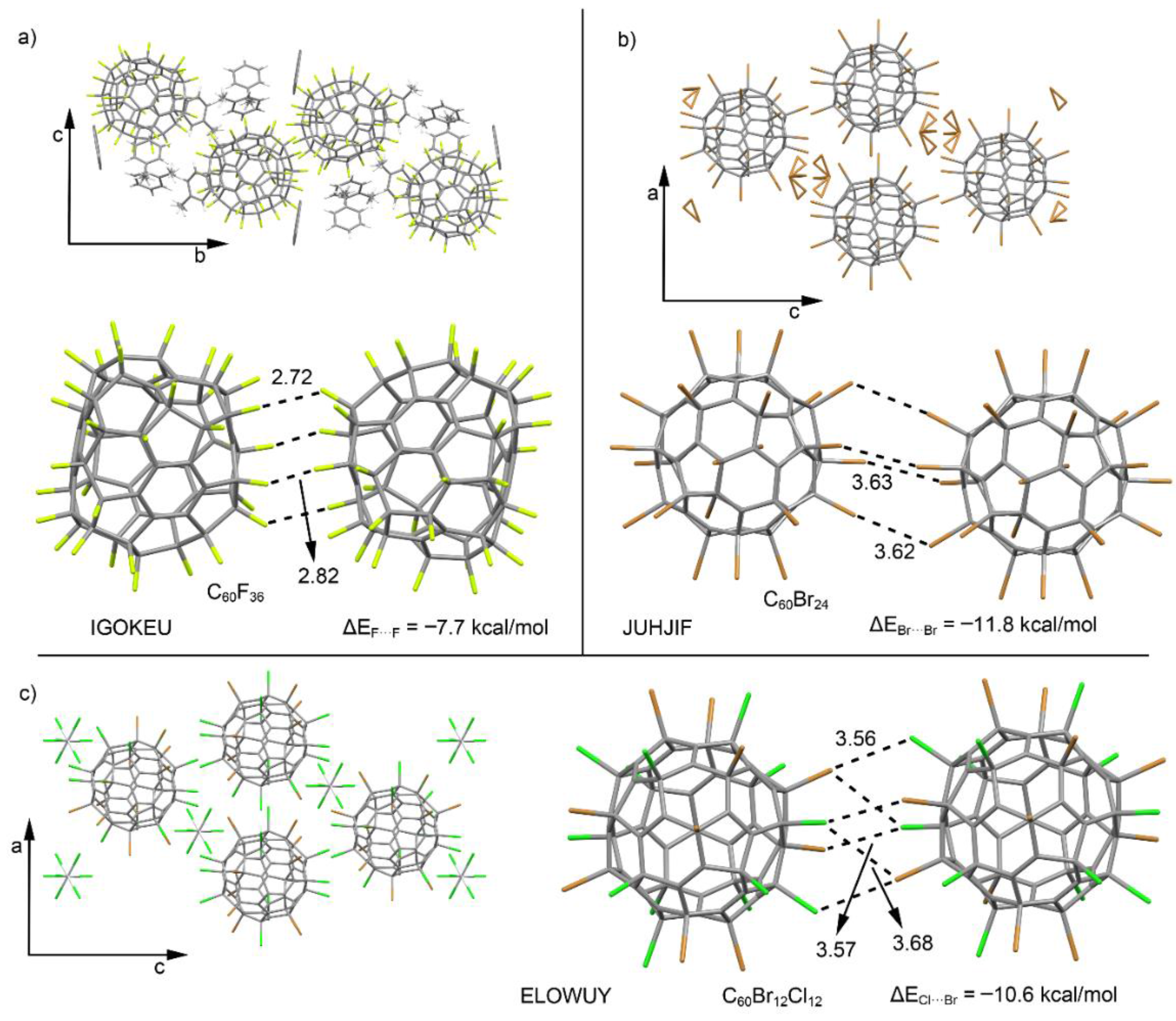
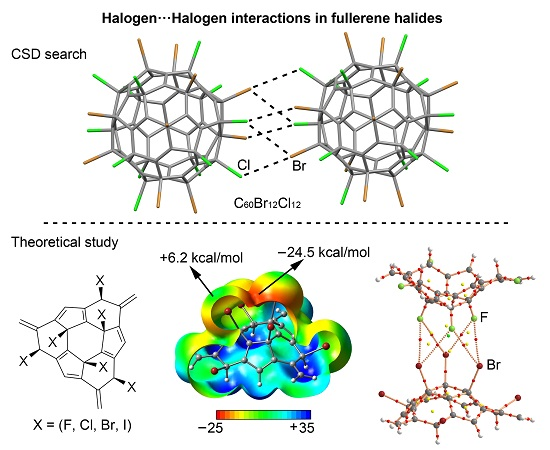
2.4. CSD Search
3. Theoretical Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| DFT | Density Functional Theory |
| AIM | Atoms in molecules |
| MEP | Molecular electrostatic potential |
| MP2 | Second order Moller-Plesset |
| BSSE | Basis Set Superposition Error |
| CSD | Cambridge Structural Database |
| CP | Critical point |
| NBO | Natural Bond Orbital |
| MINECO | Ministerio de Economía y Competitividad |
References
- Schneider, H.J. Binding mechanisms in supramolecular complexes. Angew. Chem. Int. Ed. 2009, 48, 3924–3977. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.J.; Yatsimirski, A. Principles and Methods in Supramolecular Chemistry; Wiley: Chichester, UK, 2000. [Google Scholar]
- Lehn, J.M. Supramolecular Chemistry Concepts and Perspectives; Wiley-VCH: Weinheim, Germany, 1995. [Google Scholar]
- Vögtle, F. Supramolecular Chemistry: An Introduction; Wiley: New York, NY, USA, 1993. [Google Scholar]
- Beer, P.D.; Gale, P.A.; Smith, D.K. Supramolecular Chemistry; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Steed, J.W.; Atwood, J.L. Supramolecular Chemistry; Wiley: Chichester, UK, 2000. [Google Scholar]
- Grabowski, S.J. What is the covalency of hydrogen bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Resnati, G. Halogen bonding: A paradigm in supramolecular chemistry. Chem. Eur. J. 2001, 7, 2511–2519. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Halogen Bonding: Where we are and where we are going. Cryst. Growth Des. 2012, 12, 5835–5838. [Google Scholar] [CrossRef]
- Metrangolo, P.; Neukirch, H.; Pilati, T.; Resnati, G. Halogen bonding based recognition processes: A world parallel to hydrogen bonding. Acc. Chem. Res. 2005, 38, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Resnati, G.; Pilati, T.; Biella, S. Halogen Bonding: Fundamentals and Applications; Springer: Berlin, Germany, 2008. [Google Scholar]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Meyer, F.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen bonding in supramolecular chemistry. Angew. Chem. Int. Ed. 2008, 47, 6114–6127. [Google Scholar] [CrossRef] [PubMed]
- Bertani, R.; Metrangolo, P.; Moiana, A.; Perez, E.; Pilati, T.; Resnati, G.; Rico-Lattes, I.; Sassi, A. Supramolecular route to fluorinated coatings: Self-assembly between poly(4-vinylpyridines) and haloperfluorocarbons. Adv. Mater. 2002, 14, 1197–1201. [Google Scholar] [CrossRef]
- Nguyen, H.L.; Horton, P.N.; Hursthouse, M.B.; Legon, A.C.; Bruce, D.W. Halogen bonding: A new interaction for liquid crystal formation. J. Am. Chem. Soc. 2003, 126, 16–17. [Google Scholar] [CrossRef] [PubMed]
- Praesang, C.; Nguyen, H.L.; Horton, P.N.; Whitwood, A.C.; Bruce, D.W. Trimeric liquid crystals assembled using both hydrogen and halogen bonding. Chem. Commun. 2008, 6164–6166. [Google Scholar] [CrossRef] [PubMed]
- Präsang, C.; Whitwood, A.C.; Bruce, D.W. Halogen-bonded cocrystals of 4-(N,N-Dimethylamino)pyridine with fluorinated iodobenzenes. Cryst. Growth Des. 2009, 9, 5319–5326. [Google Scholar] [CrossRef]
- Roper, L.C.; Prasang, C.; Kozhevnikov, V.N.; Whitwood, A.C.; Karadakov, P.B.; Bruce, D.W. Experimental and theoretical study of halogen-bonded complexes of DMAP with Di- and triiodofluorobenzenes. A complex with a very short N···I halogen bond. Cryst. Growth Des. 2010, 10, 3710–3720. [Google Scholar] [CrossRef]
- Murrayrust, P.; Motherwell, W.D.S. Computer retrieval and analysis of molecular geometry. 4. Intermolecular interactions. J. Am. Chem. Soc. 1979, 101, 4374–4376. [Google Scholar] [CrossRef]
- Bauzá, A.; Quiñonero, D.; Deyà, P.M.; Frontera, A. Halogen bonding versus chalcogen and pnicogen bonding: A combined Cambridge structural database and theoretical study. CrystEngComm 2013, 15, 3137–3144. [Google Scholar] [CrossRef]
- Brown, A.; Beer, P.D. Halogen bonding anion recognition. Chem. Commun. 2016, 52, 8645–8658. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. Halogen bonding: An interim discussion. ChemPhysChem 2013, 14, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Phys. Chem. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. Supramolecular nanotubes based on halogen bonding interactions: Cooperativity and interaction with small guests. Phys. Chem. Chem. Phys. 2017, 19, 12936–12941. [Google Scholar] [CrossRef] [PubMed]
- Awwadi, F.; Willett, R.; Peterson, K.; Twamley, B. The nature of halogen⋅⋅⋅halogen synthons: crystallographic and theoretical studies. Chem. Eur. J. 2006, 12, 8952–8960. [Google Scholar] [CrossRef] [PubMed]
- Trokowski, R.; Akine, S.; Nabeshima, T. Remarkably selective recognition of iodobenzene derivatives by a macrocyclic bis-PtII metallohost. Chem. Eur. J. 2011, 17, 14420–14428. [Google Scholar] [CrossRef] [PubMed]
- Neve, F.; Crispini, A. N,N′-Dodecamethylene-bis(pyridinium) goes lamellar. Role of C–H⋯I, C–H⋯M, and I⋯I interactions in the crystal structure of its hexaiododipalladate(II) derivative. CrystEngComm 2003, 5, 265–268. [Google Scholar] [CrossRef]
- Felsmann, M.; Eissmann, F.; Schwarzer, A.; Weber, E. Competitive interactions in the crystal structures of benzils effected by different halogen substitution. Cryst. Growth Des. 2011, 11, 982–989. [Google Scholar] [CrossRef]
- Saha, B.K.; Nangia, A.; Nicoud, J.F. Using halogen···halogen interactions to direct concentrosymmetric crystal packing in dipolar organic molecules. Cryst. Growth Des. 2006, 6, 1278–1281. [Google Scholar] [CrossRef]
- Yamada, M.; Kanazawa, R.; Hamada, F. Halogen–halogen interactions and halogen bonding in thiacalixarene systems. CrystEngComm 2014, 16, 2605–2614. [Google Scholar] [CrossRef]
- Baldrighi, M.; Bartesaghi, D.; Cavallo, G.; Chierotti, M.R.; Bogetto, R.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Polymorphs and co-crystals of haloprogin: an antifungal agent. CrystEngComm 2014, 16, 5897–5904. [Google Scholar] [CrossRef]
- Ochoa, M.E.; Aguilar-Granda, A.; Ramirez-Montes, P.I.; Barba, V.; López, Y.; Santillan, R.; Farfán, N. Designed synthesis of “L” shaped 17-halo-aryl-ethynyl steroids. CrystEngComm 2016, 18, 6830–6840. [Google Scholar] [CrossRef]
- Reddy, C.M.; Kirchner, M.T.; Gundakaram, R.C.; Padmanabhan, K.A.; Desiraju, G.R. Isostructurality, polymorphism and mechanical properties of some hexahalogenated benzenes: the nature of halogen⋅⋅⋅halogen interactions. Chem. Eur. J. 2006, 12, 2222–2234. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.; Roy, S.; Naskar, K.; Sinha, C.; Alam, S.M.; Kundu, S.; Vittal, J.J.; Mir, M.H. Halogen···halogen interactions in the supramolecular assembly of 2D coordination polymers and the CO2 sorption behavior. Cryst. Growth Des. 2016, 16, 5514–5519. [Google Scholar] [CrossRef]
- Mukherjee, A.; Desiraju, G.R. Halogen bonds in some dihalogenated phenols: applications to crystal engineering. IUCrJ 2014, 1, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R. Lecture Notes on Fullerene Chemistry, Imper; College Press: London, UK, 1999; p. 268. [Google Scholar]
- Birkett, P.R.; Hitchcock, P.B.; Kroto, H.W.; Taylor, R.; Walton, D.R.M. Preparation and characterization of C60Br6 and C60Br8. Nature 1992, 357, 479–481. [Google Scholar] [CrossRef]
- Tebbe, F.N.; Harlow, R.L.; Chase, D.B.; Thorn, D.L.; Campbell, G.C.; Calabrese, J.C.; Herron, N.; Young, R.J.; Wasserman, E. Synthesis and single-crystal X-ray structure of a highly symmetrical C60 derivative, C60Br24. Science 1992, 256, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Birkett, P.R.; Avent, A.G.; Darwish, A.D.; Kroto, H.W.; Taylor, R.; Walton, D.R.M. Preparation and 13C NMR spectroscopic characterization of C60Cl6. J. Chem. Soc. Chem. Commun. 1993, 1230–1232. [Google Scholar] [CrossRef]
- Birkett, P.R.; Avent, A.G.; Darwish, A.D.; Kroto, H.W.; Taylor, R.; Walton, D.R.M. Formation and characterization of C70Cl10. J. Chem. Soc. Chem. Commun. 1995, 683–684. [Google Scholar] [CrossRef]
- Taylor, R. Progress in fullerene fluorination. Russ. Chem. Bull. 1998, 47, 823–832. [Google Scholar] [CrossRef]
- Taylor, R. Fluorinated fullerenes. Chem. Eur. J. 2001, 7, 4074–4083. [Google Scholar] [CrossRef]
- Boltalina, O.V. Fluorination of Fullerenes and their derivatives. J. Fluorine Chem. 2000, 101, 273–278. [Google Scholar] [CrossRef]
- Sidorov, L.N.; Boltalina, O.V. Endohedral metal derivatives and exohedral fluorine derivatives of fullerenes. Russ. Chem. Rev. 2002, 71, 535–561. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Avent, A.G.; Clare, B.W.; Hitchcock, P.B.; Kepert, D.L.; Taylor, R. C60F36: There is a third isomer and it has C1 symmetry. Chem. Commun. 2002, 2370–2371. [Google Scholar] [CrossRef]
- Troyanov, S.I.; Burtsev, A.V.; Kemnitz, E. Synthesis and structure of fullerene halides in the C60-(TiCl4 + Br2) system: Molecular structures of (C60Cl5)2, C60X6, C60X8, and C60X24 (X = Cl, Br). Kristallografiya 2009, 54, 268–275. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Hacer, M.; Horn, H.; Kömel, C. Electronic structure calculations on workstation computers: The program system Turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.B.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar]
- AIMAll. Version 13.05.06. TK Gristmill Software: Overland Park, KS, USA, 2013. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision B.01, Guassian Inc.: Wallingford, CT, USA, 2009.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | ΔE | R1 | R2 | 102 x ρ |
|---|---|---|---|---|
| 5 (F···F) | +0.7 | 2.988 | 3.155 | 0.48 |
| 6 (Cl···Cl) | –3.5 | 3.661 | 3.787 | 0.49 |
| 7 (Br···Br) | –6.9 | 3.856 | 3.907 | 0.55 |
| 8 (I···I) | –11.7 | 4.068 | 4.175 | 0.67 |
| 9 (F···Cl) | –1.0 | 3.311 | 3.512 | 0.49 |
| 10 (F···Br) | –1.9 | 3.836 | 3.548 | 0.50 |
| 11 (F···I) | –3.0 | 3.504 | 3.630 | 0.59 |
| 12 (Cl···Br) | –5.0 | 3.747 | 3.851 | 0.53 |
| 13 (Cl···I) | –6.3 | 3.880 | 3.985 | 0.56 |
| 14 (Br···I) | –8.6 | 3.960 | 4.072 | 0.61 |
| Complex | Donora | Acceptor | E(2) |
|---|---|---|---|
| 5 | –b | –b | –b |
| 6 | LP Cl | BD* C–Cl | 1.50 |
| 7 | LP Br | BD* C–Br | 3.18 |
| 8 | LP I | BD* C–I | 5.88 |
| 9 | LP F LP Cl | BD* C–Cl BD* C–F | 0.15 0.27 |
| 10 | LP Br | BD* C–F | 0.54 |
| 11 | LP F LP I | BD* C–I BD* C–F | 0.39 0.77 |
| 12 | LP Cl LP Br | BD* C–Br BD* C–Cl | 2.40 1.05 |
| 13 | LP Cl LP I | BD* C–I BD* C–Cl | 1.35 1.56 |
| 14 | LP Br LP I | BD* C–I BD* C–Br | 1.95 2.34 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauzá, A.; Frontera, A. On the Importance of Halogen–Halogen Interactions in the Solid State of Fullerene Halides: A Combined Theoretical and Crystallographic Study. Crystals 2017, 7, 191. https://doi.org/10.3390/cryst7070191
Bauzá A, Frontera A. On the Importance of Halogen–Halogen Interactions in the Solid State of Fullerene Halides: A Combined Theoretical and Crystallographic Study. Crystals. 2017; 7(7):191. https://doi.org/10.3390/cryst7070191
Chicago/Turabian StyleBauzá, Antonio, and Antonio Frontera. 2017. "On the Importance of Halogen–Halogen Interactions in the Solid State of Fullerene Halides: A Combined Theoretical and Crystallographic Study" Crystals 7, no. 7: 191. https://doi.org/10.3390/cryst7070191
APA StyleBauzá, A., & Frontera, A. (2017). On the Importance of Halogen–Halogen Interactions in the Solid State of Fullerene Halides: A Combined Theoretical and Crystallographic Study. Crystals, 7(7), 191. https://doi.org/10.3390/cryst7070191