Conduction and Electrostriction of Polymers Induced by High Electric Fields

Abstract
: After reviewing the new physics and chemistry in high electrostatic fields we use density functional theory to show that in fields around 1.5 V/Å the bandgap of polythiophene reduces to zero leading to field-induced metallization. In poly(ethylene glycol), on the other hand, such fields lead to giant electrostriction of over 20% in length. Lastly, we give two examples of field-induced polymerization: (1) the closure of sulfur molecules Sn at n = 8 is suppressed remaining linear up to n ∼ 20. (2) This also happens to water which forms linear whiskers up to n ∼ 11.1. Introduction
Large electrostatic fields, much larger than the range of electric breakdown in air (F ∼ 3 × 106 V/m) occur over macroscopic distances at the electrode-electrolyte interface (F ∼ 108 V/m) due to charge separation, in zeolite cavities (F ∼ 109 V/m) due to the presence of localized charges, and at metallic or semiconducting field emission tips (F ∼ 1010 V/m ∼ V/Å) due to their extremely small radius of curvature of less than 100 Å. The maximum such fields (F < 6 V/Å) occur in the field ion mode (with the tip positively charged against a large negatively charged counter electrode) and are limited by the destruction of the tips by field evaporation. Such fields, maintained over macroscopic distances, are of the same order as the fields inside atoms and molecules. They are thus strong enough to induce the re-arrangement of electronic orbitals of atoms and molecules leading to new phenomena that can be summarily described as field-induced chemisorption and field-induced chemistry. Several review articles have been written over the years about these topics [1-5]. For reference in the remainder of this article we note that a field of 1 V/Å corresponds to a force of 0.16 nN, and 300 kB/Å is 42 pN.
Electric field effects on matter can be classified, rather arbitrarily, into two categories: (i) in low fields, i.e., below roughly 10−1 V/Å, atoms, molecules and condensed matter only get polarized; we will call such effects physical. (ii) In larger fields chemical effects come into play because the electronic orbitals get distorted to such a degree that the chemical characteristics of an atom or molecule become altered, e.g., by establishing new bonding orbitals. In this way, molecules, unstable in field-free situations, may be stabilized by a strong electric field. Also, new pathways in chemical reactions, e.g., in heterogeneous catalysis and in chemical vapor deposition, may be established. Of more importance in the context of this special issue is the fact that polymers can be rendered conducting in strong enough fields; an example discussed in the literature recently is poly(thiophene). It was predicted theoretically [6] to become conducting in fields of about 1.5 V/Å as was soon discovered in experiments as well [7].
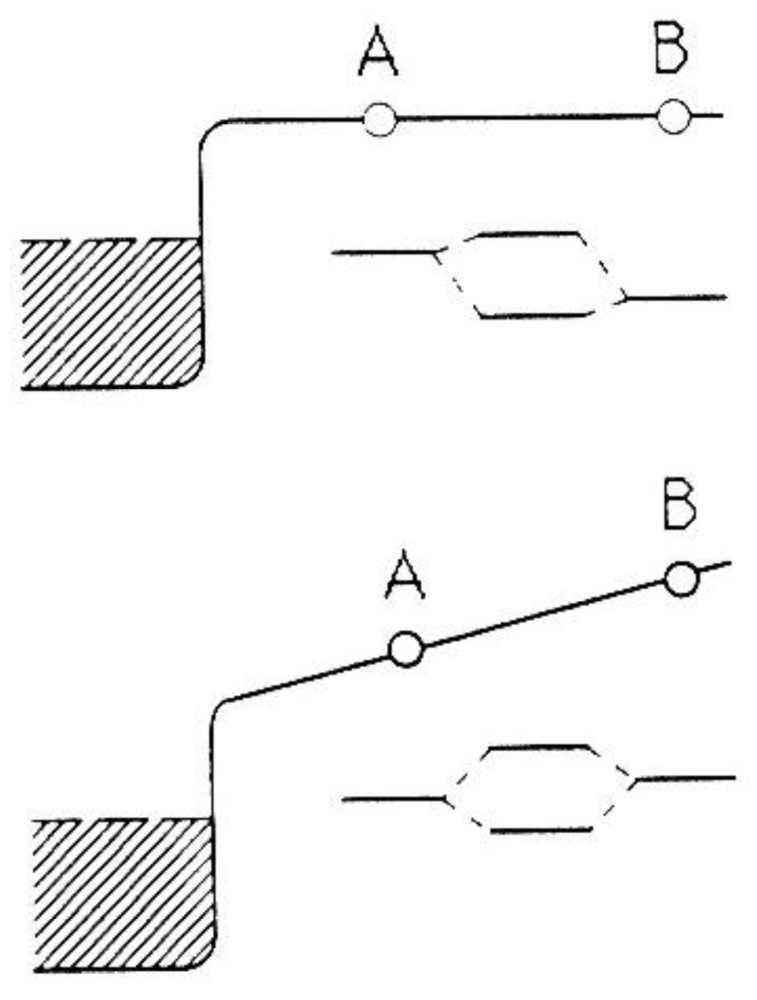
To discuss field effects qualitatively, we look, in Figure 1 at a molecule AB adsorbed on a metal. Far from the surface and in the absence of a field, the atomic orbitals of A and B hybridize into molecular orbitals, which we take to be a lower-lying bonding orbital and an empty antibonding orbital. As the molecule approaches a metal surface, additional hybridization with the conduction electrons occurs leading to shifts and broadening of these orbitals. As illustrated in Figure 1, the antibonding orbital gets partially occupied resulting in (i) bonding to the surface and (ii) weakening of the A–B bond stretch vibration. Molecules such as CO and N2 are examples. Applying an electric field, Fz, pointing away from the surface, adds the potential energy, eFzz, for the electrons outside the metal (assuming, for this simplified discussion, total expulsion of the field from the metal). This raises the atomic levels of the atoms A and B by amounts of the order eFzza and eFzzb, respectively, resulting in a substantial re-arrangement of the molecular orbitals. For the situation depicted in Figure 1, the anti-bonding orbital empties out again leading to restabilization of the molecule and probably a weakening of the surface bond. If we increase the field strength to the point where the bonding orbital is lifted above the Fermi energy of the metal, it will drain as well, leading to field-induced dissociation. Note that in the absence of the field the bonding orbital of the AB molecule is more B-like whereas the anti-bonding orbital has more A character. As the electric field is increased, these characteristics are changed in a continuous manner into a situation where the bonding orbital is more A-like and the anti-bonding orbital has B character. This possibility of changing the relative position of orbitals of the constituent atoms in a molecule with respect to each other creates a “continuous Periodic Table” and leads to new, field-induced chemistry. As an example, local electric fields, generated by a sharp metal tip, can be used to preferentially dissociate certain species that one wants to deposit locally under the tip, e.g., in field-assisted chemical vapor deposition.
Block [8], following earlier work by Inghram and Gomer [9] had developed a pulsed field technique in the field ion microscope that allows the investigation of the field effect on chemical reactions; similar work was reported by Beckey and Röllgen [10]; a detailed account had been given by Block [4,5]. Systems that have been studied by this technique are the formation of metal subcarbonyls, the polymerization of acetone, the reaction of sulphur on metal surfaces, the decomposition of methanol on metal surfaces, hydride formation on semiconductors, NO reactions on metals and many more.
In this paper we will first review our understanding of the metallization of polythiophene in high electric fields. Next we will discuss the evidence that a simple polymer like poly(ethylene glycol) will undergo a giant electrostriction in strong electrostatic fields. Our last examples are the polymerization of sulfur and water induced by electrostatic fields. The paper ends with a short summary and outlook.
2. Metallization of Polythiophene
Oligomers of thiophene and polythiophene have attracted considerable interest in recent years for direct-writing of polymer nanostructures such as nano-wires on semiconducting and insulating surfaces. They have appeared as active components in heterojunctions, photovoltaic cells, light-emitting diodes and field-effect transistors, mainly because of the ease with which the optical and electronic properties of oligothiophenes can be tuned by chemical modification [11]. More recently, the Scanning Atom Probe microscope has been used to mass analyze chemically modified polythiophene by field evaporation [12]. The dominant fragments observed were SC4Hn2+, n = 0,1,2,3, with a host of other singly and doubly charged fragments. A number of these also contained boron atoms which originated from the BF4− dopant in the electrolyte used for the polymerization of polythiophene. Related field emission studies confirm that polythiophene is semiconductive.
To shed some light on the field-induced fragmentation and dissociation of polythiophene density functional calculations were done for short chains of polythiophene with up to 8 units in electrostatic fields of the order of volts per angstrom [6]. This approach has proven successful in other studies on the intermolecular interactions in bithiophene [13] and for calculating excitation energies of terthiophene and its dioxide derivatives [14,15].
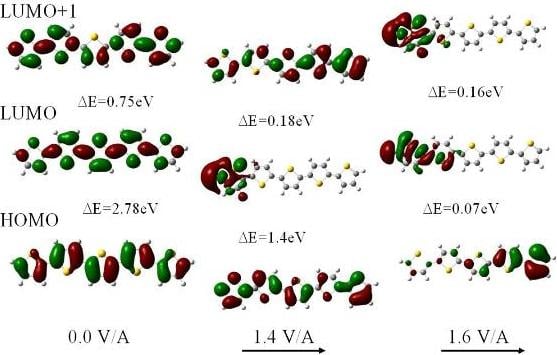
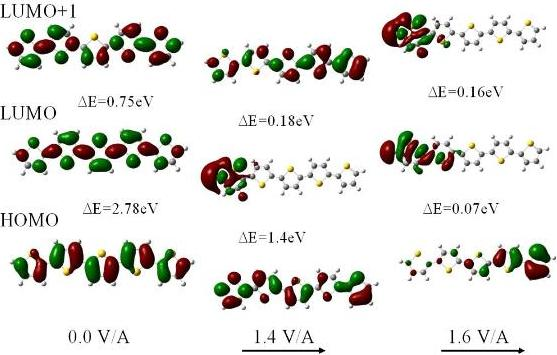
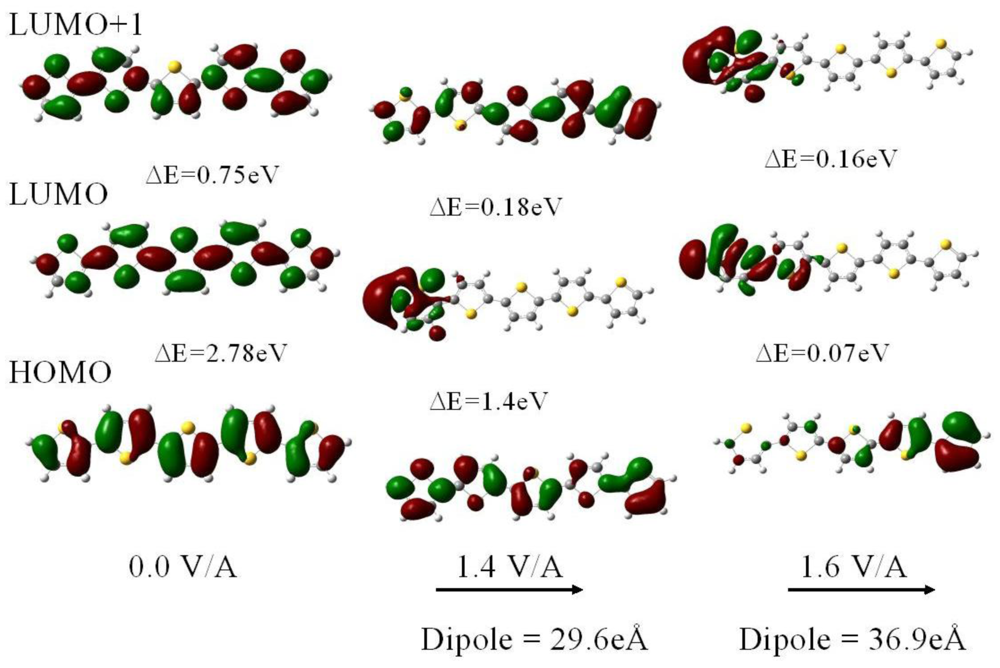
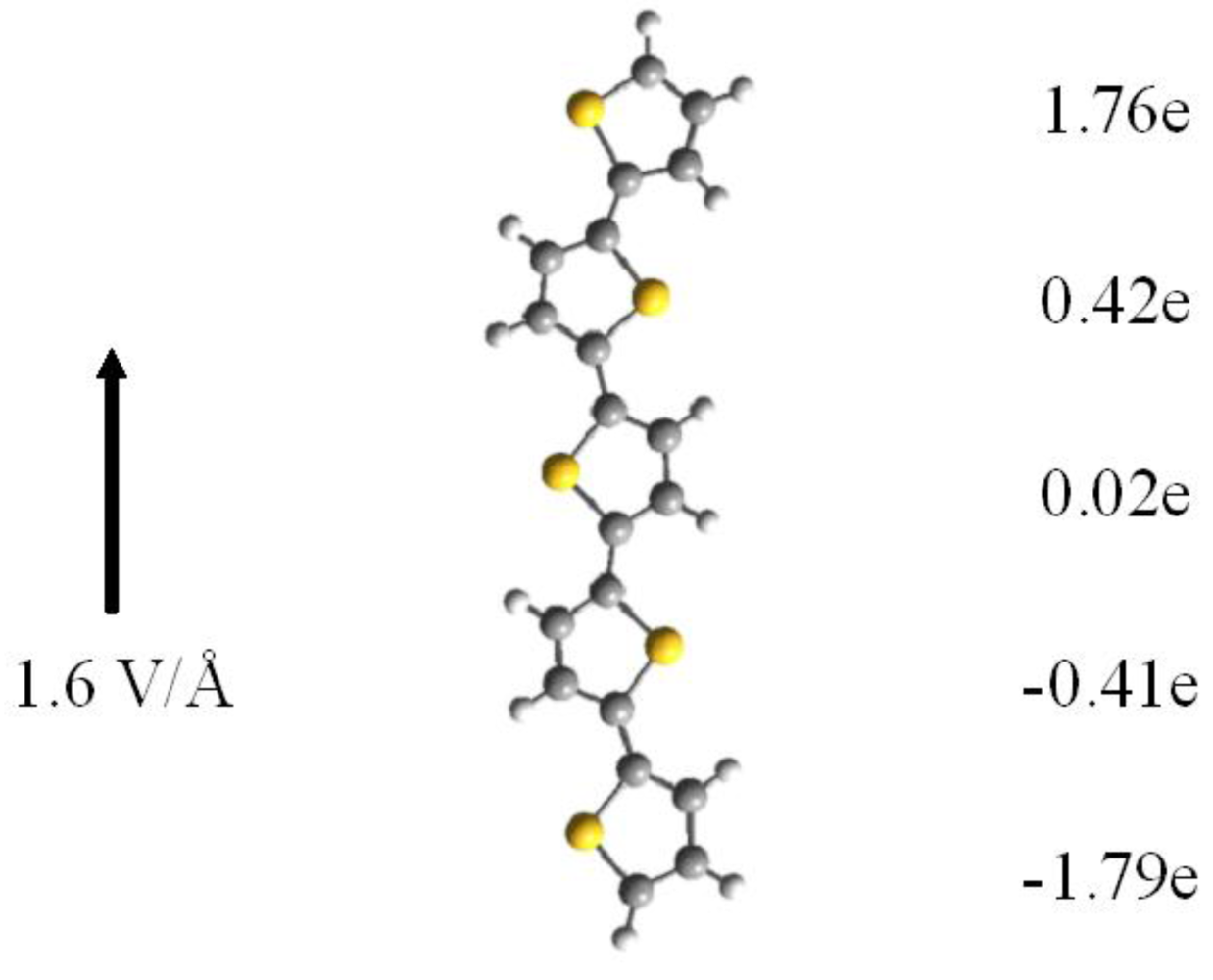
For fields below 1.6 V/Å the structure of neutral polythiophene does not change appreciably except that there is an intramolecular transfer of electrons down the electric field. In addition there is a substantial reduction in the HOMO-LUMO gap. The reason is that the field forces a concentration of the HOMO towards the end of the molecule in the field direction whereas the LUMO gets shifted towards the other end of the molecule. As a result the HOMO and LUMO energies are raised and lowered, respectively, by ±1/2 eFL with respect to the center of the molecule (of length L) This shift in the orbitals is clearly seen in the electron density plots of Figure 2. For a neutral 5-ring polythiophene the gap is 2.78 eV in zero field and only 0.07 eV for a field strength of 1.6 V/Å.
Whereas for zero field the electron density for the HOMO and the lowest two LUMOs are uniformly and symmetrically distributed along the molecule, the redistribution is already evident at 1.4 V/Å, particularly in the LUMO, and is completed at 1.6 V/Å. We note that as the HOMO-LUMO gap closes in the highest field there is still substantial overlap of the HOMO and LUMO π orbital wavefunctions suggesting that the molecule has become conductive. Although the overlap is larger at a field of 1.4 V/Å the gap is still too high, at 1.39 eV, for electron transfer along the molecule to occur at room temperature. We also note that the closure of the HOMO-LUMO gap is not monotonic as a function of field strength. We also note that these orbitals are affected differently by the field as Figure 2 clearly demonstrates.
Because for longer polymers the HOMO-LUMO gap is reduced, e.g., for an 8-ring polythiophene the gap is only 2.46 eV in zero field as compared to 2.76 eV for a 5-ring, one expects that the HOMO-LUMO gap vanishes in longer molecules at smaller fields. As long as the molecule is isolated, an external field will lead to charge separation: for very low fields the induced dipole is linear in the field but becomes progressively nonlinear as the field increases. In Figure 3 we show the accumulated charges on the various rings, obtained from a Mullikan analysis.
We briefly comment on the advantage of density functional theory over Hartree-Fock calculations in situations where an external field leads to a vanishing HOMO-LUMO gap. In the HF calculations, the self-consistent-field convergence is determined by the total energy minimization, while the electron occupation on the calculated orbitals is obtained simply by filling with electrons from the lowest energy level up, regardless of whether this leads to the lowest energy configuration or not. In most cases, there is no conflict between these two rules. However, in cases when the HOMO-LUMO gap becomes very small, it is possible that the state of total energy minimum does not correspond to the state where all unoccupied orbitals are energetically above all occupied orbitals, rather it may have an unoccupied orbital with an energy lower than the HOMO energy. As a result one finds in some systems a critical field above which self-consistent-field convergence cannot be reached if one insists on the standard Hartree-Fock rule of filling orbitals. On the other hand, density functional theory provides an equation relating the total energy, E, the occupation, ni, and the orbital energy, ei, namely ∂E/∂ni = ei. This guarantees that filling electrons with all unoccupied orbitals staying above all occupied orbitals always leads to a configuration that has the total energy at a minimum [16].
Fragmentation or dissociative field desorption of poly(thiophene) is caused foremost by the fact that in a strong electrostatic field this polymer becomes conducting so that charge transfer can occur easily along the molecule with the result that the end of the molecule in the field direction carries a substantial positive charge. This leads to the separation of the individual rings and also to their dissociation. However, as the fragments leave the high field region a partially deformed and opened ring can close again as this will minimize their energy substantially. This explains why mostly charged rings are observed in the mass spectrometer with much fewer small fragments [12].
In discussions of the structure of organic layers sandwiched between metallic contacts the point has been raised that their performance in a device might be enhanced due to tunneling across the interface layer at “microtip” structures arising from rough interface morphology [17,18]. We recall that organic layers are typically submicron thick and voltages across them are of the order of 10 V. This gives rise to fields across the polymer film of the order of mV/Å. At such strengths we do not expect significant field effects. However, if at weak spots the film thickness were reduced by an order of magnitude or more, field effects may become important. As mentioned in the previous paragraph different organic materials behave differently under high field conditions and detailed calculations have to be made for the particular material to see what constitutes a “high” field. All we can say in general is that high fields can be beneficial to reduce the band gap and may be detrimental, when they are strong enough, to lead to field-induced fragmentation.
3. Poly(ethylene glycol) in High Electric Fields
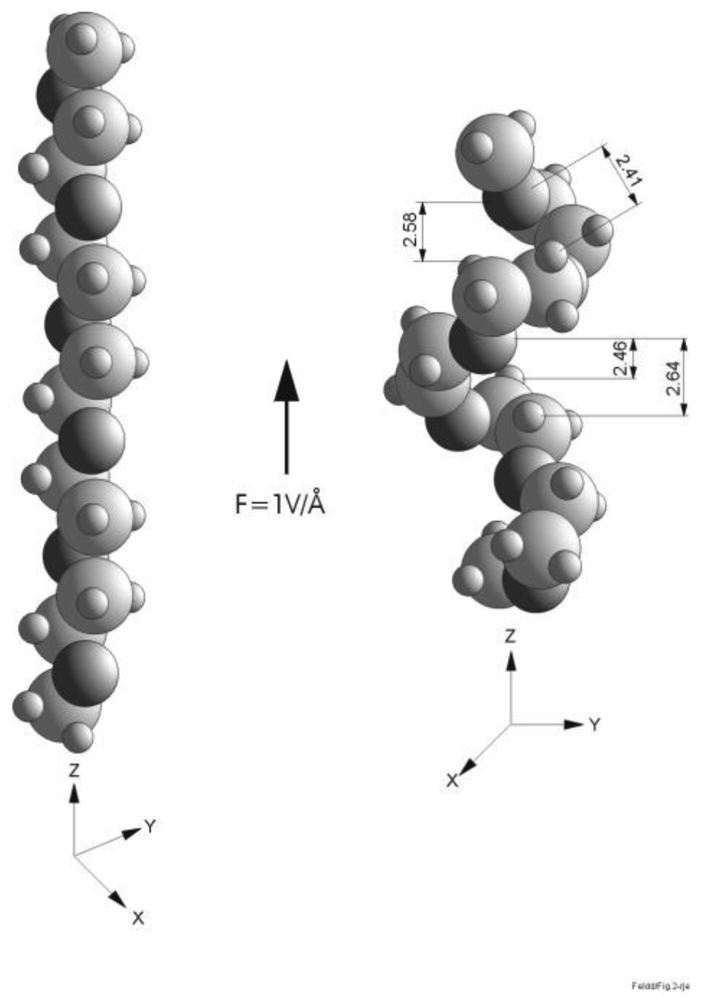
The response of polythiophene to a strong electrostatic field is very different to that of poly(ethylene glycol) or PEG. Its repeat units (which we will refer to as EG) are [–CH2–CH2–O] so that (EG)n is a polymer strand with n repeat units. The terminal groups are usually methyl or methoxy radicals. PEG retains its large HOMO-LUMO gap, severely limiting electron transfer down the molecule. All that happens is the creation of local dipole moments, more or less equal on each monomer. The result of this rather limited response is also very dramatic in that, for the helical conformer, a severe electrostriction of some 20% is observed in a field of 1.5 V/Å as we will show in this chapter.
The different conformers of PEG - such as the all-trans planar, helical or amorphous moieties differ in energy by less than 20 meV per EG unit [19,20]. As a result the environment in which these molecules are placed greatly effect their structure, stability and other properties like dipole moments and polarizabilities. For instance, it has been found that in ambient air the methoxy tri(ethylene glycol) terminal groups in a thiol self-assembled monolayer (SAM) are, on a silver surface, in the all-trans planar conformation. If the SAM is attached on a gold surface, where the packing density is lower by ca. 8–10%, helical (specified by uniform gauche rotations around the C–C bond) or amorphous (non-uniform rotations around the C–C bond) conformations are observed [21]. However, when the helical films on gold are exposed to aqueous solutions, dramatic changes are observed indicating the predominance of amorphous conformers [22].
The restructuring of the organic surface in the polar solvent indicates that the total energy of the solvated conformers is lower than the one of the dehydrated (EG) moieties. About 90% of the total solvation energy of the hydrogen bridge bonded water molecules to the oxygen atoms in the oxyethylene groups can be attributed to the interaction between the strong local electrostatic field around the water molecule (ca. 1 V/Å) and the field located around the oxygen atoms. Experimental results also suggest, that closeby charges and local electrostatic fields such as those generated by ions or a charged AFM tip effect the properties of the film, indicating that local fields generated by charged macromolecules or ions in an electrolyte can induce conformational changes in flexible organic molecules which are attached to a substrate [22].
To investigate theoretically the effect of electrostatic fields on a single PEG we must employ ab initio methods either Hartree-Fock theory plus MP2 or density functional theory with gradient corrected exchange/correlation functional [23]. We present here some of the interesting results; most of the work has been reported elsewhere [16,23]. Dipole moments and polarizabilities of a free PEG molecule in its helical and planar (all trans) conformations were calculated. Increasing the electrostatic field one finds that up to fields of the order of 1.5 V/Å the rather rigid planar conformer is not affected much in its structure. On the other hand, the easily deformable helical conformer undergoes significant electrostriction in fields between 0.5 and 1.5 V/Å, compare Figures 4 and 5, contracting by as much as 20% at 1 V/Å, accompanied by strong nonlinearities in dipole moment and polarizability. This is due largely to the establishment of additional, crosslinking hydrogen bonds between close monomers.
In contrast to poly(thiophene), poly(ethylene glycol) field-desorbs gradually with fragments such as H+, OCH+, CH2+leaving the end of the molecule in the field direction.
Electric fields of similar strength are also capable of substantially changing the orientation of the head groups making the topmost oxygen atoms either buried or exposed depending on the field polarity, structural changes starting at fields of 0.5 V/Å. At the same time the methoxy head groups twist in such a way that in the negative field direction the oxygen is exposed whereas in the positive direction the hydrogens are aligned rendering the ends electronegative and electropositive, respectively. The electrostriction is reversed around 1.5 V/Å when the molecule gets stretched more and more until around 2 V/Å field dissociation sets in breaking up the molecule piece by piece. To confirm these results (on a single macromolecule) experimentally one is tempted to study these molecules in a field ion microscope, using in particular Block's pulsed field technique.
Calculations of field effects on the structure of poly(ethylene glycol) molecules were only done for the planar and helical conformers. All other conformers have a larger aspect ratio of length to width and those atomic groups that are perpendicular to the field direction will not be effected much by the field because they lie on equipotential lines so that we do not expect drastic changes in these conformers as a field is applied. Rather, the helical conformer in a strong field is distorted to a point where it resembles more those conformers with mixed gauche transformations. Because the distortion and length contraction of the helical conformer in a strong electrostatic field is so large that it becomes similar to the mostly gauche conformers with mixed clockwise and anticlockwise rotations the entropic contribution to the free energy of a SAM will increase significantly as the field strength is increased. Such effects cannot be studied (yet) by ab initio (ground state, i.e., zero temperature) methods. We have therefore turned to Monte Carlo simulations of the response of an OEG-terminated SAM, which by its very nature, does not show any preference to specific conformers, i.e., accounts fully for entropic effects [16]. It was found that the structural changes predicted by the ab initio calculations for the helical conformer, in particular the re-orientation of the head groups making the topmost oxygen atoms either buried or exposed depending on the field polarity, are not qualitatively effected by entropic effects.
4. Polymerization of Sulfur in High Electric Fields
In the late 80's Block's group published a series of papers showing that high electric fields can help in the formation of large molecular or atomic cluster ions such as (H2.O)n and Sn. It was argued at the time that these ions might possibly be linear in which case one could talk of field-induced polymerization.
Using the field pulse technique Block and collaborators discovered the formation of large cluster ions of atoms and molecules such as (H2.O)n and Sn [4,5,24,25]. They exposed a tungsten tip in a field ion microscope to a beam of atoms or molecules leading to a multilayer adsorbed film. A field pulse desorbed some of these adparticles which in turn were detected in a mass spectrometer. A typical mass spectrum from an adlayer of sulfur is reproduced in Figure 6 [24].
Sulfur cluster ions up to S28+ were detected. This was very unexpected because in the absence of a field sulfur predominantly forms molecules with mostly two and eight atoms (in the gas phase). S8 terminates the series because of the formation of a stable octogonal ring structure. The formation of the larger cluster ions in a field were conditional on the strength of the intermittent reaction field but not on the pulse field. This suggests that the clusters were pre-formed in or on the sulfur film and ionized as they were field desorbed. Because the cluster ions did not terminate with eight atoms it was suggestive that the field suppresses the formation of an octogonal ring. Rather one can speculate that chains of sulfur atoms protrude from the adsorbate and pieces of these chains are field desorbed. Unfortunately not much work has been done since to test these ideas. Here are some preliminary results.
A number of theoretical studies of sulfur clusters have appeared over the past decade, employing the Hartree-Fock/MP2 method or gradient corrected density functional theory. It is found that the binding energy per sulfur atom increases with the size of the cluster and reaches the asymptotic limit at about S6 with S2, S6 and S8 particularly stable. Of interest in the context of field effects is the finding that the polarisability increases with the cluster size correlating linearly with the molecular volume [26]. The propensity of sulfur to catenation leads to over 20 crystalline allotropes all of which are composed of homocyclic rings with as much as 20 atoms. Their detailed structures have also been investigated theoretically in great detail [27]. Of more interest to our concerns here is a study of anionic sulfur clusters [28]. It is found that most of the anionic clusters have chain configurations with the most stable clusters from S14 to S20 having helical conformations, very different from neutral clusters. On the other hand, cationic clusters have been of little interest so far. We have used density functional theory with a gradient corrected exchange/correlation functional to calculate the energetics and structure of neutral and ionic sulfur clusters, both in the absence and in the presence of an electric field.
First we address the question of the relative stability of linear and cyclic S8 in the neutral state as a function of field strength; numbers are given in Table 1. We have defined a binding energy per sulfur atom as En = E(S1) – E(Sn)/n. Here E(Sn) is the total electronic energy of a cluster of n sulfur atoms.
In the absence of a field the binding energies per sulfur atom are 2.07 eV and 2.42 eV for the linear and the cyclic conformer of S8, respectively; note that 2.1 eV is about the average binding energy per sulfur in the smaller sulfur clusters. Applying a field of 0.5 V/Å reduces the stability of the cyclic conformer, increases its dipole moment by an order of magnitude and has a dramatic effect on its geometry. This is seen in selfconsistent ab initio calculations by following the evolution of the molecular structure from one iteration to the next. Indeed, already the first iteration results in a lengthening of the bond distance between the second and third sulfur by 0.2 Å, which increases by another 0.3 Å in the fourth iteration. This implies that in this field the linear S8 is already weakly unstable and dissociates into an S6 and S2. This is part of the reason why in the ion yield curves in Figure 6 the signal for S8 is reduced compared to that of S7. Increasing the field to 1 V/Å adds so much polarization energy that the linear molecule becomes more stable compared to the cyclic one but at the expense of being very unstable towards dissociation.
The relative probability of field ionization is also (but not solely) controled by the ionization potential, which we list in Table 2 for small sulfur clusters. As we can see the ionization potential decreases continuously as the cluster gets bigger with slight differences between different conformers. Noteworthy is the fact that S7 is particularly easy to ionize, thus its dominance in the ion yield, whereas cyclic S8 is rather hard to ionize. But then we just argued that linear S8 is stabilized by a field so that its low ionization potential is advantageous albeit with the further consequence of possible dissociation.
Problems that remain to be solved in our quest to understand the mass spectrum of sulfur cluster ions include foremost a more compehensive list of calculations for binding energies and ionization potentials as a function of field strength for all conformers of the clusters. Another important problem, not addressed at all in this short discussion, is the field variation through and above the proposed liquid sulfur multilayer on the tip of the field ion tip; a jellium type calculation that includes a dielectric layer would give a first idea. Most importantly, in our assessment, is the understanding of how a fluid sulfur layer under the influence of strong electric fields becomes hydrodynamically unstable to form protrusions and chainlike whiskers.
5. Polymerization of Water in High Electric Fields
Water is no doubt the most important molecule for life as well as for the careers of many scientists and its structure and dynamics have thus been explored extensively in natural selection and in the pursuit of intellectual curiosity.
An unexpected discovery was made in the late 80s by Jochen Block and his colleagues at the Fritz-Haber-Institut that, exposing Ni and W field emitter tips in the field ion mode to water vapor, lead not only to the adsorption of a water monolayer but under appropriate fields to the formation of a brush of water whiskers [29]. In other words, in high electric fields water aggregates become linear and the polymerization of water is triggered. Outside the small community of the field emission society this work remained unnoticed. The inference of polymer water was originally made on the basis of a simple argument [29]: the water molecule has a dipole moment pwater = 1.85 Debye. Thus aligning a water molecule in an electric field F at an angle γ leads to an energy gain of pFcosγ. To maintain one hydrogen bridge bond between two successive water molecules in a linear array the dipoles are at an angle of relative to the axis of the linear array. If the latter is also the direction of the electric field we need γ = 70°. Thus this alignment results in an energy gain of about 0.29 eV per molecule in a field of 1 V/Å. This is larger than a hydrogen bridge bond which is typically 0.05–0.25 eV. On the other hand, in the surface layer of a water film adsorbed on a metal the coordination is typically between one and two. Thus polymerization of water in a field of 1 V/Å about is plausible.
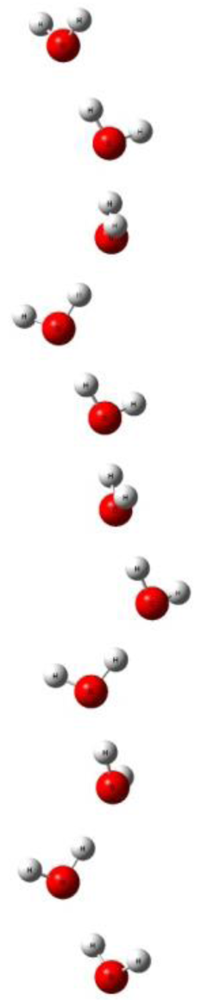
We have recently revisited this topic from a theoretical point of view with the aim to understand the details of polymerization of water answering questions such as: (1) what are the lower and upper threshold fields for water whiskers of different lengths? (2) What are the electronic mechanisms such as charge transfer that result in polymerization? (3) How valid is the dipole alignment argument presented above? At this stage we can report some preliminary results that confirm the existence of linear water whiskers. In Figure 7 we show a cluster of four water molecules which in the presence of a field of 0.5 V/Å stretches from a compact cluster into a linear one. Increasing the field beyond 1.5 V/Å leads to dissociation. We also calculated a cluster of 11 water molecules, see Figure 8. Again the linear whisker is stable around fields of 0.5 V/Å. These first results are very encouraging but more work is needed before we can address the question of whether a long water whisker in a high enough field will become conducting. This would have tremendous implications for the electric double layer in electrolytes and the local dynamics of zeolites with fields of the order of 0.1 to 0.5 V/Å.
6. Conclusions
Electric fields of the order of volts per angstrom affect the valence electrons on atoms shifting their energies by electron volts with respect to each other. In recent years a microscopic theory has emerged that looks at the new physics and chemistry from a microscopic point of view. We now understand field adsorption as field-induced chemisorption, a mechanism that affects even the lightest rare gases. We also now have a microscopic theory to describe kinetic effects in high electric fields at surfaces such as thermal field desorption and field evaporation [30,31].
Chemistry in high electric fields is an even more exciting field. Because fields of the order of V/Å affect the valence electrons of atoms, new molecular species are stabilized in high fields thus opening up new reaction pathways in heterogeneous catalysis. Most work so far has been concentrating on static electric fields; however, many new phenomena are to be expected in alternating fields as well, as the work on photon-induced field desorption suggests. In particular, the metallization of polythiophene, the electrostriction in poly(ethylene glycol) and the formation of sulfur and water polymer whisker are striking phenomena induced by high electric fields.
What is important to learn from the example of field-induced chemistry is the fact that high electric fields can significantly alter reaction pathways, and indeed promote or hinder entire reactions. The fields involved are typically less than 1 V/Å, i.e., of the order of those generated between a tunneling tip and a substrate metal or semiconductor under conditions of atomic manipulation.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F[V/Å] | ΔE[eV] | D[Debye] | D(S8-S1)[Å] | d(S2-S3)[Å] |
| 0 | −0.34 | 1.24 | 11.3 | 1.93 |
| 0.5 | −0.26 | 13.9 | 11.4 | 2.18 (1) |
| −0.24 | 16.9 | 11.7 | 2.38 (4) | |
| 1.0 | 37.0 | 11.4 | 2.18 (1) | |
| 0.195 | 43.3 | 12.2 | 2.49 (5) |
| Sn n | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8(linear) | 8(ring) |
| IP [eV] | 12.75 | 9.67 | 8.8 | 8.6 | 8.3 | 7.8 | 7.7 | 7.6 | 9.00 |
Acknowledgments
Most of the work reported here was supported over the years by grants from the Office of Naval Research and NSERC.
References
- Kreuzer, H.J. Surface Physics and Chemistry in High Electric Fields. In Chemistry and Physics of Solid Surfaces VIII; Springer Series in Surface Sciences; Vanselow, R., Howe, R., Eds.; Springer-Verlag: Berlin, Germany, 1990; Volume 22, pp. 133–158. [Google Scholar]
- Kreuzer, H.J. Physics and Chemistry in High Electric Fields. Surface Sci. 1991, 246, 336–347. [Google Scholar]
- Kreuzer, H.J. Chemical Reactions in High Electric Fields. In Surface Science of Catalysis: In-Situ Probes and Reaction Kinetics; Dwyer, D.J., Hoffmann, F.M., Eds.; ACS Symposium Series; American Chemical Society: Washington, D.C., USA, 1992; Volume 482, pp. 268–286. [Google Scholar]
- Block, J.H. Field Desorption and Photon-Induced Field Desorption. In Chemistry and Physics at Solid Surfaces IV; Vanselow, R., Howe, R., Eds.; Springer-Verlag: Berlin, Germany, 1982. [Google Scholar]
- Block, J.H. Surface Reactions on an Atomic Scale. In Surface Science of Catalysis: In-Situ Probes and Reaction Kinetics; Dwyer, D.J., Hoffmann, F.M., Eds.; ACS Symposium Series; American Chemical Society: Washington, D.C., USA, 1992; Volume 482, pp. 268–286. [Google Scholar]
- Wang, L.C.R.; Kreuzer, H.J.; Nishikawa, O. Polythiophene in Strong Electrostatic Fields. Org. Electron. 2006, 7, 99–106. [Google Scholar]
- Dhoot, A.S.; Yuen, J.D.; McCulloch, I.; Moses, D.; Heeger, A.J. Beyond the Metal-Insulator Transition in Polymer Electrolyte Gated Polymer Field-Effect Transistors. PNAS 2006, 103, 11834–11837. [Google Scholar]
- Block, J.H. Surface Structures and Surface Reactions Investigated by Field Ion Mass Spectroscopy. J. Vac. Sci. Technol. 1969, 6, 925. [Google Scholar]
- Inghram, M.G.; Gomer, R.Z. Mass Spectrometer Investigation of the Field Emission of Positive Ions. Naturforsch. Teil A 1955, 10, 864. [Google Scholar]
- Beckey, H.D.; Röllgen, F.W. Surface Reactions Induced by Field Ionization of Organic Molecules. Surf. Sci. 1970, 23, 69. [Google Scholar]
- Fichou, D. Handbook of Oligo and Polythiophene; Wiley-VCN: Weinheim, Germany, 1999. [Google Scholar]
- Nishikawa, O.; Taniguchi, M. Atom-by-Atom Analysis of Non-Metallic Materials by the Scanning Atom Probe. Chin. J. Phys. 2005, 43, 111–123. [Google Scholar]
- van Eijck, L.; Johnson, M.R.; Kearley, G.J. Intermolecular Interactions in Bithiophene as a Model for Polythiophene. J. Phys. Chem. A 2003, 107, 8980–8984. [Google Scholar]
- Della Sala, F.; Heinze, H.H.; Görling, A. Excitation Energies of Terthiophene and its Dioxide Derivative: A First-Principles Study. Chem. Phys. Lett. 2001, 339, 343. [Google Scholar]
- Telesca, R.; Bolink, H.; Yunoki, S.; Haziioannou, G; van Duijen, P.T.; Snijders, J.G.; Jonkman, H.T.; Sawatzky, G.A. Density-Functional Study of the Evolution of the Electronic Structure of Oligomers of Thiopene: Towards a Model Hamiltonian. Phys. Rev. B 2001, 63, 155112. [Google Scholar]
- Pertsin, A.J.; Grunze, M.; Kreuzer, H.J.; Wang, R.L.C. The Effect of Electrostatic Fields on an Oligo (ethylene glycol) Molecule: Dipole Moments, Polarizabilities and Field Dissociation. Phys. Chem. Chem. Phys. 2000, 2, 1729–1733. [Google Scholar]
- Kanai, H; Ichinosawa, S.; Sato, Y. Effect of Aromatic Diamines as a Cathode Interface Layer. Synth. Met. 1997, 91, 195. [Google Scholar]
- Hill, I.G.; Rajagopal, A.; Kahn, A. Energy-Level Alignment at Interfaces between Metals and the Organic Semiconductor 4,4′-N,N-dicarbazolyl-biphenyl. J. Appl. Phys. 1998, 84, 3236–3242. [Google Scholar]
- Wang, R.L.C.; Kreuzer, H.J.; Grunze, M. Molecular Conformation and Solvation of Oligo(ethylene glycol)-Terminated Self-Assembled Monolayers and Their Resistance to Protein Adsorption. J. Phys. Chem. B 1997, 101, 9767–9773. [Google Scholar]
- Wang, R.L.C.; Kreuzer, H.J.; Grunze, M. The Interaction of Oligo(ethylene oxide) with Water: A Quantum Mechanical Study. Phys. Chem. Chem. Phys. 2000, 2, 3613–3622. [Google Scholar]
- Harder, P.; Grunze, M.; Whitesides, G.M.; Laibinis, P.E. Molecular Conformation in Oligo(ethylene glycol)-Terminated Self-Assembled Monolayers on Gold and Silver Surfaces determines their Ability to Resist Protein Adsorption. J. Phys. Chem. B 1998, 102, 426436. [Google Scholar]
- Feldman, K.; Hähner, G.; Spencer, N.D.; Harder, P.; Grunze, M. Probing Resistance to Protein Adsorption of Olig(ethylene glycol)-terminated Self-Assembled Monolayers by Scanning Force Microscopy. J. Am. Chem. Soc. 1999, 121, 10134–10141. [Google Scholar]
- Wang, R.L.C.; Kreuzer, H.J.; Grunze, M.; Pertsin, A.J. The Effect of Electrostatic Fields on an Oligo (ethylene glycol) Terminated Alkanethiol Self-Assembled Monolayer. Phys. Chem. Chem. Phys. 2000, 2, 1721–1733. [Google Scholar]
- Cocke, D.L.; Abend, G.; Block, J.H. A Mass Spectrometric Technique for Observation of Chemical Kinetics in Physisorbed Layers. Int. J. Chem. Kinet. 1977, 9, 157–159. [Google Scholar]
- Dirks, J.; Drachsel, W.; Block, J.H. Photon Induced Field Desorption of Water Clusters. Coll. Phys. 1989, 50, 153–158. [Google Scholar]
- Millefiori, S.; Alperone, A. Ab initio Study of the Structure and Polarizability of Sufur Clusters Sn (n = 2–12). J. Phys. Chem. A 2001, 105, 9489–9497. [Google Scholar]
- Cioslowski, J.; Szarecka, A.; Moncrieff, D. Conformations and Thermodynamic Properties of Sulfur Homocycles, 1. The S6, S7, and S8 Molecules. J. Phys. Chem. A 2001, 105, 501–505. [Google Scholar]
- Chen, M.D.; Liu, M.L.; Zheng, L.S.; Zhang, Q.E.; Au, C.T. A Density Functional Study for the Isomers of Anionic Sulfur Clusters Sn - (n = 3–20). Chem. Phys. Lett. 2001, 350, 119–127. [Google Scholar]
- Jaenicke, J.; Ciszewski, A.; Dosselmann, J.; Drachsel, W.; Block, J.H.; Menzel, D. Field-Induced Structural Changes in Adsorbed Layers of Polar Molecules. J. Phys. Colloque C6 1988, 49, 191–195. [Google Scholar]
- Kreuzer, H.J.; Watanabe, K.; Wang, L.C. Theory of Field Desorption and Field Ionization: Thermal Field Desorption of Helium. Surface Sci. 1990, 232, 379–392. [Google Scholar]
- Wang, L.C.; Kreuzer, H.J. Kinetic Theory of Field Evaporation of Metals. Surface Sci. 1990, 237, 337–346. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Karahka, M.; Kreuzer, H.J. Conduction and Electrostriction of Polymers Induced by High Electric Fields. Polymers 2011, 3, 51-64. https://doi.org/10.3390/polym3010051
Karahka M, Kreuzer HJ. Conduction and Electrostriction of Polymers Induced by High Electric Fields. Polymers. 2011; 3(1):51-64. https://doi.org/10.3390/polym3010051
Chicago/Turabian StyleKarahka, Markus, and Hans Jürgen Kreuzer. 2011. "Conduction and Electrostriction of Polymers Induced by High Electric Fields" Polymers 3, no. 1: 51-64. https://doi.org/10.3390/polym3010051