Mild and Facile Synthesis of Multi-Functional RAFT Chain Transfer Agents

Abstract
:1. Introduction
2. Results and Discussion
2.1. R group approach for multi functional chain transfer agent synthesis





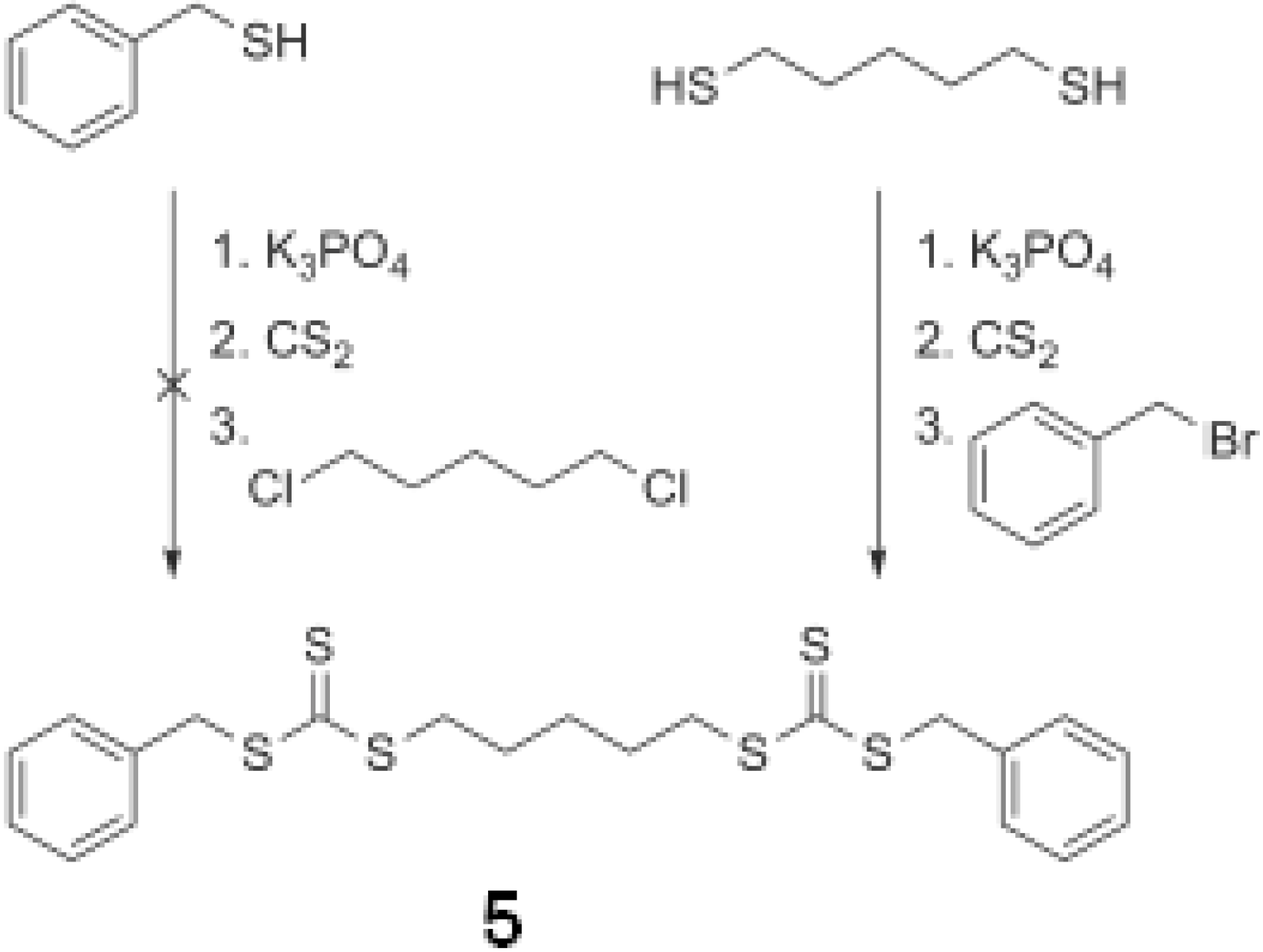
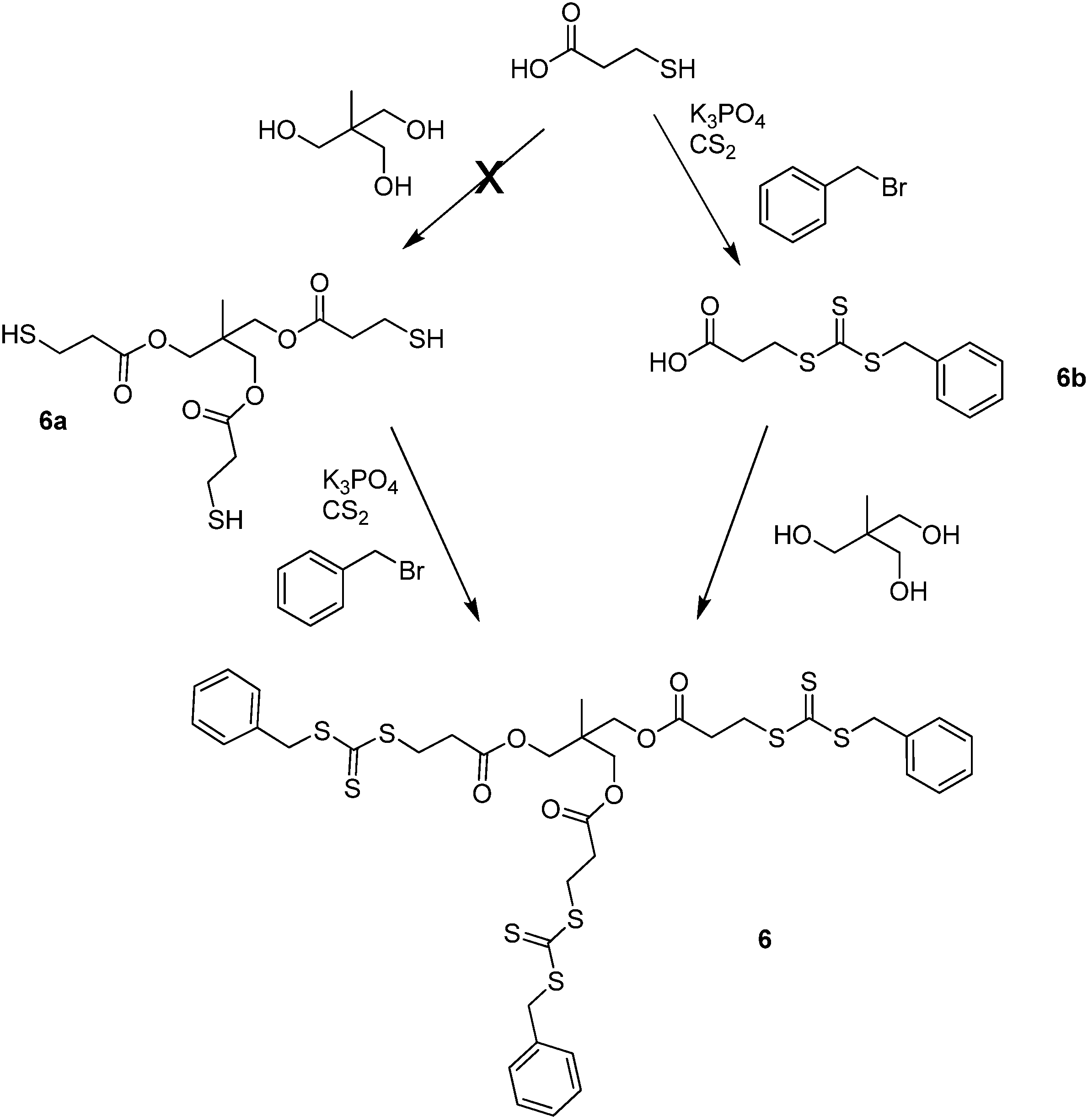
2.2. Z group approach for multi-functional chain transfer agent synthesis


2.3. Polymerization data for chain transfer agents 2-6

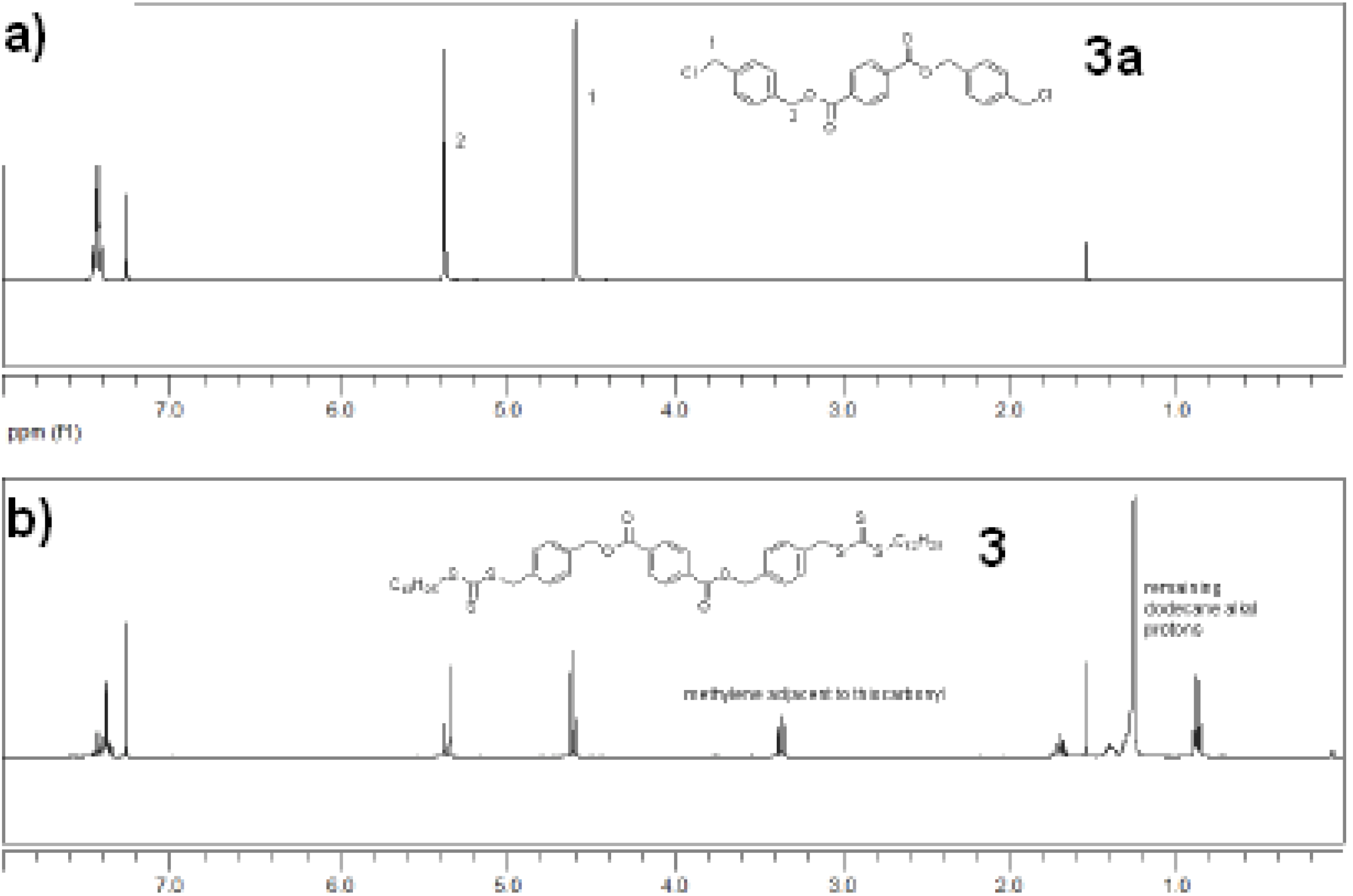{kind=link}
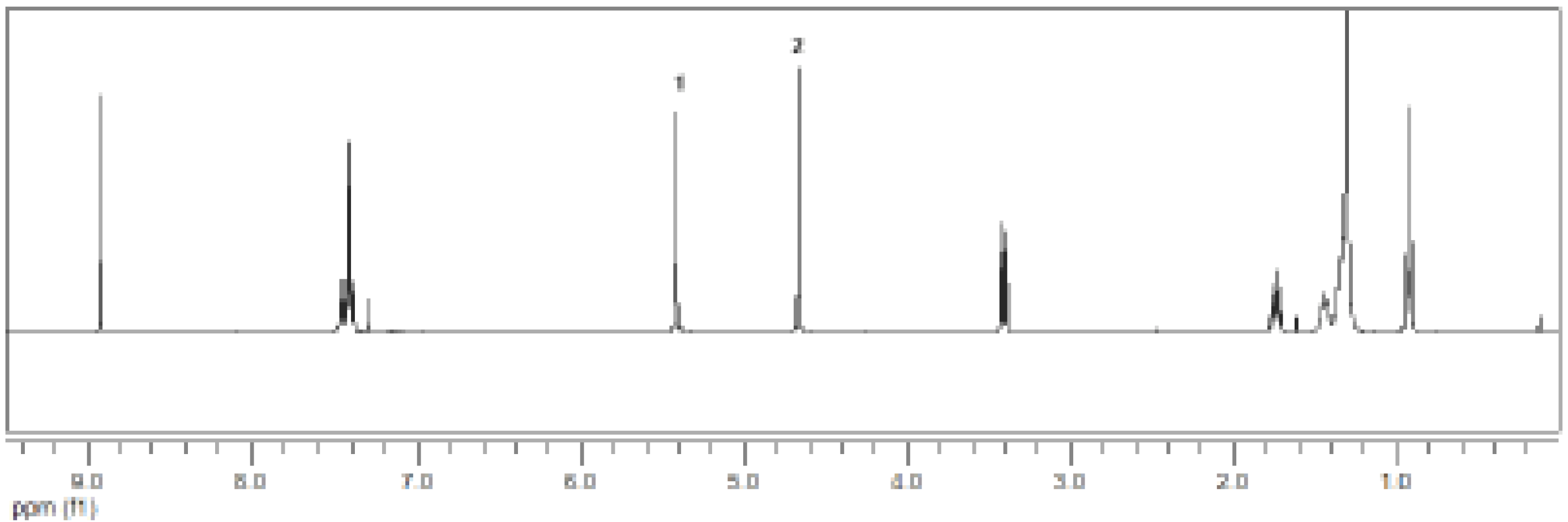{kind=link}
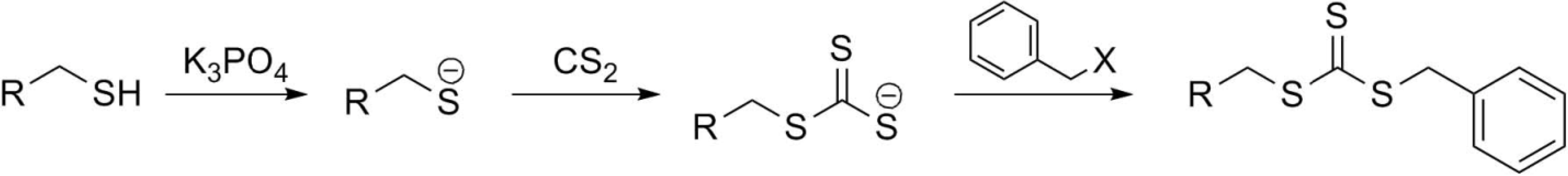{kind=link}
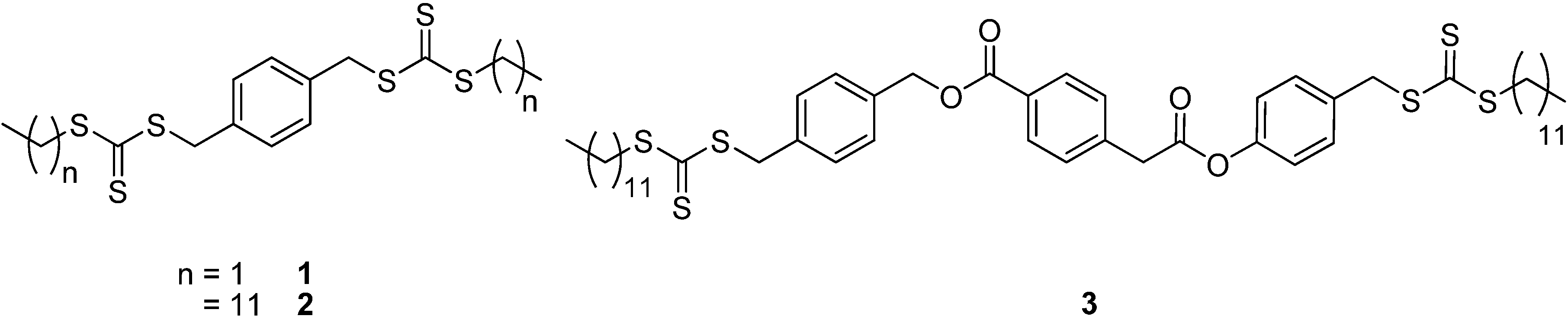{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chain transfer agent | Monomer | Time (hr) | Conv. (%) | MnGPC (Da) | Mw/Mn |
|---|---|---|---|---|---|
| 2 | St | 2 | 76 | 24,600 | 1.09 |
| 2 | tBuA | 2 | 82 | 34,000 | 1.19 |
| 3 | St | 2 | 74 | 25,300 | 1.11 |
| 3 | tBuA | 1 | 65 | 27,900 | 1.17 |
| 5 | St | 3 | 79 | 30,200 | 1.12 |
| 5 | tBuA | 4 | 84 | 35,600 | 1.07 |
3. Experimental Section
3.1. Materials and Instrumentation
3.2. Methods
3.2.1. General polymerization method
3.2.2. Attempted synthesis of 1 - 1,4-phenylenebis(methylene) diethyl dicarbonotrithiocarbonate
3.2.3. Synthesis of 2 - 1,4-phenylenebis(methylene) didodecyl dicarbonotrithiocarbonate
3.2.4. Synthesis of 3a
3.2.5. Synthesis of 3 - bis(4-((dodecylthiocarbonothioylthio)methyl)benzyl) terephthalate
3.2.6. Synthesis of 4a - tris(4-(chloromethyl)benzyl) benzene-1,3,5-tricarboxylate
3.2.7. Synthesis of 4 - 1-(3-((dodecylthiocarbonothioylthio)methyl)benzyl) 3,5-bis(4-((dodecylthiocarbonothioylthio)methyl)benzyl) benzene-1,3,5-tricarboxylate
3.2.8. Synthesis of 5 - benzyl pentane-1,5-diyl dicarbonotrithiocarbonate
3.2.9. Synthesis of 6
4. Conclusions
Acknowledgements
References and Notes
- Chiefari, J.; Chong, Y. K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T. P. T.; Mayadunne, R. T. A.; Meijs, G. F.; Moad, C. L.; Moad, G.; Rizzardo, E.; Thang, S. H. Macromolecules 1998, 31, 5559–5562.
- Moad, G.; Rizzardo, E.; Thang, S. H. Aust. J. Chem. 2006, 59, 669–692.
- Perrier, S.; Takolpuckdee, P. Journal of Polymer Science Part A-Polymer Chemistry 2005, 43, 5347–5393.
- Lowe, A. B.; McCormick, C. L. Prog. Polym. Sci. 2007, 32, 283–351.
- Moad, G.; Rizzardo, E.; Thang, S. H. Polymer 2008, 49, 1079–1131.
- Chiefari, J.; Mayadunne, R. T. A.; Moad, C. L.; Moad, G.; Rizzardo, E.; Postma, A.; Skidmore, M. A.; Thang, S. H. Macromolecules 2003, 36, 2273–2283.
- Chong, Y. K.; Krstina, J.; Le, T. P. T.; Moad, G.; Postma, A.; Rizzardo, E.; Thang, S. H. Macromolecules 2003, 36, 2256–2272.
- Wang, Y. Q.; Ge, Z. M.; Hou, X. L.; CHheng, T. M.; Li, R. T. Synthesis 2004, 675–678.
- Wood, M. R.; Duncalf, D. J.; Rannard, S. P. Org. lett. 2006, 8, 553–556.
- Skey, J.; O'Reilly, R. K. Chem. Commun. 2008, 35, 4183–4185.
- Watanbe, H.; Matsumiya, Y.; Ishida, S.; Takigawa, T.; Yamamoto, T.; Vlassopoulos, D.; Roovers, J. Macromolecules 2005, 38, 7404–7415.
- Wiltshire, J. T.; Qiao, G. G. Aust. J. Chem. 2007, 60, 699–705.
- Gao, H. F.; Matyjaszewski, K. Macromolecules 2006, 39, 4960–4965.
- Hoogenboom, R.; Moore, B. C.; Schubert, U. S. Chem. Commun. 2006, 4010.
- Hadjichristidis, N.; Pitsikalis, M.; Pispas, S.; Iatrou, H. Chem. Rev. 2001, 101, 3747–3792.
- Kanaoka, S.; Sawamoto, M.; Higashimurs, T. Macromolecules 1191, 24, 2309–2313.
- Coessens, V.; Pintauer, T.; Matyjaszewski, K. Prog. Polym. Sci. 2001, 26, 337–377.
- Dufils, P. E.; Chagneux, N.; Gigmes, D.; Trimaille, T.; Marque, S. R. A.; Bertin, D.; Tordo, P. Polymer 2007, 48, 5219–5225.
- Stenzel-Rosenbaum, M.; Davis, T. P.; Chen, V.; Fane, A. G. J. Polym. Sci. Part A Polym. Chem. 2001, 39, 2777–2783. [CrossRef]
- Bosman, A. W.; Heumann, A.; Klaerner, G.; Benoit, D.; Frechet, J. M. J.; Hawker, C. J. J. Am. Chem. Soc. 2001, 123, 6461–6462. [CrossRef]
- Zhang, X.; Xia, J. H.; Matyjaszewski, K. Macromolecules 2000, 33, 2340–2345.
- Bivigou, A. M.; Kristen, J.; Laschewsky, A.; Muller-Buschbaum, P.; Papadakis, C. M. Macromol. Chem. Phys. 2009, 210, 565–578.
- Mayadunne, R. T. A.; Jeffery, J.; Moad, G.; Rizzardo, E. Macromolecules 2003, 36, 1505–1513.
- Jesberger, M.; Barner- Kowollik, L.; Malmstrom, E.; Davis, T. P.; Barner- Kowollik, C. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 3847–3861. [CrossRef]
- Stenzel, M. H.; Davis, T. P. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 4498–4512. [CrossRef]
- Hao, X. J.; Nilsson, C.; Jesberger, M.; Stenzel, M. H.; Malmstrom, E.; Davis, T. P.; Ostmark, E.; Barner- Kowollik, C. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 5877–5890. [CrossRef]
- Barner-Kowollik, C.; Davis, T. P.; Stenzel, M. H. Aust. J. Chem. 2006, 59, 719–727.
- Samakande, A.; Sanderson, R. D.; Hartmann, P. C. Synth. Commun. 2007, 37, 3861–3872.
- Thomas, D. B.; Convertine, A. J.; Hester, R. D.; Lowe, A. B.; McCormick, C. L. Macromolecules 2004, 37, 1735–1741.
- Mertoglu, M.; Lachewsky, A.; Skrabania, K.; Wieland, C. Macromolecules 2005, 38, 3601–3614.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
O’Reilly, R.K.; Hansell, C. Mild and Facile Synthesis of Multi-Functional RAFT Chain Transfer Agents. Polymers 2009, 1, 3-15. https://doi.org/10.3390/polym1010003
O’Reilly RK, Hansell C. Mild and Facile Synthesis of Multi-Functional RAFT Chain Transfer Agents. Polymers. 2009; 1(1):3-15. https://doi.org/10.3390/polym1010003
Chicago/Turabian StyleO’Reilly, Rachel K., and Claire Hansell. 2009. "Mild and Facile Synthesis of Multi-Functional RAFT Chain Transfer Agents" Polymers 1, no. 1: 3-15. https://doi.org/10.3390/polym1010003