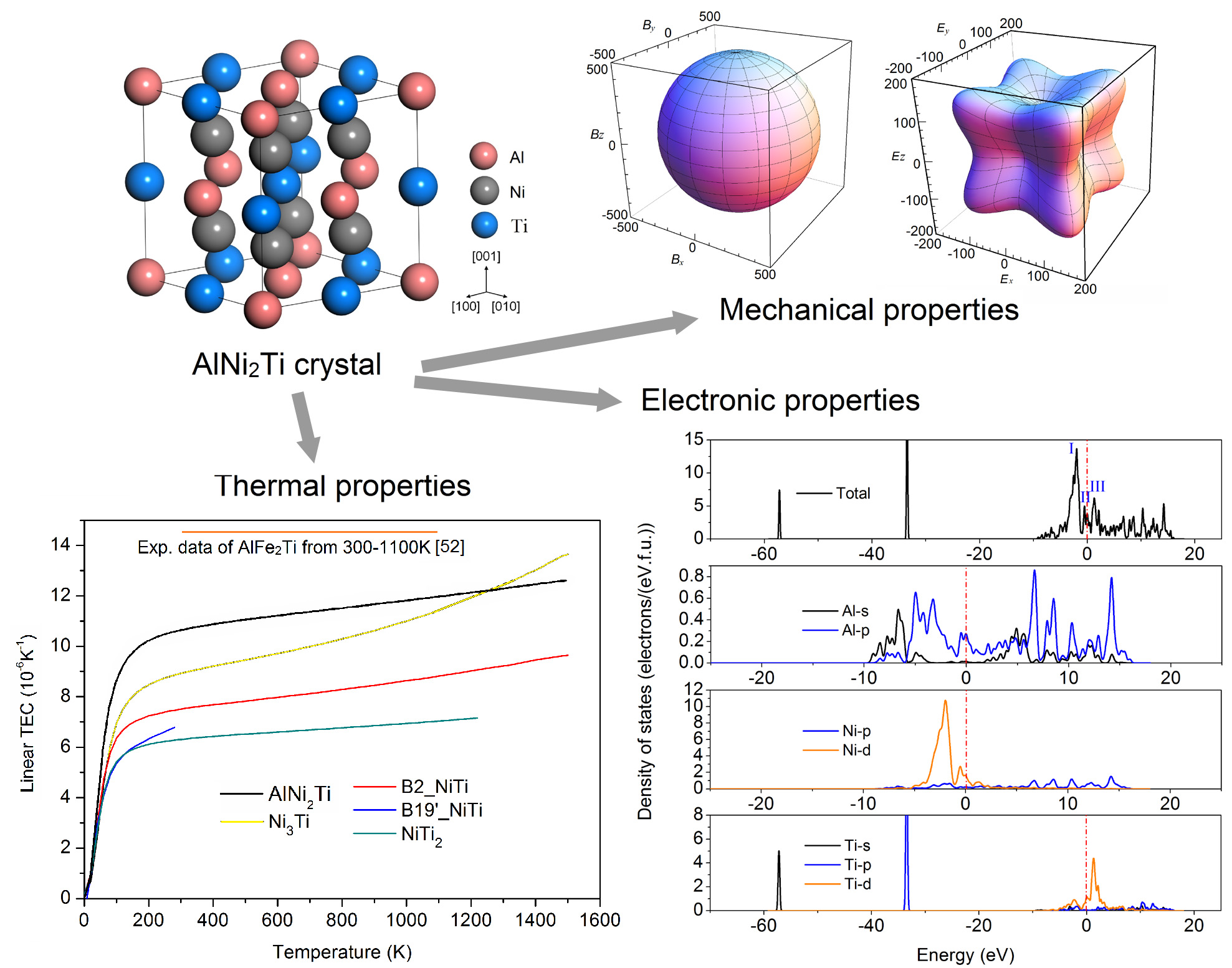
First-Principles Investigations of the Structural, Anisotropic Mechanical, Thermodynamic and Electronic Properties of the AlNi2Ti Compound

Abstract
:
1. Introduction
2. Calculation Methods
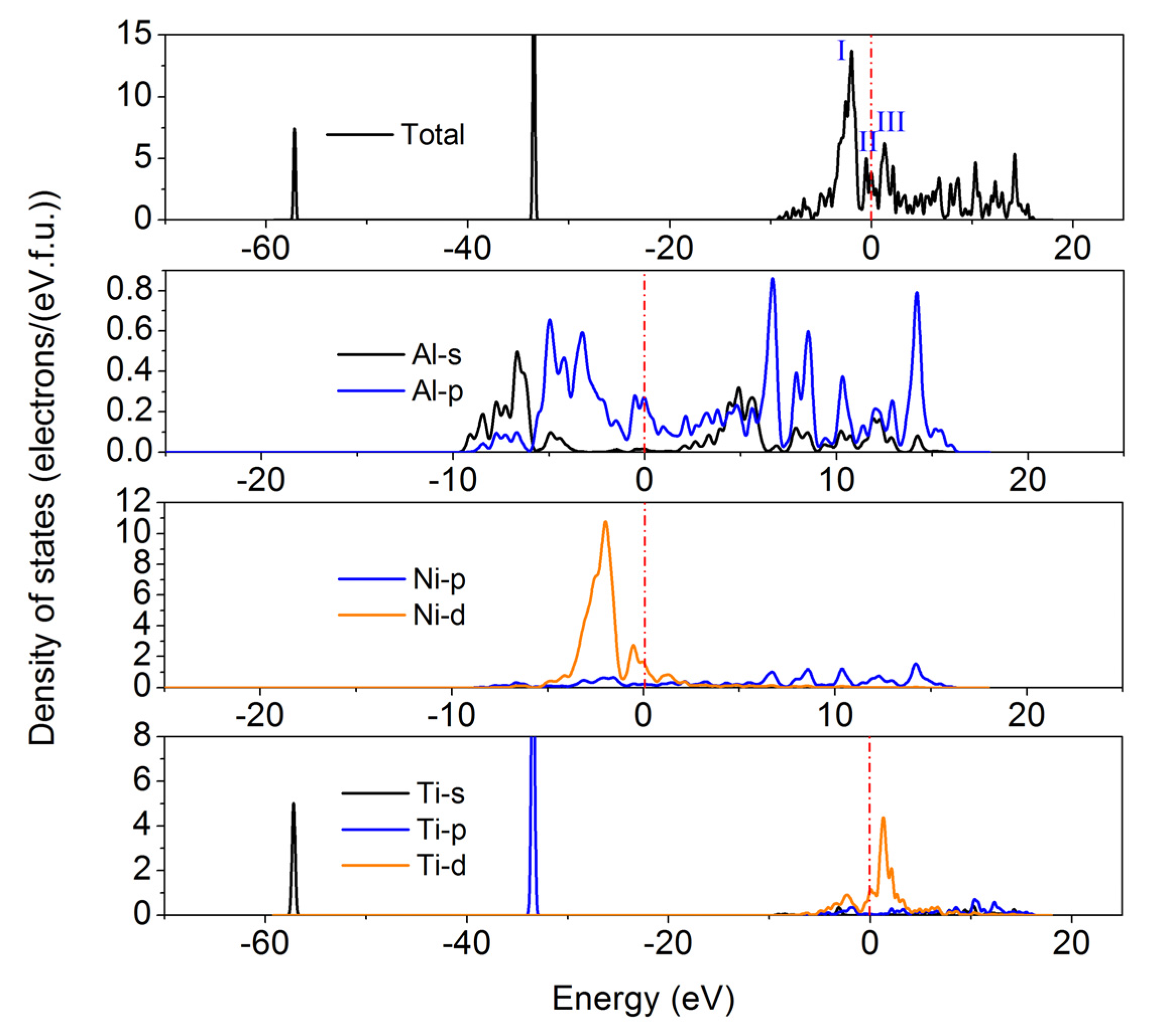
3. Results and Discussion
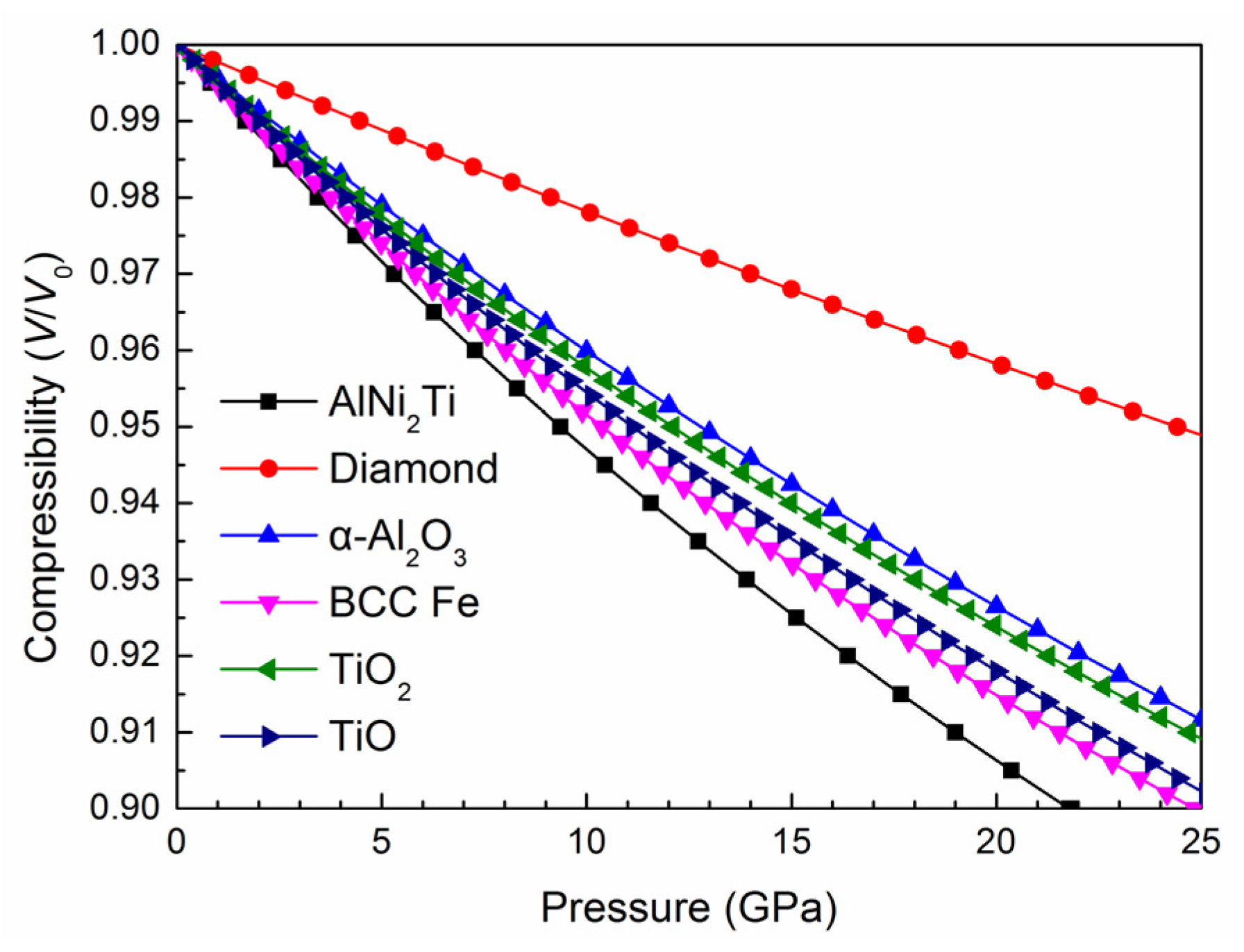
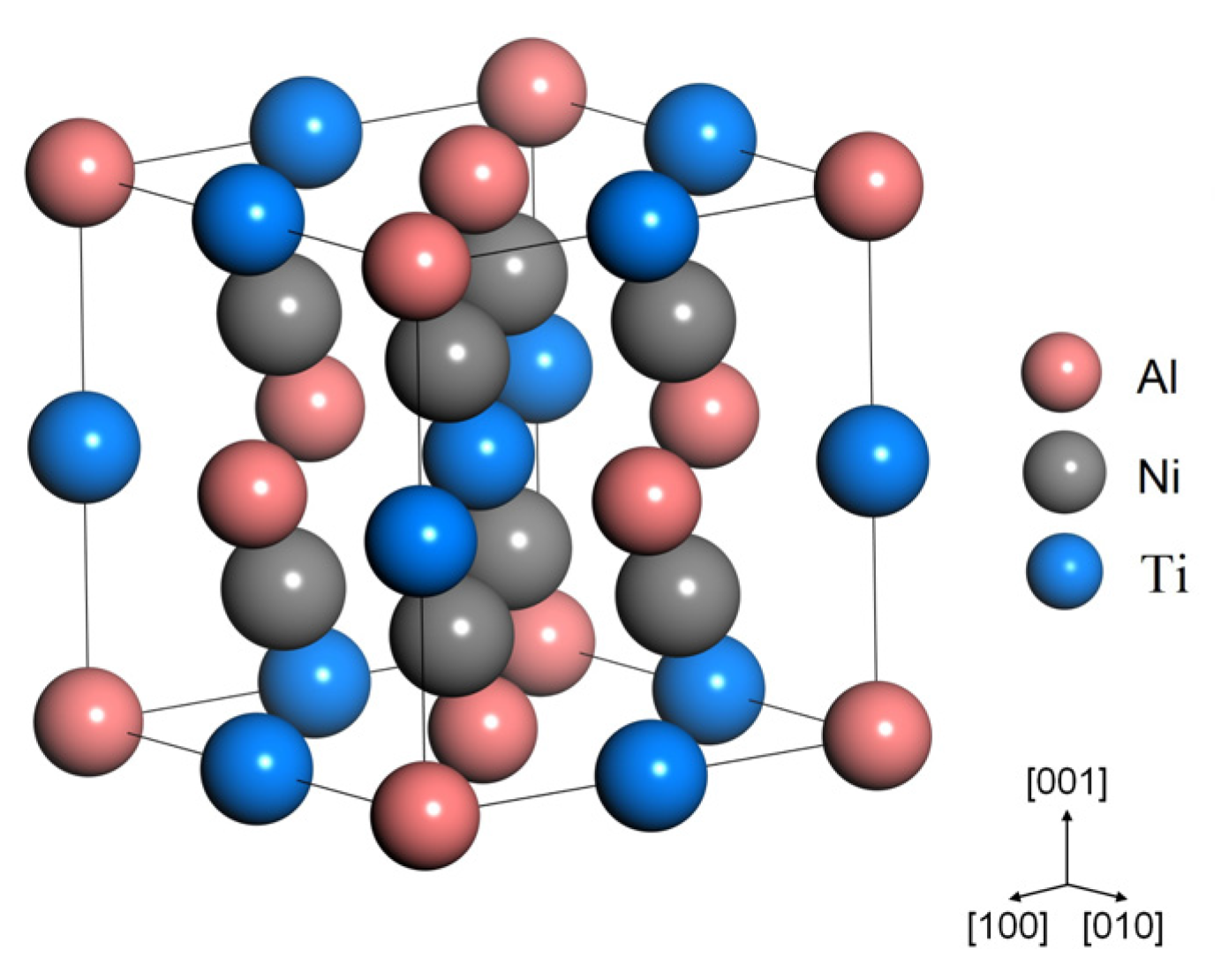
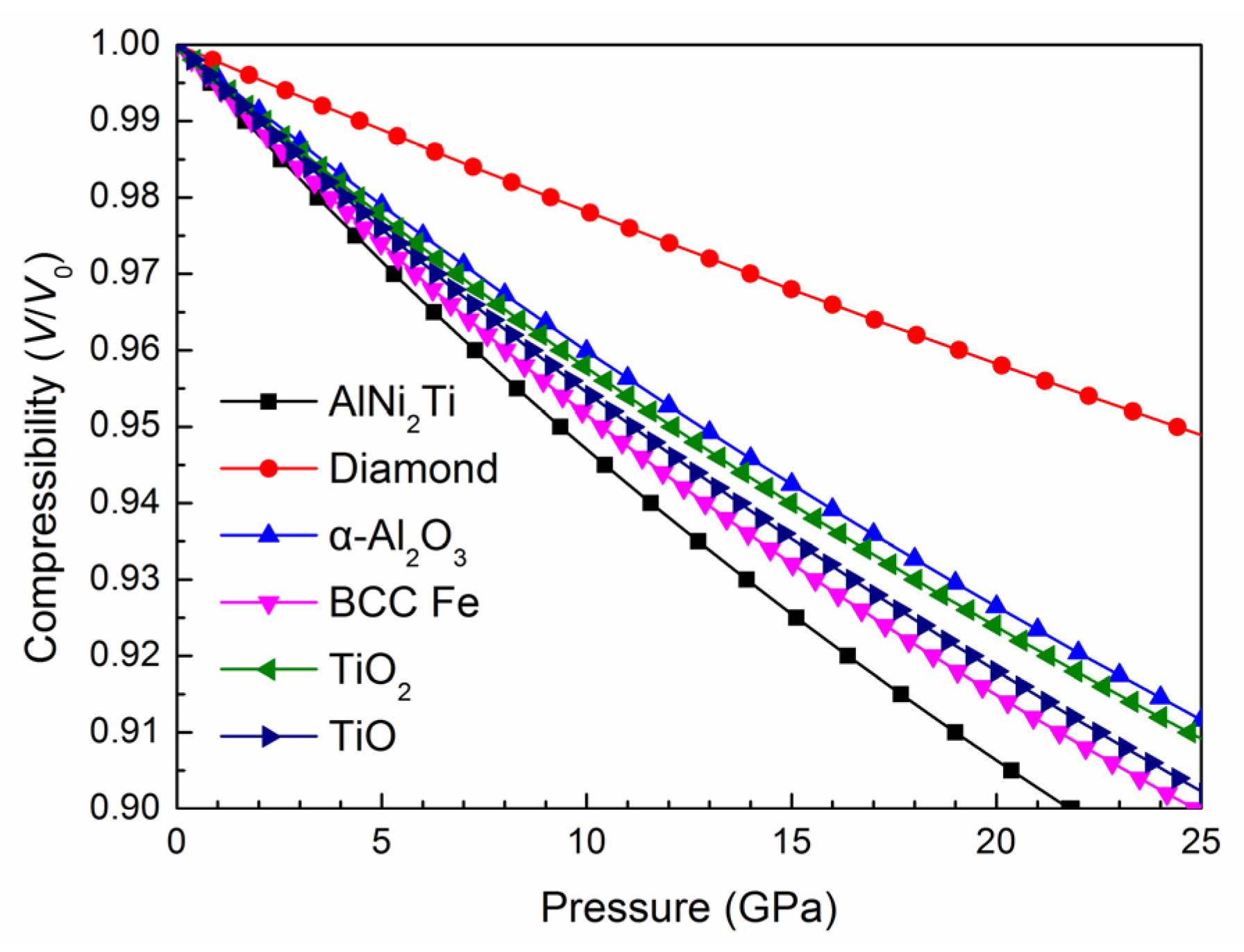
3.1. Crystal Parameters and Mechanical Properties
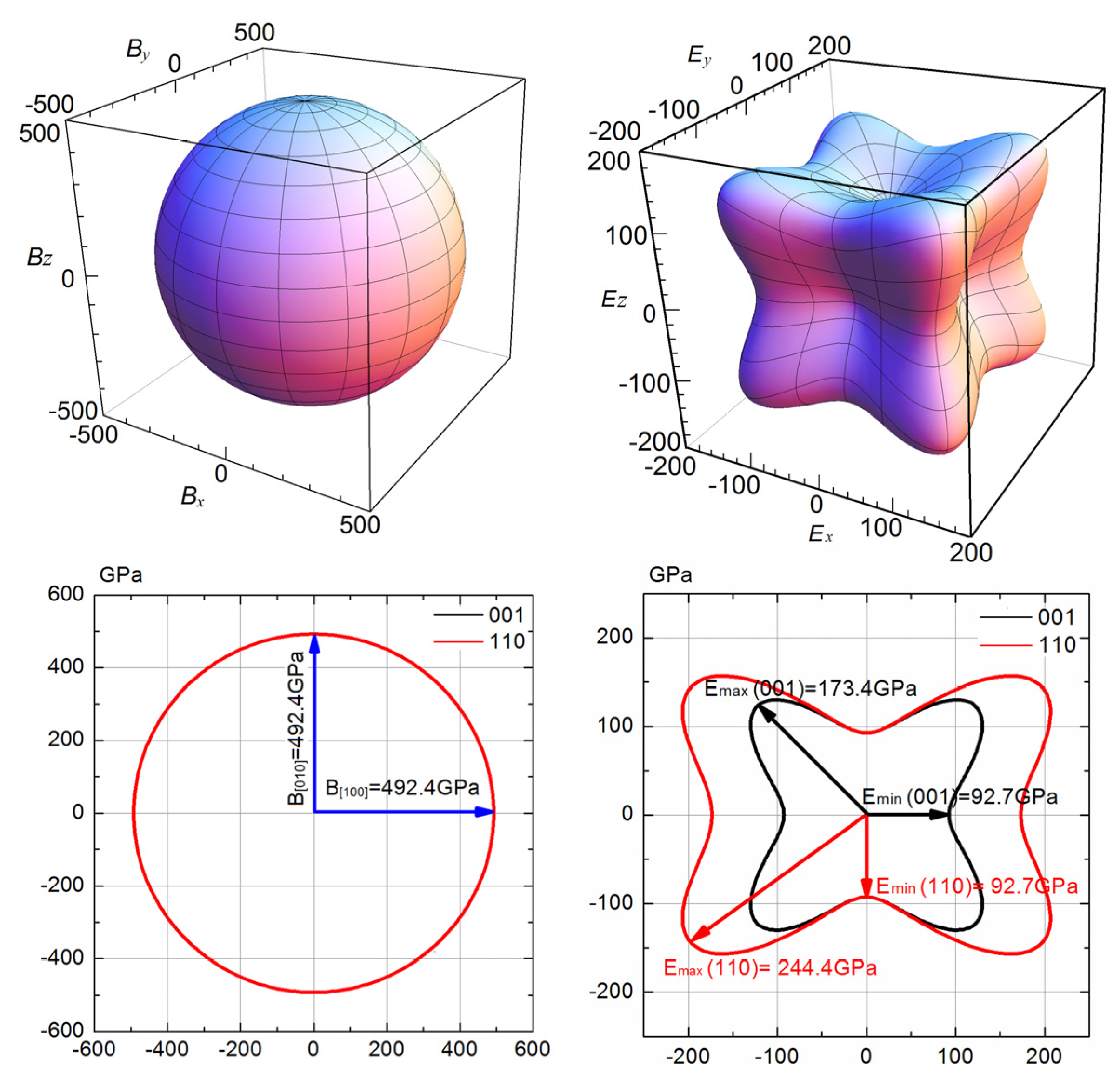
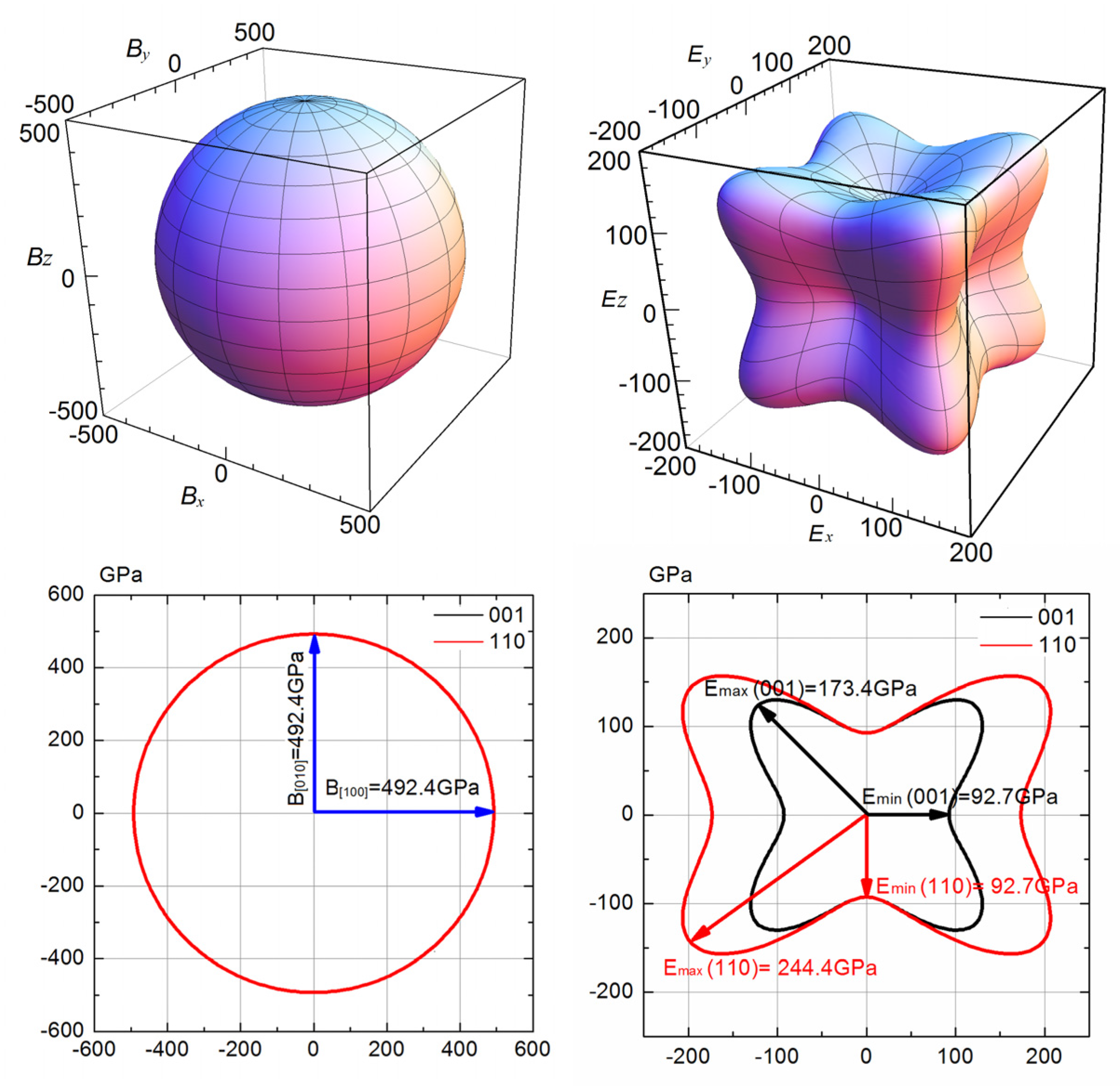
3.2. Anisotropy of Mechanical Moduli
3.3. Sound Velocities and Thermal Conductivity
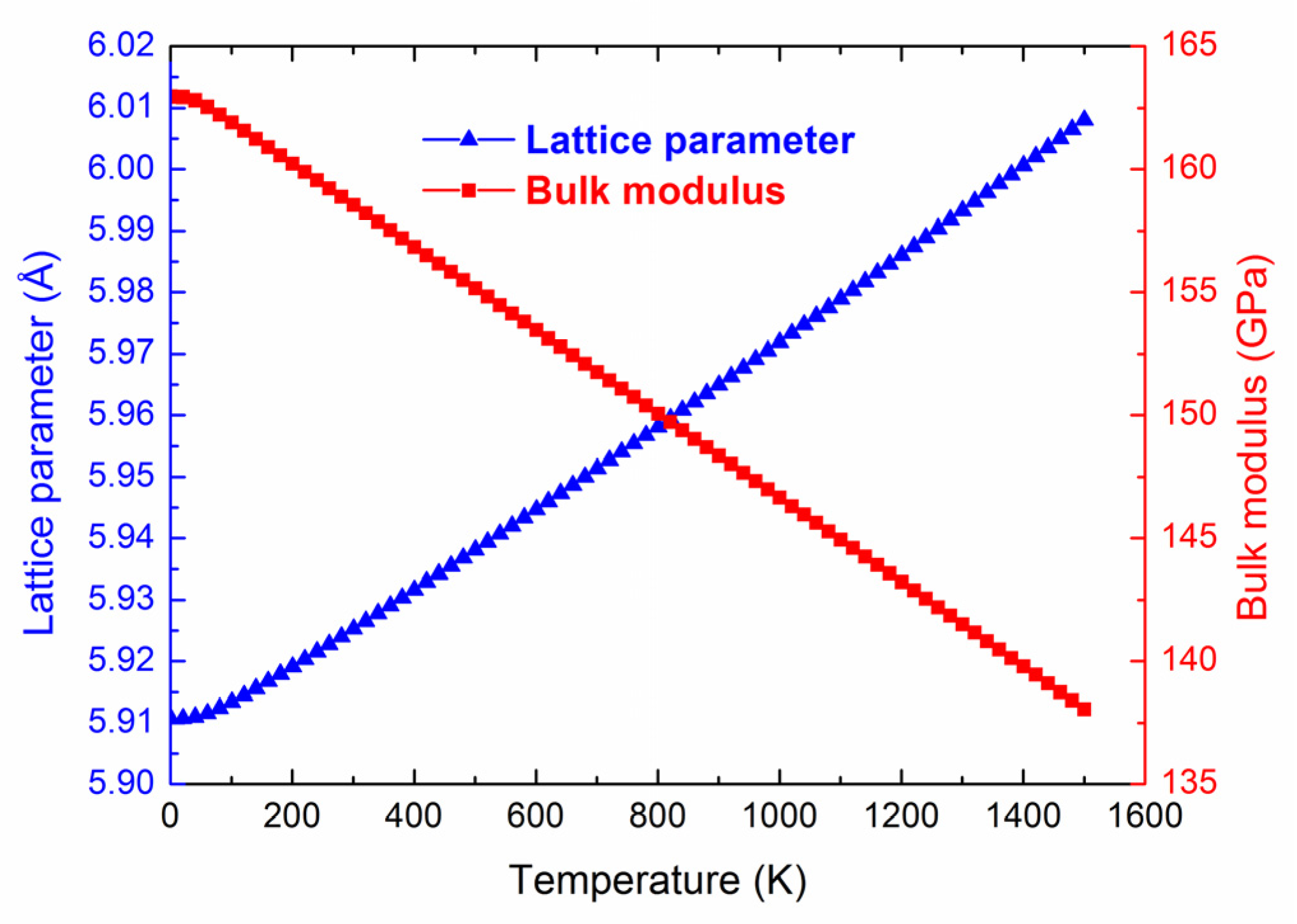
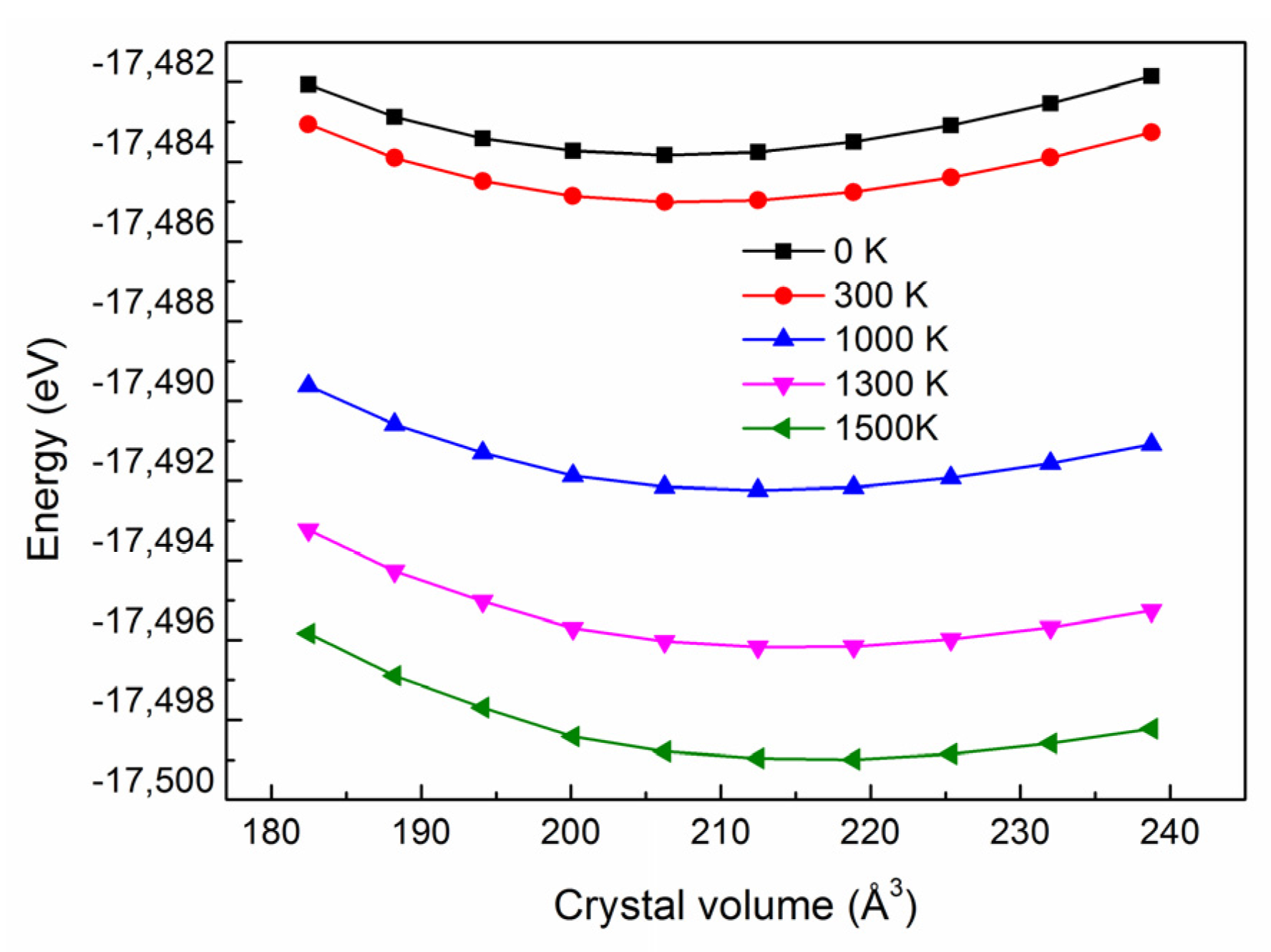
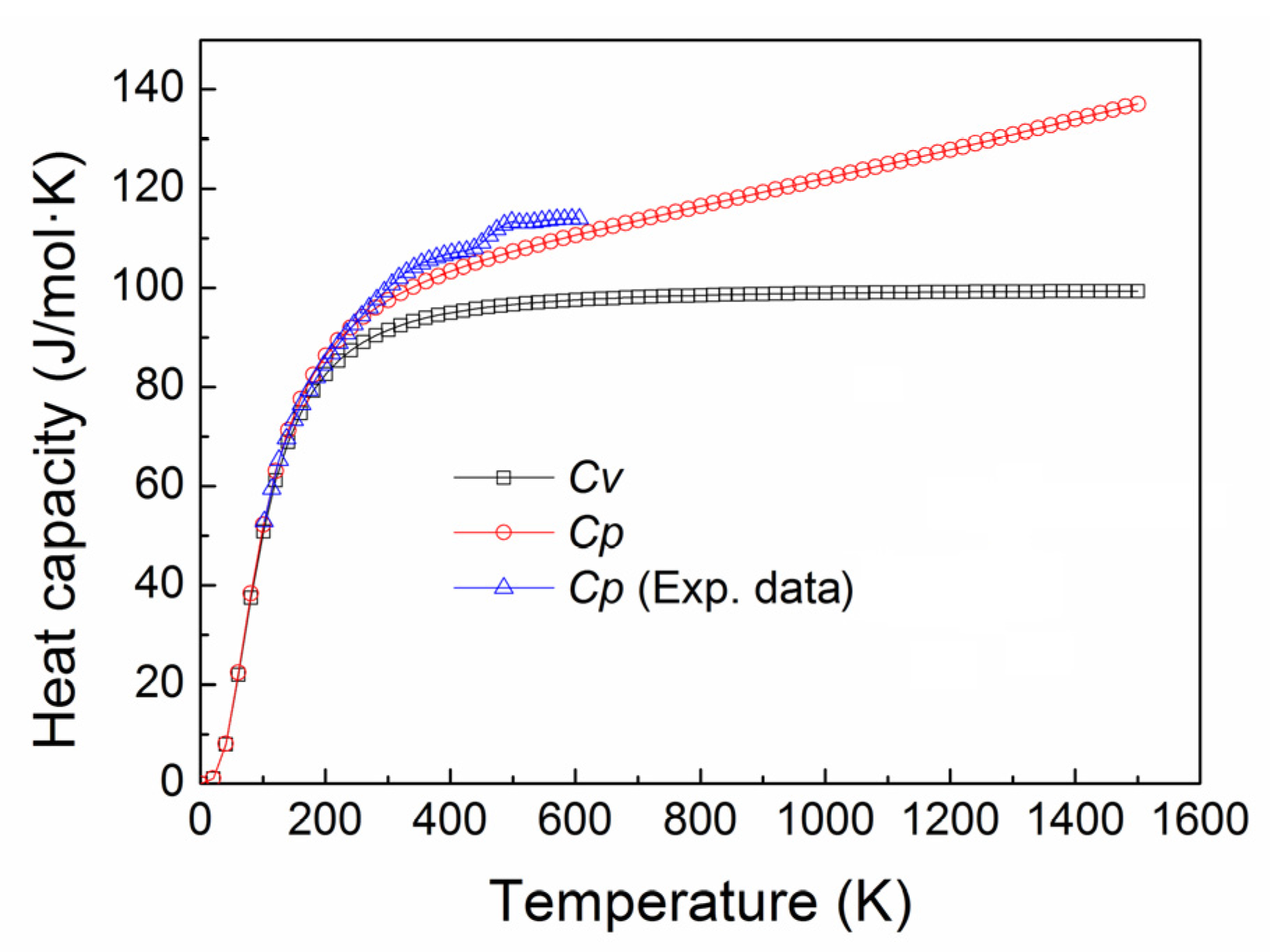
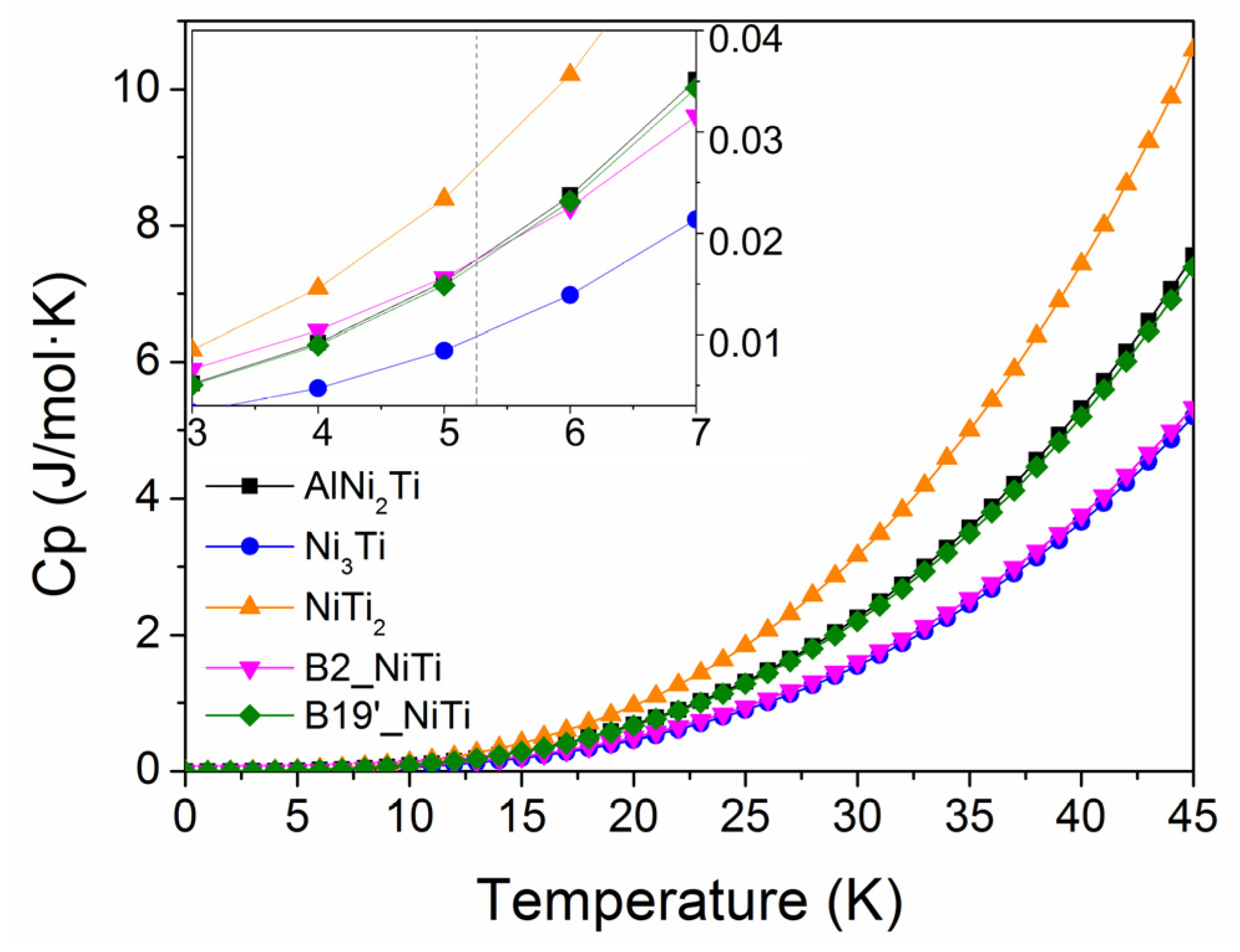
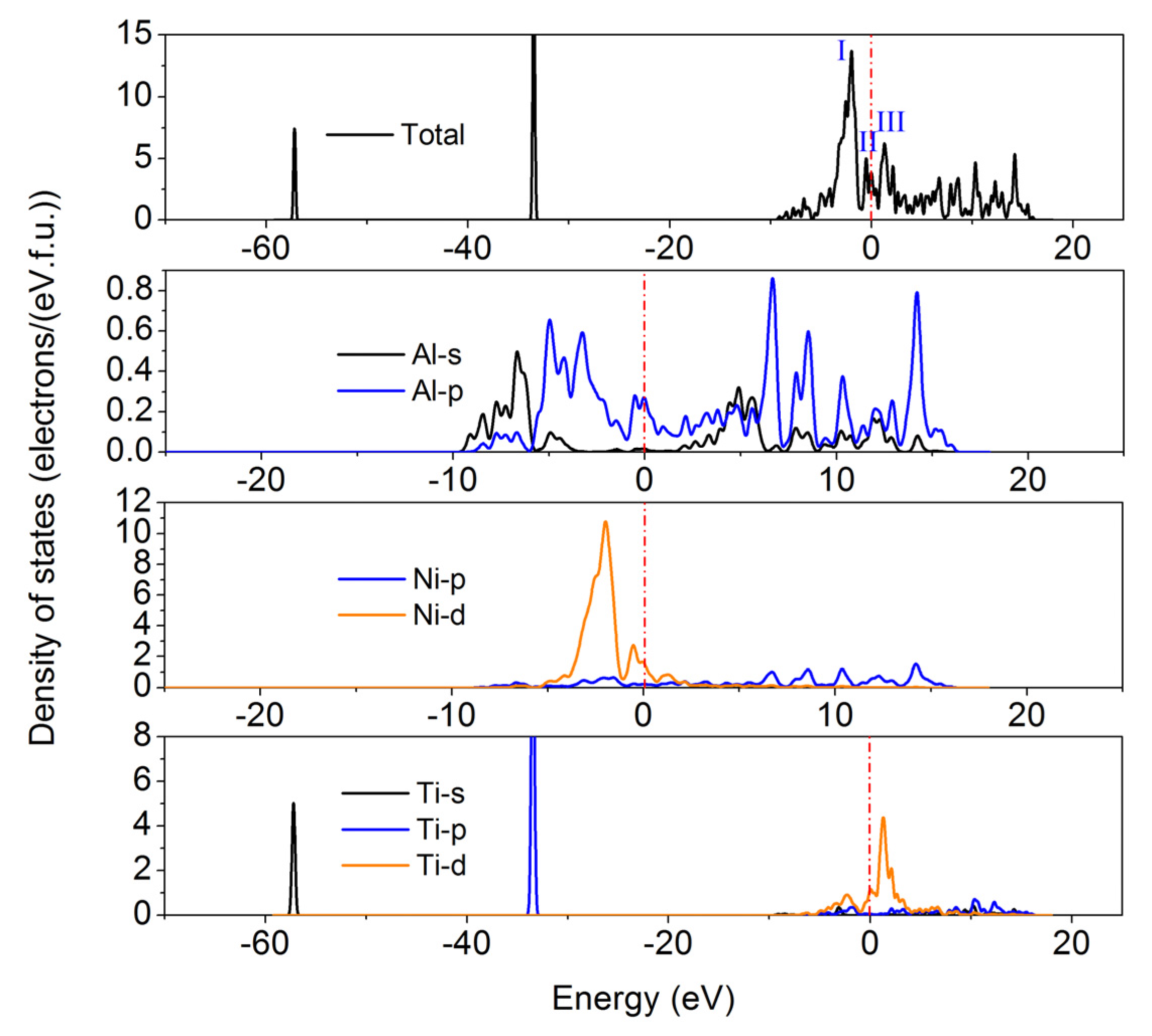
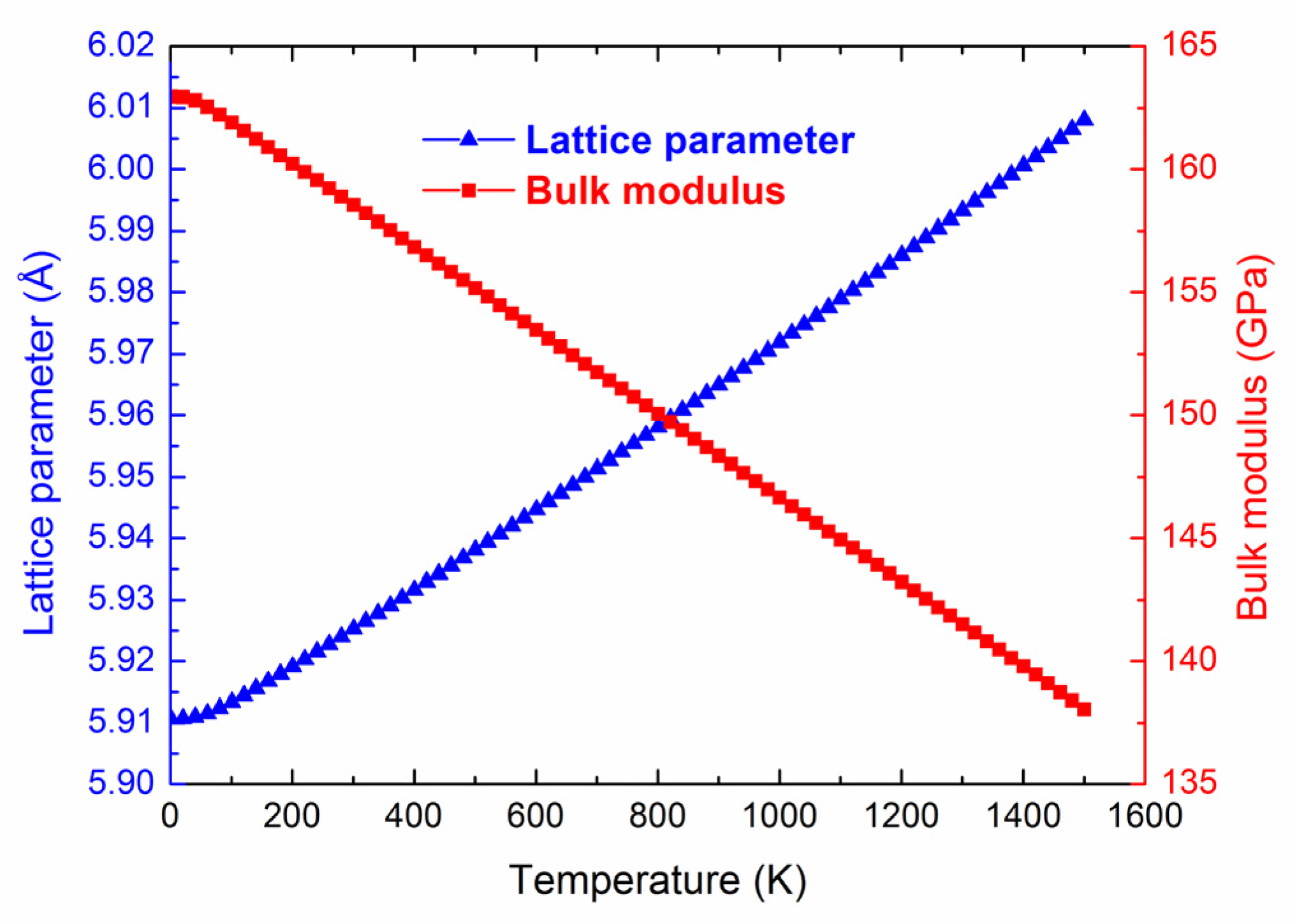
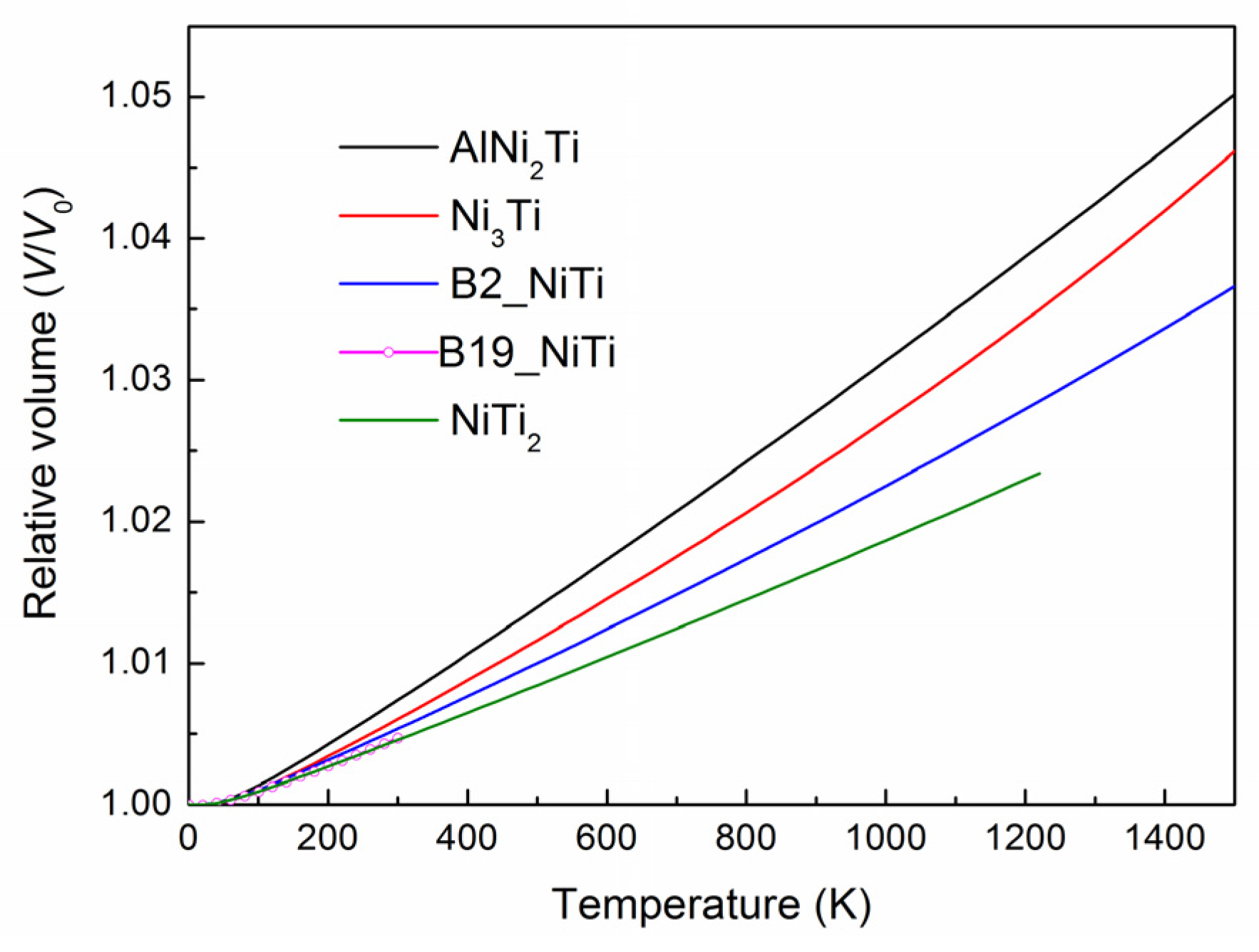
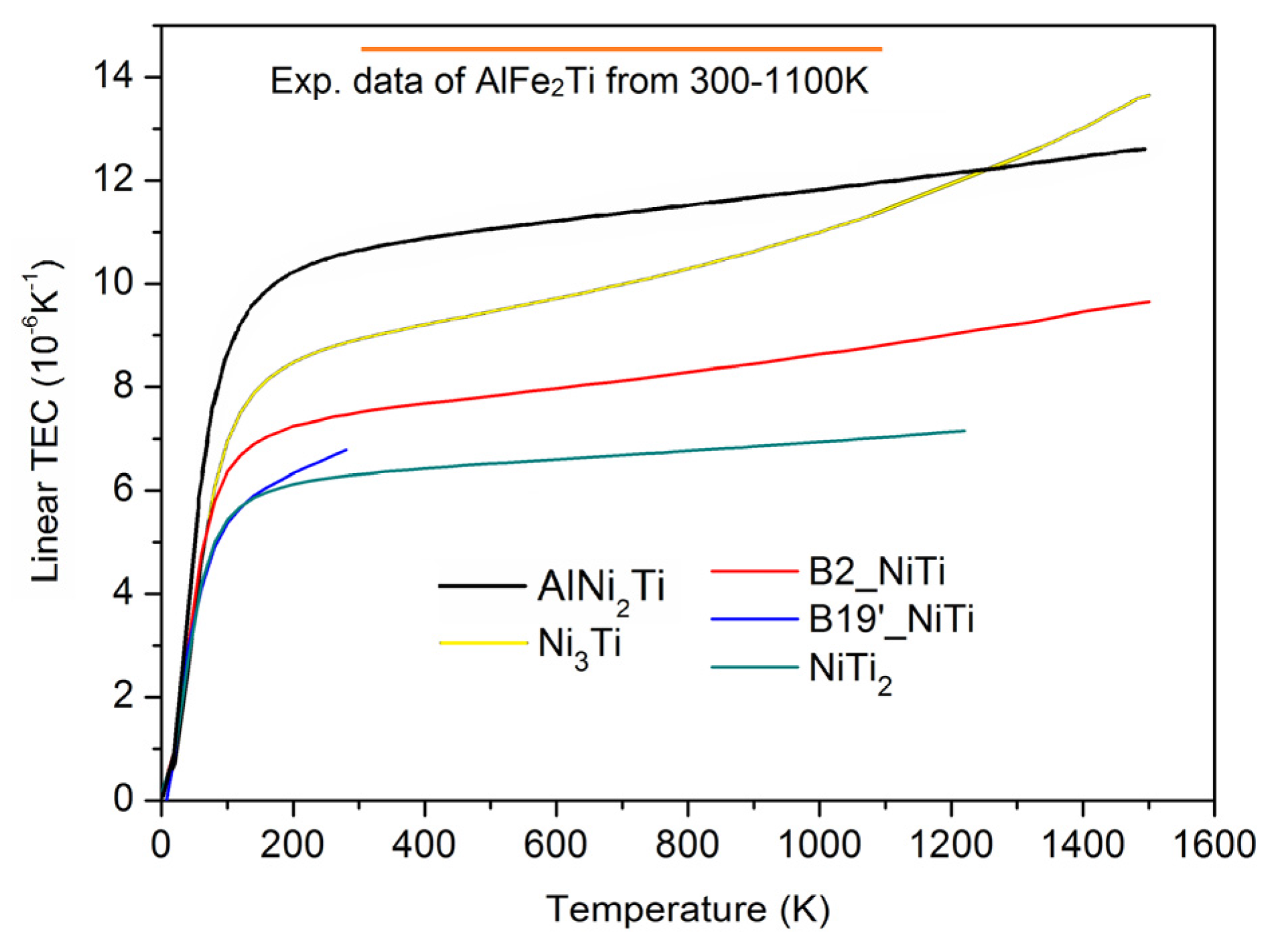
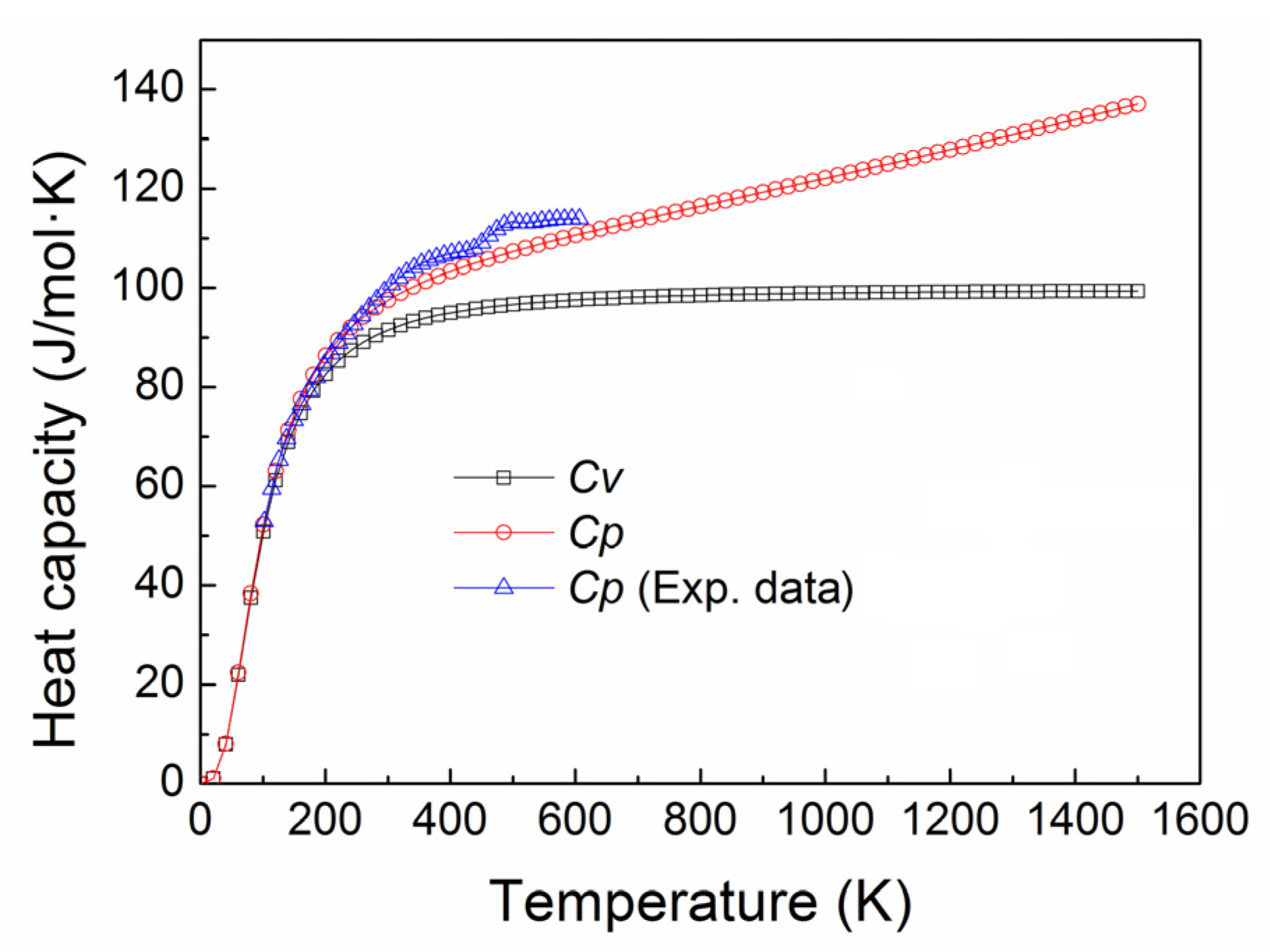
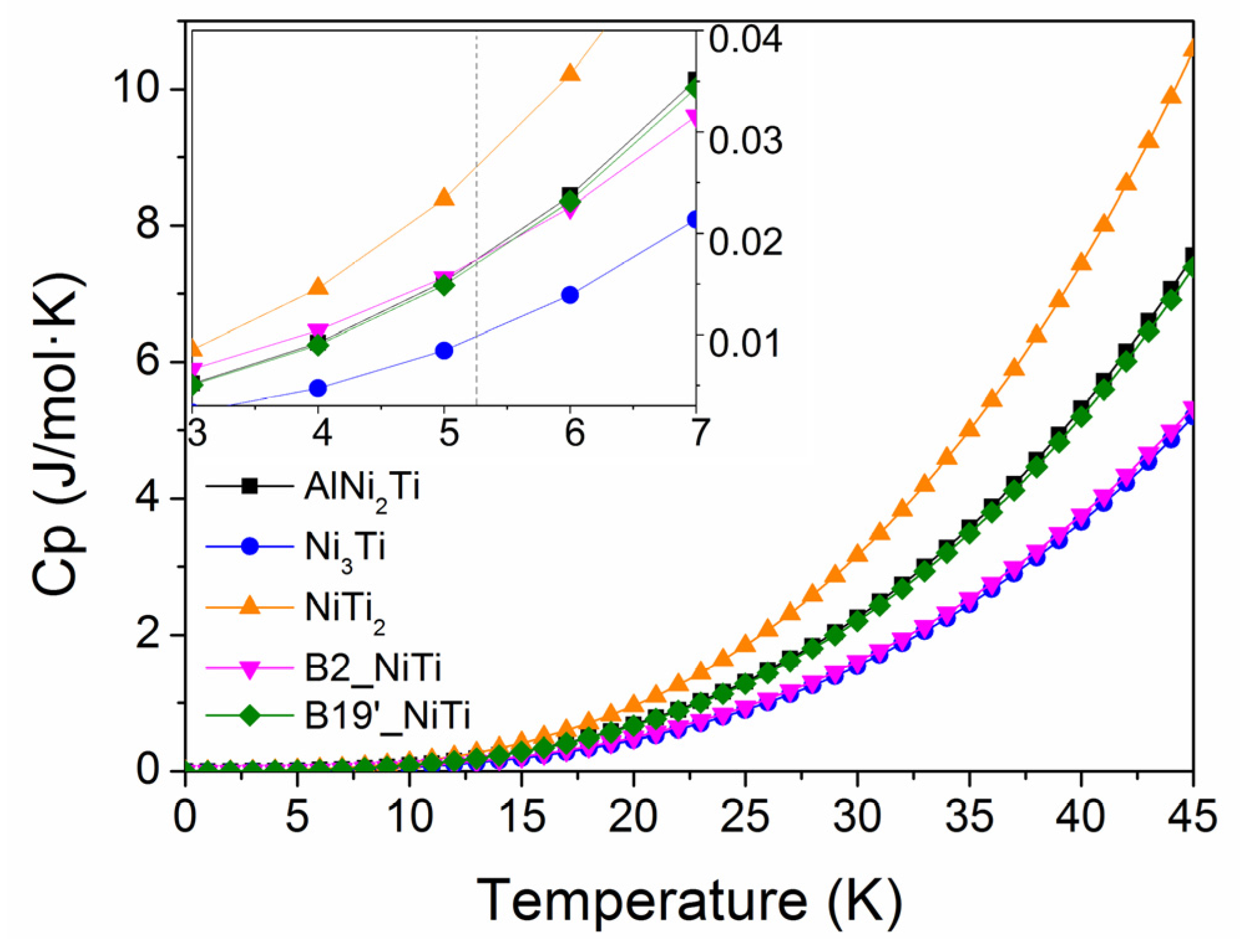
3.4. Thermal Expansion Coefficient and Heat Capacity
3.5. Formatting of Mathematical Components
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Heusler, F. Ueber magnetische manganlegierungen (on magnetic manganese alloys). Verhandlungen der Deutschen Physikalischen Gesellschaft 1903, 5, 219. (In German) [Google Scholar]
- Nakata, J.; Terada, Y.; Takizawa, S.; Ohkubo, K.; Mohri, T.; Suzuki, T. Thermal conductivity in X2YZ heusler type intermetallic compounds. Mater. Trans. JIM 1996, 37, 442–447. [Google Scholar] [CrossRef]
- Oxley, D.P.; Tebble, R.S.; Williams, K.C. Heusler alloys. J. Appl. Phys. 1963, 34, 1362–1363. [Google Scholar] [CrossRef]
- Bradley, A.J.; Rogers, J.W. The crystal structure of the Heusler alloys. Proc. R. Soc. 1934, A144, 340–359. [Google Scholar] [CrossRef]
- Wilson, A.W.; Howe, J.M. Effect of alloying additions on beta’ precipitation in NiAl-Ti base alloys. Acta Mater. 2001, 49, 2653–2660. [Google Scholar] [CrossRef]
- Koizumi, Y.; Ro, Y.; Nakazawa, S.; Harada, H. NiTi-base intermetallic alloys strengthened by Al substitution. Mater. Sci. Eng. A 1997, 223, 36–41. [Google Scholar] [CrossRef]
- Lin, W.; Freeman, A.J. Cohesive properties and electronic-structure of Heusler L21-phase compounds Ni2XAl (X = Ti, V, Zr, Nb, Hf and Ta). Phys. Rev. B 1992, 45, 61–68. [Google Scholar] [CrossRef]
- Ramos, A.S.; Vieira, M.T.; Simoes, S.; Viana, F.; Vieira, M.F. Reaction-assisted diffusion bonding of advanced materials. Defect. Diffus. Forum 2010, 297–301, 972–977. [Google Scholar] [CrossRef]
- Bozzolo, G.; Khalil, J.; Bartow, M.R.; Noebe, D. Atomistic modeling of ternary and quaternary ordered intermetallic alloys. In Materials Research Society Symposium Proceedings Vol. 646, Proceedings of the High-Temperature Ordered Intermetallic Alloys IX. Symposium, Boston, MA, USA, 27–29 November 2000; Schneibel, J.H., Hemker, K.J., Noebe, R.D., Hanada, S., Sauthoff, G., Eds.; Materials Research Society: Warrendale, PA, USA, 2001; p. N6.2.1-8. [Google Scholar]
- Bozzolo, G.; Noebe, R.B.; Ferrante, J. BFS simulation and experimental analysis of the effect of Ti additions on the structure of NiAl. J. Comput. Aided Mater. Des. 1999, 6, 33–68. [Google Scholar] [CrossRef]
- Bozzolo, G.; Noebe, R.B.; Ferrante, J.; Garg, A. Atomistic simulations of alloying additions to NiAl. Mater. Sci. Eng. A 1997, 239–240, 769–776. [Google Scholar] [CrossRef]
- Gale, W.F.; Abdo, Z.A.M. Microstructural development in cast alloys based on the β-NiAl–β′-Ni2AlTi–γ′-Ni3Al–α-Cr system. J. Mater. Sci. 1998, 33, 2299–2304. [Google Scholar] [CrossRef]
- Field, R.D.; Darolia, R.; Lahrman, D.F. Precipitation in NiAl/Ni2AlTi alloys. Scr. Metall. 1989, 23, 1469–1474. [Google Scholar] [CrossRef]
- Cahn, R.W. Nickel Alumina-Titanium Alloy Contg. Epitaxially Related Phases-Having Combined Strength, High Temp. Creep Resistance and Room Temp. Ductility or Plastic Deformability, for Gas Turbine Components. Patent WO9209712-A1, 11 June 1992. [Google Scholar]
- Matano, T.; Kimura, Y.; Miura, S.; Mishima, Y. Microstructure and mechanical properties of the L12/L21 two-phase alloys in the quaternary Co-Al-Ni-Ti system. In High-Temperature Ordered Intermetallic Alloys VI. Symposium, Vol. 2, Proceedings of the High-Temperature Ordered Intermetallic Alloys VI. Symposium, Boston, MA, USA, 28 November–1 December 1994; Horton, J., Baker, I., Hanada, S., Noebe, R.D., Schwartz, D.S., Eds.; Materials Research Society: Pittsburgh, PA, USA, 1995; pp. 1377–1382. [Google Scholar]
- Mishima, Y.; Lee, E.H.; Liu, C.T. Microstructure, phase constitution and tensile properties of Co-Ni-Ti-Al base multiphase intermetallic alloys. Mater. Trans. 1995, 36, 1031–1040. [Google Scholar] [CrossRef]
- Song, G.; Sun, Z.Q.; Poplawsky, J.D.; Xu, X.D.; Chen, M.W.; Liaw, P.K. Primary and secondary precipitates in a hierarchical-precipitate-strengthened ferritic alloy. J. Alloys Compd. 2017, 706, 584–588. [Google Scholar] [CrossRef]
- Song, G.; Sun, Z.Q.; Li, L.; Clausen, B.; Zhang, S.Y.; Gao, Y.F.; Liaw, P.K. High temperature deformation mechanism in hierarchical and single precipitate strengthened ferritic alloys by in situ neutron diffraction studies. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Sun, Z.Q.; Clausen, B.; Liaw, P.K. Microstructural characteristics of a Ni2TiAl-precipitate-strengthened ferritic alloy. J. Alloys Compd. 2017, 693, 921–928. [Google Scholar] [CrossRef]
- Song, G.L.; Sun, Z.Q.; Poplawsky, J.D.; Gao, Y.F.; Liaw, P.K. Microstructural evolution of single Ni2TiAl or hierarchical NiAl/Ni2TiAl precipitates in Fe-Ni-Al-Cr-Ti ferritic alloys during thermal treatment for elevated-temperature applications. Acta Mater. 2017, 127, 1–16. [Google Scholar] [CrossRef]
- Song, G.; Sun, Z.Q.; Li, L.; Xu, X.D.; Rawlings, M.; Liebscher, C.H.; Clausen, B.; Poplawsky, J.; Leonard, D.N.; Huang, S.Y. Ferritic alloys with extreme creep resistance via coherent hierarchical precipitates. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Sharp, J.; Rainforth, W.M. Microstructural evolution of Mn-based maraging steels and their influences on mechanical properties. Mater. Sci. Eng. A 2016, 674, 286–298. [Google Scholar] [CrossRef]
- Qian, F.; Sharp, J.; Rainforth, W.M. Characterisation of L21-ordered Ni2TiAl precipitates in Fe-Mn maraging steel. Mater. Charact. 2016, 118, 199–205. [Google Scholar] [CrossRef]
- Liebscher, C.H.; Radmilovic, V.R.; Dahmen, U.; Vo, N.Q.; Dunand, D.C.; Asta, M.; Ghosh, G. A hierarchical microstructure due to chemical ordering in the bcc lattice: Early stages of formation in a ferritic Fe-Al-Cr-Ni-Ti alloy. Acta Mater. 2015, 92, 220–232. [Google Scholar] [CrossRef]
- Sun, Z.; Liebscher, C.H.; Huang, S.; Teng, Z.; Song, G.; Wang, G.; Asta, M.; Rawlings, M.; Fine, M.E.; Liaw, P.K. New design aspects of creep-resistant NiAl-strengthened ferritic alloys. Scr. Mater. 2013, 68, 384–388. [Google Scholar] [CrossRef]
- Raqlings, M.J.S.; Liebscher, C.H.; Asta, M.; Dunand, D.C. Effect of titanium additions upon microstructure and properties of precipitation-strengthened Fe-Ni-Al-Cr ferritic alloys. Acta Mater. 2017, 128, 103–112. [Google Scholar] [CrossRef]
- Jung, J.; Ghosh, G.; Isheim, D.G.; Olson, B. Design of nanodispersion strengthened TiNi-base shape memory alloys. In Proceedings of the International Conference on Shape Memory and Superelastic Technologies, Pacific Grove, CA, USA, 5–8 May 2003; Pelton, A.R., Duerig, T., Eds.; SMST Society Inc.: Menlo Park, CA, USA, 2004; pp. 23–32. [Google Scholar]
- Hsu, D.H.D.; Hornbuckle, B.C.; Valderrama, B.; Barrie, F.; Henderson, H.B.; Thompson, G.B.; Manuel, M.V. The effect of aluminum additions on the thermal, microstructural, and mechanical behavior of NiTiHf shape memory alloys. J. Alloys Compd. 2015, 638, 67–76. [Google Scholar] [CrossRef]
- Shi, J.; Gao, Z.H.; Hu, K.; Pan, G.J.; Wei, M.Z.; Xu, L.J.; Meng, X.K. High pseudoelasticity of nanoscale L21 phase-Ni43Ti38Al19 thin films. Mater. Lett. 2014, 115, 79–81. [Google Scholar] [CrossRef]
- Peters, M.A.; Botton, G.A.; Humphreys, C.J. The precipitation of β’ Ni2TiAl from Al-doped β Ni-Ti alloys. In Institute of Physics Conference Series Vol. 147: Electron Microscopy and Analysis 1995, Proceedings of the Institute of Physics Electron Microscopy and Analysis Group Conference, Birmingham, England, 12–15 September 1995; Cherns, D., Ed.; IOP Publishing Ltd.: Bristol, UK, 1995; pp. 451–454. [Google Scholar]
- Jung, J.; Ghosh, G.; Isheim, D.; Olson, G.B. Precipitation of heusler phase (Ni2TiAl) from B2-TiNi in Ni-Ti-Al and Ni-Ti-Al-X (X = Hf, Zr) alloys. Metall. Mater. Trans. A 2003, 34, 1221–1235. [Google Scholar] [CrossRef]
- Jung, J.; Ghosh, G.; Olson, G.B. A comparative study of precipitation behavior of Heusler phase (Ni2TiAl) from B2-TiNi in Ni-Ti-Al and Ni-Ti-Al-X (X = Hf, Pd, Pt, Zr) alloys. Acta Mater. 2003, 51, 6341–6357. [Google Scholar] [CrossRef]
- Tang, S.L.; Gao, Y.M.; Li, Y.F.; Zheng, Q.L. Preparation and interface investigation of iron matrix composite reinforced by alumina particles sintered with Ti and Ni. Adv. Eng. Mater. 2016, 18, 1913–1920. [Google Scholar] [CrossRef]
- Tang, S.L.; Gao, Y.M.; Li, Y.F. Investigation on wear behavior of coated ZTA particles reinforced iron matrix composite. In Proceedings of the 3rd China International Congress on Composite Materials, Hangzhou, China, 21–23 October 2017. (In Chinese). [Google Scholar]
- Wen, Z.Q.; Zhao, Y.H.; Hou, H.; Wang, B.; Han, P.D. The mechanical and thermodynamic properties of Heusler compunds Ni2XAl (X = Sc, Ti, V) under pressure and temperature: A first-principles study. Mater. Des. 2017, 114, 398–403. [Google Scholar] [CrossRef]
- Reddy, P.V.S.; Kanchana, V. Ab initio study of Fermi surface and dynamical properties of Ni2XAl (X = Ti, V, Zr, Nb, Hf and Ta). J. Alloys Compd. 2014, 616, 527–534. [Google Scholar] [CrossRef]
- Waki, S.; Yamaguchi, Y.; Mitsugi, K. Superconductivity of Ni2NbX (X = Al, Ga and Sn). J. Phys. Soc. Jpn. 1985, 54, 1673–1676. [Google Scholar] [CrossRef]
- Durajski, A.P. Quantitative analysis of nonadiabatic effects in dense H3S and PH3 superconductors. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Szczȩśniak, D.; Zemła, T.P. On the high-pressure superconducting phase in platinum hydride. Supercond. Sci. Technol. 2015, 28, 085018. [Google Scholar] [CrossRef]
- Sahariya, J.; Ahuja, B.L. Electronic structure of Ni2TiAl: Theoretical aspects and Compton scattering measurement. Physica B 2012, 407, 4182–4185. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Y.; Feng, Y.; Yuan, H.K.; Chen, H. First-principles study on the effect of defects on the electronic and magnetic properties of the Ti2NiAl inverse Heusler alloy. Eur. Phys. J. B 2014, 87, 290. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulations: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Mattsson, A.E.; Schultz, P.A.; Desjarlais, M.P.; Mattsson, T.R.; Leung, K. Designing meaningful density functional theory calculations in materials science—A primer. Modell. Simul. Mater. Sci. Eng. 2005, 13, R1–R31. [Google Scholar] [CrossRef]
- Cao, L.Z.; Shen, J.; Chen, N.X. Theoretical study of the phase stability and site preference for R3(Fe, T)29 (R = Nd, Sm; T = V, Ti, Cr, Cu, Nb, Mo, Ag). J. Alloys Compd. 2002, 336, 18–28. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef]
- Debski, A.; Gasior, W.; Sypien, A.; Goral, A. Enthalpy of formation of intermetallic phases from Al-Ni and Al-Ni-Ti systems. Intermetallics 2013, 42, 92–98. [Google Scholar] [CrossRef]
- Huneau, B.; Rogl, P.; Zeng, K.; Schmid-Fetzer, R.; Bohn, M.; Bauer, J. The ternary system Al-Ni-Ti Part I: Isothermal section at 900 °C; Experimental investigation and thermodynamic calculation. Intermetallics 1999, 7, 1337–1345. [Google Scholar] [CrossRef]
- Yin, M.; Nash, P.; Chen, W.; Chen, S. Standard enthalpies of formation of selected Ni2YZ Heusler compounds. J. Alloys Compd. 2016, 660, 258–265. [Google Scholar] [CrossRef]
- Pearson, W.B. Handbook of Lattice Spacings and Structures of Metals; Pergamon: New York, NY, USA, 1967; Volume 2. [Google Scholar]
- Jiang, D.T.; Guo, J.T.; Shi, C.X.; Lin, D.L. Microstructure and compressive properties of in situ composite (Ni-40Al-10Ti)-(0, 20%)TiC. J. Mater. Sci. Lett. 2000, 19, 115–117. [Google Scholar] [CrossRef]
- Yan, X.L.; Grytsiv, A.; Rogl, P.; Pomjakushin, V.; Palm, M. The Heusler phase Ti25(Fe50-xNix)Al25 (0 ≤ x ≤ 50); structure and constitution. J. Phase Equilib. Diffus. 2008, 29, 500–508. [Google Scholar] [CrossRef]
- Sridharan, S.; Nowotny, H.; Wayne, S.F. Investigations within the quaternary system titanium—nickel—alumina—carbon. Monatsh. Chem. 1983, 114, 127–135. [Google Scholar] [CrossRef]
- Hu, R.X.; Nash, P.; Chen, Q. Enthalpy of formation in the Al-Ni-Ti system. J. Phase Equilib. Diffus. 2009, 30, 559–563. [Google Scholar] [CrossRef]
- Li, Y.F.; Tang, S.L.; Gao, Y.M.; Ma, S.Q.; Zheng, Q.L.; Cheng, Y.H. Mechanical and thermodynamic properties of intermetallic compounds in the Ni-Ti system. Int. J. Mod. Phys. B 2017, 31. [Google Scholar] [CrossRef]
- Zhao, J.J.; Winey, J.M.; Gupta, Y.M. First-principles calculations of second- and third-order elastic constants for single crystals of arbitrary symmetry. Phys. Rev. B 2007, 75. [Google Scholar] [CrossRef]
- Patil, S.K.R.; Khare, S.V.; Tuttle, B.R.; Bording, J.K.; Kodambaka, S. Mechanical stability of possible structures of PtN investigated using first-principles calculations. Phys. Rev. B 2006, 73. [Google Scholar] [CrossRef]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Oxford University Press: London, UK, 1954. [Google Scholar]
- Wu, Z.J.; Zhao, E.J.; Xiang, H.P.; Hao, X.F.; Liu, X.J.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, L.L.; Wan, B.; Zhang, J.W.; Li, Z.H.; Gou, H.Y. Structural evolution, mechanical properties, and electronic structure of Al-Mg-Si compounds from first principles. J. Mater. Sci. 2015, 50, 6498–6509. [Google Scholar] [CrossRef]
- Zhou, W.; Liu, L.J.; Li, B.L.; Wu, P.; Song, Q.G. Structural, elastic and electronic properties of intermetallics in the Pt-Sn system: A density functional investigation. Comput. Mater. Sci. 2009, 46, 921–931. [Google Scholar] [CrossRef]
- Chung, D.H.; Buessem, W.R. The elastic anisotropy of crystals. J. Appl. Phys. 1967, 38, 2010–2012. [Google Scholar] [CrossRef]
- Chung, D.H.; Buessem, W.R. The Voigt-Reuss-Hill approximation and elastic moduli of polycrystalline MgO, CaF2, β-ZnS, ZnSe, and CdTe. J. Appl. Phys. 1967, 38, 2535–2540. [Google Scholar] [CrossRef]
- Chung, D.H.; Buessem, W.R. The Voigt-Reuss-Hill (VRH) approximation and the elastic moduli of polycrystalline ZnO, TiO2 (rutile), and α-Al2O3. J. Appl. Phys. 1968, 39, 2777–2782. [Google Scholar] [CrossRef]
- Xiang, H.P.; Wu, Z.J. Ab initio study on the electronic, magnetic, and mechanical properties of CaCu3V4O12. Inorg. Chem. 2008, 47, 2706–2709. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Niu, H.Y.; Li, D.Z.; Li, Y.Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [Google Scholar] [CrossRef]
- Li, Y.F.; Gao, Y.M.; Xiao, B.; Min, T.; Fan, Z.J.; Ma, S.Q.; Xu, L.L. Theoretical study on the stability, elasticity, hardness and electronic structures of W-C binary compounds. J. Alloys Compd. 2010, 502, 28–37. [Google Scholar] [CrossRef]
- Teter, D.M. Computional alchemy: The search for new superhard materials. MRS Bull. 1998, 23, 22–27. [Google Scholar] [CrossRef]
- Chen, K.; Zhao, L. Elastic properties, thermal expansion coefficients and electronic structures of Ti0.75X0.25C carbides. J. Phys. Chem. Solids 2007, 68, 1805–1811. [Google Scholar] [CrossRef]
- Šimůnek, A.; Vackář, J. Hardness of covalent and ionic crystals: First-principle calculations. Phys. Rev. Lett. 2006, 96. [Google Scholar] [CrossRef] [PubMed]
- Pabst, W.; Tichá, G.; Gregorová, E. Effective elastic properties of alumina-zirconia composite ceramics part 3. Calculation of elastic moduli of polycrystalline alumina and zirconia from monocrystal data. J. Ceram.-Silikáty 2004, 48, 41–48. [Google Scholar]
- Birch, F. Finite elastic strain of cubic crystals. Phys. Rev. 1947, 71, 809–824. [Google Scholar] [CrossRef]
- Manghnani, M.H.; Fisher, E.S.; Brower, W.S., Jr. Temperature dependence of the elastic constants of single-crystal rutile. J. Phys. Chem. Solids 1972, 33, 2149–2159. [Google Scholar] [CrossRef]
- Otero-de-da-Roza, A.; Luaña, V. GIBBS2: A new version of the quasi-harmonic model code. I. Robust treatment of the static data. Comput. Phys. Commun. 2011, 182, 1708–1720. [Google Scholar] [CrossRef]
- Nye, J.F. Physical Properties of Crystals; Oxford University Press: London, UK, 1985. [Google Scholar]
- Ravindran, P.; Fast, L.; Korzhavyi, P.A.; Johansson, B.; Wills, J.; Eriksson, O. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2. J. Appl. Phys. 1998, 84, 4891–4904. [Google Scholar] [CrossRef]
- Xiang, H.M.; Feng, Z.H.; Zhou, Y.C. Theoretical investigations on mechanical anisotropy and intrinsic thermal conductivity of YbAlO3. J. Eur. Ceram. Soc. 2015, 35, 1549–1557. [Google Scholar] [CrossRef]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal elastic anisotropy index. Phys. Rev. Lett. 2008, 101. [Google Scholar] [CrossRef] [PubMed]
- Mersol, S.A.; Lynch, C.T.; Vahldiek, F.W. Anisotropy in Single Crystal Refractory Compounds; Plenum Press: New York, NY, USA, 1968. [Google Scholar]
- Brugger, K. Determination of third-order elastic coefficients in crystals. J. Appl. Phys. 1965, 36, 768–773. [Google Scholar] [CrossRef]
- Grimvall, G. Thermophysical Properties of Materials; Elsevier: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Feng, J.; Xiao, B.; Wan, C.L.; Qu, Z.X.; Huang, Z.C.; Chen, J.C.; Zhou, R.; Pan, W. Electronic structure, mechanical properties and thermal conductivity of Ln2Zr2O7 (Ln = La, Pr, Nd, Sm, Eu and Gd). Acta. Mater. 2011, 59, 1742–1760. [Google Scholar] [CrossRef]
- Chong, X.Y.; Jiang, Y.H.; Zhou, R.; Zhu, H.; Feng, J. Electronic structure, anisotropic elastic and thermal properties of the η phase Fe6W6C. Comput. Mater. Sci. 2015, 108, 205–211. [Google Scholar] [CrossRef]
- Ding, Y.C.; Xiao, B. Anisotropic elasticity, sound velocity and thermal conductivity of TiO2 polymorphs from first principles calculations. Comput. Mater. Sci. 2014, 82, 202–218. [Google Scholar] [CrossRef]
- Anderson, O.L. A simplified method for calculating the Debye temperature from elastic constants. J. Phys. Chem. Solids 1963, 24, 909–917. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004. [Google Scholar]
- Rocha, F.S.D.; Fraga, G.L.F.; Brandao, D.E.; Silva, C.M.D.; Gomes, A.A. Specific heat and electronic structure of Heusler compounds Ni2TAl (T = Ti, Zr, Hf, V, Hb, Ta). Physica B 1999, 269, 154–162. [Google Scholar] [CrossRef]
- Clarke, D.R. Materials selection guidelines for low thermal conductivity thermal barrier coatings. Surf. Coat. Technol. 2003, 163, 67–74. [Google Scholar] [CrossRef]
- Cahill, D.G.; Watson, S.K.; Pohl, R.O. Lower limit to the thermal conductivity of disordered crystals. Phys. Rev. B 1992, 46, 6131–6140. [Google Scholar] [CrossRef]
- Zheng, K.H.; Gao, Y.M.; Li, Y.F.; Zhao, S.M.; Wang, J. Three-body abrasive wear resistance of iron matrix composites reinforced with ceramic particles. Proc. Inst. Mech. Eng. Part J J. Eng. Tribol. 2014, 228, 3–10. [Google Scholar] [CrossRef]
- ASM International Handbook Committee. ASM Handbook, Vol. 1: Properties and Selection: Irons, Steels and High-Performance Alloys; ASM International: Geauga County, OH, USA, 1993. [Google Scholar]
- Onderka, B.; Sypien, A.; Wierzbicka-Miernik, A.; Czeppe, T.; Zabdyr, L.A. Specific heat capacities of some ternary aluminides. J. Phase Equilib. Diffus. 2011, 32, 39–41. [Google Scholar] [CrossRef]
- Drulis, M.K.; Czopnik, A.; Drulis, H.; Spanier, J.E.; Ganguly, A.; Barsoum, M.W. On the heat capacity of Ti3GeC2. Mater. Sci. Eng. B 2005, 119, 159–163. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Lattice Parameters | Vcell | ρ | Atomic Site | Ecoh | |||
|---|---|---|---|---|---|---|---|---|
| a = b = c | Al | Ni | Ti | |||||
| GGA-PBE | 5.908 (5.865 a) | 206.242 (201.75 a) | 6.193 (6.33 a) | 4a (0, 0, 0) | 8c (0.25, 0.25, 0.25) | 4b (0.5, 0.5, 0.5) | −0.671 | −27.602 |
| GGA-PW91 | 5.903 | 205.676 | 6.210 | −0.643 | −33.173 | |||
| Other cal. data | 5.906 b, 5.90 c, 5.828 d | −0.658 b, −0.662 m | ||||||
| Exp. data | 5.906 e, 5.883–5.910 f, 5.819 g, 5.876 i, 5.942 j, 5.889 k, 5.865–5.886 l | 203.61–206.40 f | −0.747 e, −0.578 h, −0.579 n | |||||
| EOS | 5.911 | 206.499 | 6.186 | |||||
| Method | Elastic Constants | B | G | BH/GH | E | v | HV | Shear Anisotropic Factors | Anisotropic Index | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c11 | c12 | c44 | BV | BR | BH | GV | GR | GH | A1 | A2 | A3 | AB | AG | AU | |||||
| GGA-PBE | 208.1 | 142.2 | 97.6 | 164.2 | 164.2 | 164.2 | 71.8 | 54.7 | 63.2 | 2.6 | 168.1 | 0.32 | 4.4 | 2.96 | 2.96 | 2.96 | 0.00 | 0.14 | 1.56 |
| GGA-PW91 | 209.3 | 143.4 | 94.8 | 165.4 | 165.4 | 165.4 | 70.1 | 54.2 | 62.1 | 2.7 | 165.6 | 0.33 | 4.1 | 2.88 | 2.88 | 2.88 | 0.00 | 0.13 | 1.47 |
| Cal. data a | 215.8 a, 223 b | 139.4 a, 135 b | 98.7 a, 104 b | 164.9 a, 164 b | 67.5 a, 74 b | 2.4 a | 178.1 a, 193 b | 8.1 a | 2.36 b | 0.104 a | 1.165 a | ||||||||
| Exp. data | 120 c | 97 c | 56 c | ||||||||||||||||
| EOS | 162.9 | ||||||||||||||||||
| Species | [100] | [010] | [001] | [110] | [111] | νl | νt | νm | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| νl | νt1 | νt2 | νl | νt1 | νt2 | νl | νt1 | νt2 | νl | νt1 | νt2 | νl | νt1 | νt2 | ||||
| AlNi2Ti | 5797 | 3971 | 3971 | 5797 | 3971 | 3971 | 5797 | 3971 | 3971 | 6637 | 3262 | 3971 | 6894 | 2967 | 2967 | 6334 | 3195 | 3583 |
| Ni3Ti | 6565 | 3277 | 3625 | 6565 | 3277 | 3625 | 6565 | 3277 | 3277 | 6565 | 3277 | 3625 | 7243 | 1172 | 3513 | 6427 | 3550 | 3955 |
| Species | Model | ΘD | (10−26) | P (1028) | [100] kmin | [010] kmin | [001] kmin | [110] kmin | [111] kmin | kmin | k (Exp.) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AlNi2Ti | Clark | 455.1 (462 a, 411 b) | 7.97 | 0.85 | 0.85 | 0.85 | 1.16 | 1.37 | 1.14 | 21.4 c | |
| Cahill | 7.76 | 1.39 | 1.39 | 1.39 | 1.40 | 1.30 | 1.29 | ||||
| Ni3Ti | Clark | 516.6 | 9.29 | 1.36 | 1.36 | 1.45 | 1.36 | 1.26 | 1.31 | ||
| Cahill | 8.43 | 1.44 | 1.44 | 1.40 | 1.44 | 1.28 | 1.45 |
| Species | Df | γ | β | Cp (3 K) | Cp (30 K) |
|---|---|---|---|---|---|
| AlNi2Ti | 0.416 | 0.981 | 0.0825 | 0.00517 | 2.26 |
| Ni3Ti | 0.113 | 0.267 | 0.0569 | 0.00234 | 1.54 |
| B2_NiTi | 0.714 | 1.68 | 0.0577 | 0.00661 | 1.61 |
| B19’_NiTi | 0.406 | 0.957 | 0.0806 | 0.00505 | 2.21 |
| NiTi2 | 0.763 | 1.80 | 0.115 | 0.00850 | 3.16 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, S.; Li, Y.; Gao, Y.; Zheng, Q.; Liu, Z.; Ren, X. First-Principles Investigations of the Structural, Anisotropic Mechanical, Thermodynamic and Electronic Properties of the AlNi2Ti Compound. Crystals 2018, 8, 93. https://doi.org/10.3390/cryst8020093
Tang S, Li Y, Gao Y, Zheng Q, Liu Z, Ren X. First-Principles Investigations of the Structural, Anisotropic Mechanical, Thermodynamic and Electronic Properties of the AlNi2Ti Compound. Crystals. 2018; 8(2):93. https://doi.org/10.3390/cryst8020093
Chicago/Turabian StyleTang, Shuli, Yefei Li, Yimin Gao, Qiaoling Zheng, Zhiwei Liu, and Xiangyi Ren. 2018. "First-Principles Investigations of the Structural, Anisotropic Mechanical, Thermodynamic and Electronic Properties of the AlNi2Ti Compound" Crystals 8, no. 2: 93. https://doi.org/10.3390/cryst8020093