Temperature Dependence of Crystal Structures and Band Parameters in Quantum Spin Liquid β′-EtMe3Sb[Pd(dmit)2]2 and Related Materials


1
Condensed Molecular Materials Laboratory, RIKEN, 2-1, Hirosawa, Wako-shi, Saitama 351-0198, Japan
2
Computational Materials Science Unit, National Institute for Materials Science, Tsukuba-shi, Ibaraki 305-0047, Japan
*
Author to whom correspondence should be addressed.
Crystals 2018, 8(3), 138; https://doi.org/10.3390/cryst8030138
Submission received: 27 February 2018
/
Revised: 14 March 2018
/
Accepted: 15 March 2018
/
Published: 17 March 2018
(This article belongs to the Special Issue Advances in Organic Conductors and Superconductors)
Abstract
:In an isostructural series of anion radical salts β′-(Me4-xEtxZ)[Pd(dmit)2]2 (Z = P, As, Sb; x = 0, 1, 2), [Pd(dmit)2]2− units form a two-dimensional Mott insulator layer with a quasi-isosceles triangular lattice. The anisotropy of the triangular lattice is characterized by a ratio of interdimer transfer integrals, t′/t. The crystal structures of EtMe3Sb, Me4Sb, Me4As, and Et2Me2As salts were determined in the range of 5–295 K by the single crystal X-ray diffraction technique. Interdimer transfer integrals, Fermi surface, and band structures at low temperatures were calculated by the tight binding method and the first-principles density-functional theory (DFT) method based on experimentally obtained crystal structures. Interdimer transfer integrals increased with lowering temperature. At 5 K, the ratio t′/t decreased by about 15% from the room temperature value in every salt. The relationship between the transfer integrals and interdimer S⋯S distances indicated that the change of the t′/t value with temperature was due to a thermal contraction, rather than the arch-shaped molecular distortion of the Pd(dmit)2 molecule associated with the cation dependence of t′/t.

1. Introduction
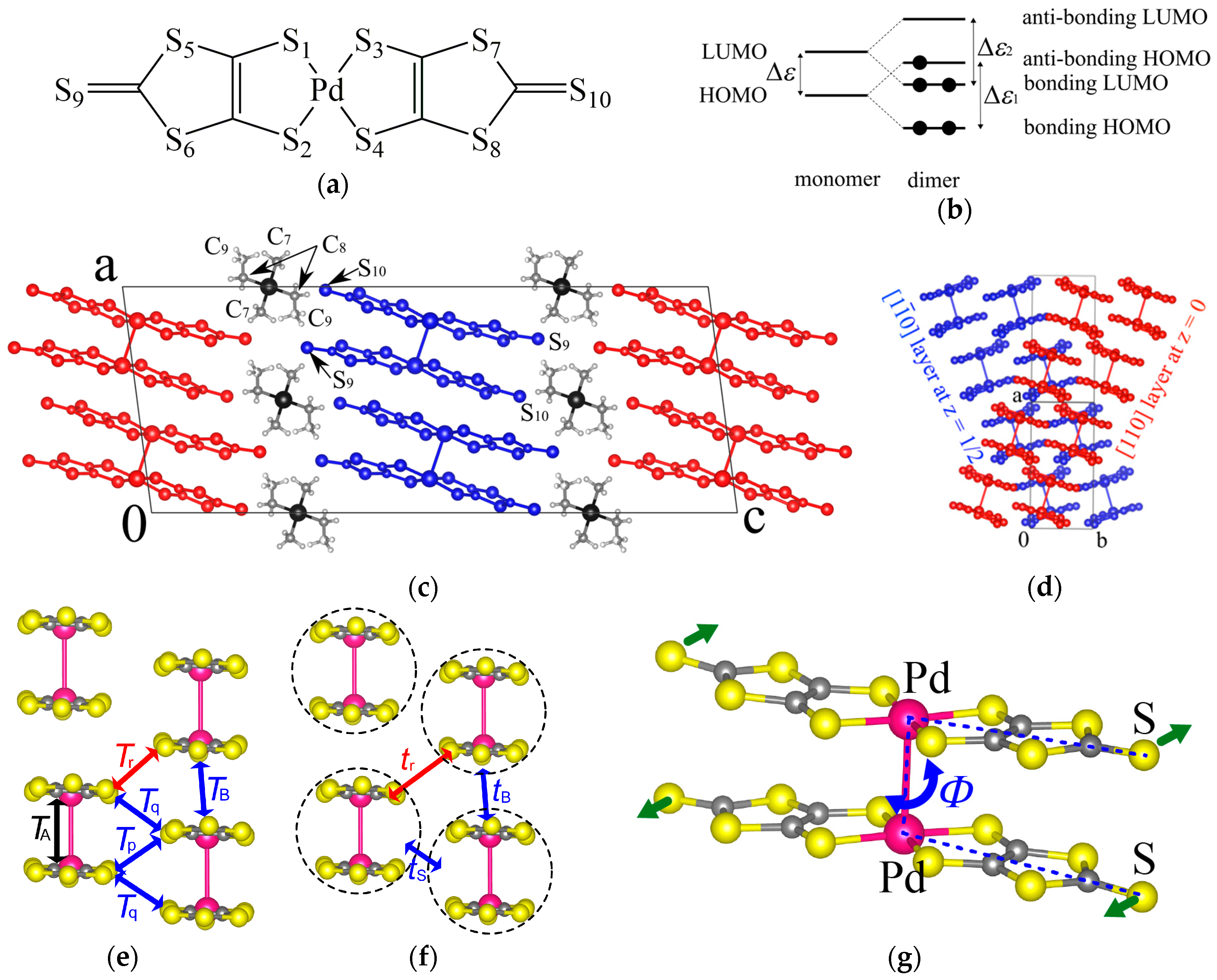
The metal dithiolene complex Pd(dmit)2 (dmit: 1,3-dithiole-2-thione-4,5-dithiolate, Figure 1a) forms various anion radical salts with closed shell monovalent cations: (Cation)[Pd(dmit)2]2 [1,2,3,4]. Many of them are categorized to a two-dimensional (2D) Mott insulator with localized S = 1/2 spin on a strongly dimerized unit, [Pd(dmit)2]2−. In a series of isostructural salts β′-(Me4-xEtxZ)[Pd(dmit)2]2 (Z = P, As, Sb; x = 0, 1, 2) with the space group of C2/c, cation and anion layers are repeated alternately (Figure 1c). The unit cell contains two crystallographically equivalent anion layers interrelated by the glide plane (Figure 1d). Within the anion layer, the dimer units form a triangular lattice that can induce geometrical frustration (Figure 1f). The cation is located on the two-fold axis and EtMe3Z-type cations without the two-fold symmetry show two possible orientations with the occupancy of 50% for each orientation.
The electronic structure of molecular conductors is simple and can be explained by tight-binding approximation [1,3,4,5,6]. We can describe the band structure of the β′-Pd(dmit)2 salts using a dimer model where electrons are considered to move from dimer to dimer [1,3,4]. The metal dithiolene complex is characterized by a small energy gap between the Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO; ∆ε in Figure 1b). In the dimer, each HOMO and LUMO in the monomer forms a bonding pair and an anti-bonding pair with a large energy gap (dimerization gap, ∆ε1 and ∆ε2). The dimerization gap is governed by the intradimer transfer integral TA (Figure 1e). When the dimerization gaps are large enough, the energy level of the anti-bonding HOMO pair can be located higher than that of the bonding LUMO pair (Figure 1b) [7]. Since the dimer unit has one negative charge, the anti-bonding HOMO pair has one unpaired electron. When each energy level forms an energy band in the crystal, the conduction band originates from the HOMO of the monomer. The conduction band is two-dimensional, half-filled, and narrow. Therefore, most of the β′-Pd(dmit)2 salts are Mott insulators at ambient pressure. The interdimer transfer integrals, tB, tS, and tr (Figure 1f) were obtained by the following equations using the intermolecular transfer integrals, TB, Tr, Tp, and Tq:tB = TB/2, ts = (Tp + 2Tq)/2, and tr = Tr/2 [3]. In the β′-Pd(dmit)2 salts, the tB and ts values are nearly the same and the tr is smaller, thus, the dimer units form an isosceles triangular lattice characterized by t′/t where t = (tB + ts)/2 and t′ = tr.
Temperature dependence of the magnetic susceptibility for the β′-Pd(dmit)2 salts can be described by the model of a spin-1/2 Heisenberg antiferromagnet on the triangular lattice with J = 200–300 K [8]. The magnetic ground state of the β′-Pd(dmit)2 salts can be classified into three types depending on the counter cation: antiferromagnetic long-range order (AFLO), quantum spin liquid (QSL), and charge ordered nonmagnetic state (CO) [8,9,10,11,12,13,14,15,16]. The CO phase transition in the Et2Me2Sb salt is accompanied by a large structural transition [15], where the coupling between the lattice and the electron plays an important role. The difference in the ground state can be understood by the anisotropy of the isosceles triangular lattice t′/t that can be tuned by the choice of counter cation [1,2,3,4]. The QSL state, where spin fluctuation prevents the spin ordering/freezing and provides liquid-like properties among the spins even at zero temperature, is realized in the EtMe3Sb salt with t′/t = 0.91 (at room temperature). The AFLO phase is observed in salts with smaller t′/t, namely closer to the square lattice. Moreover, the Néel temperature TN becomes higher with decreasing t′/t [1,2,4,8]. The transfer integrals between dimers are governed mostly by overlaps between π-electrons on sulfur atoms. The cation dependence of t′/t is not simply explained by the cation size, but by a cation-dependent distortion of the Pd(dmit)2 molecule [3]. In the dimer unit located at the center of symmetry, the Pd(dmit)2 molecules are not completely eclipsed and the Pd(dmit)2 molecule is slightly arched within the molecular plane. The degree of the arch-shaped distortion is described by the torsion angle of S–Pd–Pd–S, Φ, as shown in Figure 1g. The Φ value shows a systematic change depending on the counter cation (from −1.1° for Me4P+ to +2.2° for Et2Me2Sb+) and linearly correlates to the three interdimer transfer integrals, tB, tS, and tr.
The triangular lattice of the dimer units is not a result of the crystal symmetry but is formed through the combination of two larger interdimer transfer integrals along the side-by-side (tS) and stacking (tB) directions and the one along the diagonal direction (tr). A classical antiferromagnetic ordered structure, where two adjacent spins form an antiparallel configuration, is constricted on a triangular lattice. Such geometrical frustration becomes stronger near the regular triangular lattice, namely t′/t = 1. The QSL state is realized in some molecular conductors with a slightly distorted triangular lattice [13,14,17,18] including the EtMe3Sb salt. The reason why the QSL is observed in the distorted triangular lattice system in molecular conductors remains an open question. Since molecular conductors are characterized by a soft crystal lattice, intermolecular distances and molecular arrangement can be largely affected by temperature. Therefore, interdimer transfer integrals and the anisotropy of the triangular lattice t′/t also change with temperature. In the early stage, crystal structures of some β′-Pd(dmit)2 salts were determined at 8 K [19]. As for the EtMe3Sb salt with the QSL ground state, the detailed temperature dependence of the geometry of the triangular lattice and interdimer transfer integrals remain unknown, whereas some calculations of the DFT-derived interdimer transfer integrals for previous structural data at 4.5 K have been reported [20,21]. The knowledge of crystal and electronic structures at low temperatures is indispensable for understanding the nature of QSL.
In this study, we investigated low-temperature crystal structures of the QSL material, the EtMe3Sb salt, and the Me4Sb, Et2Me2As, and Me4As salts with the AFLO ground state (TN = 12–35 K) as a reference by using the single crystal X-ray diffraction technique down to 5 K from room temperature. Based on the structural data, we calculated low-temperature electronic structures with the tight binding method [5,6] and the first principles method [22,23,24,25]. We also discuss the temperature dependence of structural and band parameters including the anisotropy of the triangular lattice t′/t.
2. Materials and Methods
Single crystals of all salts were grown by aerial oxidation of (Cation)2[Pd(dmit)2] in acetone containing acetic acid [3]. Single crystal x-ray diffraction data were collected by a Rigaku UltraX-6-E Imaging Plate system with monochromated Mo Kα radiation (Rigaku, Akishima-shi, Japan). Low temperature experiments were carried out in the cryostat, cooled by a GM refrigerator, with a carbon fiber window down to 5 K. Initial structures were determined by the direct method (SHELXS or SHELXD) [26], and then were refined on F2 by the full-matrix least-squares method (SHELXL) [26]. Atomic displacements of all non-H atoms were determined with the anisotropic model. The H atom coordinates were placed on calculated positions and refined with the riding model. The ethyl group in the EtMe3Sb cation was treated by taking two equivalent positions around the two-fold axis.
Intermolecular transfer integrals were calculated by using extended Hückel calculations with semi-empirical parameters (shown in Table 1) based on the obtained crystal structures.
The first-principles density-functional theory (DFT) calculations were performed using the full potential linearized augmented plane wave (FLAPW) method as implemented in the QMD-FLAPW12 code [22,23]. Self-consistent-field (SCF) calculations were performed with the scalar-relativistic scheme and the improved tetrahedron integration method [24] up to 7 × 7 × 2 k-mesh points in the Brillouin zone (BZ). The exchange correlation contribution to the potential was included using the generalized gradient approximation (GGA) by Perdew-Burke-Ernzerhof [25]. We used a plane-wave cutoff of |k + G| < 4.5 a.u.−1 (~20 Ry) and 11.6 a.u.−1 (~135 Ry) for LAPW basis functions, and charge density and potential, respectively. Muffin-tin sphere radii were assumed to be 1.48 Å for Sb, 1.27 Å for Pd, 0.35 Å for H, 0.65 Å for C, and 0.90 Å for S. For the calculations of the Fermi surface, a band structure for 28 × 28 × 4 k-mesh points (1097 k-points in the irreducible wedge of BZ) was fitted with symmetrized star functions by a spline method. In the symmetry group of C2/c, cations were located on the two-fold axis, but the EtMe3Z-type cation did not have a two-fold symmetry. In this case, the cation was described by a superposition of two possible orientations with an occupancy of 0.5 for each orientation. In this study, the EtMe3Sb cation was replaced by the Et2Me2Sb cation to perform the DFT calculations.
3. Results and Discussions
3.1. Temperature Dependence of Band Parameters
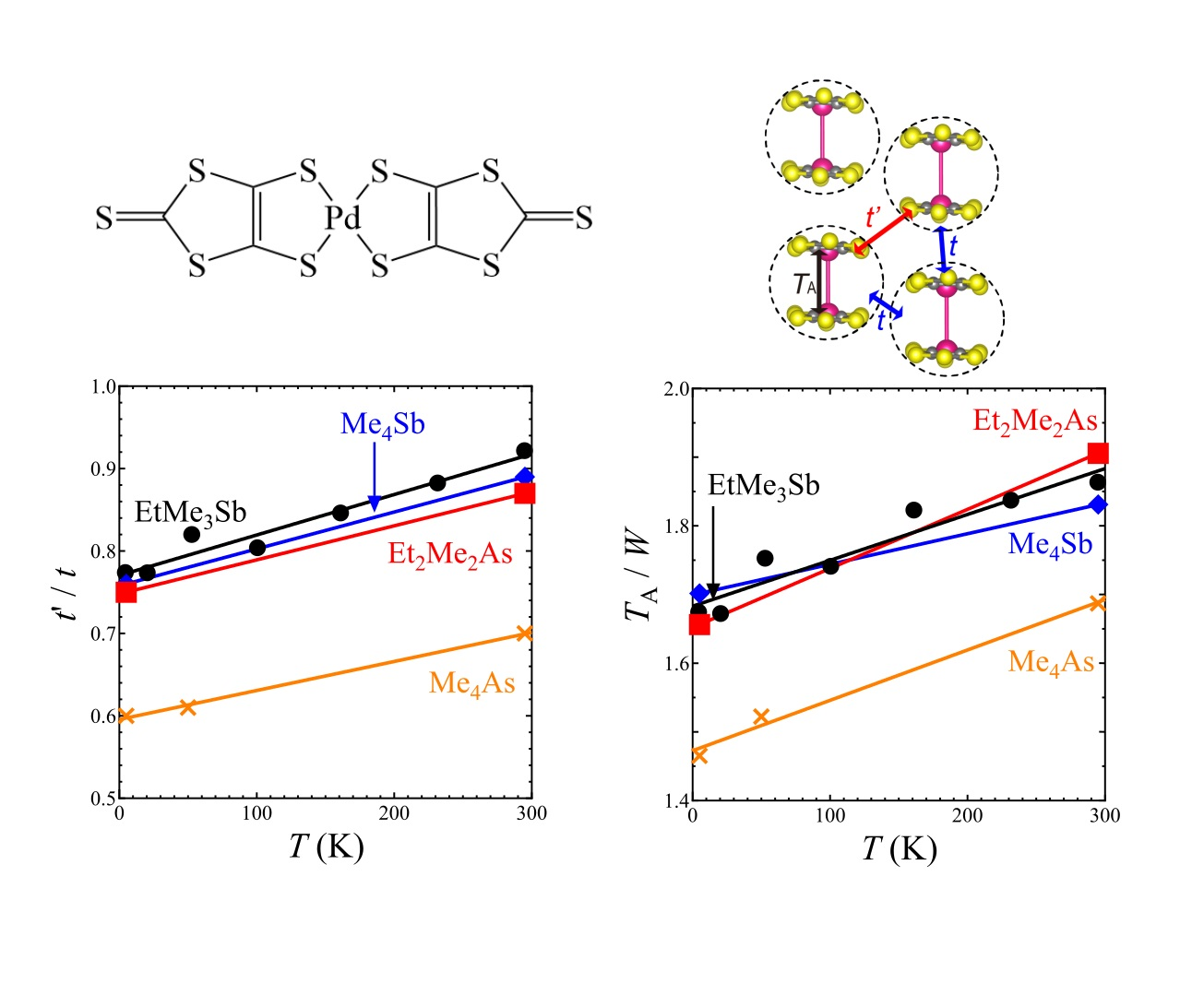
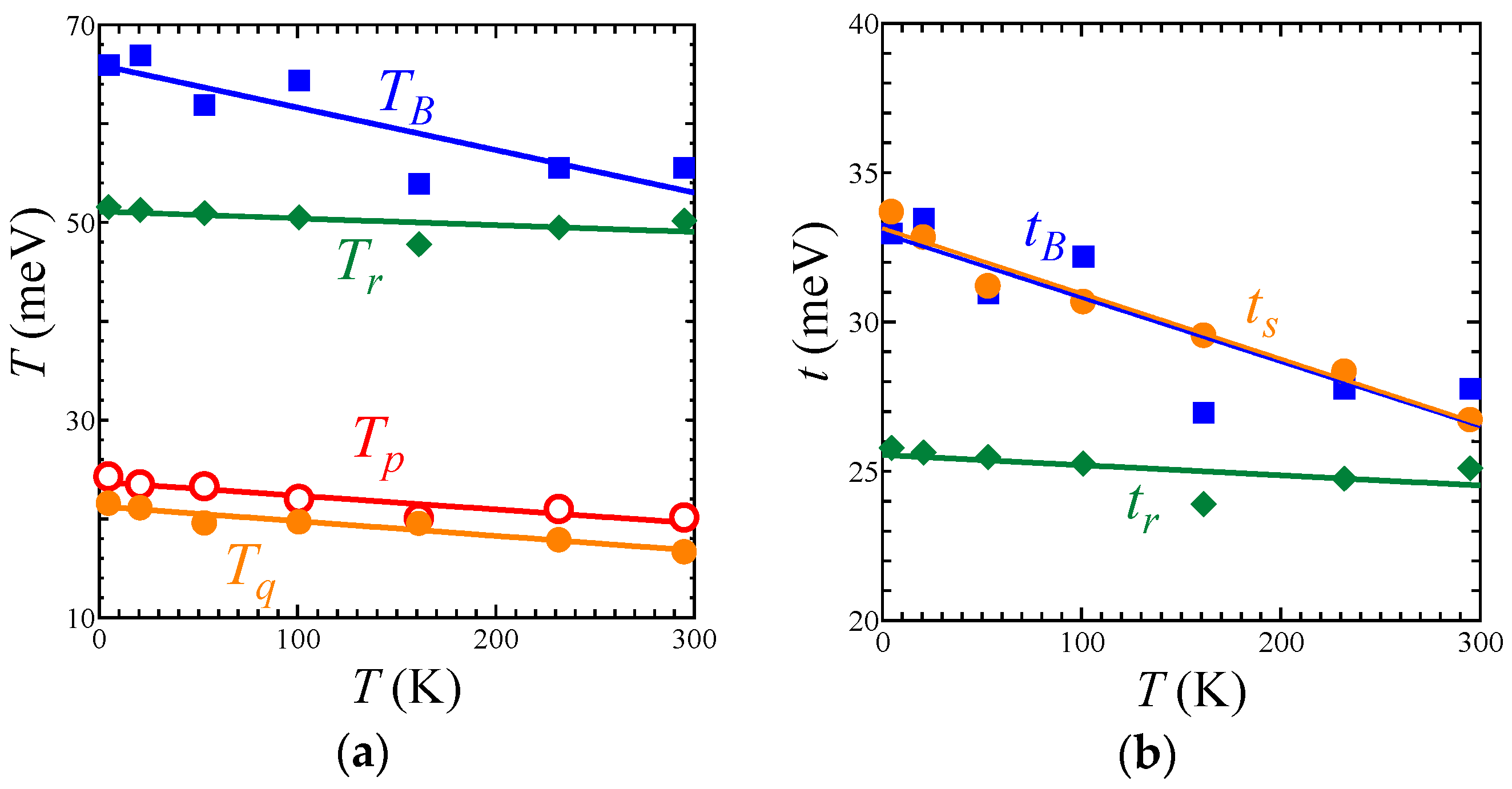
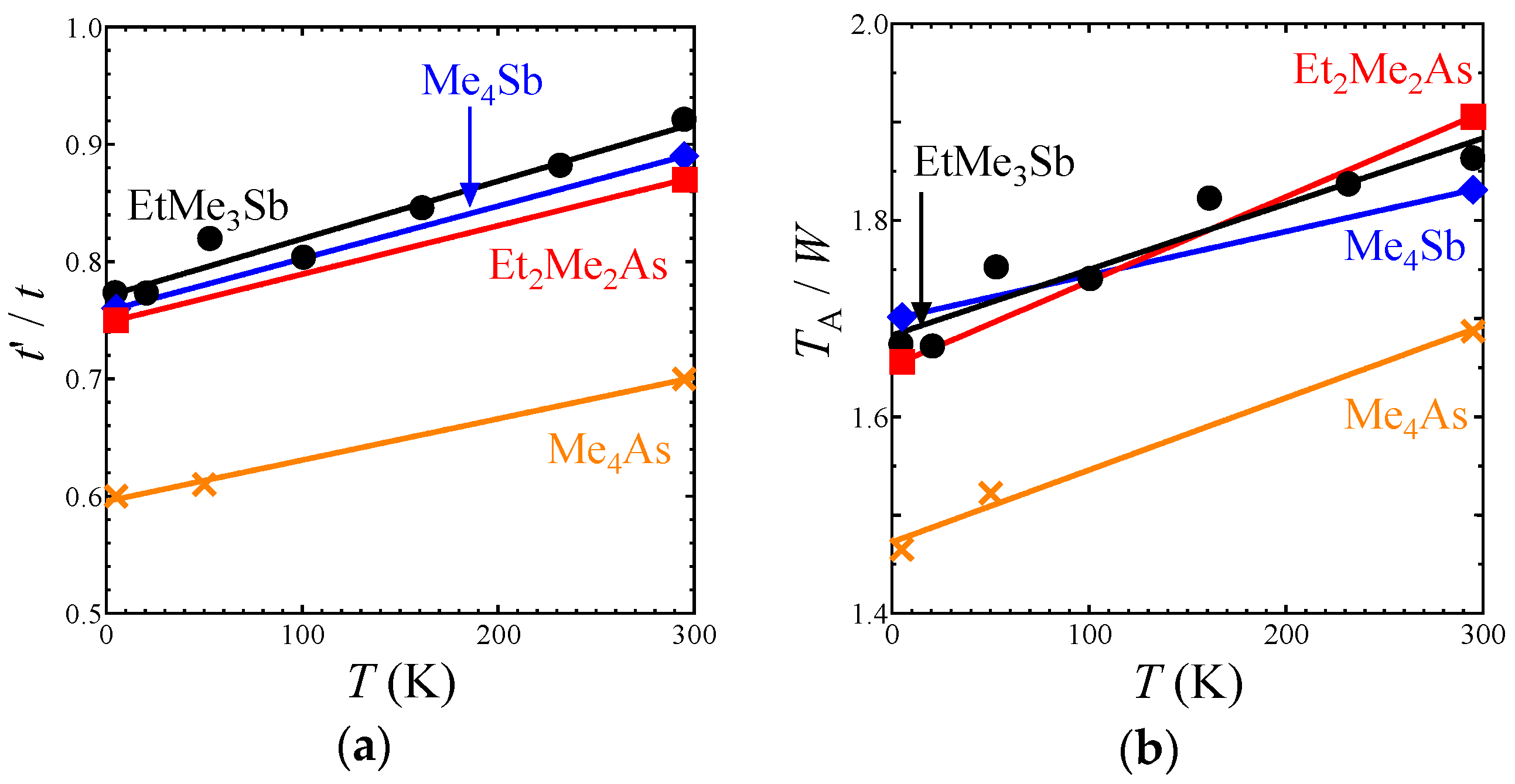
The EtMe3Sb salt showed no structural transition down to 5 K, that is, the space group and the essential molecular arrangement remained unchanged in the experimental temperature range. From room temperature to 5 K, the unit cell volume underwent a reduction by 3.2%. The temperature dependence of lattice parameters of the EtMe3Sb salt is shown in Figure S1. Crystal structures of the EtMe3Sb salt were determined with the R values of 5–9% at each temperature: 5.6% at 295 K, 5.7% at 232 K, 7.0% at 161 K, 6.4% at 101 K, 7.6% at 53 K, 8.1% at 21 K, and 8.9% at 5 K. Figure 2 shows the temperature dependence of the intermolecular and interdimer transfer integrals for the EtMe3Sb salt calculated by the extended Hückel method using obtained crystal structures. Every transfer integral increased almost linearly with lowering temperature. The tB, tS, and tr values changed similarly to each other, keeping the triangular lattice down to the lowest temperature. In contrast, the reduction of the cell volume by the cation effect resulted in a decrease of the tr value [3]. This suggested that the temperature effect on the interdimer transfer integrals differed from the cation effect. Among these three interdimer transfer integrals, the temperature dependence of tr was more moderate than those of others, and thus t′/t decreased by about 16% from the room temperature to 5 K (Figure 3a). That is, the triangular lattice in the EtMe3Sb salt became anisotropic with lowering temperature.
Intermolecular and interdimer transfer integrals for all salts are shown in Tables S1 and S2. Other salts with the AFLO ground state also showed no structural transition down to 5 K. The crystal structures of these salts were determined with R values of 4–7% at each temperature: 5.3% at 295 K and 6.0% at 5 K for the Me4Sb salt; 5.9% at 295 K, 4.0% at 53 K, and 5.7% at 5 K for the Me4As salt; and 5.7% at 295 K and 7.1% at 5 K for the Et2Me2As salt. Temperature dependences of the tB, tS, and tr values in these salts were similar to the case of the EtMe3Sb salt, and thus, the t′/t value of each salt decreased by about 15% from room temperature to 5 K (Figure 3a). This was in contrast to the case of another triangular lattice system based on κ-(ET)2X (ET = bis(ethylenedithio)tetrathiafulvalene, X = Cu2(CN)3, B(CN)4, Cu[N(NCS)2]), where the t′/t values increased with lowering temperatures [27].
The intradimer transfer integral TA is associated with the effective on-site Coulomb energy on the dimer unit as a rough approximation [28]. The ratio TA/W, where W is a bandwidth, gives an indication of the strength of the correlation effect. For all salts, the ratio decreased almost linearly with lowering temperature (Figure 3b), which suggests that the correlation effect in the β′-Pd(dmit)2 salts weakened with lowering temperature.
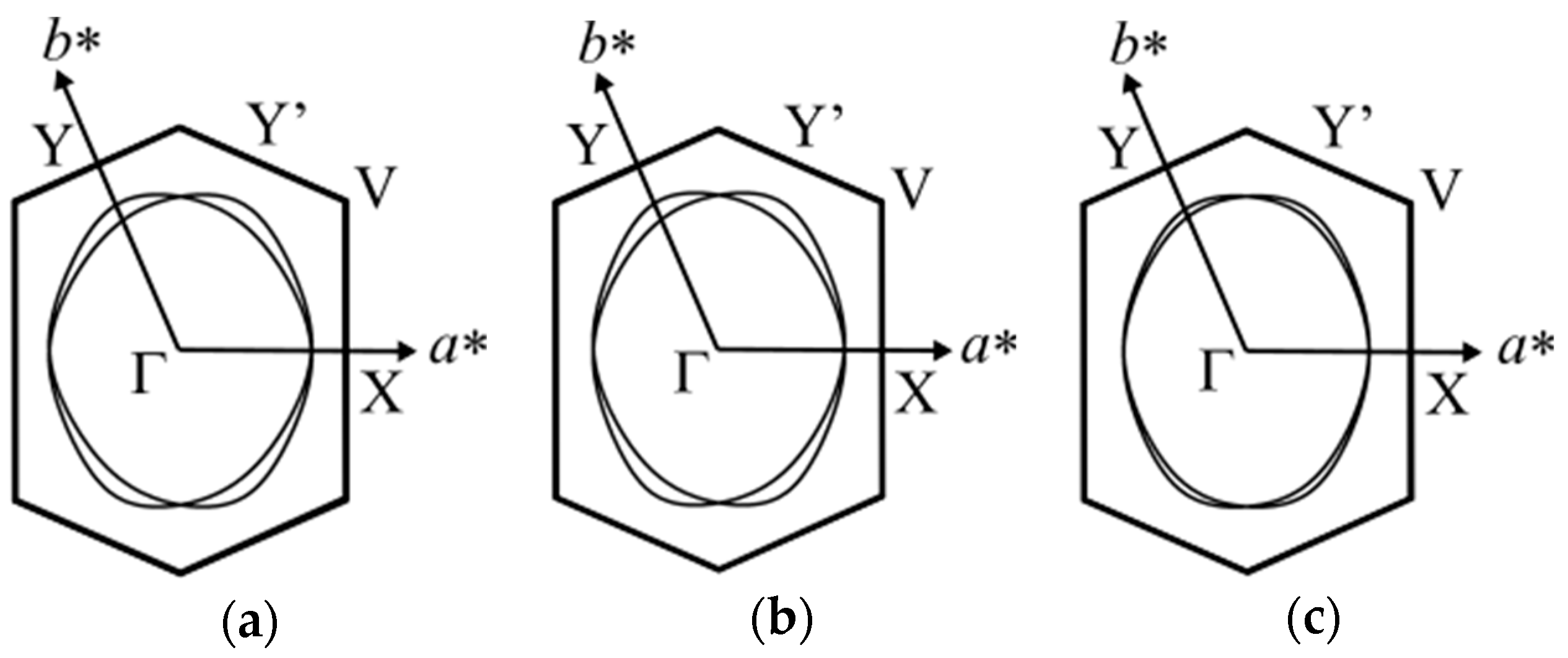
Figure 4 shows the calculated Fermi surface of the EtMe3Sb salt obtained by the tight-binding method (dimer model) using interdimer transfer integrals determined in this study at 5 K, 101 K, and 295 K [1]. This also shows the enhancement of the anisotropy of the triangular lattice in the low temperature region. Since the unit cell contains two crystallographically equivalent anion layers with different stacking directions [110] and (Figure 1d), the Fermi surface consists of two cylinders associated with these two anion layers. The Fermi surface at 295 K shows a nearly isotropic 2D nature represented by mutually superposed cylinders. As the anisotropy of the triangular lattice is enhanced due to the difference in the temperature dependence of transfer integrals (|dtS/dT|~|dtB/dT| > |dtr/dT)|, the Fermi surface becomes anisotropic with lowering temperatures as seen from a decrease in the overlapping portion of the Fermi surface.
We also performed the first-principles DFT calculations. The calculated Fermi surface and band structure are shown in Figure 5. Calculated band structures and band widths at 5 K, 101 K, and 295 K are shown in Figure S2 and Table S3. Concerning the Fermi surface, both calculation methods provided similar temperature dependence. This indicated that the dimer model using only HOMO-HOMO interactions could be used to describe the low-temperature electric structure of the EtMe3Sb salt around the Fermi level. On the other hand, since it is difficult to estimate the energy gap between HOMO and LUMO correctly with the extended Hückel method, the DFT calculations provided useful information on the arrangement of the HOMO and LUMO bands. In the tight binding model, the temperature-dependent interrelation between the HOMO and LUMO bands was explained by the dimerization gap (associated with the intradimer transfer integrals) and the band width (associated with the interdimer transfer integrals). The stronger dimerization enlarged the intradimer transfer integrals and made the anti-bonding HOMO band and the bonding LUMO band separate from each other. On the other hand, enlargement of the interdimer transfer integrals widened each band, leading to the band overlapping. The extended Hückel calculations indicated that the lowering of temperature induced the enlargement of both intra- and interdimer transfer integrals, which provided contrary effects on the band overlap. The first-principles DFT calculation showed that the Fermi level crossed the anti-bonding HOMO band that was separated from other bands at 295 K (Figure 5d). This conduction band becomes wider with lowering temperatures but remains isolated from the other bands even at the lowest temperature (5 K). That is, no band overlap occurred and the conduction band remained half-filled down to the lowest temperature. This indicated that the dimer model using only HOMO-HOMO interactions still worked to describe the electronic structure near the Fermi level of this system in the low-temperature region.
The DFT calculations provided other useful information. Figure 5e shows the partial density of states (PDOS) in the conduction band for the d-orbitals of Pd, the p-orbitals of S, and the p-orbitals of C in the dmit ligand at 5 K. This indicated that the p-orbitals of S played a dominant role in the band formation around the Fermi level. We also mention the possibility that the cations directly affected the electronic structure around the Fermi level. The energy levels of s- and p-orbitals of the C atoms in the EtMe3Sb cation were separated from the Fermi level by more than 3 eV. The PDOS of these C orbitals in the EtMe3Sb cation was less than 0.015 /eV in Figure 5e. Therefore, it is quite unlikely that the cation orbitals hybridize to the conduction band.
3.2. Temperature Dependence of Structural Parameters
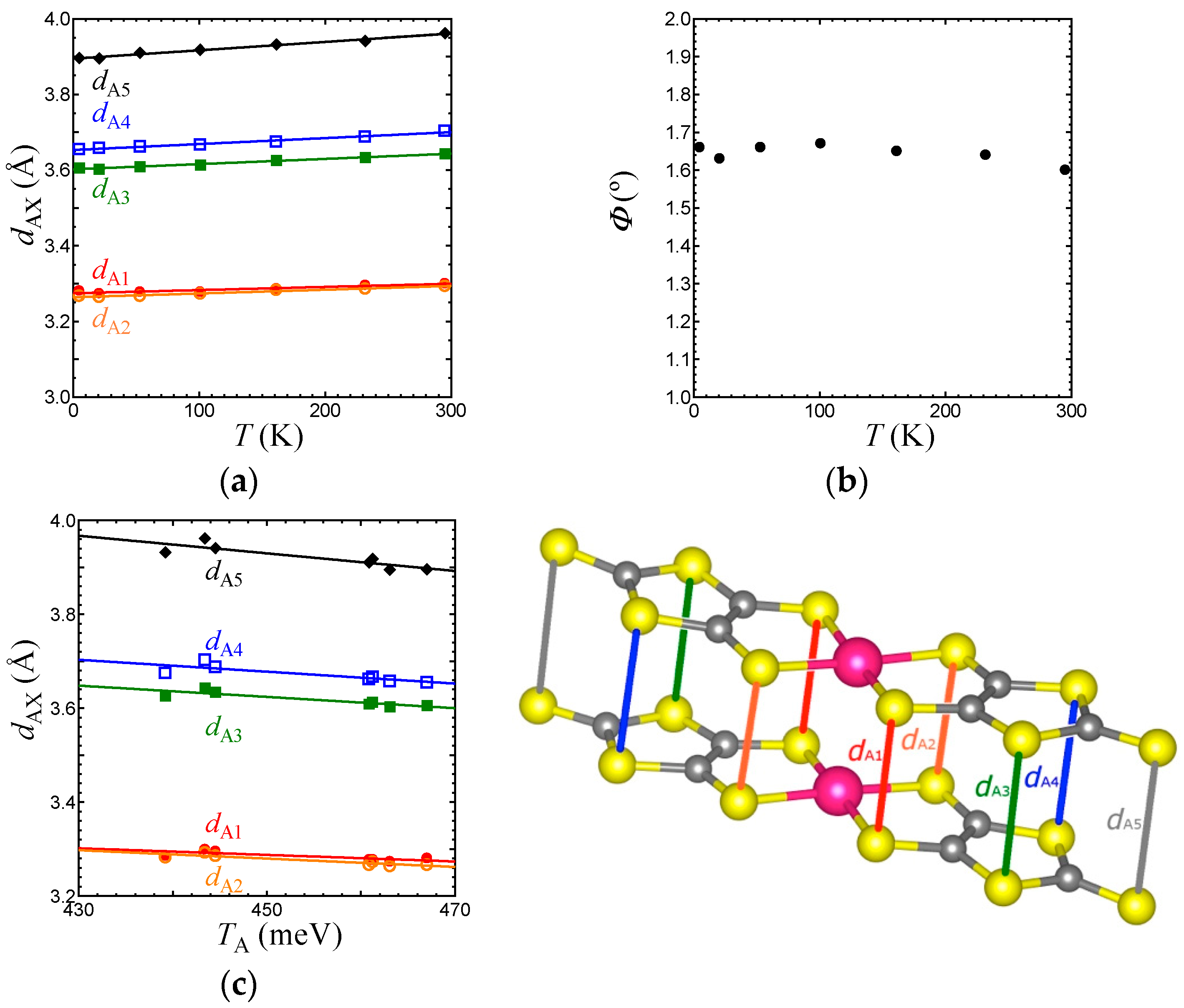
We now discuss the temperature dependence of structural parameters associated with the transfer integrals in the EtMe3Sb salt. As above-mentioned, the p-orbital in S governs the transfer integrals between HOMOs. Figure 6a,b show the temperature dependence of intradimer S⋯S distances (dA1–dA5), and the arch-shaped distortion angle Φ that characterizes the dimer structure. The S⋯S distances linearly decreased with lowering temperature. The Φ value did not show any significant changes. Figure 6c indicates a linear relation between the intradimer transfer integral TA and S⋯S distances dAX (X = 1–5), which suggests that TA is governed simply by the intradimer S⋯S distances. The arch-shaped molecular distortion plays an important role in the cation dependence of interdimer transfer integrals, tB, tS, and tr [3]. The linear relationship between Φ and interdimer transfer integrals was reported for all salts. Since Φ is independent of temperature, the temperature dependence of interdimer transfer integrals should originate in other mechanisms.
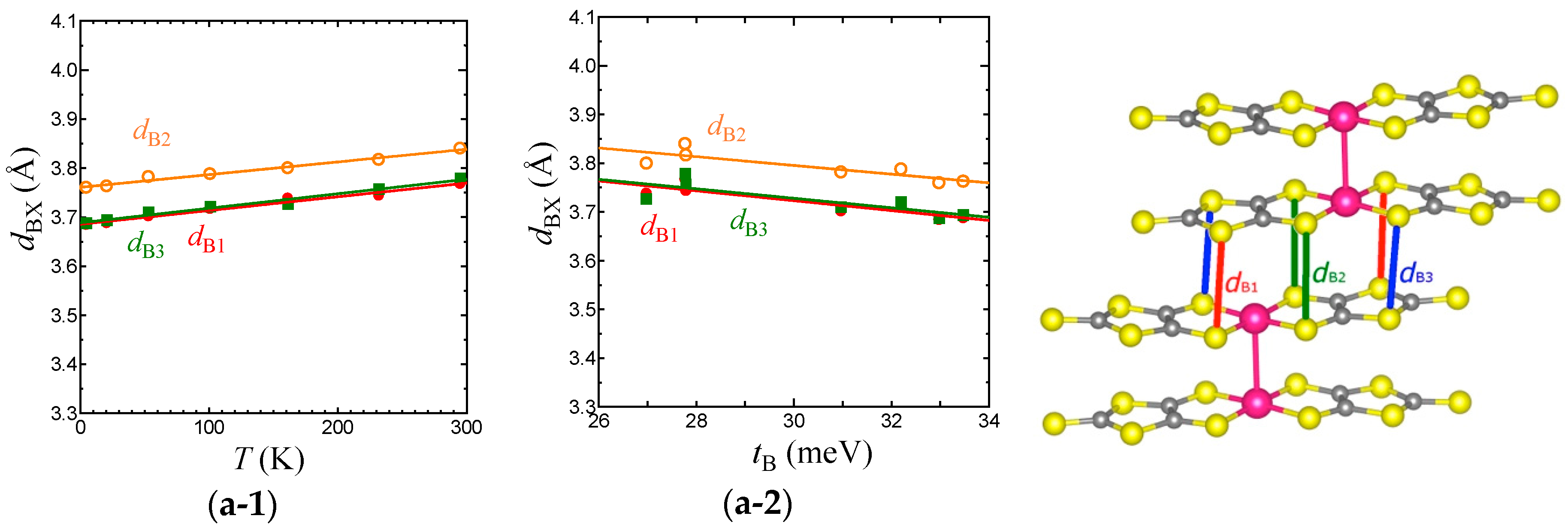
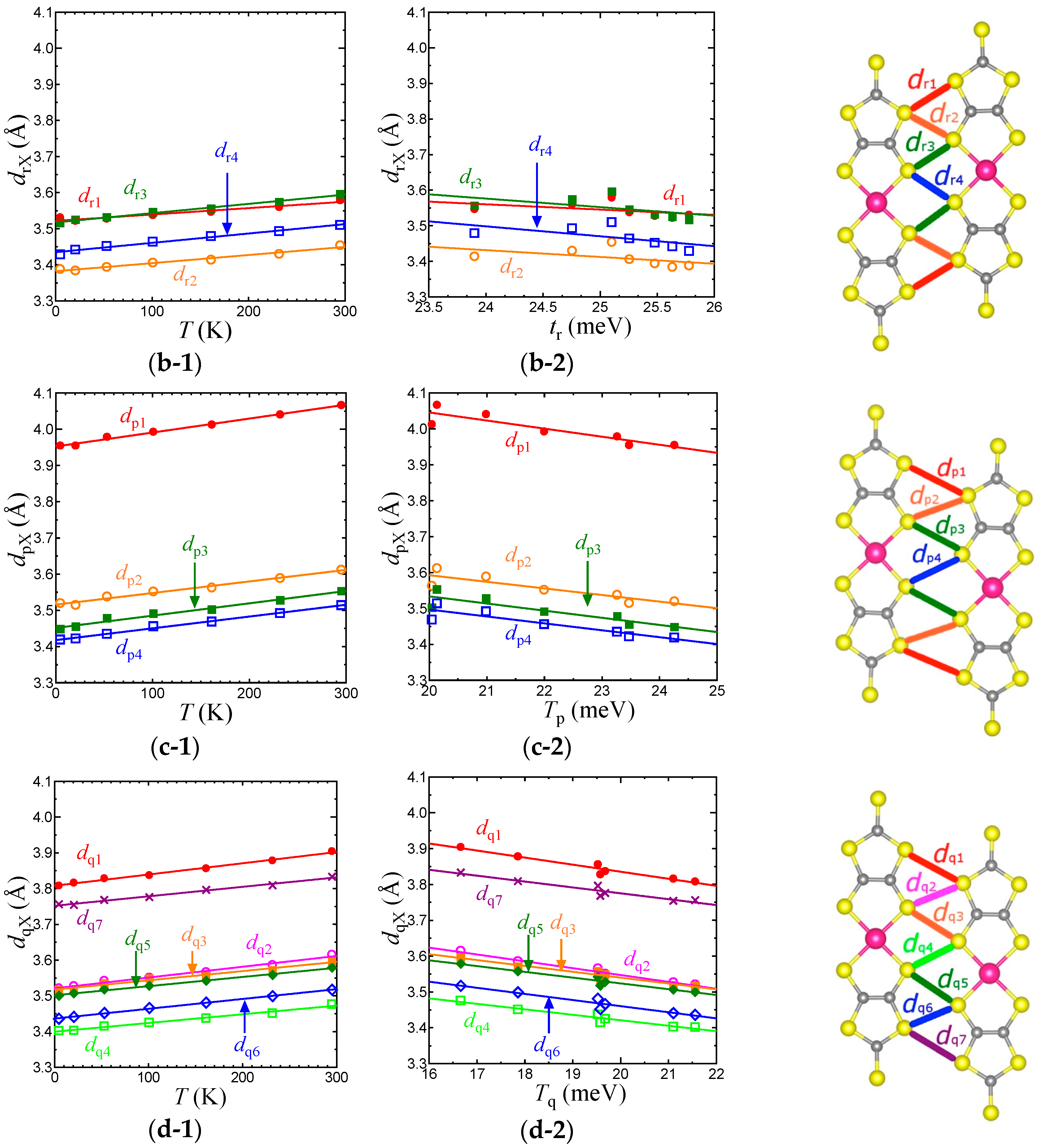
To consider the origin of the temperature dependence of the interdimer transfer integrals, tB, tS, and tr in the EtMe3Sb salt, we examined the intermolecular S⋯S distances, dBX, drX, dpX, and dqX that contribute to the intermolecular transfer integrals, TB, Tr, Tp, and Tq, respectively (Figure 7). Considering that the van der Waals radius of S is 1.85 Å, TB is associated with longer S⋯S distances (dBX) when compared with other intermolecular transfer integrals. Nevertheless, TB is the largest transfer integral among these four transfer integrals, because TB consists of mainly pσ⋯pσ contacts that give larger overlap integrals. On the other hand, all S⋯S distances that contribute to Tr (drX) are shorter than the van der Waals distance. Since Tr includes the pπ⋯pπ character as well as the pσ⋯pσ character, Tr is the second largest. Tp and Tq are associated with long and short S⋯S distances and exhibit both pσ⋯pσ and pπ⋯pπcharacters. Therefore, they are smaller than TB and Tr. The interdimer transfer integral tS, however, is expressed as a summation of Tq and Tp, tS = (2Tq + Tp)/2, and thus tS is comparable to tB (= TB/2). As a result, the triangular lattice with the relation of tB~tS > tr is formed in this system.
With lowering temperature, all intermolecular S⋯S distances were reduced linearly (Figure 7). From 295 K to 5 K, the amount of the reduction was about 0.1 Å for all cases, suggesting an almost isotropic thermal contraction of the anion layer. For each transfer integral, there existed a linear relationship with the corresponding S⋯S distances (Figure 7). Such linear relations for all the intermolecular S⋯S distances and the intermolecular transfer integrals were not observed in the case of cation dependence [3]. This also suggests that the temperature dependence of the transfer integrals was not due to the arch-shape distortion.
Each average of dBX, dpX, dqX, and drX (represented as , , , and ) decreased by 2.2%, 2.7%, 2.3%, and 1.6%, respectively, from room temperature to 5 K. The smallest variation of corresponded to a very moderate temperature dependence of tr. Although the reduction of was not so large, the enlargement of TB and thus tB was more intensive than those of other transfer integrals due to its pσ⋯pσ character. The variation of tS, which is the summation of three transfer integrals, was as large as that of tB. Consequently, the relationship between the variations, |dtS/dT|~|dtB/dT| > |dtr/dT|, led to the decrease of t′/t in the low temperature region.
Let us move on to the origin of the temperature dependence of TA/W (the indication of the electron correlation). The average of the intradimer S⋯S distances dAX () decreased by about 1.1% from room temperature to 5 K. This variation was about half as large as those of the interdimer S⋯S distances, , , and . Since the Pd(dmit)2 molecules are tightly dimerized, the intradimer S⋯S distances are supposed to change differently from the interdimer S⋯S distances with lowering temperature. On the other hand, the band width is associated with the interdimer transfer integrals. From room temperature to 5 K, the TA value increased by 5%, while the band width W increased by 17%, which resulted in the decrease of TA/W. Strong dimerization is a common feature of the β′-Pd(dmit)2 salts, thus, each salt showed a similar temperature dependence of the TA/W value.
Considering the temperature dependence of the structural parameters that related to the band parameters in the EtMe3Sb salt, we concluded that the temperature dependence of the anisotropy of the triangular lattice (t′/t), one of the most important parameters in this frustrated Mott system, primarily originated from the almost isotropic thermal contraction of the anion layer. This is a common feature of the β′-Pd(dmit)2 salts. In this frustrated Mott system with the triangular lattice of the strongly dimerized units, the electronic structure became anisotropic and the electron correlation was reduced with lowering temperatures.
3.3. Cation-Anion Interactions
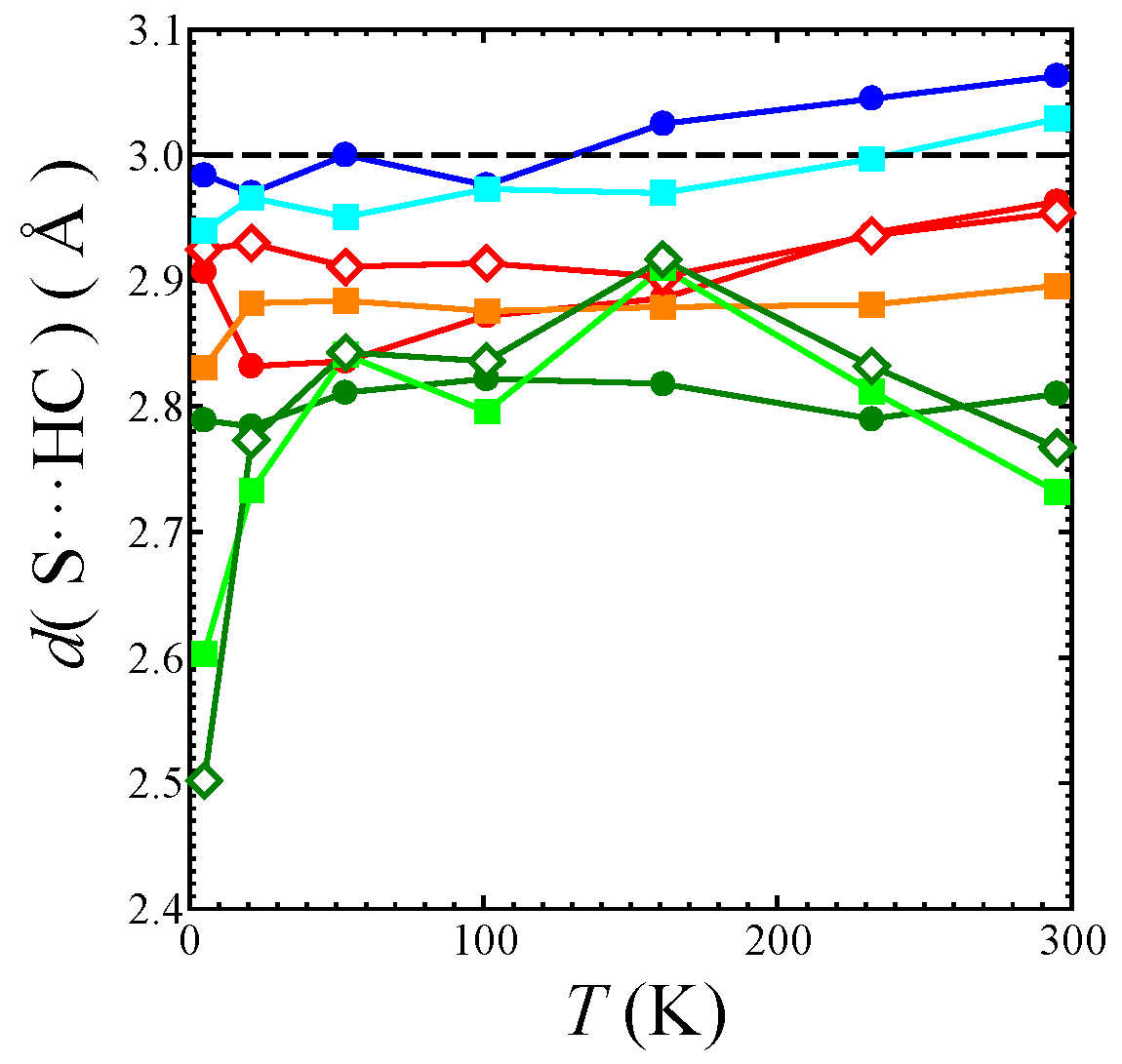
In the β′-Pd(dmit)2 salts, the counter cation strongly affects the anisotropy of the triangular lattice [3]. In addition, it has been suggested that the cation-anion interaction plays an important role in realizing the QSL state in the κ-type ET salts [29,30,31]. Therefore, we discuss the cation-anion interaction in the β′-Pd(dmit)2 salts, especially the S⋯H-C short contacts between the terminal thioketone group of the Pd(dmit)2 molecule and the hydrogen atom in the cation. In this study, the torsion angles of the -CH3 unit were refined with the AFIX 137 command in the refinement process with SHELX. Refined torsion angles are those in the staggered conformation except for in the EtMe3Sb salt at 5 K (vide infra). The number of short S⋯H-C contacts (<3.0 Å: sum of the van der Waals radii of S atom and H atom) in the Me4As, Me4Sb, Et2Me2As, and EtMe3Sb salts are summarized in Table 2. Figure 8 shows the temperature dependence of the short S⋯H-C contacts in the EtMe3Sb salt. Two S⋯H-C9 contacts were much shorter at 5 K than at other temperatures. These very short contacts are caused by an anomalous torsion angle of the -CH3 unit. The refined torsion angle of this -CH3 unit at 5 K was about 30°, which was far from the angle in the staggered conformation.
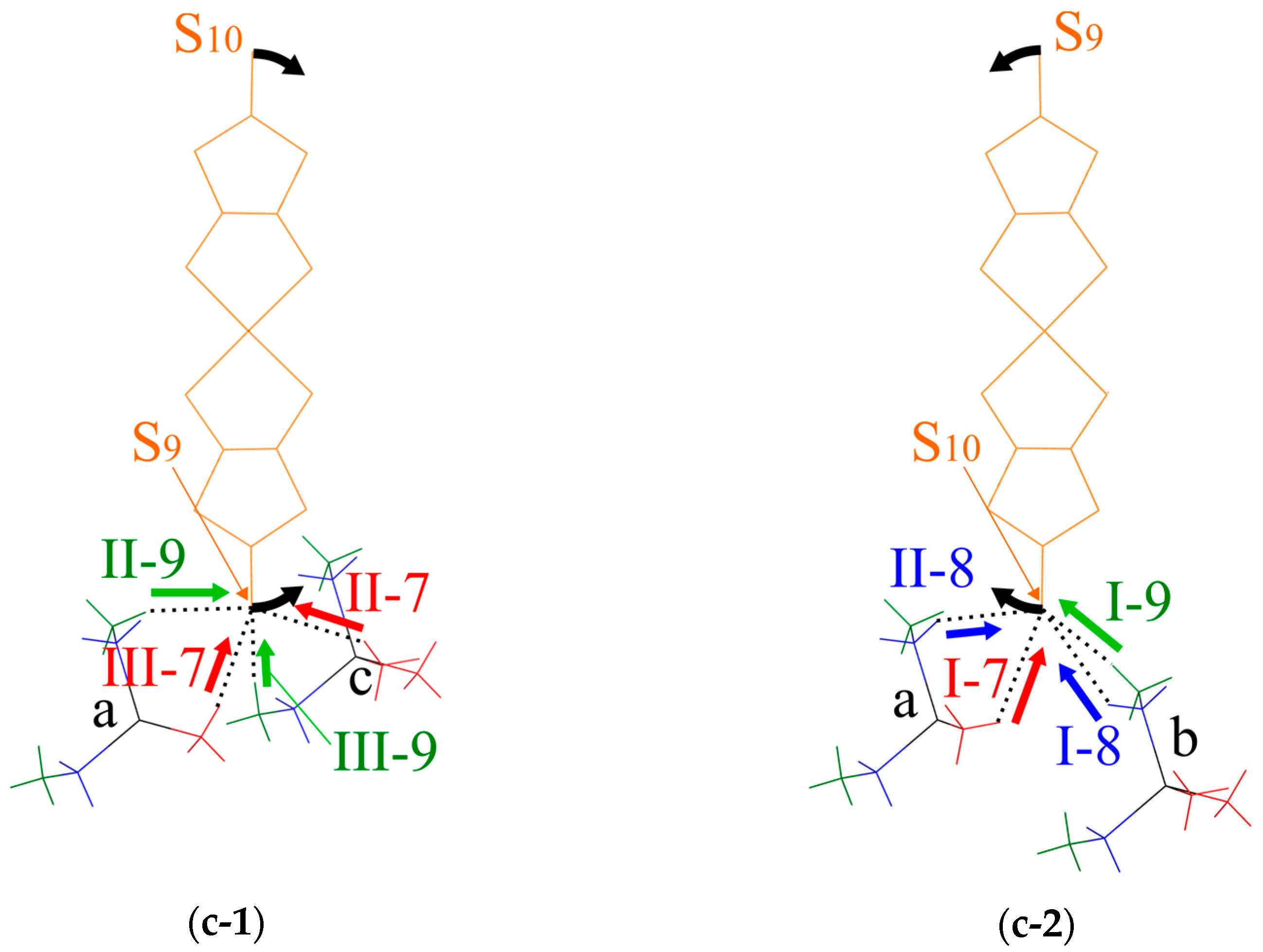
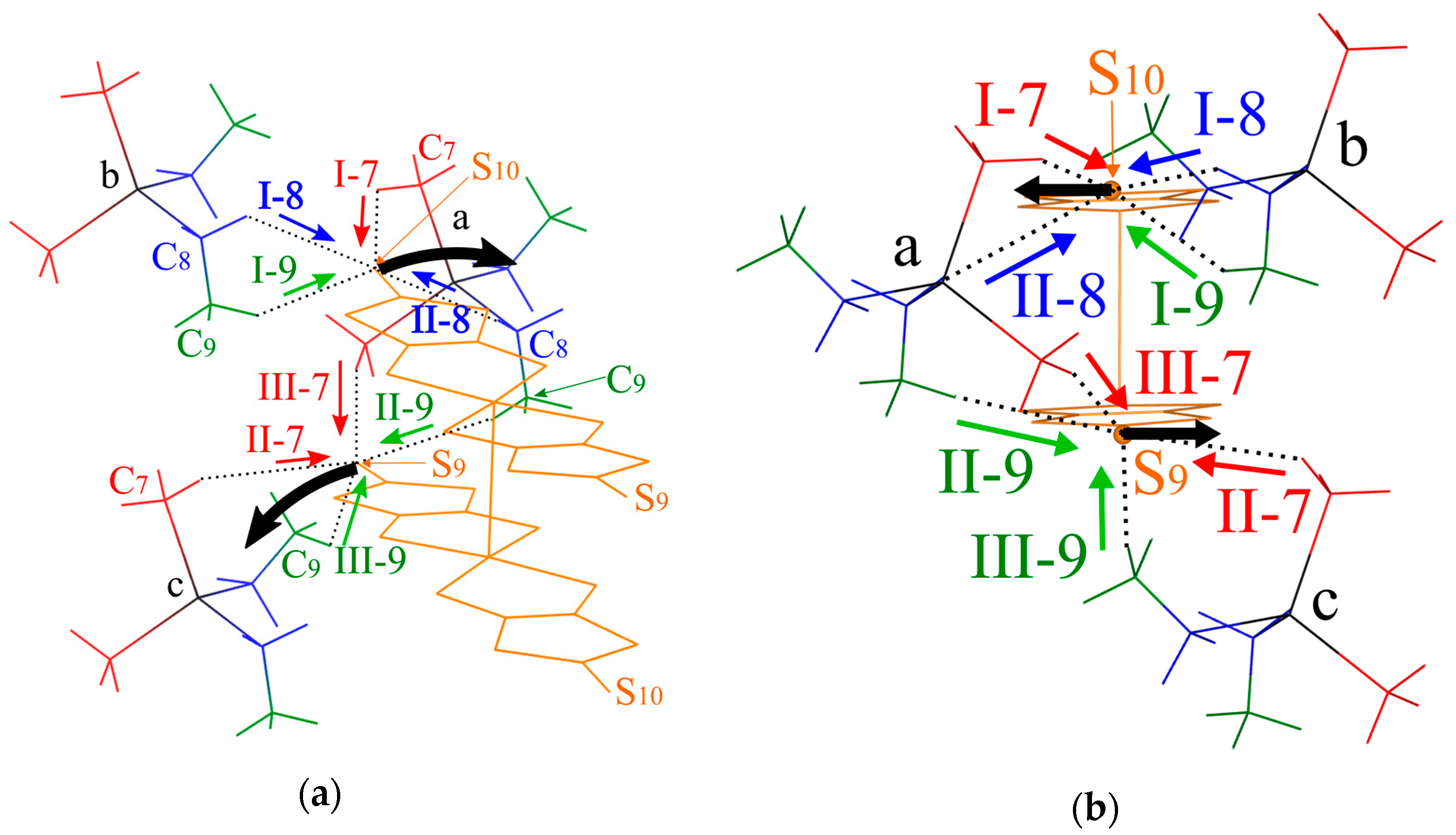
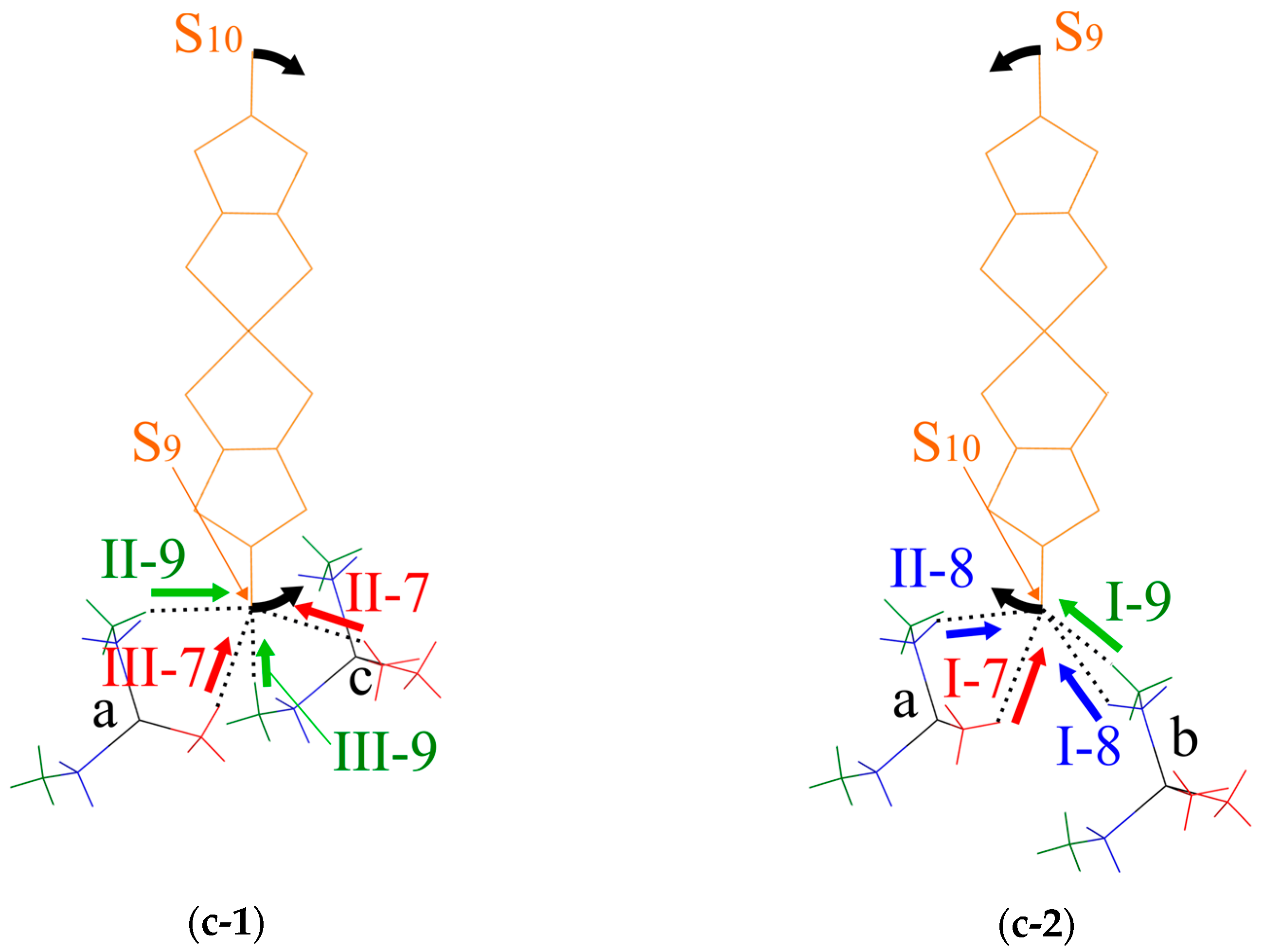
The S⋯H-C contacts in the EtMe3Sb salt are shown in Figure 9. Two Pd(dmit)2 molecules in the dimer are related by the inversion center, and there are two crystallographically independent terminal S atoms (S9 and S10). These S atoms contact three cations (labeled as Figure 9a–c) through eight S⋯H-C contacts (three S⋯H-C7 contacts, I-7, II-7, and III-7; two S⋯H-C8 contacts, I-8 and II-8; three S⋯H-C9 contacts, I-9, II-9, and III-9 in Figure 9) in the EtMe3Sb salt. Cation a contacts two terminal S atoms (Figure 9), and each of the cations b and c contact one S atom. Such features of the cation-anion contacts are commonly observed in the β′-Pd(dmit)2 salts.
We discuss how the S⋯H-C contacts affect the arch-shaped distortion of the Pd(dmit)2 molecule by considering a contact angle: the S⋯H-C interaction that can affect the Φ angle should have a component parallel to the displacement of the S atom in the distortion. The S⋯H-C9 contacts associated with the ethyl group are rather short in the EtMe3Sb salt. These tight cation-anion interactions would dominate the arch-shape distortion in the β′-Pd(dmit)2 salts containing the ethyl groups. When the S⋯H-C interaction was repulsive/attractive, two S⋯H-C9 contacts (I-9 and II-9 in Figure 9) effectively made the Φ angle large/small. As shown in Figure 9b,c-1, the III-9 contact was almost perpendicular to the displacement of the S atom in the arch-shaped distortion, thus, the effect of this contact on the Φ angle was negligible. In the EtxMe4-xZ salts, the Φ angle became larger by increasing the number of the ethyl group x [3]. Accordingly, the S⋯H-C9 contact angle and the x dependence of Φ indicated that the S⋯H-C9 contacts appeared, effectively, repulsive.
In the Me4P and Me4As salts where the cation did not contain the ethyl groups, the Φ angle was negative. Assuming that the S⋯H-C7 and S⋯H-C8 interactions were also repulsive, the I-7, II-7, and II-8 contacts decreased the Φ angle and the III-7 and I-8 contacts increased the Φ angle (Figure 9). The II-7 and III-7 contacts pushed the S9 atom, and they acted on the Φ angle oppositely. Thus, the distortion through the S9 atom was determined by the competition of these two contacts in the Me4Z salts. The II-7 contact was nearly along the displacement of the S atom, whereas the III-7 contact had less components parallel to the displacement. This difference in the contact angle was expected to provide the negative Φ value in the Me4P and Me4As salts. A similar mechanism involving the I-7, I-8. and II-8 contacts could be applied to the distortion through the S10. Note that the I-8 contact that increased the Φ angle was longer and the II-8 contact that decreased the Φ value was shorter in the Me4As salt than in the Me4Sb salt. These structural features were consistent with the smaller Φ value in the Me4As salt than that in the Me4Sb salt [3].
4. Conclusions
The crystal structures of quasi-triangular frustrated S = 1/2 system, the β′-Pd(dmit)2 salts, were determined in the range of 5–295 K by the single crystal X-ray diffraction technique. Band parameters, Fermi surface, and band structures were calculated by the tight binding calculation based on the extended Hückel method and the first-principles DFT calculation using the obtained crystal structures. In the QSL material, the EtMe3Sb salt, every interdimer transfer integral, tB, tS, and tr lineally increased with lowering temperatures. The change of tr was more moderate than those of tB and tS, thus the anisotropy of the triangular lattice was enhanced by lowering temperatures. Interdimer transfer integrals in the Me4Sb, Me4As and Et2Me2As salts also increased with lowering temperature, and the triangular lattice became anisotropic in the low temperature region. It was quite interesting that the QSL state in the EtMe3Sb salt was realized in the rather anisotropic triangular lattice system where the spin frustration would be reduced. Recently, vibrational spectroscopy has revealed that charge and lattice fluctuations associated with the formation of closed shell tetramers and octamers can suppress the magnetic order [32].
At each temperature, the Fermi surface calculated by the tight binding method reproduced the results of the first-principles DFT calculation. The conduction band, which originated from the anti-bonding HOMO pair, remained isolated from other bands and half-filled even at 5 K. These results indicated that the dimer picture based on only HOMO-HOMO interactions was still available down to the low temperature region.
Since the p-orbitals of the S atom mainly contributed to the formation of the conduction band, the intermolecular S⋯S contacts were closely related to the band parameters. In the cation effect on the intermolecular transfer integrals, the arch-shaped distortion of the Pd(dmit)2 molecule tuned the intermolecular S⋯S contacts. In the temperature effect, however, the S–Pd–Pd–S torsion angle, Φ, which characterizes the arch-shaped molecular distortion, was almost temperature-independent, and the intermolecular transfer integrals were linearly correlated to the intermolecular S⋯S distances. All these results suggested that the temperature dependence of the band parameters was mainly associated with the thermal contraction of the Pd(dmit)2 layer.
The cation-anion interactions through the short S⋯H-C contacts acted repulsively and induced the arch-shaped distortion. This was categorized as the steric effect, and we did not detect any electronic effect of the cation-anion interactions. In this sense, it should be noted that the electronic fluctuations by the rotations of the methyl and ethyl groups in the cation did not directly induce the enhancement of the dielectric constant in the EtMe3Sb salt, which was revealed by the recent NMR measurements [33].
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4352/8/3/138/s1. Figure S1: Temperature dependence of lattice constants in the EtMe3Sb salt; Figure S2: Band structures of the EtMe3Sb salt calculated with the first-principles DFT method; Table S1: Intermolecular transfer integrals in (Cation)[Pd(dmit)2]2 calculated by using the extended Hückel method; Table S2: Interdimer transfer integrals, t′/t, and band widths in (Cation)[Pd(dmit)2]2 calculated by using the extended Hückel method; Table S3: Band width of the conduction band, W, in the EtMe3Sb salt calculated with the first-principles DFT method.
Acknowledgments
This work was supported by the JSPS KAKENHI Grants, JP16H06346 and JP16K17756. The first-principles calculations were performed by the RIKEN supercomputer Hokusai GreatWave and NIMS Numerical Materials Simulator.
Author Contributions
All authors conceived and designed the experiments; Kohei Ueda performed crystal structure analyses; Kohei Ueda and Reizo Kato performed the extended Hückel and the tight binding calculations; Takao Tsumuraya performed the first-principles DFT calculations; Reizo Kato contributed reagents/materials; Kohei Ueda and Reizo Kato wrote the paper with inputs from all authors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kato, R. Conducting Metal Dithiolene Complexes: Structural and Electronic Properties. Chem. Rev. 2004, 104, 5319–5346. [Google Scholar] [CrossRef] [PubMed]
- Kanoda, K.; Kato, R. Mott Physics in Organic Conductors with Triangular Lattices. Ann. Rev. Condens. Matter Phys. 2011, 2, 167–188. [Google Scholar] [CrossRef]
- Kato, R.; Hengbo, C. Cation Dependence of Crystal Structure and Band Parameters in a Series of Molecular Conductors, β′-(Cation)[Pd(dmit)2]2 (dmit = 1,3-dithiole-2-thione-4,5-dithiolate). Crystals 2012, 2, 861–874. [Google Scholar] [CrossRef]
- Kato, R. Development of π-Electron Systems Based on [M(dmit)2] (M = Ni and Pd; dmit: 1,3-dithiole-2-thione-4,5-dithiolate) Anion Radicals. Bull. Chem. Soc. Jpn. 2014, 87, 355–374. [Google Scholar] [CrossRef]
- Kato, R.; Liu, Y.-L.; Hosokoshi, Y.; Aonuma, S.; Sawa, H. Se-Substitution and Cation Effects on the High-Pressure Molecular Superconductor, β-Me4N[Pd(dmit)2]2-A Unique Two-Band System. Mol. Cryst. Liq. Cryst. 1997, 296, 217–244. [Google Scholar] [CrossRef]
- Mori, T.; Kobayashi, A.; Sasaki, Y.; Kobayashi, H.; Saito, G.; Inokuchi, H. The Intermolecular Interaction of Tetrathiafulvalene and Bis(ethylenedithio)tetrathiafulvalene in Organic Metals. Calculation of Orbital Overlaps and Models of Energy-band Structures. Bull. Chem. Soc. Jpn. 1984, 57, 627–633. [Google Scholar] [CrossRef]
- Canadell, E.; Rachidi, I.E.-I.; Ravy, S.; Pouget, J.-P.; Brossard, L.; Legros, J.-P. On the band electronic structure of X [M (dmit)2]2 (X = TTF, (CH3)4N; M = Ni, Pd) molecular conductors and superconductors. J. Phys. 1989, 50, 2967–2981. [Google Scholar] [CrossRef]
- Tamura, M.; Kato, R. Magnetic Susceptibility of β′-[Pd(dmit)2] salts (dmit = 1,3-dithiol-2-thione-4,5-dithiolate, C3S5): Evidence for Frustration in Spin-1/2 Heisenberg Antiferromagnets on a Triangular Lattice. J. Phys. Condens. Matter 2002, 14, L729–L734. [Google Scholar] [CrossRef]
- Nakamura, T.; Takahashi, T.; Aonuma, S.; Kato, R. EPR Investigation of the Electronic States in β′-type [Pd(dmit)2]2 Compounds (where dmit is 2-thioxo-1,3-dithiole-4,5-dithiolate). J. Mater. Chem. 2001, 11, 2159–2162. [Google Scholar] [CrossRef]
- Otsuka, K.; Iikubo, H.; Kogure, T.; Takano, Y.; Hiraki, K.I.; Takahashi, T.; Cui, H.; Kato, R. Antiferromagnetic Ordering in Quasi-Triangular Localized Spin System, β′-Et2Me2P[Pd(dmit)2]2, Studied by 13C NMR. J. Phys. Soc. Jpn. 2014, 83, 054712. [Google Scholar] [CrossRef]
- Ohira, S.; Tamura, M.; Kato, R.; Watanabe, I.; Iwasaki, M. Muon Spin Relaxation Study of the Magnetic Transition in a Two-Dimensional Distorted Triangular Lattice β′-(CH3)4P[Pd(dmit)2]2. Phys. Rev. B 2004, 70, 220404. [Google Scholar] [CrossRef]
- Ohira, S.; Tamura, M.; Kato, R.; Iwasaki, M. Magnetic Order and Charge Separation in 2D Distorted Triangular Lattice Systems β′-X[Pd(dmit)2]2. Phys. B 2006, 374–375, 122–125. [Google Scholar] [CrossRef]
- Itou, T.; Oyamada, A.; Maegawa, S.; Tamura, M.; Kato, R. Quantum Spin Liquid in the Spin-1/2 Triangular Antiferromagnet EtMe3Sb[Pd(dmit)2]2. Phys. Rev. B 2008, 77, 104413. [Google Scholar] [CrossRef]
- Itou, T.; Oyamada, A.; Maegawa, S.; Kato, R. Instability of a Quantum Spin Liquid in an Organic Triangular-Lattice Antiferromagnet. Nat. Phys. 2010, 6, 673–676. [Google Scholar] [CrossRef]
- Nakao, A.; Kato, R. Structural Study of Low Temperature Charge-Separated Phases of Pd(dmit)2-Based Molecular Conductors. J. Phys. Soc. Jpn. 2005, 74, 2754–2763. [Google Scholar] [CrossRef]
- Tamura, M.; Takenaka, K.; Takagi, H.; Sugai, S.; Tajima, A.; Kato, R. Spectroscopic evidence for the low-temperature charge-separated state of [Pd(dmit)2] salts. Chem. Phys. Lett. 2005, 411, 133–137. [Google Scholar] [CrossRef]
- Shimizu, Y.; Miyagawa, K.; Kanoda, K.; Maesato, M.; Saito, G. Spin Liquid State in an Organic Mott Insulator with a Triangular Lattice. Phys. Rev. Lett. 2003, 91, 107001. [Google Scholar] [CrossRef] [PubMed]
- Isono, T.; Kamo, H.; Ueda, A.; Takahashi, K.; Kimata, M.; Tajima, H.; Tsuchiya, S.; Terashima, T.; Uji, S.; Mori, H. Gapless Quantum Spin Liquid in an Organic Spin-1/2 Triangular-Lattice κ-H3(Cat-EDT-TTF)2. Phys. Rev. Lett. 2014, 112, 177201. [Google Scholar] [CrossRef] [PubMed]
- Rouzière, S.; Yamaura, J.-I.; Kato, R. Low-temperature structural studies of molecular β′-Pd(dmit)2 conductors. Phys. Rev. B 1999, 60, 3113–3119. [Google Scholar] [CrossRef]
- Tsumuraya, T.; Seo, H.; Tsuchiizu, M.; Kato, R.; Miyazaki, T. Cation Dependence of the Electronic States in Molecular Triangular Lattice System β′-X[Pd(dmit)2]2: A First-Principles Study. J. Phys. Soc. Jpn. 2013, 82, 033709. [Google Scholar] [CrossRef]
- Nakamura, K.; Yoshimoto, Y.; Imada, M. Ab initio two-dimensional multiband low-energy models of EtMe3Sb[Pd(dmit)2]2 and κ-(BEDT-TTF)2Cu(NCS)2 with comparisons to single-band models. Phys. Rev. B 2012, 86, 205117. [Google Scholar] [CrossRef]
- Wimmer, E.; Krakauer, H.; Weinert, M.; Freeman, A.J. Full-potential self-consistent linearized-augmented-plane-wave method for calculating the electronic structure of molecules and surfaces: O2 molecule. Phys. Rev. B 1981, 24, 864–875. [Google Scholar] [CrossRef]
- Weinert, M. Solution of Poisson’s equation: Beyond Ewald-type methods. J. Math. Phys. 1981, 22, 2433–2439. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223–16234. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Ito, H.; Maesato, M.; Shimizu, Y.; Hayama, H.; Hiramatus, T.; Nakamura, Y.; Kishida, H.; Koretsune, T.; Hotta, C.; et al. Spin-disordered quantum phases in a quasi-one-dimensional triangular lattice. Nat. Phys. 2015, 11, 679–683. [Google Scholar] [CrossRef]
- Tamura, M.; Kato, R. Effective on-site repulsion in molecular conductors with dimeric structure: Is the transfer integral a good measure of correlation. J. Phys. Soc. Jpn. 2004, 73, 3108–3110. [Google Scholar] [CrossRef]
- Pinterić, M.; Čulo, M.; Milat, O.; Basletić, M.; Korin-Hamzić, B.; Tafra, E.; Hamzić, A.; Ivek, T.; Peterseim, T.; Miyagawa, K.; et al. Anisotropic charge dynamics in the quantum spin-liquid candidate κ-(BEDT-TTF)2Cu2(CN)3. Phys. Rev. B 2014, 90, 195139. [Google Scholar] [CrossRef]
- Dressel, M.; Lazić, P.; Pustogow, A.; Zhukova, E.; Gorshunov, B.; Schlueter, J.A.; Milat, O.; Gumhalter, B.; Tomić, S. Lattice vibrations of the charge-transfer salt κ-(BEDT-TTF)2Cu2(CN)3: Comprehensive explanation of the electrodynamic response in a spin-liquid compound. Phys. Rev. B 2016, 93, 081201. [Google Scholar] [CrossRef]
- Pinterić, M.; Lazić, P.; Pustogow, A.; Ivek, T.; Kuveždić, M.; Milat, O.; Gumhalter, B.; Basletić, M.; Čulo, M.; Korin-Hamzić, B.; et al. Anion effects on electronic structure and electrodynamic properties of the Mott insulator κ-(BEDT-TTF)2Ag2(CN)3. Phys. Rev. B 2016, 94, 161105. [Google Scholar] [CrossRef]
- Yamamoto, T.; Fujimoto, T.; Naito, T.; Nakazawa, Y.; Tamura, M.; Yakushi, K.; Ikemoto, Y.; Moriwaki, T.; Kato, R. Charge and Lattice Fluctuations in Molecule-Based Spin Liquids. Sci. Rep. 2017, 7, 12930. [Google Scholar] [CrossRef] [PubMed]
- Fujiyama, S.; Kato, R. Algebraic Charge Dynamics of the Quantum Spin Liquid β’-EtMe3Sb[Pd(dmit)2]2. Phys. Rev. B 2018, 97, 035131. [Google Scholar] [CrossRef]
Figure 1.
(a) Molecular structure of Pd(dmit)2. (b) Schematic illustration of orbital levels for monomer and dimer. (c) and (d) Crystal structure of β′-EtMe3Sb[Pd(dmit)2]2. End-on projections of the anion layer with notations of (e) intermolecular transfer integrals and (f) interdimer transfer integrals. (g) An arch-shaped distortion of the Pd(dmit)2 molecule. The torsion angle of S–Pd–Pd–S, Φ, characterizes the degree of the distortion.
Figure 1.
(a) Molecular structure of Pd(dmit)2. (b) Schematic illustration of orbital levels for monomer and dimer. (c) and (d) Crystal structure of β′-EtMe3Sb[Pd(dmit)2]2. End-on projections of the anion layer with notations of (e) intermolecular transfer integrals and (f) interdimer transfer integrals. (g) An arch-shaped distortion of the Pd(dmit)2 molecule. The torsion angle of S–Pd–Pd–S, Φ, characterizes the degree of the distortion.
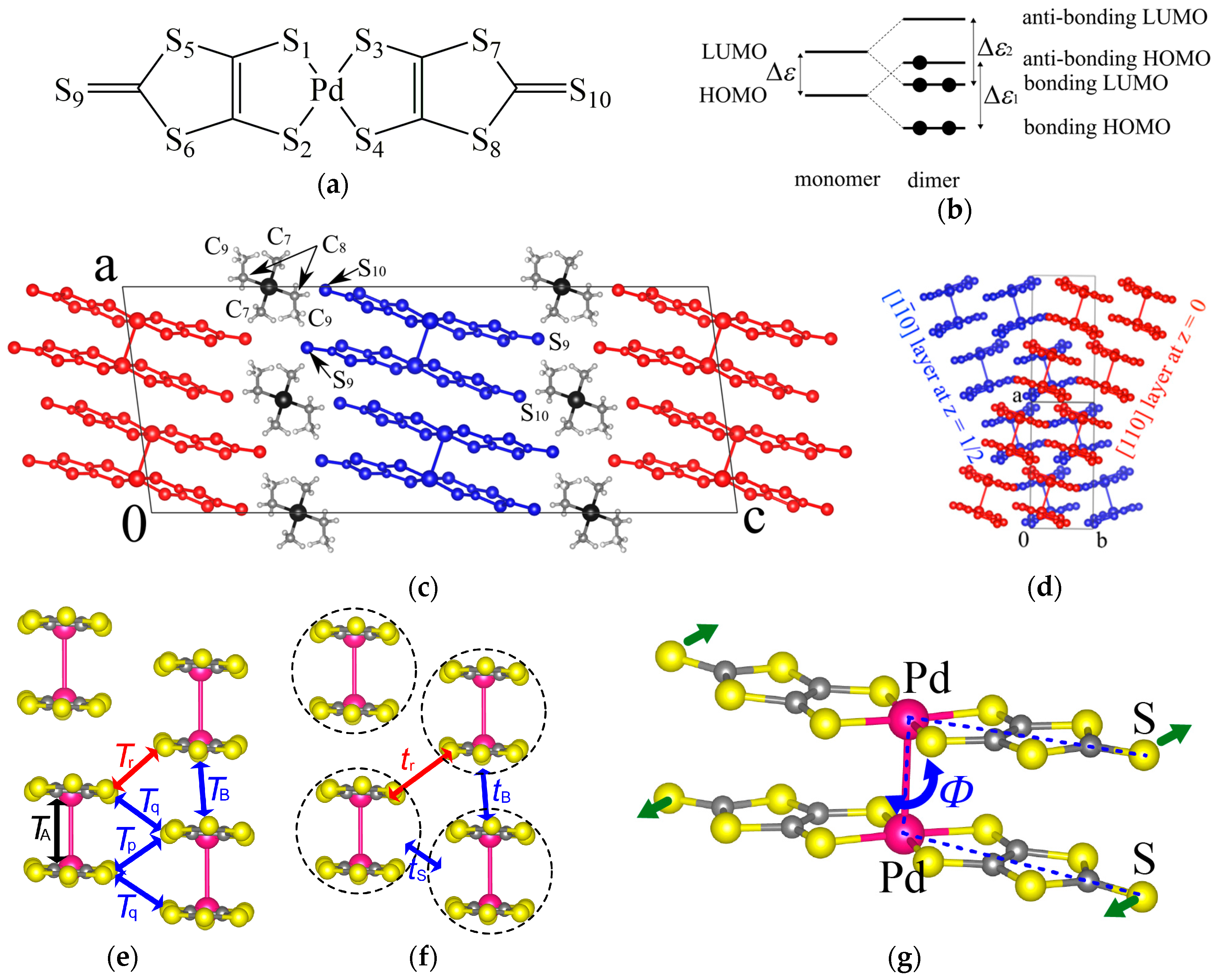
Figure 2.
Temperature dependence of (a) intermolecular transfer integrals, TB, Tr, Tp, and Tq and (b) interdimer transfer integrals, tB, ts, and tr, in the EtMe3Sb salt.
Figure 2.
Temperature dependence of (a) intermolecular transfer integrals, TB, Tr, Tp, and Tq and (b) interdimer transfer integrals, tB, ts, and tr, in the EtMe3Sb salt.
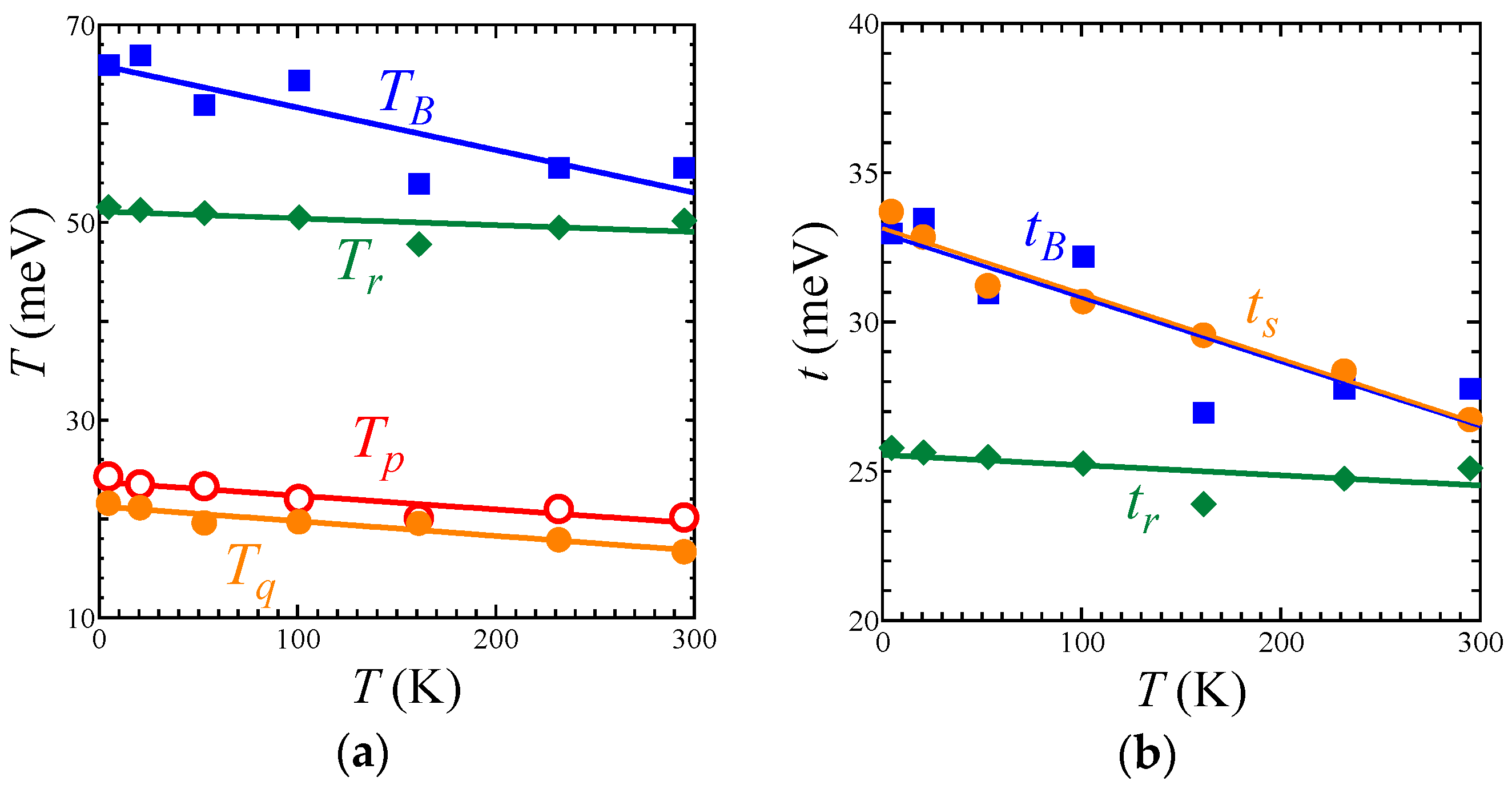
Figure 3.
Temperature dependence of (a) t′/t and (b) TA/W in the Me4Sb, Me4As, Et2Me2As, and EtMe3Sb salts.
Figure 3.
Temperature dependence of (a) t′/t and (b) TA/W in the Me4Sb, Me4As, Et2Me2As, and EtMe3Sb salts.
Figure 4.
Fermi surface of the EtMe3Sb salt calculated by the extended Hückel and tight binding methods at 5 K (a), 101 K (b), and 295 K (c). The C-centered monoclinic cell is reduced to the primitive one as ap = (a + b)/2, bp = b, cp = c.
Figure 4.
Fermi surface of the EtMe3Sb salt calculated by the extended Hückel and tight binding methods at 5 K (a), 101 K (b), and 295 K (c). The C-centered monoclinic cell is reduced to the primitive one as ap = (a + b)/2, bp = b, cp = c.
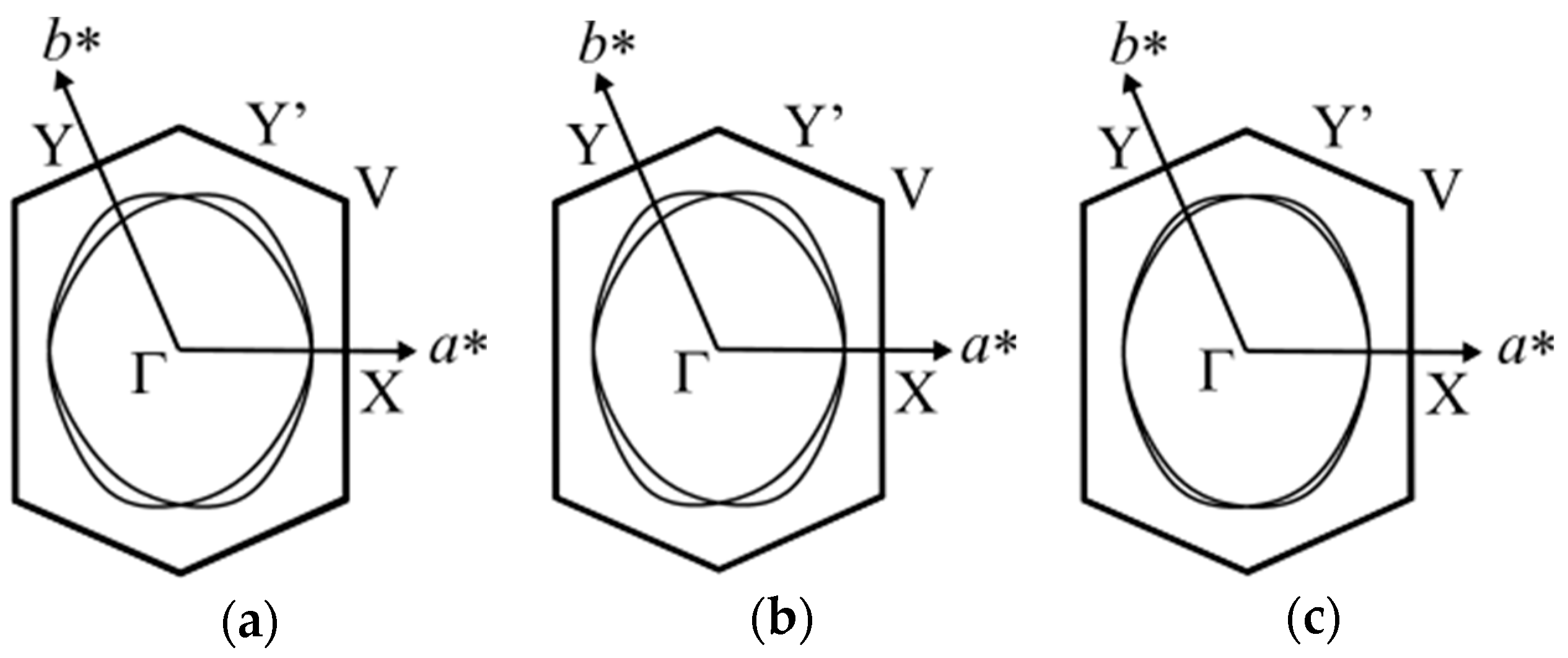
Figure 5.
Electronic structure of the EtMe3Sb salt calculated by the first-principles DFT method. Fermi surface at 5 K (a), 101 K (b), and 295 K (c). (d) Band dispersion at 5 K (red) and 295 K (black). (e) Partial density of states (PDOS) at 5 K in the conduction bands for the d-orbitals of Pd (green), the p-orbitals of S (blue), the p-orbitals of C in the dmit ligand (black), and the s- and p-orbitals of C in the EtMe3Sb cation (red). The blue and black lines each show the summation of the PDOS of all the S and C atoms belonging to the dmit ligand.
Figure 5.
Electronic structure of the EtMe3Sb salt calculated by the first-principles DFT method. Fermi surface at 5 K (a), 101 K (b), and 295 K (c). (d) Band dispersion at 5 K (red) and 295 K (black). (e) Partial density of states (PDOS) at 5 K in the conduction bands for the d-orbitals of Pd (green), the p-orbitals of S (blue), the p-orbitals of C in the dmit ligand (black), and the s- and p-orbitals of C in the EtMe3Sb cation (red). The blue and black lines each show the summation of the PDOS of all the S and C atoms belonging to the dmit ligand.
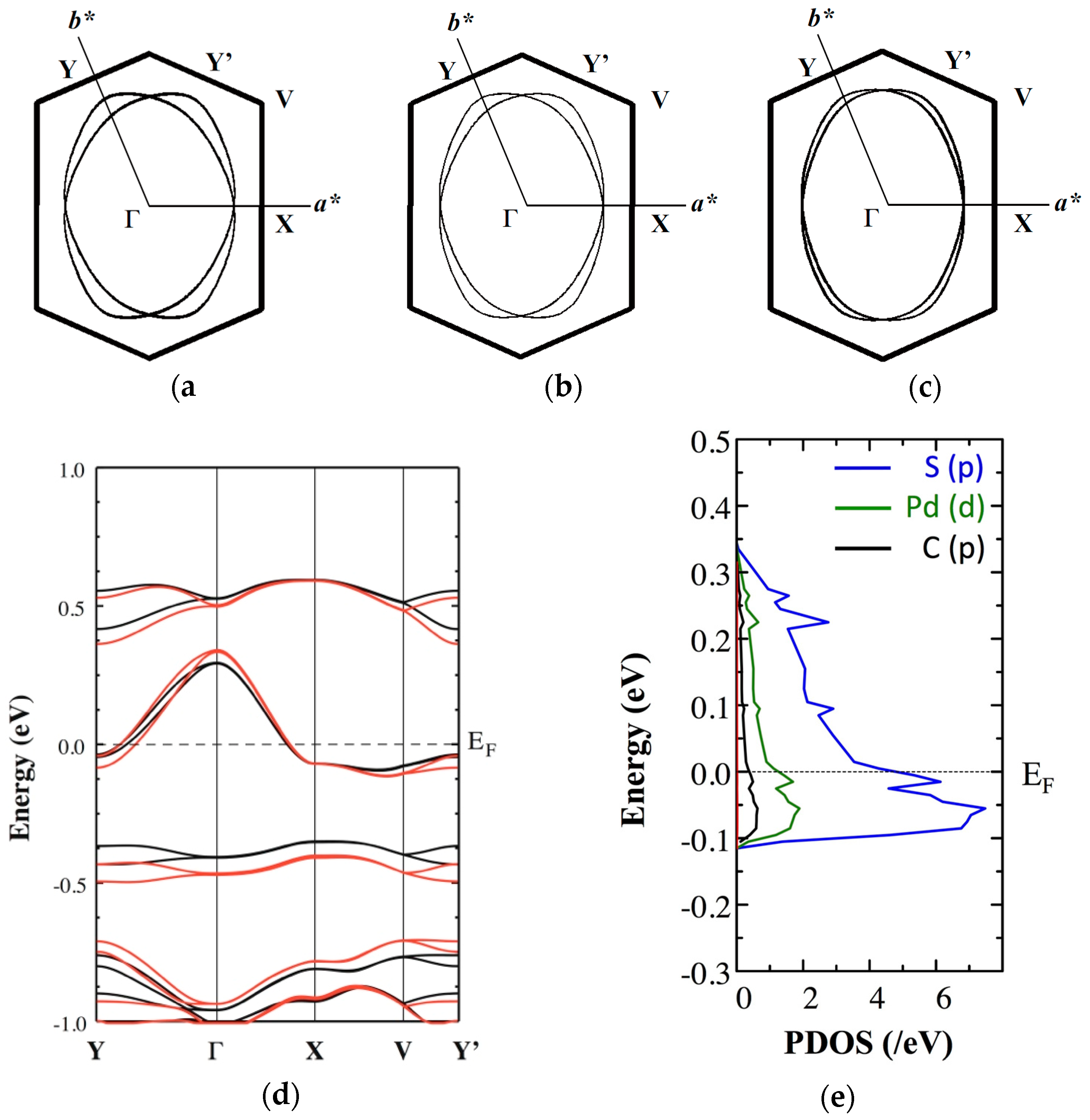
Figure 6.
Temperature dependence of (a) intradimer S⋯S distances, dAX (X = 1–5) and (b) Φ, and (c) relationship between TA and dAX in the EtMe3Sb salt.
Figure 6.
Temperature dependence of (a) intradimer S⋯S distances, dAX (X = 1–5) and (b) Φ, and (c) relationship between TA and dAX in the EtMe3Sb salt.
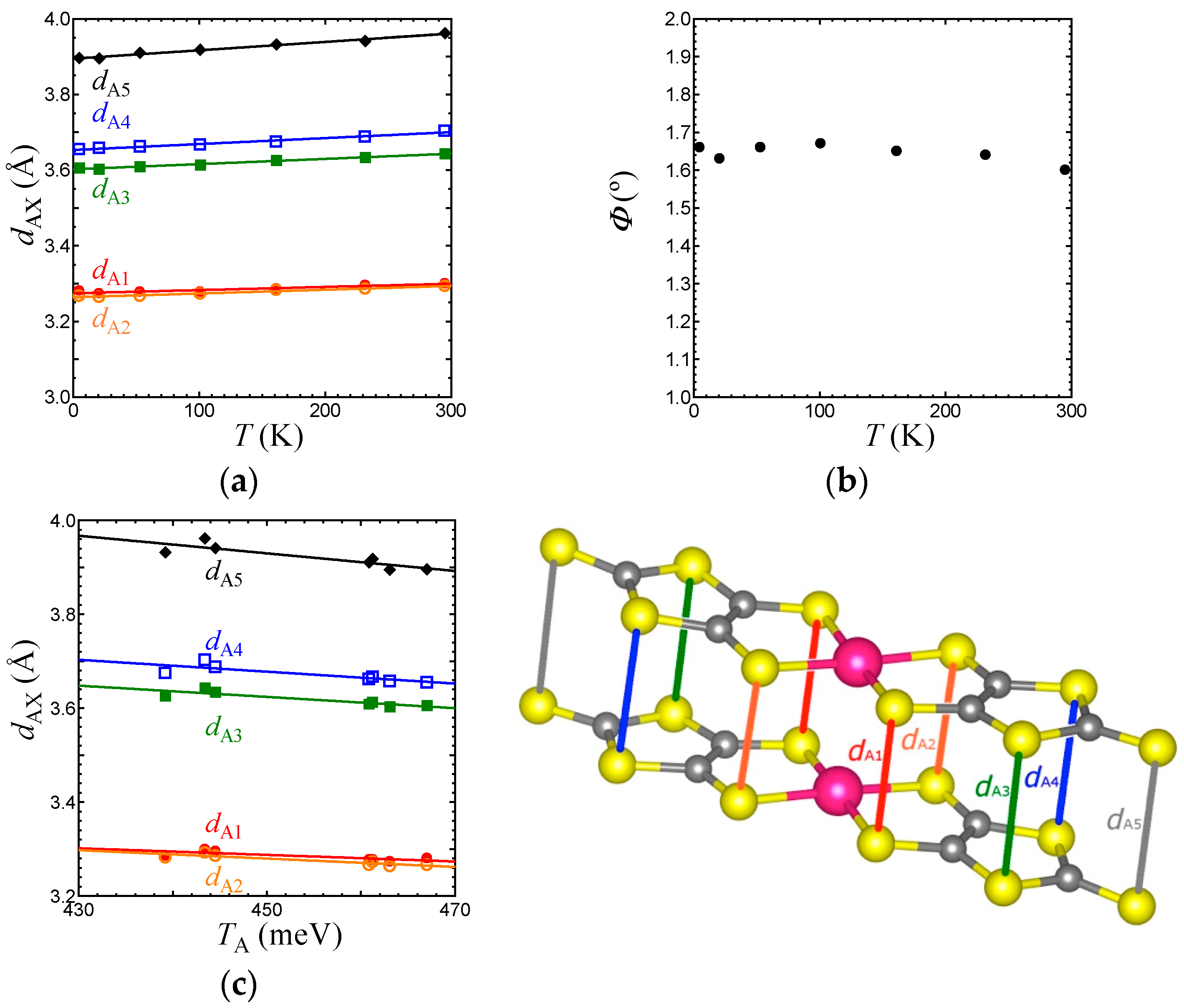
Figure 7.
Temperature dependence of intermolecular S⋯S distances associated with TB (a-1), Tr (b-1), Tp (c-1), and Tq (d-1) in the EtMe3Sb salt. Relationships between the S⋯S distances and intermolecular transfer integrals, TB (a-2), Tr (b-2), Tp (c-2), and Tq (d-2) in the EtMe3Sb salt.
Figure 7.
Temperature dependence of intermolecular S⋯S distances associated with TB (a-1), Tr (b-1), Tp (c-1), and Tq (d-1) in the EtMe3Sb salt. Relationships between the S⋯S distances and intermolecular transfer integrals, TB (a-2), Tr (b-2), Tp (c-2), and Tq (d-2) in the EtMe3Sb salt.
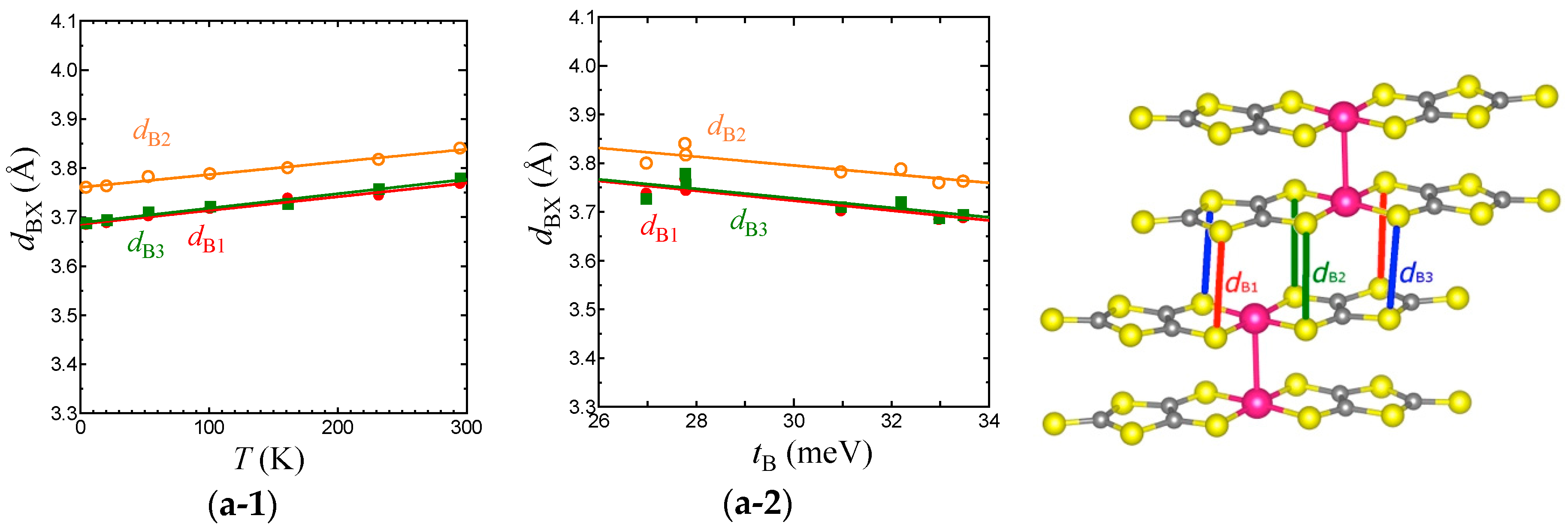
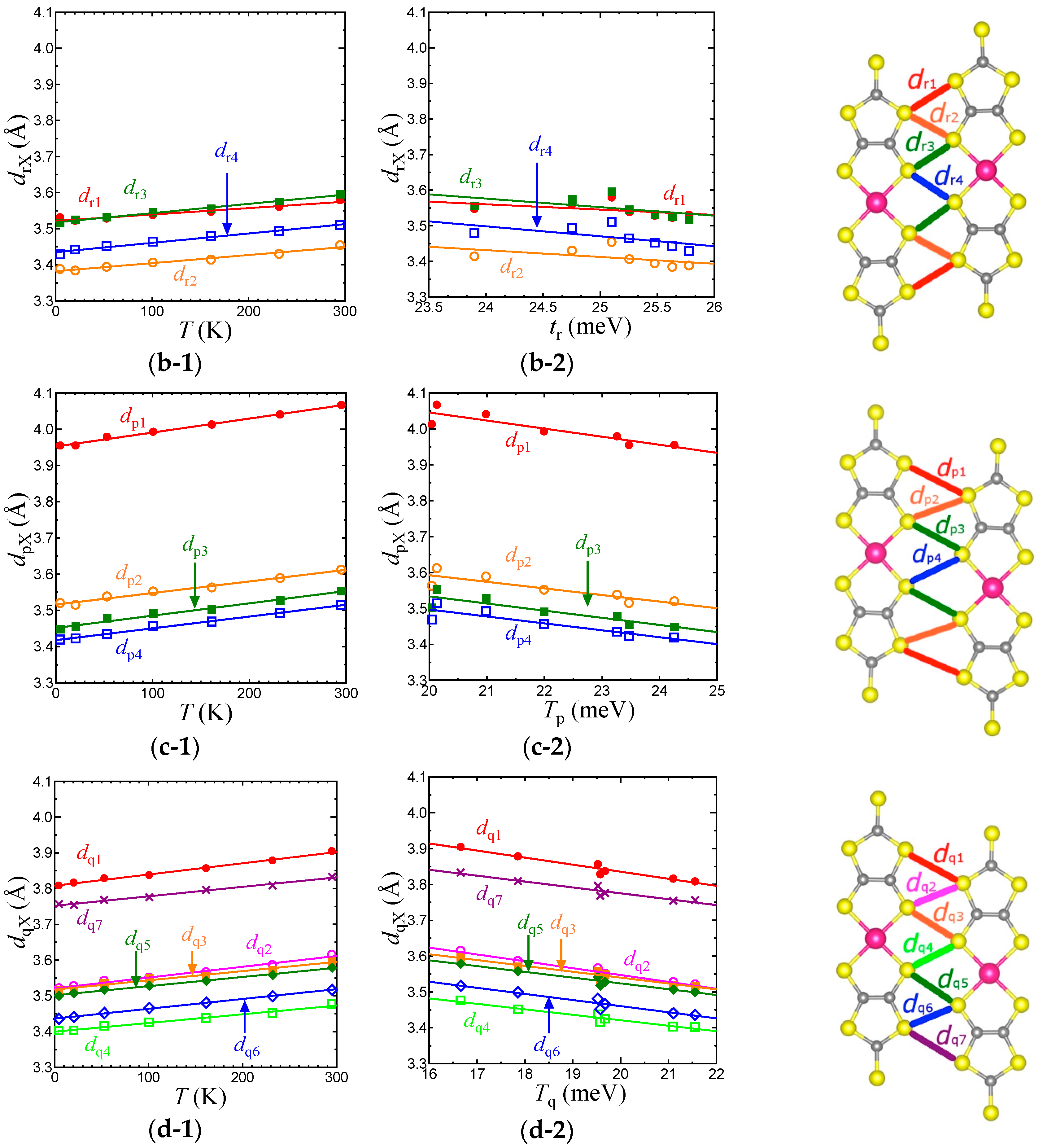
Figure 8.
Temperature dependence of short S⋯H-C distances d (S⋯H-C) in the EtMe3Sb salt: the black dashed line shows the sum of the van der Waals radii of S atom and H atom. The red filled circles, orange filled squares, and red open diamonds mark the S⋯H-C7 contacts. The blue filled circles and light blue filled squares mark the S⋯H-C8 contacts. The green filled circles, spring green filled squares, and green open diamonds mark the S⋯H-C9 contacts. See also Figure 1c and Figure 9.
Figure 8.
Temperature dependence of short S⋯H-C distances d (S⋯H-C) in the EtMe3Sb salt: the black dashed line shows the sum of the van der Waals radii of S atom and H atom. The red filled circles, orange filled squares, and red open diamonds mark the S⋯H-C7 contacts. The blue filled circles and light blue filled squares mark the S⋯H-C8 contacts. The green filled circles, spring green filled squares, and green open diamonds mark the S⋯H-C9 contacts. See also Figure 1c and Figure 9.
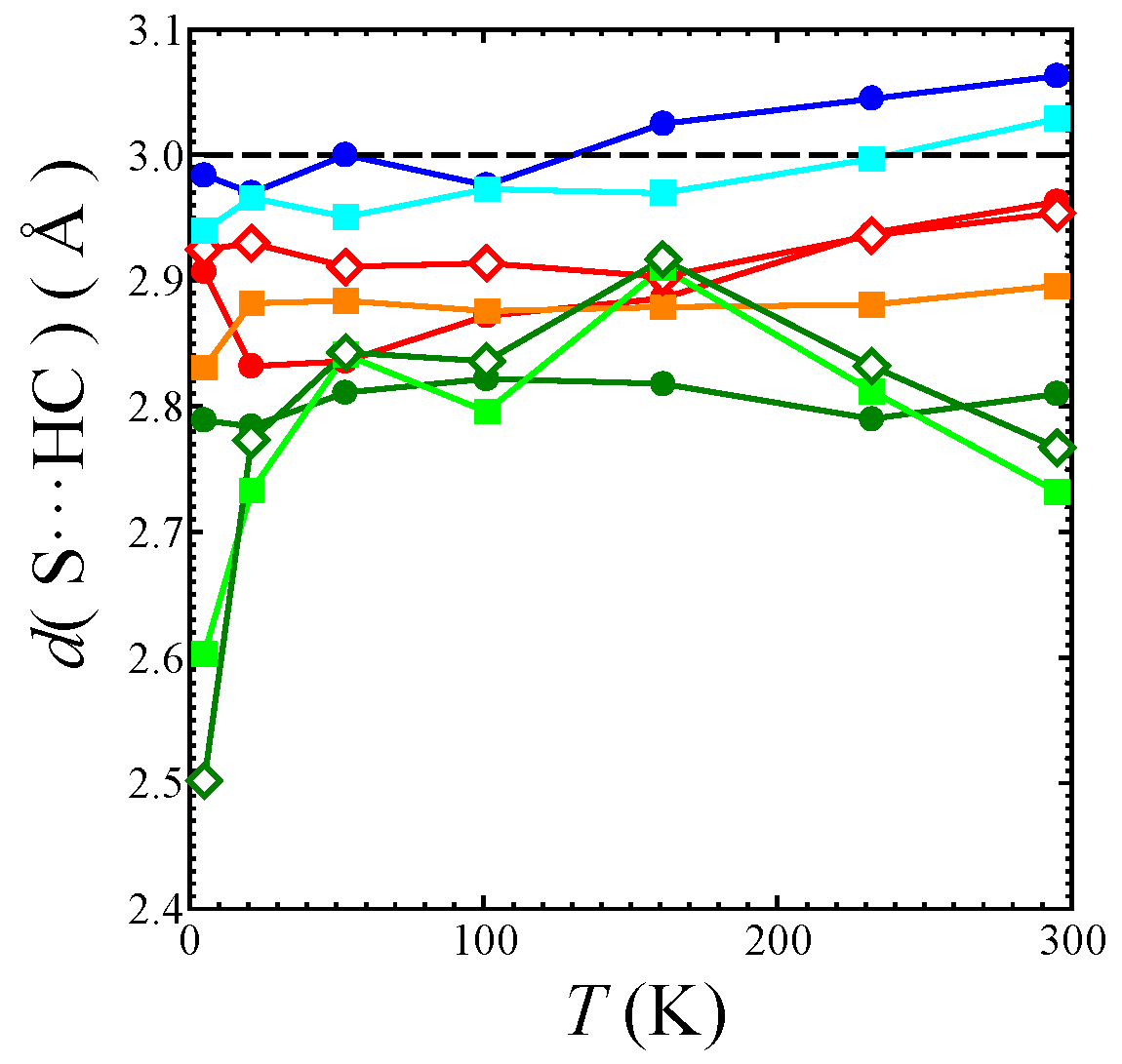
Figure 9.
Schematic illustration of the short S⋯H-C contacts. (a) Oblique view. (b) End-on projections of the dimer arrangement. (c) Views along the vector connecting the two Pd atoms in the dimer, (c-1) shows the short S⋯H-C contacts between the S9 atom in the lower Pd(dmit)2 molecule in (b) and cations a and c, (c-2) shows the S⋯H-C short contacts between the S10 atom in the upper Pd(dmit)2 molecule in (b) and cations a and b: black arrows indicate the direction of the arch-shaped distortion with a positive Φ value. The red, green and blue arrows indicate the directions of repulsive S⋯H-C7, S⋯H-C8, and S⋯H-C9 interactions, respectively.
Figure 9.
Schematic illustration of the short S⋯H-C contacts. (a) Oblique view. (b) End-on projections of the dimer arrangement. (c) Views along the vector connecting the two Pd atoms in the dimer, (c-1) shows the short S⋯H-C contacts between the S9 atom in the lower Pd(dmit)2 molecule in (b) and cations a and c, (c-2) shows the S⋯H-C short contacts between the S10 atom in the upper Pd(dmit)2 molecule in (b) and cations a and b: black arrows indicate the direction of the arch-shaped distortion with a positive Φ value. The red, green and blue arrows indicate the directions of repulsive S⋯H-C7, S⋯H-C8, and S⋯H-C9 interactions, respectively.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Semi-empirical parameters for Slater-type atomic orbitals.
| Atom | Orbital | Ip (Ryd) | ζ1 | ζ2 |
|---|---|---|---|---|
| C | 2s | 1.573 | 1.625 | |
| 2p | 0.838 | 1.625 | ||
| S | 3s | 1.620 | 2.122 | |
| 3p | 0.770 | 1.827 | ||
| Pd | 5s | 0.676 | 2.19 | |
| 5p | 0.390 | 2.15 | ||
| 4d | 0.949 | 5.98 | 2.613 | |
| (0.5264) * | (0.6372) * |
* Contraction coefficients used in the double-ζ expansion.
Table 2.
The number of short S⋯H-C contacts between the cation and terminal thioketone of the Pd(dmit)2 molecule at 295 K and 5 K.
Table 2.
The number of short S⋯H-C contacts between the cation and terminal thioketone of the Pd(dmit)2 molecule at 295 K and 5 K.
| Me4As | Me4Sb | Et2Me2As | EtMe3Sb * | |||||
|---|---|---|---|---|---|---|---|---|
| S⋯H distance: d(S⋯H-C)/Å | 295 K | 5 K | 295 K | 5 K | 295 K | 5 K | 295 K | 5 K |
| 2.95—3.00 | 2 | 4 | 4 | 12 | 8 | 4 | 2 | |
| 2.90—2.95 | 2 | 2 | 2 | 2 | 6 | |||
| 2.85—2.90 | 4 | |||||||
| 2.80—2.85 | 2 | 2 | 2 | 2 | 1 | 2 | ||
| 2.75—2.80 | 1 | 1 | ||||||
| 2.70—2.75 | 1 | |||||||
| 2.65—2.70 | ||||||||
| 2.60—2.65 | 1 | |||||||
| 2.55—2.60 | ||||||||
| 2.50—2.55 | 1 | |||||||
* The number of the short S⋯H-C9 contacts in the EtMe3Sb salt was counted as half of that in Figure 9, considering the occupancy factor of 0.5 for the ethyl group.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ueda, K.; Tsumuraya, T.; Kato, R. Temperature Dependence of Crystal Structures and Band Parameters in Quantum Spin Liquid β′-EtMe3Sb[Pd(dmit)2]2 and Related Materials. Crystals 2018, 8, 138. https://doi.org/10.3390/cryst8030138
AMA Style
Ueda K, Tsumuraya T, Kato R. Temperature Dependence of Crystal Structures and Band Parameters in Quantum Spin Liquid β′-EtMe3Sb[Pd(dmit)2]2 and Related Materials. Crystals. 2018; 8(3):138. https://doi.org/10.3390/cryst8030138
Chicago/Turabian StyleUeda, Kohei, Takao Tsumuraya, and Reizo Kato. 2018. "Temperature Dependence of Crystal Structures and Band Parameters in Quantum Spin Liquid β′-EtMe3Sb[Pd(dmit)2]2 and Related Materials" Crystals 8, no. 3: 138. https://doi.org/10.3390/cryst8030138
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.