2. Experimental Section
Materials and Instrumentation. PEG with a nominal molar mass of 1,450 g/mol, silver (I) oxide (Ag2O), p-toluenesulfonyl chloride (TsCl), methanesulfonyl chloride, triphenylphosphine (PPh3), 2,2’-dipyridyl disulfide (2-PDS), tris(2-carboxyethyl)phosphine hydrochloride (TCEP), N-hydroxysuccinimide (NHS), sodium azide (NaN3), N-ethyl-N-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), hydrochloric acid (HCl), 2-(N-morpholino) ethanesulfonic acid (MES), N,N- dimethylformamide (DMF), potassium iodide (KI), triethylamine (NEt3), ammonium hydroxide (28% in water), sodium hydrosulfide hydrate (NaSH), sodium chloride (NaCl), and calcium chloride dihydrate were purchased from Sigma-Aldrich (Sigma-Aldrich, Switzerland). Thioacetic acid, potassium bicarbonate, and acetic acid were obtained from Fluka (Fluka, Switzerland). Ammonia (7N in MeOH) was obtained from Brunschwig (Chemie Brunschwig AG, Switzerland). Na-alg (HV Kelton, lot no. 46198 A) was obtained from Kelco (San Diego, CA, USA). The intrinsic viscosity [η] in 0.1 M NaCl at 20 °C and the molar guluronic acid fraction FG have been analyzed as [η]0.1 M NaCl = 930 mL/g and FG = 0.41. Unless otherwise mentioned, all reagents were analytical grade and were used without further purification. Other chemicals and their suppliers were toluene (VWR, Switzerland), dichloromethane (DCM), methanol (MeOH) and diethyl ether (Fisher Scientific, Switzerland), filter cell cake (Macherey-Nagel, Switzerland), and sodium sulfate anhydrous (AppliChem, Germany). 1H and 13C-NMR spectra were recorded on a Bruker AV-400 NMR spectrometer. NMR data were processed by Mnova software (Mestrelab Research, Spain). Gel permeation chromatography (GPC) was carried out on a Waters system (Waters AG, Switzerland) equipped with column heater, manual injector, and refractive index and UV/Vis detector. PEG standards were used for calibration. Separation was achieved combining three columns (Waters Styragel THF HR 2, HR 3, and HR 4) at a flow rate of 1.0 mL/min with THF as the mobile phase at 40 °C. Fourier transform infrared (FTIR) spectra were recorded on a Perkin Elmer spectrometer (Spectrum One) equipped with Spectrum software (version 5.3, Perkin Elmer). Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI TOF/TOF MS) was performed in reflection mode using ABI 4800 MALDI TOF/TOF Analyzer (Applied Biosystems). The matrix solution was alpha-cyano-4-hydroxycinnamic acid (7 mg/mL in ACN/0.1%TFA (1:1 v/v)).
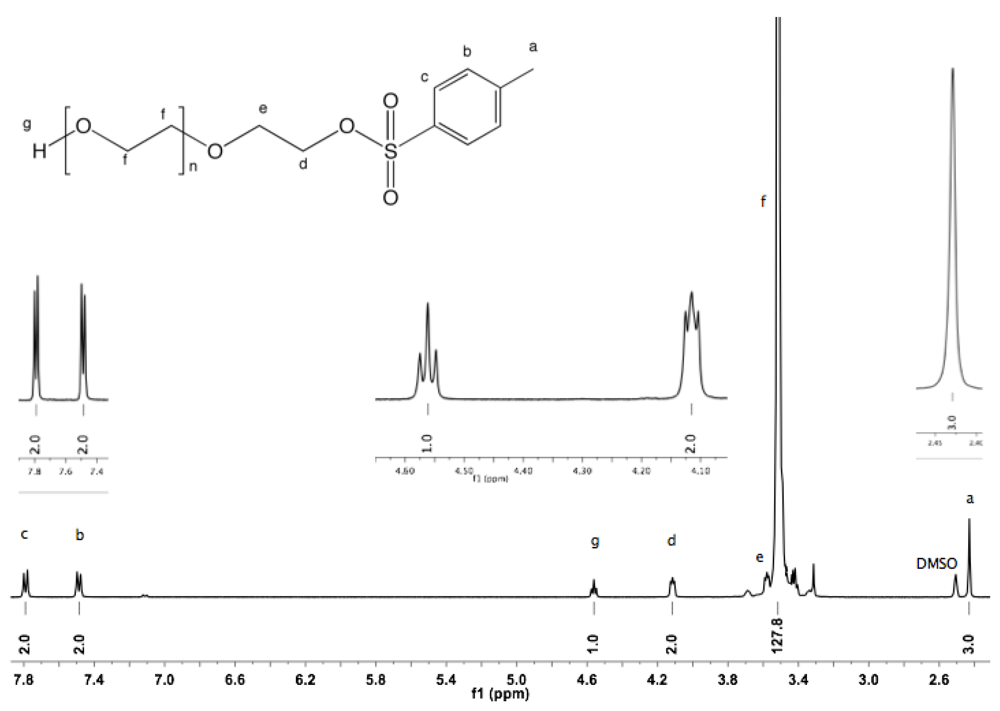
Synthesis of α-tosyl-ω-hydroxyl PEG (1). PEG (1450 g/mol, 20 g, 13.8 mmol, previously dried by azeotropic distillation in toluene using a Dean-Stark trap) was dissolved in 250 mL of dry toluene. Ag
2O (1.5 eq, 4.8 g, 20.7 mmol) and KI (0.2 eq, 458 mg, 2.76 mmol) were added. To this rapidly stirred solution, TsCl (1.05 eq, 2.76 g, 14.5 mmol) was added in one portion. The reaction mixture was left at room temperature (rt) with constant stirring for 12 h before filtration over a filter cell cake and solvent removal by rotary evaporation were performed. The crude product was dissolved in 30 mL DCM and then precipitated by dropwise addition into diethyl ether. The polymer was collected by filtration (22 g, 98%).
1H-NMR (DMSO, δ in ppm): 7.79 (2H, d, J = 8 Hz), 7.49 (2H, d, J = 8 Hz), 4.56 (1H, t, J = 5.4 Hz, O
H), 4.11 (2H, t, J = 4.4 Hz, C
H2-SO
2), 3.49 (128H, s, PEG backbone), 2.43 (3H, s, C
H3).
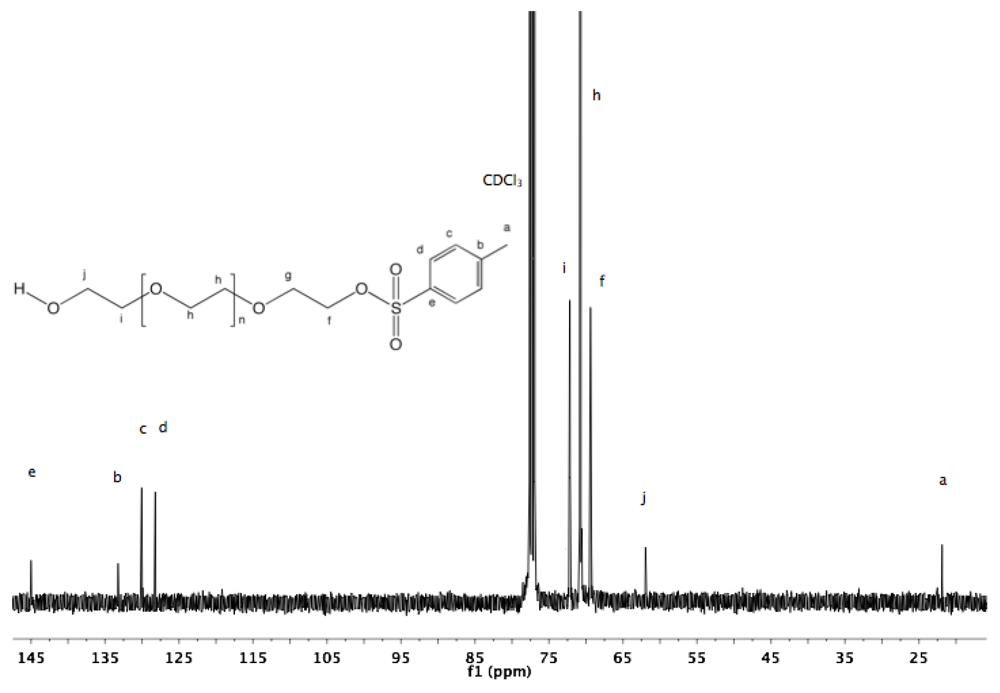
13C-NMR (CDCl
3) δ: 145, 133.24, 130.04, 128.21, 72.19, 70.79, 69.39, 61.95, 21.87. (Supporting Information (SI),
Figure S1 and
Figure S2).
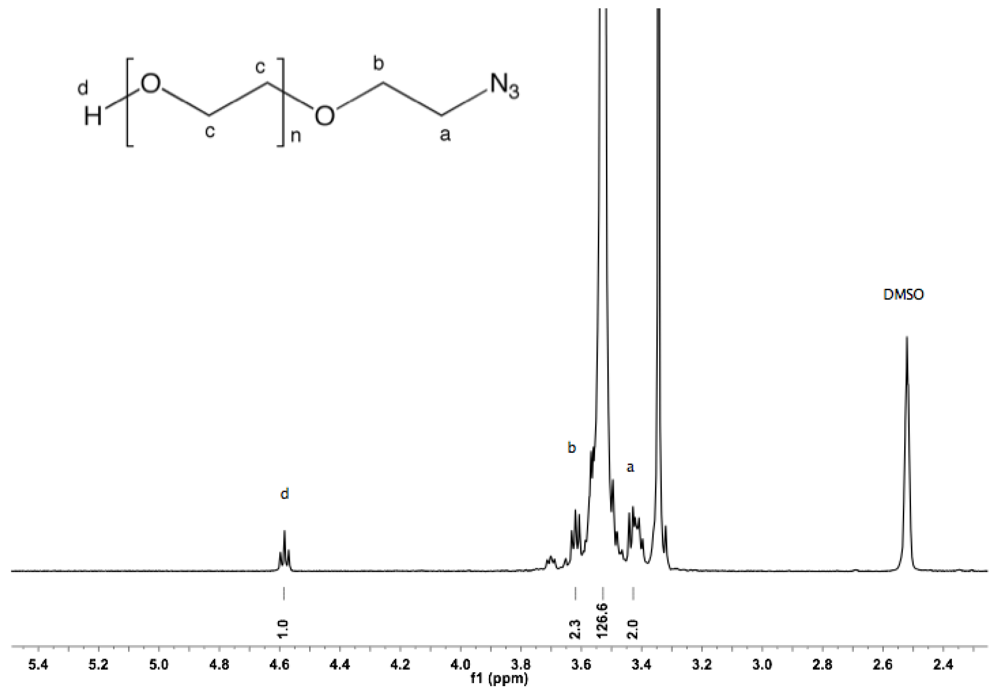
Synthesis of α-azide-ω-hydroxyl PEG (2). α-Tosyl-ω-hydroxyl PEG (1, 10 g, 6.23 mmol) and NaN
3 (2 g, 31 mmol) were dissolved in 150 mL of dry DMF, and the mixture was stirred overnight at 90 °C under argon atmosphere. After cooling down to rt and filtration, DMF was removed under vacuum. The crude product was dissolved in 100 mL DCM and washed twice with brine and twice with water. The organic layer was dried over sodium sulfate, reduced to a small volume by rotary evaporation, and finally precipitated by dropping into diethyl ether. The polymer was collected by filtration (8.7 g, 95%).
1H-NMR (DMSO, δ in ppm): 4.56 (1H, t, J = 5.6 Hz, O
H), 3.6 (2H, t, J = 5 Hz, C
H2CH
2-N
3), 3.5 (127H, s, PEG backbone), 3.4 (2H, t, J = 5 Hz, C
H2-N
3).
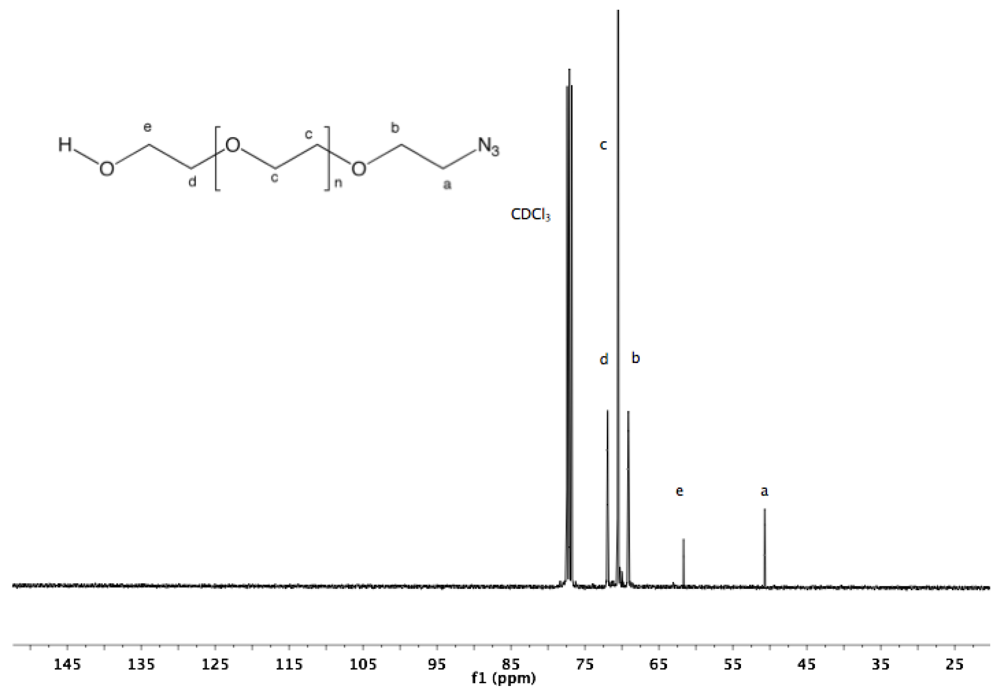
13C-NMR (CDCl
3, δ in ppm): 71.9, 70.53, 69.13, 61.65, 50.64 (
Figure S3 and
Figure S4).
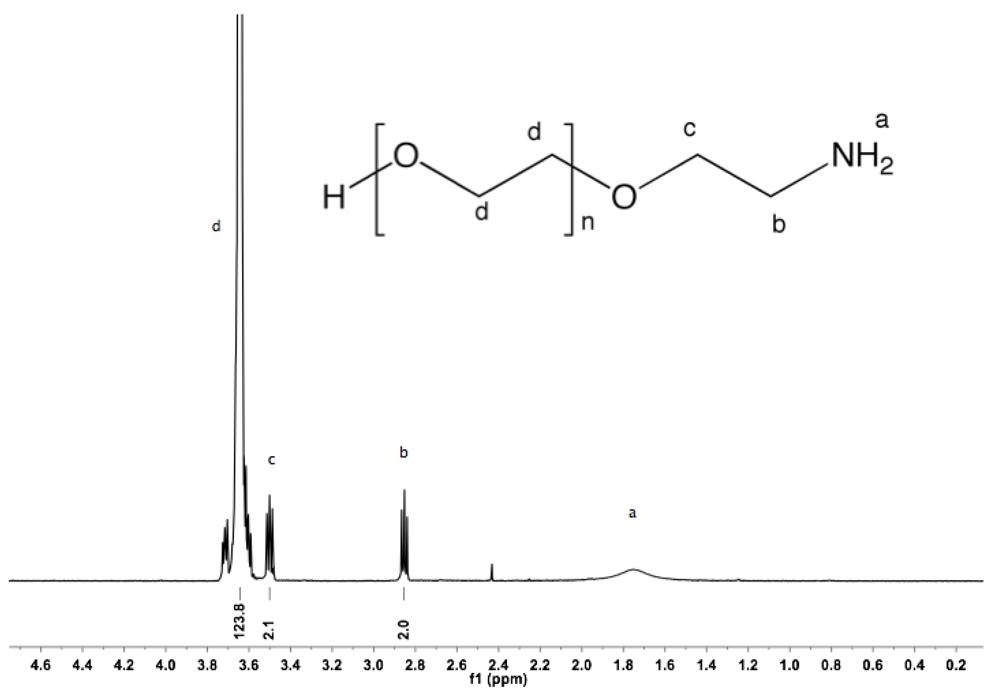
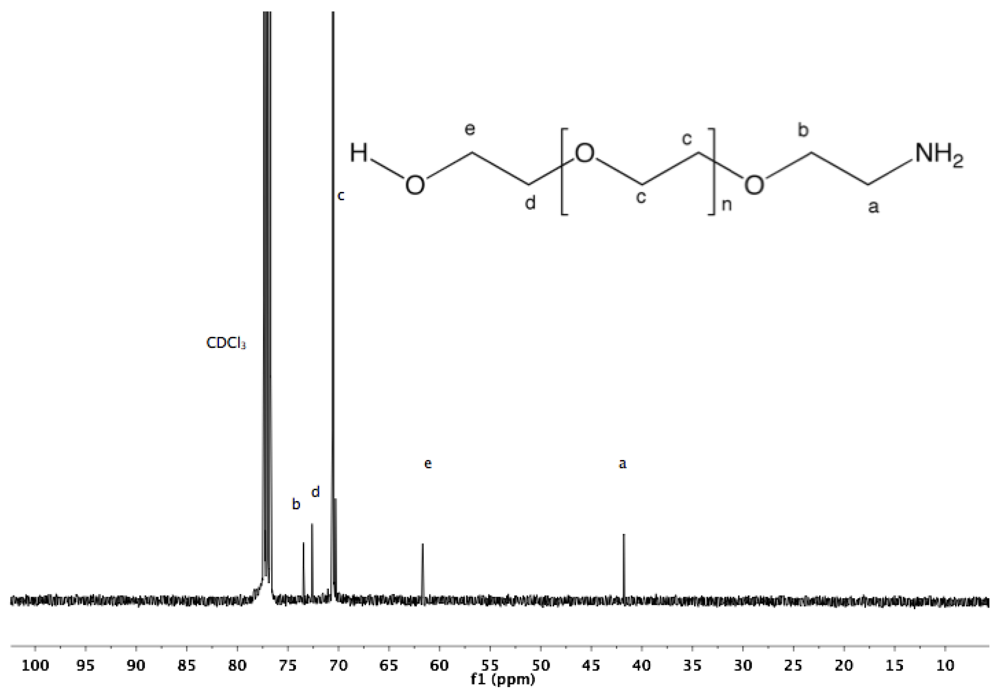
Synthesis of α-amine-ω-hydroxyl PEG (3). To a solution of α-azide-ω-hydroxyl PEG (2, 5 g, 3.4 mmol) in 100 mL MeOH, PPh
3 was added (2.67 g, 10.2 mmol). The reaction mixture was refluxed overnight under argon atmosphere and then cooled down to rt. The solvent was removed by rotary evaporation. The crude product was dissolved in 20 mL DCM and added dropwise into diethyl ether. α-Amine-ω-hydroxyl PEG was collected by filtration (4.7 g, 95%).
1H-NMR (CDCl
3, δ in ppm): 3.64 (124H, s, PEG backbone), 3.5 (2H, t, J = 5.4 Hz, C
H2CH
2-NH
2), 2.85 (2H, t, J = 5.4 Hz, C
H2-NH
2).
13C-NMR (CDCl
3, δ in ppm): 73.45, 72.61, 70.55, 61.67, 41.78 (
Figure S5 and
Figure S6).
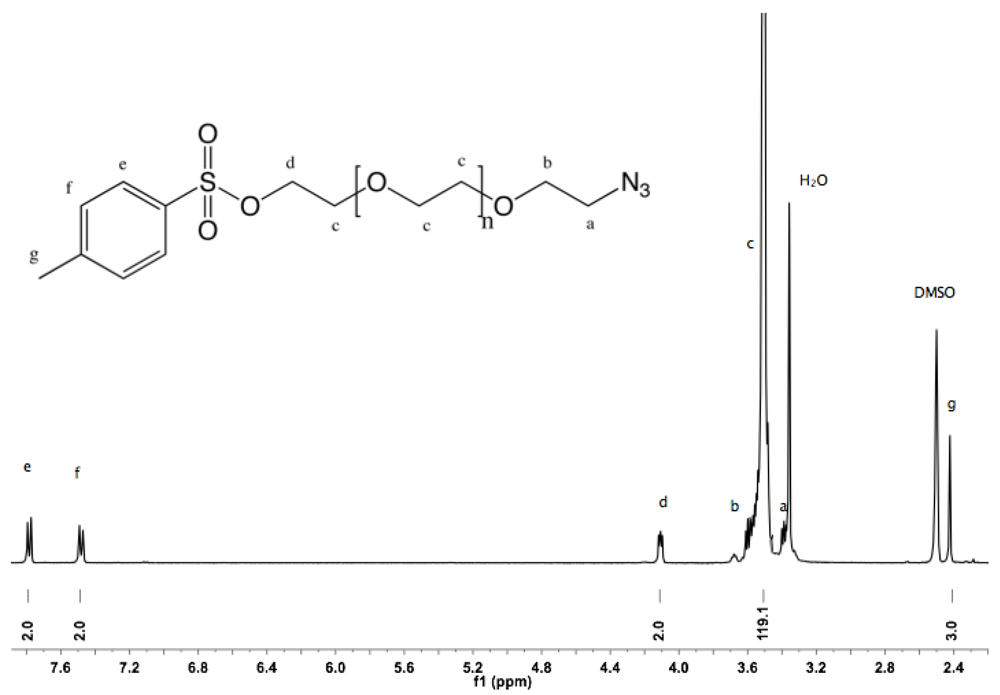
Synthesis of α-azide-ω-thioacetate PEG (4). α-Azide-ω-hydroxyl PEG (2, 10 g, 6.8 mmol) was dissolved in 100 mL DCM. NEt
3 (6 mL, 43 mmol) and TsCl (6.3 g, 33 mmol) were added. The mixture was stirred overnight at rt. The solution was then filtered, washed twice with saturated NH
4Cl solution and twice with water. The organic layer was dried over sodium sulfate, filtered, reduced to small volume by rotary evaporation, and added dropwise into diethyl ether. The α-azide-ω-tosyl PEG was collected by filtration (10.5 g, 95%).
1H-NMR (DMSO, δ in ppm): 7.78 (2H, d, J = 8 Hz), 7.48 (2H, d, J = 8 Hz), 4.1 (2H, t, J = 4.4 Hz, C
H2-SO
2), 3.5 (119H, s, PEG backbone), 3.39 (t, J = 5 Hz, C
H2-N
3), 2.41 (3H, s, C
H3) (
Figure S7). The thioacetate function was introduced by reaction with sodium carbonate/thioacetic acid. α-Azide-ω-tosyl PEG (5 g, 3.1 mmol) was dissolved in 100 mL of dry DMF, evacuated and purged with argon. To this rapidly stirred solution, potassium carbonate (2.15 g, 15.5 mmol) and thioacetic acid (1.1 mL, 15.5 mmol) were added, and the mixture was stirred at rt overnight. DMF was removed by rotary evaporation. The crude product was dissolved in 100 mL DCM before filtration and treatment with activated charcoal for 2 h. The mixture was filtered over filter cell cake, the filtrate reduced to small volume by rotary evaporation, and added into diethyl ether. α-Azide-ω-thioacetate was collected by filtration as white solid (3.9 g, 83%).
1H-NMR (CDCl
3, δ in ppm): 3.63 (129H, s, PEG backbone), 3.58 (2H, t, J= 6.4 Hz, C
H2-CH
2-S), 3.38 (2H, t, J= 5.2 Hz, C
H2-N
3), 3.07 (2H, t, J= 6.4 Hz, C
H2-S), 2.32 (3H, s, COC
H3).
13C-NMR (CDCl
3, δ in ppm): 195.59, 71.89, 69.89, 69.13, 50.65, 30.74, 28.98 (
Figure S8 and
Figure S9).
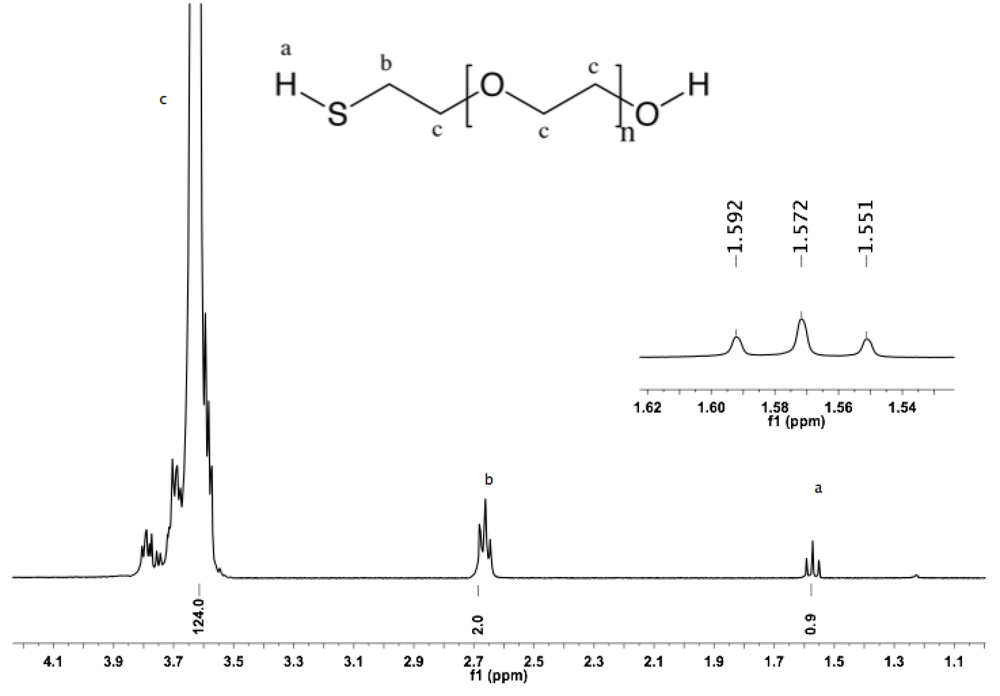
Synthesis of α-thiol-ω-hydroxyl PEG (5). α-Tosyl-ω-hydroxyl PEG (
1, 5 g, 3.1 mmol) in 80 mL of distilled water was treated with NaSH (7.5 g, 0.1 mol). The solution was kept under argon atmosphere, stirred for 6 h at rt, and subsequently at 60 °C for 2 h. The reaction mixture was neutralized (pH = 7) by slow addition of HCl, extracted three times with 50 mL DCM, and dried over sodium sulfate anhydrous. The organic phase was reduced to a small volume by rotary evaporation, and then added dropwise into diethyl ether. α-Thiol-ω-hydroxyl PEG was collected by filtration (3.8 g, 84%).
1H-NMR (DMSO: δ in ppm): 3.62 (124H, s, PEG backbone), 2.66 (2H, t, J= 8 Hz, C
H2-SH), 1.57 (t, J = 8 Hz, S
H).
13C-NMR (CDCl
3, δ in ppm): 72.78, 72.19, 70.78, 61.93, 24.48 (
Figure S10 and
Figure S11).
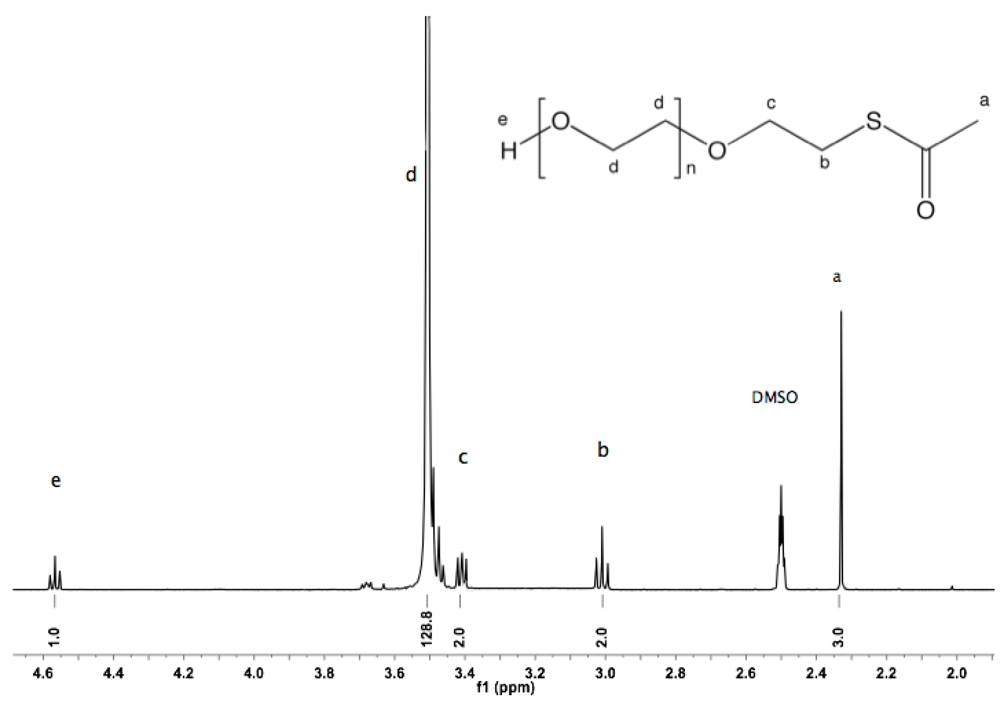
Synthesis of α-thioacetate-ω-hydroxyl PEG (6). α-Tosyl-ω-hydroxyl PEG (
1, 10 g, 6.23 mmol) was dissolved in 150 mL of dry DMF, evacuated and purged with argon. To the rapidly stirred solution, potassium carbonate (4.3 g, 31.1 mmol) and thioacetic acid (2.2 mL, 31.1 mmol) were added. The mixture was stirred at rt overnight. DMF was removed by rotary evaporation. The crude product was dissolved in 50 mL DCM before treatment with activated charcoal for 2 h, filtration over filter cell cake, reduction to small volume by rotary evaporation, and precipitation in diethyl ether. The polymer was collected by filtration (8 g, 86%).
1H-NMR (DMSO: δ in ppm): 4.56 (1H, t, J = 5.6 Hz, O
H), 3.5 (129H, s, PEG backbone), 3.01 (2H, t, J = 6.6 Hz, C
H2S), 2.33 (3H, s, SCOC
H3).
13C-NMR (CDCl
3, δ in ppm): 195.58, 72.75, 70.47, 69.89, 61.79, 30.73, 28.97 (
Figure S12 and
Figure S13).
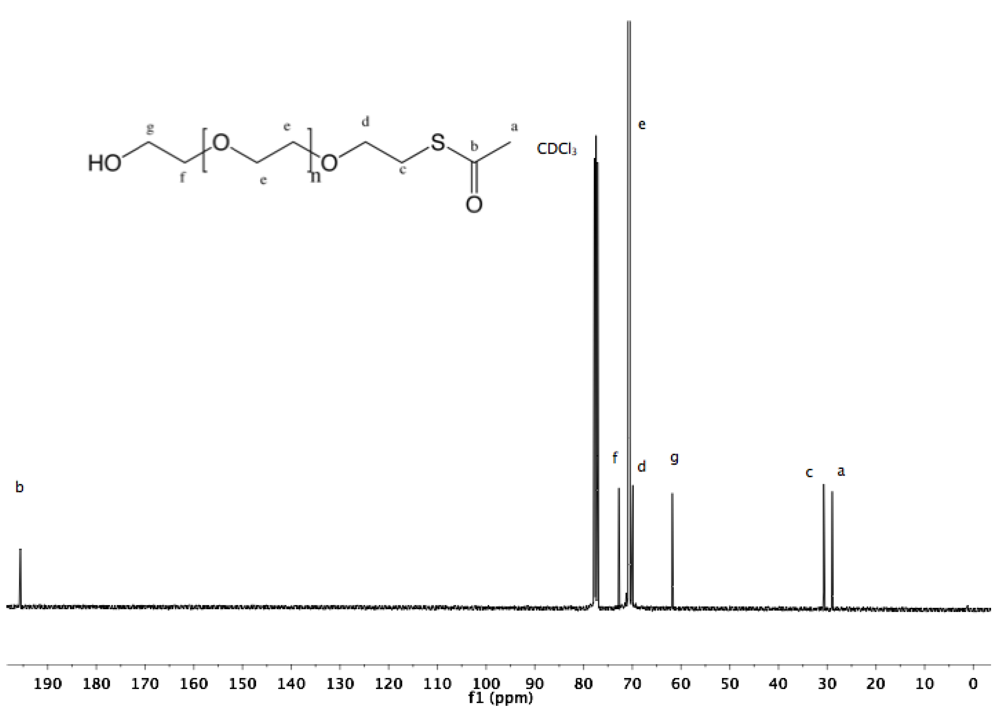
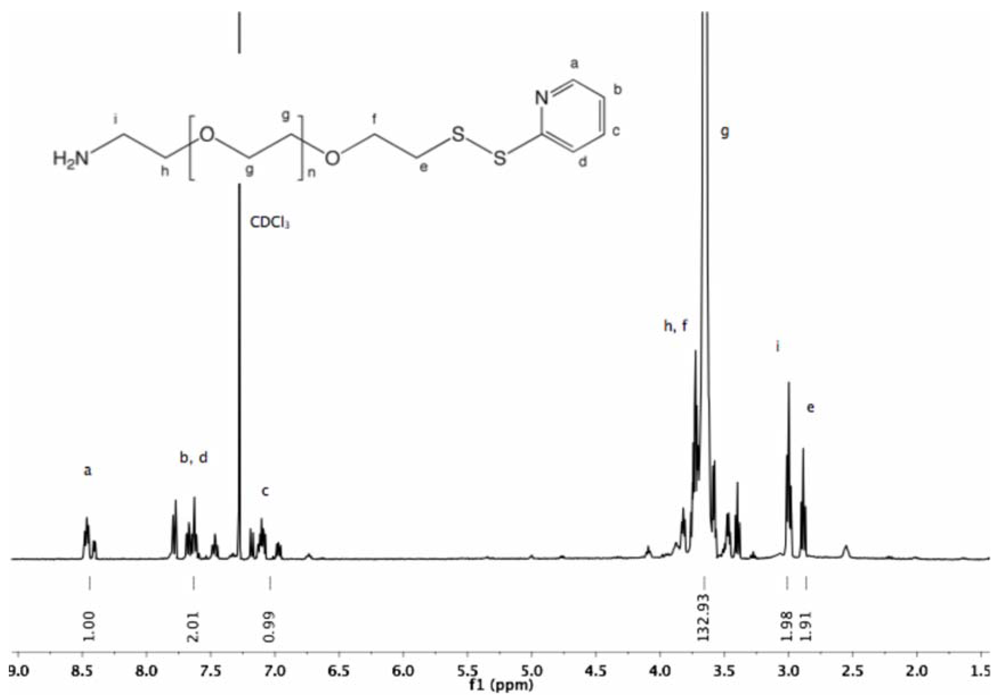
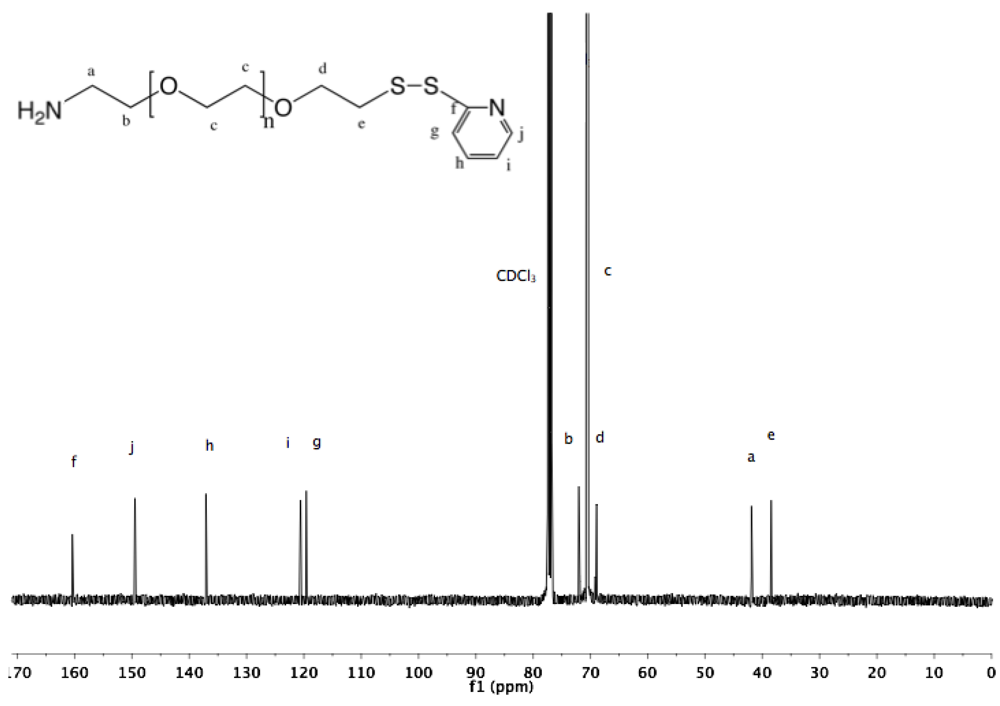
Synthesis of α-pyridyldithio-ω-hydroxyl PEG (7). α-Thioacetate-ω-hydroxyl PEG (6, 2 g, 1.3 mmol) and 2-PDS (5 eq, 1.44 g, 6.5 mmol) were dissolved in 30 mL ammonia (7 M in MeOH). The solution was stirred at rt under nitrogen for 96 h. Nitrogen was bubbled through the solution to remove ammonia, and MeOH was removed by rotary evaporation. The crude polymer was dissolved in 10 mL DCM and added dropwise into diethyl ether. α-Pyridyldithio-ω-hydroxyl PEG was recovered by filtration as slightly yellowish solid (1.97 g, 94%).
1H-NMR (DMSO, δ in ppm): 8.45–7.22 (4H, pyridyl protons), 4.55 (1H, t, J = 5.4 Hz, O
H), 3.61 (2H, t, J = 6 Hz, C
H2CH
2-S), 3.5 (126H, PEG backbone), 3.02 (2H, t, J = 6 Hz, C
H2-S).
13C-NMR (CDCl
3, δ in ppm): 160.41, 149.48, 137.10, 120.58, 119.58, 72.50, 70.55, 68.91, 61.69, 38.44 (
Figure S14 and
Figure S15).
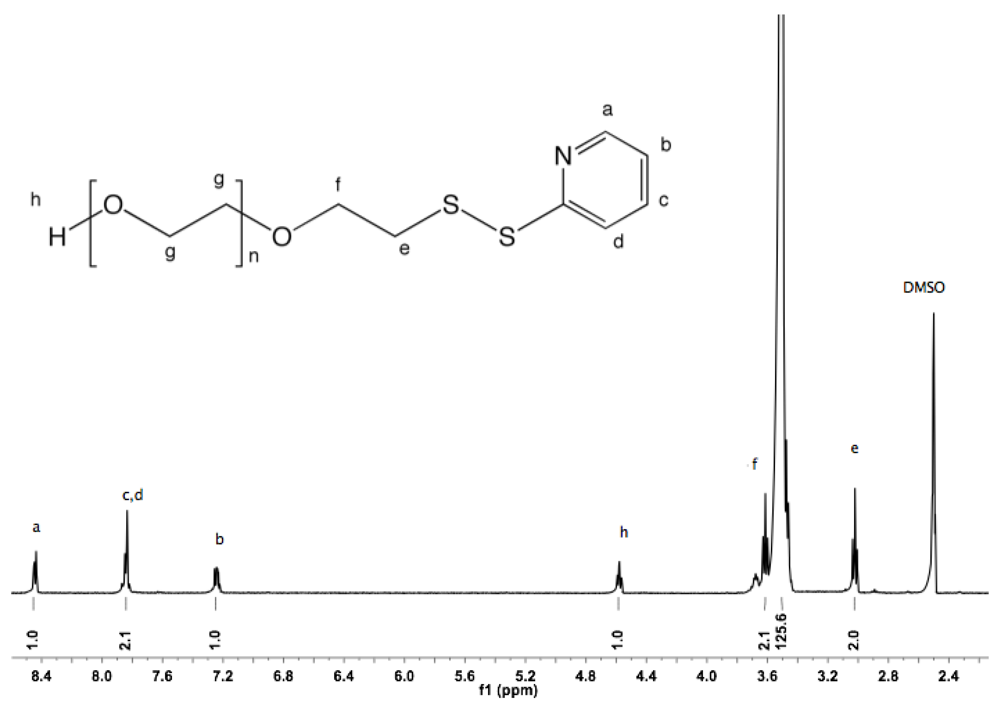
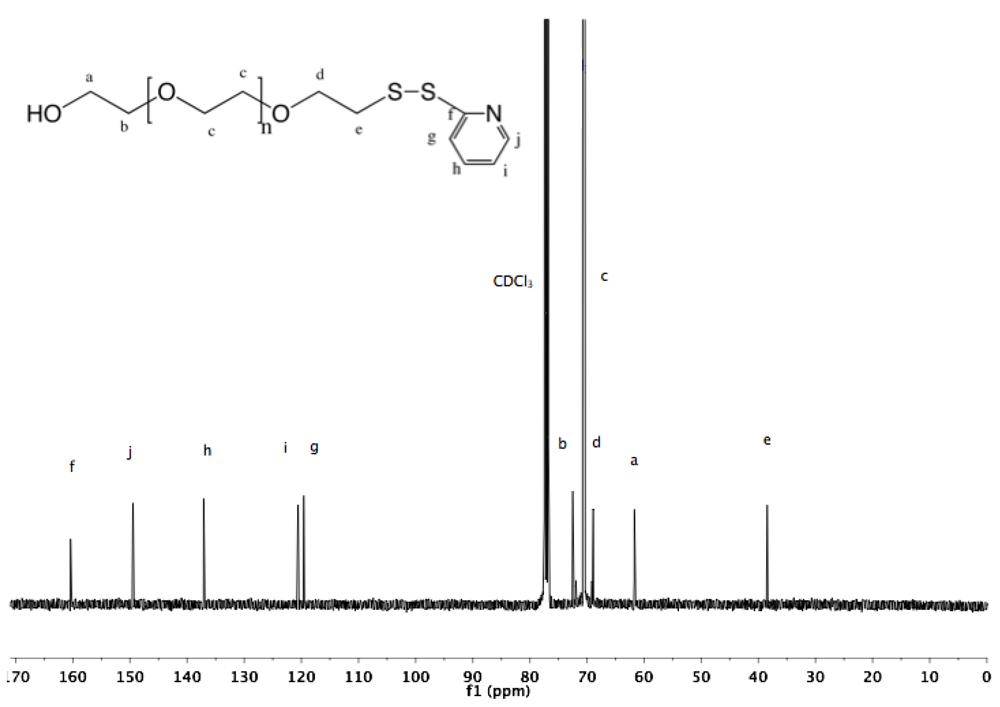
Synthesis of α-azide-ω-pyridyldithio PEG (8). α-Azide-ω-thioacetate PEG (4, 2 g, 1.3 mmol) and 2-PDS (5 eq, 1.44 g, 6.5 mmol) were dissolved in 30 mL ammonia (7 N in MeOH). The solution was stirred at rt under nitrogen for 96 h. Nitrogen gas was then bubbled through the solution to remove ammonia. MeOH was evaporated under vacuum. The crude polymer was dissolved in 10 mL DCM and added dropwise into diethyl ether. The α-azide-ω-pyridyldithio was recovered by filtration as slightly yellowish solid (1.9 g, 93%).
1H-NMR (DMSO, δ in ppm): 8.5–7.2 (4H, m, pyridyl protons), 3.64-3.59 (4H, m, C
H2CH
2-S and C
H2CH
2-N
3), 3.5 (131H, s, PEG backbone), 3.39 (t, J = 5 Hz, C
H2-N
3), 3.02 (2H, t, J = 6 Hz, CH2-S).
13C-NMR (CDCl
3): 160.4, 149.48, 137.1, 120.59, 119.59, 71.95, 70.54, 69.13, 50.66, 38.43 (
Figure S16 and
Figure S17).
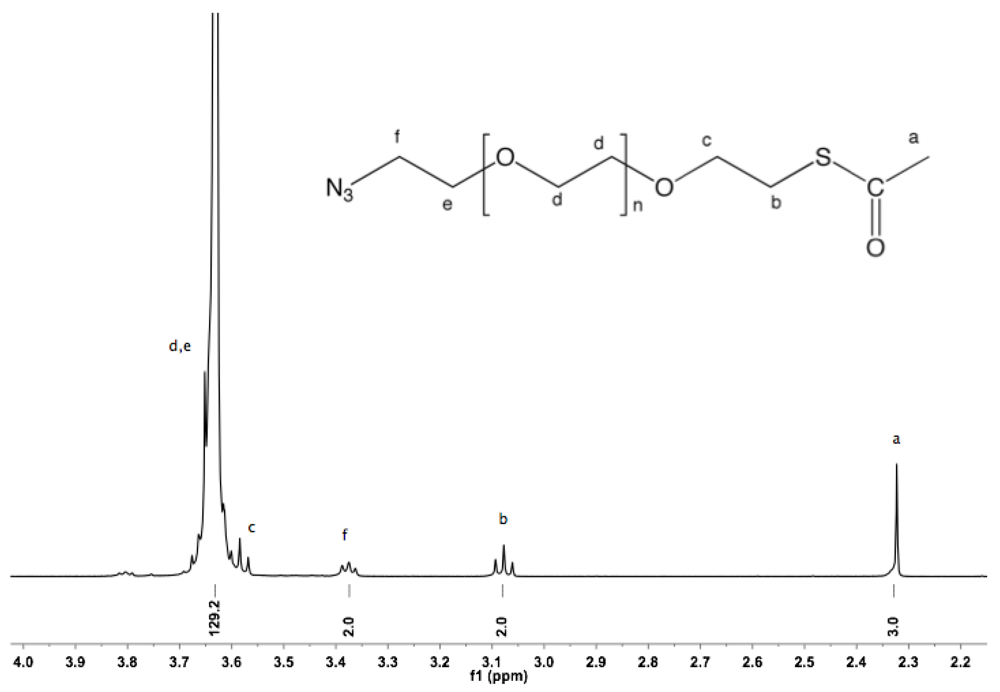
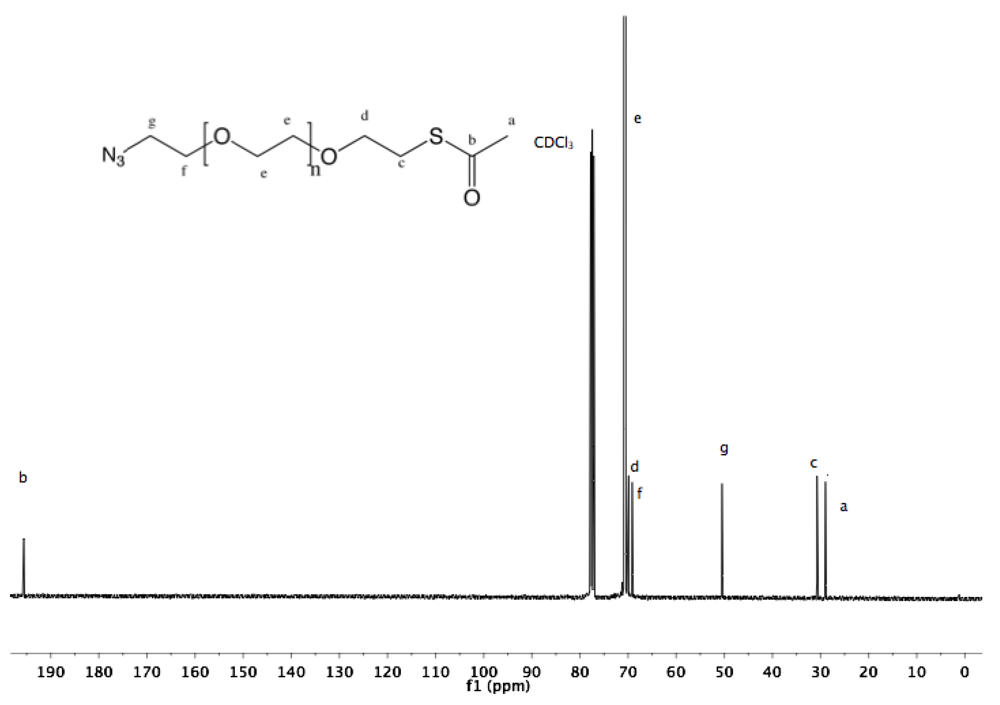
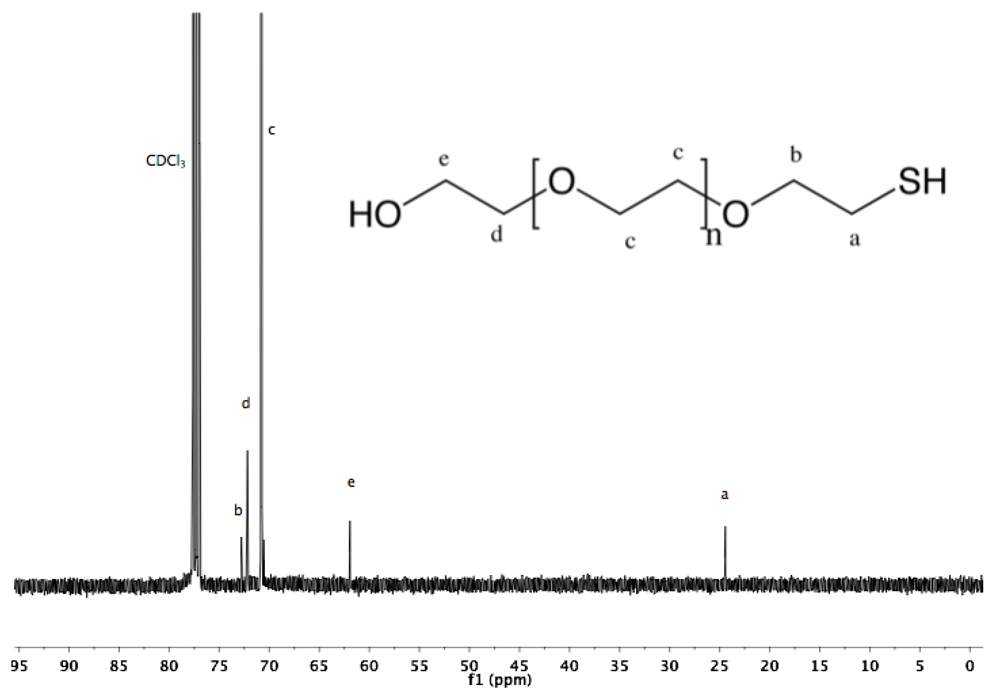
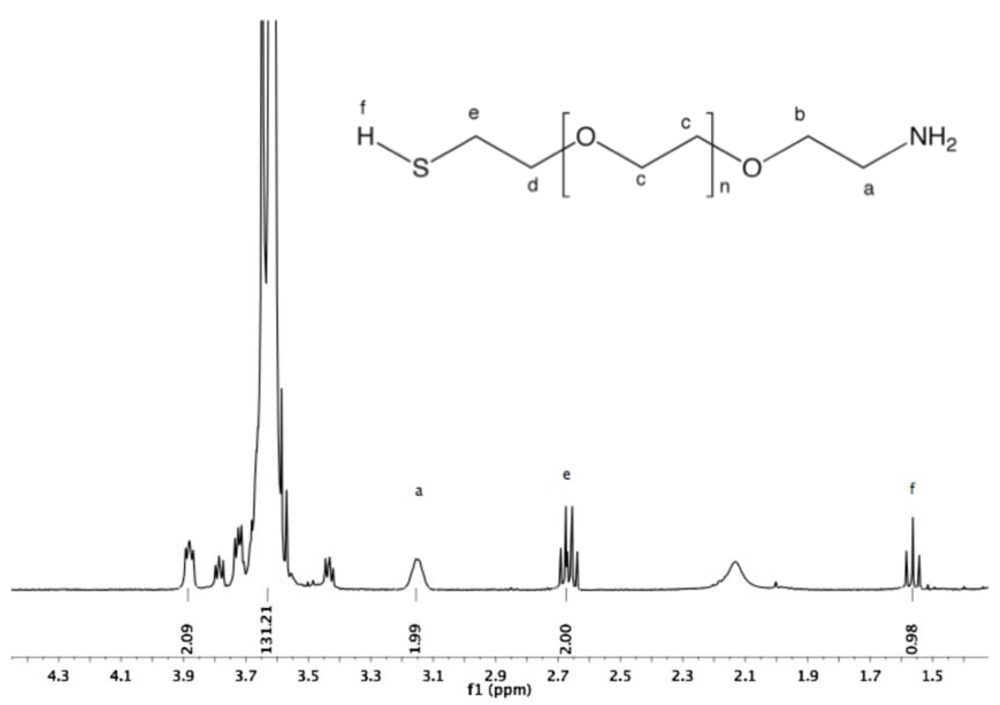
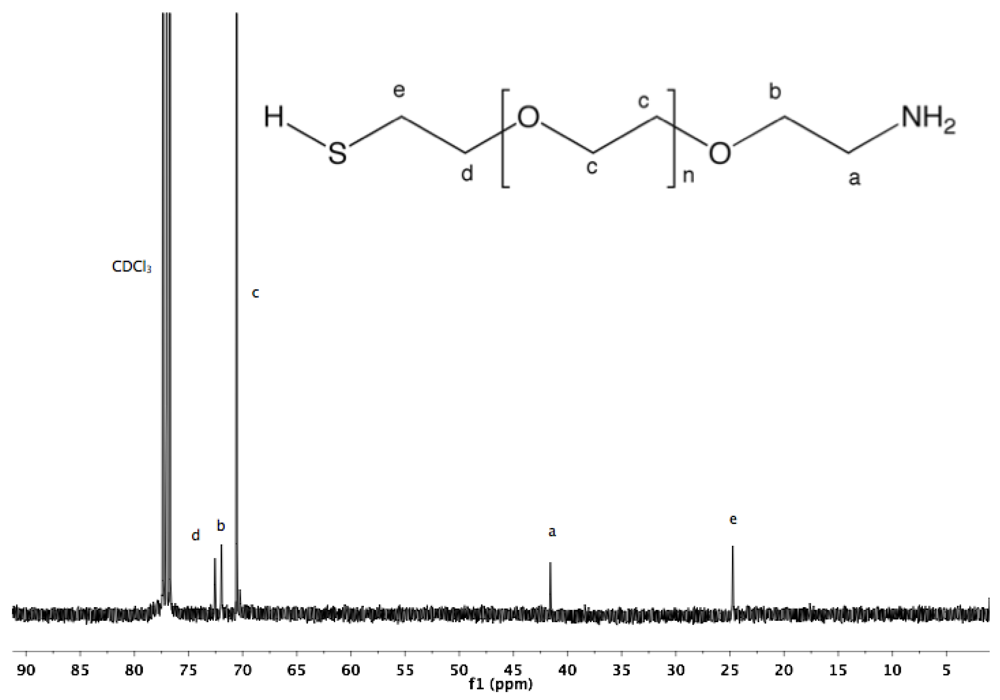
Synthesis of α-amine-ω-thiol PEG (9). To a solution of α-azide-ω-thioacetate PEG (4, 2 g, 1.3 mmol) in 50 mL dry MeOH, PPh
3 was added (1.7 g, 6 mmol), and the reaction mixture was heated to reflux overnight under argon. After cooling down to rt and solvent removal by rotary evaporation, the resulting solid was dissolved in 10 mL DCM, and then added dropwise into diethyl ether. α-Amine-ω-thiol PEG was collected by filtration (1.8 g 95%).
1H-NMR (CDCl
3, δ in ppm): 3.88 (2H, t, J = 4.8), 3.64 (131H, s, PEG backbone), 3.15 (2H), 2.67 (2H, C
H2-SH), 1.56 (1H, t, J = 8 Hz, S
H).
13C-NMR (CDCl3, δ in ppm): 71.95, 70.56, 69.64, 69.15, 41.76, 24.5 (
Figure S18 and
Figure S19).
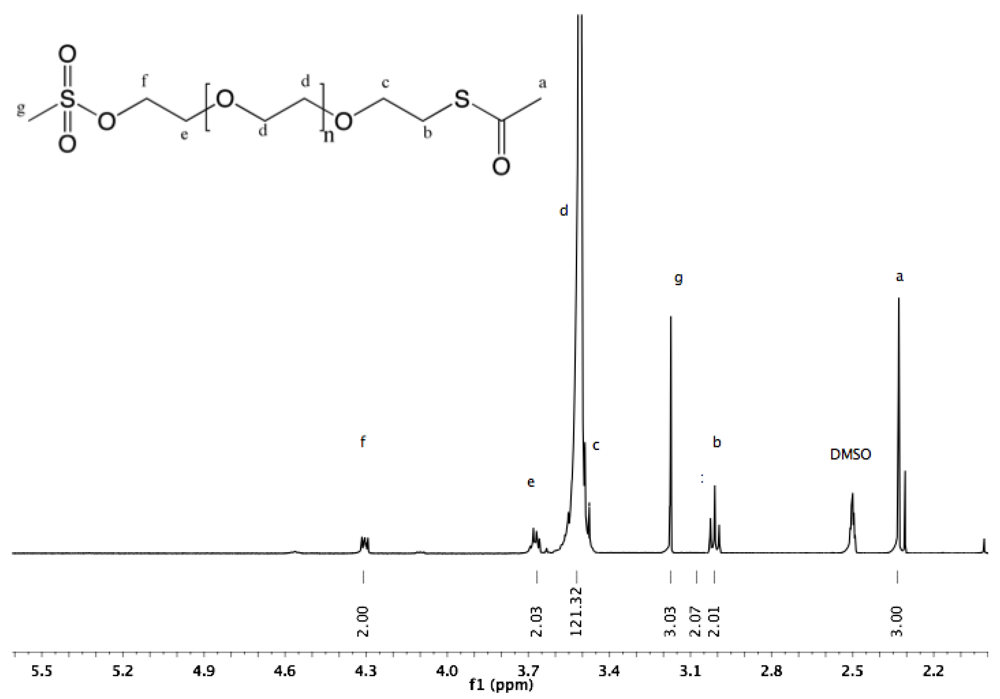
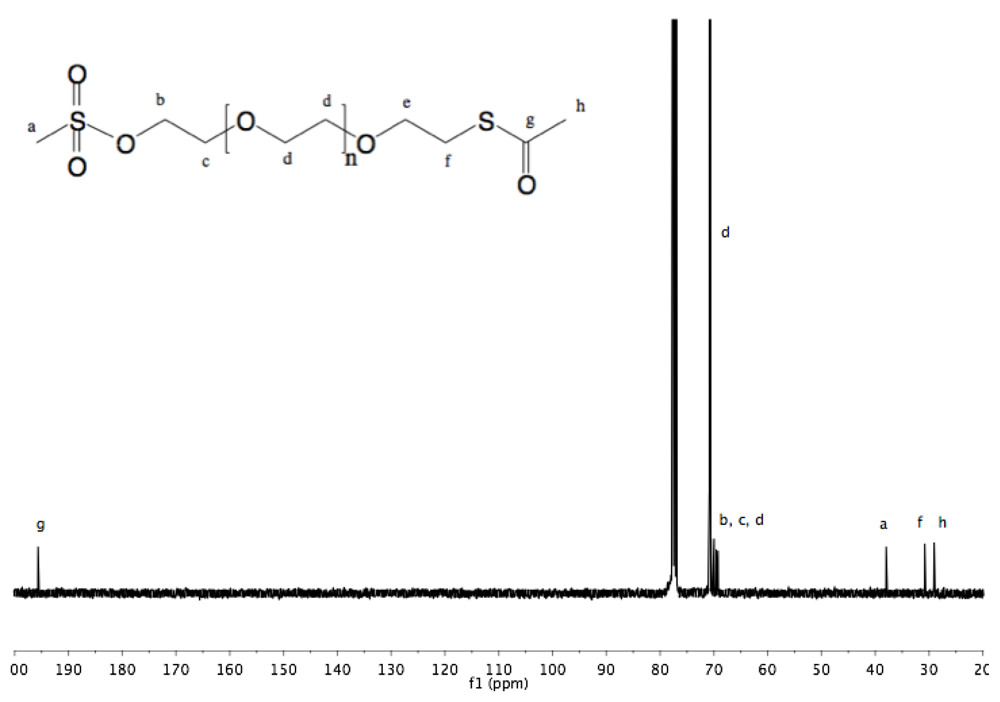
Synthesis of α-thioacetate-ω-mesyl PEG (10). To a chilled (0 °C) solution of α-thioacetate-ω-hydroxyl PEG (6, 3 g, 1.9 mmol) in dry DCM (30 mL) methanesulfonyl chloride (252 mg, 170 µL, 2.2 mmol) was added. NEt
3 (506 mg, 697 µL, 5 mmol) was slowly added to this rapidly stirred solution. The reaction was allowed to proceed for 2 h before dropwise addition of the solution into diethyl ether. White solid α-thioacetate-ω-mesyl PEG was collected by filtration and dried under vacuum (3 g, 97%).
1H-NMR (DMSO, δ in ppm): 4.3 (2H, t, J = 4.4 Hz, C
H2-O-SO
2), 3.68 (2H, t, J = 4.4 Hz, C
H2-CH
2-O-SO2), 3.52 (121H, s, PEG backbone), 3.17 (3H, s, C
H3-SO
2), 3.01 (2H, t, J = 6.4 Hz, C
H2-S-CO), 2.33 (3H, s, C
H3-CO-S).
13C-NMR (CDCl
3, δ in ppm): 195.7, 70.84, 70.52, 69.55, 69.22, 37.95, 30.79. 29.04 (
Fig S20 and
Fig S21).
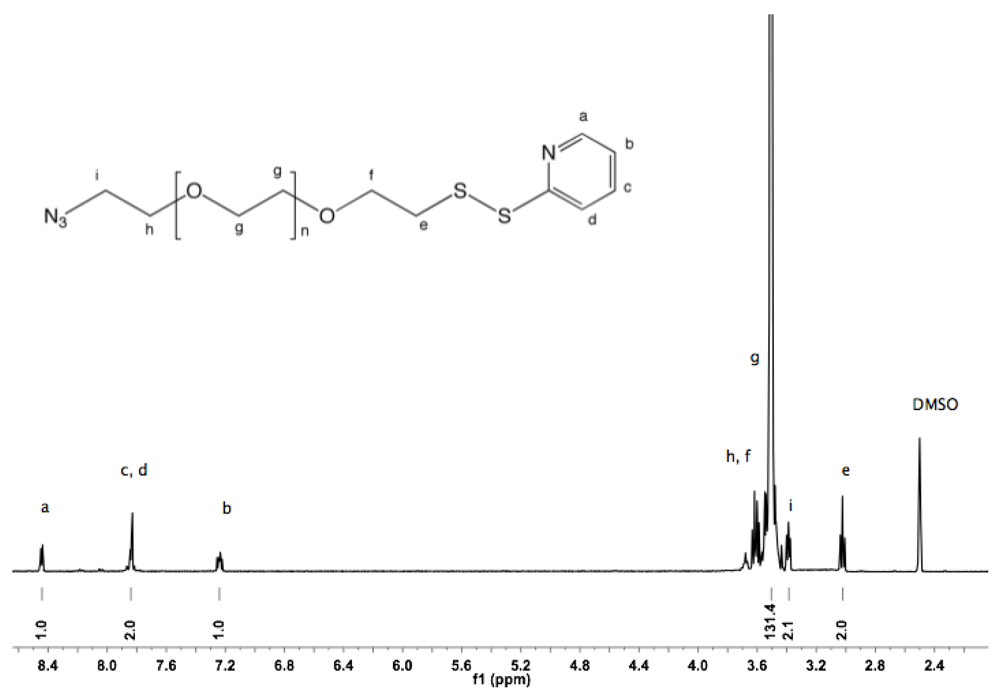
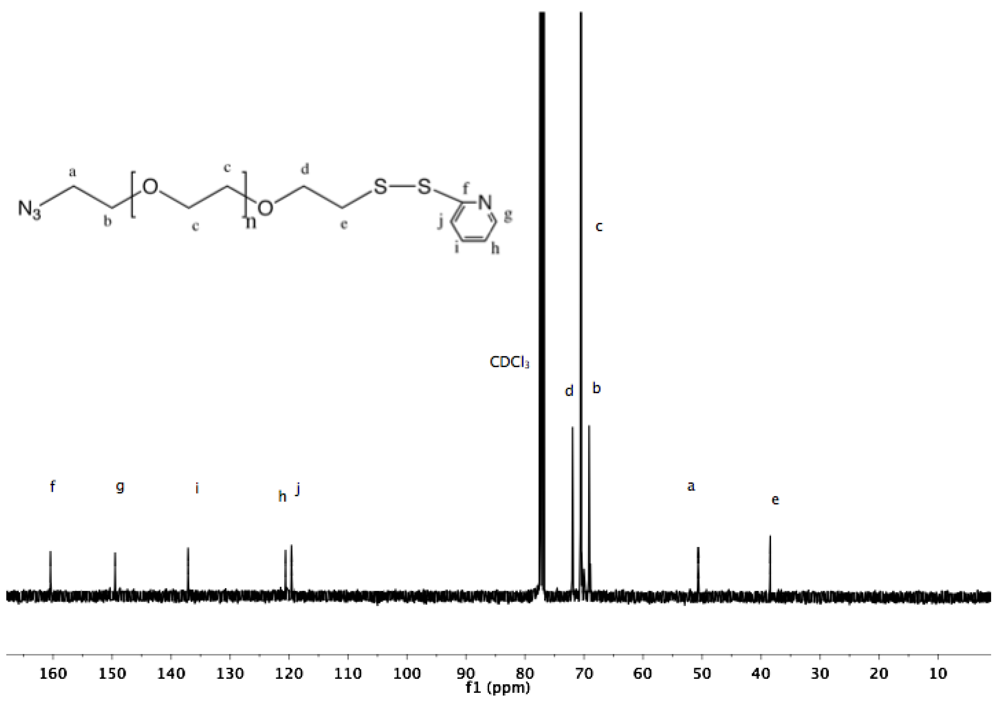
Synthesis of α-amine-ω-pyridyldithio PEG (11). α-Thioacetate-ω-mesyl PEG (10, 2 g, 1.3 mmol), and 2-PDS (1.44 g, 6.5 mmol) were dissolved in 150 mL ammonia (7 N in MeOH/ 28% in H
2O (2:1 v/v)). The solution was stirred at rt under nitrogen for 96 h. Nitrogen was then bubbled through the solution to remove ammonia. MeOH was evaporated under vacuum. The polymer was extracted from the aqueous solution three times by 30 mL DCM and dried over sodium sulfate. The organic phase was reduced to a small volume by rotary evaporation, and finally added dropwise into cold diethyl ether. α-Amine-ω-pyridyldithio PEG was collected by filtration (1.9 g, 96%).
1H-NMR (CDCl
3, δ in ppm): 8.5–7 (4H, t, pyridyl protons), 3.64 (133H, PEG backbone), 2.99 (2H, t, J = 6.6 Hz, C
H2-S), 2.89 (2H, t, J = 6.2 Hz, C
H2-NH
2).
13C-NMR (CDCl
3, δ in ppm): 160.4, 149.49, 137.11, 120.58, 119.59, 72.34, 70.54, 68.93, 41.79, 38.44 (
Figure S22 and
Figure S23).
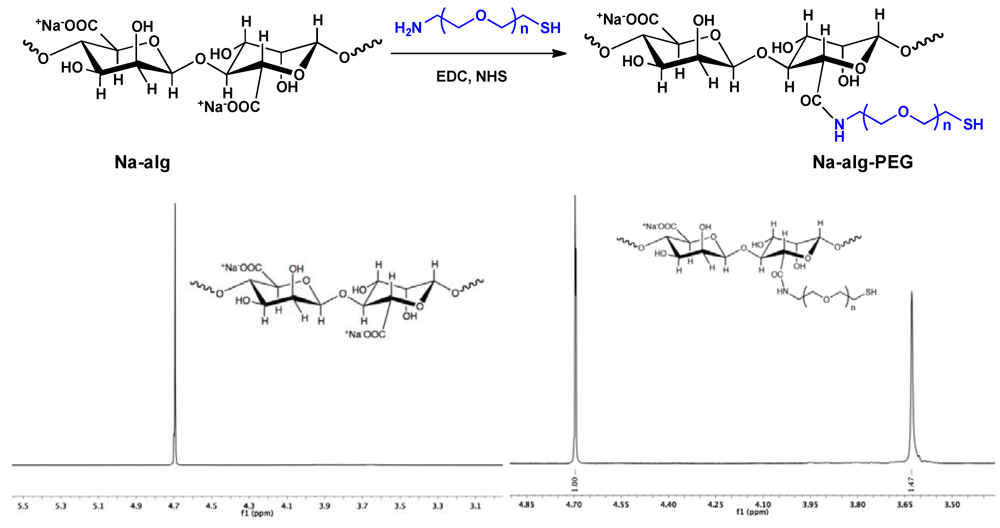
Synthesis of Na-alg-PEG. Na-alg (100 mg), NHS (28 mg, 0.24 mmol), and MES (125 mg, 0.6 mmol) were dissolved in 10 mL of distilled water. A solution of H2N-PEG-SH (9, 230 mg, 0.16 mmol in 1 mL H2O) was added. After stirring for 15 min at rt, 330 mg of EDC was added and stirring was continued for 30 min at rt, followed by addition of NaOH (120 µL, 6 M) and 10 min incubation at rt. Purification was achieved by dialysis against distilled water for 4 days. The water was replaced three times/day. During the first 3 days, NaOH (20 µL, 6 M), NaCl (800 µL, 5 M), and TCEP (1 mL, 1M) were added twice daily to the Na-alg-PEG solution inside the dialysis tube (MWCO 14,000 g/mol). The purified polymer solution was filtered (0.22 µm) and lyophilized. The degree of grafting, which refers to the percentage of reacted carboxylate groups on Na-alg, was determined by 1H-NMR (D2O, δ in ppm): 4.7 (s, alginate protons), 3.6 (PEG protons).
3. Results and Discussion
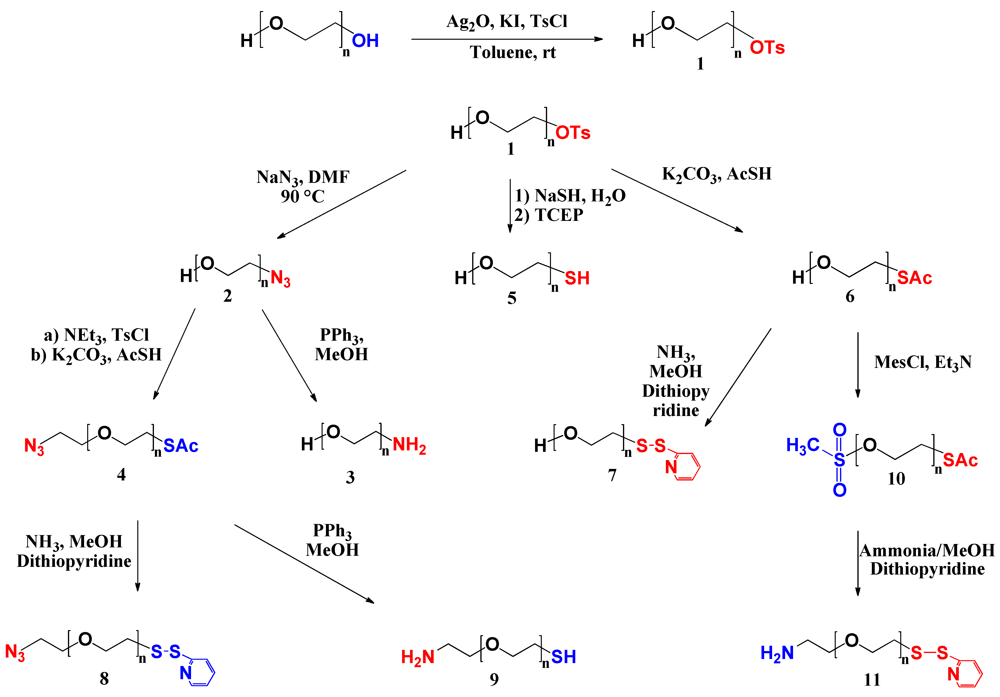
Synthesis of Heterobifunctional PEG Derivatives. A series of heterobifunctional PEG derivatives was synthesized via asymmetric activation of commercially available symmetrical PEG. First, mono-tosyl PEG was synthesized. Subsequently, the tosyl end group was converted into a variety of functional end groups, either directly or via intermediate steps as shown in
Scheme I. Synthesis efficiency, final yields and molecular characteristics are summarized in
Table 1.
Scheme I.
Syntheses pathways to heterobifunctional PEG derivatives.
Scheme I.
Syntheses pathways to heterobifunctional PEG derivatives.
Table 1.
Summary of syntheses efficiency and molecular characteristics.
Table 1.
Summary of syntheses efficiency and molecular characteristics.
| PEG label | Name | Steps | Yield (%)
a | Molar Mass (g/mol) | PDI
b |
|---|
| GPC | 1H-NMR |
|---|
| - | α, ω-dihydroxyl | - | - | 1861 | 1488 | 1.07 |
| 1 | α-tosyl-ω-hydroxyl | 1 | 98 | 2012 | 1620 | 1.07 |
| 2 | α-azide-ω-hydroxyl | 2 | 93 | 1857 | 1480 | 1.08 |
| 3 | α-amine-ω-hydroxyl | 3 | 88 | 1541 | 1423 | 1.07 |
| 4 | α-azide-ω-thioacetate | 4 | 73 | 2090 | 1610 | 1.08 |
| 5 | α-thiol-ω-hydroxyl | 2 | 82 | - | 1442 | - |
| 6 | α-thioacetate-ω-hydroxyl | 2 | 84 | 1818 | 1537 | 1.08 |
| 7 | α-pyridyldithio-ω-hydroxyl | 3 | 79 | 1890 | 1569 | 1.08 |
| 8 | α-azide-ω-pyridyldithio | 5 | 68 | 1867 | 1702 | 1.08 |
| 9 | α-amine-ω-thiol | 5 | 69 | 1862 | 1560 | 1.07 |
| 10 | α-thioacetate-ω-mesyl | 3 | 81 | 1780 | 1576 | 1.07 |
| 11 | α-amine-ω-pyridyldithio | 4 | 77 | 1842 | 1692 | 1.08 |
PEG (1). Tosylate groups are known as good substrate for substitution reactions. Their leaving group character enables the conversion to versatile types of functional groups. Thus, a tosylate end group in a PEG molecule is one of the most useful candidates for replacement by another functionality. For the preparation of a series of heterotelechelic PEG derivatives, α-tosyl-ω-hydroxyl PEG (
1) was synthesized as precursor (
Scheme I). Bouzide and Sauve reported selective monotosylation of symmetrical diols in the presence of silver oxide and a catalytic amount of potassium iodide [
21]. We adapted this approach to modify commercially available PEG. To achieve selective monotosylation also addition of an excess of symmetric diols was reported [
22]. However, in the case of PEG having a relatively high molar mass, the separation of non-modified excess from the modified product is difficult. In the present study, the synthesis was carried out at rt avoiding drawbacks such as harsh conditions or the use of an excess of diols. The degree of functionalization of α-tosyl-ω-hydroxyl PEG was calculated from
1H-NMR using deuterated dimethyl sulfoxide (DMSO) [
23]. The spectrum shows a triplet at 4.56 ppm corresponding to hydroxyl protons, which does not shift or broaden with variation of the concentration, and which is well separated from the PEG backbone peak (see Appendix,
Figure S1). The degree of functionalization was almost quantitative (≈99%). Moreover, only unimodal peaks without shoulders and indicating similar molar mass distributions were observed by GPC for the native PEG and for α-tosyl-ω-hydroxyl PEG (
Figure 1(a)).
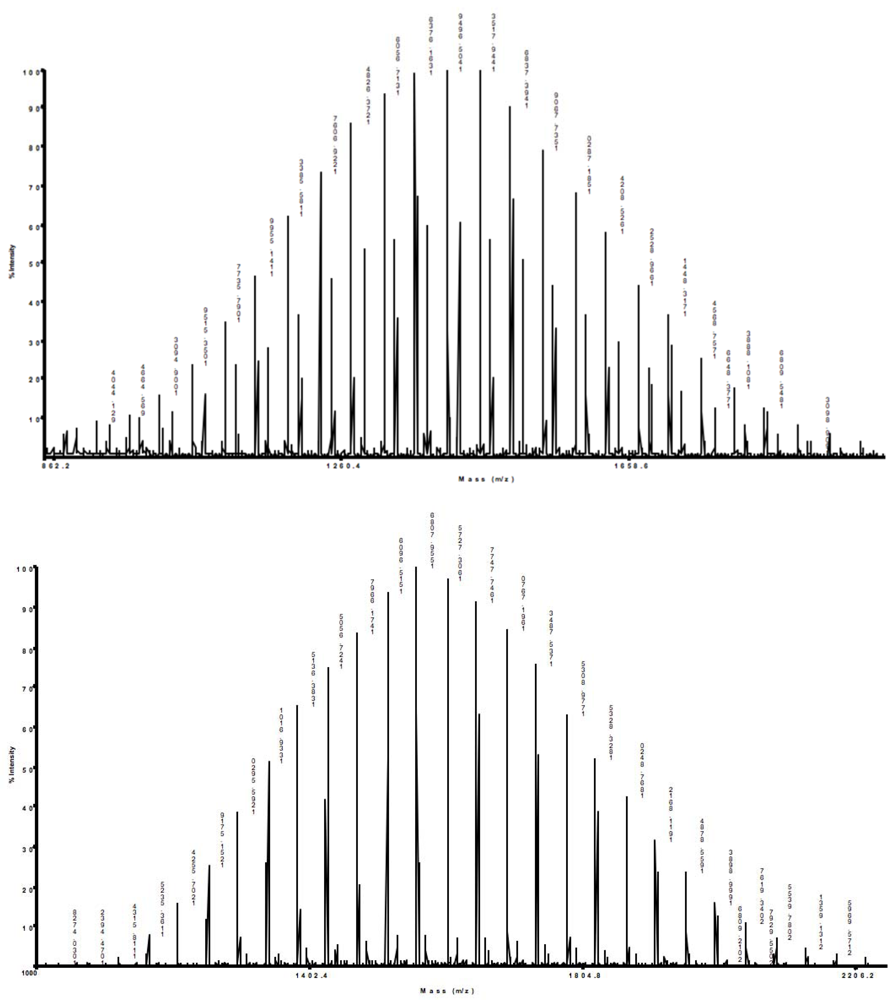
The absence of ditosylate byproduct was confirmed by MALDI-TOF/TOF MS. The main peaks for native PEG and α-tosyl-ω-hydroxyl PEG were detected at 1,449.71 and 1,603.72, respectively (see Appendix,
Figure S24). The increase of the molar mass corresponds to the introduction of the tosyl group at only one end. A peak at 1,756.71 corresponding to the ditosylate byproduct was absent. Such selective monotosylation has been attributed to internal hydrogen bonding (IHB) [
21]. The hydrogen atom involved in IHB becomes less acidic, and therefore, the second hydroxyl group will preferentially be deprotonated by silver oxide.
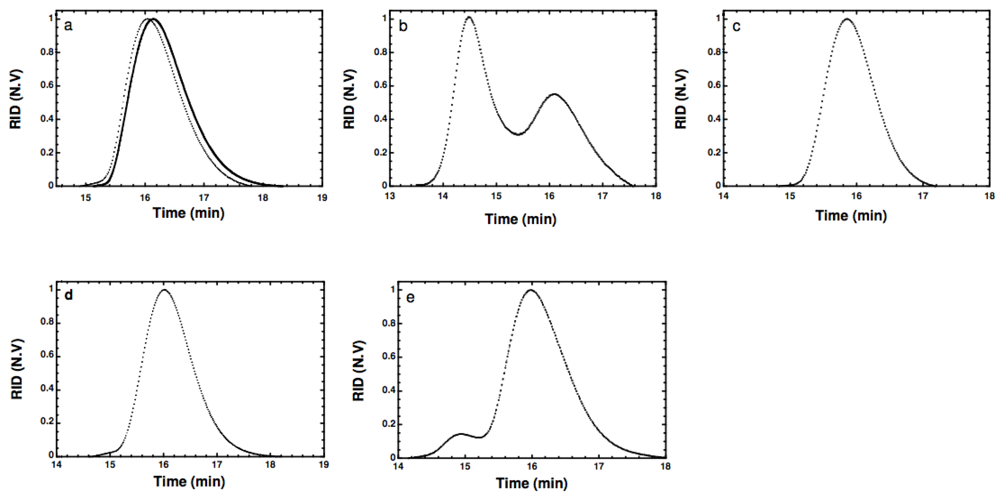
Figure 1.
(a) Gel permeation chromatograms of α-tosyl-ω-hydroxyl PEG (1) (dashed) and PEG (full); (b) α-thiol-ω-hydroxyl PEG (5); (c) α-pyridyldithio-ω-hydroxyl PEG (9); (d) Conversion of thiol-terminated PEG (9) into disulfide monitored at day 0; and (e) after 14 days storage, (refractive index detection vs. elution time).
Figure 1.
(a) Gel permeation chromatograms of α-tosyl-ω-hydroxyl PEG (1) (dashed) and PEG (full); (b) α-thiol-ω-hydroxyl PEG (5); (c) α-pyridyldithio-ω-hydroxyl PEG (9); (d) Conversion of thiol-terminated PEG (9) into disulfide monitored at day 0; and (e) after 14 days storage, (refractive index detection vs. elution time).
PEG (2). The displacement of the tosylate end group by NaN
3 yielded α-azide-ω-hydroxyl PEG (
2). Azide-terminated PEGs have received considerable attention in the field of bioconjugate chemistry, in particular for the highly chemoselective copper (I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition reaction (termed “click chemistry”). Because of its specificity, quantitative yield, and good functional group tolerance, click chemistry has shown to be a promising approach for pegylation under mild conditions in aqueous buffers within a wide pH range [
24,
25,
26,
27,
28]. The quantitative introduction of the azide function was confirmed by
1H-NMR. On the one hand, the spectra did not show any traces of tosylate signals. On the other hand, the integration of the hydroxyl group remained constant, suggesting that no hydrolysis of the tosylate group took place (see Appendix,
Figure S3). The
13C-NMR and FTIR spectra (see Appendix,
Figure S4 and
Figure 2a) confirm the quantitative introduction of the azide function. The chemical displacement of the carbon adjacent to azide and the antisymmetric stretching vibration band of azide were detected at 50.64 ppm and 2,103 cm
−1, respectively.
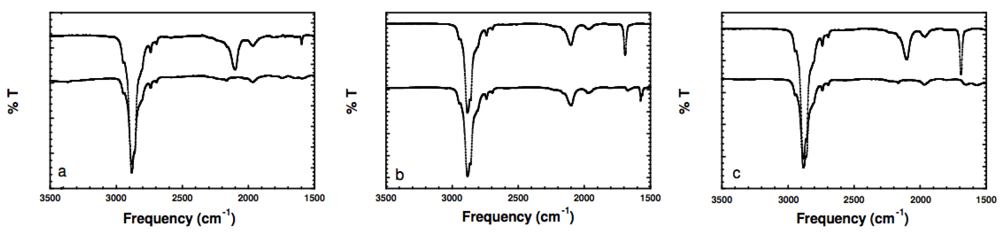
Figure 2.
(a) FTIR spectra of α-azide-ω-hydroxyl PEG (2) (upper), and α-amine-ω-hydroxyl PEG (3) (lower); (b) α-azide-ω-thioacetate PEG (4) (upper) and α-azide-ω-pyridyldithio PEG (8) (lower); (c) α-azide-ω-thioacetate PEG (4) (upper) and α-amine-ω-thiol PEG (9) (lower). (Spectra are vertically shifted for better visualization).
Figure 2.
(a) FTIR spectra of α-azide-ω-hydroxyl PEG (2) (upper), and α-amine-ω-hydroxyl PEG (3) (lower); (b) α-azide-ω-thioacetate PEG (4) (upper) and α-azide-ω-pyridyldithio PEG (8) (lower); (c) α-azide-ω-thioacetate PEG (4) (upper) and α-amine-ω-thiol PEG (9) (lower). (Spectra are vertically shifted for better visualization).
PEG (3). Besides being efficient precursors for click chemistry, azides are known to serve as synthons for the preparation of amines. The reduction of azides to amines has been widely studied and been found to be highly diverse [
29,
30,
31]. In our approach, a complete reduction of azide was achieved by Staudinger reduction. The reduction mechanism involves the formation of a linear phosphazine intermediate, which yields an iminophosphorane with concomitant loss of N
2. By spontaneous hydrolysis of iminophosphorane primary amine and the corresponding phosphine oxide are obtained [
32,
33]. The use of PPh
3 as reducing agent in MeOH leads to α-amine-ω-hydroxyl PEG (
3). The completeness of the reaction was confirmed by
13C-NMR. The spectra exhibit peaks at 41.78 and 73.45 corresponding to α- and β-amine carbons, respectively, while no traces of the characteristic peak of azide at 50.64 ppm are visible (see Appendix,
Figure S6). Furthermore, FTIR shows absence of the antisymmetric stretching vibration band of azide (
Figure 2(a)).
PEG (5). Thiol-terminated PEG is very efficient as pegylation agent. For instance, the functionalization of quantum dots and nanoparticles with PEG-thiol increased their stability and their hydrophilicity, and thus, reduced their toxicity in biological systems [
34,
35,
36]. Furthermore, the ability of thiol-terminated PEG to form hydrogels via Michael-type addition was exploited for cell encapsulation, drug delivery, and tissue engineering [
37,
38,
39]. Here, α-thiol-ω-hydroxyl PEG (
5) was prepared by reaction of α-tosyl-ω-hydroxyl PEG (
1) with an excess of sodium hydrosulfide hydrate in water. The downfield tosylate aryl peaks were not detectable by
1H-NMR (SI,
Figure S10) confirming the complete displacement of the tosylate group by the hydrosulfide ion. However, the GPC chromatogram presented in
Figure 1(b) shows two peaks, one with similar retention time as the starting α-tosyl-ω-hydroxyl PEG and a second peak with shorter retention time corresponding to twice the molar mass. The treatment of the polymer with tris(2-carboxyethyl)phosphine hydrochloride (TCEP) prior to the injection into the GPC column significantly reduces this second peak (data not shown) suggesting that the early peak corresponds to dimer molecules formed by oxidation of the thiol function to a disulfide bond, which is reduced by reaction with TCEP. The formation of disulfide bonds could not completely be avoided using a one-step process although special care was taken to eliminate air.
PEG (6) and PEG (7). In order to avoid this dimer formation and to synthesize PEG with protected thiol end groups, a two-step synthesis was adopted. The tosylate group was first displaced by reaction with the
in situ formed potassium thioacetate in DMF to yield α-thioacetate-ω-hydroxyl PEG (
6). The introduction of the thioester end group was confirmed by
1H and
13C-NMR (see Appendix,
Figure S12 and
Figure S13). The complete hydrolysis of the thioester was accomplished under mild conditions using ammonia in MeOH at rt. Furthermore, adding 2,2’-dipyridyl disulfide (2-PDS) as capping agent avoided the formation of dimer molecules during the hydrolysis. The liberated sulfhydryl end group simultaneously reacts with 2-PDS yielding a protected thiol as pyridyldithio.
1H and
13C-NMR spectra of α-pyridyldithio-ω-hydroxyl PEG (
7) exhibit the characteristic peaks of the pyridyl protons (see Appendix,
Figure S14 and
Figure S15), while no traces of the thioester bond were detected. GPC confirmed the absence of the dimer byproduct (
Figure 1(c)).
PEG (4), PEG (8), and PEG (9). The α-azide-ω-hydroxyl PEG (
2) was used as precursor for the synthesis of α-azide-ω-thioacetate PEG (
4). The first step involved the activation of the free hydroxyl into tosylate. The reaction proceeded quantitatively as the hydroxyl protons typically observed by
1H- NMR in DMSO at 4.56 ppm were not detectable (see Appendix,
Figure S7). The thioacetate function was introduced by the reaction with the
in situ formed potassium thioacetate in DMF yielding α-azide-ω-thioacetate PEG (
4).
1H and
13C-NMR confirmed the quantitative introduction of the thioacetate function (
Figure S8 and
Figure S9). Beside the antisymmetric stretching vibration band of azide detected at 2103 cm
−1, the FTIR spectrum of α-azide-ω-thioacetate PEG (
4) in
Figure 2b exhibited a strong band at 1692 cm
−1 corresponding to carbonyl stretching vibration. The reaction of (
4) with ammonia in presence of 2-PDS yielded α-azide-ω-pyridyldithio PEG (
8). FTIR spectra show complete disappearance of the peak corresponding to carbonyl stretching,
Figure 2b. Reduction of α-azide-ω-thioacetate PEG (
4) was achieved by PPh
3.
13C-NMR exhibit the characteristic peaks of α- and β-amine carbons of PEG (
9), while no traces of the characteristic peak of α-azide carbon at 50.64 ppm were detected (see Appendix,
Figure S19), suggesting complete reduction of the azide function. Furthermore, the cleavage of the thioester bond was confirmed by FTIR. The spectrum shows complete disappearance of both the antisymmetric stretching vibration band of azide at 2,103 cm
−1 and the peak corresponding to carbonyl stretching at 1,692 cm
−1 (
Figure 2(c)).
1H-NMR spectra confirm the presence of amine and thiol end groups (see Appendix,
Figure S18). The GPC curve of PEG (
9) shows a unimodal peak without any shoulder (
Figure 1(d)). This is explained by the reductive effect of the PPh
3, which reduces the formation of disulfide bonds [
40]. However, disulfide formation becomes obvious after two-weeks storage by a peak at shorter retention time corresponding to the double of the molar mass (
Figure 1(e)). This dimer formation confirms the usefulness of protecting the thiol group at the chain end. It could be removed easily by standard reducing agents such as dithiothreitol (DTT) or TCEP before further use for specific pegylation.
PEG (10) and PEG (11). Simultaneous introduction of amine and pyridyldithio end groups was accomplished by performing a two-step synthesis using α-thioacetate-ω-hydroxyl PEG (
6) as precursor. The hydroxyl group was first converted into mesylate, PEG (
10).
1H-NMR confirmed the quantitative introduction of a mesylate end group exhibiting no traces of hydroxyl groups at 4.56 ppm (
Figure S20). The integration of the thioacetate protons remained unchanged suggesting that no hydrolysis of the thioacetate end is taking place during the reaction. The absence of hydrolysis might be explained both by the low concentration of NEt
3 and/or by the short reaction time. The amine function was successfully introduced by displacing the mesylate end group by ammonia yielding PEG (
11), as shown in SI (
Figure S22 and
Figure S23). Simultaneously, the cleavage of the thioester end group was accomplished, and a pyridyldithio end group was obtained by trapping the liberated sulfhydryl by 2-PDS. Because of the low solubility of 2-PDS in water, a mixture of MeOH/H
2O was used as solvent.
Overall, the syntheses of all heterobifunctional PEG derivatives aimed at almost quantitative end group functionalization and high yields obtainable at mild reaction conditions. Optimization of the reaction conditions, in particular reaction times, seems possible.
Pegylation of Sodium Alginate. The presence of the carboxylic groups and cis-diols in the Na-alg chain units provides numerous approaches for chemical modification [
41,
42,
43,
44,
45]. Here, the modification of Na-alg by pegylation aims at improving the mechanical resistance and the durability of alginate-based hydrogels while not interfering the biocompatibility. In our approach, the conjugation of α-amine-ω-thiol PEG (
9) to Na-alg was achieved via amide bonding (
Scheme II). The carboxylate groups of Na-alg reacted with (
9) in presence of N-ethyl-N-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) in aqueous solution.
Scheme II.
Conjugation of α-amine-ω-thiol PEG (9) to Na-alg resulting in Na-alg-PEG (top). 1H-NMR in D2O of Na-alg and Na-alg-PEG (bottom).
Scheme II.
Conjugation of α-amine-ω-thiol PEG (9) to Na-alg resulting in Na-alg-PEG (top). 1H-NMR in D2O of Na-alg and Na-alg-PEG (bottom).
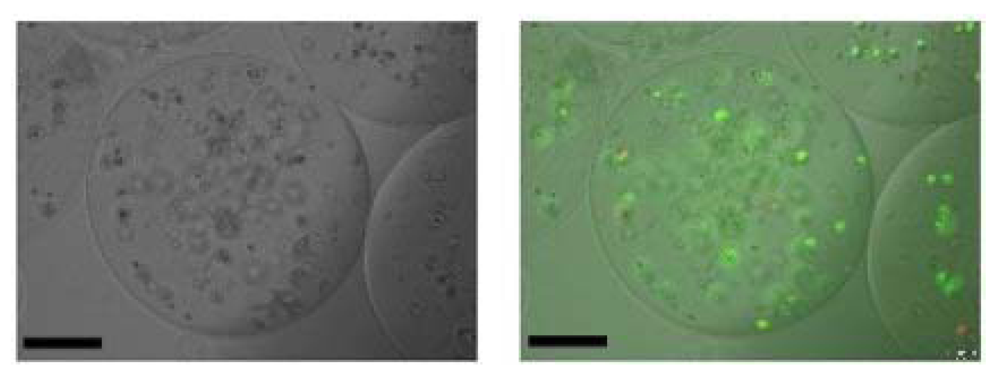
The conjugation of the hydrophilic PEG chains to Na-alg significantly increased the solubility of the resulting Na-alg-PEG. For the same concentration, a homogeneous aqueous solution of Na-alg-PEG is obtained within minutes while the unmodified Na-alg needs hours to dissolve. Thus, especially for higher molar mass Na-alg, a positive impact on the solution preparation for practical applications is achieved. With the aim to extend the materials basis for cell microencapsulation, we prepared novel calcium alginate poly(ethylene glycol) hybrid microspheres (Ca-alg-PEG) from Na-alg-PEG and studied the suitability of these microspheres for cell microencapsulation. Although the focus of this paper is not to investigate the suitability of Na-alg-PEG for cell encapsulation, we briefly show some results of the ongoing studies. It was found that Na-alg-PEG maintains the gelling capacity in presence of divalent cations, while the free thiol end groups allow for simultaneous chemical cross-linking. Stable microspheres were prepared in a one-step process and without incorporation of polycations [
46,
47]. Human hepatocellular carcinoma cells (Huh-7) were successfully encapsulated within Ca-alg-PEG (
Figure 3). They maintained their viability, proliferated and continued secreting albumin during a two-weeks study [
46,
47].
Figure 3.
Huh-7 encapsulated within Ca-alg-PEG hybrid microspheres. The cells were observed at day 3 using optical (left) and fluorescence (right) microscopy. Viable cells fluorescence green, while dead cells fluorescence bright red. Scale bar: 150 µm.
Figure 3.
Huh-7 encapsulated within Ca-alg-PEG hybrid microspheres. The cells were observed at day 3 using optical (left) and fluorescence (right) microscopy. Viable cells fluorescence green, while dead cells fluorescence bright red. Scale bar: 150 µm.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






























