
Dual Inhibition of AChE and BChE with the C-5 Substituted Derivative of Meldrum’s Acid: Synthesis, Structure Elucidation, and Molecular Docking Studies

 ,
,  ,
,  , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
2.1. NMR Studies
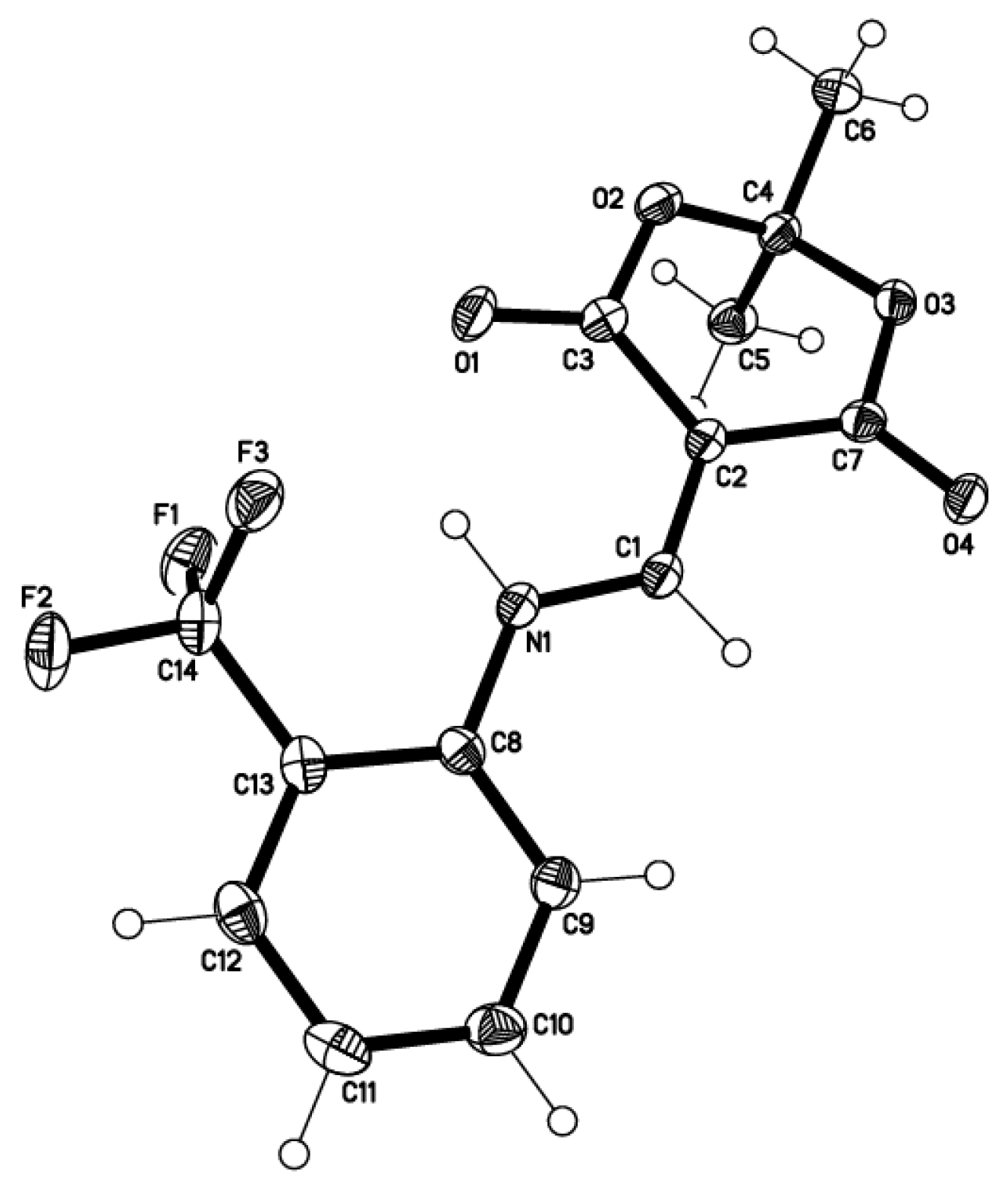
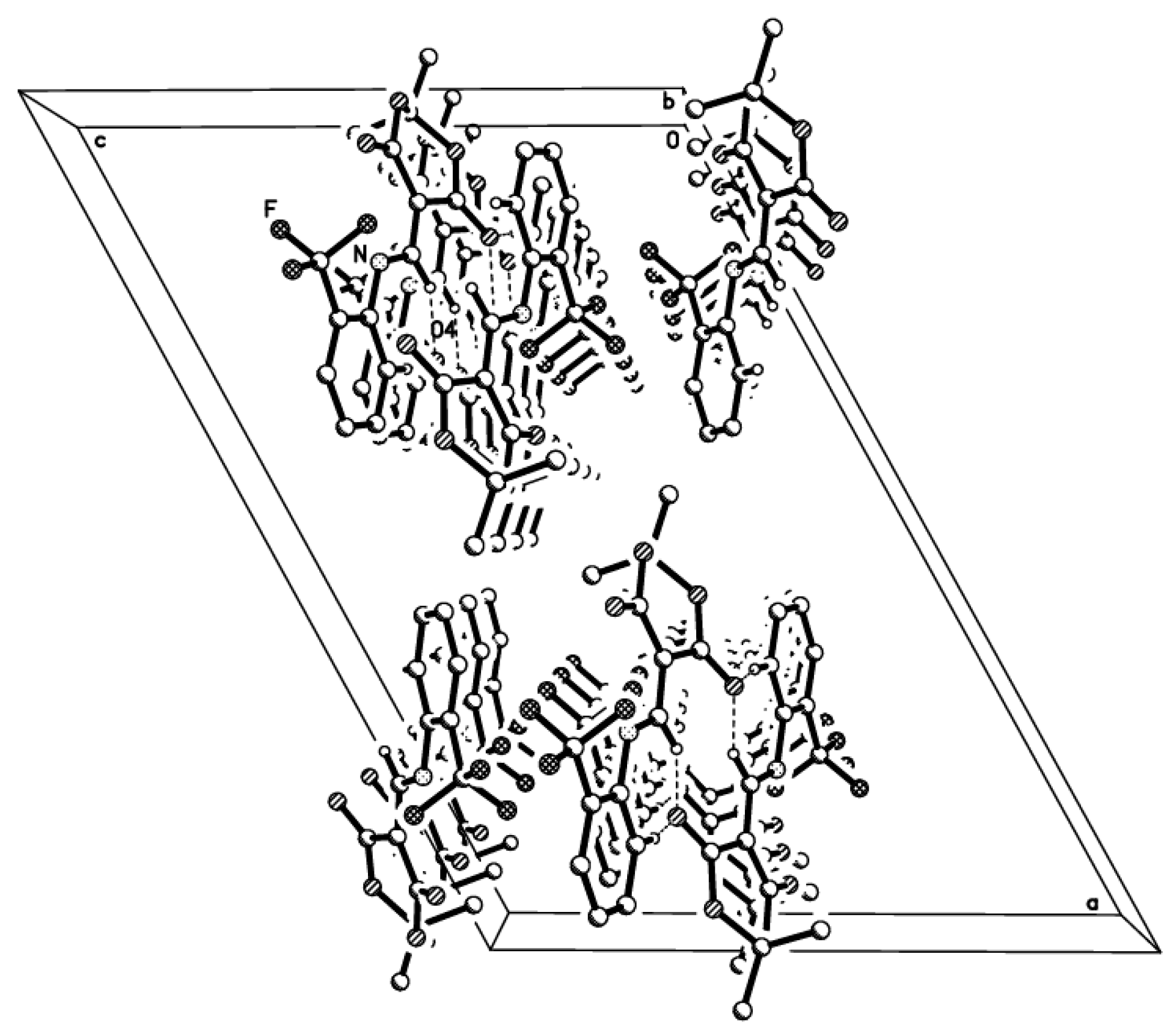
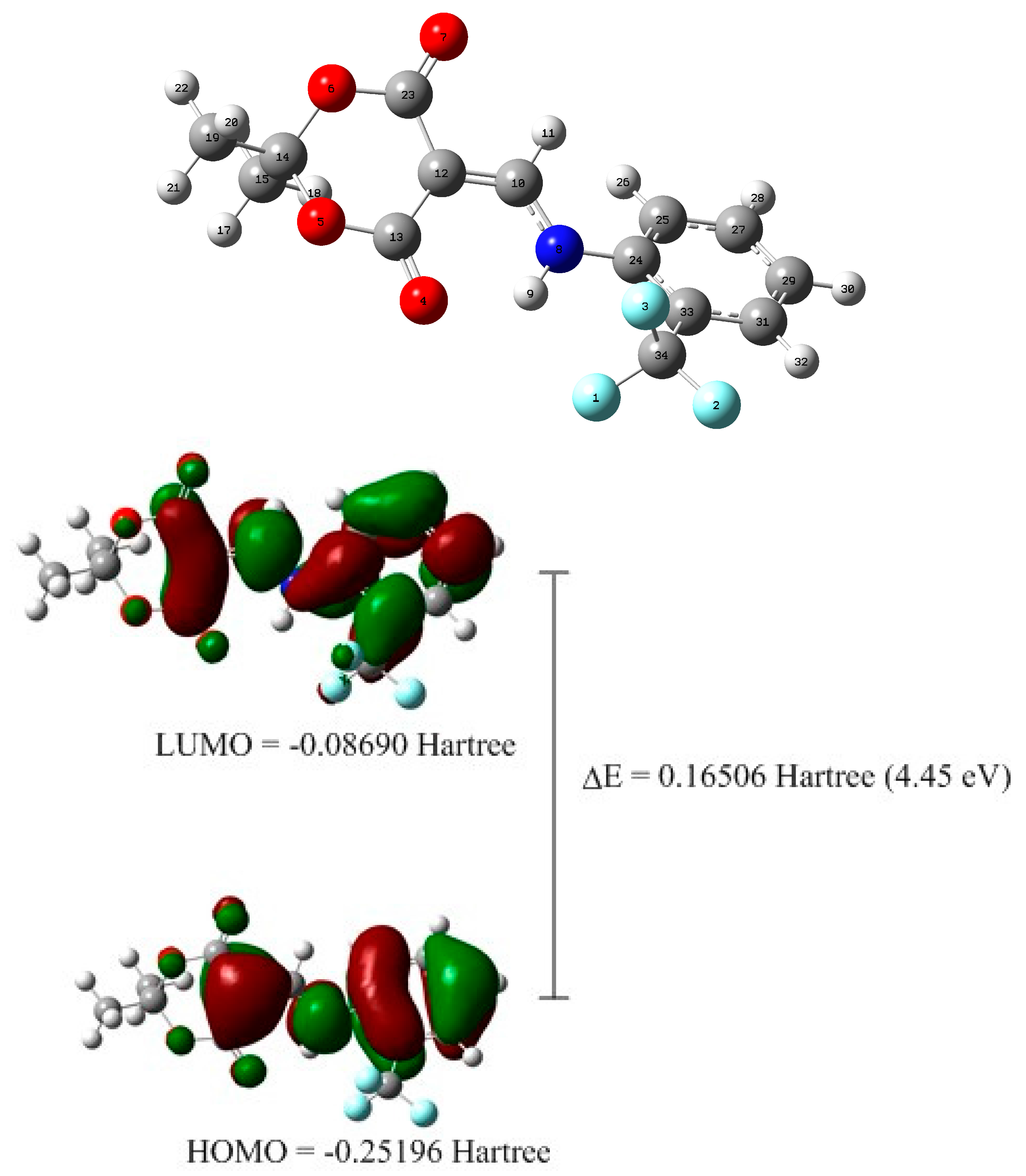
2.2. X-ray Structural Study and Theoretical Calculations
2.3. Acetylcholinesterase and Butyrylcholinesterase Inhibition Assay
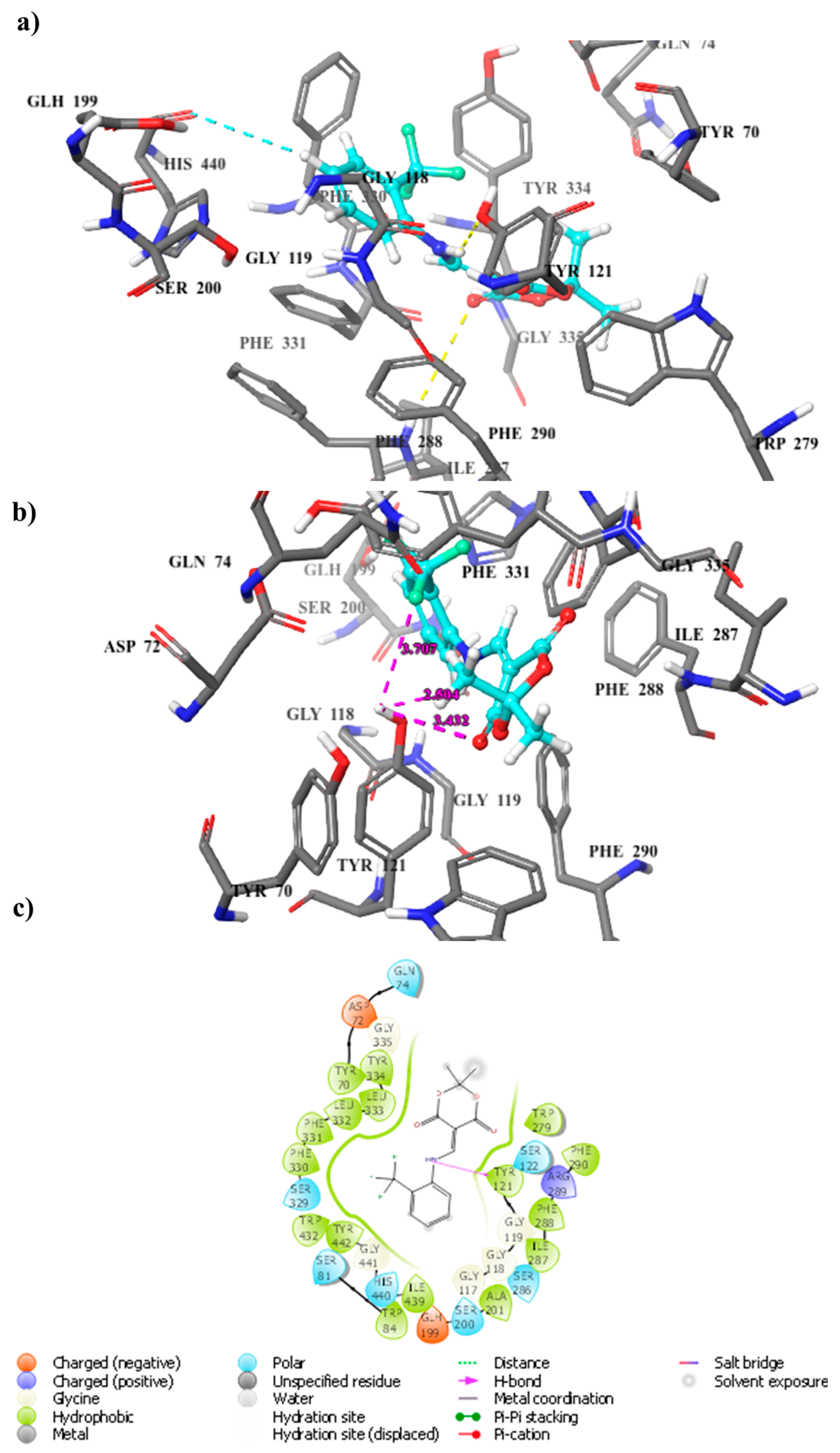
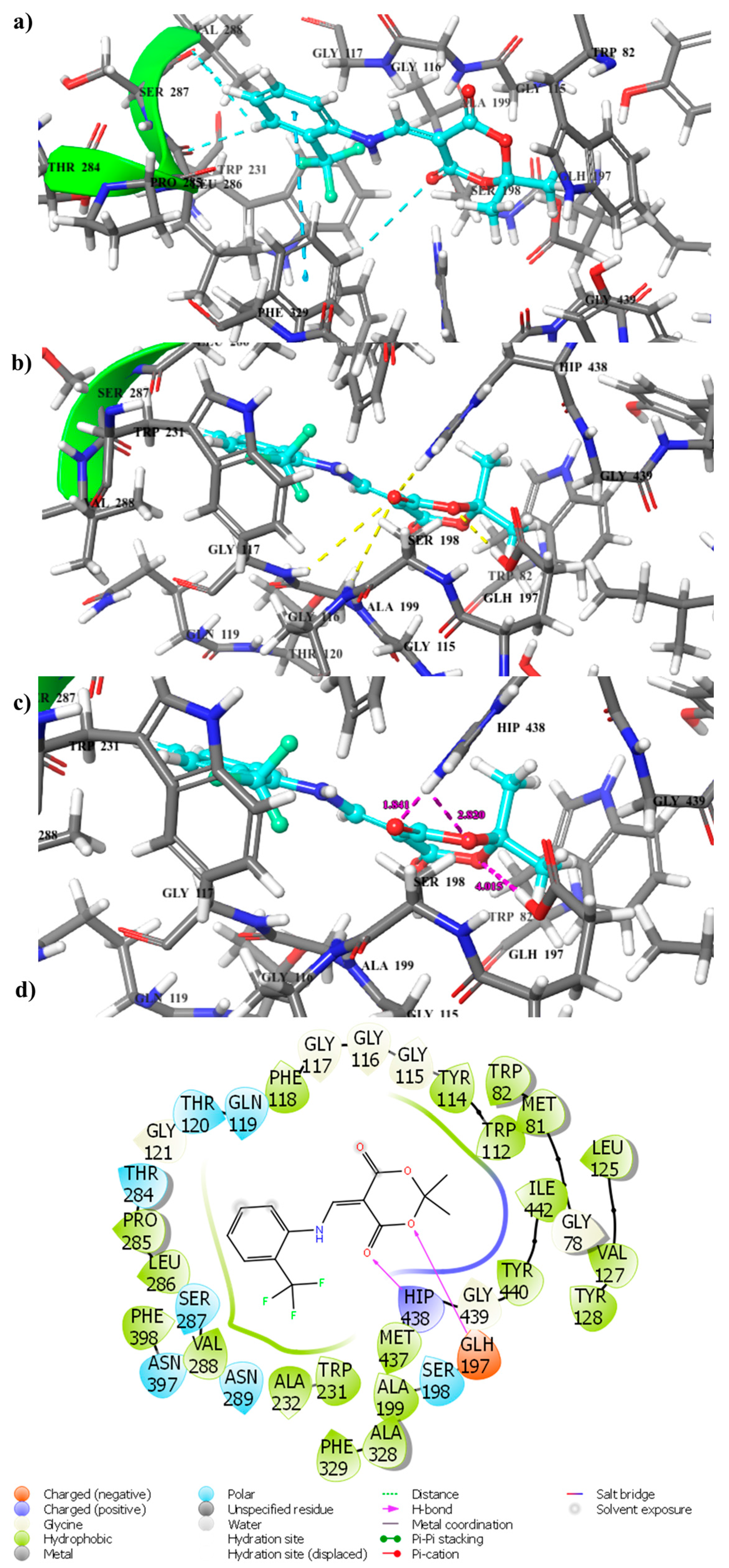
2.4. Docking Computation
2.5. Drug-Like Profiling
3. Materials and Methods
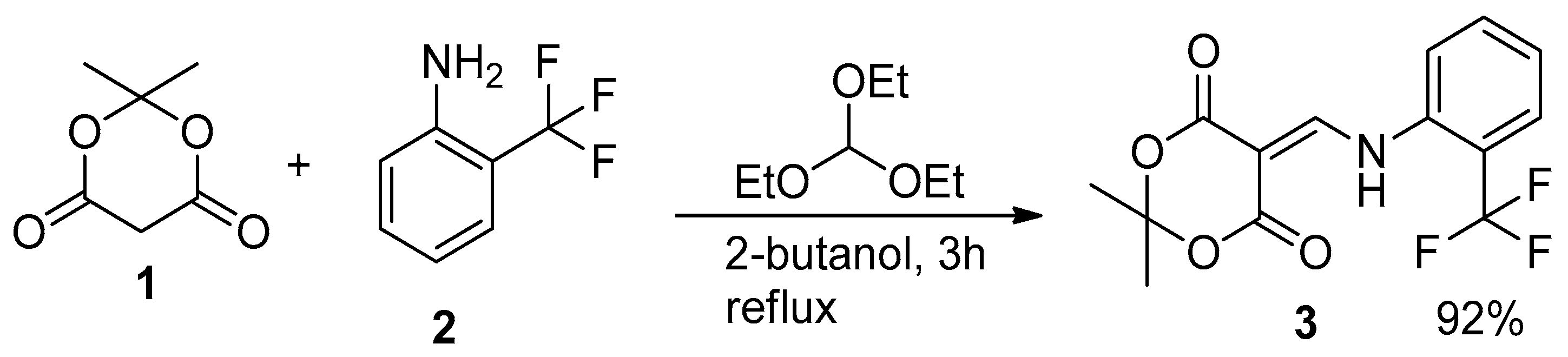
3.1. Synthesis of 2,2-Dimethyl-5-(([2-(trifluoromethyl)phenyl]Amino)methylidene)-1,3-dioxane-4,6-dione (3)
3.2. Structure Determination
3.3. DFT Calculations
3.4. Acetylcholinesterase and Butyrylcholinesterase Inhibition Assay
3.5. Molecular Docking Studies
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Small, D.H.; San Mok, S.; Bornstein, J.C. Alzheimer’s disease and aβ toxicity: From top to bottom. Nat. Rev. Neurosci. 2001, 2, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Kuruva, C.S.; Reddy, P.H. Amyloid beta modulators and neuroprotection in alzheimer’s disease: A critical appraisal. Drug Discov. Today 2017, 22, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Zaib, S.; Ashraf, S.; Iftikhar, J.; Muddassar, M.; Zhang, K.Y.; Iqbal, J. Synthesis, cholinesterase inhibition and molecular modelling studies of coumarin linked thiourea derivatives. Bioorg. Chem. 2015, 63, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Mahesar, P.A.; Zaib, S.; Khan, M.S.; Matin, A.; Shahid, M.; Iqbal, J. Synthesis, cytotoxicity and molecular modelling studies of new phenylcinnamide derivatives as potent inhibitors of cholinesterases. Eur. J. Med. Chem. 2014, 78, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi-Khanaposhtani, M.; Saeedi, M.; Zafarghandi, N.S.; Mahdavi, M.; Sabourian, R.; Razkenari, E.K.; Alinezhad, H.; Khanavi, M.; Foroumadi, A.; Shafiee, A. Potent acetylcholinesterase inhibitors: Design, synthesis, biological evaluation, and docking study of acridone linked to 1, 2, 3 triazole derivatives. Eur. J. Med. Chem. 2015, 92, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Larik, F.A.; Saeed, A.; Channar, P.A.; Ismail, H.; Dilshad, E.; Mirza, B. New 1-octanoyl-3-aryl thiourea derivatives: Solvent-free synthesis, characterization and multi-target biological activities. Bangladesh J. Pharmacol. 2016, 11, 894–902. [Google Scholar] [CrossRef]
- Raza, A.; Saeed, A.; Ibrar, A.; Muddassar, M.; Khan, A.A.; Iqbal, J. Pharmacological evaluation and docking studies of 3-thiadiazolyl-and thioxo-1, 2, 4-triazolylcoumarin derivatives as cholinesterase inhibitors. ISRN Pharmacol. 2012, 2012, 11. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tran, T.D.; Nguyen, T.C.V.; Nguyen, N.S.; Nguyen, D.M.; Nguyen, T.T.H.; Le, M.T.; Thai, K.M. Synthesis of novel chalcones as acetylcholinesterase inhibitors. Appl. Sci. 2016, 6, 198. [Google Scholar] [CrossRef]
- Saeed, A.; Shah, M.S.; Larik, F.A.; Khan, S.U.; Channar, P.A.; Flörke, U.; Iqbal, J. Synthesis, computational studies and biological evaluation of new 1-acetyl-3-aryl thiourea derivatives as potent cholinesterase inhibitors. Med. Chem. Res. 2017, 8, 1635–1646. [Google Scholar] [CrossRef]
- Sultana, N.; Sarfraz, M.; Tahir, S.; SafwanAkram, M.; Sadiq, A.; Rashid, U.; Tariq, M.I. Synthesis, crystal structure determination, biological screening and docking studies of n 1-substituted derivatives of 2, 3-dihydroquinazolin-4 (1h)-one as inhibitors of cholinesterases. Bioorg. Chem. 2017, 72, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Andrisano, V.; Bartolini, M.; Banzi, R.; Melchiorre, C. Propidium-based polyamine ligands as potent inhibitors of acetylcholinesterase and acetylcholinesterase-induced amyloid-β aggregation. J. Med. Chem. 2005, 48, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; Darvesh, K.V.; McDonald, R.S.; Mataija, D.; Walsh, R.; Mothana, S.; Lockridge, O.; Martin, E. Carbamates with differential mechanism of inhibition toward acetylcholinesterase and butyrylcholinesterase. J. Med. Chem. 2008, 51, 4200–4212. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi-Khanaposhtani, M.; Mahdavi, M.; Saeedi, M.; Sabourian, R.; Safavi, M.; Khanavi, M.; Foroumadi, A.; Shafiee, A.; Akbarzadeh, T. Design, synthesis, biological evaluation, and docking study of acetylcholinesterase inhibitors: New acridone-1, 2, 4-oxadiazole-1, 2, 3-triazole hybrids. Chem. Biol. Drug Des. 2015, 86, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Torre, P.D.L.; Saavedra, L.A.; Caballero, J.; Quiroga, J.; Alzate-Morales, J.H.; Cabrera, M.G.; Trilleras, J. A novel class of selective acetylcholinesterase inhibitors: Synthesis and evaluation of (e)-2-(benzo [d] thiazol-2-yl)-3-heteroarylacrylonitriles. Molecules 2012, 17, 12072–12085. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, S.D.; Chee, C.F.; Viswanathan, G.; Buckle, M.J.; Othman, R.; Abd Rahman, N.; Chung, L.Y. Synthesis, biological evaluation and molecular modelling of 2’-hydroxychalcones as acetylcholinesterase inhibitors. Molecules 2016, 21, 955. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Ruiz, P.; Rubio, L.; García-Palomero, E.; Dorronsoro, I.; Del Monte-Millán, M.; Valenzuela, R.; Usán, P.; De Austria, C.; Bartolini, M.; Andrisano, V. Design, synthesis, and biological evaluation of dual binding site acetylcholinesterase inhibitors: New disease-modifying agents for alzheimer’s disease. J. Med. Chem. 2005, 48, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Parveen, M.; Aslam, A.; Nami, S.A.; Malla, A.M.; Alam, M.; Lee, D.U.; Rehman, S.; Silva, P.P.; Silva, M.R. Potent acetylcholinesterase inhibitors: Synthesis, biological assay and docking study of nitro acridone derivatives. J. Photochem. Photobiol. B Biol. 2016, 161, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Piazzi, L.; Rampa, A.; Bisi, A.; Gobbi, S.; Belluti, F.; Cavalli, A.; Bartolini, M.; Andrisano, V.; Valenti, P.; Recanatini, M. 3-(4-{[benzyl (methyl) amino] methyl} phenyl)-6, 7-dimethoxy-2 h-2-chromenone (ap2238) inhibits both acetylcholinesterase and acetylcholinesterase-induced β-amyloid aggregation: A dual function lead for alzheimer’s disease therapy. J. Med. Chem. 2003, 46, 2279–2282. [Google Scholar] [CrossRef] [PubMed]
- Raza, R.; Saeed, A.; Arif, M.; Mahmood, S.; Muddassar, M.; Raza, A.; Iqbal, J. Synthesis and biological evaluation of 3-thiazolocoumarinyl schiff-base derivatives as cholinesterase inhibitors. Chem. Biol. Drug Des. 2012, 80, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Sauvaître, T.; Barlier, M.; Herlem, D.; Gresh, N.; Chiaroni, A.; Guenard, D.; Guillou, C. New potent acetylcholinesterase inhibitors in the tetracyclic triterpene series. J. Med. Chem. 2007, 50, 5311–5323. [Google Scholar] [CrossRef] [PubMed]
- Tasso, B.; Catto, M.; Nicolotti, O.; Novelli, F.; Tonelli, M.; Giangreco, I.; Pisani, L.; Sparatore, A.; Boido, V.; Carotti, A.; et al. Quinolizidinyl derivatives of bi-and tricyclic systems as potent inhibitors of acetyl-and butyrylcholinesterase with potential in Alzheimer’s disease. Eur. J. Med. Chem. 2011, 46, 2170–2184. [Google Scholar] [CrossRef] [PubMed]
- Bartorelli, L.; Giraldi, C.; Saccardo, M.; Cammarata, S.; Bottini, G.; Fasanaro, A.M.; Trequattrini, A. Effects of switching from an AChE inhibitor to a dual AChE-BuChE inhibitor in patients with Alzheimer’s disease. Curr. Med. Res. Opin. 2005, 21, 1809–1817. [Google Scholar] [CrossRef] [PubMed]
- Nicolotti, O.; Pisani, L.; Catto, M.; Leonetti, F.; Giangreco, I.; Stefanachi, A.; Carotti, A. Discovery of a Potent and Selective Hetero-Bivalent AChE Inhibitor via Bioisosteric Replacement. Mol. Inform. 2011, 30, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Li, J.S.; Da, Y.D.; Chen, G.Q.; Yang, Q.; Li, Z.W.; Yang, F.; Huang, P.M. Solvent-, and catalyst–free acylation of anilines with meldrum’s acids: Aneat access to anilides. ChemistySelect 2017, 2, 1770–1773. [Google Scholar] [CrossRef]
- Çalışkan, R.; Nohut, N.; Yılmaz, Ö.; Sahin, E.; Balci, M. Unusual manganese (iii)-mediated oxidative free-radical additions of meldrum’s acid and dimethyl malonate to benzonorbornadiene and oxabenzonorbornadiene. Tetrahedron 2017, 73, 291–297. [Google Scholar] [CrossRef]
- Gopakumar, D.A.; Pasquini, D.; Henrique, M.A.; De Morais, L.C.; Grohens, Y.; Thomas, S. Meldrum’s acid modified cellulose nanofiber-based polyvinylidene fluoride microfiltration membrane for dye water treatment and nanoparticle removal. ACS Sustain. Chem. Eng. 2017, 5, 2026–2033. [Google Scholar] [CrossRef]
- Zhang, J.W.; Xue, J.H.; Zhang, H.H.; Xu, F.; Chen, Q.G.; Li, J.H. Reaction of meldrum’s acid with an aminomethylating agent and nitroalkanes. J. Chem. Res. 2017, 41, 72–74. [Google Scholar] [CrossRef]
- Ivanov, A.S. Meldrum’s acid and related compounds in the synthesis of natural products and analogs. Chem. Soc. Rev. 2008, 37, 789–811. [Google Scholar] [CrossRef] [PubMed]
- De Toledo, T.; Da Silva, L.; Teixeira, A.; Freire, P.; Pizani, P. Characterization of meldrum’s acid derivative 5-(5-ethyl-1, 3, 4-thiadiazol-2-ylamino) methylene-2, 2-dimethyl-1, 3-dioxane-4, 6-dione by raman and ft-ir spectroscopy and dft calculations. J. Mol. Struct. 2015, 1091, 37–42. [Google Scholar] [CrossRef]
- Sandhu, H.S.; Sapra, S.; Gupta, M.; Nepali, K.; Gautam, R.; Yadav, S.; Kumar, R.; Jachak, S.M.; Chugh, M.; Gupta, M.K. Synthesis and biological evaluation of arylidene analogues of meldrum’s acid as a new class of antimalarial and antioxidant agents. Biorg. Med. Chem. 2010, 18, 5626–5633. [Google Scholar] [CrossRef] [PubMed]
- Rotzoll, S.; Reinke, H.; Fischer, C.; Langer, P. Synthesis of novel halogenated 4 (1H)-quinolones by thermolysis of arylaminomethylene-1, 3-dioxane-4, 6-diones. Synthesis 2009, 1, 69–78. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Jabeen, F.; Oliferenko, P.V.; Oliferenko, A.A.; Pillai, G.G.; Ansari, F.L.; Hall, C.D.; Katritzky, A.R. Dual inhibition of the α-glucosidase and butyrylcholinesterase studied by molecular field topology analysis. Eur. J. Med. Chem. 2014, 80, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Del. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. Shelxtl-pc (Version 5.1); Siemens Analytical Instruments, Inc.: Madison, WI, USA, 1997. [Google Scholar]
- Sheldrick, G.M. A short history of shelx. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wennmohs, F.; Hansen, A.; Beccker, U. Efficient approximate ana parallel Hartree-Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithms for the Hartree-Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Honarparvar, B.; Govender, T.; Maguire, G.E.; Soliman, M.E.; Kruger, H.G. Integrated approach to structure-based enzymatic drug design: Molecular modeling, spectroscopy, and experimental bioactivity. Chem. Rev. 2013, 114, 493–537. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–90. [Google Scholar] [CrossRef]
- Harel, M.; Schalk, I.; Ehret-Sabatier, L.; Bouet, F.; Goeldner, M.; Hirth, C.; Axelsen, P.; Silman, I.; Sussman, J. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1993, 90, 9031–9035. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L. Schrödinger Suite 2016-4 Protein Preparation Wizard, Epik, version; Schrödinger, Inc.: New York, NY, USA, 2016; Volume 2. [Google Scholar]
- ChemAxon. Marvin. Marvinn.n.n (versionnumber), 201n. Available online: http://www.chemaxon.com (accessed on 9 July 2010).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | AChE Activity | BChE Activity |
|---|---|---|
| IC50 (µM) ± SEM | ||
| 3 | 1.13 ± 0.03 | 2.12 ± 1.22 |
| Neostigmine | 22.2 ± 3.2 | 49.6 ± 6.11 |
| Donepezil | 6.4 ± 0.4 nM | 33.65 nM |
| Tacrine | 45.1 ± 7 nM | 3.2 ± 0.2 nM |
| Rivastigmine | 501 ± 3.08 µM | 19.95 ± 0.20 µM |
| Galantamine | 4 ± 0.13 µM | 7.96 ± 0.59 µM |
| Entry | Descriptors Name | Computed Values |
|---|---|---|
| 3 | Number of rotatable bonds | 3.00 |
| Hydrogen Bond Acceptor | 2.0 | |
| Hydrogen Bond Donor | 0.0 | |
| Log P (o/w) = logarithm of the (octanol/water) partition coefficient | 6.7 | |
| Total polar surface area (TPSA) | 32.67 | |
| Molecular weight | 315.05 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehfooz, H.; Saeed, A.; Sharma, A.; Albericio, F.; Larik, F.A.; Jabeen, F.; Channar, P.A.; Flörke, U. Dual Inhibition of AChE and BChE with the C-5 Substituted Derivative of Meldrum’s Acid: Synthesis, Structure Elucidation, and Molecular Docking Studies. Crystals 2017, 7, 211. https://doi.org/10.3390/cryst7070211
Mehfooz H, Saeed A, Sharma A, Albericio F, Larik FA, Jabeen F, Channar PA, Flörke U. Dual Inhibition of AChE and BChE with the C-5 Substituted Derivative of Meldrum’s Acid: Synthesis, Structure Elucidation, and Molecular Docking Studies. Crystals. 2017; 7(7):211. https://doi.org/10.3390/cryst7070211
Chicago/Turabian StyleMehfooz, Haroon, Aamer Saeed, Anamika Sharma, Fernando Albericio, Fayaz Ali Larik, Farukh Jabeen, Pervaiz Ali Channar, and Ulrich Flörke. 2017. "Dual Inhibition of AChE and BChE with the C-5 Substituted Derivative of Meldrum’s Acid: Synthesis, Structure Elucidation, and Molecular Docking Studies" Crystals 7, no. 7: 211. https://doi.org/10.3390/cryst7070211
APA StyleMehfooz, H., Saeed, A., Sharma, A., Albericio, F., Larik, F. A., Jabeen, F., Channar, P. A., & Flörke, U. (2017). Dual Inhibition of AChE and BChE with the C-5 Substituted Derivative of Meldrum’s Acid: Synthesis, Structure Elucidation, and Molecular Docking Studies. Crystals, 7(7), 211. https://doi.org/10.3390/cryst7070211