Design of Spin-Frustrated Monomer-Type C60•− Mott Insulator
by
 ,
,
Akihiro Otsuka
1,2,* ,
,
Dmitri V. Konarev
3,*,
Rimma N. Lyubovskaya
3,
Salavat S. Khasanov
4,
Mitsuhiko Maesato
2,
Yukihiro Yoshida
2,5 and
Gunzi Saito
5,6,* 1
Research Center for Low Temperature and Materials Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan
2
Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan
3
Institute of Problems of Chemical Physics RAS, Chernogolovka, Moscow Region 142432, Russia
4
Institute of Solid State Physics RAS, Chernogolovka, Moscow Region 142432, Russia
5
Department of Agriculture, Meijo University, 1-501 Shiogamaguchi, Tempaku-ku, Nagoya 468-8502, Japan
6
Toyota Physical and Chemical Research Institute, Nagakute, Aichi 480-1192, Japan
*
Authors to whom correspondence should be addressed.
Crystals 2018, 8(3), 115; https://doi.org/10.3390/cryst8030115
Submission received: 30 January 2018
/
Revised: 23 February 2018
/
Accepted: 25 February 2018
/
Published: 28 February 2018
(This article belongs to the Special Issue Advances in Organic Conductors and Superconductors)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Spin-frustrated monomer-type Mott insulator C60•− solids are discussed in this review article. For the C60•− solids, the interfullerene center-to-center distance (r) is the key parameter that controls the competition between covalent bond-formation, itinerancy, and spin frustration. Eight C60•− salts with various compositions and dimensionalities are reviewed. In all of these C60•− salts except one, neither bond-formation nor long-range magnetic ordering was observed down to low temperatures. A plot of Weiss temperature (|ΘCW|) against r shows that |ΘCW| grows rapidly below r = 10.0 Å.
1. Introduction: Quantum Spin Liquid State
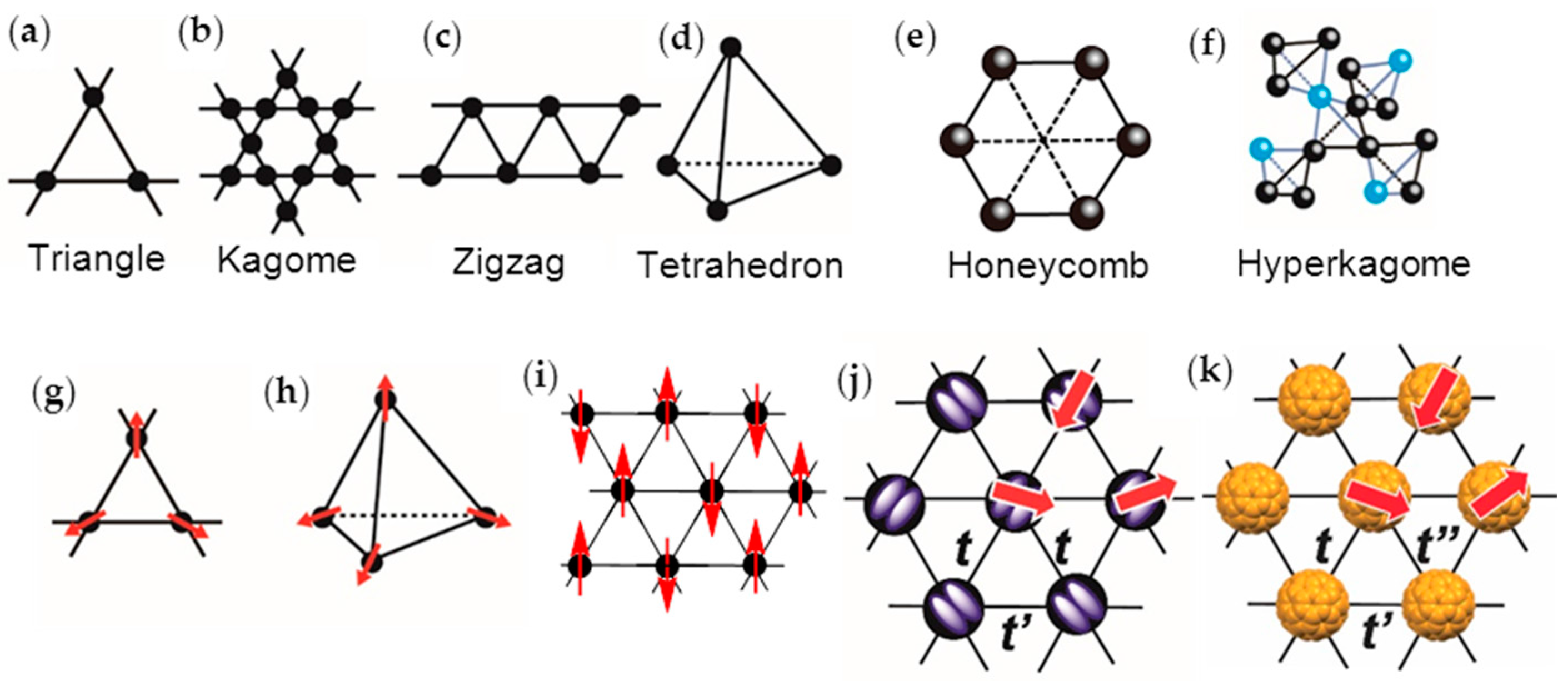
Strong geometrical spin frustration suppresses the classical long-range magnetic ordering of the Néel state, and allows the novel quantum states such as the quantum spin liquid (QSL) state for two-dimensional (2D) S = 1/2 antiferromagnets (AFs), as proposed by Anderson [1,2]. The QSL phase is a quantum-disordered insulating phase, which has been theoretically predicted to have a ground state with many degenerate states [3], and, hence, to exhibit large spin entropy, even at 0 K. To obtain spin-frustrated materials, the geometries of spin lattice subject to contradictory constraints are crucial. The spin lattices—triangle (Figure 1a), kagome (Figure 1b), zigzag (Figure 1c), tetrahedron (Figure 1d), honeycomb (Figure 1e), and hyperkagome (Figure 1f)—have been discussed with the basic building block being the triangle [4]. The magnetic exchange interaction J of spins is proportional to the square of transfer interaction t (Equation (1)), where U is the on-site Coulomb repulsion energy,
J ~ 4t2/U
A small distance between two spins (r) induces a large t and eventually, a metallic band with itinerant electrons is formed. Equation (1) suggests that the QSL state may be adjacent to such an itinerant (metallic or superconducting (SC)) phase. When the distance between the spin sites in the triangular spin lattice becomes extremely large, all of the spins may act as Curie spins. With decreasing distance between the spins, spins feel frustration to each other and form a QSL or a compromised spin configurations which do not have spin frustration with geometry of spin lattice of 120° (triangle, Figure 1g), 109° (tetrahedron, Figure 1h), or collinear AF (AFC) (Figure 1i) structures [5,6,7,8,9,10,11]. When the triangular spin lattice is distorted, the AF phase may get preferentially stabilized. Therefore, the QSL, AF, 120° structure (Figure 1g), AFC, and metallic (or SC) phases compete with each other in a triangular spin lattice based on the parameters r, t, W, U, J, outer stimuli (temperature T, pressure P, magnetic field H, etc.), and the geometry of the spin lattice, where W is the bandwidth.
The Curie-Weiss temperature ΘCW given by,
is a parameter showing the easiness to access the QSL state, where, z, S, and kB are the number of the nearest neighbour sites, spin quantum number, and Boltzmann constant, respectively. The frustration index f defined by Equation (3),
was proposed by Ramirez as a measure of the spin frustration [13,14]. Strong spin-frustrated systems are thought to be those associated with f > 10. In Equation (3), Tm is the temperature at which magnetic ordering occurs.
ΘCW = 2zS(S + 1)J/3kB
f = − ΘCW/Tm
The easiest way to design spin-frustrated systems is to arrange magnetic transition metals in a triangular or kagome geometry. Numerous inorganic compounds have been examined based on this idea. A few examples are VX2 (X = Cl, Br) [15], ABO2 [16,17,18,19,20,21] (A = monocation, B = trivalent transition metal ions: CuFeO2 [16,17,18], LiNiO2 [19], NaTiO2, [19], LiCrO2 [20,21,22]), ABX3 [23,24] (X = halogenide ions: CsCoCl3 [23], CsMnBr3 [24]), and large clusters (Mo72Fe30, Mo72V20) [25] for the triangular lattice, and KFe3(SO4)2(OH)6 [26,27,28] and Rb2SnCu3F12 [29] for the kagome lattice. Some typical magnetically frustrated inorganic systems of transition metal oxides studied prior 2000 have been summarized by Ramirez [13] and Greedan [4].
However, QSL systems are scarce in materials with spin quantum numbers S > 1/2, even in the triangular spin lattices and kagome lattices with f ≥ 102 [30,31,32]. Even for the triangular S = 1/2 spin systems with large |ΘCW| and f, no QSL materials have been prepared due to the difficulty in maintaining the precise geometry of spin-frustrated lattices at low temperatures [33,34,35].
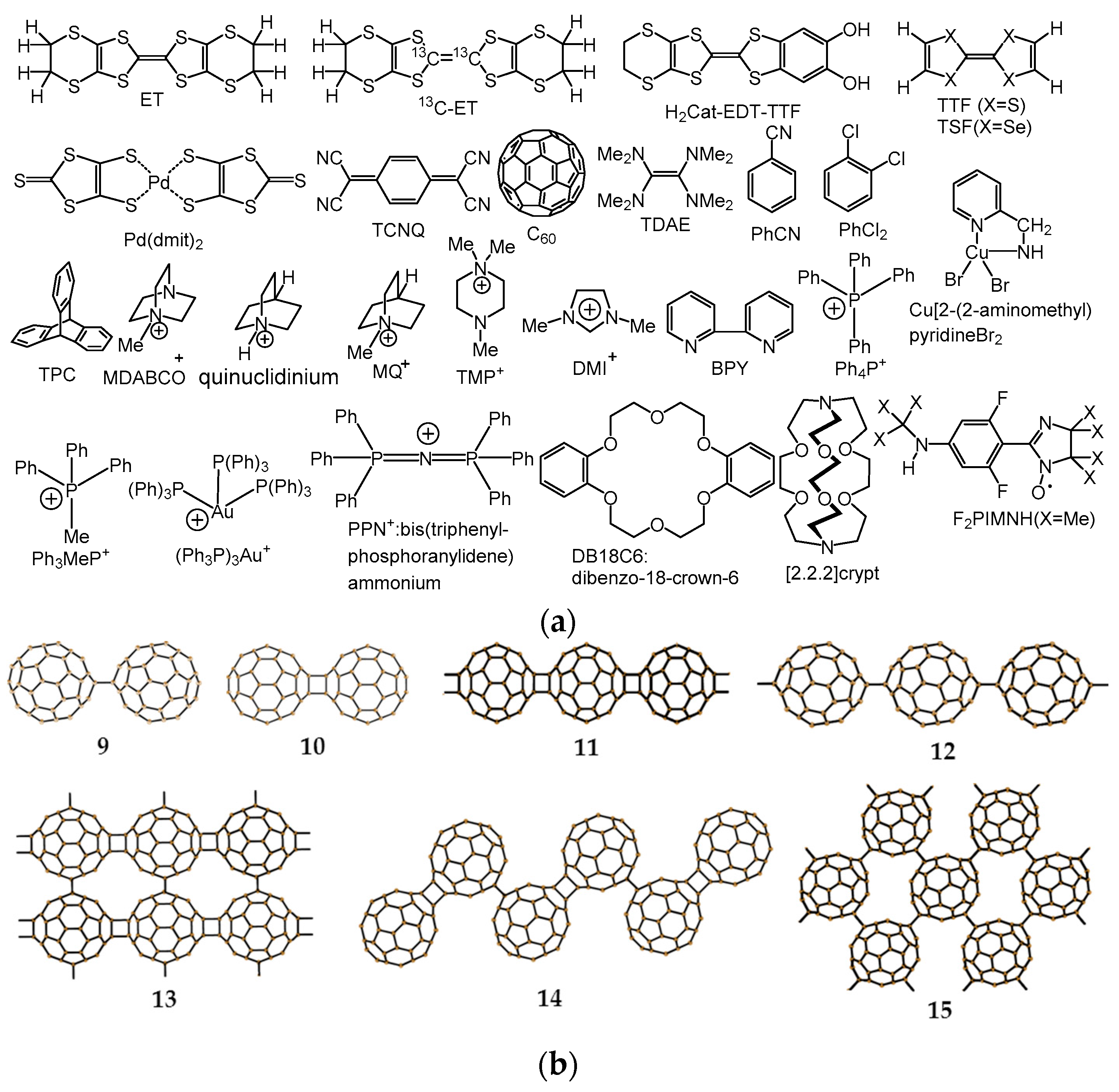
The first real QSL candidate was a charge-transfer (CT) salt of a dimer-type Mott insulator κ-(ET)2Cu2(CN)3 [36], where ET is bis(ethylenedithio)tetrathiafulvalene (chemicals in this review are shown in Figure 2) and [Cu2(CN)3−]∞ is a diamagnetic polymeric anion. The planar tridentate coordination of diamagnetic Cu(I) ions in [Cu2(CN)3−]∞ is the main driving force for the two-dimensional (2D) triangular magnetic lattice that is composed of partially charged (ET)2•+, since [Cu2(CN)3−]∞ has openings, and the arrangement of the anion openings is triangular due to the planar tridentate coordination of Cu(I) ions. The geometrical fit between a spin-site (ET)2•+ and the anion opening results in a triangular magnetic lattice (Figure 1j) according to a key-keyhole relation, where the key is the spin-site, (ET)2•+, and the keyhole is the anion opening.
The features of the Mott insulator κ-(ET)2Cu2(CN)3 are: (1) it has a nearly equilateral triangular lattice (t′/t = 1.09) with very strong electron correlation (U/W = 0.93), (2) the QSL state is experimentally confirmed down to 20 mK [36,45,46], (3) the anisotropic SC state resides directly next to the QSL state without passing through the spin ordered AF state under pressure [47,48,49], (4) the transition from the QSL state to the metallic state shows positive pressure dependence, indicating that the residual spin entropy is in the QSL state [47,48,49,50], and (5) 13C NMR measurements under hydrostatic pressure on the salt of ET with 13C enriched at the central C=C bond (13C-ET) indicate d-wave SC symmetry [51]. Thus, a competition among the localized (and frustrated), itinerant, and exotic pairing of spins is manifested in this salt [36,45,46,47,48,49,50,51,52,53]. We have proposed designing principles for a QSL candidate residing next to the SC state based on the crystal, electronic, and spin structures for selected κ-(ET)2X (X = anion) including X = Cu2(CN)3 as follows [54].
The requirements for a QSL state next to an itinerant state for κ-(ET)2X are:
- 1)
- the system has a low spin state (S = 1/2),
- 1)
- the system should be a Mott insulator in ambient conditions,
- 1)
- its Mott insulating state has both a partial CT state close to the itinerant region and a small Mott gap,
- 1)
- the spin lattice should have a geometry that affords a strong geometrical frustration, i.e., t′/t ~ 1 for a triangular spin lattice,
- 1)
- a high |ΘCW| or high |J| value to observe the QSL state at the experimentally available temperatures, and
- 1)
- the material must maintain weak energy dispersion along the weakest direction for the magnetic interactions of the 2D system, i.e., negligibly weak magnetic interaction perpendicular to the 2D magnetic layer in order to keep the geometry of spin-frustrated spin lattice down to low temperatures.
Since the discovery of the QSL state in κ-(ET)2Cu2(CN)3 (|ΘCW| = 375 K, |J|/kB = 250 K, f > 1.3 × 104), several materials based on the triangular, kagome, honeycomb, and hyperkagome spin lattices have been reported to have such a spin state [55,56,57]. Some organic and inorganic compounds with sufficiently large |ΘCW|, |J|, and f examined on single crystals include κ-H3(Cat-EDT-TTF)2 (triangle, H2Cat-EDT-TTF: catechol-fused ethylenedithio-TTF, |ΘCW| = 120–150 K, |J|/kB = 80–100 K, f > 2.4 × 103) [58], a CT solid of (ethyltrimethylantimonate)[Pd(dmit)2]2 (triangle, dmit: 4,5-dimercapto-1,3-dithiole-2-thione, |ΘCW| = 325–375 K, |J|/kB = 220–250 K, f > 1.6 × 104) [59], ZnCu3(OH)6Cl2 (kagome, |ΘCW| = 314 K, |J|/kB = 180 K, f > 5.1 × 103) [60,61,62], [NH4]2(C7H14N)[V7O6F18] (kagome, |ΘCW|= 81 K, f ≥2.0 × 103) [63] where (C7H14N) is quinuclidinium ion, and [(C2H5)3NH]2Cu2(oxalate)3 (hyperhoneycomb, |ΘCW|= 180 K, f > 3 × 103) [64].
Another organic QSL system of a dimer-type Mott insulator κ-(ET)2Ag2(CN)3 (t′/t = 0.97) was very recently developed by us (|ΘCW| = 263 K, |J|/kB = 175 K, f > 2.1 × 103) having a different key-keyhole relation from that of κ-(ET)2Cu2(CN)3 resulting in both a more robust QSL state and a higher critical temperature (Tc) of SC than the X = Cu salt [54,65], with similar relaxor ferroelectric response [66,67]. Among these QSL systems, only dimer-type ET salts κ-(ET)2Cu2(CN)3 and κ-(ET)2Ag2(CN)3 manifest competition among the itinerancy of electrons (metal), pairing of two electrons (SC), and localization (Mott insulator and QSL).
In order to have such competition between localized and itinerant spins in a monomer-type Mott insulator, the best candidate will be the C60 CT materials having triangular or hexagonal packing of C60•− molecules (Figure 1k) among several kinds of such monomer-type Mott insulators based on TTF, TSF, ET, TCNQ, etc. [67,68,69], where TTF, TSF, and TCNQ are well known donor or acceptor molecules in the CT salts [70]. In these monomer-type Mott insulators, the upper-HOMO band for the CT solid of D•+X− (D: TTF, TSF, ET) and the lower-LUMO band for the CT solid of M+A•− (A: TCNQ) are completely filled, where HOMO and LUMO are the highest occupied and lowest unoccupied molecular orbitals, respectively. The Mott gap being large makes it difficult for the competition between localization and itinerancy in the solids, thus requiring partial CT state or dimer-type Mott insulating state to satisfy the requirement 3 above mentioned. While for the C60 CT solids, a partial CT state such as ET1/2+ is not necessary and the completely ionized C60•− molecules are able to afford the itinerant state owing to the triply degenerate LUMO t1u orbital, e.g., the compound CsC60 quenched in liquid N2 is reported to exhibit metallic behavior down to low temperatures [71,72].
In this review, the preparation, crystal and electronic structures, and the physical properties of spin-frustrated monomer-type Mott insulators of C60•− CT solids, namely (MDABCO+)(C60•−) (1), (Ph3MeP+)(C60•−) (2), (TPC0)(MDABCO+)(C60•−) (3), (TPC0)(MQ+)(C60•−) (4), (PhCN0)(TMP+)(C60•−) (5), (PhCN0)(Ph3MeP+)(C60•−) (6), (PhCl20){(Ph3P)3Au+)}2(C60•−)2(C60) (7), and (DMI+)3(C60•−)(I−)2 (8), and the design of the QSL systems will be discussed.
2. Characteristic Features of C60: Superconductors and Other Functions for C60 Charge-Transfer Materials
Superconductivity (SC) is one of the most remarkable features of C60 CT materials. An icosahedral C60 molecule with Ih symmetry has triply degenerate LUMO and LUMO + 1 orbitals with t1u and t1g symmetries, respectively. Such multiple degeneracy (N) contributes to the relaxation of the Mott criterion [73,74], i.e., U/W ~ √N or an upper limit of U/W ~ 2.5, and the enhancement of the density of states at the Fermi level (D(εF)) inducing high Tc for SC [75], when C60 is placed in the highly symmetric, i.e., cubic crystal field. Typical SC materials are represented as A3C60 (A: alkali metal), e.g., Rb3C60 (Tc = 29 K [76]), Rb2CsC60 (Tc = 31 K [77]), and RbCs2C60 (Tc = 33 K [77]), with a face-centered cubic (fcc) structure. The structural and physical properties of A3C60 and the related fullerene compounds were reviewed [73,78,79,80]. The critical temperature Tc varies monotonously with the lattice constant, independent of the type of the alkali dopant [77,81]. Thus far, about 40 SC materials have been synthesized with the highest Tc of 33 K (RbCs2C60) at normal pressure and 38 K (A15 or body-centered cubic (bcc) Cs3C60) under a pressure of approximately 0.7 Gpa [82]. The fcc phase of Cs3C60 also shows SC (Tc = 35 K) under an applied hydrostatic pressure of approximately 0.8 Gpa [83].
The critical temperature Tc decreases as the valence state (n) of C60 deviates from n = −3 or lowering the symmetry of the crystal (non-cubic) such as Yb2.75C60 (n = −5.5, orthorhombic, Tc = 6 K) [84], Sm2.75C60 (n = −5.5, orthorhombic, Tc = 8 K) [85], Ba4C60 (n = −8, body-centered orthorhombic (bco), Tc = 6.7 K) [86,87], Sr4C60 (n = −8, bco, Tc = 4.4 K) [86] and K3Ba3C60 (n = −9, bcc, Tc = 5.6 K) [88]. Eu6C60 with bcc packing undergoes a ferromagnetic transition at 12 K, owing from Eu2+ cations with S = 7/2 spin [89]. CexC60 shows the coexistence of SC and ferromagnetism below 13.5 K, although its crystal structure and composition are currently not clear [90].
A3C60 SCs show a dome-shaped curve of normalized Tc versus lattice volume [82,83,91], which may be a hallmark of the competition between electron-phonon attractive and electron-electron repulsive interactions. The observation of a Hebel-Slichter peak in the relaxation rate just below Tc in NMR and μSR indicate a BCS-type isotopic gap [92,93].
Besides SCs, (TDAE+)(C60•−) is a soft ferromagnet with a Curie temperature of 16.1 K, in which only spins on C60•− contribute to the ferromagnetism, where TDAE is tetrakis(dimethylamino)ethylene [94].
3. Requirements for Spin-Frustrated Spin Lattice of C60•−
3.1. Competition among Bond-Formation, Itinerancy, Localization, and Frustration in Fulleride Solids
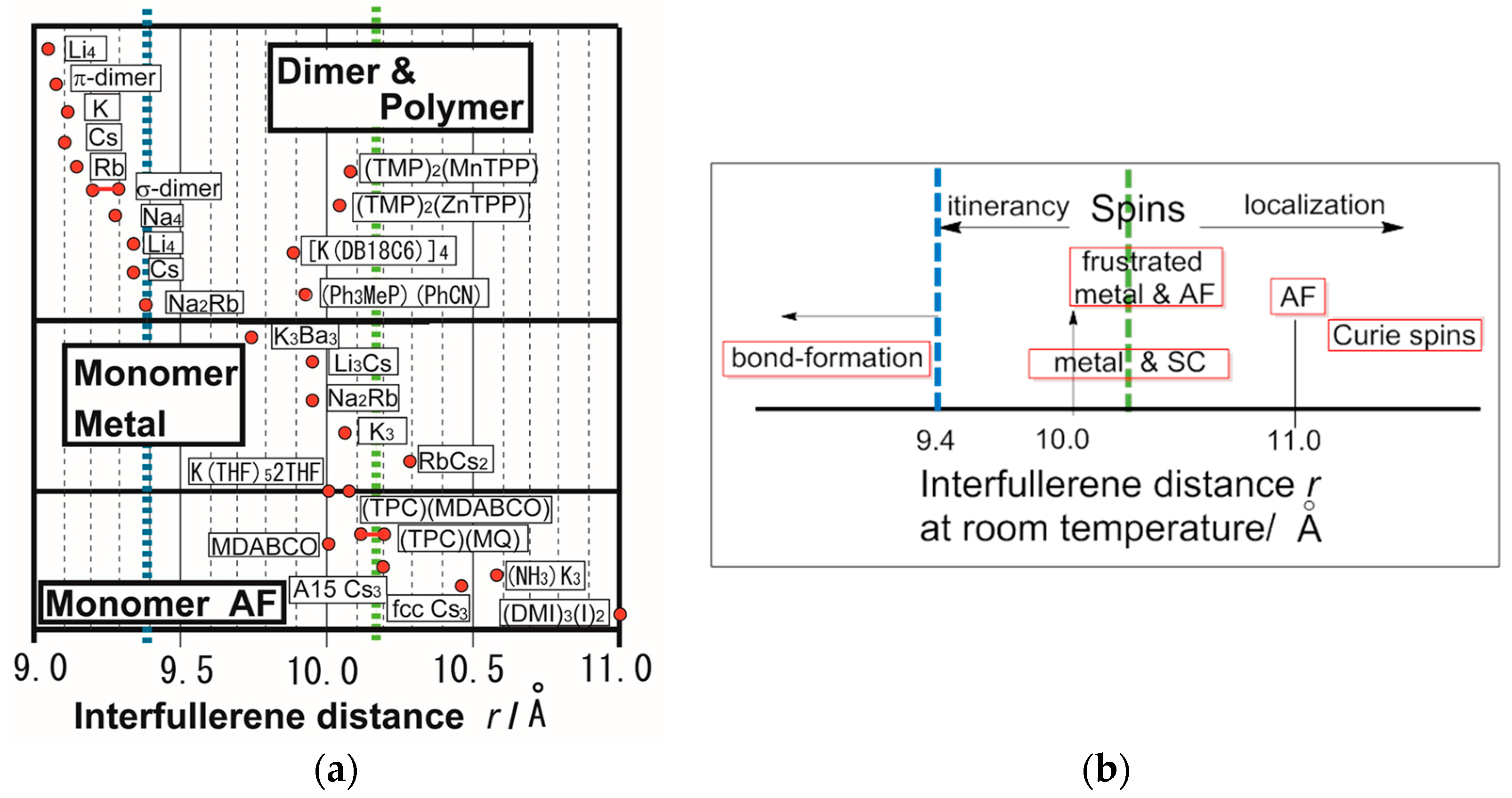
It can be easily seen that the close packing of C60•− leads to triangular (Figure 1k) or honeycomb (Figure 1e) spin lattices. In the C60 system, there is another factor, namely, the bond-formation between C60•− molecules, which leads to competition among itinerancy, localization, and frustration. Figure 3a,b summarize the electronic competition for bond-formation, such as dimers and polymers (Figure 2b) (top panel of Figure 3a), itinerancy (monomeric C60 metallic solids, middle panel in Figure 3a), and localization (monomeric AF solids, bottom panel in Figure 3a) for 27 situations in C60 CT solids as a function of center-to-center interfullerene distance r, as reported in the literatures [37,38,44,79,95,96,97,98]. The van der Waals (vdW) diameter [99] of C60 is 10.18 Å indicated by green dotted line in Figure 3a,b.
When the interfullerene distance of the C60 solids is small (r < 9.4 Å, which is indicated by blue dotted line in Figure 3a,b), the C60•− molecules tend to form dimers (9 and 10 in Figure 2b) [37,38] or polymers (11–15 in Figure 2b) by single or double bonds. Polymer 11 with K+ is a 3D metal [39], while those with Rb+ and Cs+ are one-dimensional (1D) metals and become spin-density-wave insulators at low temperatures [40,100]. Single carbon-carbon bonded polymer 12 in Na2CsC60 seems to be an SC with Tc about 3 K lower than that for the starting non-polymeric phase. Analogous lowering of Tc at the formation of polymer 12 in Na2RbC60 leads to very low Tc or the absence of SC [41,101].
The compound 13 is an insulator with high ionic conductivity [42], 14 is a diamagnetic semiconducting polymer [43], and 15 is a highly correlated metal [43].
Even when r > 9.4 Å, C60•− bond-formation proceeds for the cation species, which are not able to prevent bond-formation. The C60•− molecules in [K(DB18C6)]4(C60)512THF, where DB18C6 is dibenzo-18-crown-6 ether and THF is tetrahydrofuran, dimerize at r = 9.89 Å (at 225 K), but remain as a monomer at r = 10.1 Å [102]. For (MQ+)(CoIIOEP)(C60•−)(PhCl2), where OEP is octaethylporphyrin, no bond-formation between C60•− molecules (r = 9.88 Å at 100 K) is observed owing to the steric protection by the coordination of C60•− with CoII, that gives rise to a diamagnetic state [103]. However, for {(TMP+)2MIITPP}(C60•−)2(PhCN)2(PhCl2)2 (M = Zn, Mn, TPP: tetraphenylporphyrin), where MIITPP is weakly coordinated with TMP+, but, no coordination of C60•− with MII, with r = 10.04 Å (M = Zn at 250 K) and r = 10.08 Å (M = Mn at 270 K), singly bonded (C60−)2 dimers form at 100 K [104]. Structural analysis of (Ph3MeP+)(C60•−)(PhCN) indicates that the lowest value of r is 9.92 Å at room temperature (RT) for monomer state, and singly bonded (C60−)2 dimers appear when the temperature is reduced to 120 K for r = 9.28 Å [105]. (DMI+)2(C60•−){Cd(diethyldithiocarbamate)I−} (r = 10.03 Å at 250 K) shows reduced-temperature dependent bond-formation, such as (1) monomeric C60•− upon instant quenching below 95 K, (2) a mixture of dimerized (C60−)2 and monomeric C60•−, and (3) stable singly bonded (C60−)2 only upon slow cooling for 6 h [106].
As can be seen in Figure 3a, C60 compounds having an interfullerene distance in the range 9.4 Å < r < 10.0 Å at RT have not been extensively explored. It is highly plausible that the shorter interfullerene distance that is less than 9.7 Å instantaneously leads to bond-formation resulting in r ≤ 9.4 Å.
Above results indicate that, even at r ~ 9.9–10.1 Å, C60•− anion molecules dimerize, when the C60•− molecules are not properly protected against bond-formation.
An isolated fulleride anion structure has been observed for the interfullerene distance r ≥ 12 Å by using both bulky cation molecules (Ph4P+, PPN+, [Ru(BPY)3]2+) [107,108,109,110,111,112,113,114,115,116], and fulleride anions with a charge of −2 or −3 and alkali metal cations, which coordinate with THF, crown ether or cryptand molecules [113,114,115,116]. The preparative methods of these compounds were reviewed [95].
When r is greater than 9.73 Å and less than 12 Å, the “bond-formation”, “monomer metal”, and “monomer AF” in Figure 3a compete with each other. The “monomer metal” is observed for many C60n− solids, e.g., K3Ba3C60 (bcc, r = 9.74 Å) [88], Li3CsC60 (fcc, r = 9.98 Å) [117], Na2RbC60 (fcc, r = 9.96 Å) [118], K3C60 (fcc, r = 10.07 Å) [119], and RbCs2C60 (fcc, r = 10.19 Å) [77]. The “monomer AF” state is observed for a wide range of r. The A15 Cs3C60 (r = 10.20 Å) solid shows AF ordering below TN = 46 K (ΘCW = −68 K) [120], and the fcc phase (r = 10.44 Å) also shows AF ordering at 2.2 K (ΘCW = −105 K) at normal pressure [83,121,122], where TN is Néel temperature. The intercalation of NH3 molecules (e.g., (NH3)K3C60, r = 10.57 Å at RT) results in a phase transition from a cubic to orthorhombic lattice structure accompanied by the appearance of AF ordering instead of SC [123] and exhibits an SC state under pressure (Tc = 28 K at 15 kbar) [124].
Figure 3 will be a guiding map for the search for C60 functional materials. A phase diagram of C603− solids indicates that the SC state is in the vicinity of spin ordered AF states while the critical temperature Tc decreases as approaching to the AF phase [82,120,125]. Therefore, the C60 system is a potential candidate for the QSL state to be close to the metallic, SC, and bond-formed states.
The C60•− molecules in both the A15 and fcc phases of Cs3C60 are not in the high-spin state (S = 3/2), which prefers spin-ordering, but are in the low-spin states (S = 1/2) due to the splitting of the t1u-orbitals by the Jahn-Teller effect. The triangular spin lattices of fcc have strong spin frustration with strong AF interactions characterized by its frustration index f ~ 48 for Cs3C60. However, the Cs3C60 solid in the fcc structure prefers the AF state as compared to the QSL state. A large frustration index f and a low-spin state are necessary for QSL state formation, however, the geometry of the triangular spin lattice is also important. For example, κ-(ET)2X with a nearly equilateral triangular spin lattice (t′/t = 0.97–1.09 for X = Ag2(CN)3 and Cu2(CN)3) exhibits the QSL state [36,54], while the distorted triangular spin lattice (e.g., t′/t = 0.715 for X = Cu[N(CN)2]Cl [126,127], t′/t = 1.79 for X = CF3SO3) [128] exhibits an AF ordered spin state. Similarly a spin-frustrated system in which t′/t deviates from unity shows AF ordering, i.e., Cs2CuCl4 (t′/t = 1.71) with TN = 0.69 K [129].
The fundamental and simple prerequisites for the formation of QSL C60 system are:
- (I)
- existence of C60•− or low-spin state of C603− (S = 1/2),
- (II)
- no polymerization between C60 molecules,
- (III)
- triangular or hexagonal packing of C60 with equal interfullerene distance r, or t′/t ~ 1, and
- (IV)
- strong AF interactions.
It is believed that smaller r values lead to stronger AF interactions. The critical r between the “itinerant”, “localized spin”, and “bond-formation” has not been elucidated yet, and the competition strongly depends on the environment of the C60 molecules, pattern, and dimensionality of packing of C60 molecules, charge of C60 molecules, etc.
3.2. Charged State of C60 and Effective On-Site Coulomb Repulsion
The C60n− species (n = 0, 1, 2, 3) are well discriminated by IR and UV-Vis-NIR spectra [78,79,80,95]. The on-site Coulomb energy of free C60 molecule (U0) was calculated by DFT as 2.7–3.1 eV [130,131,132]. The effective U (Ueff = U0 − V, V is nearest neighbour Coulomb repulsion energy) values in solids are found to be 0.8–1.3 eV [130,131,132], when an electron is added to a C60 molecule surrounded by other C60 molecules in the fcc lattice, which is due to polarization by the charged C60 molecules. While, Auger spectroscopy yields Ueff in the range 1.4–1.6 eV [133,134]. The first CT band of non-metallic M+(C60•−) solid corresponds to Ueff. With organic cation or supramolecular cation in this paper, the IR and NIR absorption spectra of CT solids in KBr show CT band peaks at 0.25 and 0.68 eV for (TPC0)(MDABCO+)(C60•−) [12], 0.69 eV for (TDAE+)(C60•−) [12], 0.71 eV for (TPC0)(MQ+)(C60•−) [12], 0.74 eV for (PhCN0)(Ph3MeP+)(C60•−) [105], and 0.77 eV for (PhCN0)(TMP+)(C60•−) [12]. The lowest band of 0.25 eV in (TPC0)(MDABCO+)(C60•−) is ascribed to the intraband absorption due to the metallic nature. The estimated Ueff (Ueff = 0.68–0.77 eV) should be smaller than those for (alkali metal+)(C60•−) owing to the high polarizability of organic cation molecules, which is similar to that proposed for TCNQ CT solids [135].
The effect of orbital degeneracy on the Mott-Hubbard criterion leads to a conclusion that the Mott transition takes place at U/W = √3 or an upper limit of U/W ~ 2.5, attributed to triple degeneracy or negligible splitting of t1u orbitals [73,94]. Therefore, this relaxed Mott criterion is effective as long as the splitting of t1u orbitals by Jahn-Teller distortion is not large enough, and forms one LUMO band. The upper limit of U/W requires W to be 0.27 eV, by considering the lowest value of Ueff (calculated value—0.68 eV), which is about 2–3 times the calculated W value (0.10–0.15 eV for (TPC0)(MDABCO+)(C60•−)) by the AM1 method [12]. A preliminary DFT calculation for this CT solid indicates a total W of about 0.48 eV at 160 K [12], which is still smaller than the estimated Ueff and is not able to account for the metallic nature. Since the calculated W values in these systems are not reliable at present, only the ratio of overlap integrals or transfer interactions will be discussed in the following.
3.3. Packing of C60 and Magnetic Interactions in Fulleride Solids
So far, no structure-magnetic property relationship, especially concerning the geometrical spin frustration and the QSL state has been studied for the triangular or hexagonally packed C60•− solids. Hexagonal packing of C60•− has been suggested for (tetramethylammonium)(C60•−)∙1.5THF polycrystals based on the postulated structure, from both the calculation of total energy for various arrangements of the component molecules and the observed powder diffraction pattern [136]. The estimated r was 10.13 Å. The conductivity of the pellet sample was 10−2 S·cm−1 and the effective magnetic moment of the complex was ~1.75 μB in good agreement with the value for the system containing one S = 1/2 spin per formula unit. However, the susceptibility did not indicate AF interactions. (Na+)(C60•−)(THF)5 polycrystals [137] showed a Curie-Weiss behavior above 200 K (μ = 1.70 μB, ΘCW = −58 K) suggesting triangular or hexagonal packing of C60•−. The χ (magnetic susceptibility) value decreased sharply below 180 K (μ = ca. 0.8 μB), probably owing to the dimerization of C60•−, though no crystal structure above 200 K was found.
Recent work on needle-like single crystal (up to 6 mm) of (K+)(C60•−)(THF)5(THF)2 revealed that K+ cation coordinate with five THF molecules (by K∙∙∙O contacts) to form bulky cationic building units of [K(THF)5]+, which separates C60•− corrugated layers with a distorted square arrangement [138]. At RT, C60•− molecules display rotational disorder, and ordering takes place below 240 K with a simultaneous appearance of a Dysonian EPR line down to 40 K. In the non-conductive region in the 240–295 K range, Curie-Weiss behavior (ΘCW = −54 K) was observed with r = ~10.01 Å at 260 K. A metallic nature in the conductive region between 240 K and 40 K was suggested by the observation of the Dysonian EPR line shape, though the Dysonian line shape has been observed not only in metallic materials, but also in semiconductors with very high conductivity [139]. It should be emphasized that “localization” or “itinerancy” of spins in C60 solids is strongly associated with the rotational disorder and ordering of C60 molecules.
Crystals of [K([2.2.2]crypt)]2(C602−)∙4(toluene) exhibit a 2D distorted hexagonal layer of C602− dianion molecules separated by layers of [K([2.2.2]crypt)]+ cations [115]. The shortest interfullerene distance of 13.77 Å is too long owing to the large size of cation to have appropriate intermolecular interactions for the spin-frustrated system, regardless of the magnitude of effective spin of the dianion system.
3.4. Requirements for Triangular or Hexagonal Packing of C60•− by Key-Keyhole Relation: 2D or 3D Polycationic Template
The donor molecules that can reduce fullerene moieties are sparse because of the weak electron-accepting ability of fullerene moiety [140]. Simple use of very strong donor species, such as alkali metal has not led to 2D triangular or hexagonal packing of C60•−. A strategy to obtain new versatile ionic complexes, including polymerized fullerenes has been developed by Konarev, et al. [12,37,38,95,103,104,105,106,141,142,143,144,145,146,147,148,149], which can lead to multi-component CT complexes composed of small-sized strong donor molecules including alkali metals (D2), which are able to ionize fullerene, and a structure-forming neutral molecule including solvent molecules (D1), e.g., {[(D10)(D2+)](fullerene−)}. A variety of single crystals of [(D10)(D2+)](C60•−) with versatile packing patterns of C60•− molecules have been prepared according to this concept, which satisfies requirement (I) described in Section 3.1.
The distance between the cation species in the supramolecule formed between D1 and D2 molecules [(D10)(D2+)] is the essential parameter to control both the bond-formation and the AF interaction among C60•− molecules. Therefore, the r value is the key to satisfy requirements (II) and (IV). When the supramolecular assemblies of D1 and D2 molecules have a periodic cationic site with sufficient space to hold one C60•− molecule, C60•− molecules (spin site = key) will be arranged according to the pattern of the cationic parts of supramolecular assemblies [(D10)(D2+)] (=keyhole). This is the key-keyhole relation between C60•− molecules and polycationic template [(D10)(D2+)]. To construct triangular or hexagonal packing of C60•−, the template [(D10)(D2+)] should have a triangular or hexagonal pattern of cationic sites. It was observed that the threefold symmetry of D1 and D2 molecules satisfies requirement (III) [12,146]. Higher than threefold symmetry would be satisfactory. However, the design principle to satisfy the requirement for t′/t ~ 1 has not been developed yet for multi-component solid [(D10)(D2+)](C60•−) and two-component solid (D+)(C60•−).
A good chemical choice of D1 and/or D2 concerning the donor strength, size, shape, and symmetry satisfies all of the requirements, (I)–(III). Among the multi-component complexes {[(D10)(D2+)](C60•−)}, C60•− molecules form 2D hexagonal packing (triangular spin lattice, Figure 1k) in (TPC0)(MDABCO+)(C60•−) (3) and (TPC0)(MQ+)(C60•−) (4) and three-dimensional (3D) one in (DMI+)3(C60•−)(I−)2 (8) [12,145,146]. The coexistence of monomer metallic and monomer AF phases was observed in 3 (r = 10.07 Å at RT). Insulators 4 and 8 are AF insulators with 2D and 3D packing of C60•−, respectively, with r = 10.12–10.18 Å at 250 K and 10.06–10.12 Å at 100 K for 4 and 11.05 and 13.36 Å at 100 K for 8. Even in two-component materials, (D+)(C60•−), the bond-formation sometimes is prevented when r is large enough and the spins on C60•− interact antiferromagnetically. In (MDABCO+)(C60•−) (1), 3D close packing of C60•− and high |ΘCW| were realized and the geometry of the spin lattice is 3D distorted bipyramid with r = 10.01–10.11 Å at 250 K and 9.91–10.12 Å at 100 K [147]. The solid (Ph3MeP+)(C60•−) (2) has double chains of C60•− with zigzag spin lattice having weak interchain interactions with r = 10.08–10.10 Å at 100 K [105].
4. Key-Keyhole Relations in C60•− Charge-Transfer Solids
4.1. Three-Component Materials
4.1.1. Coexistence of Itinerant and Frustrated Spins in 2D Hexagonal Packing of C60•− in (TPC0)(MDABCO+)(C60•−)
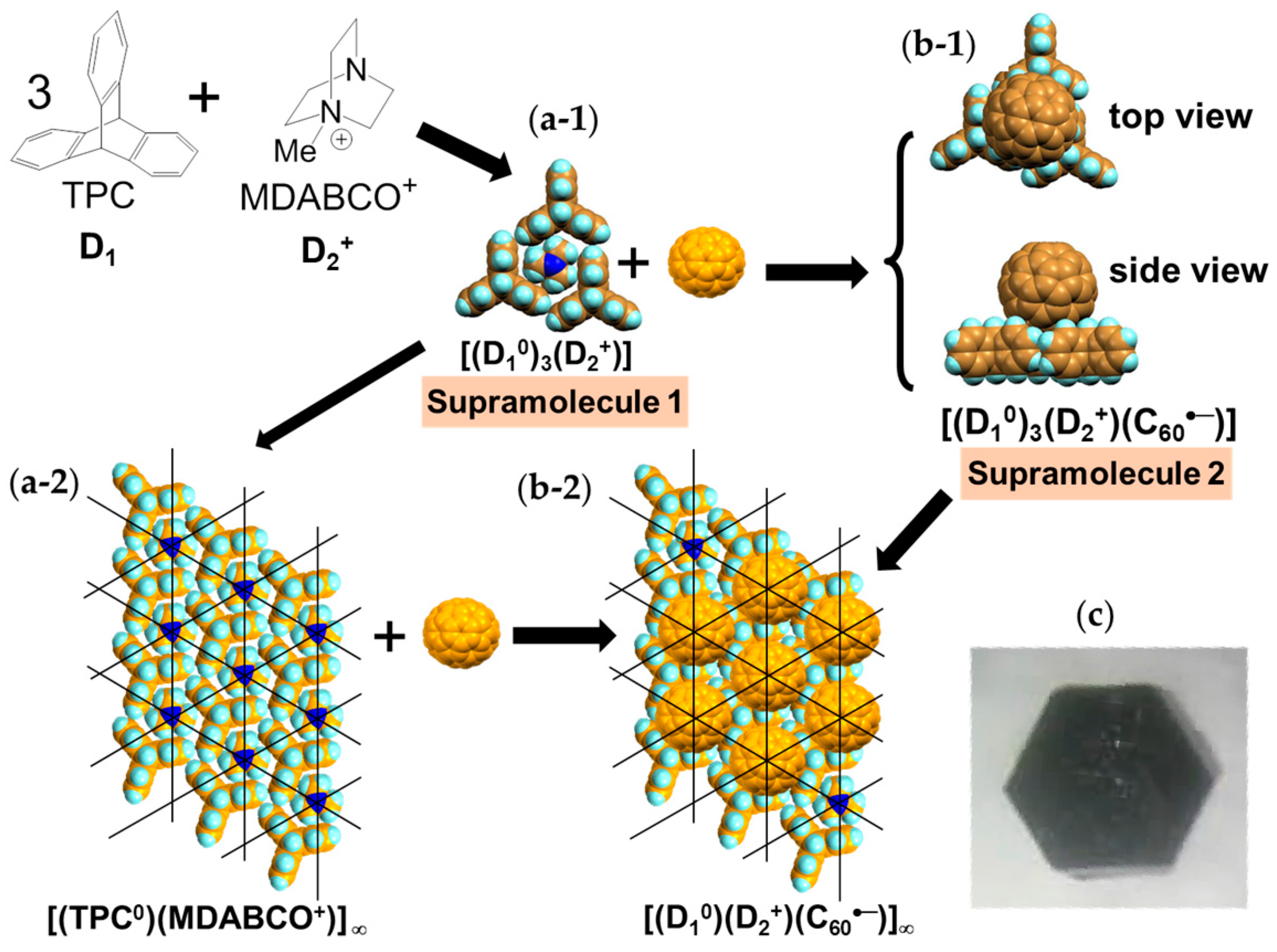
Single crystals of (TPC0)(MDABCO+)(C60•−) (3) were obtained by the diffusion method. C60, reductant CH3CH2SNa, and MDABCO∙I were stirred in a PhCl2/PhCN mixture. TPC was dissolved in the obtained solution and n-hexane was layered. The diffusion was carried out over a period of two months to give black hexagonal prisms on the walls of the tube of sizes up to 0.5 × 2 × 2 mm3 (Figure 4c) [146].
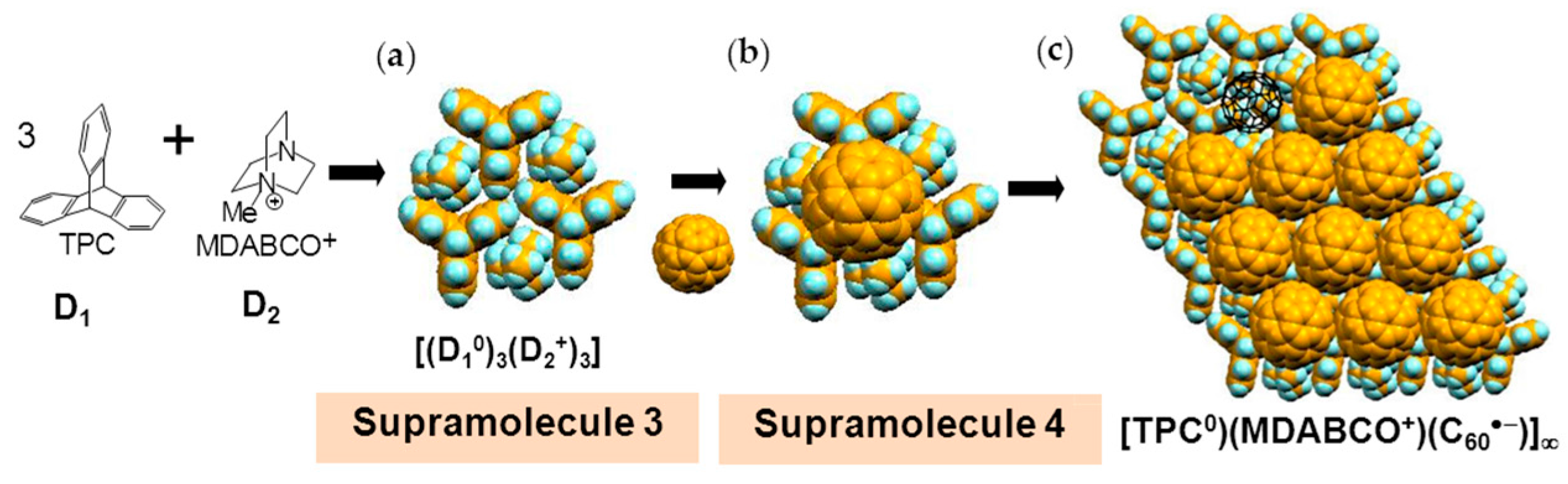
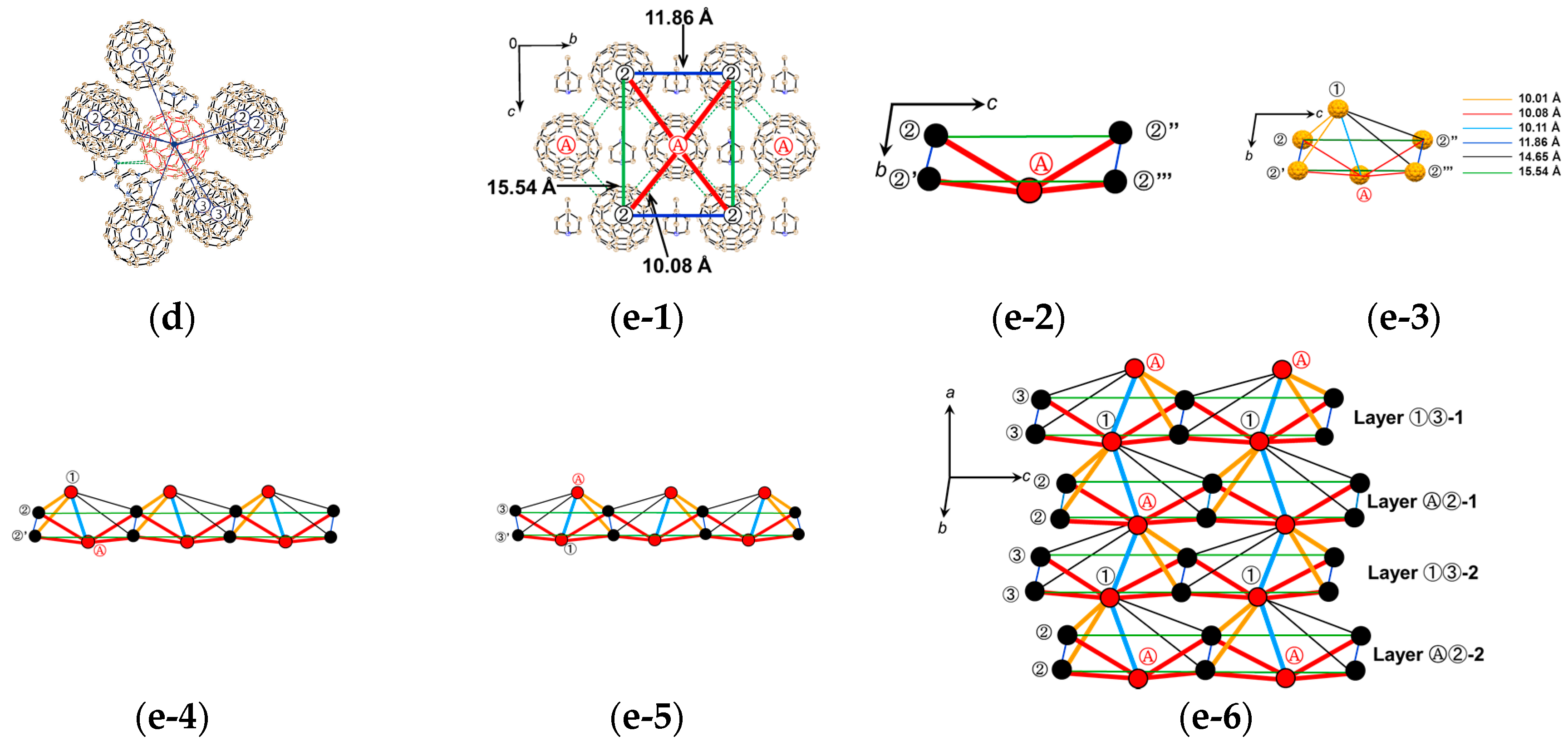
The formation of 3 can be well interpreted by two kinds of key-keyhole relation (Figure 4). In the first step, three TPC molecules (D1) surround MDABCO+ molecule (D2+), both molecules have threefold symmetry, to form cationic supramolecular unit (Supramolecule 1: [(TPC0)3(MDABCO+)]) as shown in Figure 4(a-1).
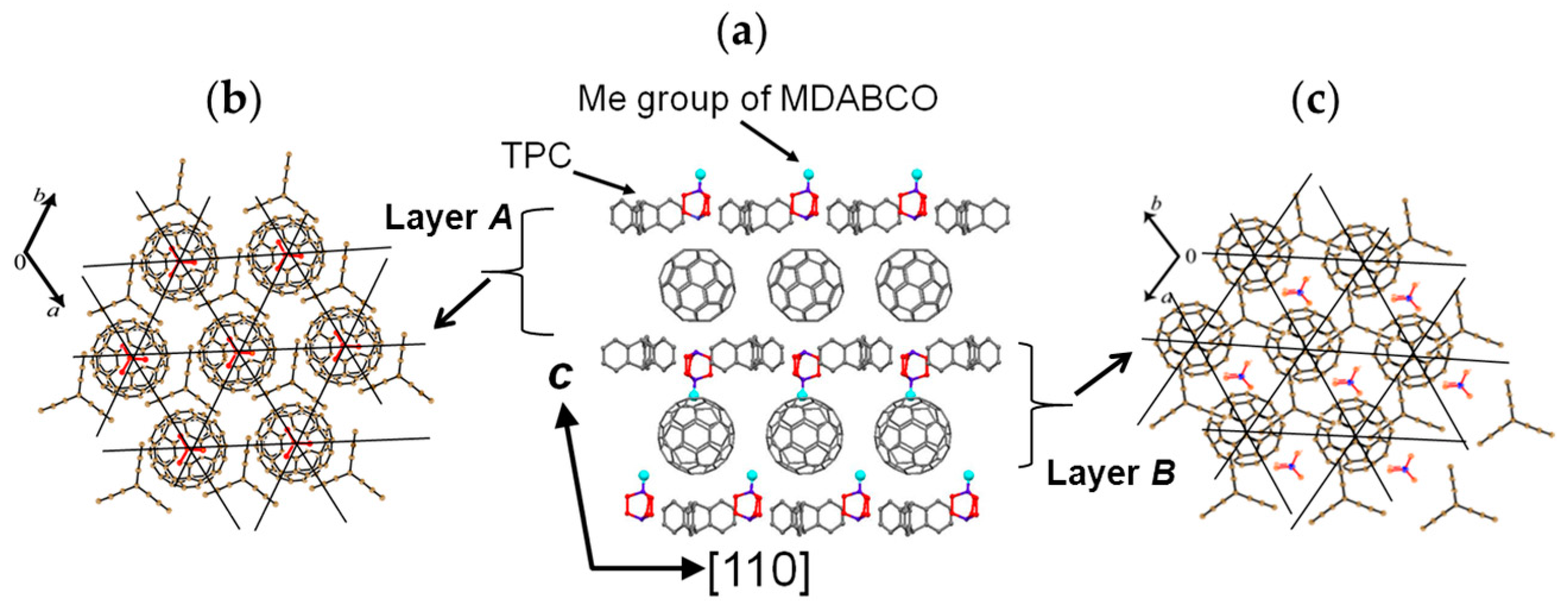
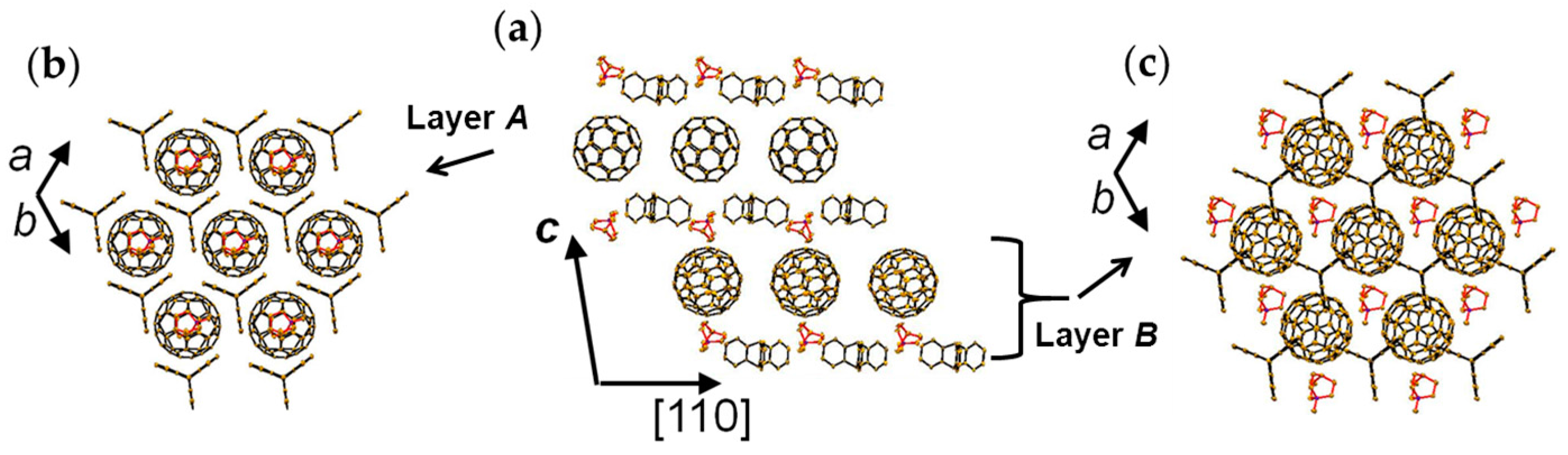
A C60•− molecule fit into the concave of the supramolecular unit to form a unit of {Supramolecule 2: [(TPC0)3(MDABCO+)](C60•−)} according to the second key-keyhole relation (Figure 4(b-1)). The Supramolecule 2 units assemble to form 2D layer of hexagonally packed C60•− in the ab plane ([(TPC0)(MDABCO+)(C60•−)]∞, Figure 4(b-2)). In another view, C60•− molecules were assembled according to the 2D sheet of the polycationic template [(TPC0)(MDABCO+)]∞ shown in Figure 4(a-2) to form crystal 3. Such packing of C60•− was generated on one side of the polycationic template and is denoted as Layer A.
Figure 5 shows the size of MDABCO+ (Figure 5a,b), the height of TPC (Figure 5c), and some parts of Supramolecule 1 (Figure 5d) at 200 K. The neighboring TPC molecules are separated by 9.82 Å, so that it is able to prevent the bond-formation between C60•− molecules when they pack on the template in a hexagonal lattice structure, where the intermolecular distance of MDABCO+ molecules corresponds to N···N (blue points in Figure 4(a-2)) with separation distance of 9.99 Å. The MDABCO+ molecule is thicker (7.27 Å) than the thickness of the holder, which is composed of three TPC molecules (7.05 Å). Consequently, one side of Supramolecule 1 has a concave shape where N atoms are centered and the Me of N-Me+ group of MDABCO+ extrudes from the holder on the opposite side. Therefore, the C60•− hexagonal layer on the top of the polycationic template in Figure 4(a-2), namely Layer A (Figure 6a,b) has different steric and electronic environment than the C60•− hexagonal layer at the bottom of the polycationic template (Layer B, Figure 6a,c).
Figure 7 demonstrates the key-keyhole relation for layer B. Three TPC0 and three MDABCO+ molecules constitute Supramolecule 3, [TPC0]3[MDABCO+]3 (Figure 7a), according to the first key-keyhole relation. Here, six methyl groups of six MDABCO+ molecules, three from the top (TPC0)(MDABCO+) layer and three from the bottom layer, constitute an octopore to hold one C60•− molecule to afford {(TPC0)6(MDABCO+)6(C60•−)}, according to the second key-keyhole relation.
Figure 7b shows one Supramolecule 4, (TPC0)3(MDABCO+)3(C60•−). The 2D assembly of Supramolecule 4 generates the hexagonally packed C60•− Layer B (Figure 7c), which corresponds to Figure 6c.
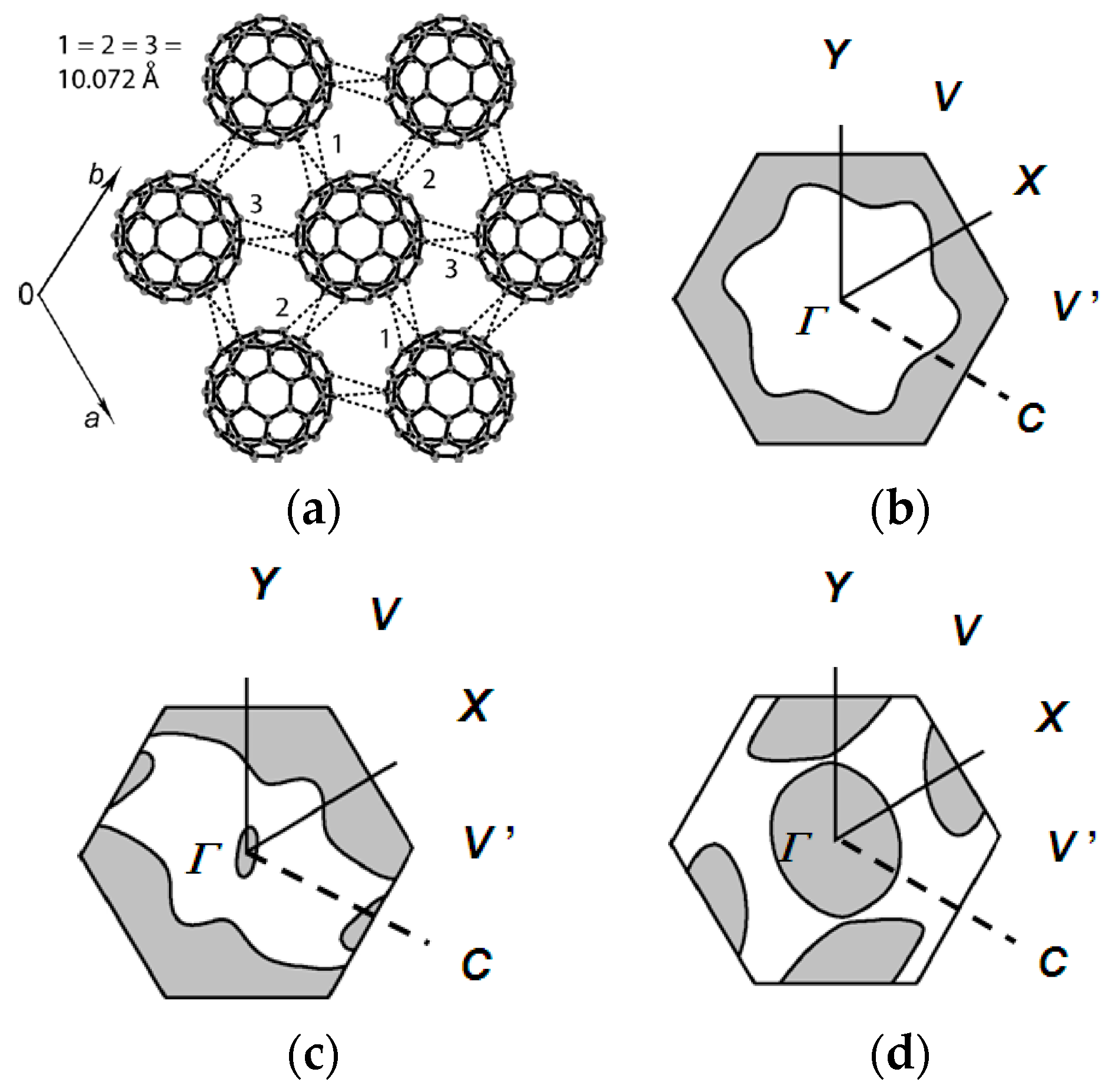
At 300 K, C60•− molecules are ordered in Layer A (r = 10.07 Å, overlap integral s = 1.91 × 10−3, Figure 8a), while C60•− molecules in Layer B are disordered. At the same temperature, half of the MDABCO+ cations are disordered between three orientations that are linked by their rotation about the lattice threefold axis. On Layer A, the r value decreases monotonously to 9.97 Å at 183 K at which temperature a transition from rhombohedral to triclinic occurs. Assuming linear shrinkage of the interfullerene distance along the a axis below 160 K, r = 9.54 Å is evaluated at around 4 K, where no dimerization was experimentally detected. The calculated Fermi surfaces of both C60•− layers have a closed 2D pocket at Γ point and suggest 2D metallic nature in the ab plane (Figure 8b).
With decreasing temperature, ordering of the orientations of C60•− and MDABCO+ started below 200 K. A complete ordering of all three of the component molecules was found in the crystal structure at 160 K (Figure 6). These observations suggest that the orientation disorder of C60•− is closely linked with that of MDABCO+. The calculated overlap integrals at 160 K are s = 2.57 × 10−3 (//a), 2.03 × 10−3 (//b), and 2.76 × 10−3 (//a + b) for Layer A and 2.45 × 10−3 (//a), 2.21 × 10−3 (//b), and 1.61 × 10−3 (//a + b) for Layer B. The calculated bandwidth W is 0.103 eV at 300 K for Layer A, 0.150 eV and 0.133 eV for Layers A and B at 160 K, respectively. The calculated anisotropy of the transfer interactions ta:tb:ta+b = 1:1:1 for Layer A above 183 K changed to ta:tb:ta+b = 1.27:1:1.36 for Layer A and ta:tb:ta+b = 1.52:1.37:1 for Layer B at 160 K. The ratio of the triangular spin lattice is defined as 2ta/(tb + ta+b), 2tb/(ta + ta+b), and 2ta+b/(ta + tb). However, the last two definitions provide inadequate t′/t values of 0.60–0.76 for layer A and 0.54–0.69 for Layer B at 160 K that suggests a much enhanced 2D nature than that at RT. So using the ratio t′/t = 2ta/(tb + ta+b), the calculated anisotropy is 1.00 (300 K) and 1.07 (160 K) for Layer A and 0.99 (185 K) and 1.28 (160 K) for Layer B. The spin frustration of this spin lattice is comparable to that of κ-(ET)2Cu2(CN)3 (t′/t = 1.09 at RT, 1.07 at 100 K) [54] and stronger than a Mott insulator κ-(ET)2B(CN)4 [150] (t′/t = 1.42 at RT, 1.61 at 100 K, ground state is valence-bond solid). However, the increase of W together with the ordering of C60•− molecules in Layer B below 200 K gave rise to a superior itinerancy (metallic state) than localization (QSL state) for this salt.
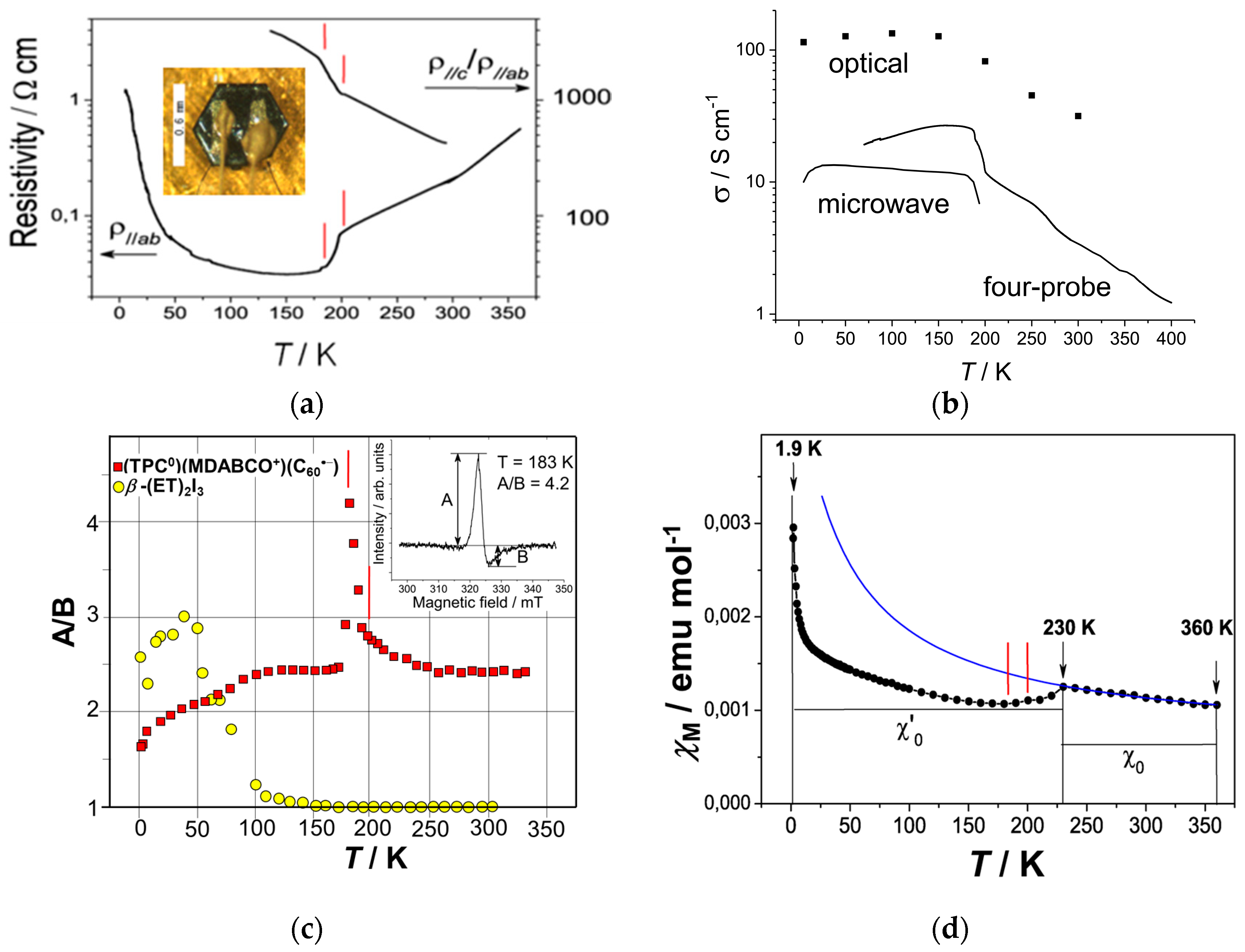
The resistivity measurements carried out using the four probe method indicate that salt 3 is metallic within the ab plane from 360 (1.8 S·cm−1) to 200 K (14 S·cm−1), after which a rapid enhancement of the metallic nature occurs from 200 K to 185 K (33 S·cm−1) (Figure 9a).
The temperature range of this anomaly between 200 K and 185 K, indicated by two red lines, coincides well with that of an ordering of C60•− in Layer B, showing that the ordered C60•− radical anions in Layer B start to participate in the metallic transport below 200 K. The resistivity could not be measured correctly below 70 K because of a large increase in the contact resistance, due to its air-sensitivity, even though it was measured in an inert atmosphere. The contactless microwave and optical measurements revealed that the conductivity increased down to 100–25 K and the metallic state is preserved down to 5 K (Figure 9b) [12]. The optical conductivity is nearly flat below 150 K down to 5 K.
The asymmetry ratio of the Dysonian EPR line shape between the maximum and minimum of the absorption derivative (A/B, Figure 9c inset) of 3 is compared with that of β-(ET)2I3 (Figure 9c) [151], which is highly metallic and shows SC with Tc = 1.5 K [152]. For β-(ET)2I3, the EPR signal is Lorentzian (A/B = 1) at RT and the peak ratio A/B increases below 130 K to about A/B = 3.0 (at approximately 50 K) followed by a gradual decrease down to 5 K. In comparison, the A/B values of 3 are considerably large even at RT (A/B = 2.4). It exhibits an abrupt increase below 200 K reaching a maximum of 4.2 at 183 K, which coincides well with the rapid conductivity increase, then falls to 2.3–2.4 below 183 K. The A/B ratio slowly decreases below 100 K, but the Dysonian shape is observed even at 4 K (A/B = 1.64), thus confirming the existence of a highly conducting state down to 4 K.
At 230–330 K, molar magnetic susceptibility (χM) can be fitted by a combination of the Pauli and Curie-Weiss terms: χM = χ0 + C/(T − ΘCW) with a constant χ0 = 6.5 × 10−4 emu∙mol−1, C = 0.160 emu∙K∙mol−1, and ΘCW = −31 K (blue curve in Figure 9d). C of 0.160 emu∙K∙mol−1 corresponds to the contribution of about 43% of the spins from the total amount of C60 (C = 0.374 emu∙K∙mol−1 for 100% of spins). Consequently, the spins in one layer (Layer B) are treated as localized ones and they interact antiferromagnetically with ΘCW of −31 K. A reversible decrease in χM is observed at 200–230 K. Below 200 K, the temperature-independent susceptibility (χ′0) of about 10.0 × 10−4 emu∙mol−1 is attributed to the Pauli paramagnetic contribution, implying a metallic state down to 1.9 K. The scenario is that ordering of both C60•− in layer B and MDABCO+ triggered a transition from a non-metallic and antiferromagnetically frustrated state to a metallic state for spins of C60•− in Layer B, while ordered C60•− in Layer A kept its 2D itinerancy over the entire temperature range. The strong coupling between the ordering of C60 and physical properties is intriguing and was previously observed in some fullerene salts [153,154,155,156].
Salt 3 is the first 2D monomer-type C60•− organic metal composed of only light elements (C, H, N). Even though rapidly cooled AC60•− (A = Cs and Rb) were reported to be monomer-type metals below 150 K and 125 K, respectively, definitive information is needed concerning the stoichiometry, metallic behavior, dimensionality, and crystal structure to confirm a monomer-type metal [71,72,157].
Summarizing the information concerning the geometry of spin lattice of 3, the C60•− molecules form hexagonal stacking according to the geometry of cationic template (TPC0)(MDABCO+), both of the component molecules have threefold symmetry, by key-keyhole relation. Layer B has t′/t = 0.99 (185 K), which indicates strong spin frustration and is close to those of QSL candidates κ-(ET)2M2(CN)3 (M = Cu; t′/t = 1.09, Ag; t′/t = 0.97), |ΘCW| = 31 K is estimated in the range of 260−300 K, and r = 10.07 Å (RT). No dimerization of C60•− occurred down to 1.9 K.
4.1.2. Only Frustrated Spins in 2D Hexagonal Packing of C60•− in (TPC0)(MQ+)(C60•−)
By a using MQ+ instead of MDABCO+, where MQ+ is N-methylquinuclidinium cation, both of which have threefold symmetry, an AF insulator (TPC0)(MQ+)(C60•−) (4) was obtained [12]. The C60 molecule, a 10-fold molar excess of CH3CH2SNa, and a 5-fold molar excess of MQ∙I were reacted in a PhCl2/PhCN mixture. Into a filtered solution, TPC was dissolved and filtered. n-Hexane was layered over the obtained solution. Black hexagonal prisms up to 0.2 × 0.5 × 0.5 mm3 were harvested after two months.
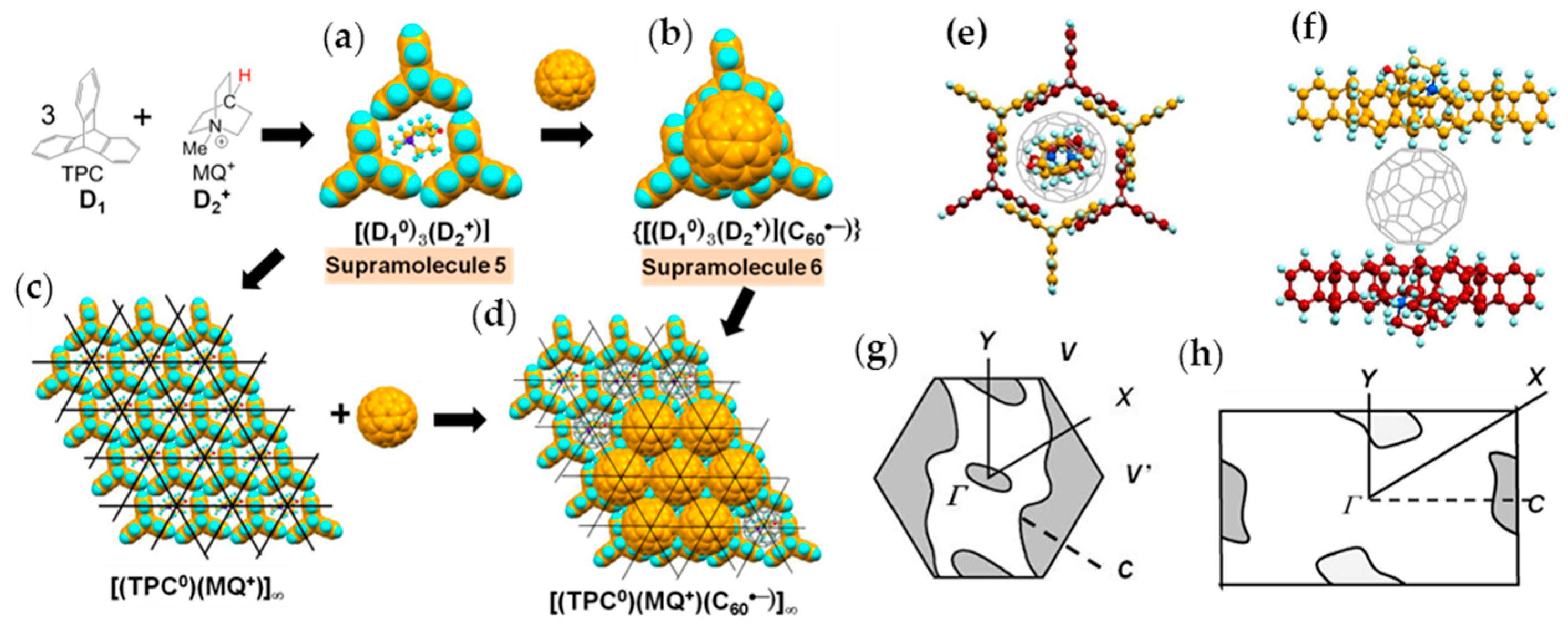
A major difference between MQ+ (Figure 10a–c) and MDABCO+ is that the vertical size of MQ+ (7.87 Å) is larger than that of MDABCO+ cation (7.27 Å) since the carbon atom with hydrogen in MQ+ instead of uncharged nitrogen atom in MDABCO+ that is caused some kind of distortion in the layered packing of C60 molecules. The size of MQ+ is 5.86 Å × 6.40 Å (Figure 10c), which is very close to that of MDABCO+. Figure 10d,e show the sizes of the fundamental units at 250 K. Similar to the MDABCO+ salt, three TCP and one MQ+ molecules form Supramolecule 5; (TPC0)3(MQ+), with a periodicity of TPC molecules of an average of 10.15 Å (Figure 10d) and periodicity of N-site in MQ+ molecules of an average of 10.04 Å (Figure 10e).
Crystal 4 has lower symmetry (triclinic unit cell) than 3 at RT. Similar to 3, crystal 4 has layered packing in which hexagonal fullerene layers alternate with the (TPC0)(MQ+) layers along the с axis (Figure 11a) in the sequence of (TPC0)(MQ+) layer/C60•− Layer A/(TPC0)(MQ+) layer/C60•− Layer B. Figure 11b,c show the view along the c axis for Layer A and Layer B, respectively.
The key-keyhole relation between C60 in Layer A and (TPC0)(MQ+) is presented in Figure 12. Three TPC molecules and one MQ+ form Supramolecule 5, (TPC0)3(MQ+), via the first key-keyhole relation. Then, C60•− molecule fits into the concave of Supramolecule 5 to give Supramolecule 6, [(TPC0)3(MQ+)](C60•−), similar to the case of {[(TPC0)3(MDABCO+)](C60•−)} (Figure 4(b-1)) by the second key-keyhole relation. The Supramolecule 6 units assembled to form (Supramolecule 6)∞ generating Layer A of (TPC0)(MQ+)(C60•−) (Figure 11b and Figure 12d).
From a different point of view, C60•− molecules fit into the concaves in the polycationic template of layered unit of [(TPC0)(MQ+)]∞ (Figure 12c) to form Layer A in 4 (Figure 12d). Figure 12e,f illustrate how one C60•− molecule in Layer A is embedded between two layers of (TPC0)(MQ+) where the TPC molecules in the upper and lower layers are shown in different colors. C60•− molecules in Layer A are well fitted in the TPC hole formed by the six TPC molecules in the upper and lower (TPC0)(MQ+) layers (Figure 12e). In Supramolecule 5 [(TPC0)3(MQ+)] (Figure 10d), the extra hydrogen atom (red circle in Figure 10a) in MQ+ prevents the MQ+ cation from arranging vertically relative to the fullerene layers resulting in a lowered crystal symmetry. Figure 13g,h show the calculated Fermi surfaces by the tight-binding method combined with the semiempirical (AM1) molecular orbital calculations based on crystal structures at 250 K and 100 K, respectively. The band calculation of Layer A at 250 K indicates that the salt has 2D Fermi surfaces. However, due to the doubling of the unit cell along the b axis, a semi-metallic Fermi surface was estimated at 100 K.
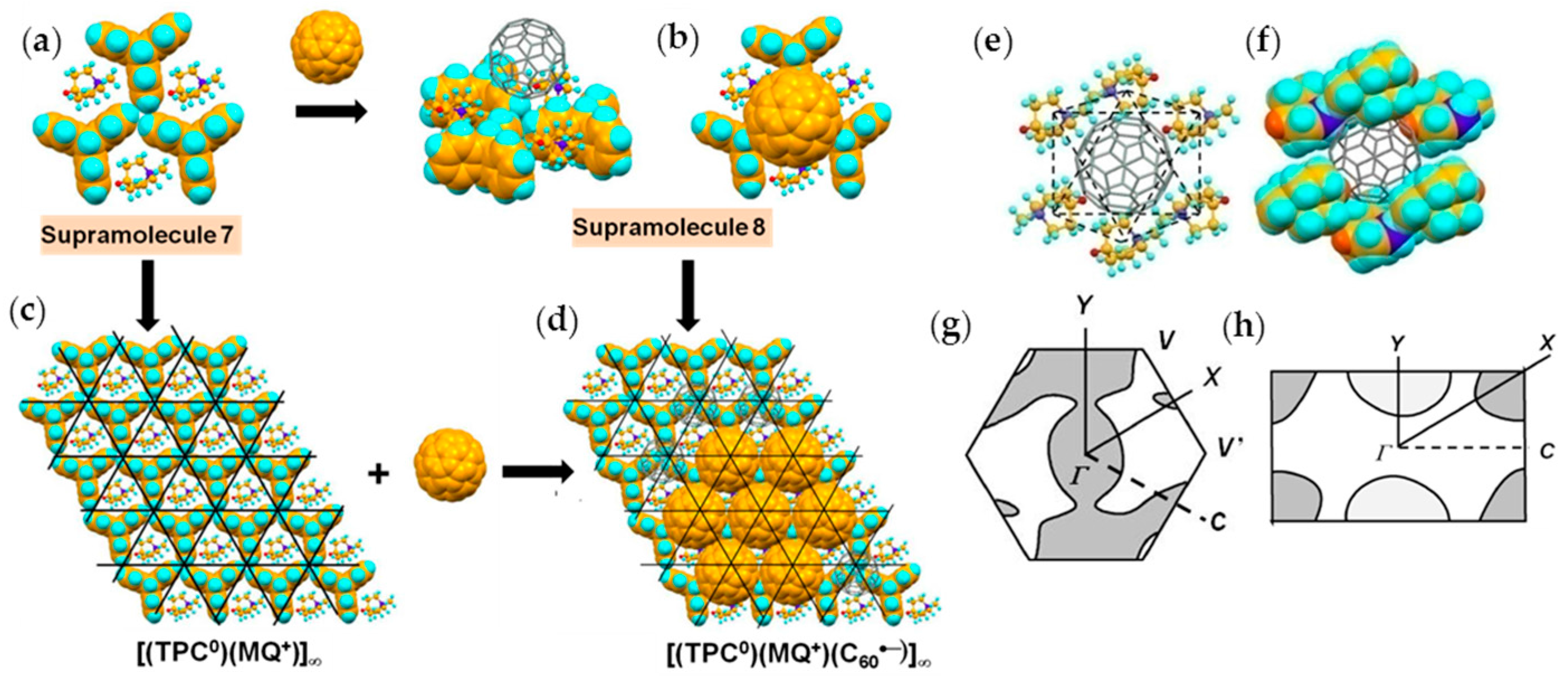
Surprisingly, Layer B also has a hexagonal packing of C60•− molecules in spite of the use of longer cation molecule MQ+ by 0.60 Å than MDABCO+. Figure 13 shows the formation of Layer B similar to that in Figure 7. Figure 13a shows Supramolecule 7 by the first key-keyhole relation made of three TPC and three MQ+ molecules. The C60 molecule fit into the hollow site where three MQ+ molecules formed that corresponds to the crossing points of black lines in Figure 13c to form Supramolecule 8 [(TPC0)3(MQ+)3(C60•−)] (Figure 13b). This is the second key-keyhole relation.
The 2D assembly of supramolecular units [(TPC0)3(MQ+)3(C60•−)] leads to Layer B on the (TPC0)(MQ+) layer (Figure 13d). From another view, the C60•− molecules assemble using polycationic template of 2D assembly of the first supramolecular units (Figure 13c) and leads to Layer B on the (TPC0)(MQ+) layer (Figure 13d).
In Layer B, C60•− molecules are sandwiched between 2D layer of (TPC0)(MQ+) formed by Supramolecule 7 (Figure 13e). Here, the C60•− molecules are more in contact with MQ+ than with the TPC molecules. The intermolecular interactions between C60•− and MQ+ are dominant factors that determine the packing of (TPC0)(MQ+) and C60•−. C60•− molecules in Layer B, which are allocated at each hollow site where the six MQ+ cation molecules combine: three MQ+ molecules in one (TPC0)(MQ+) layer and three MQ+ in the neighboring (TPC0)(MQ+) layer constitute an octopore for C60•− molecule in Layer B (Figure 13e,f). The fusion of the assemblies of (TPC0)(MQ+) and C60•− molecules while maintaining the relation of octopores in Figure 13e,f leads to the packing pattern of C60•− in Layer B in the ab plane (Figure 13d). Similar to Layer A, the band calculation of Layer B indicates that the salt has 2D Fermi surfaces at 250 K and a semi-metallic Fermi surface at 100 K (Figure 13g,h).
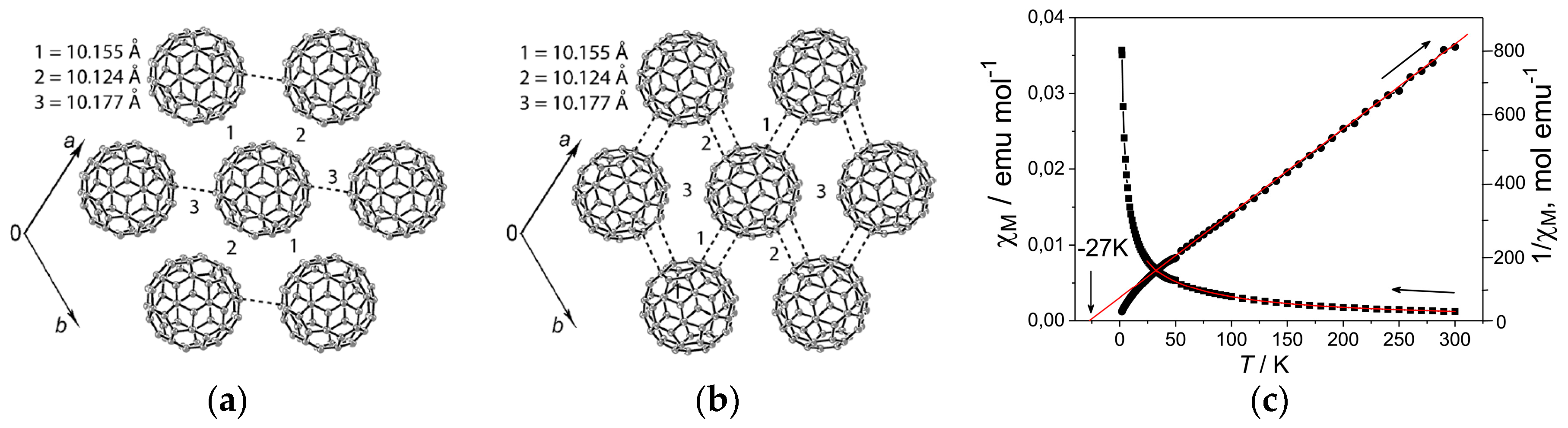
The MQ+ cations are disordered between two orientations at 250 K and even 100 K. In spite of the disorder of MQ+, the fullerene anions are ordered in 4 at both temperatures. The center-to-center interfullerene distances of 10.12, 10.16, and 10.18 Å at 250 K (Figure 14a,b) are noticeably larger than the distance of 10.07 Å in 3 at 300 K. Owing to the increased interfullerene distance in 4, the average overlap integrals are smaller than those of Layer A in 3, giving rise to localized nature of spins (Mott insulator) in 4 with distorted triangular spin lattice.
The calculated overlap integrals at 100 K are s = 0.78 × 10−3 (//a), 1.82 × 10−3 (//b), and 2.24 × 10−3 (//a + b) for Layer A and 2.81 × 10−3 (//a), 1.97 × 10−3 (//b), and 1.51 × 10−3 (//a + b) for Layer B. The calculated bandwidths are 0.103 (0.112) and 0.097 (0.113) eV for Layer A and Layer B at 250 K (100 K), respectively. Similar to 3, the ratios 2tb/(ta + ta+b) and 2(ta + tb)/(ta + tb) are 0.85 and 1.17 for Layer A and 0.91 and 0.64 for Layer B at 100 K. Since the calculated Fermi surface shows 1D properties, it is more appropriate to use 2ta/(ta + ta+b) instead of 2tb/(ta + ta+b) and 2ta+b/(ta + tb). The calculated anisotropy of the transfer interactions at 250 K is ta:tb:ta+b = 1.04:1:1 (t′/t = 1.04) and 1:1.40:1.23 (t′/t = 0.76) for Layer A and Layer B, respectively. The anisotropy changed to 1:0.90:1.11 (t′/t = 0.99) for Layer A and 1:0.70:0.54 (t′/t = 1.61) for Layer B at 100 K. The anisotropy of Layer A is close to that of κ-(ET)2Cu2(CN)3, and the geometrical spin frustration is comparable to that of 3.
The relatively large distances between C60•− prevent their dimerization but allow for the manifestation of a magnetic interaction between them. Reciprocal molar magnetic susceptibility is described well by the Curie-Weiss law in the 30–300 K range with negative Weiss temperature of ΘCW = −27 K (Figure 14c), indicating AF interaction of spins in the fullerene layers. The |ΘCW| is small, owing to the weaker AF interactions than that in 3 because of the larger interfullerene distance. In spite of the strong AF interaction of spins, magnetic ordering is not observed down to 1.9 K in this distorted triangular spin lattice system (f > 14). The resistivity measurements were impossible owing to small size of the crystals.
In summary, concerning the geometry of spin lattice of 4, the C60•− molecules form hexagonal stacking according to the geometry of cationic template (TPC0)(MQ+) by key-keyhole relation similar to that for 3. There are two fulleride layers and both have distorted hexagonal arrangement of C60•− with t′/t = 0.99 for Layer A and t′/t = 1.61 for Layer B. Owing to large r (10.12–10.18 Å at 250 K), small |ΘCW| (27 K) is estimated in the range of 30–300 K. No dimerization of C60•− occurred down to 1.9 K (f > 14).
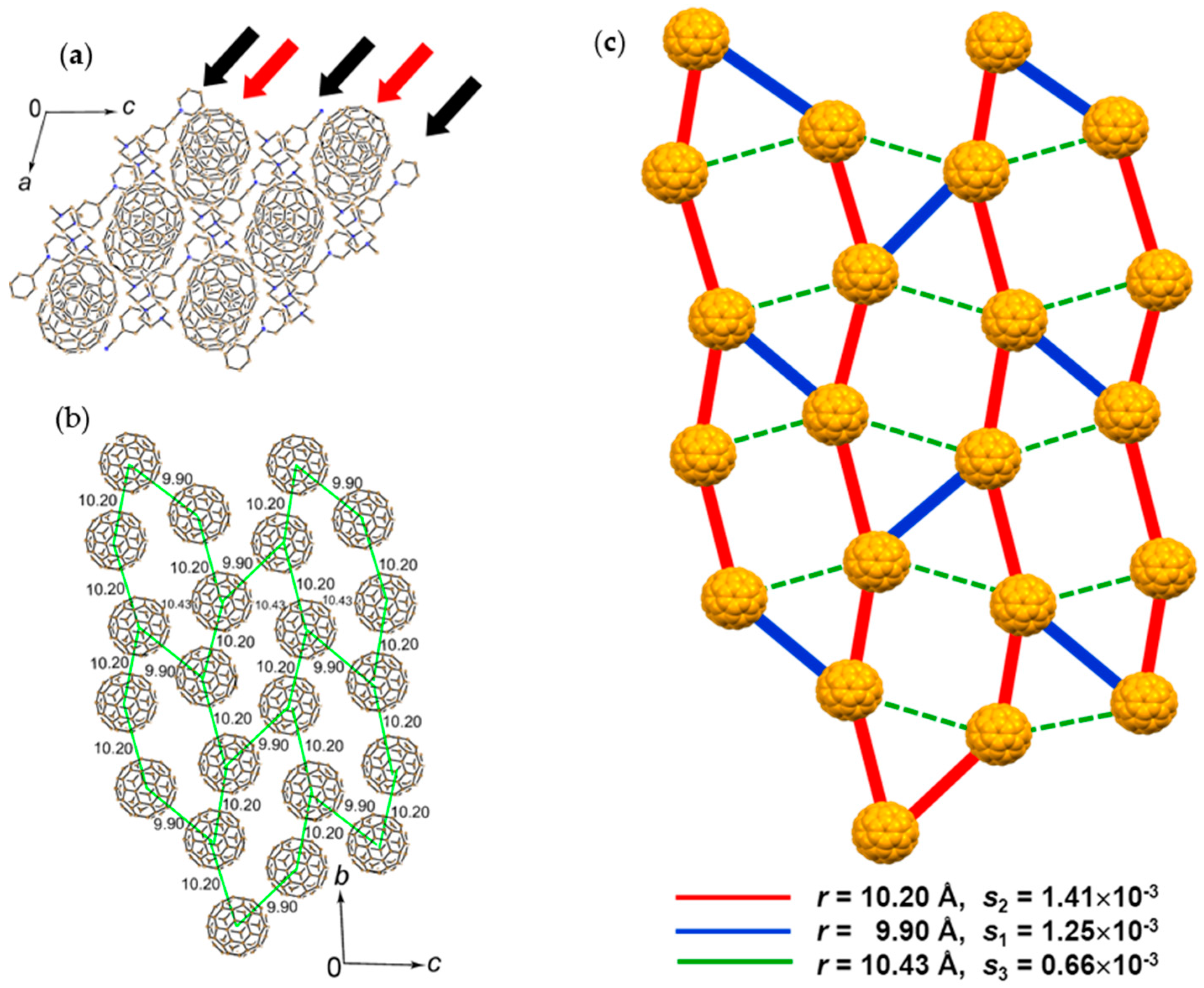
4.1.3. Distorted Hexagonal Packing of C60•− in (PhCN0)(TMP+)(C60•−), (PhCN0)(Ph3MeP+)(C60•−), and (PhCl20)[(Ph3P)3Au+]2(C60•−)2(C60)
(PhCN0)(TMP+)(C60•−)
Single crystals of the three-component salt (PhCN0)(TMP+)(C60•−) (5) were unintentionally obtained, where TMP+ is N,N,N′-trimethylpiperazinium cation (D2+) and solvent molecule PhCN is neutral (D10) [148]. The crystal structure was solved at 120 K and 90 K. The C60 molecules are disordered at 120 K and ordered at 90 K with trebling of the unit cell, which made the calculation of band parameters difficult.
The 2D C60•− hexagonal layer in 5 is sandwiched between the layers composed of (PhCN0)(TMP+) (Figure 15a). Since the components molecules D1 and D2 do not have threefold symmetry, the C60•− layer shows distorted hexagonal packing (Figure 15b). Further, the TMP+ cations form pairs and are deeply embedded in the C60•− layers to deform the C60•− packing and are found near the center of the hexagonally arranged C60•− molecules. Methyl groups of TMP+ and PhCN molecules work to prevent the close approach of C60•− molecules and no bond-formation between C60•− was detected down to 2 K. The shortest r value along the interlayer direction is 10.39 Å at 120 K, indicating weak interlayer interactions, where the spin lattice should be 2D.
Only three types of interfullerene interactions with different r (9.90, 10.20, and 10.43 Å at 120 K) are essential within the C60 layer and the ratio of the transfer interactions are t1:t2:t3 = 0.9:1:0.5 for the major C60 orientation. In spite of the hexagonal environment of the fullerenes in the layers, vdW C∙∙∙C contacts are formed with only three fullerene neighbors. Therefore, the geometry of model spin lattice is not triangular owing to weak magnetic interaction shown by dashed green lines (for r = 10.43 Å, s3 = 0.66 × 10−3) in Figure 15c. Blue (for r = 9.90 Å, s1 = 1.25 × 10−3) and red (for r = 10.20 Å, s2 = 1.41 × 10−3) lines represent the main interactions J1:J2:J3 = 0.79:1:0.22. Even though the r value for blue line is much shorter than that of the red line, s2 is larger than s1 owing to more favorable orientation of C60 for s2. The main magnetic interactions indicated by red lines extend along the b axis, and such 1D zigzag magnetic chains are connected by magnetic interactions along the c axis by blue lines. The unit of the spin lattice is edge-shared hexagonal, which is composed of four red lines and two blue lines (Figure 15c) and forms a 2D layer.
The temperature dependence of the reciprocal magnetic susceptibility for salt 5 is linear in the 70–300 K range with ΘCW = −11 K (f = 5.5, no dimerization of C60•− occurred). The resistivity at RT is ρ = 7 × 107 Ω·cm.
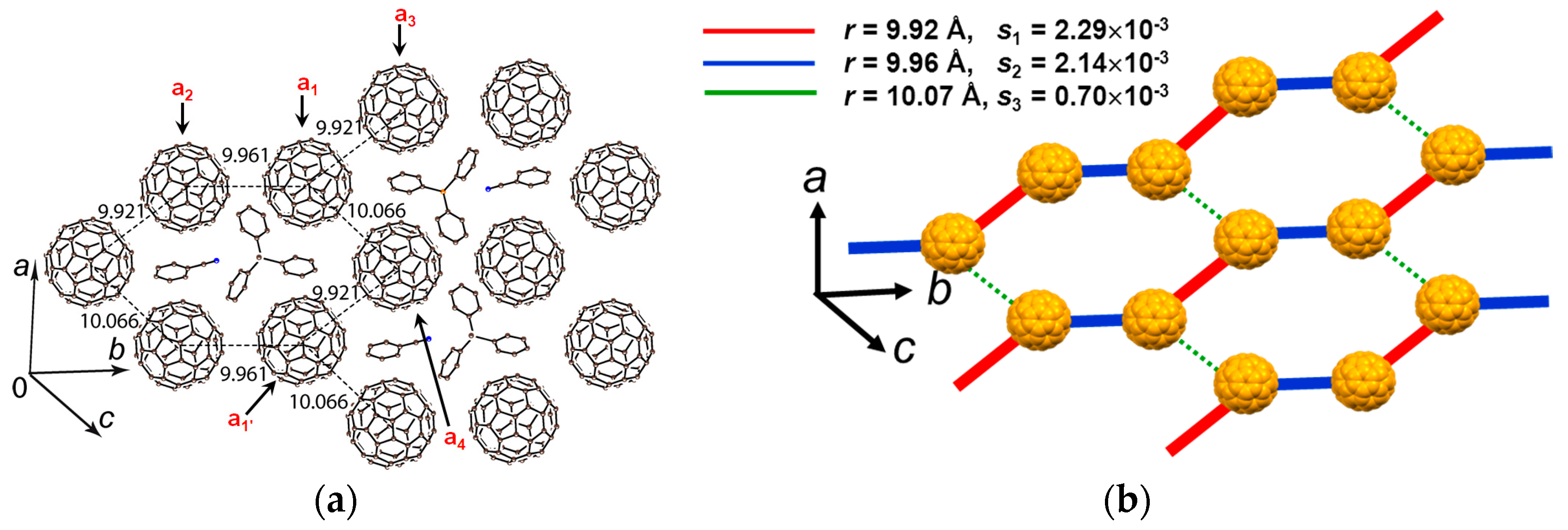
(PhCN0)(Ph3MeP+)(C60•−)
Insoluble precipitates obtained by the reduction of C60 with sodium fluorenone ketyl in PhCl2 in the presence of Ph3PMeBr were dissolved by the addition of PhCN. n-Hexane was layered on the filtered solution to grow single crystals of (PhCN0)(Ph3MeP+)(C60•−) (6) where Ph3MeP+ has a threefold symmetry [105]. The crystal structure at 250 K (Figure 16) shows hexagonal packing of C60•− and supramolecules (PhCN0)(Ph3PMe+) are located in the centers of fullerene hexagon.
Hence, C60•− has only three negatively charged fullerene neighbors similar to that observed in 5 (Figure 15b). The somewhat low value of r = 9.92, 9.96, and 10.07 Å may result in higher spin frustration than for 5. The lowest interlayer r is 10.15 Å suggesting strong 2D nature within the C60 layer in Figure 16. The C60•− molecules a1-a4 in Figure 16 form a flat layer, where the overlap integrals are s(a1−a2) = 2.14 × 10−3, s(a1−a3) = 2.29 × 10−3, s(a1−a4) = 0.70 × 10−3, and s(a2−a3) ~ s(a3–a4) ~ s(a2–a4) ~ s(a1–a1′) = 0. The spin lattice geometry (Figure 16b) is approximated as the 1D nonuniform zigzag chain along the b axis with alternating red and blue lines and the lines are connected by weak magnetic interactions by green lines (ratio of J values = 1:0.87:0.09). Salt 6 shows much stronger 1D properties than those of salt 5.
The EPR intensity decreases from 295–220 K, smoothly followed by a rapid decrease due to reversible dimerization of C60•− below 220 K. Upon cooling down to 120 K, the C60•− radical anion pairs, which has r = 9.92 Å at 250 K, form singly bonded (C60−)2 dimers with r = 9.28 Å. Therefore, no ΘCW value is determined in this system. The steric protection to avoid the bond-formation is not sufficient in this solid. The solvent free crystal (Ph3MeP+)(C60•−) (2) exhibits completely different structural and physical properties (vide infra) without the dimerization down to 1.9 K.
(PhCl20){(Ph3P)3Au+}2(C60•−)2(C60)
By using a very bulky cation (Ph3P)3Au+ with C3v symmetry, single crystals of (PhCl20){(Ph3P)3Au+}2(C60•−)2(C60) (7) with a highly symmetric trigonal lattice were obtained, in which the supramolecule {(PhCl20)[(Ph3P)3Au+]2} is a cationic template that accommodates C60 molecules hexagonally [149]. The crystal structure was solved for a crystal slowly cooled down to 100 K.
Hexagonal corrugated C60 layer is sandwiched between the layers of {(PhCl20)[(Ph3P)3Au+]2} along the c axis (Figure 17a).
Fullerenes and PhCl2 molecules located on the C3v symmetry axes are statistically disordered between three orientations. The (Ph3P)3Au+ cations are ordered and located on the C3v symmetry axis. The (Ph3P)3Au+ cations are too large in size to fit into the size of a C60 molecule, but the size of supramolecule {(PhCl20)[(Ph3P)3Au+]2} approximately corresponds to that of three C60 molecules. The C60 layer and {(PhCl20)[(Ph3P)3Au+]2} layer, which is indicated by black arrows in Figure 17b, alternate along the c axis. C60 molecules move from the layers toward planar PhCl2 molecules to form strongly corrugated C60 layers.
The fullerene layer consists of different charged C60 molecules with −1 and 0 denoted as I and II, respectively, in Figure 17a–c). Interestingly, C60•− molecules are sandwiched between a (Ph3P)3Au+ cation molecule and a PhCl2 molecule while C600 molecules are sandwiched between two (Ph3P)3Au+ molecules along the c axis, as shown in Figure 17b. Negatively charged and neutral C60 molecules are closely packed within hexagonal layers with r (I∙∙∙II) = 10.02 Å, while between C60•−, it is long with r (I∙∙∙I) = 10.37 Å due to corrugation. The magnetic interactions between C60•− molecules in the neighboring fullerene layers are expected to be small based on its r value (r ~ 13.9 Å). As a result, the magnetic interactions are 2D. Each C60•− has only three negatively charged fullerene neighbors within a fullerene layer, namely C60•− molecule a1 is surrounded by C60•− molecules a2–a4 in Figure 17c. They form distorted tetrahedral spin lattice composed of C60•− molecules a1-a4 (red lines in Figure 17d). The a1 molecule projects out of the a2–a4 plane in Figure 17d by only 3.03 Å. The tetrahedral units are arranged in the 2D plane by apex-sharing. The overlap integrals have not been obtained due to severe disorder of fullerene molecules. Owing to the very large center-to-center distance between the C60•− molecules, the AF interaction is weak (ΘCW = −5 K, f = 2.6). In order to enhance the magnetic interactions, smaller sized cationic supramolecules than [(PhCl20){(Ph3P)3Au+}2] well matched with two C60 molecules would be preferable. The resistivity at RT is approximately ρ = 4 × 105 Ω·cm. EPR measurements confirmed no dimerization of C60•− down to 4.2 K.
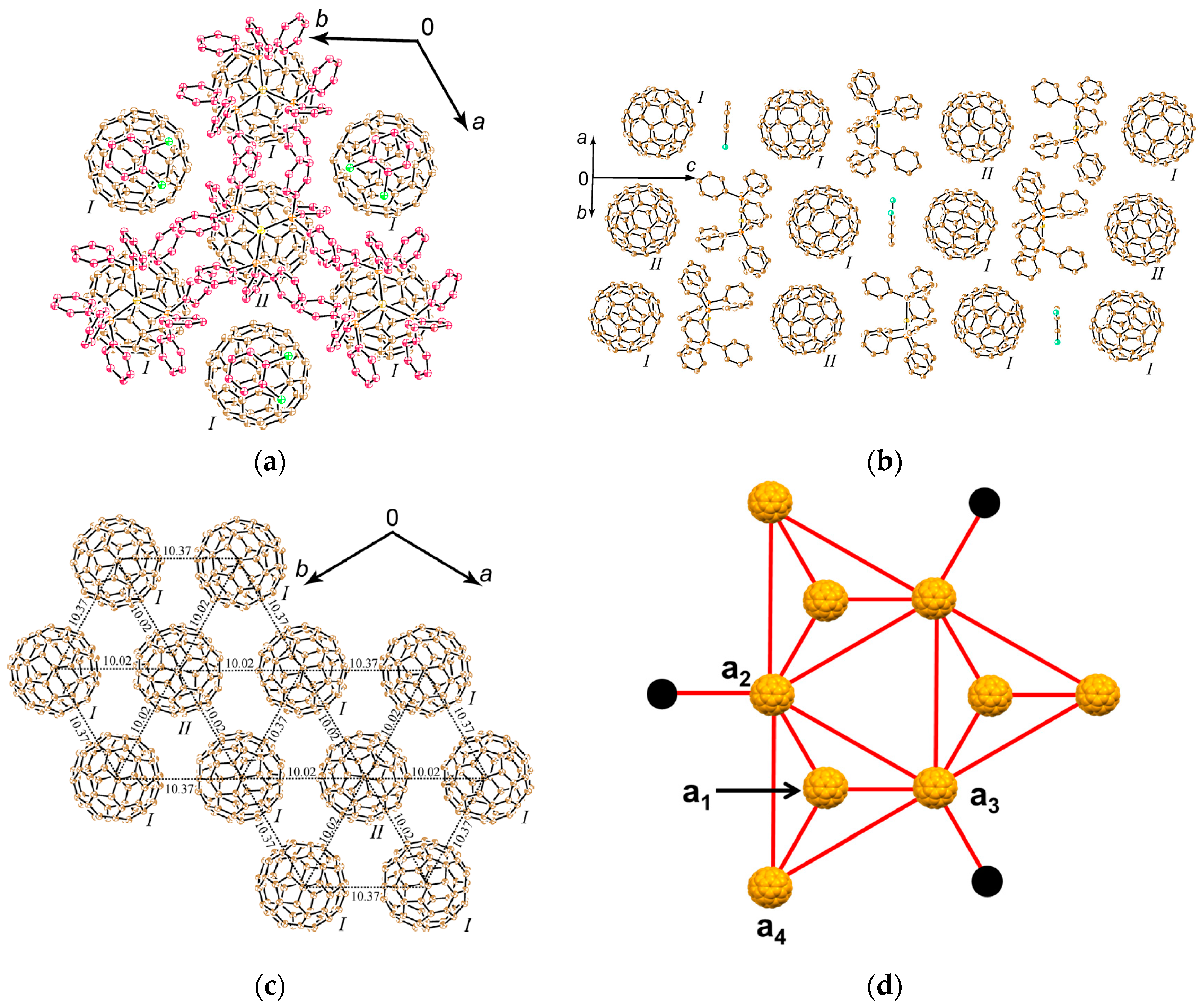
4.1.4. Frustrated Spins in 3D Hexagonal Packing of C60•− in (DMI+)3(C60•−)(I−)2
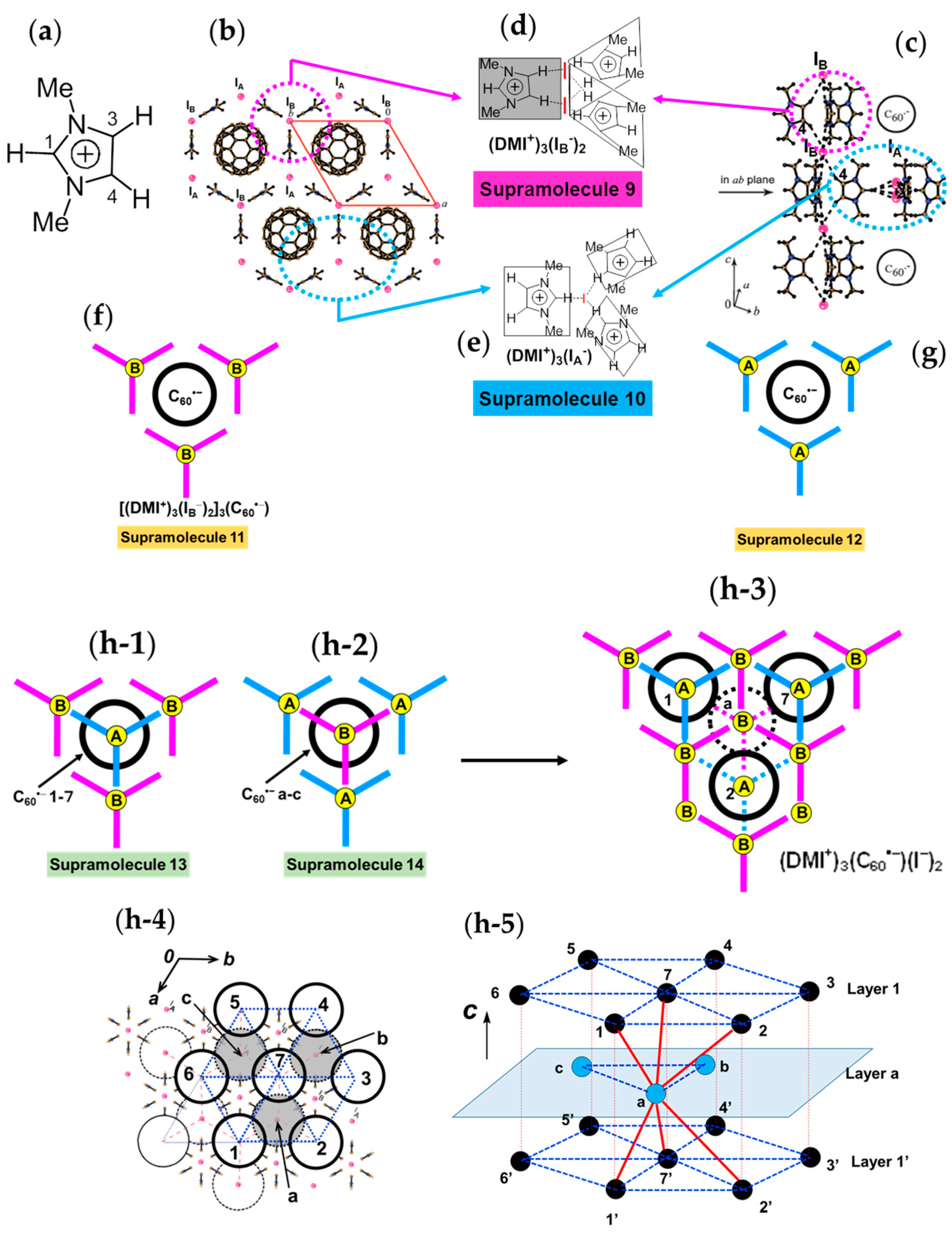
Hexagonal packing of C60•− molecules is also formed in (DMI+)3(C60•−)(I−)2 (8) [145], where cationic template is delivered by a three-dimensional (3D) network of [(DMI+)3(I−)2] (Figure 18), where DMI+ is N,N’-dimethylimidazolium cation with no threefold symmetry. This system is not within the (D10)(D2+)(C60•−) scheme, but the supramolecular cationic template [(DMI+)3(I−)2] has threefold symmetry.
The single crystals were obtained by a diffusion method. C60, an excess of DMI∙I and reductant CH3CH2SNa were stirred in a PhCl2/PhCN mixture. The mixture was cooled and n-hexane was layered over the solution. The diffusion was carried out during one month to give the single crystals on the wall of the tube with the size of 1 × 1 × 0.5 mm3.
The 3D network of [(DMI+)3(I−)2] is held together by the hydrogen (H)-bonds between the H atoms of DMI+ cation (Figure 18a) and I− anion. The crystal structures at 100 K are shown in Figure 18b,c. The H atoms at 3- and 4-positions of DMI+ have H-bonds with one kind of I− anion, denoted as IB−. One IB− anion is surrounded by six DMI+ molecules; namely, IB− anion in red color in Figure 18b,c has H-bonds with six H atoms at 4-position (three up and three down along the c axis, see Figure 18c). The supramolecular unit composed of three DMI+ molecules and two IB− anion molecules; Supramolecule 9 [(DMI+)3(IB−)2], which corresponds to the unit encircled by magenta dotted circle in Figure 18b,c, has a similar threefold symmetry like TPC molecule as shown in Figure 18d. Three Supramolecular 9 units encircle one C60•− molecules to form Supramolecule 11 [(DMI+)3(IB−)2]3(C60•−) which is schematically seen in Figure 18f where magenta figure Y represents Supramolecule 9 and B represents an IB− anion.
H atoms at 1-position have contacts with different kind of I− anions (IA−), and three DMI+ molecules and one IA− anion molecule form another supramolecular unit [(DMI+)3(IA−)] in the ab plane encircled by blue circle (Supramolecule 10, Figure 18e) in Figure 18b,c, which has also threefold symmetry like TPC. Similar to Supramolecule 11, one C60•− molecule is surrounded by three Supramolecular 10 units [(DMI+)3(IA−)] in the ab plane to form Supramolecule 12, where blue figure Y represents Supramolecule 10 and A represents an IA− anion (Figure 18g).
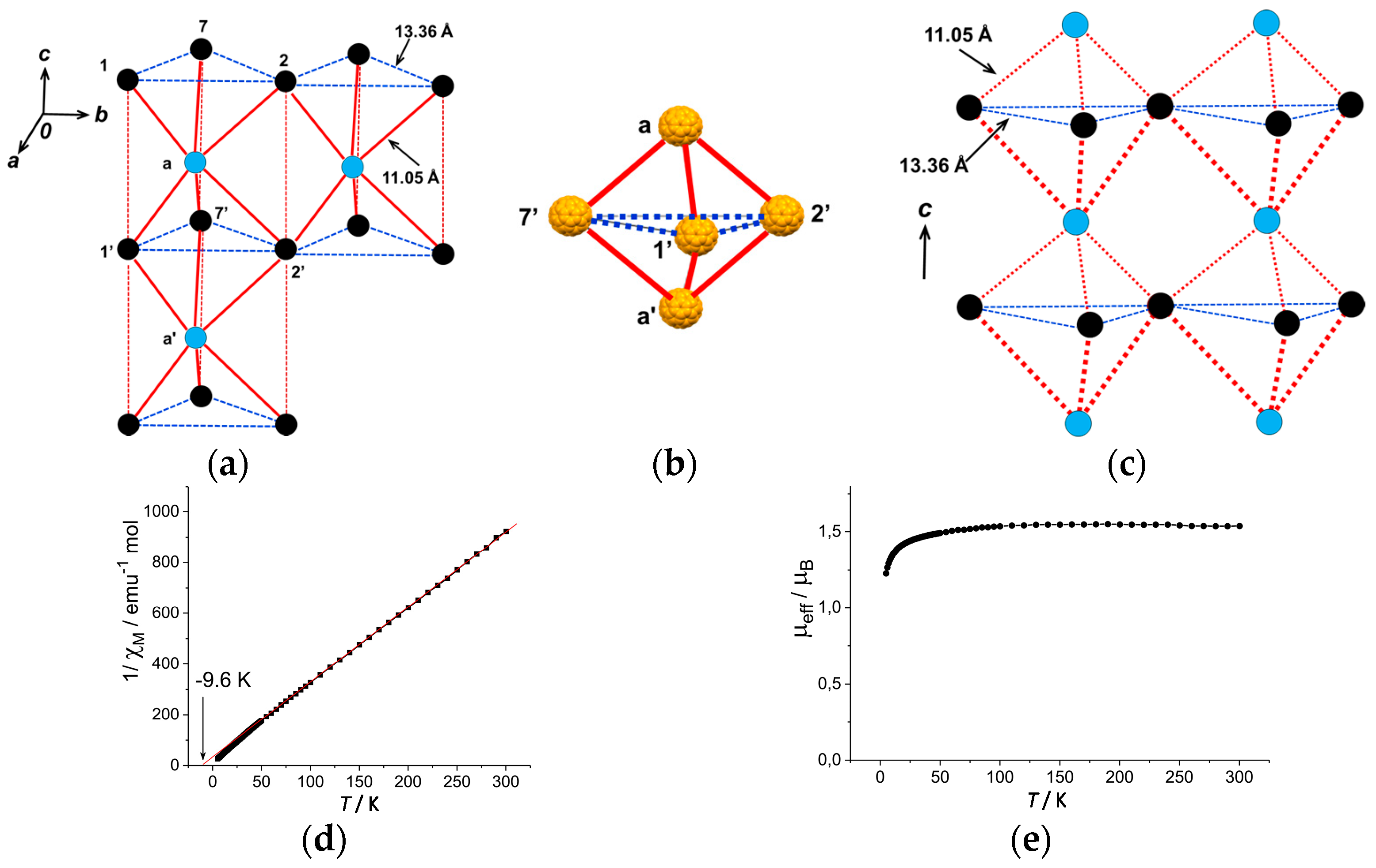
The top and bottom of Supramolecule 11 are capped by Supramolecule 10 along the c axis, yielding Supramolecule 13, [(Supramolecule 9)3(Supramolecule 10)(C60•−)], as schematically shown in Figure 18(h-1), where C60•− molecules labeled 1–7 form one layer (Layer 1). Similar capping occurs for Supramolecule 12 by Supramolecule 9 to form Supramolecule 14, [(Supramolecule 9)(Supramolecule 10)3(C60•−)], in Figure 18(h-2), where C60•− molecules labeled a-c form one layer (Layer a). Two kinds of Supramolecule units 13 and 14 stack alternately along the c axis to give the single crystal of 8. Figure 18(h-3) schematically illustrates the molecular packing. In the crystal viewed along the c axis (Figure 18(h-4,h-5)), four C60•− molecules, namely three molecules from Layer 1 (drawn in black in Figure 18(h-5)) and one molecule from Layer a (drawn in blue in Figure 18(h-5)) form tetrahedral [(1,2,7,a), (3,4,7,b) and (5,6,7,c)] structure. The labeling of C60•− molecules is the same as that in Figure 18(h-1–h-4)) Center-to-center distances between C60•− of 13.36 Å are marked by blue dotted lines and those of 11.05 Å (between blue and black circles) are marked by red lines in Figure 18(h-5).
As a result, geometry of the unit of model spin lattice is not triangular, but distorted tetragonal, i.e., tetrahedron with t′/t ~ 0. The AF spin configuration is expected within a 1D Mott insulating C60•− chain of black-blue-black-blue- fullerenes along the c axis, while within a layer of blue or black fullerenes in the ab plane parallel spin configurations are expected. Such spin units form a column along the c axis and the columns are arranged in the bc plane (Figure 19a). The other spin unit composed of C60•− molecules [(3,4,7,b) and (5,6,7,c)] has the same distorted tetrahedral geometry and form similar 2D packing to that in Figure 19a. These 2D spin sheets are connected, for example, through 7 and 7′ to form 3D network of magnetic interactions. It is emphasized here that according to the 3D template network composed of DMI+ and I− molecules, the C60•− radical anions provide the 2H-hexagonal 3D packing by a 3D key-keyhole relation between cation [(DMI+)3(I−)2]+ and anion C60•− molecules.
The geometry of the spin lattice of 8 is expected to be 3D apex sharing triangular bipyramid based on the r values. We calculated overlap integrals based on the two main orientations of C60 molecules at 100 K and the mean square of each is s = 0.12 × 10−3. Based on the overlap integrals, the model spin lattice is shown in Figure 19b, which are arranged in the ab plane to form 3D apex sharing triangular bipyramid.
The effective magnetic moment of 8 is 1.64 μB, slightly smaller than the value of 1.73 μB for the system containing one S = 1/2 spin per formula unit. The Curie-Weiss temperature of −9.6 K in the 40–300 K range indicates AF coupling of spins (Figure 19d). Effective magnetic moment also decreases below about 50 K (Figure 19e) due to AF coupling of spins. Because of the large interfullerene distance, 11.05 Å at 100 K, and small overlap integrals, the AF interaction is not strong. The long-range magnetic ordering is not observed down to 1.9 K.
In summary, concerning the geometry of the spin lattice of 8, the cationic supramolecule [(DMI+)3(I−)2] forms threefold assemblies that act as a template for the C60•− molecules to achieve hexagonal stacking of (C60•−). The overlap integrals lead to a 3D apex sharing triangular bipyramid with r = 11.05 Å at 100 K. Owing to a large r, a small |ΘCW| (9.6 K) is estimated in the range of 50–300 K. No dimerization of C60•− takes place down to 1.9 K (f ~ 5).
4.2. Two-Component Materials
4.2.1. Frustrated Spins in 3D Corrugated Packing of C60•− in (MDABCO+)(C60•−) 3D Spin Lattice
3D close packing of C60•− and high |ΘCW| are realized in the two-component CT solid of (MDABCO+)(C60•−) (1) where cation molecules MDABCO+ show threefold symmetry [147]. Black block single crystals of 1 (~0.4 × 0.4 × 0.5 mm3) were obtained by the reduction of C60 with slight excess of strong reductant sodium fluorenone ketyl in PhCl2 in the presence of stoichiometric amount of MDABCO∙I. Then, n-hexane was layered to precipitate CT solids. The crystal structures were solved at 250 K and 100 K. At 250 K, there is one independent C60•− and one ordered MDABCO+ molecules. The ordering of C60•− is observed below 160 K, with a trebling of the unit cell b axis.
The C60•− radical anions form square corrugated fullerene layers in the bc plane and the C60 layers alternate with the MDABCO+ cations along the a direction (Figure 20a at 100 K). 2D C60 layer in the bc plane is corrugated along the c axis (Figure 20b at 100 K). The corrugation of C60 layer is clearly shown in Figure 20c (at 100 K) and C60 columns extend along the b axis.
A C60•− molecule (drawn in red in Figure 20d at 250 K, labeled as Ⓐ) is surrounded by eight neighboring C60•− molecules labeled as ①, ②, and ③). There are three kinds of small r values: 10.01 Å with molecule ③, 10.08 Å with molecule ②, and 10.11 Å with molecule ① at 250 K. Four C60•− molecules, all of them are molecules ② among the eight neighbors, are located in the bc plane to form corrugated C60•− layer with vdW C∙∙∙C contacts in the 3.17–3.30 Å. Four other C60•− molecules, two ① and two ③, are located in the adjacent fullerene layers without short vdW C∙∙∙C contacts, though the r between molecules Ⓐ and ③ is shorter than that between molecules Ⓐ and ②.
The ordering of fullerenes is observed below 160 K with trebling of the unit cell b axis. As a result, the packing of C60•− molecules becomes denser and strongly anisotropic; namely r changes from Ⓐ−② = 10.08 Å at 250 K to (10.01, 10.04, 10.05 Å) at 100 K in the bc plane. Similarly, Ⓐ−③ and ①−② reduce to 9.91 and 9.96 Å, but Ⓐ−① becomes a little longer, 10.12 Å. Additional vdW interfullerene C∙∙∙C contacts are formed for each C60•− within the bc plane and between fullerene layers to account for the total number of such contacts as 18 for each C60•− at 100 K.
Since the structure at 100 K is complicated by the appearance of one and a half independent molecules both for C60•− and MDABCO+ molecules, at first the schematic of a possible spin lattice is examined based on the crystal structure at 250 K. Figure 20(e-1) shows the packing of C60•− viewed along the a axis, that corresponds to Figure 20a at 100 K. The square corrugated C60•− packing composed of C60•− molecules (one Ⓐ and four ②) form a pyramidal shape with r(Ⓐ−②) = 10.08 Å, which is schematically shown in Figure 20(e-2). The other combination of C60•− molecules ① and ③ form the equivalent pyramid and these two pyramids have short contacts with r values of 10.01 Å for 1−2 and 10.11 Å for ①−Ⓐ. As a consequence, the unit of intermolecular interactions is approximated as a distorted bipyramid, as shown in Figure 20(e-3). The plane of ②−②′−②′′−②′′′ bisects the bond ①−Ⓐ by 6.91 Å and 3.20 Å. The distorted bipyramids are connected to each other by sharing edge in the bc plane to form a 2D sheet. Figure 20(e-4) shows the part of the sheet extending along the c axis. The other unit of bipyramidal spin lattice composed of C60•− molecules ①, Ⓐ, and ③ forms equivalent 2D layer with different orientation (Figure 20(e-5)). The model geometry of the possible spin lattice of 1 is obtained by sharing apexes of the layers in Figure 20(e-4) and Figure 20(e-5) along the a axis to form 3D distorted bipyramidal spin lattice in which corrugated layers alternate as Layer Ⓐ②/Layer ①③ along the a axis (Figure 20(e-6)).
At low temperatures, the C60•− environment is strongly anisotropic in terms of r and overlap integrals s. The r values inside corrugated Layer Ⓐ② or Layer ①③, namely bonds Ⓐ−② and ①−, show shrinkage by 0.3–0.7%, while those bonds that are connecting neighboring corrugated layers, namely bonds ①−② and Ⓐ−③, show larger shrinkage by 0.5–1.0%. Therefore, though the corrugated square nature shown by Figure 20(e-2) is important to account for the spin interactions, the spin interactions between corrugated layers along the a axis becomes more significant at low temperatures. The model spin lattice geometry keeps the distorted bipyramidal one down to low temperatures. No dimerization was detected down to 1.9 K.
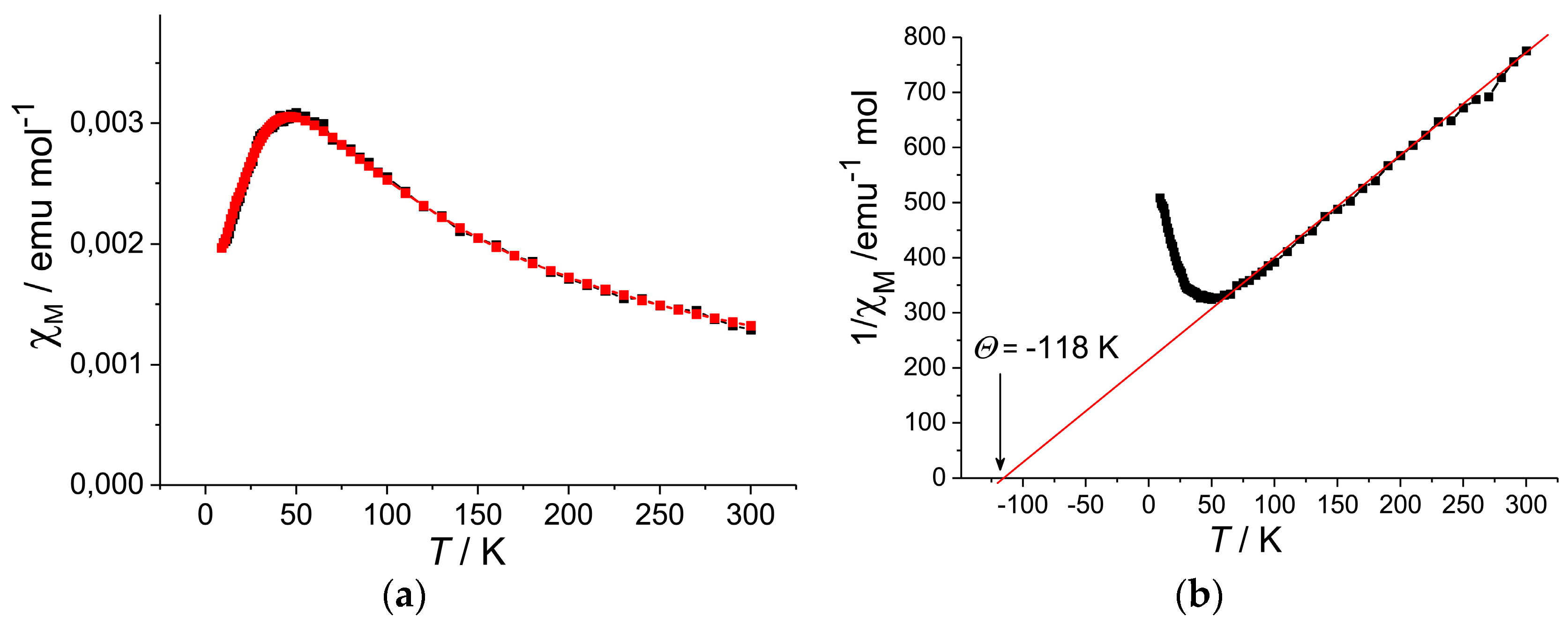
The temperature dependence of the molar magnetic susceptibility χM of 1 showed a maximum at 46 K, followed by a decrease but then an increase below 10 K owing to the Curie impurity of about 2.7% of total amount of C60•−. Figure 21 shows the temperature dependence of χM and 1/χM after the correction of Curie impurity. The magnetic susceptibility clearly indicates a characteristic peak near 50 K. Such peak in χM has been usually detected in the low-D (1D-2D) Mott insulators with strong spin frustration, such as κ-(ET)2X (X = Cu2(CN)3, Ag2(CN)3, B(CN)4, CF3SO3) [36,45,46,47,48,49,50,51,54,65,66,128,150,158]. The ΘCW temperature of −118 K was derived in the 70–300 K range (Figure 21b). The temperature dependence of χM was fitted by the Heisenberg model for square 2D AF coupling of spins [159] to give J/kB = −25.3 K. The long-range magnetic ordering is not observed down to 1.9 K (f > 62).
The overlap integrals between C60•− radical anions based on the real crystal structures are helpful to understand the situation of intermolecular interaction. At 250 K, 1 contains only one crystallographically independent C60•− and one MDABCO+, however the C60•− radical anions are disordered between three orientations with 49.4/25.3/25.3 (%) occupancies. On the other hand, 1 contains one and a half crystallographically independent C60•− and MDABCO+ at 100 K. Half of C60•− is well ordered at 100 K, while another C60•− is rotationally disordered between two orientations with 91.3/8.7 (%) occupancies. Although it is not reasonable to discuss the overlap integrals at 250 K due to the severe orientational disorder, we can calculate the overlap integrals in 1 at 100 K, assuming that all of the C60•− are well ordered ignoring the minor orientation (8.7%) in one of the two kinds of C60•−.
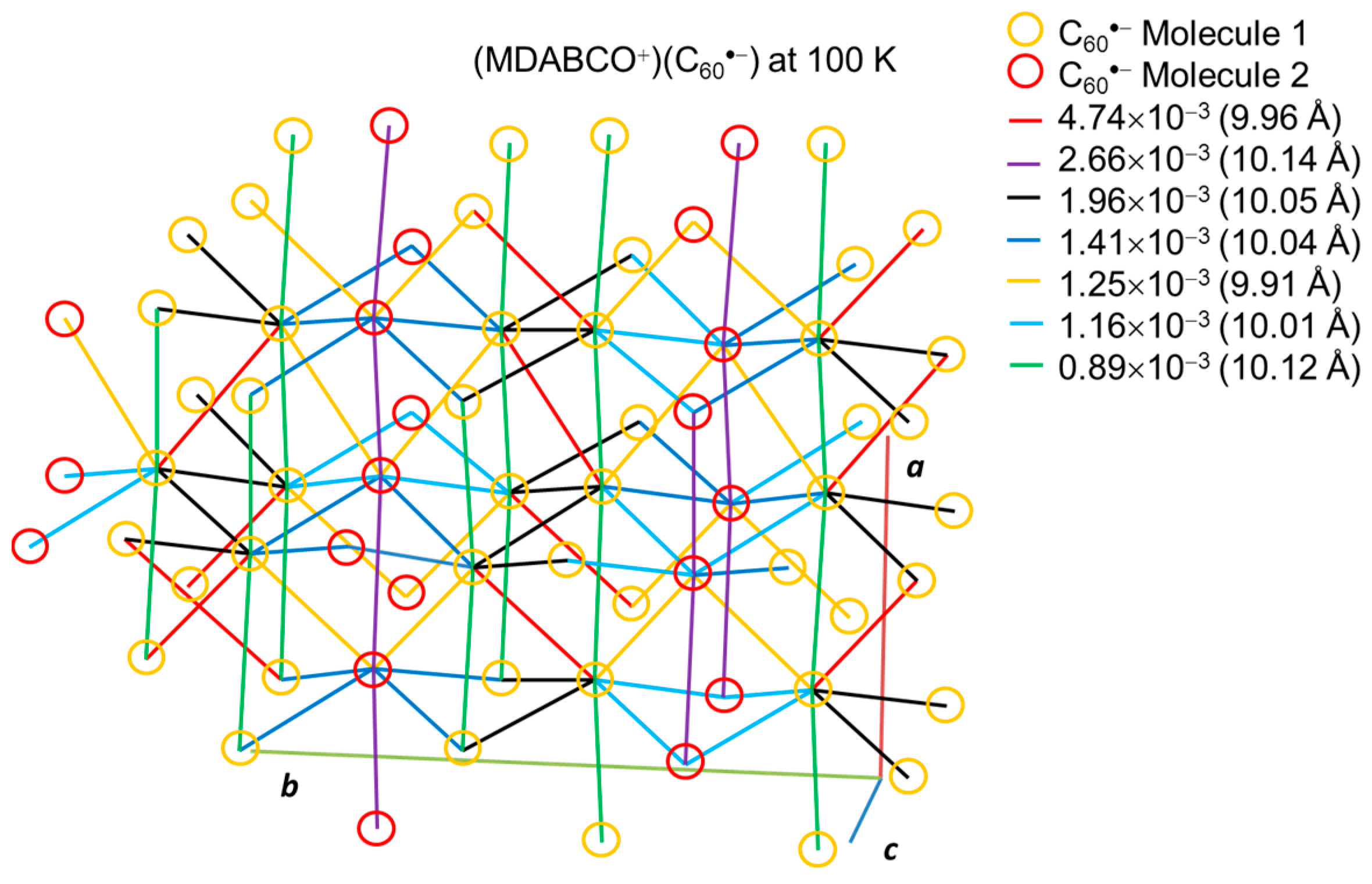
Figure 22 shows the network between the two kinds of C60•− radical anions at 100 K that are connected by several magnitudes of overlap integrals. The dominant interaction (red line) is 4.74 × 10−3 between the molecules 1 with 9.96 Å distance that forms a pair of C60•− between the adjacent layers. The second largest interaction (purple line) is 2.66 × 10−3 between the molecules 2 with 10.14 Å uniform distance that extends linearly along the a axis. The third one (black line) is 1.96 × 10−3 between the molecules 1 with 10.05 Å distance which forms zig-zag path ways along the c axis. Note that the shortest distance of 9.91 Å (orange line) between the different kinds of molecules 1 and 2 resulted in only the fifth largest interaction of 1.25 × 10−3. As demonstrated also in 2 and 5, the magnitude of overlap integrals between C60•− anion radicals does not necessarily scale with the closeness between them. The relative orientation of the molecular orbitals in the nearest neighbors as well as the center-to-center distances between them plays an important role to characterize the molecular interactions in the crystal.
The small size of the MDABCO+ cations with threefold symmetry and the absence of solvent molecules induced a densely packed 3D bipyramidal C60•− packing in 1 resulting in strong AF interactions. However, the strong anisotropic packing of C60•− may reduce the AF interaction considerably. The key-keyhole relation between MDABCO+ and C60•− is not clear in 1, however, it should be emphasized that MDABCO+ cations work to prevent the bond-formation even though the r values became small (①-② = 9.91 Å, Ⓐ-③ = 9.96 Å at 100 K).
4.2.2. Frustrated Spins in Double Chains of Triangles from C60•− in (Ph3MeP+)(C60•−): Weakly Coupled Zigzag Chains
Single crystals of (Ph3MeP+)(C60•−) (2), where Ph3MeP+ is a triphenylmethylphosphonium cation with threefold symmetry, were prepared by the reduction of C60 by (Ph3MeP+)(vanadyl(IV) phthalocyanine) in PhCl2 and slow mixing of the obtained PhCl2 solution in n-hexane [105].
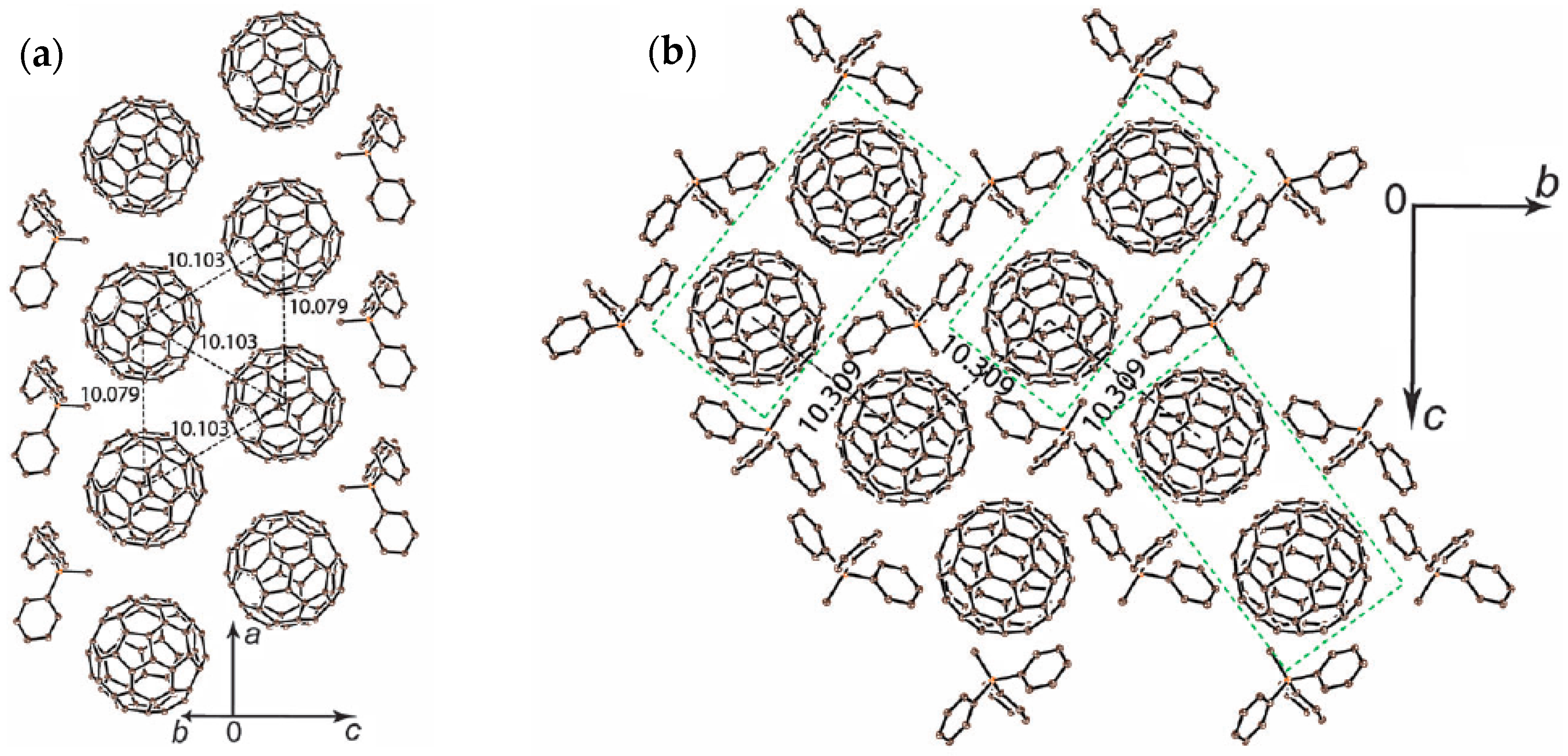
The crystal structure determined at 100 K indicates nearly ordered state of C60•− molecules. Crystal 2 involves a nearly isolated double chain with nearly equivalent fullerene triangles with small r values 10.08 Å, 10.103 Å, and 10.103 Å at 100 K (Figure 23a). The overlap integral is s2 = 1.19 × 10−3 for r = 10.079 Å while that with longer r = 10.103 Å has larger s1 = 1.79 × 10−3 owing to favorable orientation of C60•− molecules. Even though the double chain can be characterized as a zigzag chain, the chains are not isolated but are coupled weakly with a separation of r = 10.309 Å (s3 = 0.58 × 10−3) within the bc plane (Figure 23b). Therefore, the possible spin lattice of 2 consists of pseudo 3D weakly coupled zigzag chains (Figure 23c) with t′/t = 1.50 and J1:J2:J3 = 1:0.44:0.10.
Several zigzag-chain spin systems, which are the simplest frustrated magnet and treated by J1-J2 model or zigzag chain model, have been developed, such as CaV2O4 (S = 1, V3+, TN = 69 K) [160], Cu[2-(2-aminomethyl)pyridine]Br2 (S = 1/2, Cu2+) [161,162], (VO)(μ3-MoO4)(BPY) (S = 1/2, V4+) [163,164], and F2PIMNH [165]. A theoretical study predicted that the S = 1/2 zigzag chain has a gapless phase for J1/J2 < 0.241 [166,167], as exemplified for Cu[2-(2-aminomethyl)pyridine]Br2 (J1/J2 = 0.2, J1/kB = 8.5 K) [162] and (VO)(μ3-MoO4)(BPY) (J′/J = 0.2, J1/kB = 51 K) [164]. It is known that the interchain magnetic interactions are critical for the spin-ladder system either to manifest a Néel ordered or disordered spin-frustrated state, and the critical value is reported to be J/J′ = 0.11, where J and J′ are intraladder and interladder interactions, respectively [168]. Even though the actual J3 values for the above zigzag systems were not estimated in these reports, it is likely that the J3 values are very small according to their crystal structures. If we are able to expand the separation between the zigzag chains for 2 using more bulky cation molecules than Ph3MeP+, we may have a real zigzag system of C60•−.
The temperature dependence of the molar magnetic susceptibility χM shows an increase below 10 K, owing to the Curie impurity of about 1.2% of total amount of C60•−. Figure 23d showed the temperature dependence of χM and 1/χM after the correction of Curie impurity. No peak of χM was observed even though 2 has strong spin frustration. The magnetic behavior is described well by the Curie-Weiss law in the 30–300 K range with ΘCW = −60 K, and no AF ordering was observed down to 1.9 K (f > 30). Between C60•− molecules arranged along the a axis, Me groups of Ph3MeP+ cation molecules penetrate and prevent the dimerization even at r = 10.08–10.10 Å.
5. Summary
Geometrical spin frustration is discussed for monomer-type Mott insulators of C60 CT solids. When compared with the ET QSL system, bond-formation between C60 molecules and disorder of C60 molecule additionally participate in the competition among the itinerancy, localization, and spin frustration. The donor ability, size, shape, and symmetry of donor molecules in multi-component approach provide suitable geometrical space and spatial regulation for C60•− molecules by forming versatile supramolecules through the key-keyhole relation. A hexagonal packing of C60•− is achieved by the multi-component concept using cations (MDABCO+ or MQ+) and structure defining molecule (TPC) with threefold symmetry. C60•− molecules are packed according to the pattern of the polycationic supramolecular template of [(TPC0)(MDABCO+)] or [(TPC0)(MQ+)]. (TPC0)(MDABCO+)(C60•−) has uniform close packed hexagonal layers of two types, with an ordered C60•− layer (Layer A) and disordered C60•− layer (Layer B). The Layer A with t′/t = 1.00 at 300 K shows 2D metallic conductivity, whereas AF interaction of spins is observed in nonmetallic Layer B above 200 K. This AF layer has monomer-type Mott insulating state with t′/t = 0.99 (at 185 K) and ΘCW = −31 K. The disordered layer becomes metallic below 200 K through the ordering of C60•−. (TPC0)(MQ+)(C60•−) also has a 2D hexagonal packing of C60•− and shows relatively longer interfullerene distances (10.12–10.18 Å at 250 K) than that in (TPC0)(MDABCO+)(C60•−) (10.07 Å at 300 K). (TPC0)(MQ+)(C60•−) has a monomer-type Mott insulating state with t′/t = 0.99 (Layer A at 100 K), t′/t = 1.61 (Layer B at 100 K), and ΘCW = −27 K (f ~ 14). Solvent molecules participate in the formation of three-component C60 solids to achieve C60 hexagonal packing in (PhCN0)(TMP+)(C60•−) and (PhCN0)(Ph3MeP+)(C60•−). The former has a distorted edge-shared honeycomb 2D spin lattice with J1:J2:J3 = 0.79:1:0.22 and ΘCW = −11 K (f ~ 5), and the latter has a non-uniform 1D zigzag chain of C60•− as an effective spin geometry and forms singly bonded dimers that are below 220 K. (PhCl20)[(Ph3P)3Au+]2(C60•−)2(C600) has a charge-disproportionated hexagonal 2D layer and the C60•− molecules form an apex-sharing tetrahedral spin lattice with large r = 10.37 Å at 100 K and small ΘCW = −5 (f ~ 3). 3D hexagonal packing is realized in (DMI+)3(C60•−)(I−)2 in which H-bonds between DMI+ and I− formed a polycationic template [(DMI+)3(I−)2] with threefold symmetry. A possible geometry of spin lattice is an apex-sharing bipyramid (t′/t = 0) and the long interfullerene distance of 11.05 Å at 100 K resulted in weak magnetic interaction ΘCW = −9.6 K (f ~ 5). For two-component systems, a triangular unit of C60•− is observed in (MDABCO+)(C60•−) and (Ph3MeP+)(C60•−) both cation molecules have threefold symmetry. 3D close packing of C60•− was observed in (MDABCO+)(C60•−) with a possible geometry of spin lattice of deformed 3D edge-shared bipyramid. The AF interaction with high ΘCW of −118 K (f ~ 62) was observed though the bipyramidal geometry becomes distorted at low temperatures. (Ph3MeP+)(C60•−) has double chains that are composed of a triangular arrangement of C60•−, resulting in weakly coupled zigzag chains with t′/t = 1.50, J1:J2:J3 = 1:0.44:0.10, and ΘCW = −60 K (f > 30).
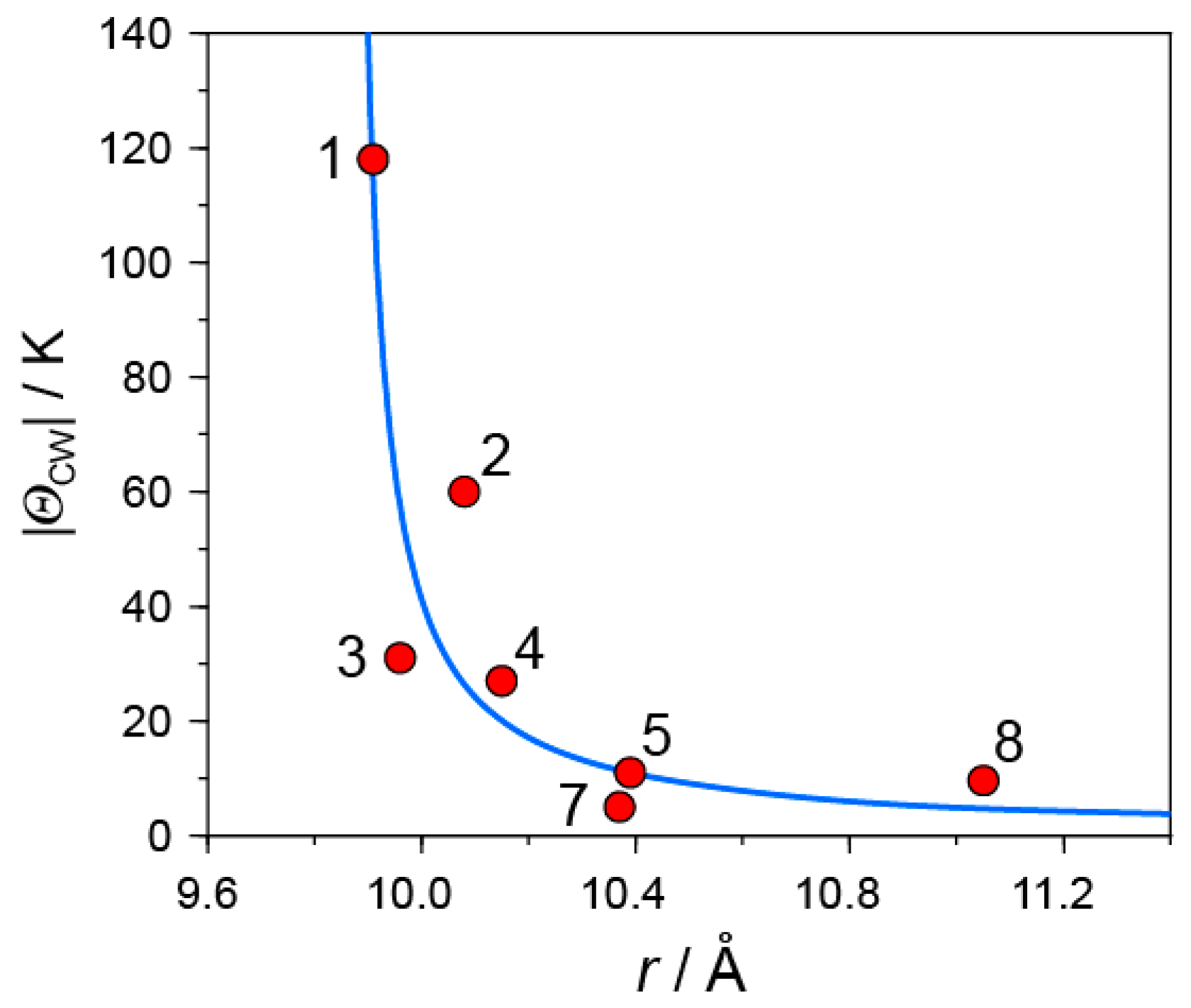
Even though the overlap integrals between C60 molecules depend on the molecular orientation of C60, the center-to-center distance between C60•− molecules r is the key parameter that determines the competition among the bond-formation, itinerancy, and spin frustration. Figure 24 shows the relation between |ΘCW| values and interfullerene distances r in this study. In the following, the summary and the perspective developed are presented.
- |ΘCW| seems to increase rapidly when r < 10 Å and magnetic dimensionality is 3D. Such low values of r were realized for two-component CT solids with cation molecules of small size with threefold symmetry. However, it is difficult to find a good key-keyhole relation to provide uniform triangular or hexagonal packing of C60•− for the two-component case. Furthermore, single crystals of CT solids were not always obtainable. For example, a quinuclidinium cation, which is smaller than MDABCO+ and MQ+, gave no CT solids so far.
- The cationic supramolecular template with threefold symmetry leads to uniform triangular or hexagonal packing of C60•− for three-component case based on the key-keyhole relation. For three-component case, it is critical to decrease the r value and increase the magnetic dimensionality.
In all of these monomer-type C60•− Mott insulator except 6, no long-range magnetic ordering nor bond-formation were observed down to low temperatures. However, their |ΘCW| values in the range 10–118 K, are not large enough to detect QSL state at experimentally available temperatures. This is apparent from the comparison with the |ΘCW| values of the QSL systems obtained; |ΘCW| = 180, 263, 314, 375, and 325-375 K for [(C2H5)3NH]2Cu2(oxalate)3, κ-(ET)2Ag2(CN)3, ZnCu3(OH)6Cl2, κ-(ET)2Cu2(CN)3, and EtMe3Sb[Pd(dmit)2], respectively [36,59,60,61,62,64,65]. It is important to explore a C60•− Mott insulator with r = 9.4–10 Å to detect a QSL state neighboring metallic and SC states, as a decrease in r increases |t|, |J|, and |ΘCW| values drastically according to their relations (Equations (1) and (2)).
Acknowledgments
This work was supported by Russian Science Foundation RSF-18-13-00292 and JSPS KAKENHI Grant Number JP23225005 “Development of multi-electronic-functions based on spin triangular lattice”.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Anderson, P.W. Resonating valence bonds: A new kind of insulator? Mater. Res. Bull. 1973, 8, 153–160. [Google Scholar] [CrossRef]
- Fazekas, P.; Anderson, P.W. On the ground state properties of the anisotropic triangular antiferromagnet. Philos. Mag. 1974, 30, 423–440. [Google Scholar] [CrossRef]
- Wannier, G.H. Antiferromagnetism. The triangular ising net. Phys. Rev. 1950, 79, 357–364. [Google Scholar] [CrossRef]
- Greedan, J.E. Geometrically frustrated magnetic materials. J. Mater. Chem. 2001, 11, 37–53. [Google Scholar] [CrossRef]
- Anderson, P.W. Ordering and antiferromagnetism in ferrites. Phys. Rev. 1956, 102, 1008–1013. [Google Scholar] [CrossRef]
- Hirakawa, K.; Yoshizawa, H.; Ubukoshi, K. Magnetic and neutron scattering study of one-dimensional heisenberg antiferromagnet CsVCl3. J. Phys. Soc. Jpn. 1982, 51, 1119–1122. [Google Scholar] [CrossRef]
- Ferey, G.; De Pape, R.; Leblanc, M.; Pannetier, J. Ordered magnetic frustration: VIII. Crystal and magnetic structures of the pyrochlore form of iron trifluoride between 2.5 and 25 K from powder neutron diffraction. Comparison with the other varieties of FeF3. Rev. Chim. Miner. 1986, 23, 474–484. [Google Scholar]
- Huse, D.; Elser, V. Simple variational wave functions for two-dimensional heisenberg spin-1/2 antiferromagnets. Phys. Rev. Lett. 1988, 60, 2531–2534. [Google Scholar] [CrossRef] [PubMed]
- Tocchio, L.F.; Gros, C.; Valentif, R.; Becca, F. One-dimensional spin liquid, collinear, and spiral phases from uncoupled chains to the triangular lattice. Phys. Rev. B 2014, 89, 235107. [Google Scholar] [CrossRef]
- Yamada, A. Magnetic properties and Mott transition in the Hubbard model on the anisotropic triangular lattice. Phys. Rev. B 2014, 89, 195108. [Google Scholar] [CrossRef]
- Yamada, A. Magnetic properties and Mott transition of the Hubbard model for weakly coupled chains on the anisotropic triangular lattice. Phys. Rev. B 2014, 90, 235138. [Google Scholar] [CrossRef]
- Konarev, D.V.; Khasanov, S.S.; Otsuka, A.; Maesato, M.; Uruichi, M.; Yakushi, K.; Shevchun, A.F.; Yamochi, H.; Saito, G.; Lyubovskaya, R.N. Metallic and Mott insulating spin-frustrated antiferromagnetic states in ionic fullerene complexes with a two-dimensional hexagonal C60•− packing motif. Chem. Eur. J. 2014, 20, 7268–7277. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.P. strongly geometrically frustrated magnets. Annu. Rev. Mater. Sci. 1994, 24, 453–480. [Google Scholar] [CrossRef]
- Schiffer, P.; Ramirez, A.P. Recent experimental progress in the study of geometrical magnetic frustration. Comments Condens. Matter Phys. 1996, 18, 21–50. [Google Scholar]
- Hirakawa, K.; Kadowaki, H.; Ubukoshi, K. Study of frustration effects in two-dimensional triangular lattice antiferromagnets-neutron powder diffraction study of VX2, X≡Cl, Br and I. J. Phys. Soc. Jpn. 1983, 52, 1814–1824. [Google Scholar] [CrossRef]
- Ajiro, Y.; Asano, T.; Takagi, T.; Mekata, M.; Katori, H.A.; Goto, T. High-field magnetization process in the triangular lattice antiferromagnet CuFeO2 up to 100 T. Physica B 1994, 201, 71–74. [Google Scholar] [CrossRef]
- Kimura, T.; Lashley, J.C.; Ramirez, A.P. Inversion-symmetry breaking in the noncollinear magnetic phase of the triangular-lattice antiferromagnet CuFeO2. Phys. Rev. B 2006, 73, 220401. [Google Scholar] [CrossRef]
- Mitamura, H.; Mitsuda, S.; Kanetsuki, S.; Katori, H.A.; Sakakibara, T.; Kindo, K. Dielectric polarization measurements on the antiferromagnetic triangular lattice system CuFeO2 in pulsed high magnetic fields. J. Phys. Soc. Jpn. 2007, 76, 094709. [Google Scholar] [CrossRef]
- Hirakawa, K.; Kadowaki, H.; Ubukoshi, K. Experimental studies of triangular lattice antiferromagnets with S = 1/2: NaTiO2 and LiNiO2. J. Phys. Soc. Jpn. 1985, 54, 3526–3536. [Google Scholar] [CrossRef]
- Tauber, A.; Moller, W.M.; Banks, E. Magnetic ordering in LiCr1−xFexO2. J. Solid State Chem. 1972, 4, 138–152. [Google Scholar] [CrossRef]
- Ajiro, Y.; Kikuchi, H.; Sugiyama, S.; Nakashima, T.; Shamoto, S.; Nakayama, N.; Kiyama, M.; Yamamoto, N.; Oka, Y. Z2 Vortex-induced broadening of the EPR linewidth in the two-dimensional triangular lattice antiferromagnets, HCrO2 and LiCrO2. J. Phys. Soc. Jpn. 1988, 57, 2268–2271. [Google Scholar] [CrossRef]
- Moreno, N.O.; Israel, C.; Pagliuso, P.G.; Garcia-Adeva, A.J.; Rettori, C.; Sarrao, J.L.; Thompson, J.D.; Oseroff, S.B. Magnetic properties of the frustrated antiferromagnet LiCrO2. J. Mag. Mag. Mater. 2004, 272–276, e1023–e1024. [Google Scholar] [CrossRef]
- Mekata, M. Antiferro-ferrimagnatic transition in triangular ising lattice. J. Phys. Soc. Jpn. 1977, 42, 76–82. [Google Scholar] [CrossRef]
- Plakhty, V.P.; Kulda, J.; Visser, D.; Moskvin, E.V.; Wosnitza, J. Chiral critical exponents of the triangular-lattice antiferromagnet CsMnBr3 as determined by polarized neutron scattering. Phys. Rev. Lett. 2000, 85, 3942–3945. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Kögerler, P.; Dress, A.W.M. Giant metal-oxide-based spheres and their topology: From pentagonal building blocks to keplerates and unusual spin systems. Coord. Chem. Rev. 2001, 222, 193–218. [Google Scholar] [CrossRef]
- Takano, M.; Shinjo, T.; Kiyama, M.; Takada, T. Magnetic properties of jarosites, RFe3(OH)6(SO4)2(R = NH4, Na or K). J. Phys. Soc. Jpn. 1968, 25, 902. [Google Scholar] [CrossRef]
- Townsend, M.G.; Longworth, G.; Roudaut, E. Triangular-spin, kagome plane in jarosites. Phys. Rev. B 1986, 33, 4919–4926. [Google Scholar] [CrossRef]
- Maegawa, S.; Nishiyama, M.; Tanaka, N.; Oyamada, A.; Takano, M. Observation of successive phase transitions in kagomé lattice antiferromagnets RFe3(OH)6(SO4)2 [R = NH4, Na, K]. J. Phys. Soc. Jpn. 1996, 65, 2776–2778. [Google Scholar] [CrossRef]
- Matan, K.; Ono, T.; Fukumoto, Y.; Sato, T.J.; Yamaura, J.; Yano, M.; Morita, K.; Tanaka, H. Pinwheel valence-bond solid and triplet excitations in the two-dimensional deformed kagome lattice. Nat. Phys. 2010, 6, 865–869. [Google Scholar] [CrossRef]
- Nakatsuji, S.; Nambu, Y.; Tonomura, H.; Sakai, O.; Jonas, S.; Broholm, C.; Tsunetsugu, H.; Qiu, Y.; Maeno, Y. Spin Disorder on a triangular lattice. Science 2005, 309, 1697–1700. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.L.; Ressouche, E.; Miller, J.S. Spin Frustration in MII[C(CN)3]2 (M = V, Cr). A magnetism and neutron diffraction study. Inorg. Chem. 2000, 39, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.P.; Espinosa, G.P.; Cooper, A.S. Strong Frustration and dilution-enhanced order in a quasi-2D spin glass. Phys. Rev. Lett. 1990, 64, 2070–2073. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Nohara, M.; Aruga-Katori, H.; Takagi, H. Spin-liquid state in the S = 1/2 hyperkagome antiferromagnet Na4Ir3O8. Phys. Rev. Lett. 2007, 99, 137207. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Yoshida, H.; Hiroi, Z. Vesignieite BaCu3V2O8(OH)2 as a candidate spin-1/2 kagome antiferromagnet. J. Phys. Soc. Jpn. 2009, 78, 033701. [Google Scholar] [CrossRef]
- Yoshida, H.; Yamaura, J.; Isobe, M.; Okamoto, Y.; Nilsen, G.J.; Hiroi, Z. Orbital switching in a frustrated magnet. Nat. Commun. 2012, 3, 860. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Miyagawa, K.; Kanoda, K.; Maesato, M.; Saito, G. Spin liquid state in an organic mott insulator with a triangular lattice. Phys. Rev. Lett. 2003, 91, 107001. [Google Scholar] [CrossRef] [PubMed]
- Konarev, D.V.; Khasanov, S.S.; Otsuka, A.; Saito, G. The Reversible Formation of a single-bonded (C60−)2 dimer in ionic charge transfer complex: Cp*2Cr·C60(C6H4Cl2)2. The molecular structure of (C60−)2. J. Am. Chem. Soc. 2002, 124, 8520–8521. [Google Scholar] [CrossRef] [PubMed]
- Konarev, D.V.; Khasanov, S.S.; Otsuka, A.; Saito, G.; Lyubovskaya, R.N. Negatively charged π-(C60−)2 Dimer with biradical state at room temperature. J. Am. Chem. Soc. 2006, 128, 9292–9293. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.W.; Bortel, G.; Faigel, G.; Tegze, M.; Jánossy, A.; Pekker, S.; Oszlanyi, G.; Forró, L. Polymeric fullerene chains in RbC60 and KC60. Nature 1994, 370, 636–639. [Google Scholar] [CrossRef]
- Bommeli, F.; Degiorgi, L.; Wachter, P.; Legeza, Ö.; Jánossy, A.; Oszlanyi, G.; Chauvet, O.; Forro, L. Metallic conductivity and metal-insulator transition in (AC60)n (A = K, Rb, and Cs) linear polymer fullerides. Phys. Rev. B 1995, 51, 14794–14797. [Google Scholar] [CrossRef]
- Bendele, G.M.; Stephens, P.W.; Prassides, K.; Vavekis, K.; Kortados, K.; Tanigaki, K. Effect of charge state on polymeric bonding geometry: The ground state of Na2RbC60. Phys. Rev. Lett. 1998, 80, 736–739. [Google Scholar] [CrossRef]
- Margadonna, S.; Pontiroli, D.; Belli, M.; Shiroka, T.; Riccò, M.; Brunelli, M. Li4C60: A polymeric fulleride with a two-dimensional architecture and mixed interfullerene bonding motifs. J. Am. Chem. Soc. 2004, 126, 15032–15033. [Google Scholar] [CrossRef] [PubMed]
- Riccò, R.; Pontiroli, D.; Mazzani, M.; Gianferrari, F.; Pagliari, M.; Goffredi, A.; Brunelli, M.; Zandomeneghi, G.; Meier, B.H.; Shiroka, T. Fullerenium salts: A new class of C60-based compounds. J. Am. Chem. Soc. 2010, 132, 2064–2068. [Google Scholar] [CrossRef] [PubMed]
- Oszlányi, G.; Baumgartner, G.; Faigel, L.; Forró, L. Na4C60: An alkali intercalated two-dimensional polymer. Phys. Rev. Lett. 1997, 78, 4438–4441. [Google Scholar] [CrossRef]
- Ohira, S.; Shimizu, Y.; Kanoda, K.; Saito, G. Spin liquid state in κ-(BEDT-TTF)2Cu2(CN)3 studied by muon spin relaxation method. J. Low Temp. Phys. 2006, 142, 153–158. [Google Scholar] [CrossRef]
- Pratt, F.L.; Baker, P.J.; Blundell, S.J.; Lancaster, T.; Ohira-Kawamura, S.; Baines, C.; Shimizu, Y.; Kanoda, K.; Watanabe, I.; Saito, G. Magnetic and non-magnetic phases of a quantum spin liquid. Nature 2011, 471, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Maesato, M.; Saito, G.; Drozdova, O.; Ouahab, L. Transport properties of a Mott insulator κ-(ET)2Cu2(CN)3 under the uniaxial strain. Synth. Met. 2003, 133, 225–226. [Google Scholar] [CrossRef]
- Saito, G.; Maesato, M. Organic superconductors. Mol. Cryst. Liq. Cryst. 2006, 455, 31–46. [Google Scholar] [CrossRef]
- Shimizu, Y.; Maesato, M.; Saito, G. Uniaxial strain effects on mott and superconducting transitions in κ-(ET)2Cu2(CN)3. J. Phys. Soc. Jpn. 2011, 80, 074702. [Google Scholar] [CrossRef]
- Kurosaki, Y.; Shimizu, Y.; Miyagawa, K.; Kanoda, K.; Saito, G. Mott transition from a spin liquid to a fermi liquid in the spin-frustrated organic conductor κ-(ET)2Cu2(CN)3. Phys. Rev. Lett. 2005, 95, 177001. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Kasahara, H.; Furuta, T.; Miyagawa, K.; Kanoda, K.; Maesato, M.; Saito, G. Pressure-induced superconductivity and Mott transition in spin-liquid κ-(ET)2Cu2(CN)3 probed by 13C NMR. Phys. Rev. B 2010, 81, 224508. [Google Scholar] [CrossRef]
- Saito, G.; Yoshida, Y. Development of conductive organic molecular assemblies: Organic metals, superconductors, and exotic functional materials. Bull. Chem. Soc. Jpn. 2007, 80, 1–137. [Google Scholar] [CrossRef]
- Saito, G.; Yoshida, Y. Organic superconductors. Chem. Rec. 2011, 11, 124–145. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, T.; Yoshida, Y.; Saito, G.; Otsuka, A.; Yamochi, H.; Maesato, M.; Shimizu, Y.; Ito, H.; Kishida, H. Quantum spin liquid: Design of a quantum spin liquid next to a superconducting state based on a dimer-type ET Mott insulator. J. Mater. Chem. C 2015, 3, 1378–1388. [Google Scholar] [CrossRef]
- Balents, L. Spin liquids in frustrated magnets. Nature 2010, 464, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Norman, M.R. The Challenge of unconventional superconductivity. Science 2011, 332, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Powell, B.J.; McKenzie, R.H. Quantum frustration in organic Mott insulators: From spin liquids to unconventional superconductors. Rep. Prog. Phys. 2011, 74, 056501. [Google Scholar] [CrossRef]
- Isono, T.; Kamo, H.; Ueda, A.; Takahashi, K.; Kimata, M.; Tajima, H.; Tsuchiya, S.; Terashima, T.; Uji, S.; Mori, H. Gapless quantum spin liquid in an organic spin-1/2 triangular-lattice κ-H3(Cat-EDT-TTF)2. Phys. Rev. Lett. 2014, 112, 177201. [Google Scholar] [CrossRef] [PubMed]
- Itou, T.; Oyamada, A.; Maegawa, S.; Tamura, M.; Kato, R. Spin-liquid state in an organic spin-1/2 system on a triangular lattice, EtMe3Sb[Pd(dmit)2]2. J. Phys. Condens. Matter 2007, 19, 145247. [Google Scholar] [CrossRef]
- Shores, M.P.; Nytko, E.A.; Bartlett, B.M.; Nocera, D.G. A structurally perfect S = 1/2 kagomé antiferromagnet. J. Am. Chem. Soc. 2005, 127, 13462–13463. [Google Scholar] [CrossRef] [PubMed]
- Mandels, P.; Bert, F. Quantum kagome antiferromagnet ZnCu3(OH)6Cl2. J. Phys. Soc. Jpn. 2010, 79, 011001. [Google Scholar] [CrossRef]
- Han, T.-H.; Helton, J.S.; Chu, S.; Nocera, D.G.; Rodriguez-Rivera, J.A.; Broholm, C.; Lee, Y.S. Fractionalized excitations in the spin-liquid state of a kagome-lattice antiferromagnet. Nature 2012, 492, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.; Orain, J.C.; Bert, F.; De Vries, M.A.; Aidoudi, F.H.; Morris, R.E.; Lightfoot, P.; Lord, J.S.; Telling, M.T.F.; Bonville, P.; et al. Gapless spin liquid ground State in the S = 1/2 vanadium oxyfluoride kagome antiferromagnet [NH4]2[C7H14N][V7O6F18]. Phys. Rev. Lett. 2013, 110, 207208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Baker, P.J.; Zhang, Y.; Wang, D.; Wang, Z.; Su, S.; Zhu, D.; Pratt, F.L. Quantum spin liquid from a three-dimensional copper-oxalate framework. J. Am. Chem. Soc. 2018, 140, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Hiramatsu, T.; Maesato, M.; Otsuka, A.; Yamochi, H.; Ono, A.; Itoh, M.; Yoshida, M.; Takigawa, M.; Yoshida, Y.; et al. Pressure-tuned exchange coupling of a quantum spin liquid in the molecular triangular lattice κ-(ET)2Ag2(CN)3. Phys. Rev. Lett. 2016, 117, 107203. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Jawad, M.; Terasaki, I.; Sasaki, T.; Yoneyama, N.; Kobayashi, N.; Uesu, Y.; Hotta, C. Anomalous dielectric response in the dimer Mott insulator κ-(BEDT-TTF)2Cu2(CN)3. Phys. Rev. B 2010, 82, 125119. [Google Scholar] [CrossRef]
- Hiramatsu, H.; Yoshida, Y.; Saito, G.; Otsuka, A.; Yamochi, H.; Shimizu, Y.; Hattori, Y.; Nakamura, Y.; Kishida, H.; Ito, H.; et al. Spin frustration in antiperovskite systems: (TTF•+ or TSF•+)3[(Mo6X14)2−Y−]. J. Mater. Chem. C 2015, 3, 11046–11054. [Google Scholar] [CrossRef]
- Bray, J.W.; Interrante, L.V.; Jacobs, I.S.; Bonner, J.C. The Spin-Peierls Transition. In Extended Linear Chain Compounds: Volume 3; Miller, J.S., Ed.; Plenum Press: New York, NY, USA, 1983; pp. 353–415. [Google Scholar]
- Batail, P.; Livage, C.; Parkin, S.S.P.; Coulon, C.; Martin, J.D.; Canadell, E. Antiperovskite structure with ternary tetrathiafulvalenium salts: construction, distortion, and antiferromagnetic ordering. Angew. Chem. Int. Ed. Engl. 1991, 30, 1498–1500. [Google Scholar] [CrossRef]
- Conwell, E. (Ed.) Semiconductors and Semimetals: Volume 27, Highly Conducting Quasi-One-Dimensional Organic Crystals; Academic Press: Boston, MA, USA, 1988. [Google Scholar]
- Kosaka, M.; Tanigaki, K.; Tanaka, T.; Atake, T.; Lappas, A.; Prassides, K. Conducting phase of rapidly cooled AC60 (A = Cs and Rb). Phys. Rev. B 1995, 51, 12018–12021. [Google Scholar] [CrossRef]
- Brouet, V.; Alloul, H.; Forró, L. Coexistence of spin singlets and metallic behavior in simple cubic CsC60. Phys. Rev. B 2002, 66, 155123. [Google Scholar] [CrossRef]
- Gunnarsson, O.; Koch, E.; Martin, R.M. Mott transition in degenerate Hubbard models: Application to doped fullerenes. Phys. Rev. B 1996, 54, R11026–R11029. [Google Scholar] [CrossRef]
- Gunnarsson, O. Superconductivity in fullerides. Rev. Mod. Phys. 1997, 69, 575–606. [Google Scholar] [CrossRef]
- Tinkham, M. Introduction to Superconductivity, 2nd ed.; McGraw-Hill: New York, NY, USA, 1996. [Google Scholar]
- Rosseinsky, M.J.; Ramirez, A.P.; Glarum, S.H.; Murphy, D.W.; Haddon, R.C.; Hebard, A.F.; Palstra, T.T.M.; Kortan, A.R.; Zahurak, S.M.; Makhija, A.V. Superconductivity at 28 K in RbxC60. Phys. Rev. Lett. 1991, 66, 2830–2832. [Google Scholar] [CrossRef] [PubMed]
- Tanigaki, K.; Ebbesen, T.W.; Saito, S.; Mizuki, J.; Tsai, J.S.; Kubo, Y.; Kuroshima, S. Superconductivity at 33 K in CsxRbyC60. Nature 1991, 352, 222–223. [Google Scholar] [CrossRef]
- Rosseinsky, M.J. Recent developments in the chemistry and physics of metal fullerides. Chem. Mater. 1998, 10, 2665–2685. [Google Scholar] [CrossRef]
- Andreoni, W. (Ed.) The Physics of Fullerene-Based and Fullerene-Related Materials; Springer: Dordrecht, The Netherlands, 2000. [Google Scholar]
- Reed, C.A.; Bolskar, R.D. Discrete fulleride anions and fullerenium cations. Chem. Rev. 2000, 100, 1075–1120. [Google Scholar] [CrossRef] [PubMed]
- Fleming, R.M.; Ramirez, A.P.; Rosseinsky, M.J.; Murphy, D.W.; Haddon, R.C.; Zahurak, S.M.; Makhija, A.V. Relation of structure and superconducting transition temperatures in A3C60. Nature 1991, 352, 787–788. [Google Scholar] [CrossRef]
- Ganin, A.Y.; Takabayashi, Y.; Khimyak, Y.Z.; Margadonna, S.; Tamai, A.; Rosseinsky, M.J.; Prassides, K. Bulk superconductivity at 38 K in a molecular system. Nat. Mater. 2008, 7, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Ganin, A.Y.; Takabayashi, Y.; Jeglič, Y.; Arčon, D.; Potočnik, A.; Baker, P.J.; Ohishi, Y.; McDonald, M.T.; Tzirakis, M.D.; McLennan, A.; et al. Polymorphism control of superconductivity and magnetism in Cs3C60 close to the Mott transition. Nature 2010, 466, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Özdaş, E.; Kortan, A.R.; Kopylov, N.; Ramirez, A.P.; Siegrist, T.; Rabe, K.M.; Bair, H.E.; Schuppler, S.; Citrin, P.H. Superconductivity and cation-vacancy ordering in the rare-earth fulleride Yb2.75C60. Nature 1995, 375, 126–129. [Google Scholar] [CrossRef]
- Chen, X.H.; Roth, G. Superconductivity at 8 K in samarium-doped C60. Phys. Rev. B 1995, 52, 15534–15536. [Google Scholar] [CrossRef]
- Brown, C.M.; Taga, S.; Gogia, B.; Kordatos, K.; Margadonna, S.; Prassides, K.; Iwasa, Y.; Tanigaki, K.; Fitch, A.N.; Pattison, P. Structural and electronic properties of the noncubic superconducting fullerides A’4C60 (A’ = Ba, Sr). Phys. Rev. Lett. 1999, 83, 2258–2261. [Google Scholar] [CrossRef]
- Baenitz, M.; Heinze, M.; Lüders, K.; Werner, H.; Schlögl, R.; Weiden, M.; Sparn, G.; Steglich, F. Superconductivity of Ba doped C60—Susceptibility results and upper critical field. Solid State Commun. 1995, 96, 539–544. [Google Scholar] [CrossRef]
- Iwasa, Y.; Hayashi, H.; Furudate, T.; Mitani, T. Superconductivity in K3Ba3C60. Phys. Rev. B 1996, 54, 14960–14962. [Google Scholar] [CrossRef]
- Ksari-Habiles, Y.; Claves, D.; Chouteau, G.; Touzain, P.; Jeandey, C.; Oddou, J.L.; Stepanov, A. Superexchange and magnetic relaxation in novel Eu-DOPED C60 phases. J. Phys. Chem. Solids 1997, 58, 1771–1778. [Google Scholar] [CrossRef]
- Maruyama, Y.; Motohashi, S.; Sakai, N.; Watanabe, K.; Suzuki, K.; Ogata, H.; Kubozono, Y. Possible competition of superconductivity and ferromagnetism in CexC60 compounds. Solid State Commun. 2002, 123, 229–233. [Google Scholar] [CrossRef]
- Zadik, R.H.; Takabayashi, Y.; Klupp, G.; Colman, R.H.; Ganin, A.Y.; Potočnik, A.; Jeglič, P.; Arčon, D.; Matus, P.; Kamarás, K.; et al. Optimized unconventional superconductivity in a molecular Jahn-Teller metal. Sci. Adv. 2015, 1, e1500059. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Matsuda, A.; Chu, C.W. Fermi-liquid behavior and BCS s-wave pairing of K3C60 observed by 13C-NMR. J. Phys. Soc. Jpn. 1994, 63, 1670–1673. [Google Scholar] [CrossRef]
- Kiefl, R.F.; MacFarlane, W.A.; Chow, K.H.; Dunsiger, S.; Duty, T.L.; Johnston, T.M.S.; Schneider, J.W.; Sonier, J.; Brard, L.; Strongin, R.M.; et al. Coherence Peak and Superconducting energy gap in Rb3C60 observed by muon spin relaxation. Phys. Rev. Lett. 1993, 70, 3987–3990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allemand, P.-M.; Khemani, K.C.; Koch, A.; Wudl, F.; Holczer, K.; Donovan, S.; Grüner, G.; Thompson, J.D. Organic Molecular Soft ferromagnetism in a fullerene C60. Science 1991, 253, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Konerav, D.V.; Lyubovskaya, R.N. Molecular design, study of the structures and properties of ionic fullerene compounds. Rus. Chem. Rev. 2012, 81, 336–366. [Google Scholar] [CrossRef]
- Prassides, K. Polymer and Dimer Phases in Doped Fullerenes. In The Physics of Fullerene-Based and Fullerene-Related Materials; Andreoni, W., Ed.; Kluwer Academic Publishers: Boston, MA, USA, 2000; pp. 175–202. [Google Scholar]
- Eklund, P.C.; Rao, A.M. (Eds.) Fullerene Polymers and Fullerene Polymer Composites; Springer: Berlin, Germany, 2000. [Google Scholar]
- Rosseinsky, M.J. Fullerene intercalation chemistry. J. Mater. Chem. 1995, 5, 1497–1513. [Google Scholar] [CrossRef]
- Bondi, A. Van der waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Brouet, V.; Alloul, H.; Yoshinari, Y.; Forro, L. NMR Evidence for 1D Antiferromagnetic Properties in Cs1C60 and Rb1C60 Polymers. Phys. Rev. Lett. 1996, 76, 3638–3641. [Google Scholar] [CrossRef] [PubMed]
- Schirber, J.E.; Morosin, B.; Kwei, G.H.; Yildirim, T.; Fischer, J.E.; Jorgensen, J.D. Superconductivity in the polymeric phase of Na2CsC60. Physica C 2001, 353, 207–212. [Google Scholar] [CrossRef]
- Kozhemyakina, N.V.; Amsharov, K.Y.; Nuss, J.; Jansen, M. Synthesis and Structure analysis of (K[DB18 C6])4(C60)5∙12THF containing C60 in three different bonding states. Chem. Eur. J. 2011, 17, 1798–1805. [Google Scholar] [CrossRef] [PubMed]
- Konarev, D.V.; Khasanov, S.S.; Saito, G.; Lyubovskaya, R.N. Design of molecular and ionic complexes of fullerene C60 with metal(II) octaethylporphyrins, MIIOEP (M = Zn, Co, Fe, and Mn) containing coordination M−N(ligand) and M−C(C60–) bonds. Cryst. Growth Des. 2009, 9, 1170–1181. [Google Scholar] [CrossRef]
- Konarev, D.V.; Khasanov, S.S.; Mukhamadieva, G.R.; Zorina, L.V.; Otsuka, A.; Yamochi, H.; Saito, G.; Lyubovskaya, R.N. Magnetic and structural transitions at dimerization of C60•− in ionic fullerene complexes with metalloporphyrins: {(TMP+)2·MIITPP}·(C60−)2·(C6H4Cl2)2·(C6H5CN)2 (M = Zn and Mn). Inorg. Chem. 2010, 49, 3881–3887. [Google Scholar] [CrossRef] [PubMed]
- Konarev, D.V.; Khasanov, S.S.; Kuzmin, A.V.; Otsuka, A.; Yamochi, H.; Saito, G.; Lyubovskaya, R.N. Effective magnetic coupling with strong spin frustration in (Ph3MeP+)(C60•−) and reversible C60•− dimerization in (Ph3MeP+)(C60•−)·C6H5CN. Effect of solvent on structure and properties. New J. Chem. 2016, 40, 2792–2798. [Google Scholar] [CrossRef]
- Konarev, D.V.; Khasanov, S.S.; Otsuka, A.; Yamochi, H.; Saito, G.; Lyubovskaya, R.N. Effect of the cooling rate on dimerization of C60•− in fullerene salt (DMI+)2·(C60•−)·{Cd(Et2NCS2)2I−}. Inorg. Chem. 2012, 51, 3420–3426. [Google Scholar] [CrossRef] [PubMed]
- Pénicaud, A.; Peréz-Benítez, A.; Gleason, V.R.; Muñoz, P.E.; Escudero, R. Electrocrystallizing C60: Synthesis, single crystal X-ray structure, and magnetic (ESR, SQUID) characterization of [(C6H5)4P]2[C60][I]x. J. Am. Chem. Soc. 1993, 115, 10392–10393. [Google Scholar] [CrossRef]
- Bilow, U.; Jansen, M. Electrocrystallisation and crystal structure determination of Ph4PC60·Ph4PCl. J. Chem. Soc. Chem. Commun. 1994, 403–404. [Google Scholar] [CrossRef]
- Bilow, U.; Jansen, M. Electrocrystallization of fullerides. Z. Anorg. Allg. Chem. 1995, 621, 982–986. [Google Scholar] [CrossRef]
- Gritsenko, V.V.; Dyachenko, O.A.; Shilov, G.V.; Spitsyna, N.G.; Yagubskii, E.B. Crystal and molecular structures of new fullerides, (Ph4P)2C60Hal (Hal = Br or I) and (Ph4As)2C60Cl. Rus. Chem. Bull. 1997, 46, 1878–1882. [Google Scholar] [CrossRef]
- Kobayashi, H.; Moriyama, H.; Kobayashi, A.; Watanabe, T. Synthesis and characterization of some fulleride compounds by electrocrystallization of C60. Synth. Met. 1995, 70, 1451–1452. [Google Scholar] [CrossRef]
- Hong, J.; Shores, M.P.; Elliott, C.M. Establishment of structure-conductivity relationship for Tris(2,2′-bipyridine) ruthenium ionic C60 salts. Inorg. Chem. 2010, 49, 11378–11385. [Google Scholar] [CrossRef] [PubMed]
- Fässler, T.F.; Hoffmann, R.; Hoffmann, S.; Wörle, M. Triple-decker type coordination of a fullerene trianion in [K([18]crown-6)]3[η6,η6-C60](η3-C6H5CH3)2—Single crystal structure and magnetic properties. Angew. Chem. Int. Ed. 2000, 39, 2091–2094. [Google Scholar] [CrossRef]
- Boeddinghaus, M.B.; Salzinger, M.; Fässler, T.F. Synthesis, X-ray single-crystal structure determination, and magnetic properties of [Rb(benzo[18]crown-6)]+ salts containing well-ordered fulleride trianions C603−. Chem. Eur. J. 2009, 15, 3261–3267. [Google Scholar] [CrossRef] [PubMed]
- Fässler, T.F.; Spiekermann, A.; Spahr, M.E.; Nesper, R. Unprecedented layered structure of a fulleride: synthesis, structure, and magnetic properties of a potassium-containing salt with a C602− counterion. Angew. Chem. Int. Ed. Engl. 1997, 36, 486–488. [Google Scholar] [CrossRef]
- Janiak, C.; Mühle, S.; Hemling, H.; Köhler, K. The solid-state structure of K3C60(THF)14. Polyhedron 1996, 15, 1559–1563. [Google Scholar] [CrossRef]
- Kosaka, M.; Tanigaki, K.; Prassides, K.; Margadonna, S.; Lappas, A.; Brown, C.M.; Fitch, A.N. Superconductivity in LixCsC60 fullerides. Phys. Rev. B 1999, 59, R6628–R6630. [Google Scholar] [CrossRef]
- Tanigaki, K.; Prassides, K. Conducting and superconducting properties of alkali-metal C60 fullerides. J. Mater. Chem. 1995, 5, 1515–1527. [Google Scholar] [CrossRef]
- Tanigaki, K.; Hirosawa, I.; Ebbesen, T.W.; Mizuki, J.; Shimakawa, Y.; Kubo, Y.; Tsai, J.S.; Kuroshima, S. Superconductivity in sodium- and lithium-containing alkali-metal fullerides. Nature 1992, 356, 419–421. [Google Scholar] [CrossRef]
- Takabayashi, Y.; Ganin, A.Y.; Jeglič, P.; Arčon, D.; Takano, T.; Iwasa, Y.; Ohishi, Y.; Takata, M.; Takeshita, N.; Prassides, K.; et al. The disorder-free Non-BCS superconductor Cs3C60 emerges from an antiferromagnetic insulator parent state. Science 2009, 323, 1585–1590. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Shibasaki, S.; Kubozono, Y.; Kambe, T. Antiferromagnetic resonance in the Mott insulator fcc-Cs3C60. J. Phys. Condens. Matter 2013, 25, 366001. [Google Scholar] [CrossRef] [PubMed]
- Ihara, Y.; Alloul, H.; Wzietek, P.; Pontiroli, D.; Mazzani, M.; Riccò, M. NMR Study of the Mott transitions to superconductivity in the two Cs3C60 Phases. Phys. Rev. Lett. 2010, 104, 256402. [Google Scholar] [CrossRef] [PubMed]
- Takenobu, T.; Muro, T.; Iwasa, Y.; Mitani, T. Antiferromagnetism and phase diagram in ammoniated alkali fulleride salts. Phys. Rev. Lett. 2000, 85, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Zhou, O.; Palstra, T.T.M.; Iwasa, Y.; Fleming, R.M.; Hebard, A.F.; Sulewski, P.E.; Murphy, D.W.; Zegarski, B.R. Structural and electronic properties of (NH3)xK3C60. Phys. Rev. B 1995, 52, 483–489. [Google Scholar] [CrossRef]
- Iwasa, Y.; Takenobu, T. Superconductivity, Mott-Hubbard states, and molecular orbital order in intercalated fullerides. J. Phys.: Condens. Matter 2003, 15, R495–R519. [Google Scholar] [CrossRef]
- Welp, U.; Fleshler, S.; Kwok, W.K.; Crabtree, G.W.; Carlson, K.D.; Wang, H.H.; Geiser, U.; Williams, J.M.; Hitsman, V.M. Weak Ferromagnetism in κ-(ET)2Cu[N(CN)2]Cl, where (ET) is Bis(ethylenedithio)tetrathiafulvalene. Phys. Rev. Lett. 1992, 69, 840–843. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, K.; Kawamoto, A.; Nakazawa, Y.; Kanoda, K. antiferromagnetic ordering and spin structure in the organic conductor, κ-(BEDT-TTF)2Cu[N(CN)2]Cl. Phys. Rev. Lett. 1995, 75, 1174–1177. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Asai, T.; Shimizu, Y.; Hayama, H.; Yoshida, Y.; Saito, G. Pressure-induced superconductivity in the antiferromagnet κ-(ET)2CF3SO3 with quasi-one-dimensional triangular spin lattice. Phys. Rev. B 2016, 94, 020503. [Google Scholar] [CrossRef]
- Coldea, R.; Tennant, D.A.; Tsvelik, A.M.; Tylczynski, Z. Experimental realization of a 2D fractional quantum spin liquid. Phys. Rev. Lett. 2001, 86, 1335–1338. [Google Scholar] [CrossRef] [PubMed]
- Antropov, P.V.; Gunnarsson, O.; Jepsen, O. Coulomb integrals and model Hamiltonians for C60. Phys. Rev. B 1992, 46, 13647–13650. [Google Scholar] [CrossRef]
- Pederson, M.R.; Quong, A.A. Polarizabilities, charge states, and vibrational modes of isolated fullerene molecules. Phys. Rev. B 1992, 46, 13584–13591. [Google Scholar] [CrossRef]
- Martin, R.L.; Ritchie, J.P. Coulomb and exchange interactions in C60n−. Phys. Rev. B 1993, 48, 4845–4849. [Google Scholar] [CrossRef]
- Brühwiler, P.A.; Maxwell, A.J.; Nilsson, A.; Mårtensson, N.; Gunnarsson, O. Auger and photoelectron study of the Hubbard U in C60, K3C60, and K6C60. Phys. Rev. B 1993, 48, 18296–18299. [Google Scholar] [CrossRef]
- Lof, R.W.; van Veenendaal, M.A.; Koopmans, B.; Jonkman, H.T.; Sawatzky, G.A. Band gap, excitons, and coulomb interaction in solid C60. Phys. Rev. Lett. 1992, 68, 3924–3927. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, O.H., Jr. On the Electrical conductivities of tetracyanoquinodimethan anion-radical salt. J. Chem. Phys. 1965, 42, 4307–4308. [Google Scholar] [CrossRef]
- Douthwaite, R.E.; Green, M.A.; Green, M.L.H.; Rosseinsky, M.J. Synthesis, reactivity, structure and electronic properties of [N(CH3)4]C60·1.5thf: Fullerides with simple hexagonal packing. J. Mater. Chem. 1996, 6, 1913–1920. [Google Scholar] [CrossRef]
- Douthwaite, R.E.; Brough, A.R.; Green, M.L.H. Synthesis and characterisation of NaC60·5thf. J. Chem. Soc. Chem. Commun. 1994, 3, 267–268. [Google Scholar] [CrossRef]
- Kromer, A.; Wedig, U.; Roduner, E.; Jansen, M.; Amsharov, K.Y. Counterintuitive anisotropy of electron transport properties in KC60(THF)5∙2THF fulleride. Angew. Chem. Int. Ed. 2013, 52, 12610–12614. [Google Scholar] [CrossRef] [PubMed]
- Kodera, H. Dyson effect in the electron spin resonance of phosphorus doped silicon. J. Phys. Soc. Jpn. 1970, 28, 89–98. [Google Scholar] [CrossRef]
- Saito, G.; Teramoto, T.; Otsuka, A.; Sugita, Y.; Ban, T.; Kusunoki, M.; Sakaguchi, K. Preparation and ionicity of C60 charge transfer complexes. Synth. Met. 1994, 64, 359–368. [Google Scholar] [CrossRef]
- Konarev, D.V.; Khasanov, S.S.; Vorontsov, I.I.; Saito, G.; Antipin, M.Y.; Otsuka, A.; Lyubovskaya, R.N. The formation of a single-bonded (C70−)2 dimer in a new ionic multicomponent complex of cyclotriveratrylene: (Cs+)2(C70−)2·CTV·(DMF)7(C6H6)0.75. Chem. Commun. 2002, 21, 2548–2549. [Google Scholar] [CrossRef]
- Konarev, D.V.; Khasanov, S.S.; Otsuka, A.; Yoshida, Y.; Saito, G. Synthesis and crystal structure of ionic multicomponent complex: {[CrI(PhH)2]•+}2[CoIITPP(C60(CN)2)]− [C60(CN)2]•−·3(o-C6H4Cl2) containing C60(CN)2•− radical anion and σ-bonded diamagnetic CoIITPP(C60(CN)2)− anion. J. Am. Chem. Soc. 2002, 124, 7648–7649. [Google Scholar] [CrossRef] [PubMed]
- Konarev, D.V.; Khasanov, S.S.; Saito, G.; Otsuka, A.; Yoshida, Y.; Lyubovskaya, R.N. Formation of single-bonded (C60−)2 and (C70−)2 dimers in crystalline ionic complexes of fullerenes. J. Am. Chem. Soc. 2003, 125, 10074–10083. [Google Scholar] [CrossRef] [PubMed]
- Konarev, D.V.; Neretin, I.S.; Saito, G.; Slovokhotov, Y.L.; Otsuka, A.; Lyubovskaya, R.N. Multicomponent ionic complexes of cobalt(ii) tetraphenylporphyrin with c60 fullerides—Transition from the σ-bonded [(CoIITPP)·(C60−)] anion to nonbonded CoIITPP and C60•− components. Eur. J. Inorg. Chem. 2004, 2004, 1794–1798. [Google Scholar] [CrossRef]
- Konarev, D.V.; Khasanov, S.S.; Otsuka, A.; Saito, G.; Lyubovskaya, R.N. Ionic fullerene complex (DMI+)3·(C60•−)·(I−)2 with 2H-hexagonal fullerene packing and 3-D DMI+-I− network. CrystEngComm 2009, 11, 811–816. [Google Scholar] [CrossRef]
- Konarev, D.V.; Khasanov, S.S.; Otsuka, A.; Maesato, M.; Saito, G.; Lyubovskaya, R.N. A two-dimensional organic metal based on fullerene. Angew. Chem. Int. Ed. 2010, 49, 4829–4832. [Google Scholar] [CrossRef] [PubMed]
- Konarev, D.V.; Khasanov, S.S.; Otsuka, A.; Yamochi, H.; Saito, G.; Lyubovskaya, R.N. Strong Antiferromagnetic coupling of spins in the (MDABCO+)(C60•−) salt with 3D close packing of the C60•− radical anions (MDABCO+: N-Methyldiazabicyclooctanium Cation). Chem. Asian J. 2014, 9, 1629–1635. [Google Scholar] [CrossRef] [PubMed]
- Konarev, D.V.; Kuzmin, A.V.; Khasanov, S.S.; Ishikawa, M.; Otsuka, A.; Yamochi, H.; Saito, G.; Lyubovskaya, R.N. Structure and magnetic properties of the ionic fullerene salt (TMP+)·(C60•−)·C6H5CN containing layers of monomeric C60•− radical anions. New J. Chem. 2013, 37, 2521–2527. [Google Scholar] [CrossRef]
- Konarev, D.V.; Khasanov, S.S.; Otsuka, A.; Ishikawa, M.; Yamochi, H.; Saito, G.; Lyubovskaya, R.N. Formation of hexagonal fullerene layers from neutral and negatively charged fullerenes in {(Ph3P)3Au+}2(C60•−)2(C60)·C6H4Cl2 containing gold cations with the C3v symmetry. Inorg. Chem. 2014, 53, 6850–6855. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Ito, H.; Maesato, M.; Shimizu, Y.; Hayama, H.; Hiramatsu, T.; Nakamura, Y.; Kishida, H.; Koretsune, T.; Hotta, C.; et al. Spin-disordered quantum phases in a quasi-one-dimensional triangular lattice. Nat. Phys. 2015, 11, 679–683. [Google Scholar] [CrossRef]
- Sugano, T.; Saito, G.; Kinoshita, M. Conduction-electron-spin resonance in organic conductors: α and β phases of di[bis(ethylenedithiolo)tetrathiafulvalene]triiodide [(BEDT-TTF)2I3]. Phys. Rev. B 1986, 34, 117–125. [Google Scholar] [CrossRef]
- Yagubskii, E.B.; Shchegolev, I.F.; Laukhin, V.N.; Kononovich, P.A.; Karstovnik, M.V.; Zvarykina, A.V.; Buravov, L.I. Normal-pressure superconductivity in an organic metal (BEDT-TTF)2I3 [bis (ethylene dithiolo) tetrathiof ulvalene triiodide]. JETP Lett. 1984, 39, 12–16. [Google Scholar]
- Yildirim, T.; Fischer, J.E.; Harris, A.B.; Stephens, P.W.; Liu, D.; Brard, L.; Strongin, R.M.; Smith, A.B. Orientational phase transition in NaxC60 (1 <x <3). Phys. Rev. Lett. 1993, 71, 1383–1386. [Google Scholar] [PubMed]
- Prassides, K.; Christides, C.; Thomas, I.M.; Mizuki, J.; Tanigaki, K.; Hirosawa, I.; Ebbesen, T.W. Crystal structure, bonding, and phase transition of the superconducting Na2CsC60 fulleride. Science 1994, 263, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Mihailovic, D.; Arcon, D.; Verturini, P.; Blinc, R.; Omerzu, A.; Cevc, P. Orientational and magnetic ordering of buckyballs in TDAE-C60. Science 1995, 268, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Akada, M.; Yamamoto, T.; Kumashiro, R.; Hojyo, A.; Matsui, H.; Toyota, N.; Lu, J.P.; Tanigaki, K. Erectric transport and modulated density of states in rotational order and disorder in Na2CsC60. In Multifunctional Conducting Molecular Materials; Saito, G., Wudl, F., Haddon, R.C., Tanigaki, K., Enoki, T., Katz, H.E., Maesato, M., Eds.; RSC Publishing: Cambridge, UK, 2007; pp. 191–197. [Google Scholar]
- Brouet, V.; Alloul, H.; Quéré, F.; Baumgartner, G.; Folló, L. Detection by NMR of a “Local Spin Gap” in quenched CsC60. Phys. Rev. Lett. 1999, 82, 2131–2134. [Google Scholar] [CrossRef]
- Pinterić, M.; Lazić, P.; Pustogow, A.; Ivek, T.; Kuveždić, M.; Milat, O.; Gumhalter, B.; Basletić, M.; Čulo, M.; Korin-Hamzić, B.; et al. Anion effects on electronic structure and electrodynamic properties of the Mott insulator κ-(BEDT-TTF)2Ag2(CN)3. Phys. Rev. B 2016, 94, 161105. [Google Scholar] [CrossRef]
- Lines, M.E. The quadratic-layer antiferromagnet. J. Phys. Chem. Solids 1970, 31, 101–116. [Google Scholar] [CrossRef]
- Zong, X.; Suh, B.J.; Niazi, A.; Yan, J.Q.; Schlagel, D.L.; Lograsso, T.A.; Johnston, D.C. 17O and 51V NMR for the zigzag spin-1 chain compound CaV2O4. Phys. Rev. B 2008, 77, 014412. [Google Scholar] [CrossRef]
- Helis, H.M.; Goodman, W.H.; Wilson, R.B.; Morgan, J.A.; Hodgson, D.J. A novel halogen-bridged system: Synthesis and structures of dibromo[2-(2-aminomethyl)pyridine]copper(II) and dibromo(2-methyl- 1,2-diaminopropane)copper(II). Inorg. Chem. 1977, 16, 2412–2416. [Google Scholar] [CrossRef]
- Kikuchi, H.; Nagasawa, H.; Ajiro, Y.; Asano, T.; Goto, T. Susceptibility and high-field magnetization of one-dimensional S = 1/2 Heisenberg antiferromagnet with next-nearest exchange interaction. Physica B 2000, 284–288, 1631–1632. [Google Scholar] [CrossRef]
- Khan, M.I.; Giri, S.; Ayesh, S.; Doedens, R.J. Synthesis and characterization of a new hybrid chain derived from oxovanadyl and molybdate moieties [VIVO(μ3-MoO4)(2,2′-bpy)]: Topological equivalence of {MoO4} and {SO4} motifs. Inorg. Chem. Commun. 2004, 7, 721–724. [Google Scholar] [CrossRef]
- Kikuchi, H.; Ishikawa, Y.; Fujii, Y.; Matsuo, A.; Kindo, K. Magnetic properties of S = 1/2 J1-J2 one-dimensional magnets, VO(XO4)(2,2′-bpy) (X = S, Mo; bpy = bipyridine). J. Phys. Conf. Ser. 2014, 568, 042017. [Google Scholar] [CrossRef]
- Hosokoshi, Y.; Katoh, K.; Inoue, K.; Goto, T. Construction of a quantum-spin system of S = 1/2 antiferromagnetic chain with the next-nearest-neighbor interactions. J. Phys. Soc. Jpn. 1999, 68, 2910–2913. [Google Scholar] [CrossRef]
- Nomura, K.; Okamoto, K. Phase diagram of S = 1/2 antiferromagnetic XXZ chain with next-nearest-neighbor interactions. J. Phys. Soc. Jpn. 1993, 62, 1123–1126. [Google Scholar] [CrossRef]
- Maeshima, N.; Okunishi, K. Antiferromagnetic zigzag spin chain in magnetic fields at finite temperatures. Phys. Rev. B 2000, 62, 934–939. [Google Scholar] [CrossRef]
- Troyer, M.; Zhitomirsky, M.E.; Ueda, K. Nearly critical ground state of LaCuO2.5. Phys. Rev. B 1997, 55, R6117–R6120. [Google Scholar] [CrossRef]
Figure 1.
Geometries of spin lattices having strong spin-frustration (a–f), non-frustrated spin-configurations (g) 120°, (h) 109°, and (i) collinear AF (AFC): (j,k) are schematic view of triangular spin lattice of dimer-type Mott insulator of κ-(ET)2X (j) and monomer-type Mott insulator of C60•− (k). Purple ellipsoid in (j) is an ET molecule and the black circle represents one spin site (ET)2. In (j), t and t′ are interdimer transfer interactions between parallel dimers and perpendicular dimers, respectively, and t′/t represents the shape of the isosceles triangular spin lattice. In (k), t, t′, and t″ are interfullerene transfer interactions. Red arrows indicate spins. (j,k) were reproduced from [12].
Figure 1.
Geometries of spin lattices having strong spin-frustration (a–f), non-frustrated spin-configurations (g) 120°, (h) 109°, and (i) collinear AF (AFC): (j,k) are schematic view of triangular spin lattice of dimer-type Mott insulator of κ-(ET)2X (j) and monomer-type Mott insulator of C60•− (k). Purple ellipsoid in (j) is an ET molecule and the black circle represents one spin site (ET)2. In (j), t and t′ are interdimer transfer interactions between parallel dimers and perpendicular dimers, respectively, and t′/t represents the shape of the isosceles triangular spin lattice. In (k), t, t′, and t″ are interfullerene transfer interactions. Red arrows indicate spins. (j,k) were reproduced from [12].
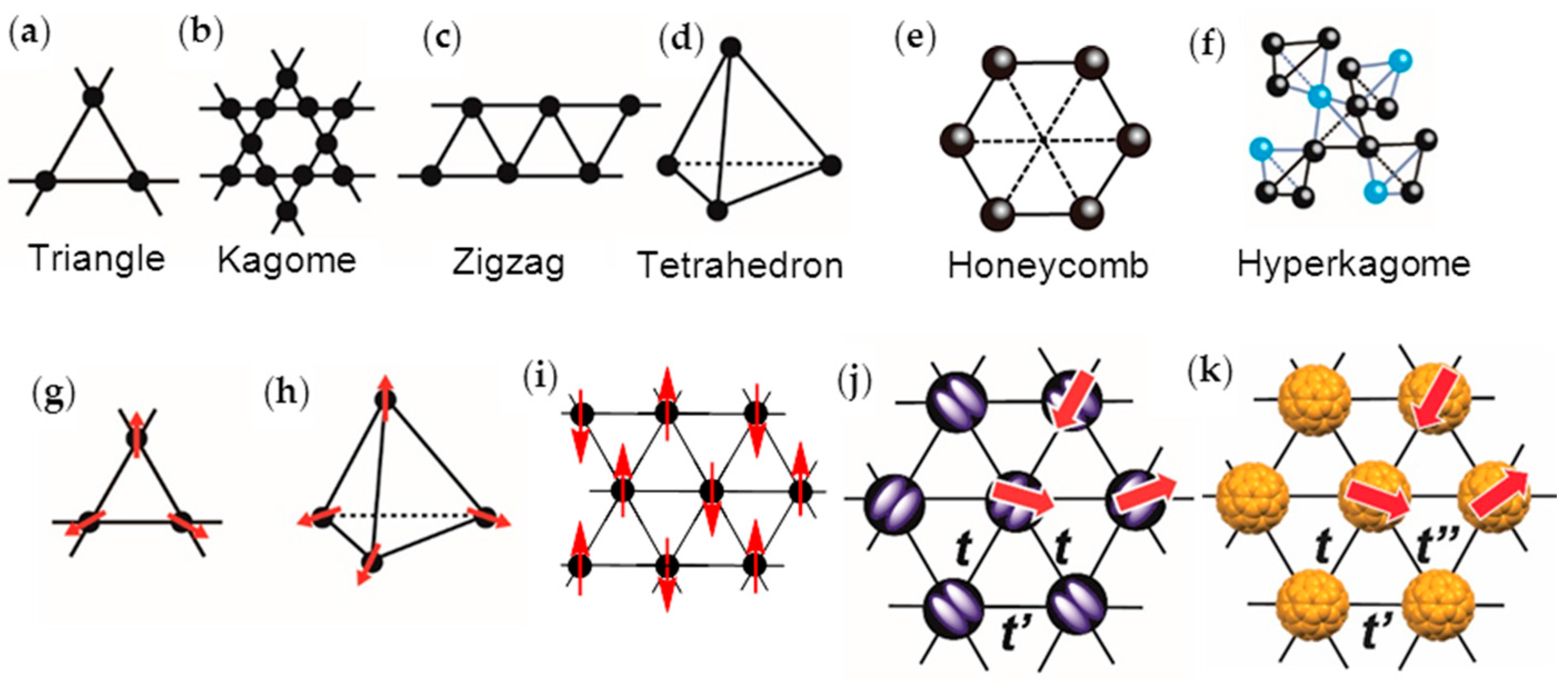
Figure 2.
(a) Chemicals in this review. The abbreviations are, TDAE: tetrakis(dimethylamino)ethylene, TPC: triptycene, MDABCO+: N-methyldiazabicyclooctane cation, MQ+: N-methylquinuclidinium cation, DMI+: N,N′-dimethylimidazolium cation, TMP+: N,N,N′-trimethylpiperazinium cation, BPY: 2,2′-bipyridine, PhCN: benzonitrile (Ph = phenyl group), PhCl2: o-dichlorobenzene, Ph4P+: tetraphenylphosphonium cation, Ph3MeP+: methyltriphenylphosphonium cation, (Ph3P)3Au+: tris(triphenyl)phosphine Au cation, PPN+: bis(triphenylphosphoranylidene)ammonium cation, DB18C6: dibenzo-18-crown-6, and [2.2.2]crypt: 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo-[8.8.8] hexacosane; (b) Some examples of fullerene dimers and polymers. C60 σ-dimer (9) [37], C60 π-dimer (10) [38], (C60−)n polymer in MC60 (M = K, Rb, and Cs) (11) [39,40], linear polymer of (C603−)n trianion in Na2RbC60 (12) [41], polymeric layer of (C604−)n tetraanion in Li4C60 (13) [42], zigzag polymer of (C602+)n dication in (C602+)(AsF6−)2 (14) [43], polymeric layer of (C604−)n tetraanion in Na4C60 (15) [44].
Figure 2.
(a) Chemicals in this review. The abbreviations are, TDAE: tetrakis(dimethylamino)ethylene, TPC: triptycene, MDABCO+: N-methyldiazabicyclooctane cation, MQ+: N-methylquinuclidinium cation, DMI+: N,N′-dimethylimidazolium cation, TMP+: N,N,N′-trimethylpiperazinium cation, BPY: 2,2′-bipyridine, PhCN: benzonitrile (Ph = phenyl group), PhCl2: o-dichlorobenzene, Ph4P+: tetraphenylphosphonium cation, Ph3MeP+: methyltriphenylphosphonium cation, (Ph3P)3Au+: tris(triphenyl)phosphine Au cation, PPN+: bis(triphenylphosphoranylidene)ammonium cation, DB18C6: dibenzo-18-crown-6, and [2.2.2]crypt: 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo-[8.8.8] hexacosane; (b) Some examples of fullerene dimers and polymers. C60 σ-dimer (9) [37], C60 π-dimer (10) [38], (C60−)n polymer in MC60 (M = K, Rb, and Cs) (11) [39,40], linear polymer of (C603−)n trianion in Na2RbC60 (12) [41], polymeric layer of (C604−)n tetraanion in Li4C60 (13) [42], zigzag polymer of (C602+)n dication in (C602+)(AsF6−)2 (14) [43], polymeric layer of (C604−)n tetraanion in Na4C60 (15) [44].
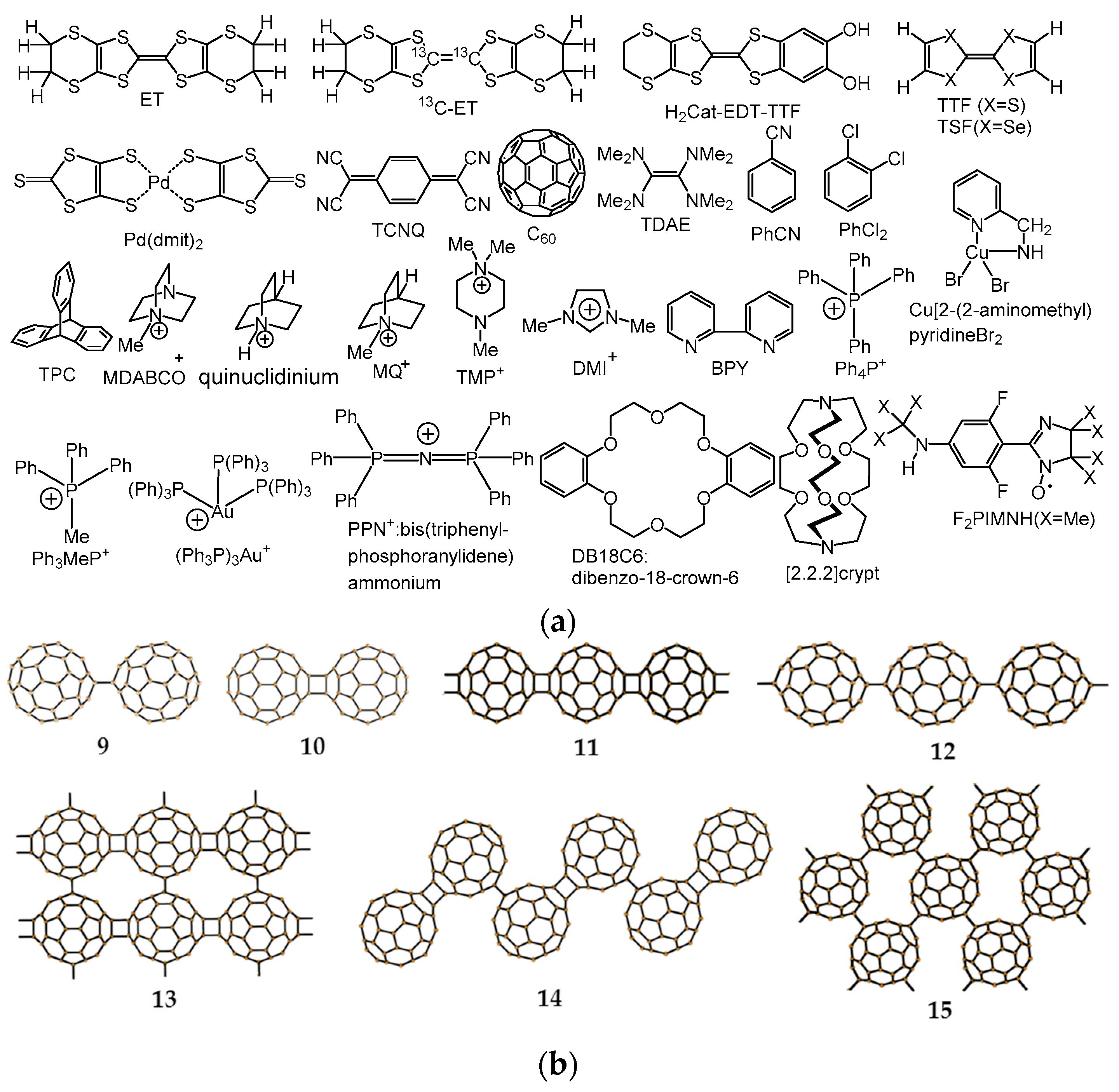
Figure 3.
(a,b) Bond formation (dimer and polymer), monomer metal, and monomer AF regions of C60n− with the center-to-center interfullerene distance r at RT except [K(DB18C6)]4(C60)5∙12THF (225 K indicated as [K(DB18C6)]4), {(TMP+)2MIITPP}(C60•−)2(PhCN)2(PhCl2)2 (270 K indicated as (TMP)2(MnTPP) and 250 K indicated as (TMP)2(ZnTPP), K(C60•−)(THF)5∙2THF (260 K indicated as K(THF)52THF) and (MDABCO)(C60•−) (250 K indicated as MDABCO). Cation or donor species are shown in boxes and Figure 2a. Li4 and Cs are plotted twice according to different interfullerene distances in the 2D polymer and in different phases (polymer or dimer), respectively. K(THF)52THF and (TPC)(MDABCO) are located on the borderline between monomer metal and monomer AF. Blue dotted line and green one indicate r = 9.4 Å and van der Waals diameter of C60 (10.18 Å), respectively.
Figure 3.
(a,b) Bond formation (dimer and polymer), monomer metal, and monomer AF regions of C60n− with the center-to-center interfullerene distance r at RT except [K(DB18C6)]4(C60)5∙12THF (225 K indicated as [K(DB18C6)]4), {(TMP+)2MIITPP}(C60•−)2(PhCN)2(PhCl2)2 (270 K indicated as (TMP)2(MnTPP) and 250 K indicated as (TMP)2(ZnTPP), K(C60•−)(THF)5∙2THF (260 K indicated as K(THF)52THF) and (MDABCO)(C60•−) (250 K indicated as MDABCO). Cation or donor species are shown in boxes and Figure 2a. Li4 and Cs are plotted twice according to different interfullerene distances in the 2D polymer and in different phases (polymer or dimer), respectively. K(THF)52THF and (TPC)(MDABCO) are located on the borderline between monomer metal and monomer AF. Blue dotted line and green one indicate r = 9.4 Å and van der Waals diameter of C60 (10.18 Å), respectively.

Figure 4.
The formation of two-dimensional (2D) C60•− layer (Layer A) on the polycationic template of [(TPC0)(MDABCO+)]∞: Three TPC (D1) and one MDABCO+ (D2+) molecules form the first supramolecular unit ((a-1): Supramolecule 1 [(D10)3(D2+)]), and a C60•− molecule fits into the concave to form the second supramolecular unit ((b-1): Supramolecule 2 [(D10)3(D2+)](C60•−)). They assemble to form a 2D layer (Layer A) of hexagonal packing of C60•− ((b-2): [(TPC0)(MDABCO+)(C60•−)]∞). In another view, the 2D arrangement of the first supramolecular units (a-2), where N atoms of MDABCO agglomerate at the crossing points of lines, works as a cationic template to arrange C60•− molecules at the crossing points to form hexagonal layer of C60•− (Layer A) (b-2). (N: blue, H: pale blue, C: yellow). The thin lines in (a-2,b-2) are a guide to the eye. (c) Single crystal showing the ab face.
Figure 4.
The formation of two-dimensional (2D) C60•− layer (Layer A) on the polycationic template of [(TPC0)(MDABCO+)]∞: Three TPC (D1) and one MDABCO+ (D2+) molecules form the first supramolecular unit ((a-1): Supramolecule 1 [(D10)3(D2+)]), and a C60•− molecule fits into the concave to form the second supramolecular unit ((b-1): Supramolecule 2 [(D10)3(D2+)](C60•−)). They assemble to form a 2D layer (Layer A) of hexagonal packing of C60•− ((b-2): [(TPC0)(MDABCO+)(C60•−)]∞). In another view, the 2D arrangement of the first supramolecular units (a-2), where N atoms of MDABCO agglomerate at the crossing points of lines, works as a cationic template to arrange C60•− molecules at the crossing points to form hexagonal layer of C60•− (Layer A) (b-2). (N: blue, H: pale blue, C: yellow). The thin lines in (a-2,b-2) are a guide to the eye. (c) Single crystal showing the ab face.
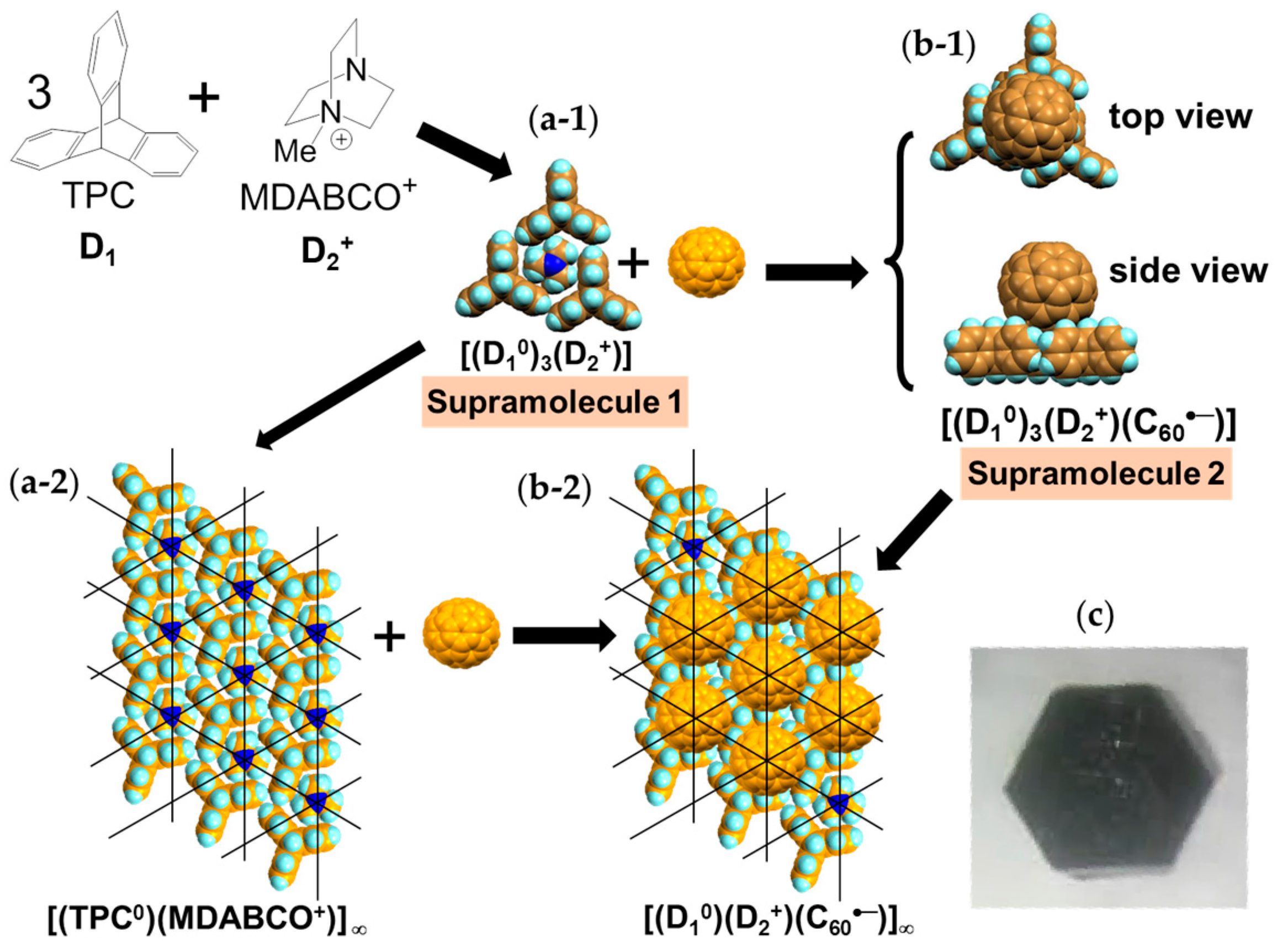
Figure 5.
Size of the component molecules and parts of Supramolecule 1 (TPC0)(MDABCO+), at 200 K. C: yellow, H: white-blue, N: blue: (a,b) Molecular size of MDABCO+ molecule; (c) The height of TPC molecule (Schematic figure); (d) Supramolecule 1 made of three TPC0 molecules and one MDABCO+ molecule.
Figure 5.
Size of the component molecules and parts of Supramolecule 1 (TPC0)(MDABCO+), at 200 K. C: yellow, H: white-blue, N: blue: (a,b) Molecular size of MDABCO+ molecule; (c) The height of TPC molecule (Schematic figure); (d) Supramolecule 1 made of three TPC0 molecules and one MDABCO+ molecule.
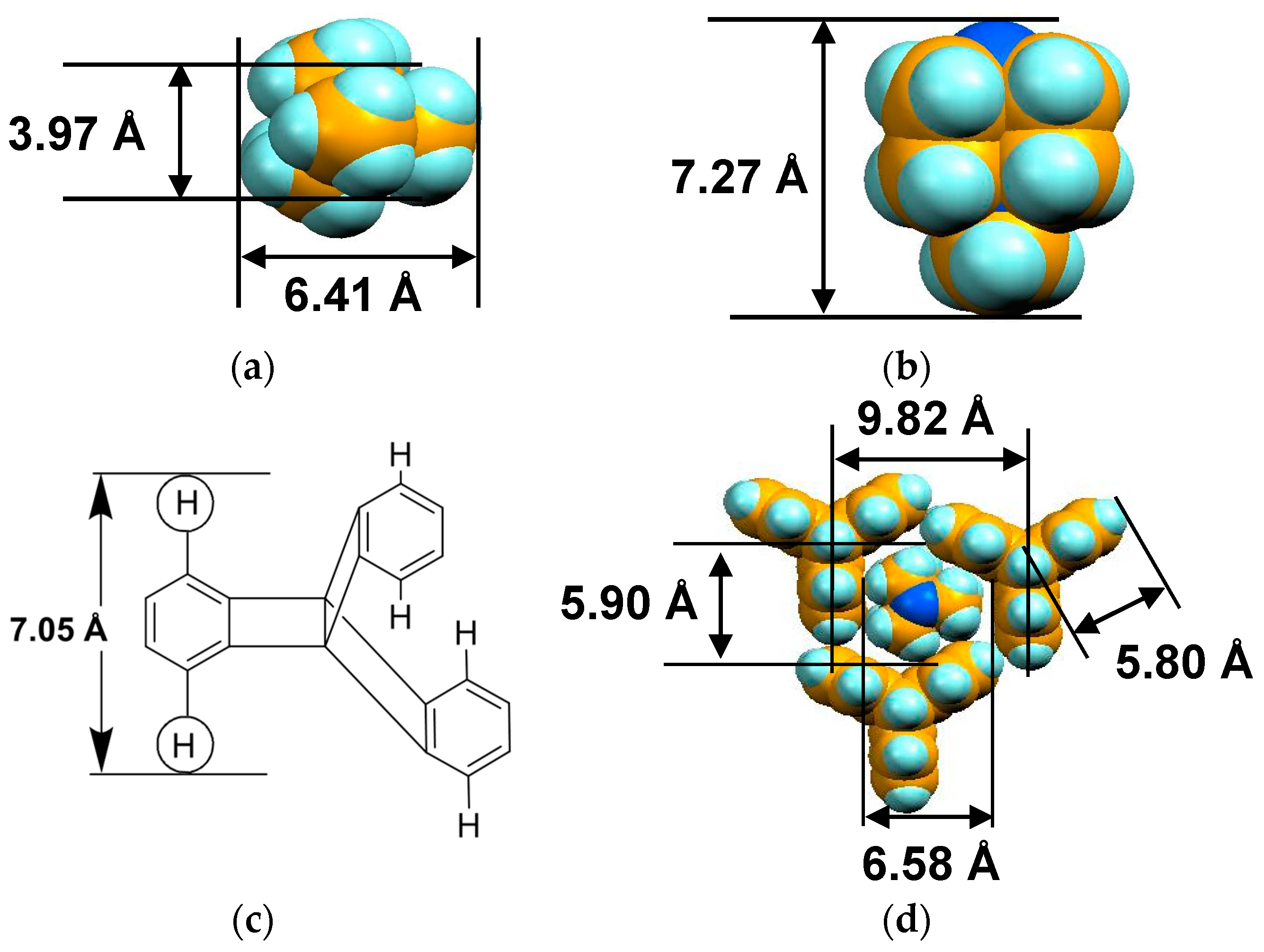
Figure 6.
Crystal structure of (TPC0)(MDABCO+)(C60•−) at 160 K: The (TPC0)(MDABCO+) layer and C60•− layer stack along the c axis with the sequence of the Layer A of C60•−/(TPC0)(MDABCO+) layer/Layer B of C60•−. (a) C60•− molecules in Layer A are arranged between the N atoms of MDABCO (drawn in red and N atoms and methyl groups are drawn in dark and bright blue, respectively) when viewed along the c axis. The methyl groups of MDABCO molecules are arranged towards the C60 molecules in Layer B and outline an octopore around C60. Configuration of the molecules in the slabs A (b) and B (c). The thin lines show the geometry (triangular lattice) connected between the centers of C60 molecules.
Figure 6.
Crystal structure of (TPC0)(MDABCO+)(C60•−) at 160 K: The (TPC0)(MDABCO+) layer and C60•− layer stack along the c axis with the sequence of the Layer A of C60•−/(TPC0)(MDABCO+) layer/Layer B of C60•−. (a) C60•− molecules in Layer A are arranged between the N atoms of MDABCO (drawn in red and N atoms and methyl groups are drawn in dark and bright blue, respectively) when viewed along the c axis. The methyl groups of MDABCO molecules are arranged towards the C60 molecules in Layer B and outline an octopore around C60. Configuration of the molecules in the slabs A (b) and B (c). The thin lines show the geometry (triangular lattice) connected between the centers of C60 molecules.
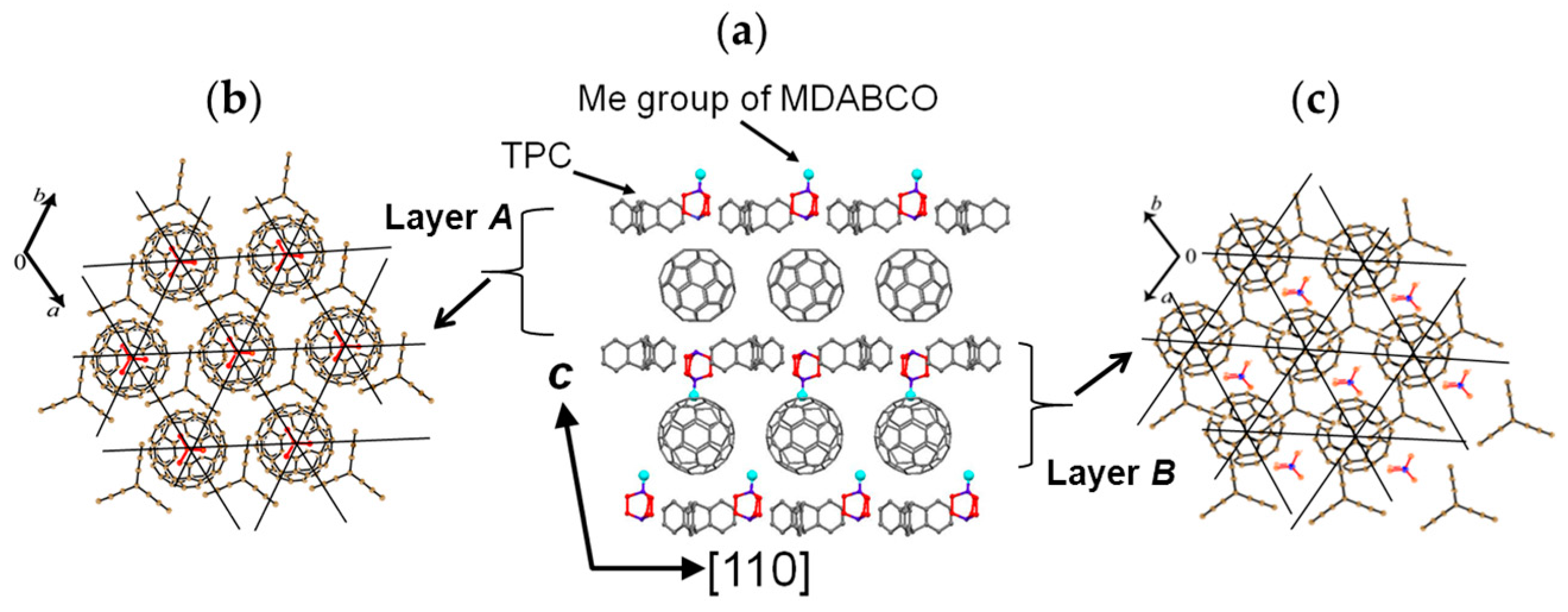
Figure 7.
Formation of a 2D C60•− layer (layer B) on the polycationic template of [(TPC0)(MDABCO+)]∞: (a) Three TPC0 (D1) and three MDABCO+ (D2+) molecules form Supramolecule 3 [(D10)3(D2+)3]; (b) A C60•− molecule fits into the concave to form Supramolecule 4 {[(D10)3(D2+)3]C60•−}; (c) 2D layer (Layer B) of hexagonal packing of C60•− by assembly of Supramolecule 4.
Figure 7.
Formation of a 2D C60•− layer (layer B) on the polycationic template of [(TPC0)(MDABCO+)]∞: (a) Three TPC0 (D1) and three MDABCO+ (D2+) molecules form Supramolecule 3 [(D10)3(D2+)3]; (b) A C60•− molecule fits into the concave to form Supramolecule 4 {[(D10)3(D2+)3]C60•−}; (c) 2D layer (Layer B) of hexagonal packing of C60•− by assembly of Supramolecule 4.
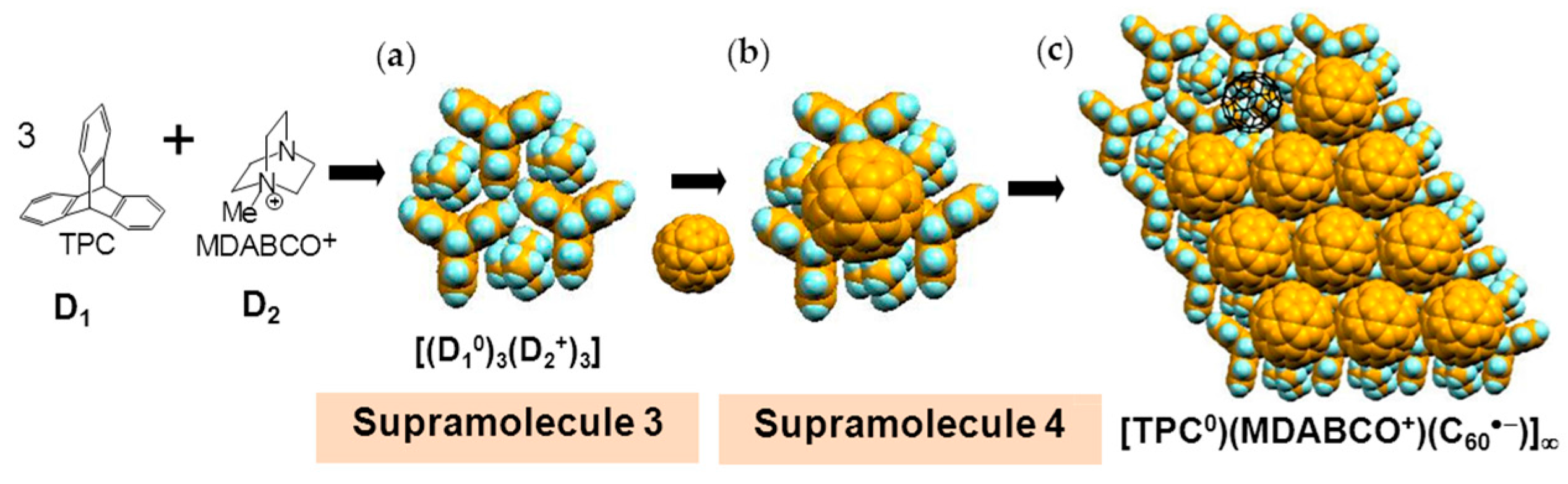
Figure 8.
(a) View of the ab plane of fullerene Layer A in (TPC0)(MDABCO+)(C60•−) at 300 K. The van der Waals C∙∙∙C contacts shorter than 3.42 Å are shown by dashed lines. Numbers 1–3 indicate the r values; (b) The calculated Fermi surface of Layer A at 200 K, (c) that of Layers A at 160 K, and (d) that of Layer B at 160 K. (a) was reproduced from [12] and (b−d) were from [146].
Figure 8.
(a) View of the ab plane of fullerene Layer A in (TPC0)(MDABCO+)(C60•−) at 300 K. The van der Waals C∙∙∙C contacts shorter than 3.42 Å are shown by dashed lines. Numbers 1–3 indicate the r values; (b) The calculated Fermi surface of Layer A at 200 K, (c) that of Layers A at 160 K, and (d) that of Layer B at 160 K. (a) was reproduced from [12] and (b−d) were from [146].
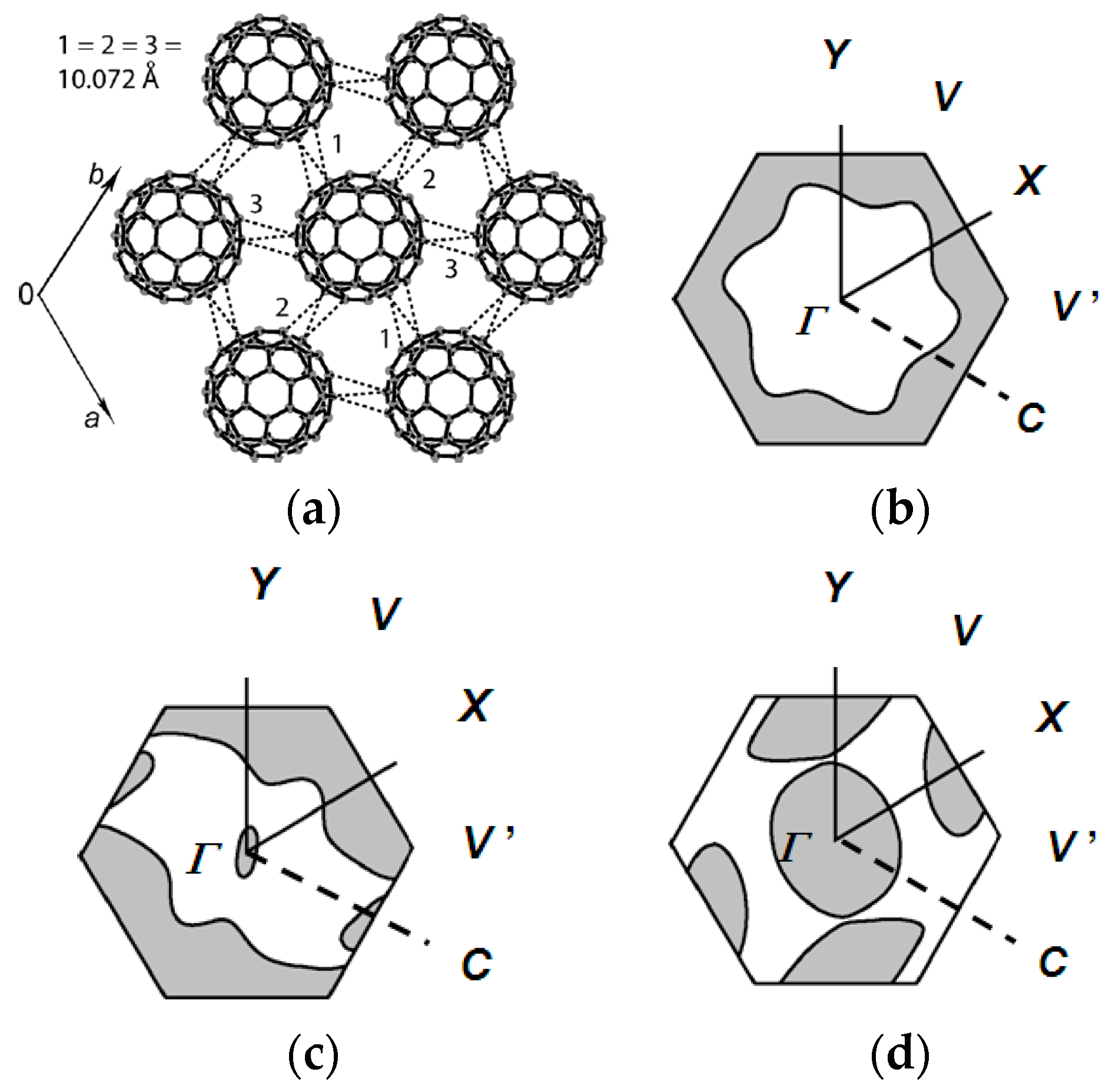
Figure 9.
(a) Temperature dependent resistivity in the ab plane and the ratio σ//c/σ//ab of (TPC0) (MDABCO+)(C60•−) by the four probe method (inset: single crystal with gold wires and paste) (from [146]); (b) Temperature dependence of conductivity by the four probe method, microwave and optical conductivity (from [12]); (c) Temperature dependent EPR peak ratio A/B for the Dysonian line from the single crystal (inset: the spectrum at 183 K) (from [146]) is compared with that of a superconductor β-(ET)2I3 (Tc = 1.5 K); (d) Temperature dependent molar magnetic susceptibility (χM) (closed circles, and blue curve fits in χ0 + C/(T − ΘCW) (χ0 = 6.5 × 10−4 emu∙mol−1, C = 0.160 emu∙K∙mol−1, and ΘCW = −31 K). Red bars for (a,c,d) show temperature interval from 185 to 200 K (from [146]).
Figure 9.
(a) Temperature dependent resistivity in the ab plane and the ratio σ//c/σ//ab of (TPC0) (MDABCO+)(C60•−) by the four probe method (inset: single crystal with gold wires and paste) (from [146]); (b) Temperature dependence of conductivity by the four probe method, microwave and optical conductivity (from [12]); (c) Temperature dependent EPR peak ratio A/B for the Dysonian line from the single crystal (inset: the spectrum at 183 K) (from [146]) is compared with that of a superconductor β-(ET)2I3 (Tc = 1.5 K); (d) Temperature dependent molar magnetic susceptibility (χM) (closed circles, and blue curve fits in χ0 + C/(T − ΘCW) (χ0 = 6.5 × 10−4 emu∙mol−1, C = 0.160 emu∙K∙mol−1, and ΘCW = −31 K). Red bars for (a,c,d) show temperature interval from 185 to 200 K (from [146]).

Figure 10.
(a–c) Shape and size of cation molecule MQ+ in (TPC0)(MQ+)(C60•−) at 250 K. The H atom attached to one N atom is shown in red; (d) The distance of the neighboring TPC molecules in Supramolecule 5 (TPC0)3(MQ+); (e) Intermolecular distance between neighboring MQ+ molecules (N∙∙∙N distance) in the polycationic template of (Supramolecule 5)∞.
Figure 10.
(a–c) Shape and size of cation molecule MQ+ in (TPC0)(MQ+)(C60•−) at 250 K. The H atom attached to one N atom is shown in red; (d) The distance of the neighboring TPC molecules in Supramolecule 5 (TPC0)3(MQ+); (e) Intermolecular distance between neighboring MQ+ molecules (N∙∙∙N distance) in the polycationic template of (Supramolecule 5)∞.
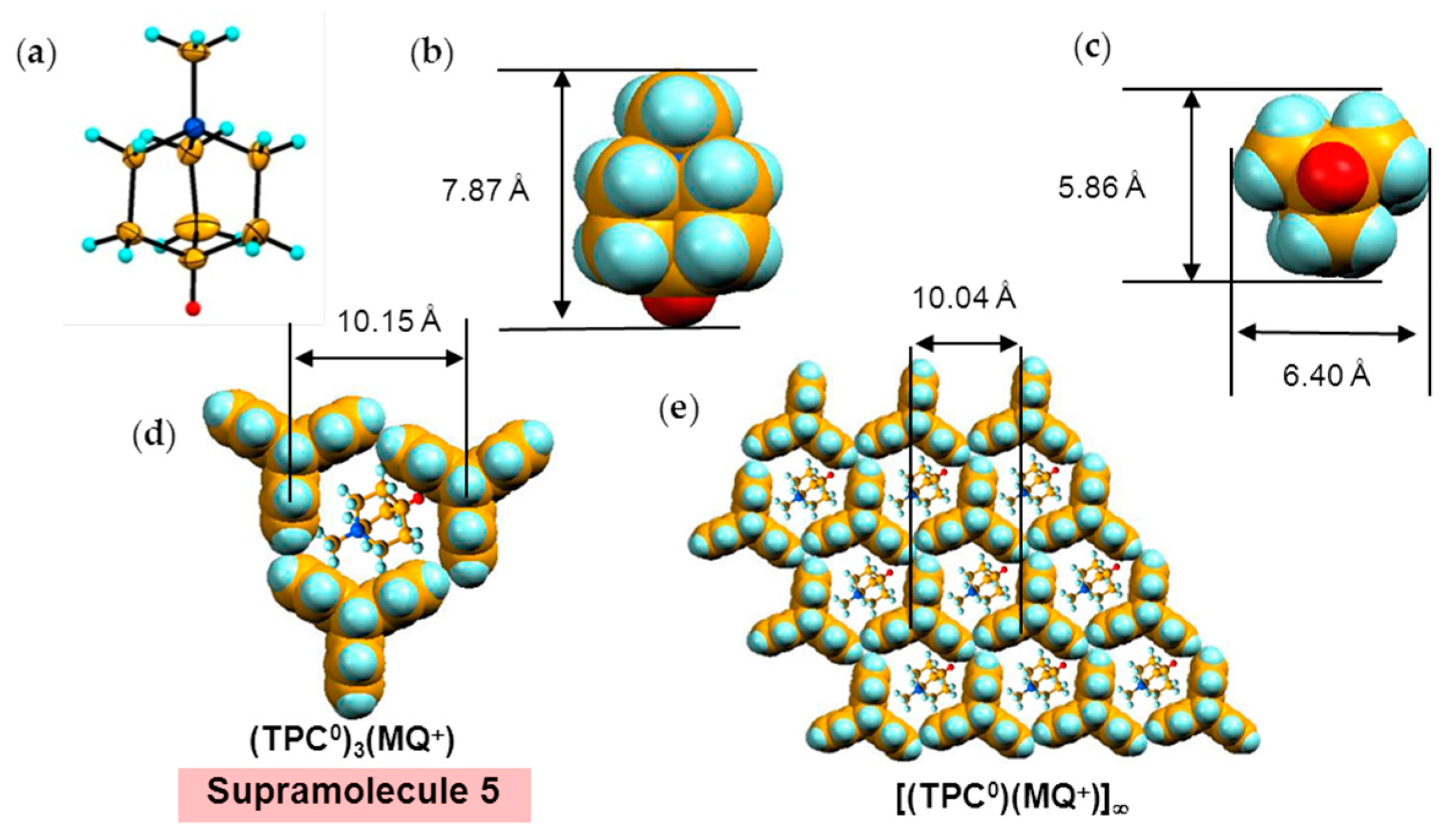
Figure 11.
(a) Crystal structure (250 K) projected along the [11(–)0] axis. The (TPC0)(MQ+) layer and C60•− layer stack along the c axis with the sequence of (TPC0)(MQ+) layer/C60•− Layer A/(TPC0)(MQ+) layer/C60•− Layer B. The MQ+ cations are shown in major occupied orientation in red color; (b,c) Projected views perpendicular to the layers, A layer slab (b) and B layer slab (c) [12].
Figure 11.
(a) Crystal structure (250 K) projected along the [11(–)0] axis. The (TPC0)(MQ+) layer and C60•− layer stack along the c axis with the sequence of (TPC0)(MQ+) layer/C60•− Layer A/(TPC0)(MQ+) layer/C60•− Layer B. The MQ+ cations are shown in major occupied orientation in red color; (b,c) Projected views perpendicular to the layers, A layer slab (b) and B layer slab (c) [12].
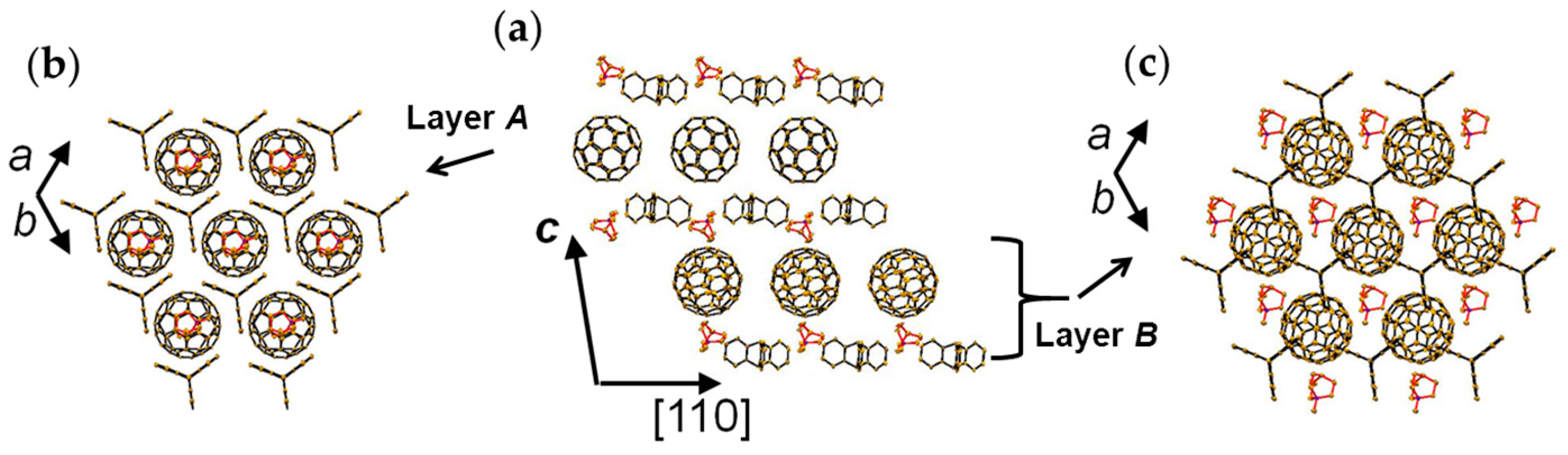
Figure 12.
Key-keyhole relation for the Layer A in (TPC0)(MQ+)(C60•−): Asymmetric placing of MQ+ in the (TPC0)3 hole to form Supramolecule 5 by first key-keyhole relation (a) and docking of C60•− into the pit of Supramolecule 5 gives Supramolecule 6 (second key-keyhole relation) (b); (c) A layer of [(TPC0)(MQ+)]∞ composed of Supramolecule 5; (d) A layer of [(TPC0)(MQ+)]∞ and C60•− molecules are assembled by fitting the C60 molecules into the concaves in the layer of [(TPC0)(MQ+)]∞ to form the Layer A of (TPC0)(MQ+)(C60•−). Black lines are guides to the eye. C60 molecules are arranged at the crossing points of black lines; (e,f) show the relation among the C60 molecule in Layer A and upper and lower layers of (TPC0)(MQ+). Upper and lower layers of (TPC0)(MQ+) are drawn in different colors: top down view (e) and side view (f); (g,h) Calculated Fermi surface of C60 assemble in Layer A at 250 K (g) and 100 K (h) [12].
Figure 12.
Key-keyhole relation for the Layer A in (TPC0)(MQ+)(C60•−): Asymmetric placing of MQ+ in the (TPC0)3 hole to form Supramolecule 5 by first key-keyhole relation (a) and docking of C60•− into the pit of Supramolecule 5 gives Supramolecule 6 (second key-keyhole relation) (b); (c) A layer of [(TPC0)(MQ+)]∞ composed of Supramolecule 5; (d) A layer of [(TPC0)(MQ+)]∞ and C60•− molecules are assembled by fitting the C60 molecules into the concaves in the layer of [(TPC0)(MQ+)]∞ to form the Layer A of (TPC0)(MQ+)(C60•−). Black lines are guides to the eye. C60 molecules are arranged at the crossing points of black lines; (e,f) show the relation among the C60 molecule in Layer A and upper and lower layers of (TPC0)(MQ+). Upper and lower layers of (TPC0)(MQ+) are drawn in different colors: top down view (e) and side view (f); (g,h) Calculated Fermi surface of C60 assemble in Layer A at 250 K (g) and 100 K (h) [12].
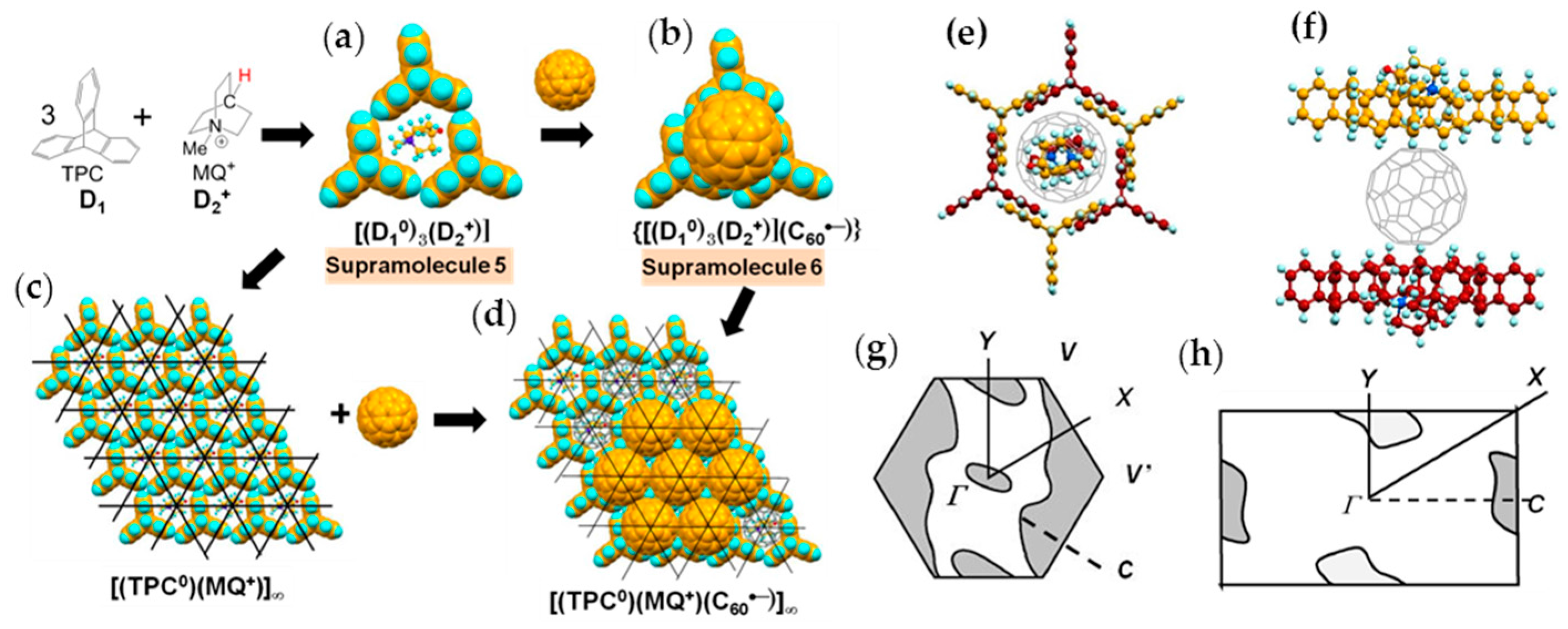
Figure 13.
Schematic key-keyhole relation to form Layer B of (TPC0)(MQ+)(C60•−): (a) Supramolecule 7: an assembly of three TPC and three MQ+ molecules for Layer B by the first key-keyhole relation; (b) C60•− molecule fit into Supramolecule 7 to form Supramolecule 8 [(TPC0)3(MQ+)3(C60•−)] in the Layer B; (c) Polycationic template of 2D layer (TPC0)(MQ+) composed of Supramolecule 7; (d) C60•− molecules are assembled by fitting the C60 molecules into the concaves in the layer of (TPC0)(MQ+) to form the Layer B of (TPC0)(MQ+)(C60•−). Dark thin lines are guides to the eye; (e,f) Six MQ+ molecules from the adjacent (TPC0)(MQ+) layers form octopores for C60•− in Layer B; (g,h) Calculated Fermi surface of C60 assembly in Layer B at 250 K (g) and 100 K (h) [12].
Figure 13.
Schematic key-keyhole relation to form Layer B of (TPC0)(MQ+)(C60•−): (a) Supramolecule 7: an assembly of three TPC and three MQ+ molecules for Layer B by the first key-keyhole relation; (b) C60•− molecule fit into Supramolecule 7 to form Supramolecule 8 [(TPC0)3(MQ+)3(C60•−)] in the Layer B; (c) Polycationic template of 2D layer (TPC0)(MQ+) composed of Supramolecule 7; (d) C60•− molecules are assembled by fitting the C60 molecules into the concaves in the layer of (TPC0)(MQ+) to form the Layer B of (TPC0)(MQ+)(C60•−). Dark thin lines are guides to the eye; (e,f) Six MQ+ molecules from the adjacent (TPC0)(MQ+) layers form octopores for C60•− in Layer B; (g,h) Calculated Fermi surface of C60 assembly in Layer B at 250 K (g) and 100 K (h) [12].
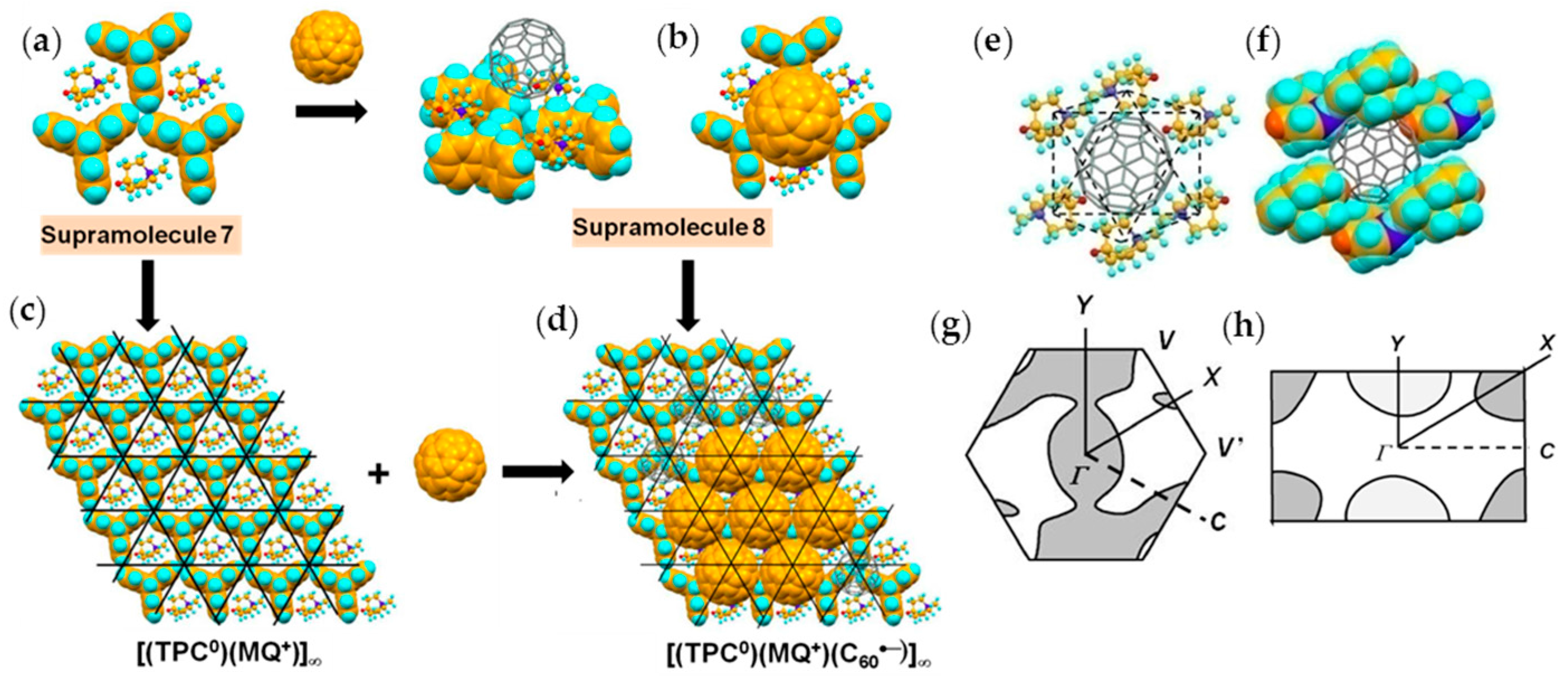
Figure 14.
Packing of C60•− at 250 K and magnetic behavior of (TPC0)(MQ+)(C60•−) [12]: (a) Packing pattern of C60 in Layer A in the ab plane; (b) Packing pattern of C60 in Layer B in the ab plane. Van der Waals C∙∙∙C contacts shorter than 3.42 Å are shown by dashed lines. Numbers 1–3 indicate the center-to-center distances between C60•−; (c) Temperature dependence of molar magnetic susceptibility and reciprocal molar magnetic susceptibility of (TPC0)(MQ+)(C60•−). Red curve shows the fitting of the molar magnetic susceptibility data in the 50–300 K by the Curie-Weiss law with Weiss temperature of −27 K.
Figure 14.
Packing of C60•− at 250 K and magnetic behavior of (TPC0)(MQ+)(C60•−) [12]: (a) Packing pattern of C60 in Layer A in the ab plane; (b) Packing pattern of C60 in Layer B in the ab plane. Van der Waals C∙∙∙C contacts shorter than 3.42 Å are shown by dashed lines. Numbers 1–3 indicate the center-to-center distances between C60•−; (c) Temperature dependence of molar magnetic susceptibility and reciprocal molar magnetic susceptibility of (TPC0)(MQ+)(C60•−). Red curve shows the fitting of the molar magnetic susceptibility data in the 50–300 K by the Curie-Weiss law with Weiss temperature of −27 K.
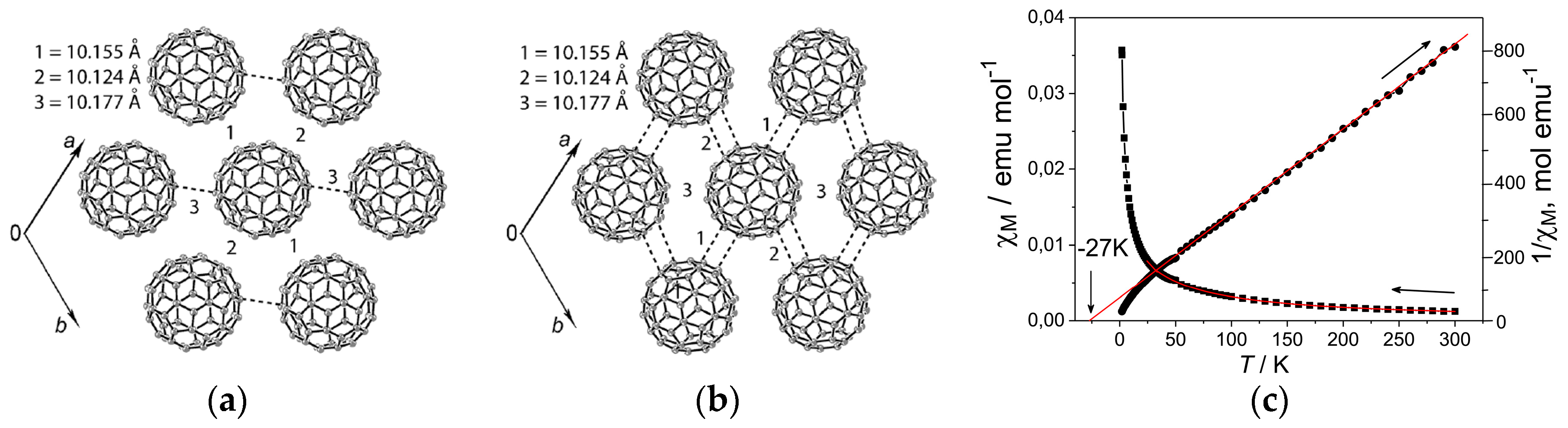
Figure 15.
(a) Crystal structure of (PhCN0)(TMP+)(C60•−) at 90 K viewed along the b axis. Black and red arrows indicate the layer of (PhCN0)(TMP+) and (C60•−), respectively; (b) View of the C60 layer at 120 K showing the r ≤ 10.20 Å by green lines; (c) Possible spin lattice geometry corresponding to (b) with overlap integrals; red line: s2 = 1.41 × 10−3 (r = 10.20 Å), blue line: s1 = 1.25 × 10−3 (r = 9.90 Å) and green line: s3 = 0.66 × 10−3 (r = 10.43 Å) giving J1:J2:J3 = 0.79:1:0.22 for the major C60 orientation. Only one C60•− orientation is shown in (c). (a,b) were reproduced from [148].
Figure 15.
(a) Crystal structure of (PhCN0)(TMP+)(C60•−) at 90 K viewed along the b axis. Black and red arrows indicate the layer of (PhCN0)(TMP+) and (C60•−), respectively; (b) View of the C60 layer at 120 K showing the r ≤ 10.20 Å by green lines; (c) Possible spin lattice geometry corresponding to (b) with overlap integrals; red line: s2 = 1.41 × 10−3 (r = 10.20 Å), blue line: s1 = 1.25 × 10−3 (r = 9.90 Å) and green line: s3 = 0.66 × 10−3 (r = 10.43 Å) giving J1:J2:J3 = 0.79:1:0.22 for the major C60 orientation. Only one C60•− orientation is shown in (c). (a,b) were reproduced from [148].
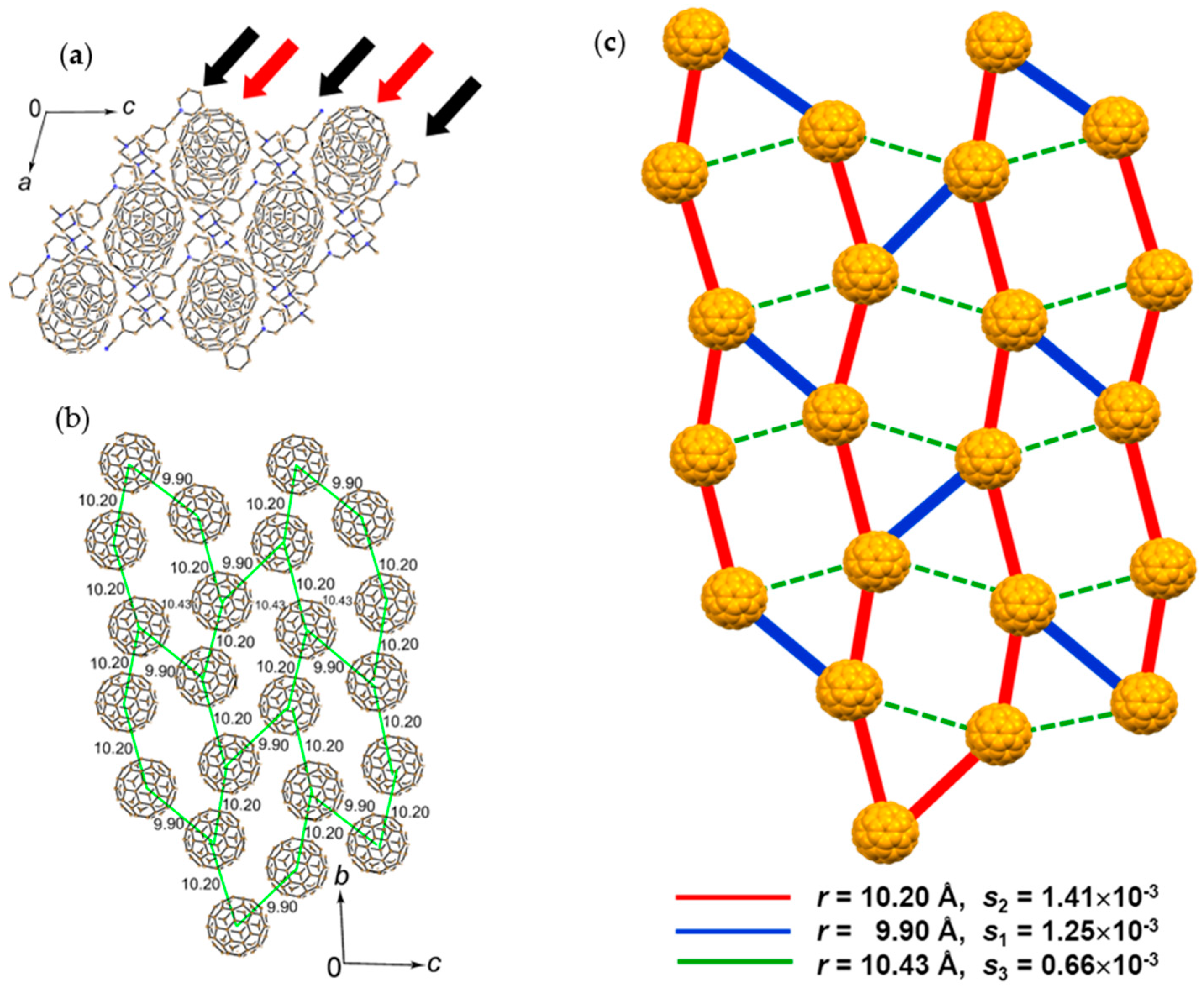
Figure 16.
(a) Crystal structure of (PhCN0)(Ph3MeP+)(C60•−) at 250 K. Four C60•− molecules (a1–a4) form a flat layer [105]; (b) Schematic of possible spin lattice with magnetic interactions with overlap integrals s1= 2.29 × 10−3 (red, r = 9.921 Å) > s2 = 2.14 × 10−3 (blue, r = 9.961 Å) > s3 = 0.70 × 10−3 (green, r = 10.066 Å), giving J1:J2:J3 = 1:0.87:0.09. Only one major orientation is shown for C60•− in (a,b).
Figure 16.
(a) Crystal structure of (PhCN0)(Ph3MeP+)(C60•−) at 250 K. Four C60•− molecules (a1–a4) form a flat layer [105]; (b) Schematic of possible spin lattice with magnetic interactions with overlap integrals s1= 2.29 × 10−3 (red, r = 9.921 Å) > s2 = 2.14 × 10−3 (blue, r = 9.961 Å) > s3 = 0.70 × 10−3 (green, r = 10.066 Å), giving J1:J2:J3 = 1:0.87:0.09. Only one major orientation is shown for C60•− in (a,b).
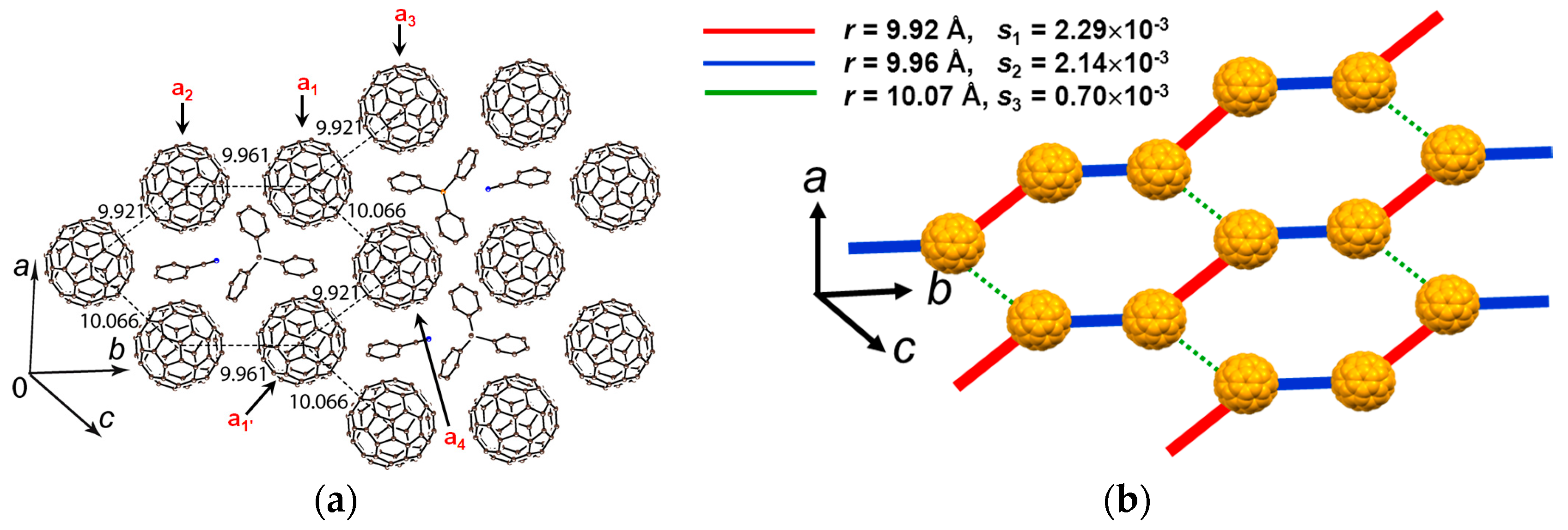
Figure 17.
(a) Crystal structure of (PhCl20)[(Ph3P)3Au+]2(C60•−)2(C60) at 100 K viewed along the c axis (in the cationic supramolecule, C: red-violet, Cl: green, P: orange, Au: yellow). Only one of three orientations is shown for C60 and PhCl2 molecules. I: C60•−, II: C600; (b) View along hexagonal C60 layers and the diagonal of the ab plane. Black arrows indicate the (PhCl20)[(Ph3P)3Au+]2 supramolecule layer (ra = 10.37 Å, rb ~ 13.92 Å); (c) View of the C60 layer at 100 K for (PhCl20)[(Ph3P)3Au+]2(C60•−)2(C60) showing r in Å. Black dashed lines link C60 centers. Four C60•− molecules (a1–a4) form tetrahedral spin lattice with r = 10.37 Å and the a1 molecule projects out of the a2-4 plane by 3.03 Å; (d) Possible spin lattice geometry (red lines). Only one orientation of C60•− is shown. Black ball is C60•−. (a–c) were reproduced from [149].
Figure 17.
(a) Crystal structure of (PhCl20)[(Ph3P)3Au+]2(C60•−)2(C60) at 100 K viewed along the c axis (in the cationic supramolecule, C: red-violet, Cl: green, P: orange, Au: yellow). Only one of three orientations is shown for C60 and PhCl2 molecules. I: C60•−, II: C600; (b) View along hexagonal C60 layers and the diagonal of the ab plane. Black arrows indicate the (PhCl20)[(Ph3P)3Au+]2 supramolecule layer (ra = 10.37 Å, rb ~ 13.92 Å); (c) View of the C60 layer at 100 K for (PhCl20)[(Ph3P)3Au+]2(C60•−)2(C60) showing r in Å. Black dashed lines link C60 centers. Four C60•− molecules (a1–a4) form tetrahedral spin lattice with r = 10.37 Å and the a1 molecule projects out of the a2-4 plane by 3.03 Å; (d) Possible spin lattice geometry (red lines). Only one orientation of C60•− is shown. Black ball is C60•−. (a–c) were reproduced from [149].
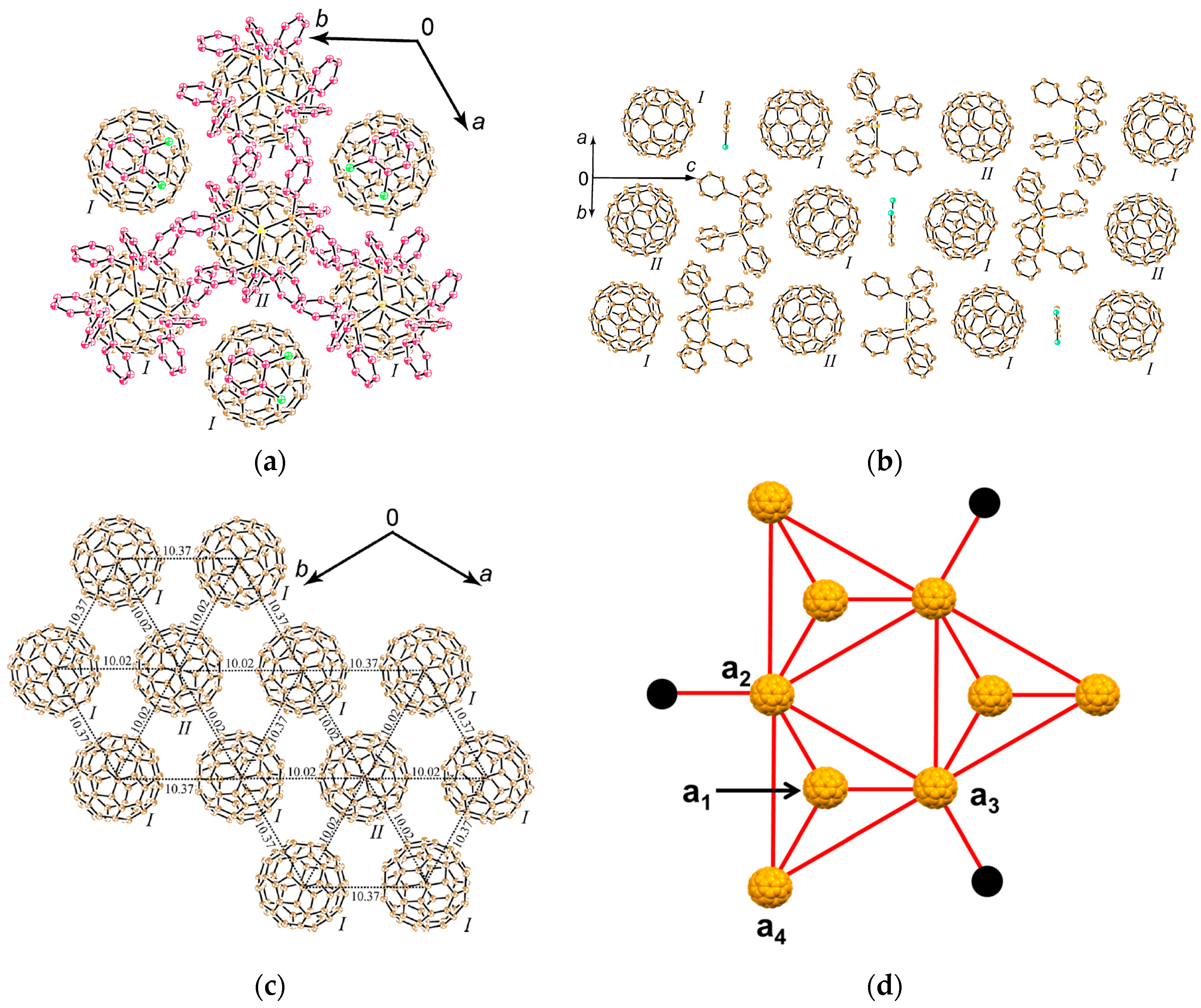
Figure 18.
(a) DMI+ molecule; (b) View of the honeycomb DMI+-I− layer in the ab plane of (DMI+)3(C60•−)(I−)2 at 100 K; (c) The formation of the H-bonds (dashed lines) between H atoms of DMI+ and I− (IA and IB) in the ab plane and along the c axis. The positions of C60•− are shown by black circles; (d) The supramolecular unit (DMI+)3(IB−)2 (Supramolecule 9) within the ab plane which is indicated by magenta circle in (b,c) and has a threefold symmetry like TPC; (e) The supramolecular unit (DMI+)3(IA−) (Supramolecule 10) within the ab plane which is indicated by blue circle in (b,c) and has a threefold symmetry like TPC; (f) Three units of Supramolecule 9 (magenta figure Y) form a wall to enclose one C60•− to form Supramolecule 11, where B represents an IB− anion; (g) Three units of Supramolecule 10 (blue figure Y) form a wall to enclose one C60•− (Supramolecule 12), where A represents an IA− anion; (h-1–h-4) Supramolecules 11 and 12 are capped along the c axis by Supramolecules 10 and 9, respectively, to form Supramolecule 13 [(DMI+)3(I−)3]3[(DMI+)3(I−)](C60•−) (h-1) and Supramolecule 14 [(DMI+)3(I−)]3[(DMI+)3(I−)3](C60•−) (h-2). Their combination leads to a crystal of (DMI+)3(C60•−)(I−)2. Packing of units of Supramolecule 13 (including C60•− molecules labeled 1, 2, and 7) above the unit of Supramolecule 14 (including C60•− molecule labeled a) leads to a crystal (h-3); (h-4) A schematic figure showing a layer (Layer 1) of hexagonal packing of C60•− molecules (labeled as 1–7) above a layer (Layer a) of the hexagonal packing of C60•− molecules (labeled as a–c) viewed along the c axis; (h-5) shows alternating stacking of Layer 1, Layer a, and Layer 1′ along the c axis with r = 11.05 Å (red line) and 13.36 Å (blue dotted line).
Figure 18.
(a) DMI+ molecule; (b) View of the honeycomb DMI+-I− layer in the ab plane of (DMI+)3(C60•−)(I−)2 at 100 K; (c) The formation of the H-bonds (dashed lines) between H atoms of DMI+ and I− (IA and IB) in the ab plane and along the c axis. The positions of C60•− are shown by black circles; (d) The supramolecular unit (DMI+)3(IB−)2 (Supramolecule 9) within the ab plane which is indicated by magenta circle in (b,c) and has a threefold symmetry like TPC; (e) The supramolecular unit (DMI+)3(IA−) (Supramolecule 10) within the ab plane which is indicated by blue circle in (b,c) and has a threefold symmetry like TPC; (f) Three units of Supramolecule 9 (magenta figure Y) form a wall to enclose one C60•− to form Supramolecule 11, where B represents an IB− anion; (g) Three units of Supramolecule 10 (blue figure Y) form a wall to enclose one C60•− (Supramolecule 12), where A represents an IA− anion; (h-1–h-4) Supramolecules 11 and 12 are capped along the c axis by Supramolecules 10 and 9, respectively, to form Supramolecule 13 [(DMI+)3(I−)3]3[(DMI+)3(I−)](C60•−) (h-1) and Supramolecule 14 [(DMI+)3(I−)]3[(DMI+)3(I−)3](C60•−) (h-2). Their combination leads to a crystal of (DMI+)3(C60•−)(I−)2. Packing of units of Supramolecule 13 (including C60•− molecules labeled 1, 2, and 7) above the unit of Supramolecule 14 (including C60•− molecule labeled a) leads to a crystal (h-3); (h-4) A schematic figure showing a layer (Layer 1) of hexagonal packing of C60•− molecules (labeled as 1–7) above a layer (Layer a) of the hexagonal packing of C60•− molecules (labeled as a–c) viewed along the c axis; (h-5) shows alternating stacking of Layer 1, Layer a, and Layer 1′ along the c axis with r = 11.05 Å (red line) and 13.36 Å (blue dotted line).
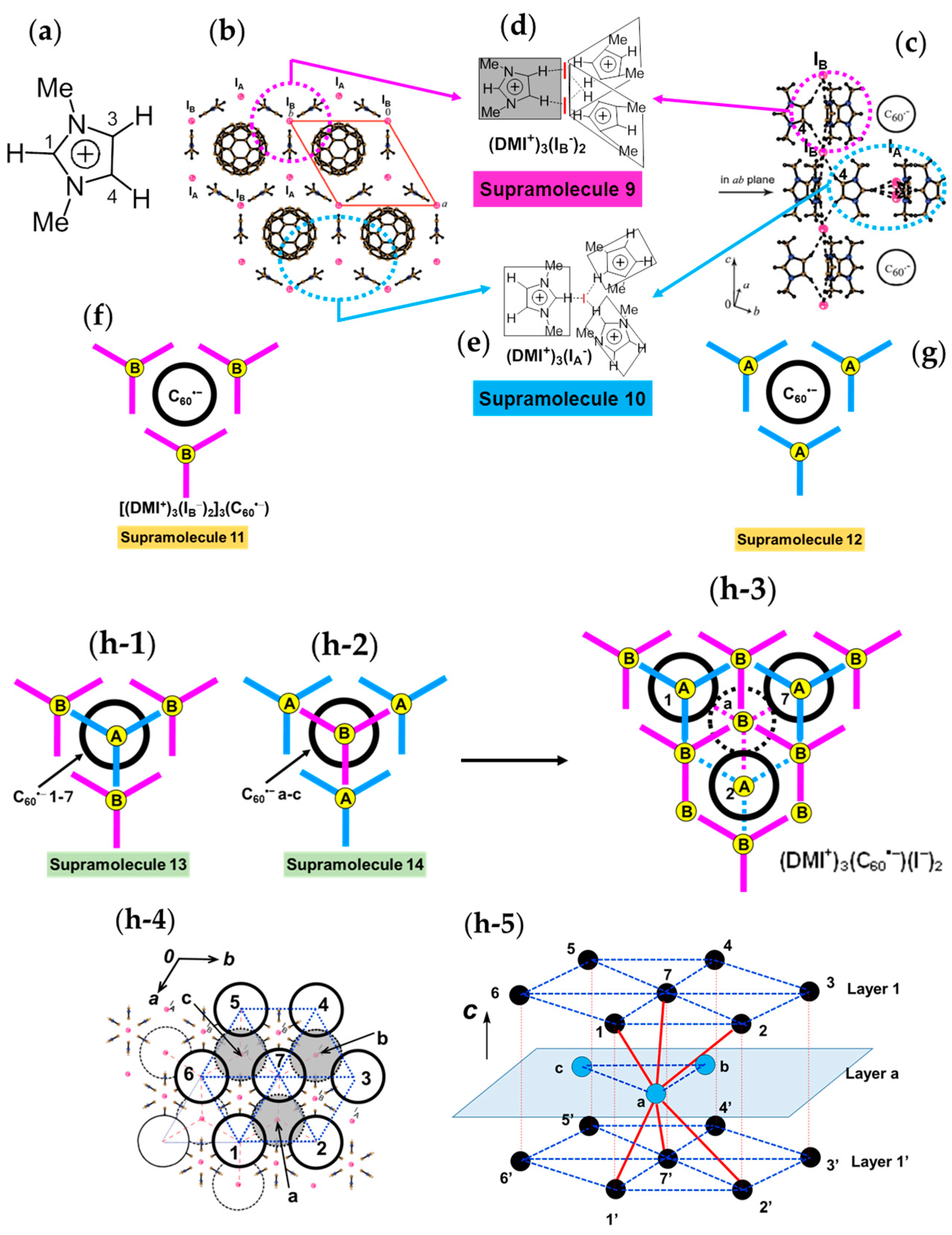
Figure 19.
(a) Schematic figure of 2H-hexagonal packing of C60•− molecules in (DMI+)3(C60•−)(I−)2 at 100 K showing apex-sharing bipyramid; (b) Model spin unit of triangular bipyramid with only one C60•− orientation. Labeling of C60 molecules correspond to those in Figure 18; (c) Model spin lattice based on overlap integrals; (d) Temperature dependence of reciprocal molar magnetic susceptibility of (DMI+)3(C60•−)(I−)2. Red curve shows the fitting of the molar magnetic susceptibility data in the 50–300 K by the Curie-Weiss law with Weiss temperature of −9.6 K [145]; (e) Temperature dependence of magnetic moment of (DMI+)3(C60•−)(I−)2.
Figure 19.
(a) Schematic figure of 2H-hexagonal packing of C60•− molecules in (DMI+)3(C60•−)(I−)2 at 100 K showing apex-sharing bipyramid; (b) Model spin unit of triangular bipyramid with only one C60•− orientation. Labeling of C60 molecules correspond to those in Figure 18; (c) Model spin lattice based on overlap integrals; (d) Temperature dependence of reciprocal molar magnetic susceptibility of (DMI+)3(C60•−)(I−)2. Red curve shows the fitting of the molar magnetic susceptibility data in the 50–300 K by the Curie-Weiss law with Weiss temperature of −9.6 K [145]; (e) Temperature dependence of magnetic moment of (DMI+)3(C60•−)(I−)2.
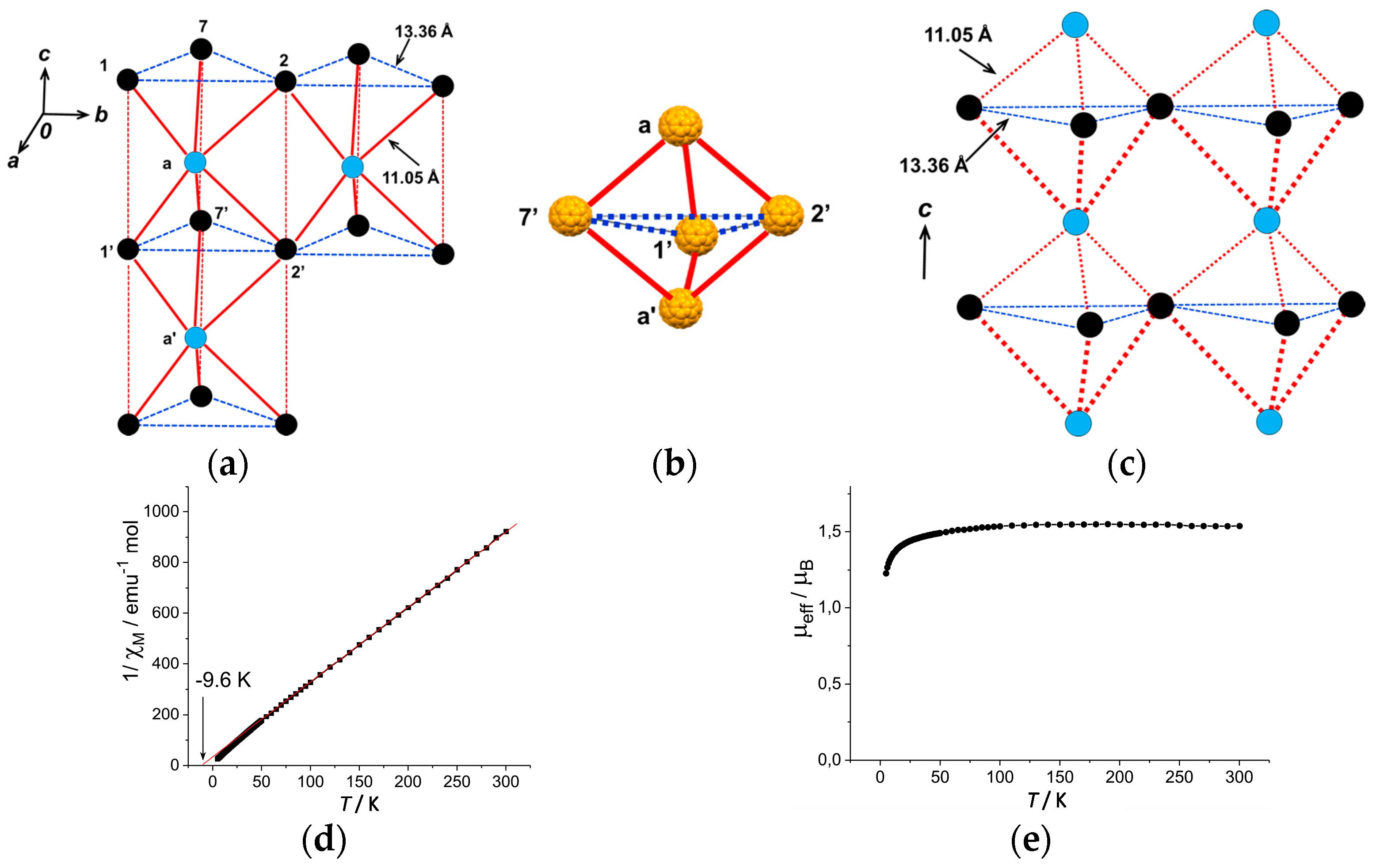
Figure 20.
(a) View of crystal structure of (MDABCO+)(C60•−) along the a axis at 100 K; (b) View along the b axis at 100 K. The van der Waals contacts between fullerenes are shown in green dashed lines in (a,b); (c) C60•− molecules stack along the b axis to form columns as shown with (c-1) or without MDABCO+ (c-2, for simplicity). Yellow and red C60•− columns are arranged alternately along the c axis to form a corrugated sheet in the bc plane; (d) Environment of one C60•− radical anion (shown by red color) from eight neighboring C60•− and five MDABCO+ cations at 250 K. Center-to-center distances between red fullerene (Ⓐ) and surrounding C60•− (marked by numbers ①–③): Ⓐ−①: 10.107, Ⓐ−②: 10.081 and Ⓐ−③: 10.008 Å. Van der Waals contacts between C60•− and nitrogen atom of the MDABCO+ cation are shown by green dashed lines. Only the major orientation of C60•− is shown; (e) (e-1) Corrugated layer composed of molecules Ⓐ and ② at 250 K; (e-2) Geometry of a unit of possible spin lattice made of Ⓐ and ②; (e-3) Schematic of bipyramidal spin lattice composed of molecules Ⓐ, ①, and ②; (e-4) The unit shown by (e-3) forms 2D layer by sharing an edge. Only the ribbon extending along the c axis is shown; (e-5) The other unit of bipyramidal spin lattice composed of ①, Ⓐ, and ③ forms equivalent 2D layer; (e-6) The model geometry of the possible spin lattice obtained by sharing apexes of the layer in (e-4,e-5) along the a axis to form a 3D distorted bipyramidal spin lattice (a,b,d), (e-1) from [147]).
Figure 20.
(a) View of crystal structure of (MDABCO+)(C60•−) along the a axis at 100 K; (b) View along the b axis at 100 K. The van der Waals contacts between fullerenes are shown in green dashed lines in (a,b); (c) C60•− molecules stack along the b axis to form columns as shown with (c-1) or without MDABCO+ (c-2, for simplicity). Yellow and red C60•− columns are arranged alternately along the c axis to form a corrugated sheet in the bc plane; (d) Environment of one C60•− radical anion (shown by red color) from eight neighboring C60•− and five MDABCO+ cations at 250 K. Center-to-center distances between red fullerene (Ⓐ) and surrounding C60•− (marked by numbers ①–③): Ⓐ−①: 10.107, Ⓐ−②: 10.081 and Ⓐ−③: 10.008 Å. Van der Waals contacts between C60•− and nitrogen atom of the MDABCO+ cation are shown by green dashed lines. Only the major orientation of C60•− is shown; (e) (e-1) Corrugated layer composed of molecules Ⓐ and ② at 250 K; (e-2) Geometry of a unit of possible spin lattice made of Ⓐ and ②; (e-3) Schematic of bipyramidal spin lattice composed of molecules Ⓐ, ①, and ②; (e-4) The unit shown by (e-3) forms 2D layer by sharing an edge. Only the ribbon extending along the c axis is shown; (e-5) The other unit of bipyramidal spin lattice composed of ①, Ⓐ, and ③ forms equivalent 2D layer; (e-6) The model geometry of the possible spin lattice obtained by sharing apexes of the layer in (e-4,e-5) along the a axis to form a 3D distorted bipyramidal spin lattice (a,b,d), (e-1) from [147]).

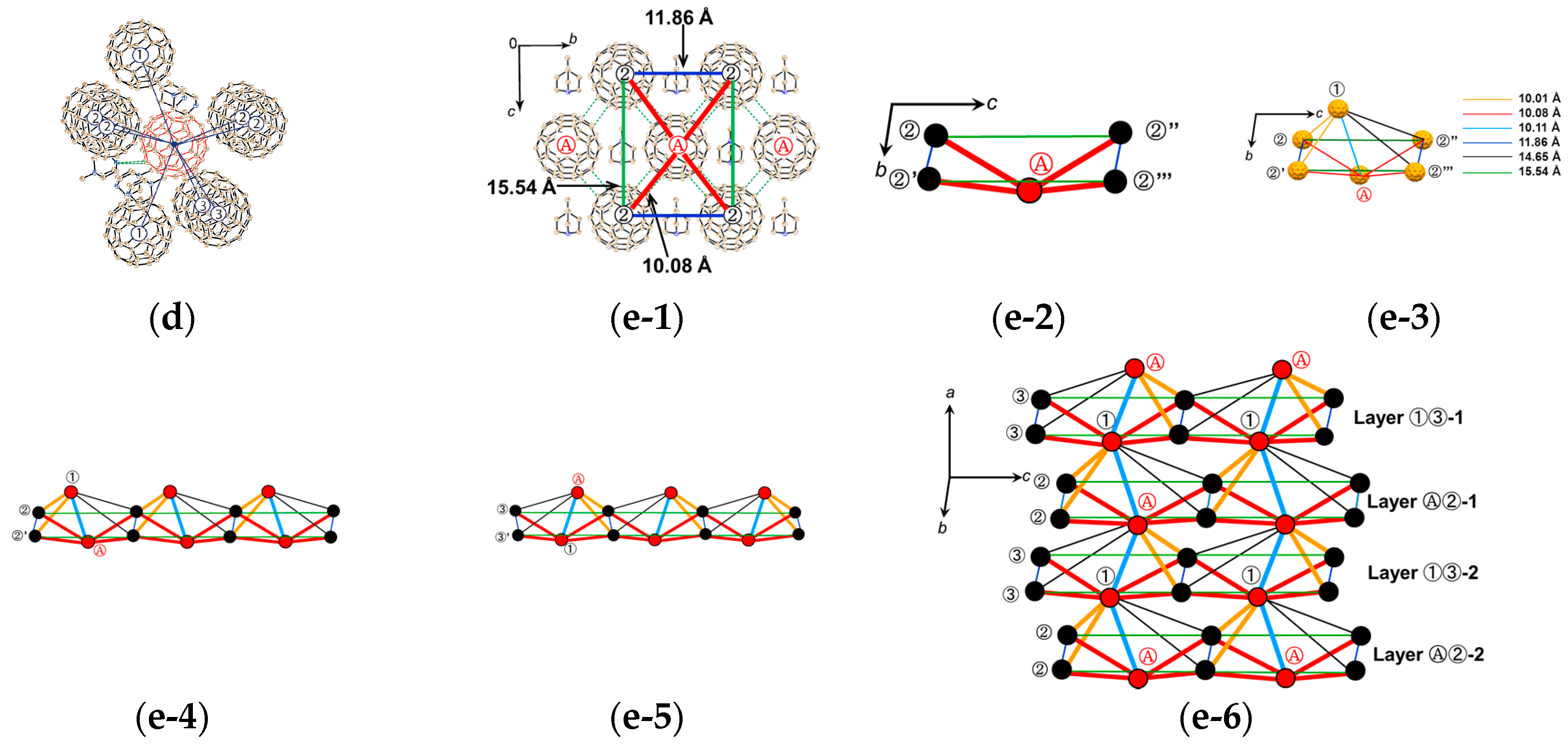
Figure 21.
Temperature dependence of molar magnetic susceptibility χM (a) and 1/χM (b) of (MDABCO+)(C60•−) after correction of Curie impurity. Red curve in (a) is the fit by the Heisenberg model for square 2D AF coupling of spins with J/kB = −25.3 K. Red line in (b) is the Curie-Weiss fit with ΘCW = −118 K [147].
Figure 21.
Temperature dependence of molar magnetic susceptibility χM (a) and 1/χM (b) of (MDABCO+)(C60•−) after correction of Curie impurity. Red curve in (a) is the fit by the Heisenberg model for square 2D AF coupling of spins with J/kB = −25.3 K. Red line in (b) is the Curie-Weiss fit with ΘCW = −118 K [147].

Figure 22.
Overlap integrals between C60•− anion radicals in (MDABCO+)(C60•−) at 100 K. One (molecule 1) and a half (molecule 2) of C60•− spheres are crystallograpically independent.
Figure 22.
Overlap integrals between C60•− anion radicals in (MDABCO+)(C60•−) at 100 K. One (molecule 1) and a half (molecule 2) of C60•− spheres are crystallograpically independent.
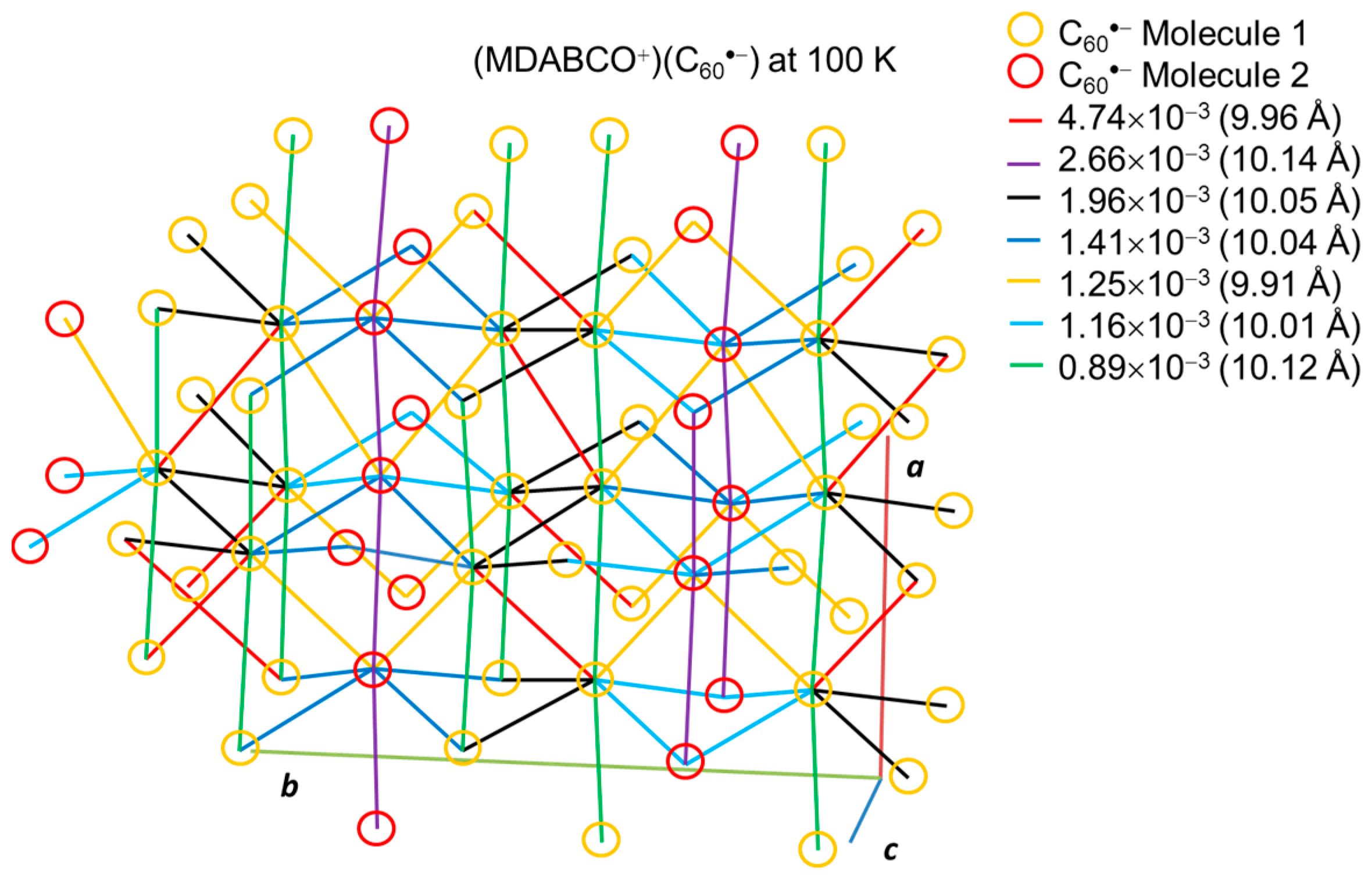
Figure 23.
(a) Crystal structure of (Ph3MeP+)(C60•−) showing the double chains containing triangles from C60•−; (b) view along the a axis showing the arrangement of double chains in the bc plane; (c) Schematic of possible zigzag spin lattice with weak interchain interactions (J1:J2:J3 = 1:0.44:0.10) together with r and s; (d) Temperature dependence of molar magnetic susceptibility χM and 1/χM after correction of Curie impurity. Red curve in (d) is the Curie-Weiss fit with ΘCW = −60 K. (a,b,d) were reproduced from [105].
Figure 23.
(a) Crystal structure of (Ph3MeP+)(C60•−) showing the double chains containing triangles from C60•−; (b) view along the a axis showing the arrangement of double chains in the bc plane; (c) Schematic of possible zigzag spin lattice with weak interchain interactions (J1:J2:J3 = 1:0.44:0.10) together with r and s; (d) Temperature dependence of molar magnetic susceptibility χM and 1/χM after correction of Curie impurity. Red curve in (d) is the Curie-Weiss fit with ΘCW = −60 K. (a,b,d) were reproduced from [105].
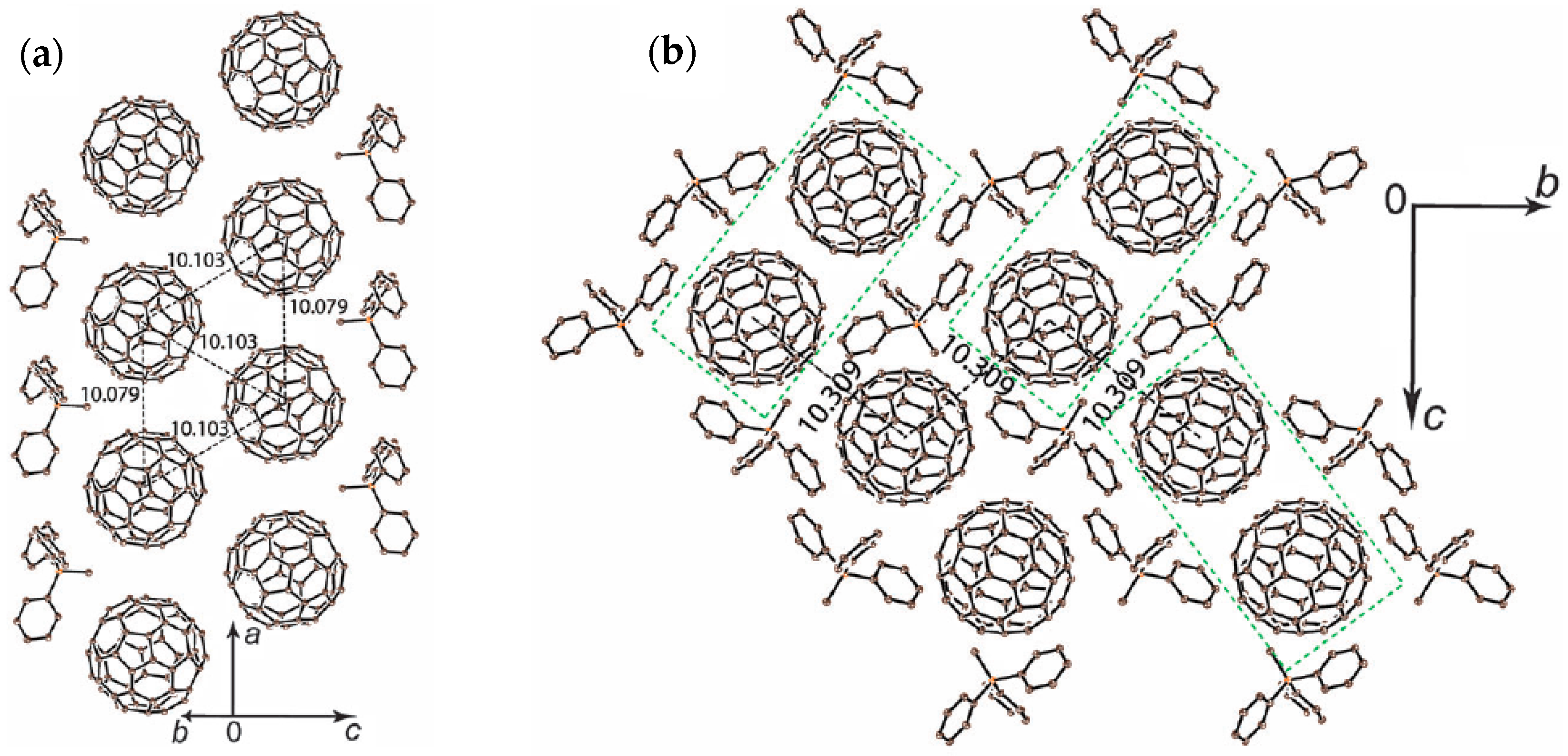
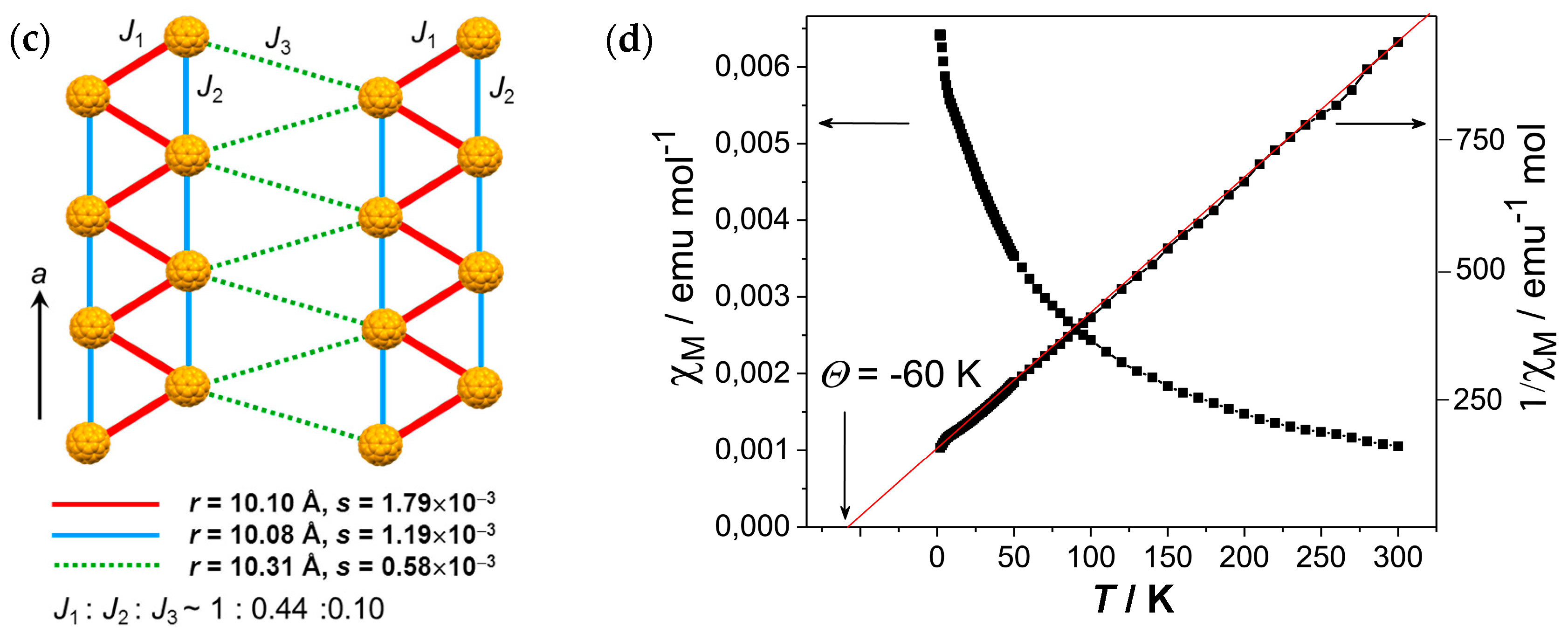
Figure 24.
Relation between Curie-Weiss temperature |ΘCW| and interfullerene center-to-center distance r for C60•− CT solids in this study. 1: (MDABCO+)(C60•−) (|ΘCW| = 118 K, 3D, r = 9.91 Å at 100 K), 2: (Ph3MeP+)(C60•−) (|ΘCW| = 60 K, pseudo 3D, coupled double chains, r = 10.08 Å at 100 K), 3: (TPC0)(MDABCO+)(C60•−) (|ΘCW| = 31 K, 2D layer r = 10.06 Å at 300 K, 9.97 Å at 185 K), 4: (TPC0)(MQ+)(C60•−) (|ΘCW| = 27 K, 2D layer, r = 10.12–10.18 Å at 250 K), 5: (PhCN0)(TMP+)(C60•−) (|ΘCW| = 11 K, 2D hexagon, r = 10.39 Å at 120 K), 7:{(Ph3P)3Au+}2(C60•−)2(C60)(PhCl2) (|ΘCW| = 5 K, 2D tetragonal, r = 10.37 Å at 100 K), 8: (DMI+)3(C60•−)(I−)2 (|ΘCW| = 9.6 K, 3D triangular bipyramid, r = 11.05 Å at 100 K). The blue line is a guide to the eye.
Figure 24.
Relation between Curie-Weiss temperature |ΘCW| and interfullerene center-to-center distance r for C60•− CT solids in this study. 1: (MDABCO+)(C60•−) (|ΘCW| = 118 K, 3D, r = 9.91 Å at 100 K), 2: (Ph3MeP+)(C60•−) (|ΘCW| = 60 K, pseudo 3D, coupled double chains, r = 10.08 Å at 100 K), 3: (TPC0)(MDABCO+)(C60•−) (|ΘCW| = 31 K, 2D layer r = 10.06 Å at 300 K, 9.97 Å at 185 K), 4: (TPC0)(MQ+)(C60•−) (|ΘCW| = 27 K, 2D layer, r = 10.12–10.18 Å at 250 K), 5: (PhCN0)(TMP+)(C60•−) (|ΘCW| = 11 K, 2D hexagon, r = 10.39 Å at 120 K), 7:{(Ph3P)3Au+}2(C60•−)2(C60)(PhCl2) (|ΘCW| = 5 K, 2D tetragonal, r = 10.37 Å at 100 K), 8: (DMI+)3(C60•−)(I−)2 (|ΘCW| = 9.6 K, 3D triangular bipyramid, r = 11.05 Å at 100 K). The blue line is a guide to the eye.
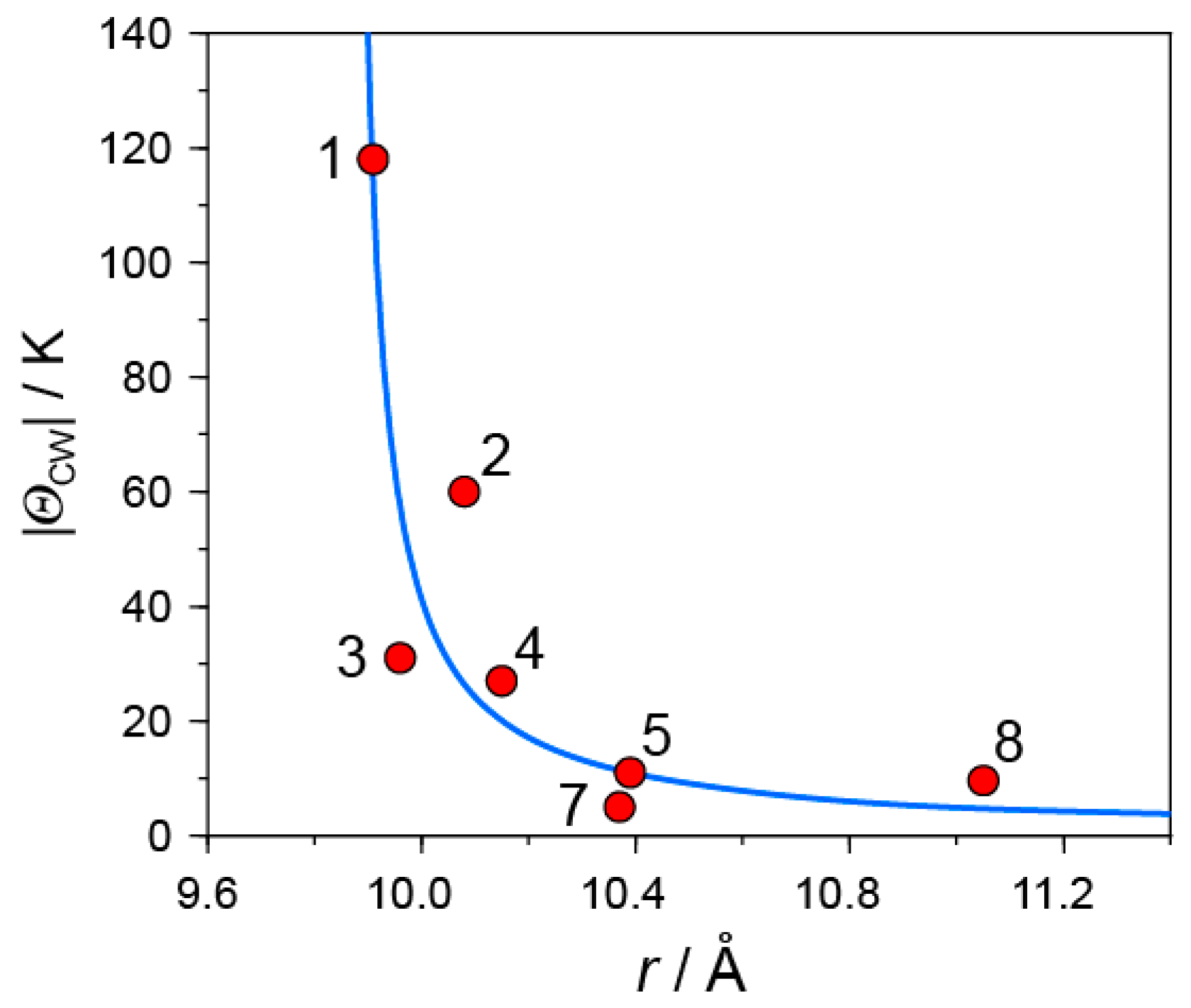
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Otsuka, A.; Konarev, D.V.; Lyubovskaya, R.N.; Khasanov, S.S.; Maesato, M.; Yoshida, Y.; Saito, G. Design of Spin-Frustrated Monomer-Type C60•− Mott Insulator. Crystals 2018, 8, 115. https://doi.org/10.3390/cryst8030115
AMA Style
Otsuka A, Konarev DV, Lyubovskaya RN, Khasanov SS, Maesato M, Yoshida Y, Saito G. Design of Spin-Frustrated Monomer-Type C60•− Mott Insulator. Crystals. 2018; 8(3):115. https://doi.org/10.3390/cryst8030115
Chicago/Turabian StyleOtsuka, Akihiro, Dmitri V. Konarev, Rimma N. Lyubovskaya, Salavat S. Khasanov, Mitsuhiko Maesato, Yukihiro Yoshida, and Gunzi Saito. 2018. "Design of Spin-Frustrated Monomer-Type C60•− Mott Insulator" Crystals 8, no. 3: 115. https://doi.org/10.3390/cryst8030115
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.