Electrocrystallization of CaCO3 Crystals Obtained through Phosphorylated Chitin




Abstract
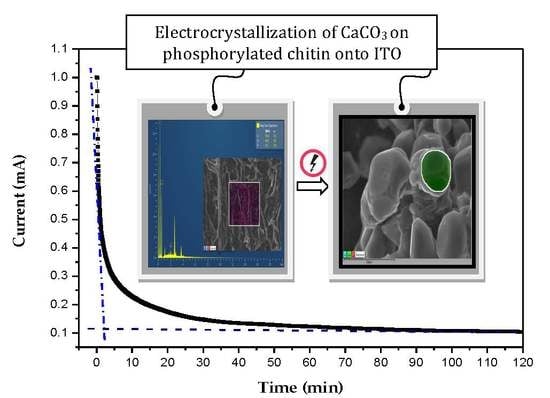
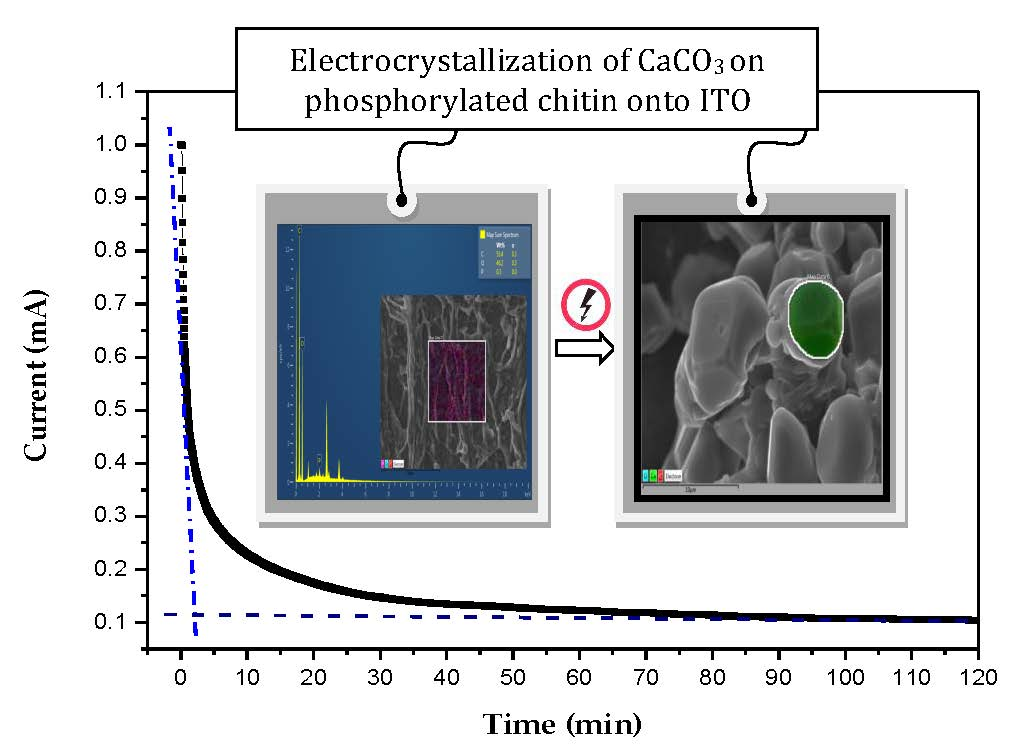
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reactants
2.2. Gas Diffusion (GD) Method
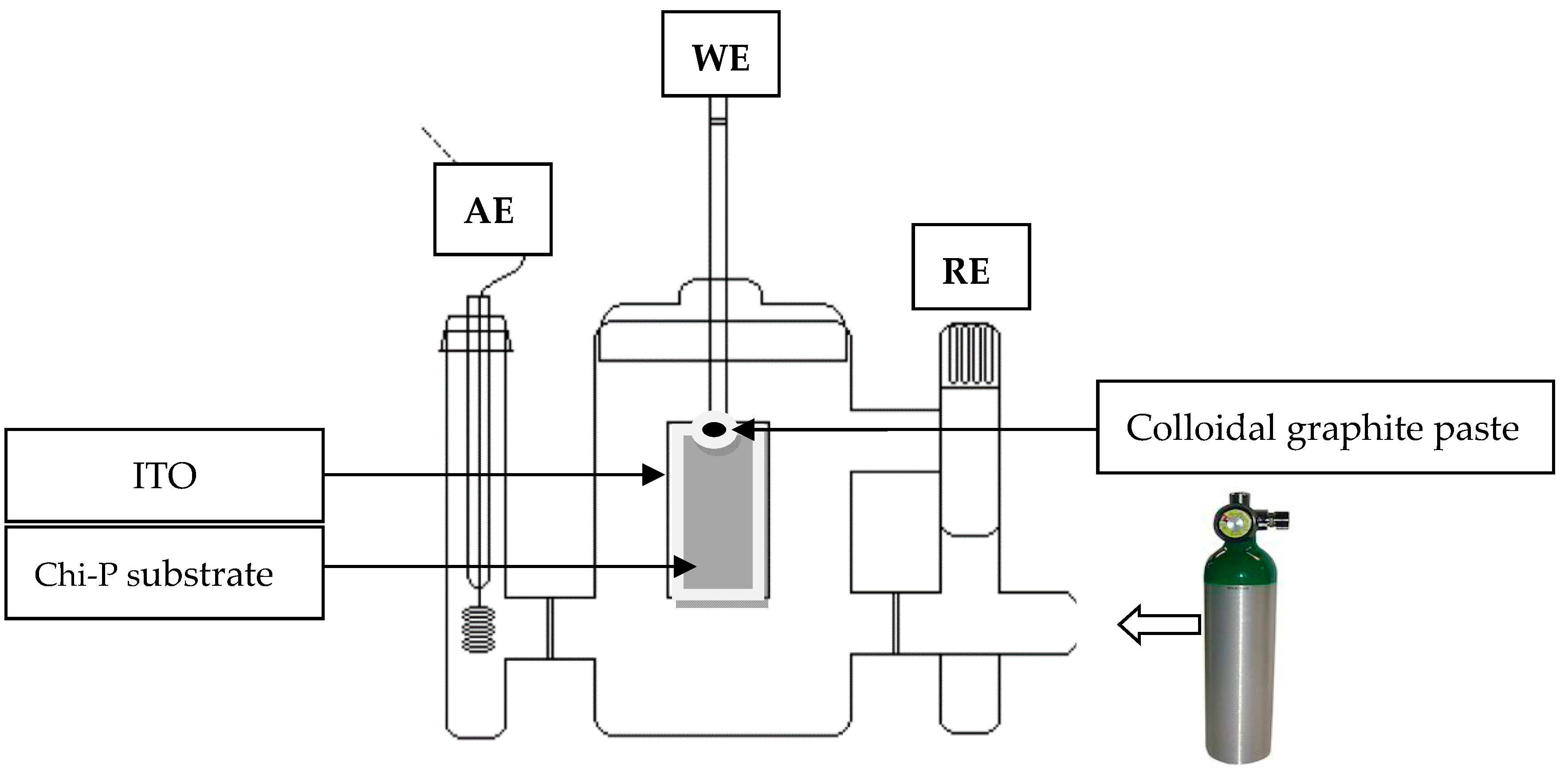
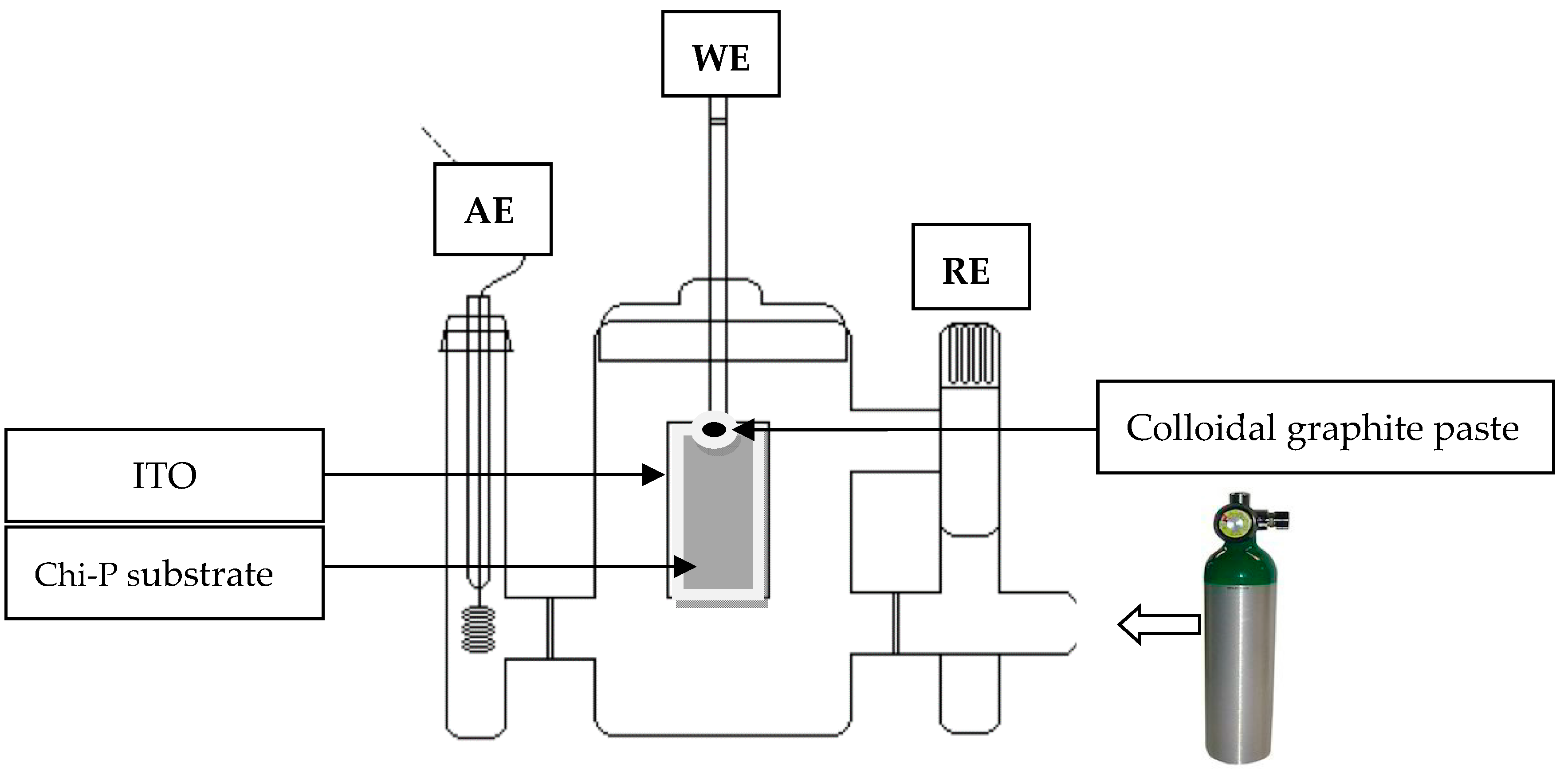
2.3. Electrocrystallization (EC) Method
2.4. Characterization
3. Results
4. Discussion
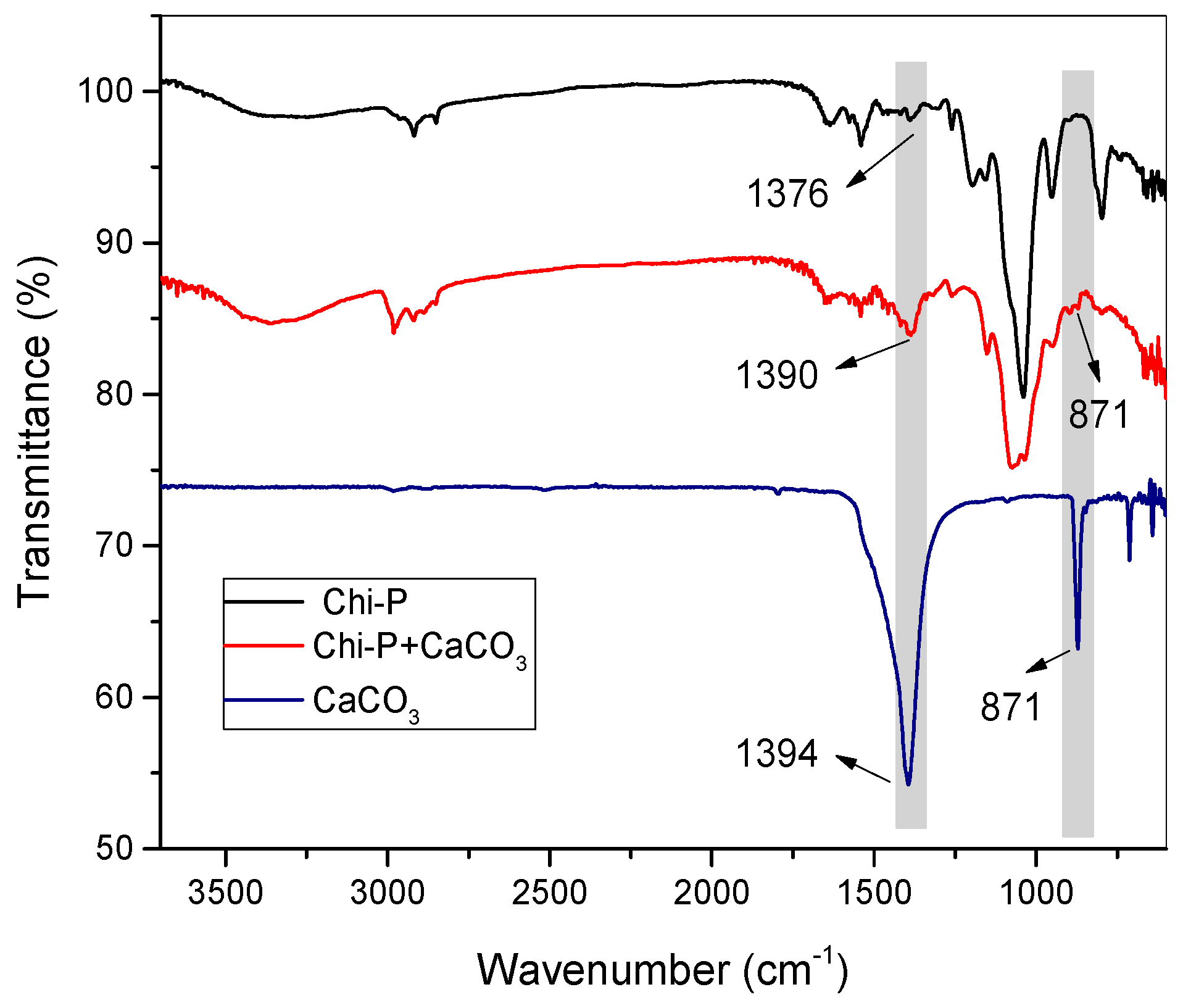
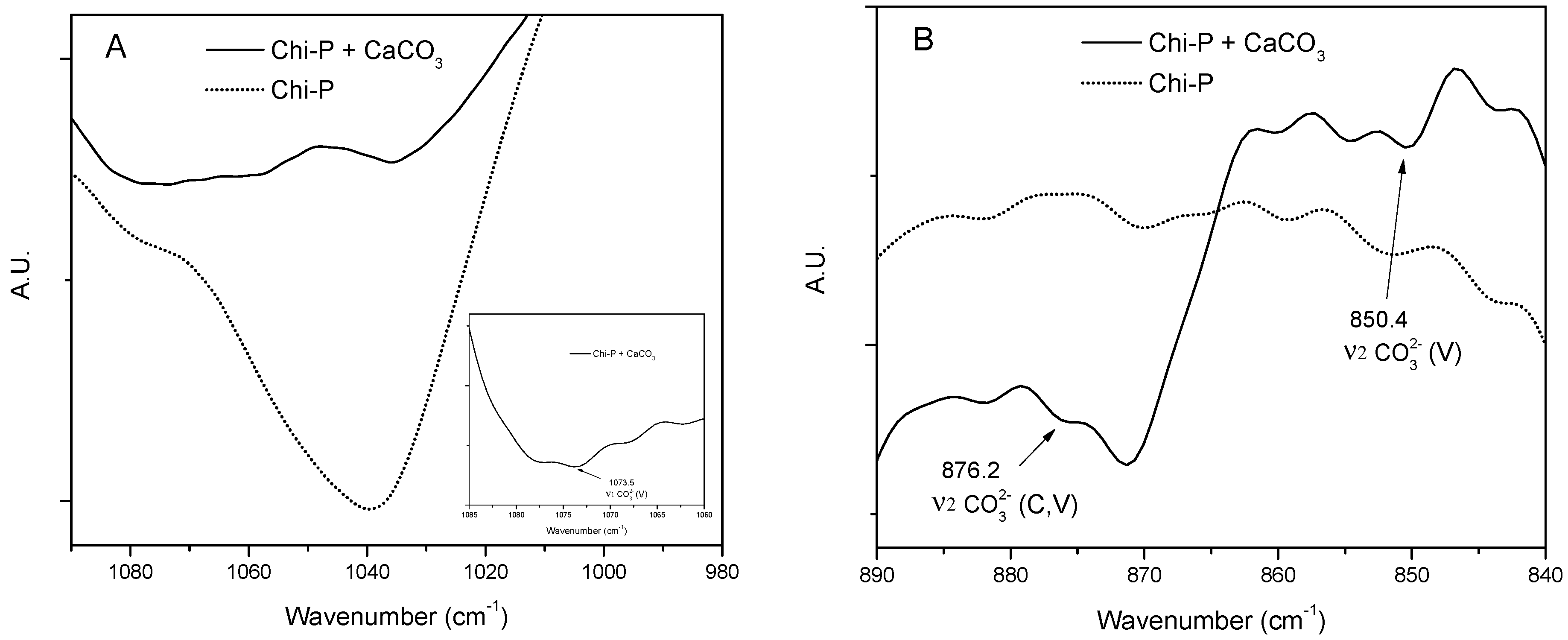
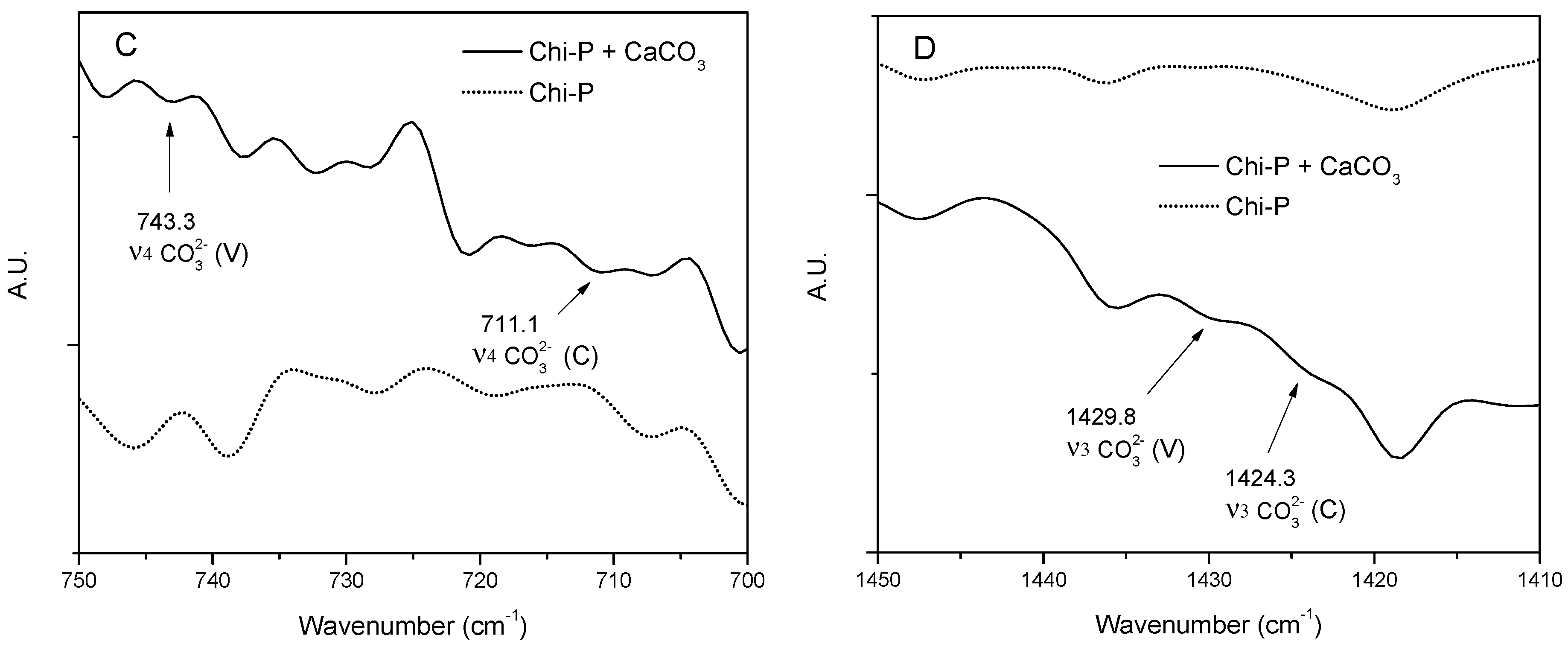
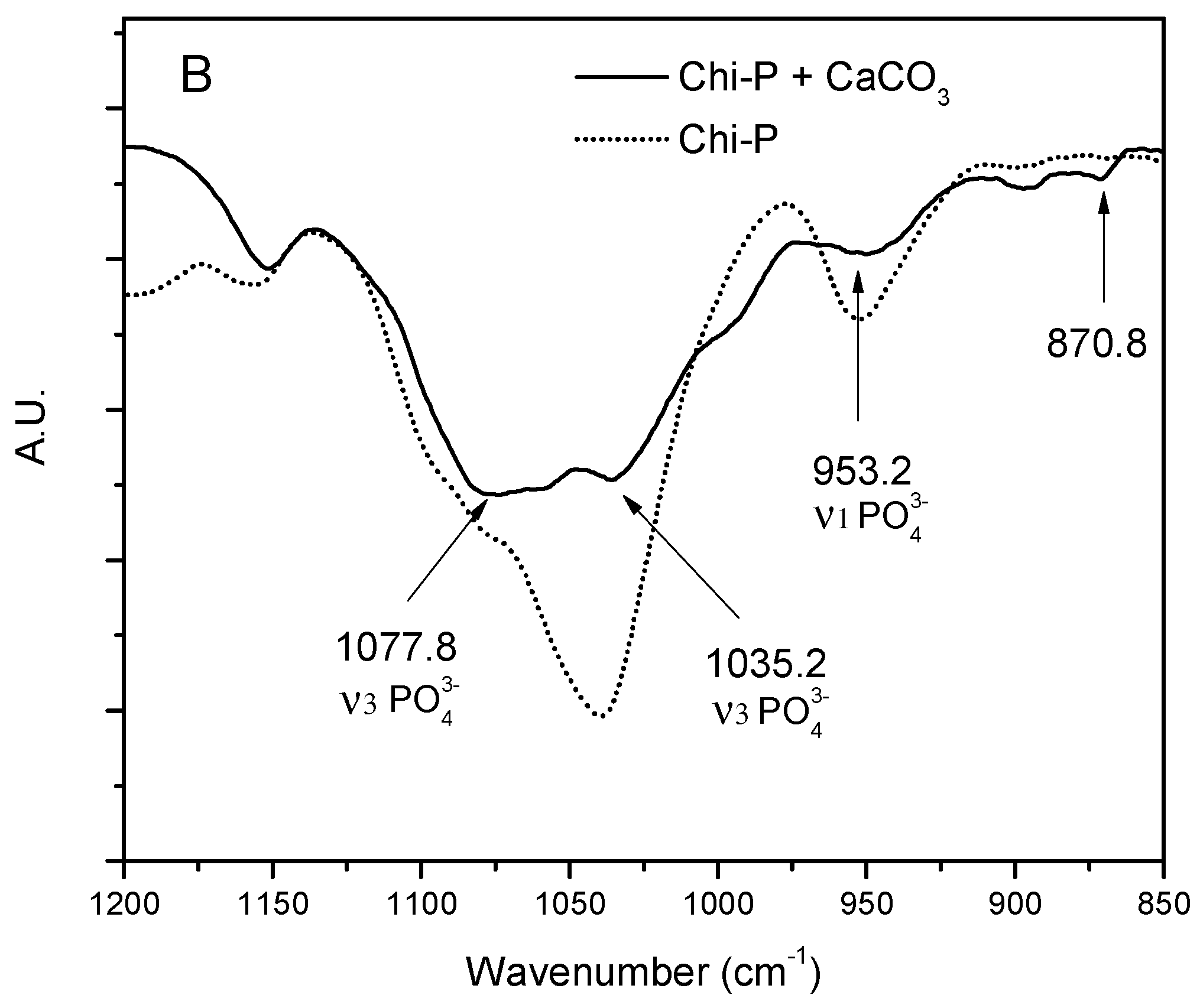
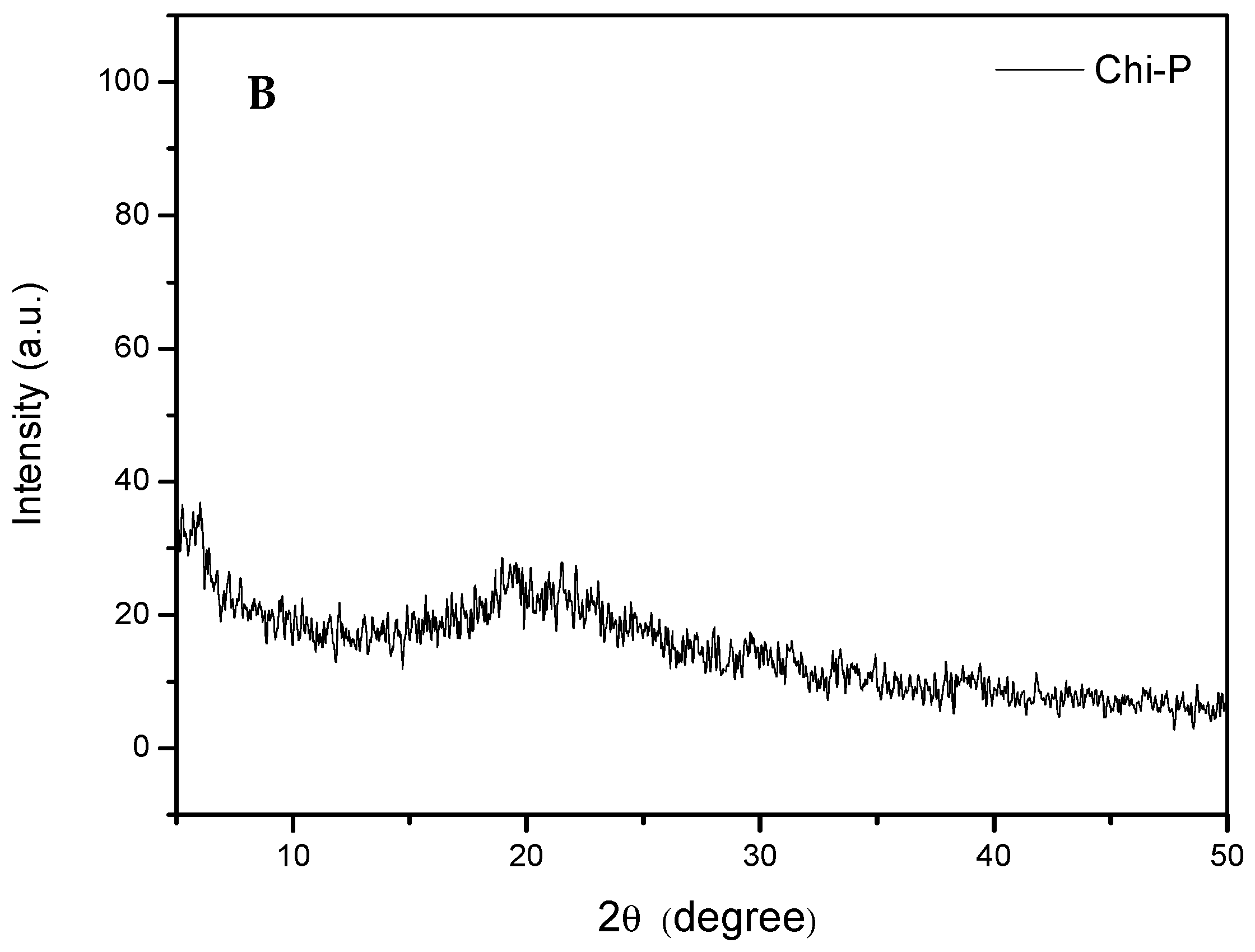
4.1. Chemical Characterization of Chitin (Chi) and Chitin Phosphate (Chi-P)
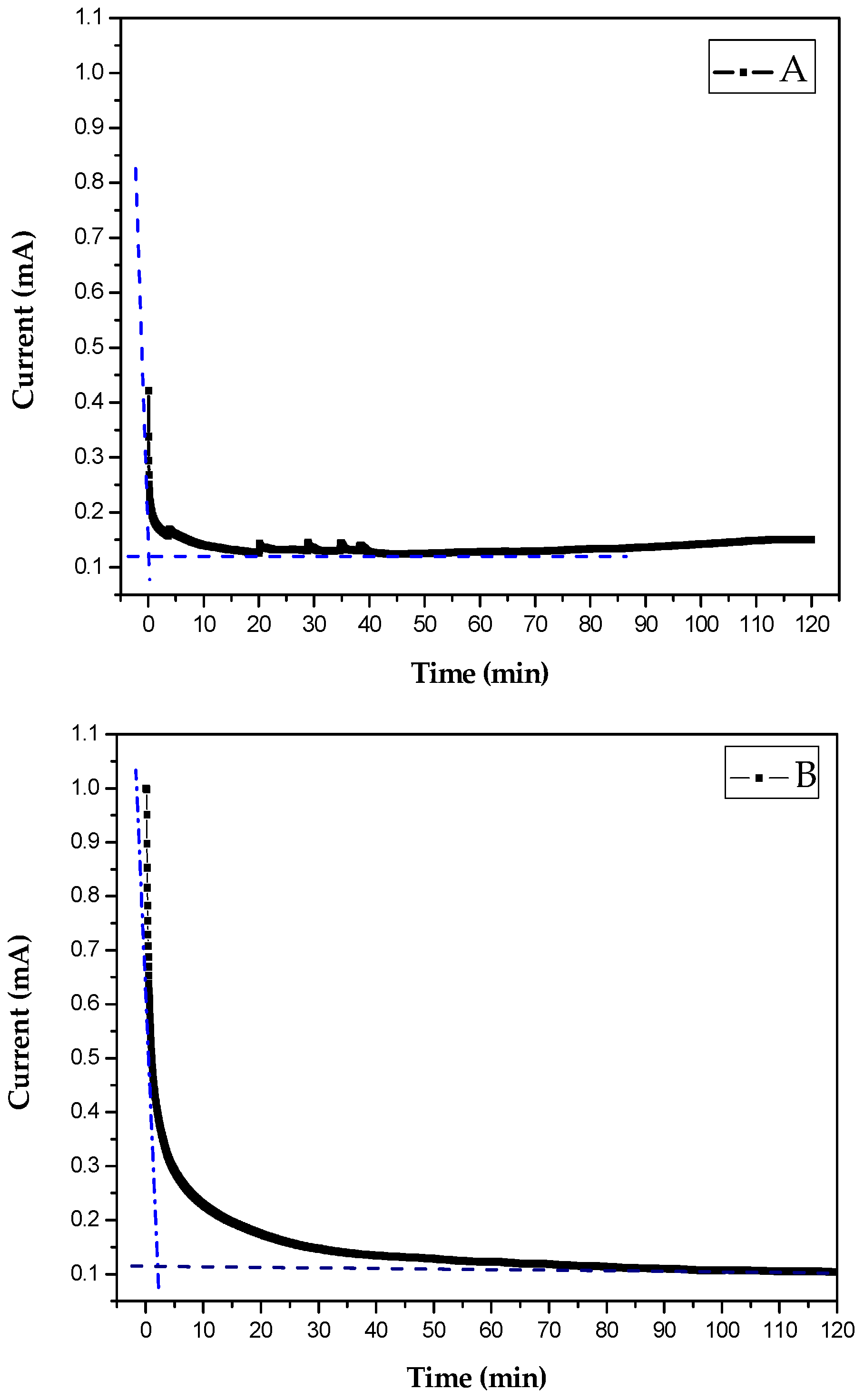
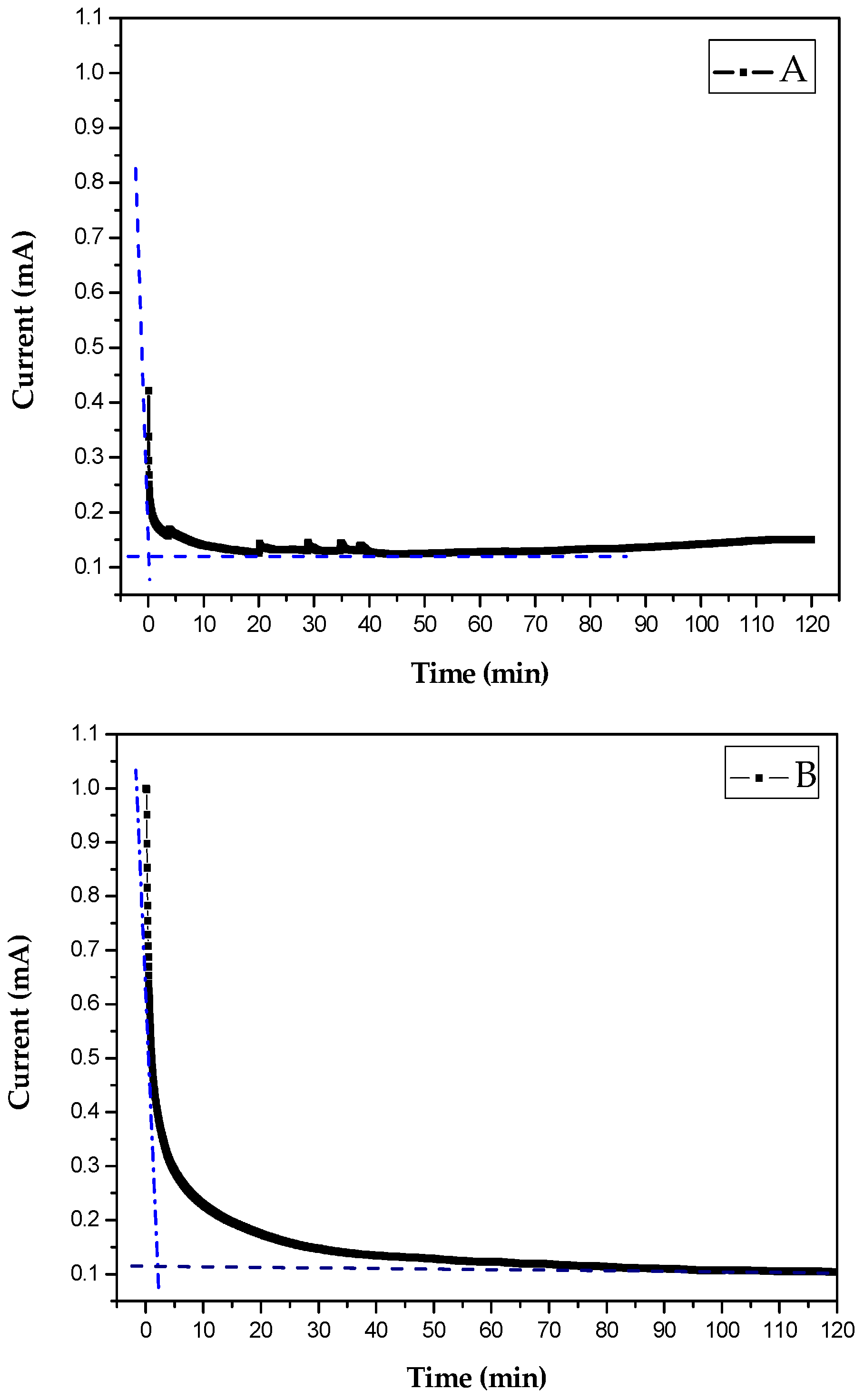
4.2. Study of CaCO3 Crystals Deposition by EC and GD Methods
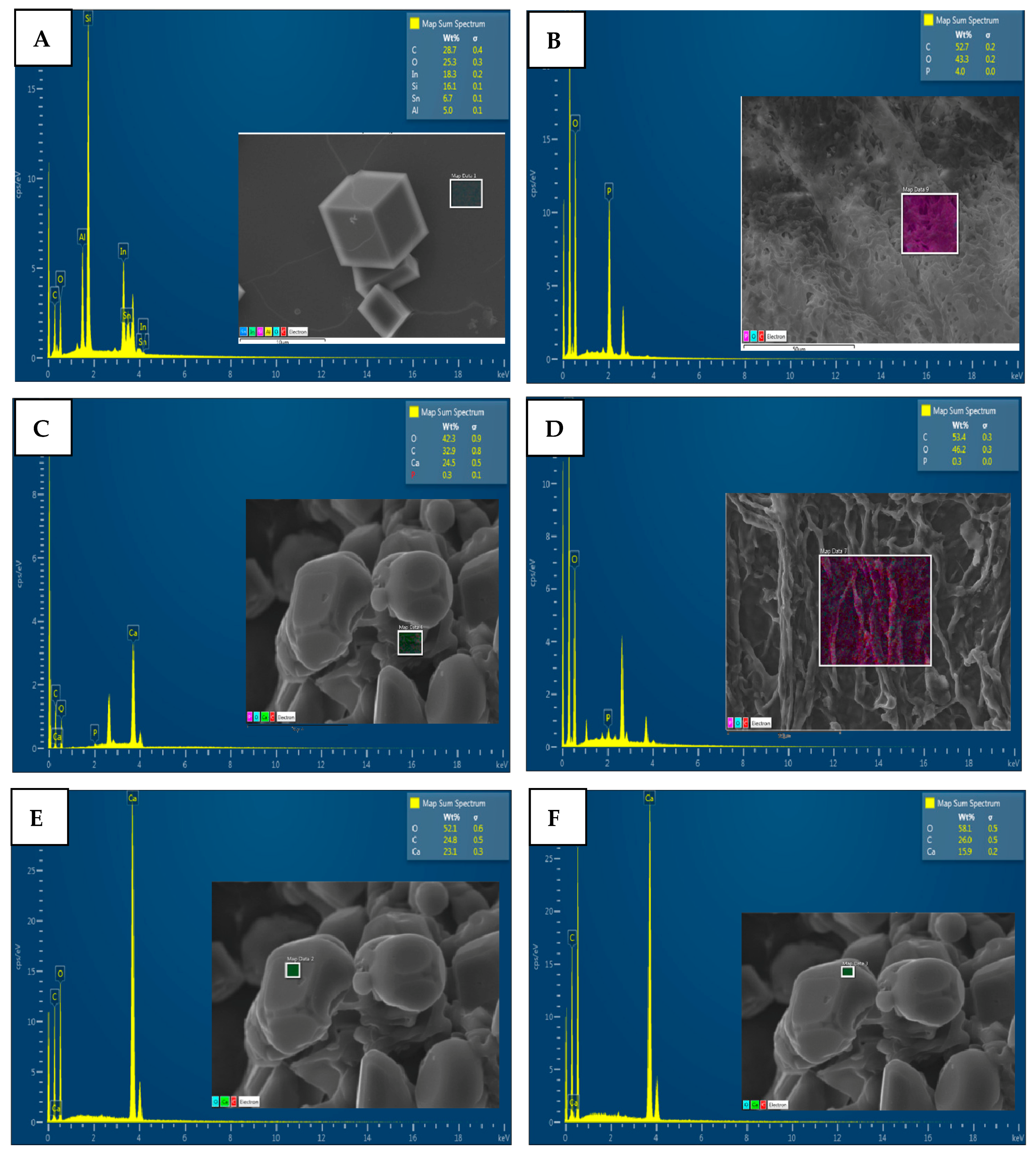
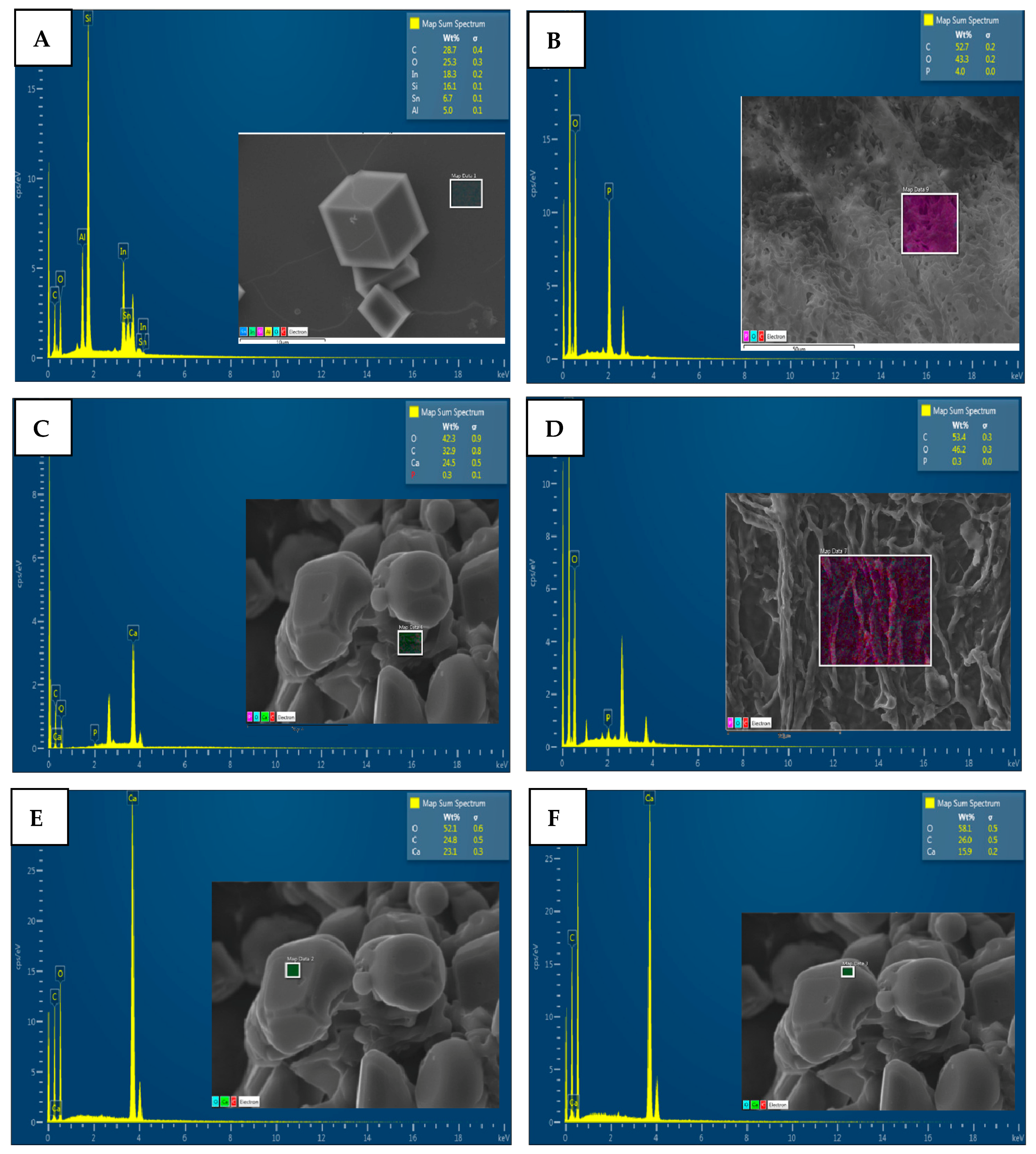
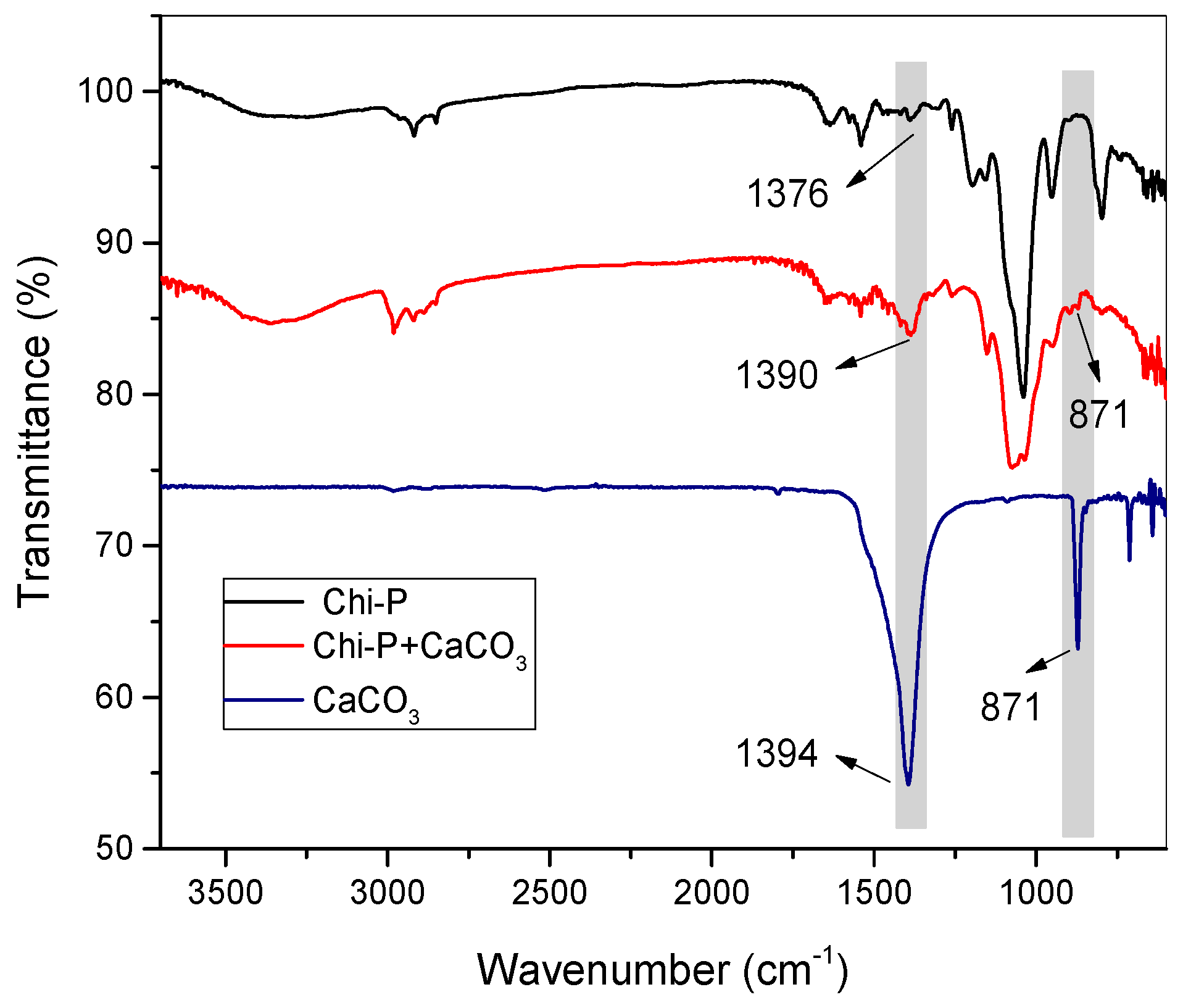
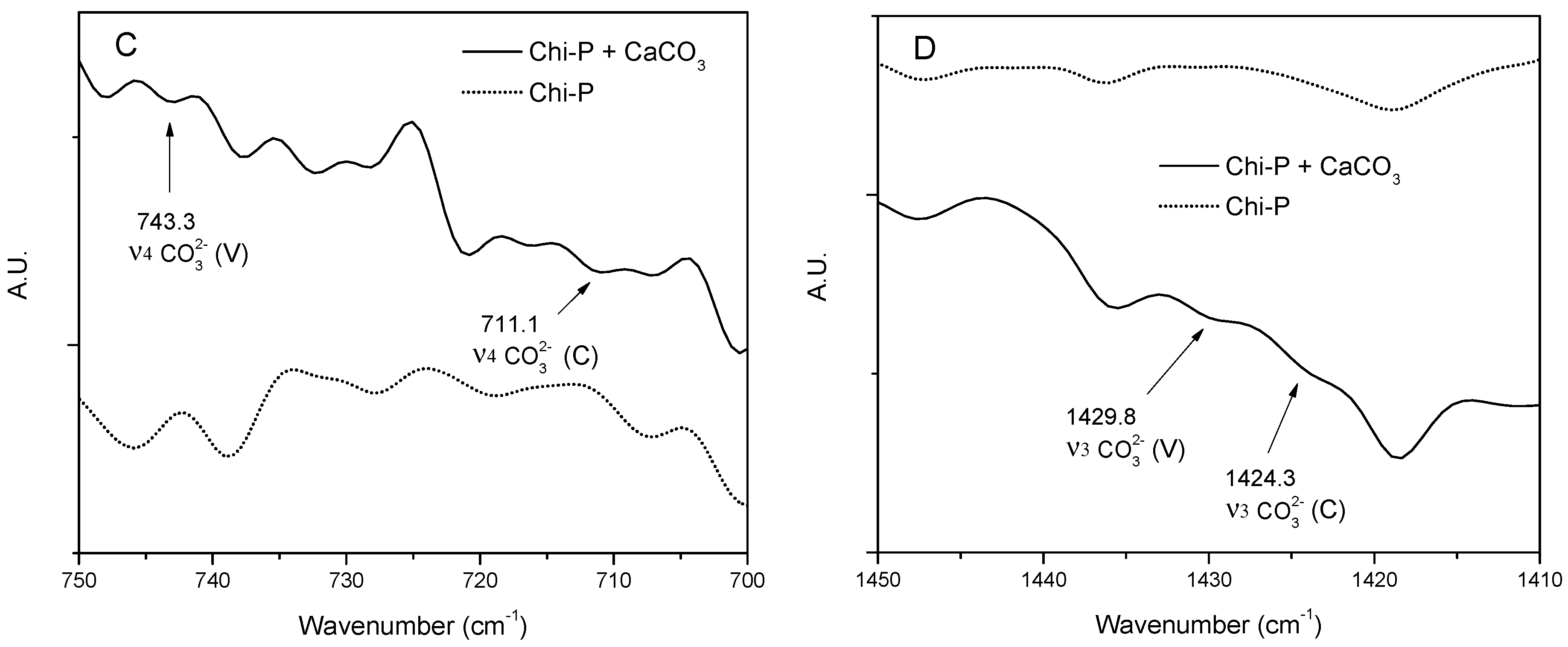
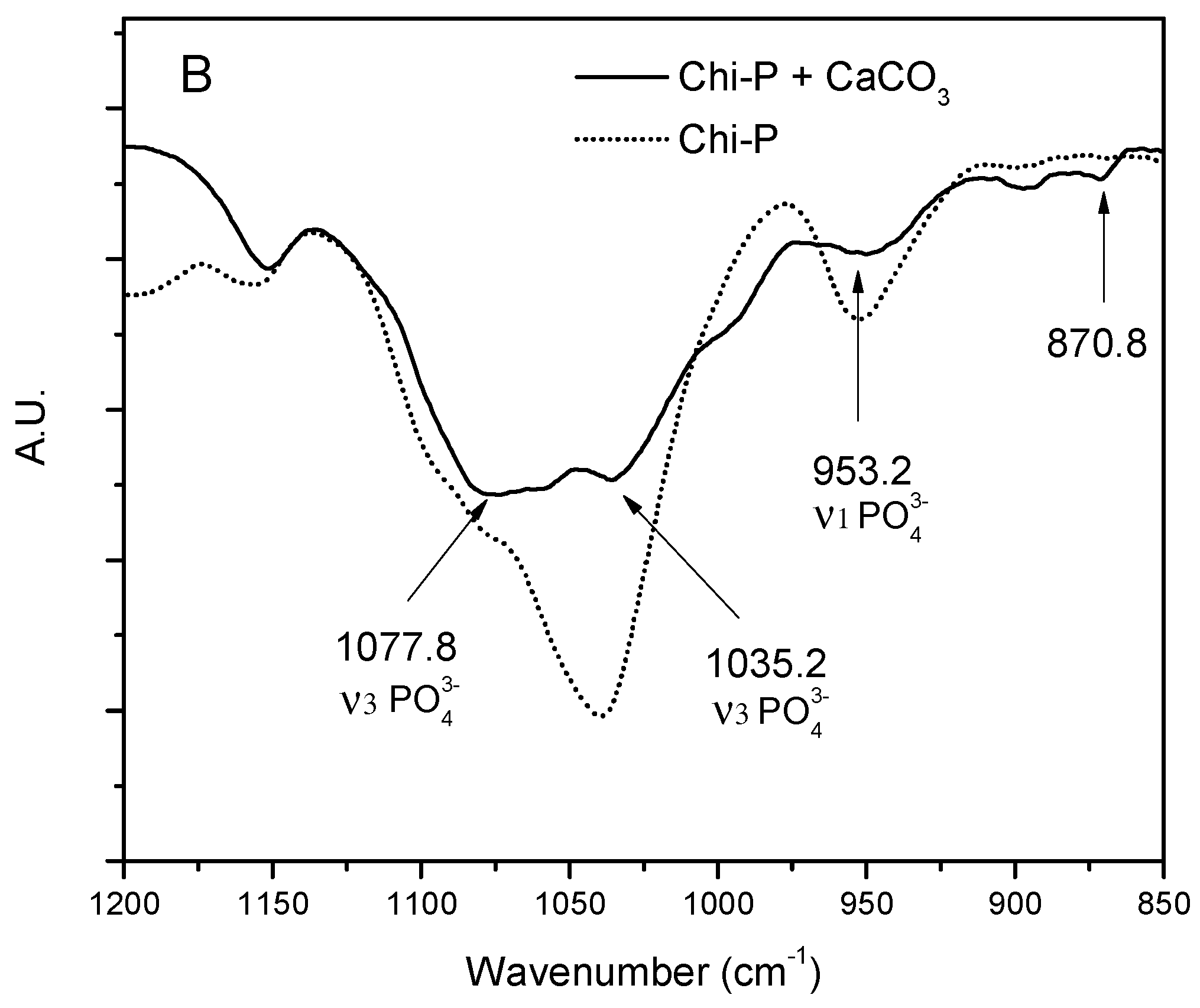
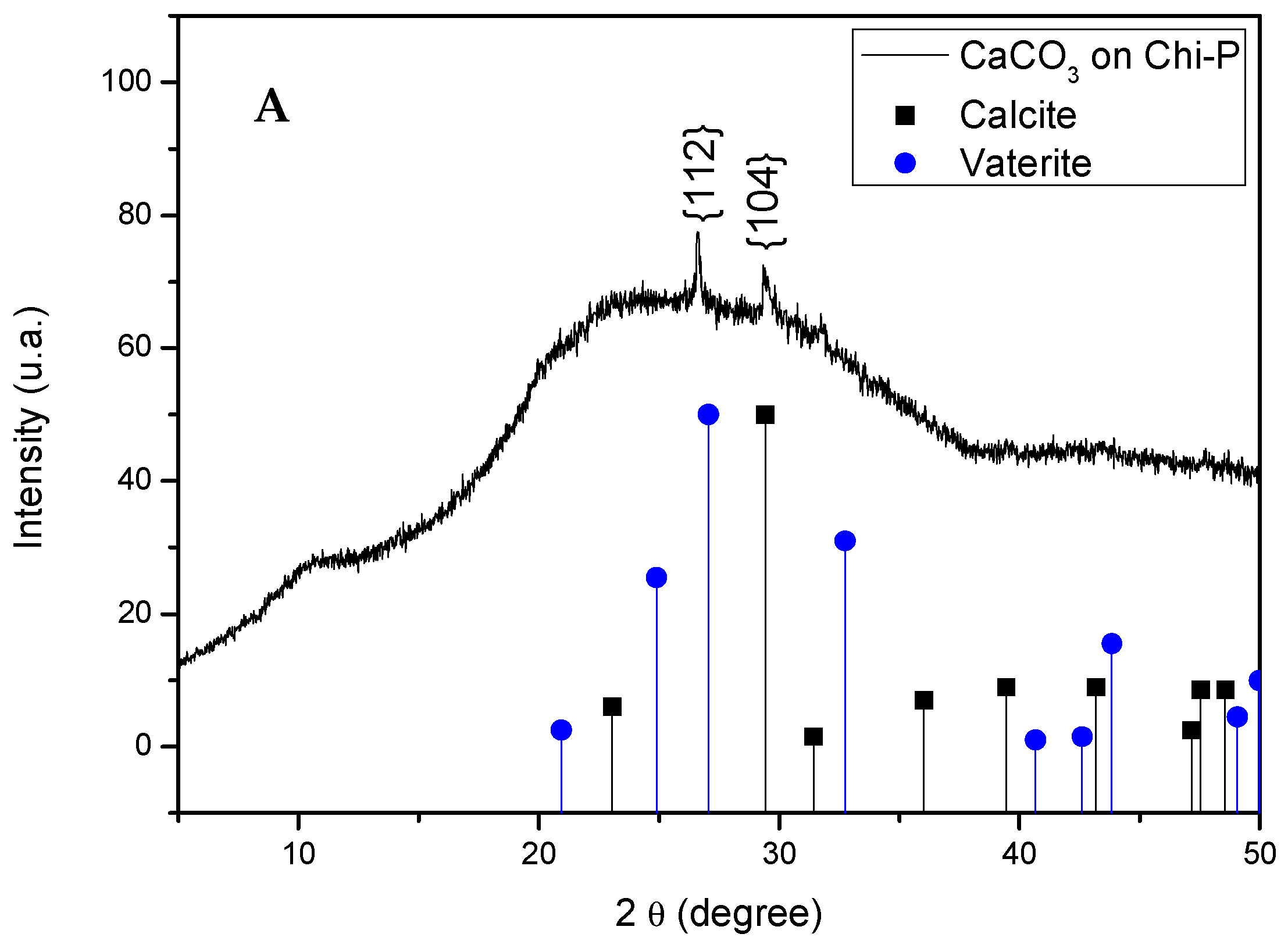
4.3. Characterization of CaCO3 Crystals through Amperometry, FTIR, SEM-EDS, and XRD Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
Appendix A.1. Synthesis of Phosphorylated Chitin
Appendix A.2. FTIR Analysis
Appendix A.3. Elemental Analysis
References
- Lowenstam, H.A. Minerals formed by organisms. Science 1981, 211, 1126–1131. [Google Scholar] [CrossRef] [PubMed]
- Lowenstam, H.A.; Weiner, S. On Biomineralization; Oxford University Press: Oxford, UK, 1989; p. 336. ISBN 0-19-504977-2. [Google Scholar]
- Sanchez, C.; Arribart, H.; Giraud Guille, M.M. Biomimetism and bioinspiration as tools for the design of innovative materials and systems. Nat. Mater. 2005, 4, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Simkiss, K.; Wilbur, K.M. Biomineralization: Cell Biology and Mineral Deposition; Academic Press: San Diego, CA, USA, 1989; p. 337. ISBN 0-12-643830-7. [Google Scholar]
- Addadi, L.; Weiner, S. Control and design principles in biological mineralization. Angew. Chem. Int. Ed. Engl. 1992, 31, 153–169. [Google Scholar] [CrossRef]
- Cölfen, H.; Mann, S. Higher-order organization by mesoscale self-assembly and transformation of hybrid nanostructures. Angew. Chem. Int. Ed. 2003, 42, 2350–2365. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Wu, G.; Qing, C. Biomimetic synthesis of hollow calcium carbonate with the existence of the agar matrix and bovine serum albumin. Mater. Sci. Eng. C 2016, 58, 409–411. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.L.; Neira-Carrillo, A.; Arias, J.I.; Escobar, C.; Bodero, M.; David, M.; Fernández, M.S. Sulfated polymers in biological mineralization: A plausible source for bio-inspired engineering. J. Mater. Chem. 2004, 14, 2154–2160. [Google Scholar] [CrossRef]
- Addadi, L.; Raz, S.; Weiner, S. Taking advantage of disorder: Amorphous calcium carbonate and its roles in biomineralization. Adv. Mater. 2003, 15, 959–970. [Google Scholar] [CrossRef]
- Gower, L.B. Biomimetic model systems for investigating the amorphous precursor pathway and its role in biomineralization. Chem. Rev. 2008, 108, 4551–4627. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, F.C. Calcium carbonate in biomineralisation and biomimetic chemistry. Int. Mater. Rev. 2003, 48, 187–224. [Google Scholar] [CrossRef]
- Cartwright, J.H.E.; Checa, A.G.; Gale, J.D.; Gebauer, D.; Sainz-Díaz, C.I. Calcium carbonate polyamorphism and its role in biomineralization: How many amorphous calcium carbonates are there? Angew. Chem. Int. Ed. 2012, 51, 11960–11970. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qian, X.; An, X. Using calcium carbonate whiskers as papermaking filler. Bioresources 2011, 6, 2435–2447. [Google Scholar]
- Falini, G.; Fermani, S. The strategic role of adsorption phenomena in biomineralization. Cryst. Res. Technol. 2013, 48, 864–876. [Google Scholar] [CrossRef]
- Neira-Carrillo, A.; Yazdani-Pedram, M.; Retuert, J.; Díaz-Dosque, M.; Gallois, S.; Arias, J.L. Selective crystallization of calcium salts by poly(acrylate)-grafted chitosan. J. Colloid Interface Sci. 2005, 286, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, D.; Cö̈lfen, H.; Verch, A.; Antonietti, M. The multiple roles of additives in CaCO3 crystallization: A quantitative case study. Adv. Mater. 2009, 21, 435–439. [Google Scholar] [CrossRef]
- Gao, Y.X.; Yu, S.H.; Guo, X.H. Double hydrophilic block copolymer controlled growth and self-assembly of CaCO3 multilayered structures at the air/water interface. Langmuir 2006, 22, 6125–6129. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.N.; Feng, J.D.; Wang, X.Y.; Wang, Z.C. Synthesis of self-assembled needle-shaped calcium carbonate superstructures. Chin. Sci. Bull. 2010, 55, 2131–2135. [Google Scholar] [CrossRef]
- Laskshminarayanan, R.; Valiyaveettil, S.; Loy, G.L. Selective nucleation of calcium carbonate polymorphs: Role of surface functionalization and poly (vinyl alcohol) additive. Cryst. Growth Des. 2003, 3, 953–958. [Google Scholar] [CrossRef]
- Butler, M.F.; Glaser, N.; Weaver, A.C.; Kirkland, M.; Heppenstall-Butler, M. Calcium carbonate crystallization in the presence of biopolymers. Cryst. Growth Des. 2006, 6, 781–794. [Google Scholar] [CrossRef]
- Neira, A.C.; Fernandez, M.S.; Retuert, J.; Arias, J.L. Effect of the crystallization chamber design on the polymorphs of calcium carbonate using the sitting-drop method. In Architecture and Application of Biomaterials and Biomolecular Materials; EXS-1:321-326; Barron, A.E., Klok, H.-A., Deming, T.J., Eds.; Materials Research Society: Warrendale, PA, USA, 2004. [Google Scholar]
- Ma, Y.F.; Gao, Y.H.; Feng, Q.L. Effects of pH and temperature on CaCO3 crystallization in aqueous solution with water soluble matrix of pearls. J. Cryst. Growth 2010, 312, 3165–3170. [Google Scholar] [CrossRef]
- Pai, R.K.; Pillai, S. Nanoparticles of amorphous calcium carbonate by miniemulsion: Synthesis and mechanism. CrystEngComm 2008, 10, 865–872. [Google Scholar] [CrossRef]
- Gomez-Morales, J.; Hernández-Hernández, A.; Sazaki, S.; García-Ruiz, J.M. Nucleation and Polymorphism of calcium carbonate by a vapor diffusion sitting drop crystallization technique. Cryst. Growth Des. 2010, 10, 963–969. [Google Scholar] [CrossRef]
- Song, R.-Q.; Colfen, H. Additive controlled crystallization. CrystEngComm 2011, 13, 1249–1276. [Google Scholar] [CrossRef]
- Mann, K.; Olsen, J.V.; Macek, B.; Gnad, F.; Mann, M. Phosphoproteins of the chicken eggshell calcified layer. Proteomics 2007, 7, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Falini, G.; Fermani, S. Chitin mineralization. Tissue Eng. 2004, 10, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, H. Chitin and collagen as universal and alternative templates in biomineralization. Int. Geol. Rev. 2010, 52, 661–669. [Google Scholar] [CrossRef]
- Arias, J.L.; Fernández, M.S. Biomimetic processes through the study of mineralized shells. Mater. Charact. 2003, 50, 189–195. [Google Scholar] [CrossRef]
- Jimenez-Lopez, C.; Rodríguez-Navarro, A.; Domínguez-Vera, J.M.; Garcia-Ruiz, J.M. Influence of lysozyme on the precipitation of calcium carbonate: A kinetic and morphologic study. Geochim. Cosmochim. Acta 2003, 67, 1667–1676. [Google Scholar] [CrossRef]
- Fernández, M.S.; Passalacqua, K.; Arias, J.I.; Arias, J.L. Partial biomimetic reconstitution of avian eggshell formation. J. Struct. Biol. 2004, 148, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Hernández, A.; Rodríguez-Navarro, A.B.; Gomez-Morales, J.; Jiménez-Lopez, C.; Nys, Y.; García-Ruiz, J.M. Influence of model globular proteins with different isoelectric points on the precipitation of calcium carbonate. Cryst. Growth Des. 2008, 8, 1495–1502. [Google Scholar] [CrossRef]
- Hernández-Hernández, A.; Vidal, M.L.; Gomez-Morales, J.; Rodríguez-Navarro, A.B.; Labas, V.; Gautron, J.; Nys, Y.; García Ruiz, J.M. Influence of eggshell matrix proteins on the precipitation of calcium carbonate (CaCO3). J. Cryst. Growth 2008, 310, 1754–1759. [Google Scholar] [CrossRef]
- Hernández-Hernández, A.; Gomez-Morales, J.; Rodríguez-Navarro, A.B.; Gautron, J.; Nys, Y.; García-Ruiz, J.M. Identification of some active proteins in the process of hen eggshell formation. Cryst. Growth Des. 2008, 8, 4330–4339. [Google Scholar] [CrossRef]
- Rao, A.; Vásquez-Quitral, P.; Drechsler, M.; Fernández, M.S.; Neira-Carrillo, A.; Arias, J.L.; Berg, J.K.; Sánchez, M.; Gebauer, D.; Cölfen, H. pH-dependent schemes of calcium carbonate formation in the presence of alginates. Cryst. Growth Des. 2016, 16, 1349–1359. [Google Scholar] [CrossRef]
- Neira-Carrillo, A.; Retuert, J.; Martínez, F.; Arias, J.L. Effect of crosslinked chitosan as a constrained volume on the in vitro calcium carbonate crystallization. J. Chil. Chem. Soc. 2008, 53, 1367–1372. [Google Scholar] [CrossRef]
- Neira-Carrillo, A.; Fernández, M.S.; Poblete Hevia, G.; Gebauer, D.; Cölfen, H.; Arias, J.L. Retrosynthesis of CaCO3 via amorphous precursor particles using gastroliths of the Red Claw lobster (Cherax quadricarinatus). J. Struct. Biol. 2017, 199, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Lédion, J.; Leroy, J.P.; Labbé, J.P. Détermination du caractère incrustant D’une eau par un essai D’entartrage accélère. TSM L’eau 1985, 80, 323–328. [Google Scholar]
- Pavez, J.; Silva, J.; Melo, F. Homogeneous calcium carbonate coating obtained by electrodeposition: In Situ atomic force microscope observations. Electrochim. Acta 2005, 50, 3488–3494. [Google Scholar] [CrossRef]
- Neira-Carrillo, A.; Sánchez, M.; Vásquez-Quitral, P.; Vargas-Fernández, A.; Silva, J.F. Control of calcium oxalate morphology through electrocrystallization as an electrochemical approach for preventing pathological disease. Ionics 2015, 21, 3141–3149. [Google Scholar] [CrossRef]
- Hau, N.; Chang, Y.; Feng, C. Kinetics study of silver Electrocrystallization on (3-mercaptopropyl) trimethoxysilane-grafted indium tin oxide plastic substrate. Electrochim. Acta 2015, 158, 121–128. [Google Scholar] [CrossRef]
- Gozalvo, R. Efecto de un Extracto de Origen Vegetal en la Mineralizacion Patologica de Oxalato de Calcio Sobre Fibras Polimericas Electrohiladas. Bachelor´s Thesis, Facultad de Ciencias de la Salud, Universidad San Sebastián, Santiago, Chile, 2017. [Google Scholar]
- Cardenas, G.; Cabrera, G.; Taboada, E.; Miranda, S.P. Chitin characterization by SEM, FTIR, XRD, and 13C cross polarization/mass angle spinning NMR. J. Appl. Polym. Sci. 2004, 93, 1876–1885. [Google Scholar] [CrossRef]
- Neira-Carrillo, A.; Retuert, J.; Martínez, F.; Arias, J.L. Calcium carbonate growth modification by constituents released from porous filter membranes. J. Chil. Chem. Soc. 2008, 53, 1469–1473. [Google Scholar] [CrossRef]
- Neira-Carrillo, A.; Vásquez-Quitral, P.; Díaz, M.P.; Fernández, M.S.; Arias, J.L.; Yazdani-Pedram, M. Control of calcium carbonate crystallization by using anionic polymethylsiloxanes as templates. J. Solid State Chem. 2012, 194, 400–408. [Google Scholar] [CrossRef]
- Neira-Carrillo, A.; Krishna Pai, R.; Fernández, M.S.; Carreño, E.; Vásquez-Quitral, P.; Arias, J.L. Synthesis and characterization of sulfonated polymethylsiloxane polymer as template for Crystal growth of CaCO3. Colloid Polym. Sci. 2009, 287, 385–393. [Google Scholar] [CrossRef]
- Neira-Carrillo, A.; Vásquez Quitral, P.; Yazdani-Pedram, M.; Arias, J.L. Crystal growth of CaCO3 induced by new carboxylated monomethylitaconate grafted polymethylsiloxane. Eur. Polym. J. 2010, 46, 1184–1193. [Google Scholar] [CrossRef]
- Neira-Carrillo, A.; Acevedo, D.F.; Miras, M.C.; Barbero, C.A.; Gebauer, D.; Cölfen, H.; Arias, J.L. Influence of conducting polymers based on carboxylated polyaniline on in vitro CaCO3 crystallization. Langmuir 2008, 24, 12496–12507. [Google Scholar] [CrossRef] [PubMed]
- Castañeda, E. Efecto de los Ácidos Poliacrílico y Fítico y de Quitosano Sobre la Electrocristalización de Carbonato Cálcico. Bachelor´s Thesis, Facultad de Ciencias Veterinarias y Pecuarias, Universidad de Chile, Santiago, Chile, 2015. [Google Scholar]
- Vagenas, N.V.; Gatsouli, A.; Kontoyannis, C.G. Quantitative analysis of synthetic calcium carbonate polymorphs using FT-IR spectroscopy. Talanta 2003, 59, 831–836. [Google Scholar] [CrossRef]
- Rodriguez-Blanco, J.D.; Shaw, S.; Benning, L.G. The kinetics and mechanisms of amorphous calcium carbonate (ACC) crystallization to calcite, via vaterite. Nanoscale 2011, 3, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xiang, L. Controllable synthesis of calcium carbonate polymorphs at different temperatures. Powder Technol. 2009, 189, 64–69. [Google Scholar] [CrossRef]
- Destainville, A.; Champion, E.; Bernache-Assollante, D.; Laborde, E. Synthesis, characterization and thermal behavior of apatite tricalcium phosphate. Mater. Chem. Phys. 2003, 80, 269–277. [Google Scholar] [CrossRef]
- Han, J.-K.; Song, H.-Y.; Saito, F.; Lee, B.-T. Synthesis of height purity nano-sized hydroxyapatite powder by microwave-hydrothermal method. Mater. Chem. Phys. 2006, 99, 235–239. [Google Scholar] [CrossRef]
- Berzina-Cimdina, L.; Borodajenko, N. Research of calcium phosphates using Fourier transform infrared spectroscopy. In Infrared Spectroscopy-Materials Science, Engineering and Technology; InTech: London, UK, 2012; ISBN 978-953-51-0537-4. [Google Scholar]
- Jastrzębski, W.; Sitarz, M.; Rokita, M.; Bułat, K. Infrared spectroscopy of different phosphates structures. Spectroc. Acta A Mol. Biomol. Spectrosc. 2011, 79, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, R.; Egawa, T.; Furuike, T.; Nair, S.V.; Tamur, H. Synthesis, characterization, and thermal properties of phosphorylated chitin for biomedical applications. Polym. Eng. Sci. 2009, 49, 844–849. [Google Scholar] [CrossRef]
- Stawski, D.; Rabiej, S.; Herczynska, L.; Draczynski, Z. Thermogravimetric analysis of chitins of different origin. J. Therm. Anal. Calorim. 2008, 93, 489–494. [Google Scholar] [CrossRef]
- Juhasz-Bortuzzo, J.A.; Myszka, B.; Silva, R.; Boccaccini, A.R. Sonosynthesis of vaterite-Type calcium carbonate. Cryst. Growth Des. 2017, 17, 2351–2356. [Google Scholar] [CrossRef]
- Nishi, N.; Ebina, A.; Nishimura, S.; Tsutsumi, A.; Hasegawa, O.; Tokura, S. Highly phosphorylated derivatives of chitin, partially deacetylated chitin and chitosan as new functional polymers: Preparation and characterization. Int. J. Biol. Macromol. 1986, 8, 311–317. [Google Scholar] [CrossRef]
- He, Z.; Honeycutt, C.W. A modified molybdenum blue method for orthophosphate determination suitable for investigating enzymatic hydrolysis of organic phosphates. Commun. Soil Sci. Plant Anal. 2005, 36, 1373–1383. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butto, N.; Cabrera-Barjas, G.; Neira-Carrillo, A. Electrocrystallization of CaCO3 Crystals Obtained through Phosphorylated Chitin. Crystals 2018, 8, 82. https://doi.org/10.3390/cryst8020082
Butto N, Cabrera-Barjas G, Neira-Carrillo A. Electrocrystallization of CaCO3 Crystals Obtained through Phosphorylated Chitin. Crystals. 2018; 8(2):82. https://doi.org/10.3390/cryst8020082
Chicago/Turabian StyleButto, Nicole, Gustavo Cabrera-Barjas, and Andrónico Neira-Carrillo. 2018. "Electrocrystallization of CaCO3 Crystals Obtained through Phosphorylated Chitin" Crystals 8, no. 2: 82. https://doi.org/10.3390/cryst8020082
APA StyleButto, N., Cabrera-Barjas, G., & Neira-Carrillo, A. (2018). Electrocrystallization of CaCO3 Crystals Obtained through Phosphorylated Chitin. Crystals, 8(2), 82. https://doi.org/10.3390/cryst8020082