Fifty Years of Hydrosilylation in Polymer Science: A Review of Current Trends of Low-Cost Transition-Metal and Metal-Free Catalysts, Non-Thermally Triggered Hydrosilylation Reactions, and Industrial Applications


Abstract
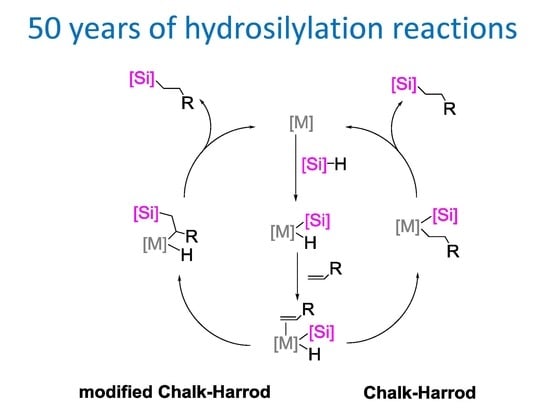
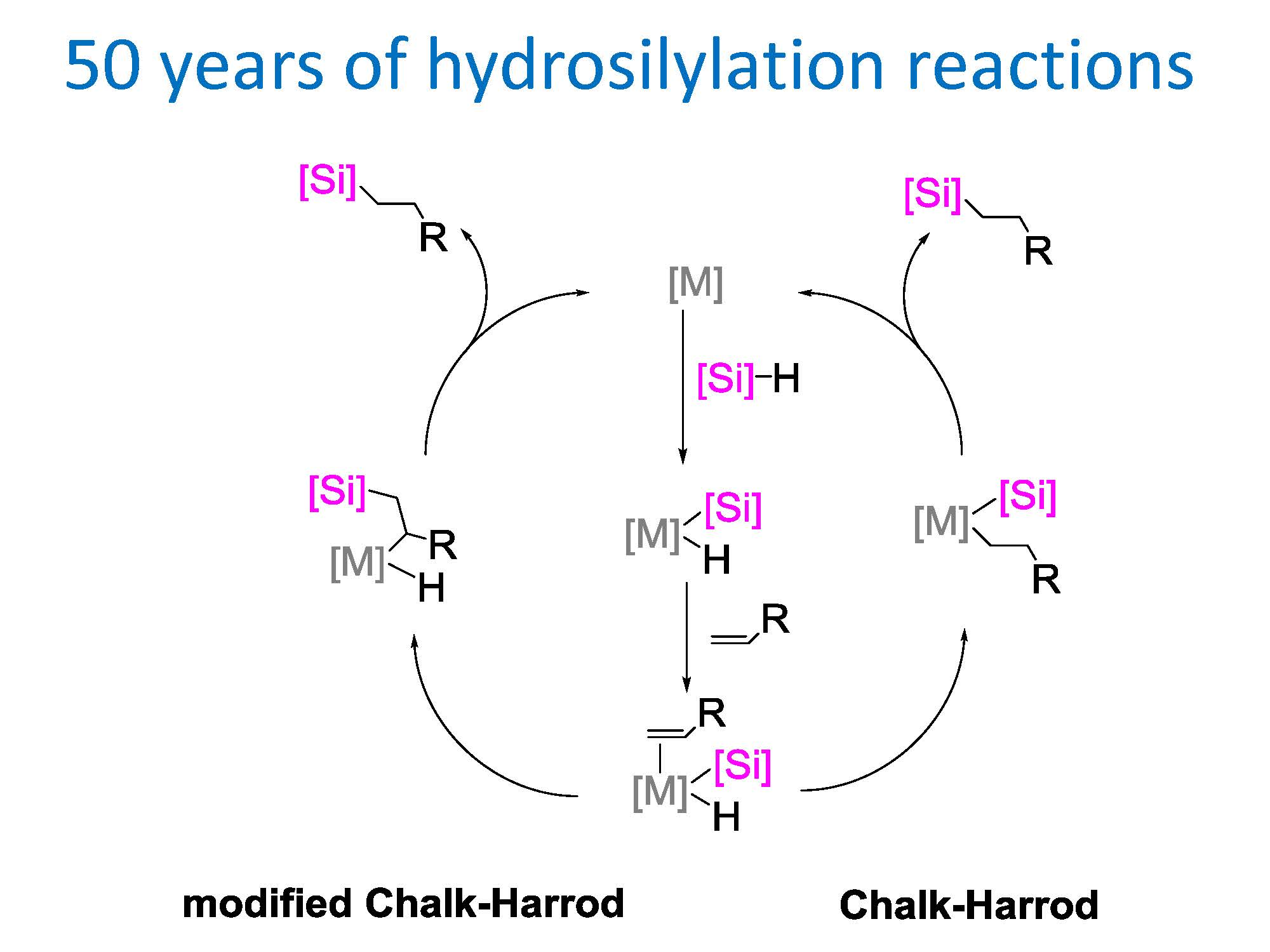
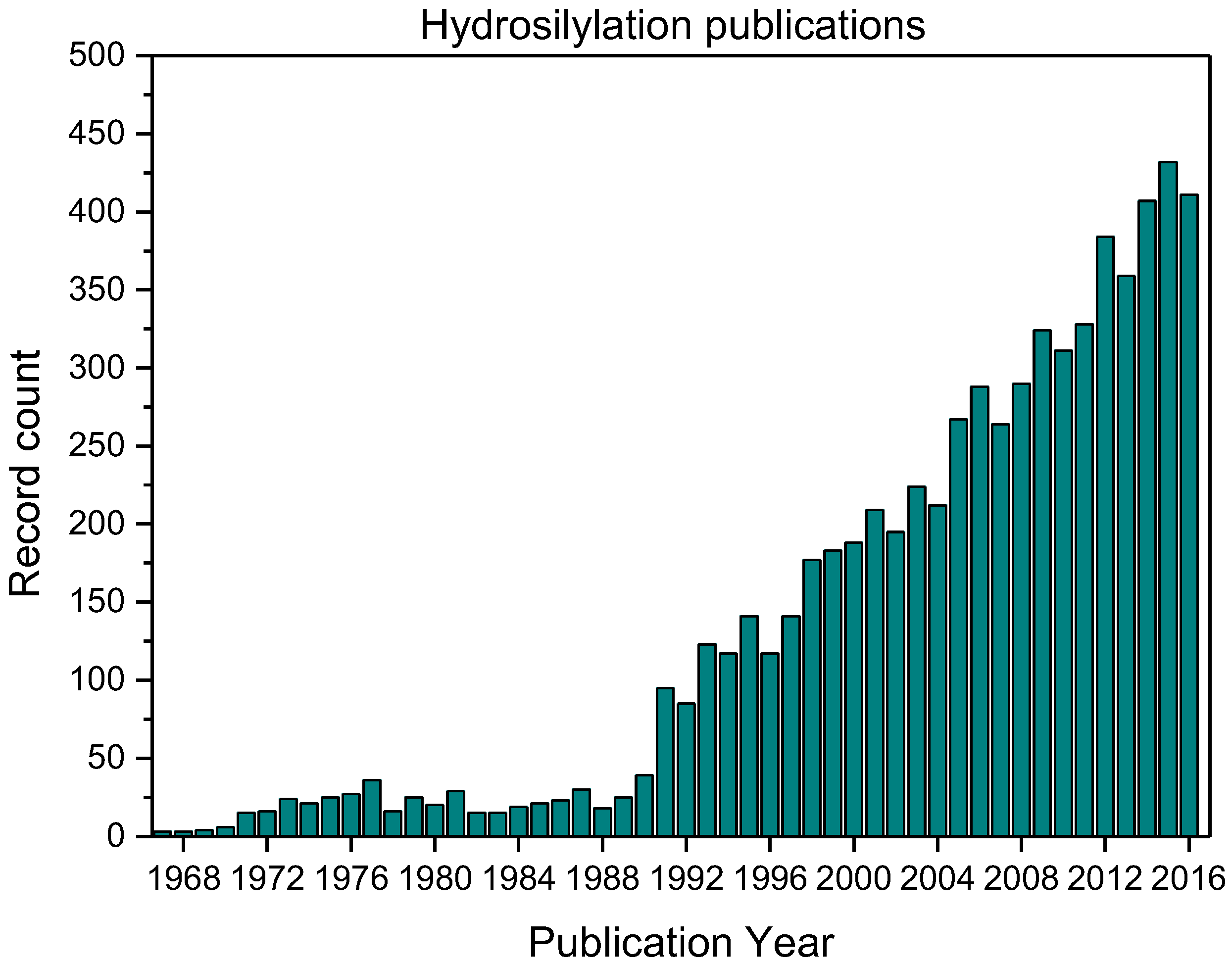
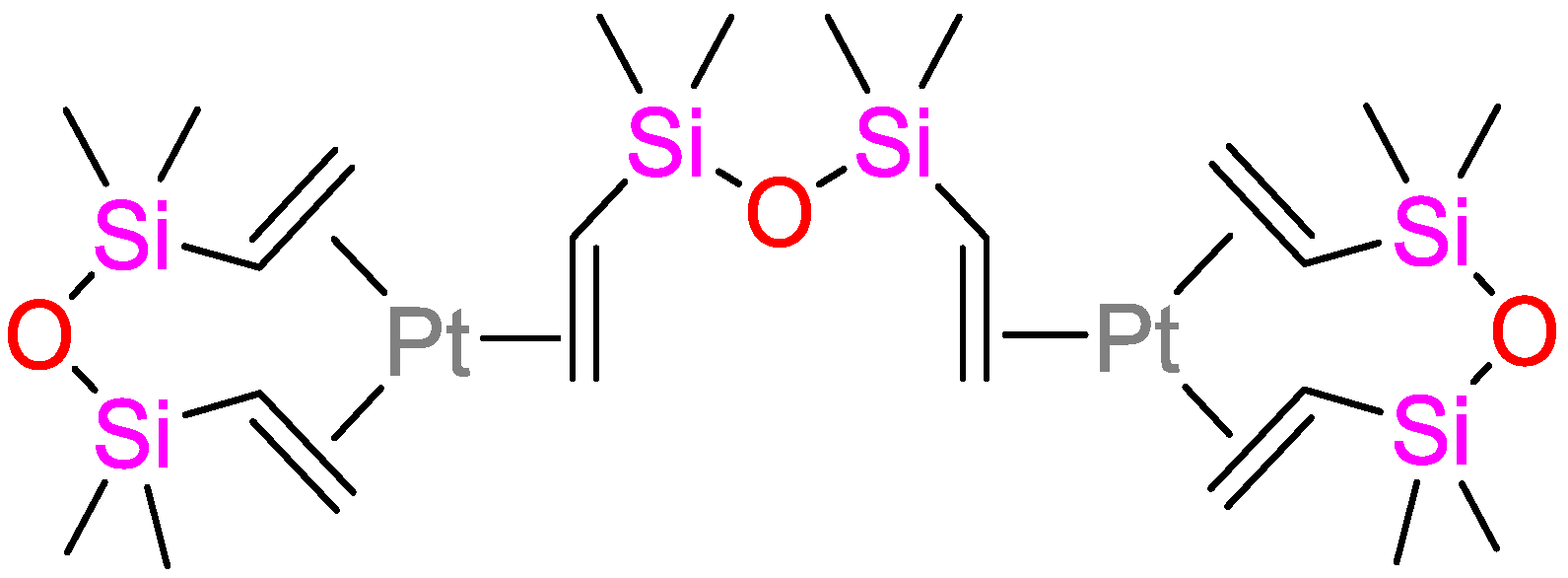
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
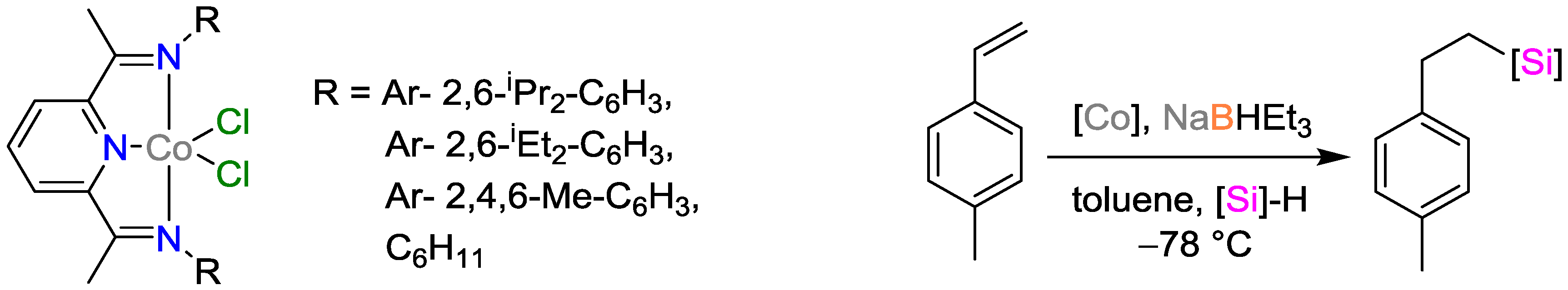{kind=link}
{kind=link}
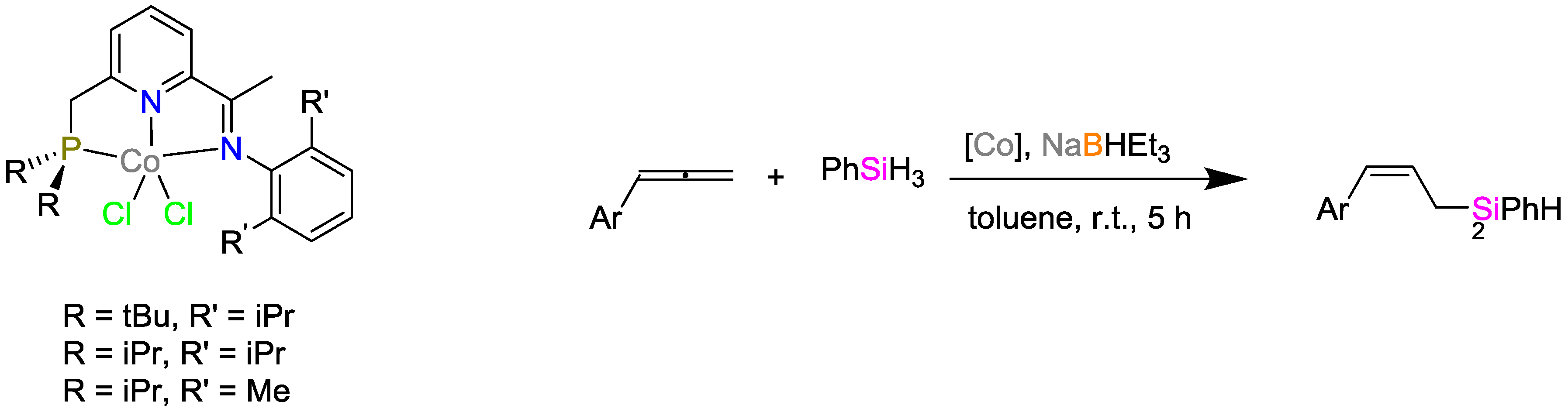{kind=link}
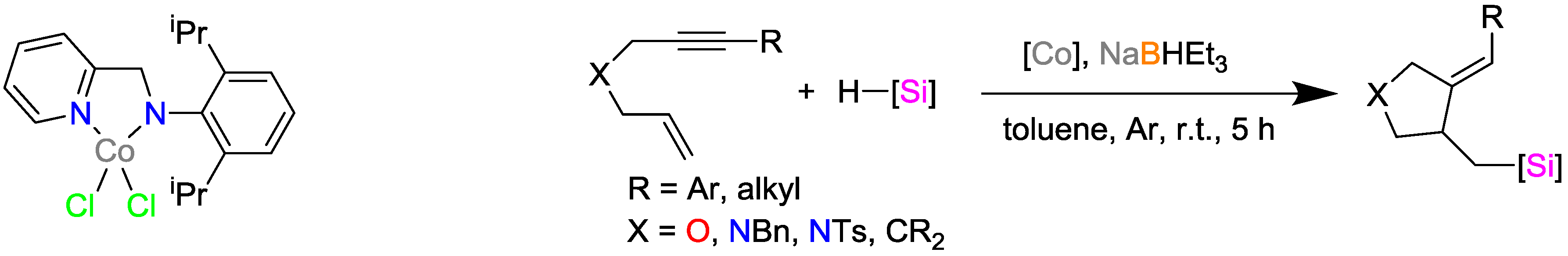{kind=link}
{kind=link}
{kind=link}
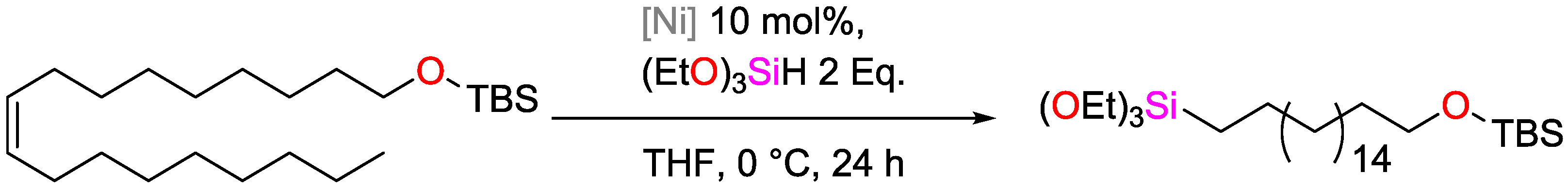{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
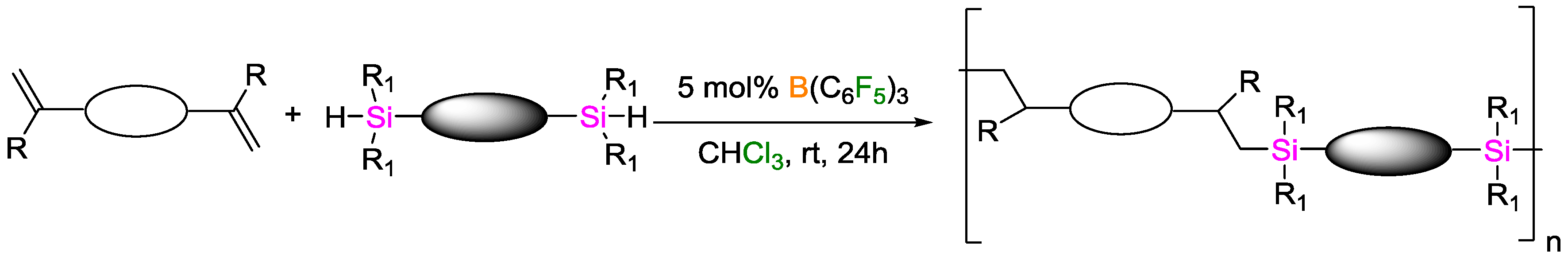{kind=link}
{kind=link}
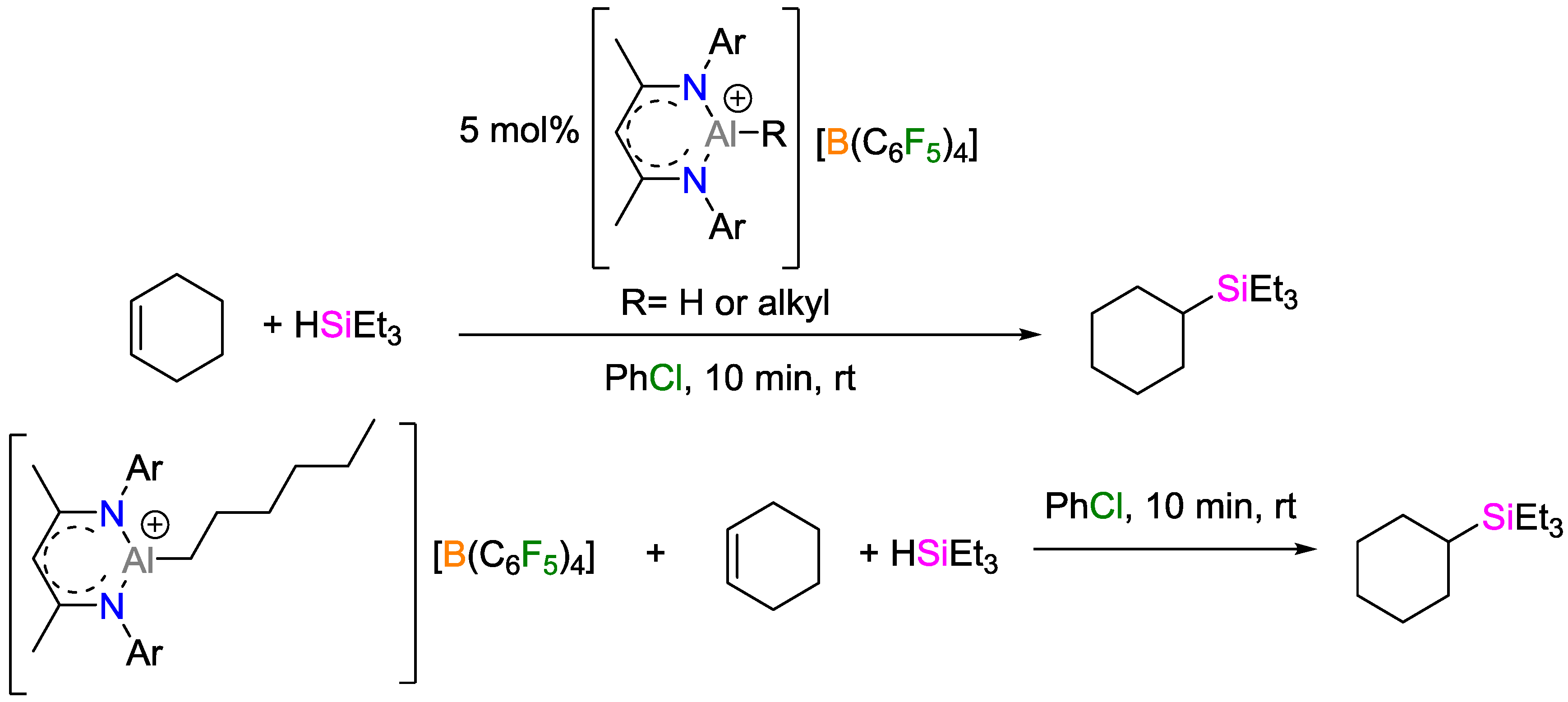{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
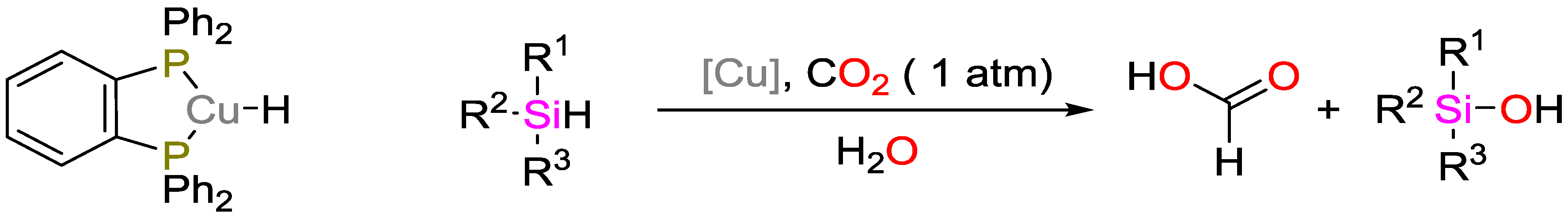{kind=link}
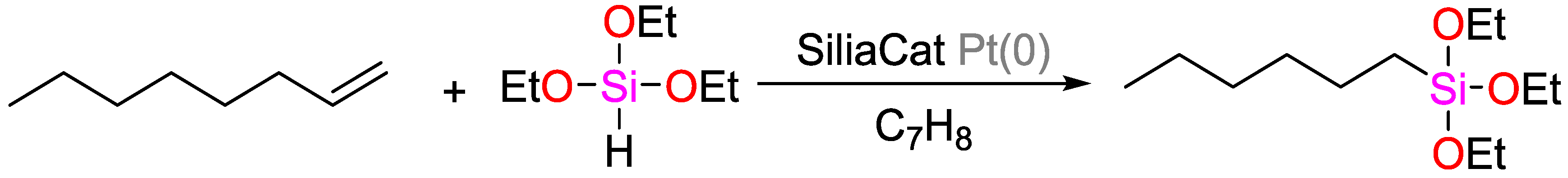{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Low-Cost Transition Metal Catalysts for Hydrosilylation
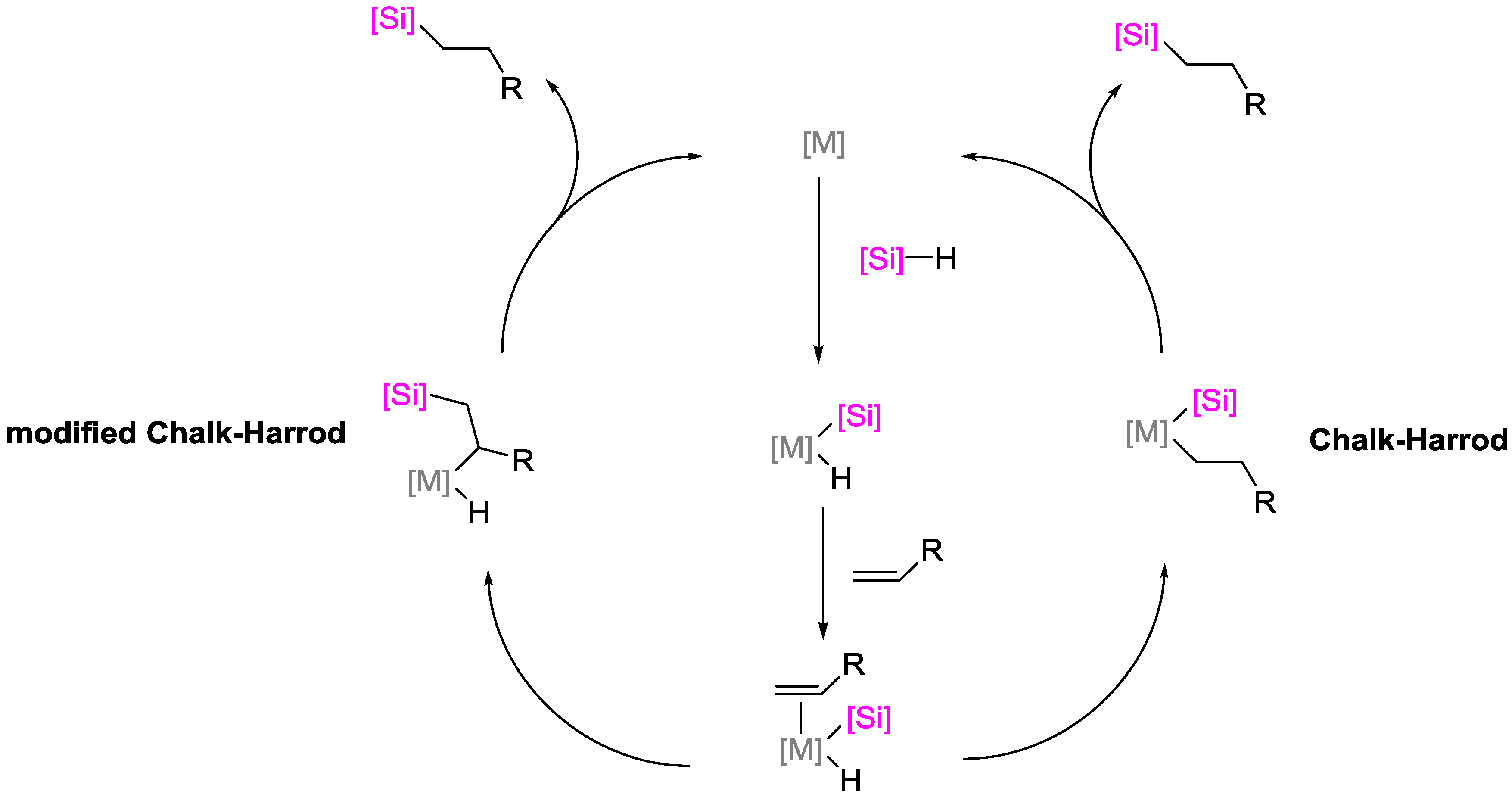
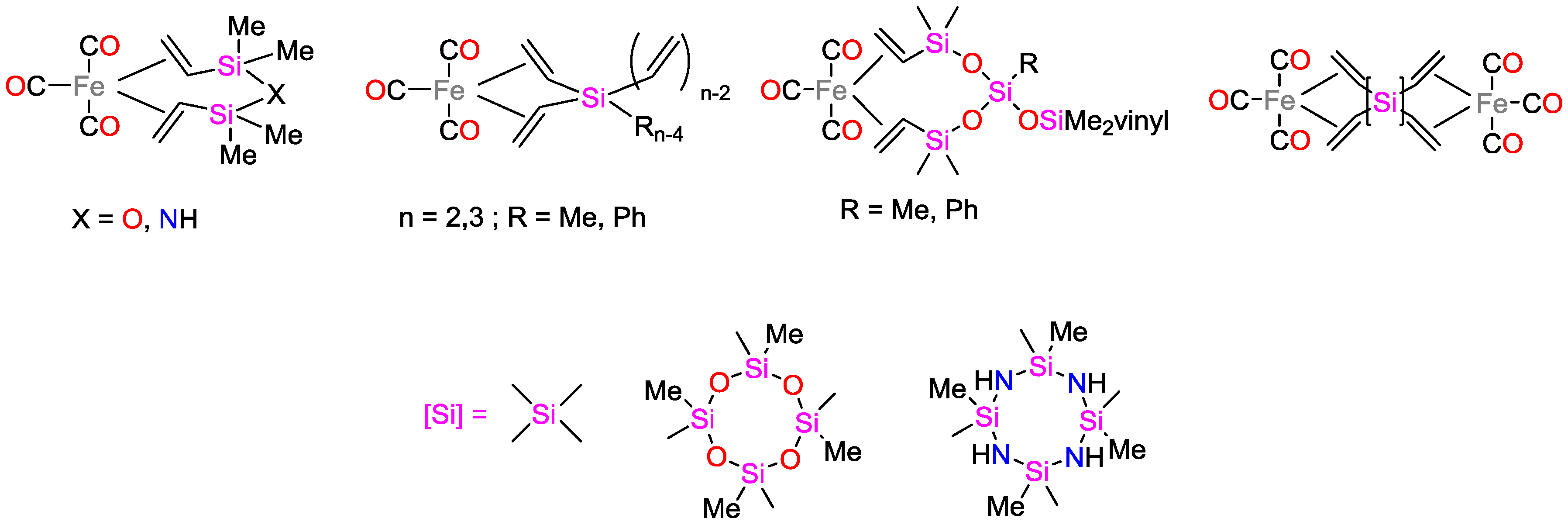
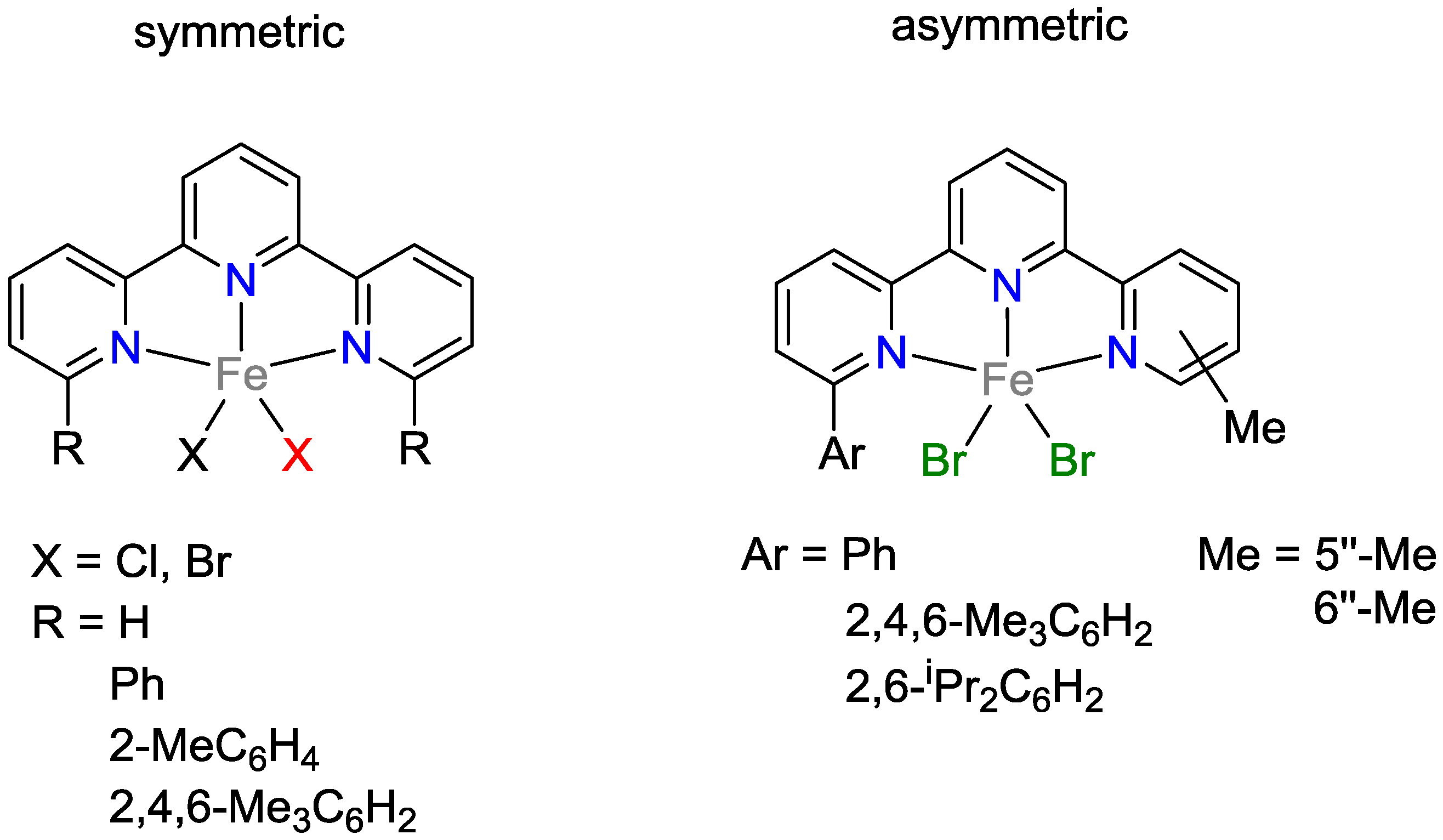
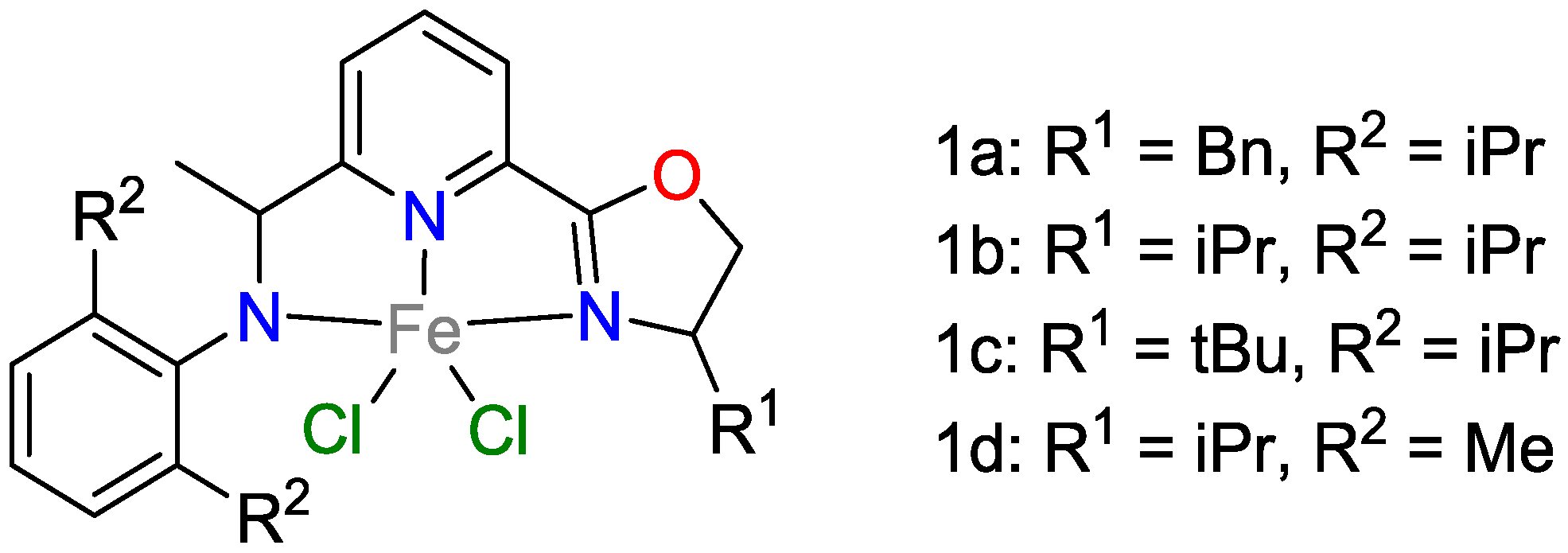
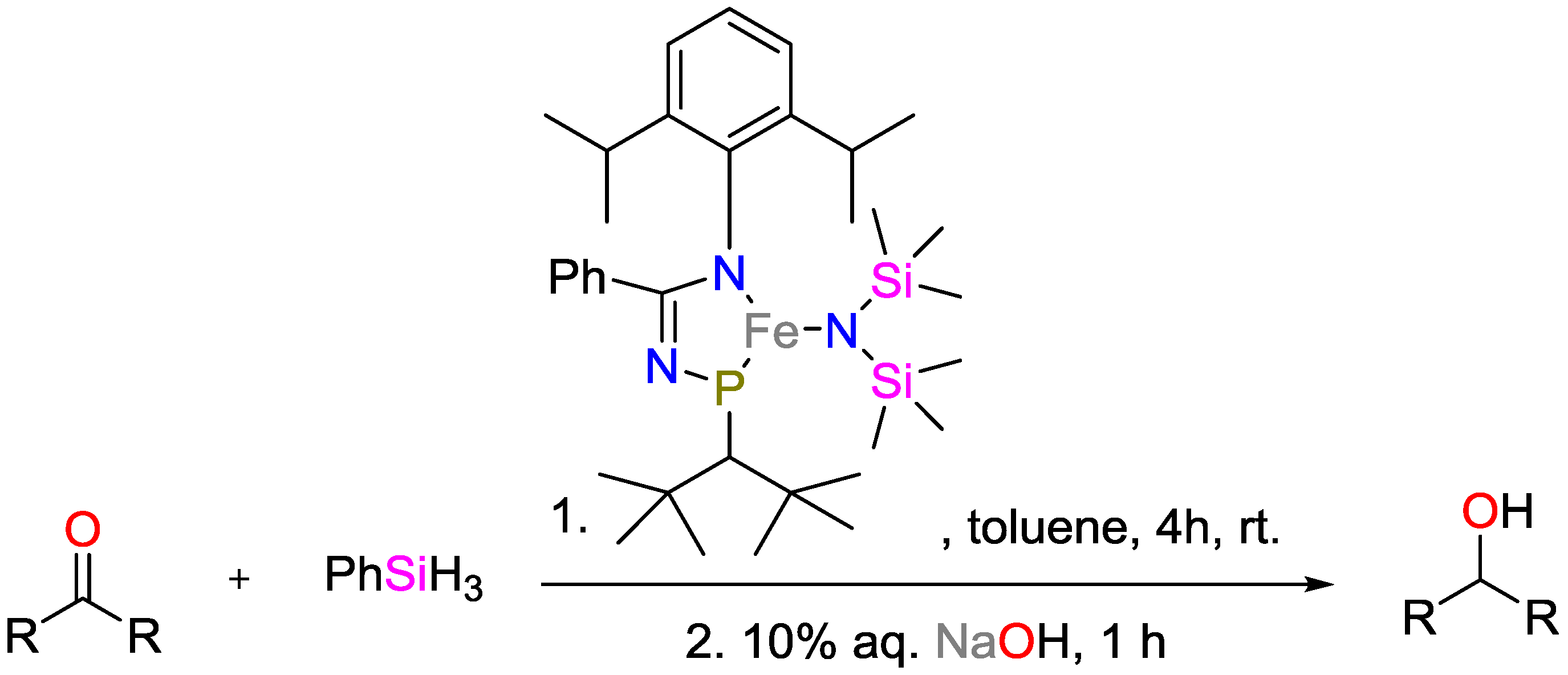
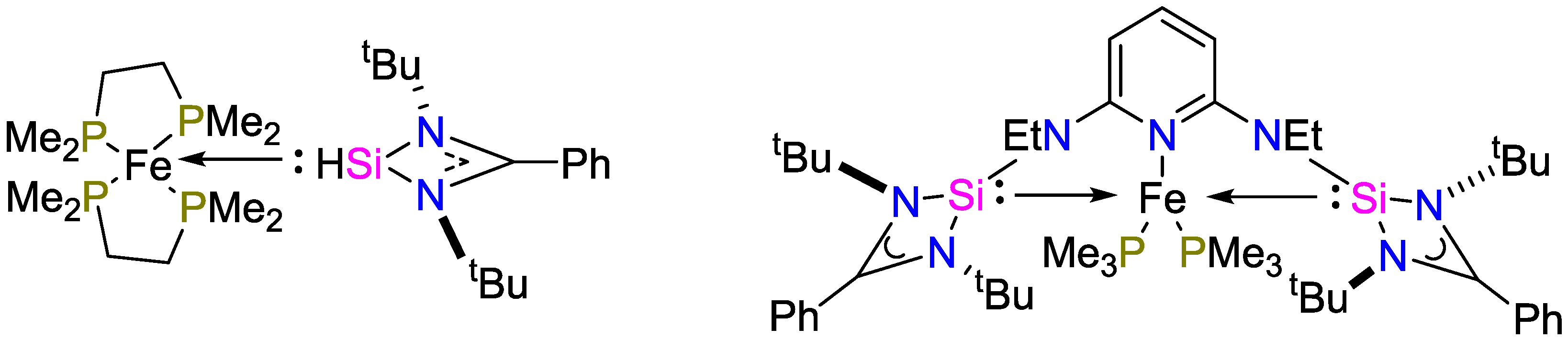
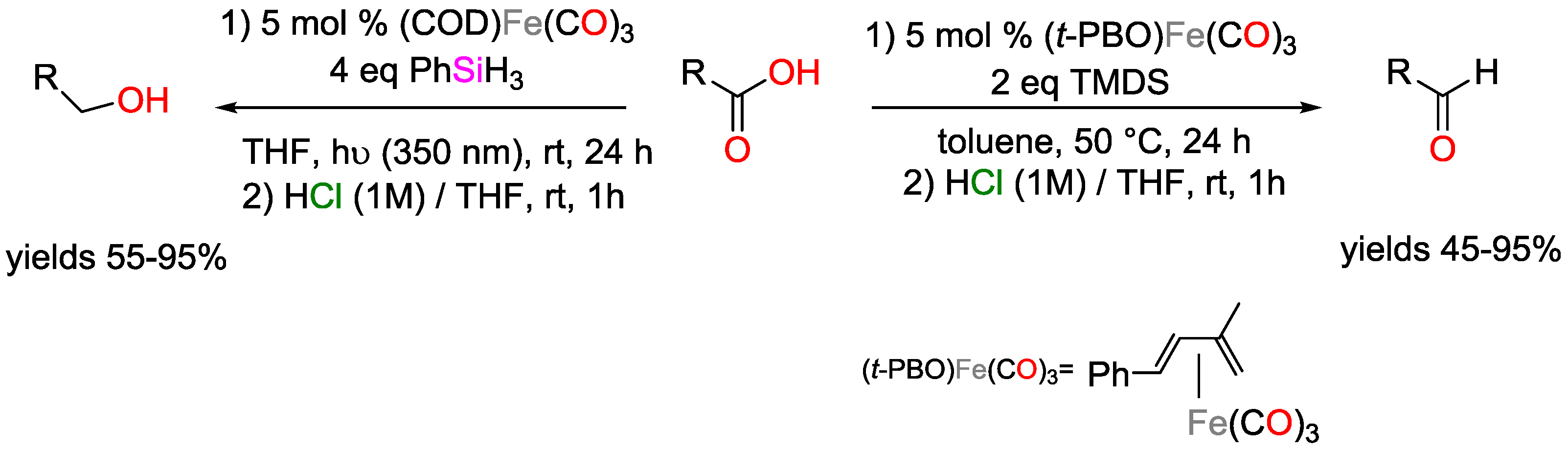
2.1. Iron Catalysts
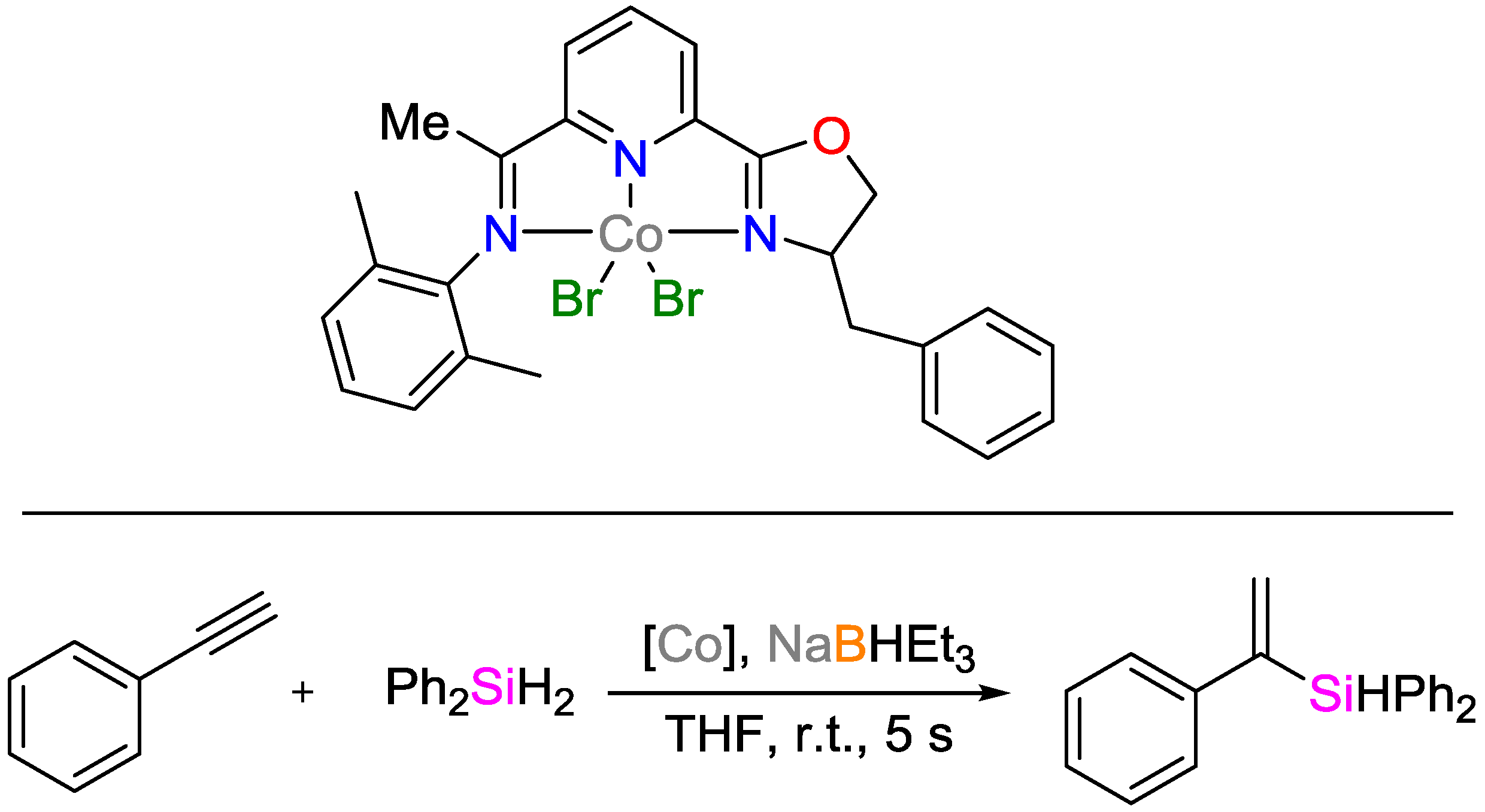
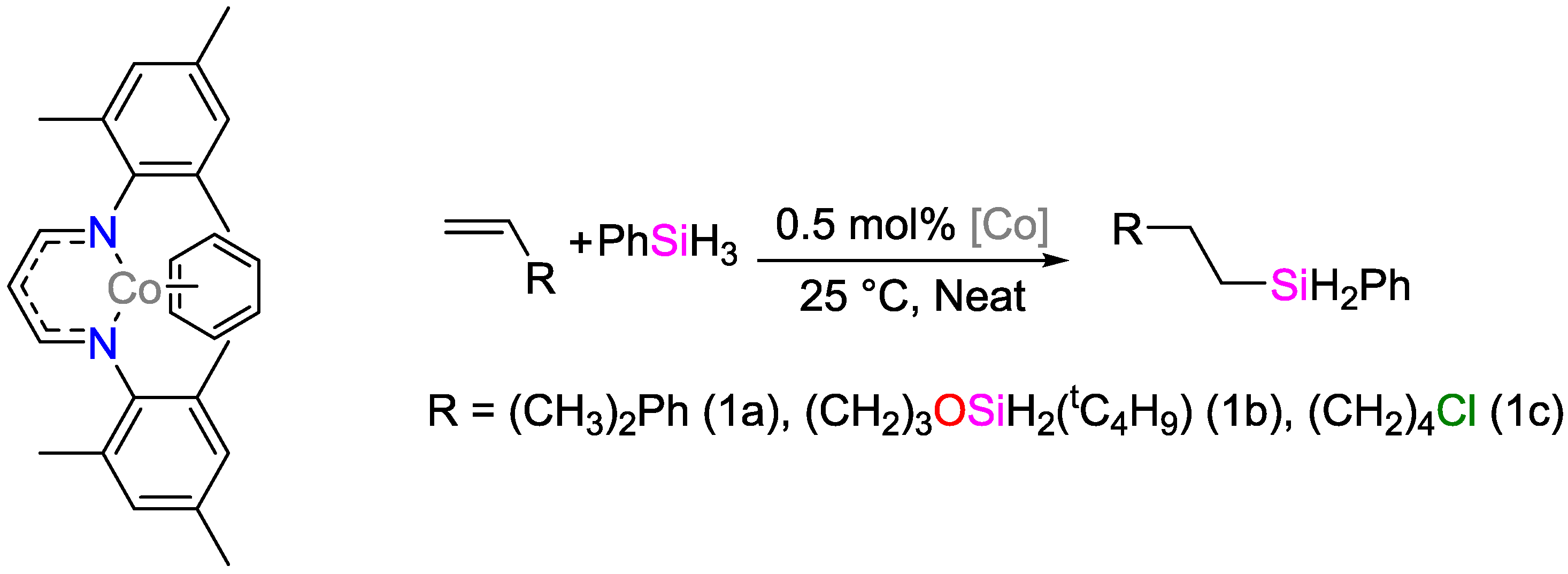
2.2. Cobalt Catalysts
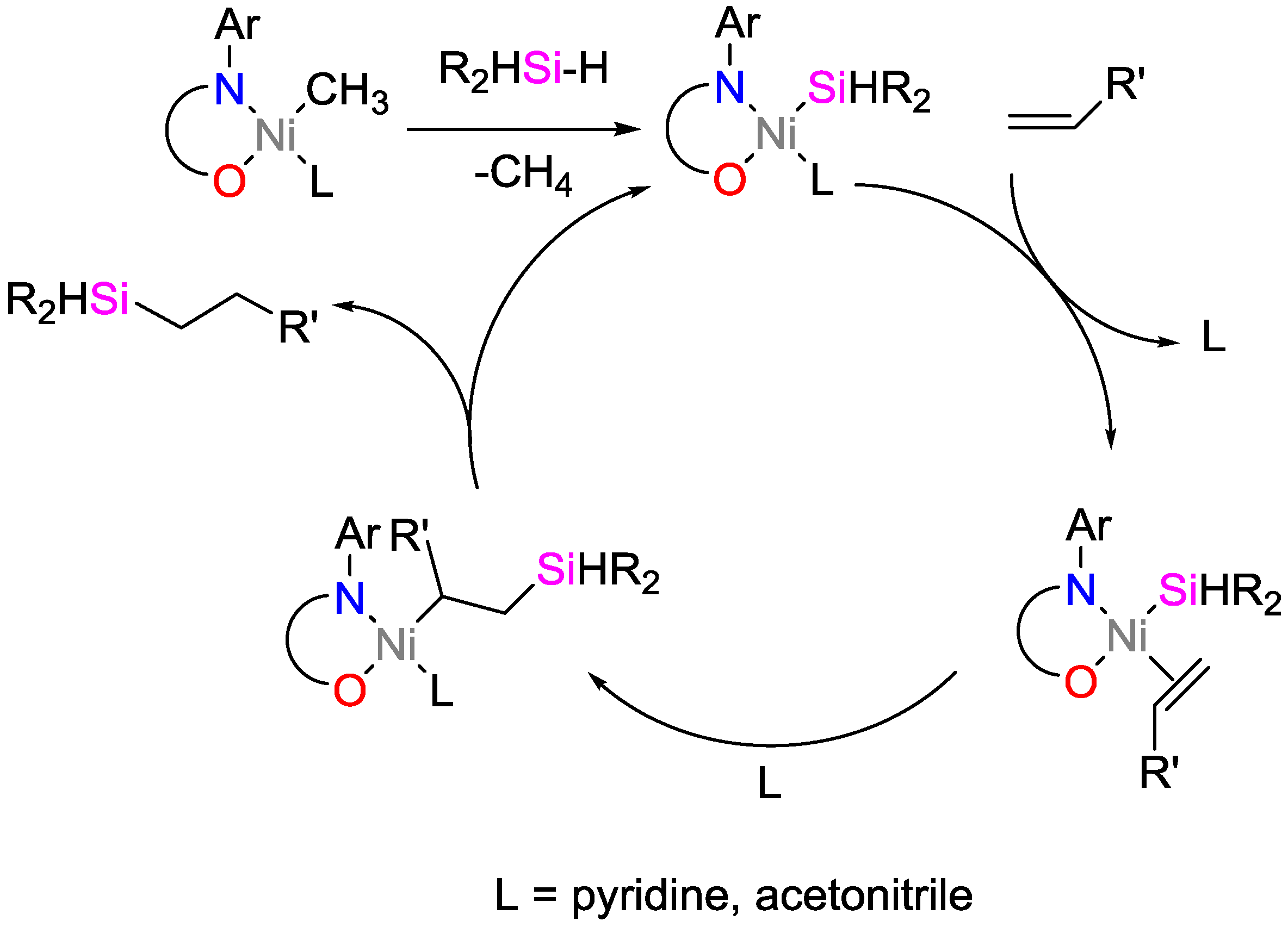
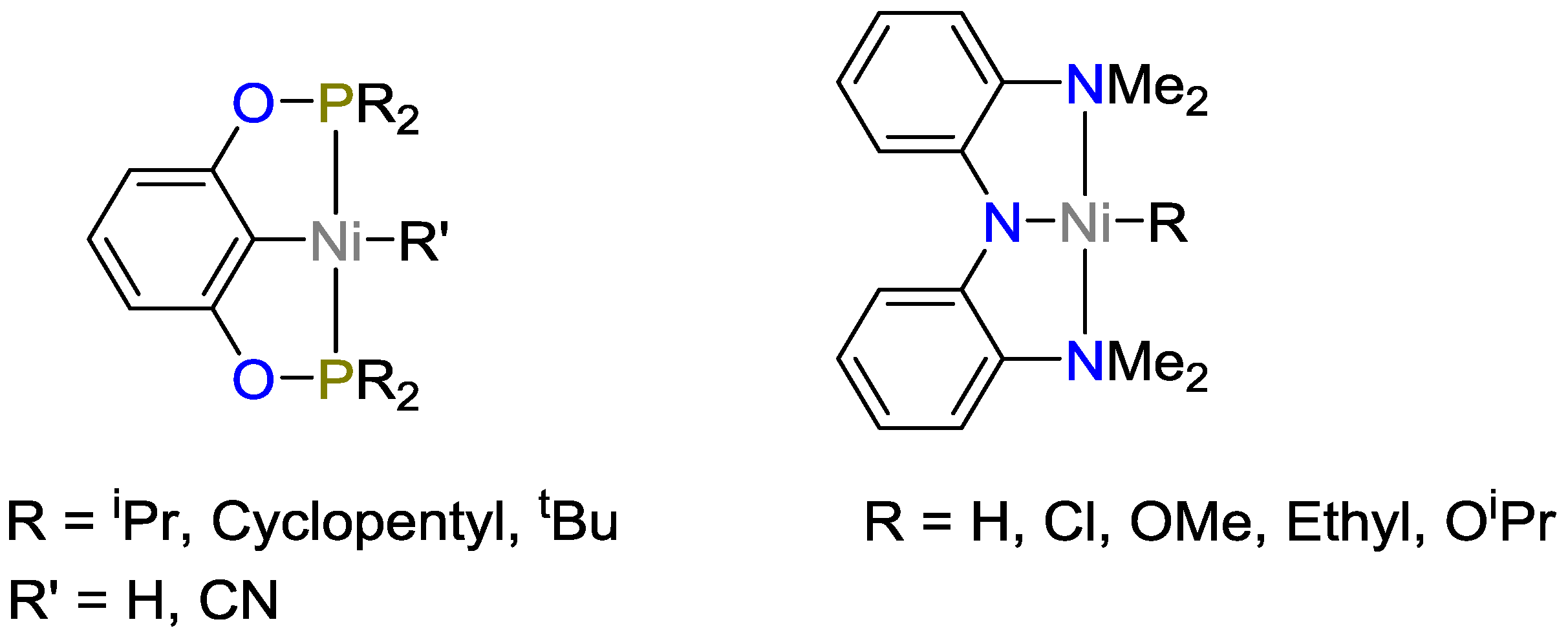
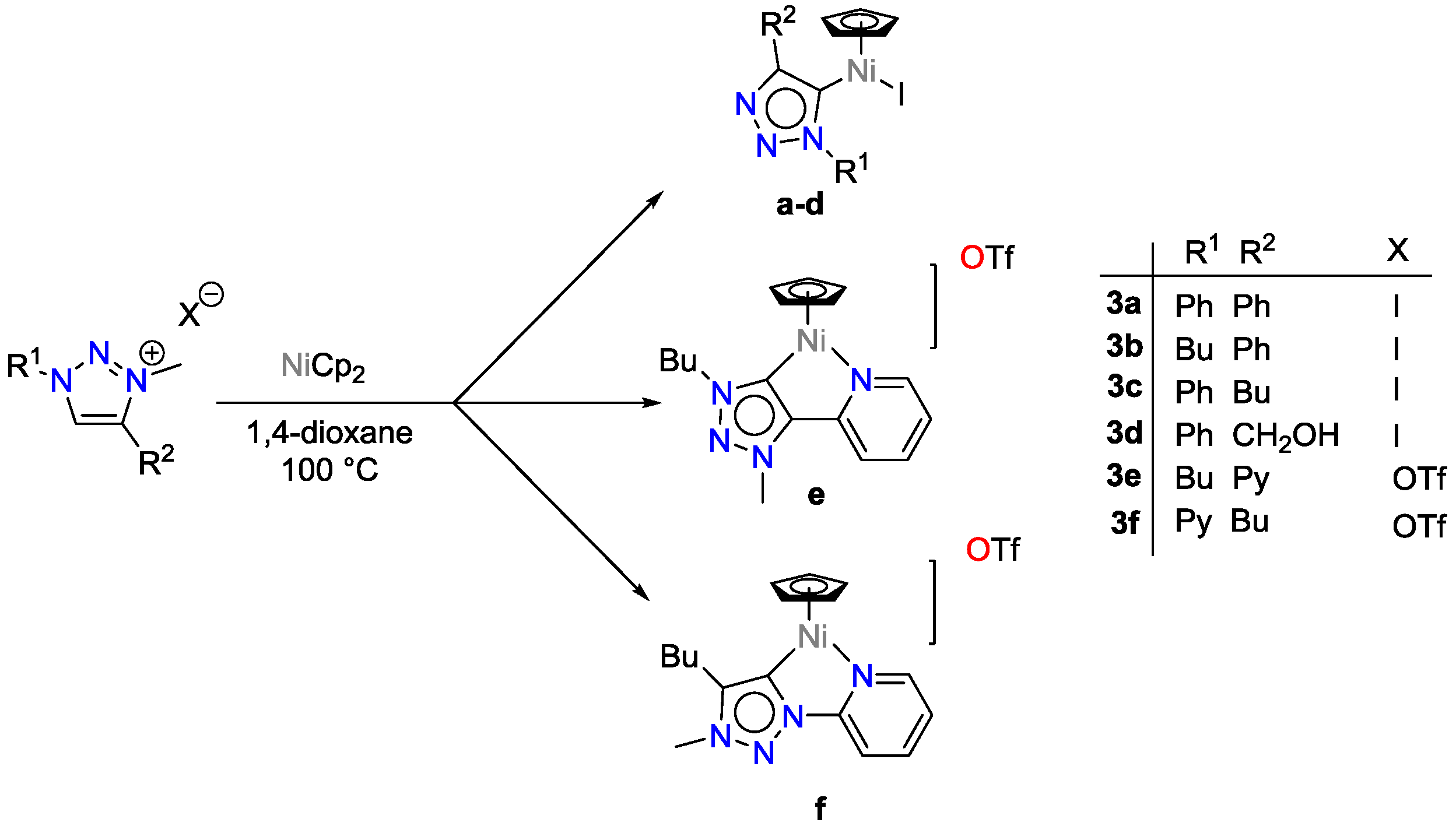
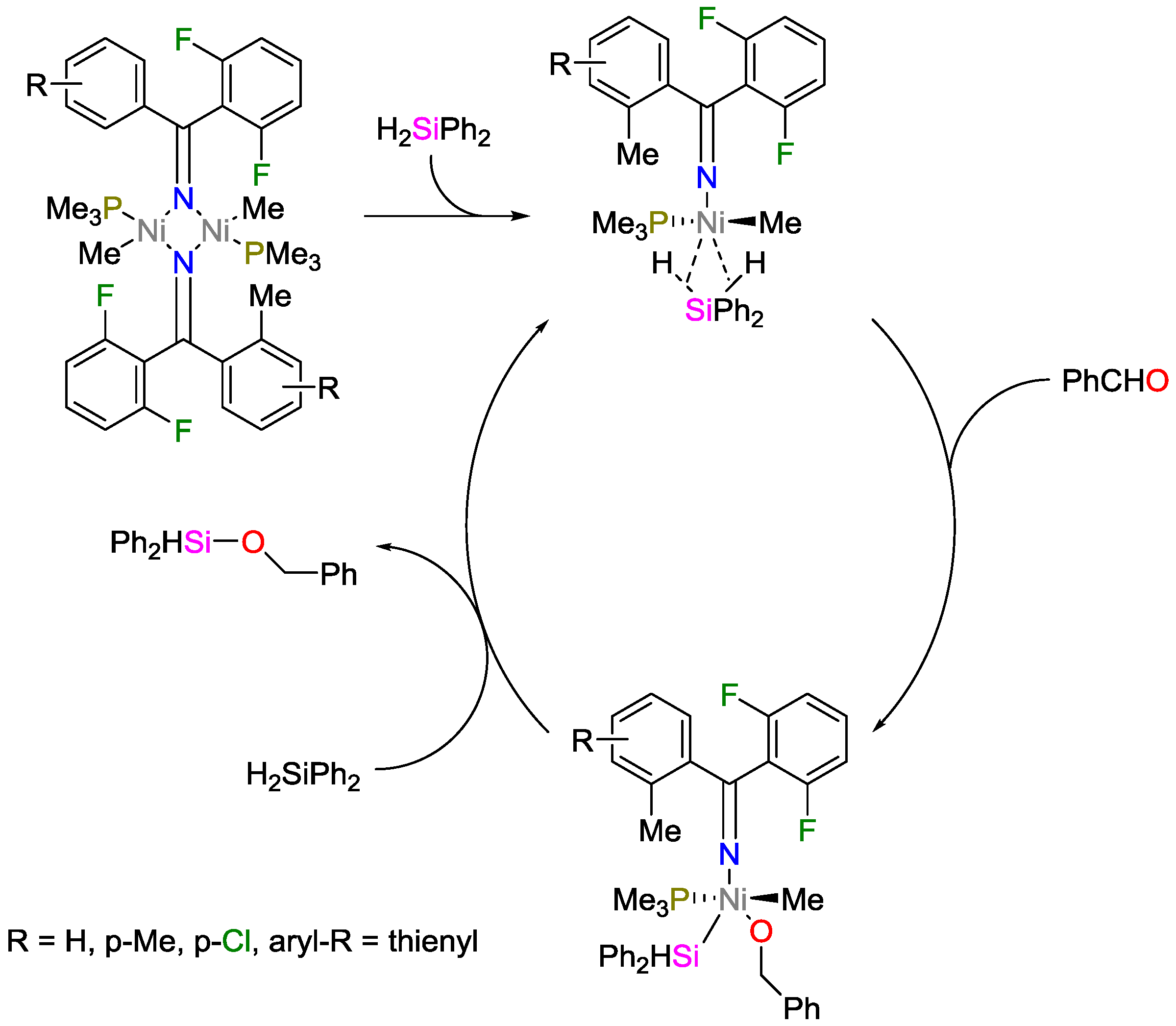
2.3. Nickel Catalysts
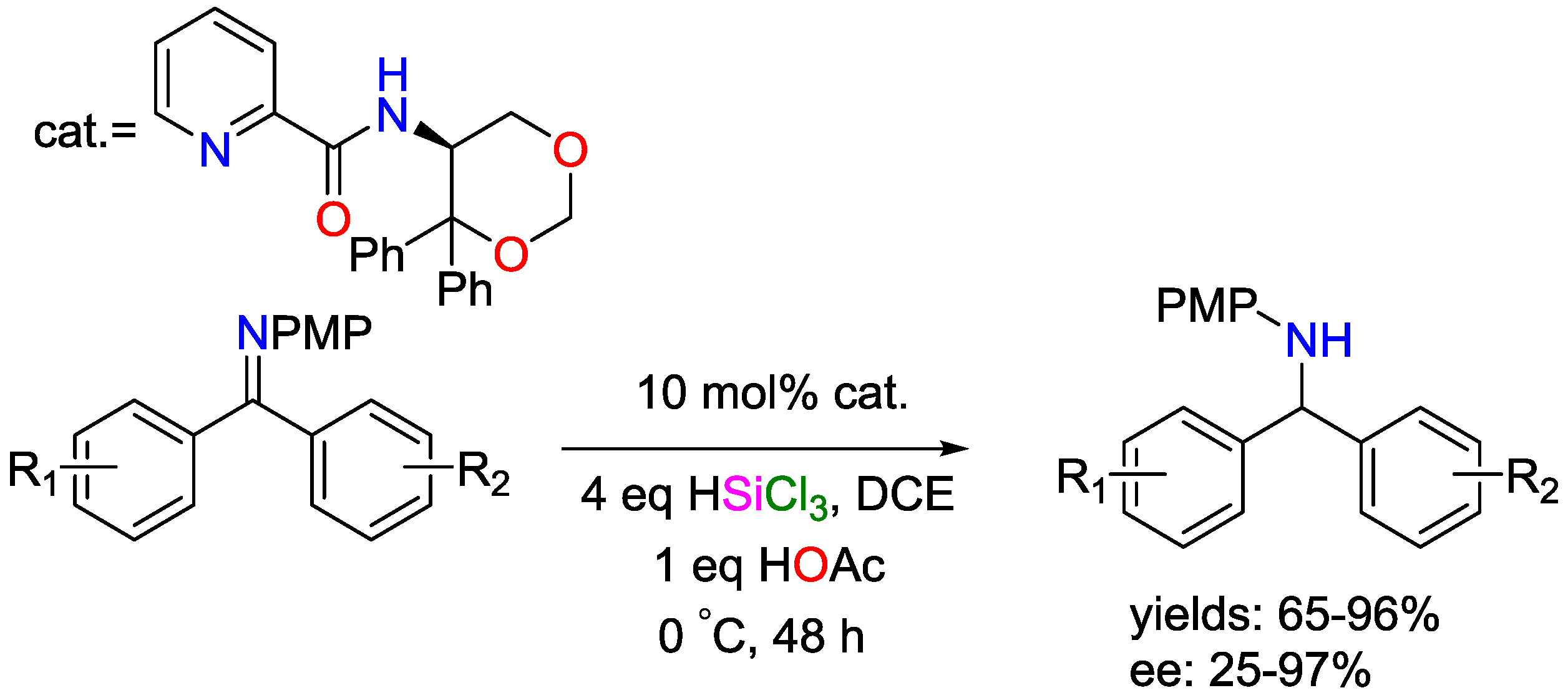
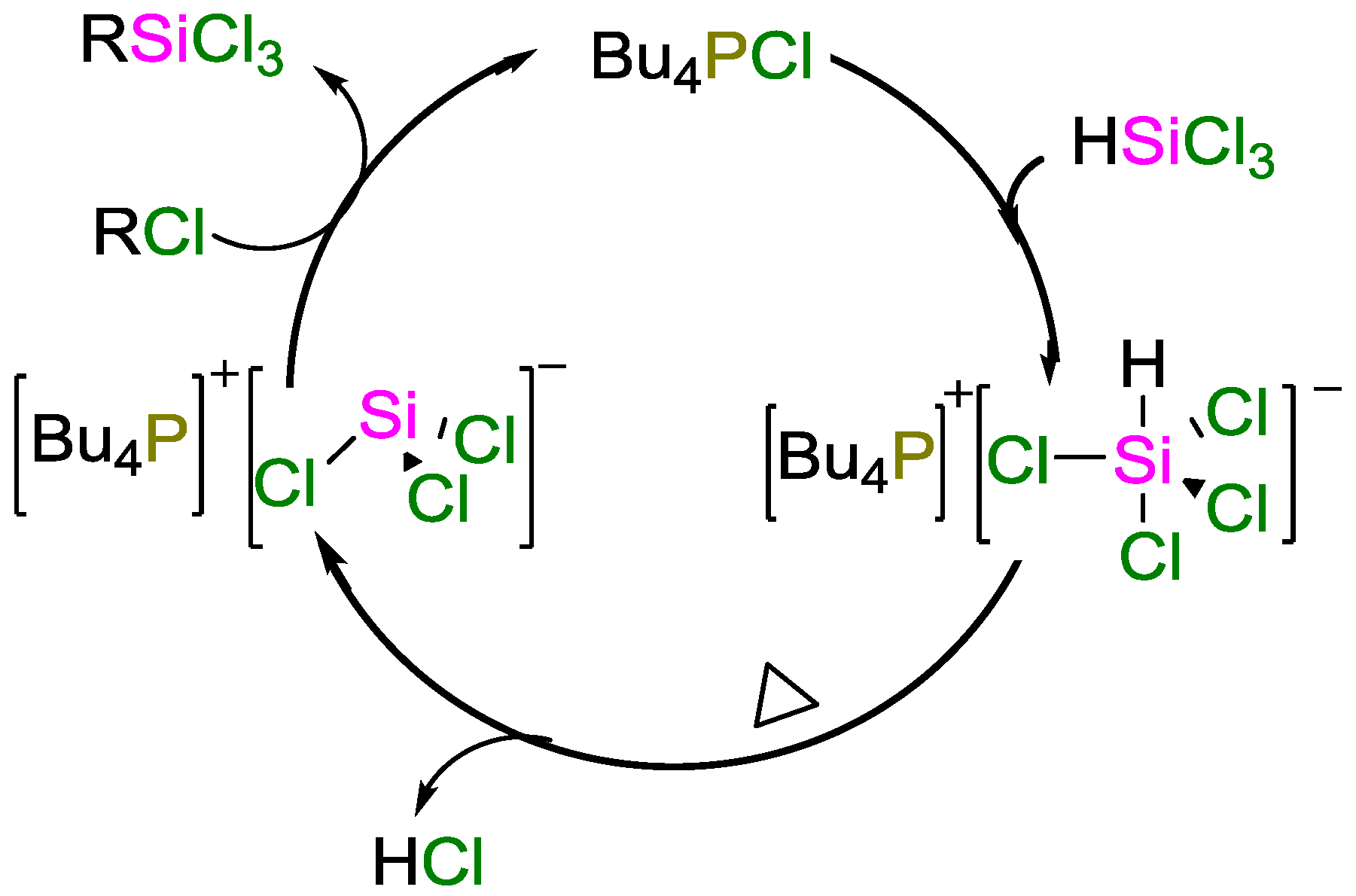
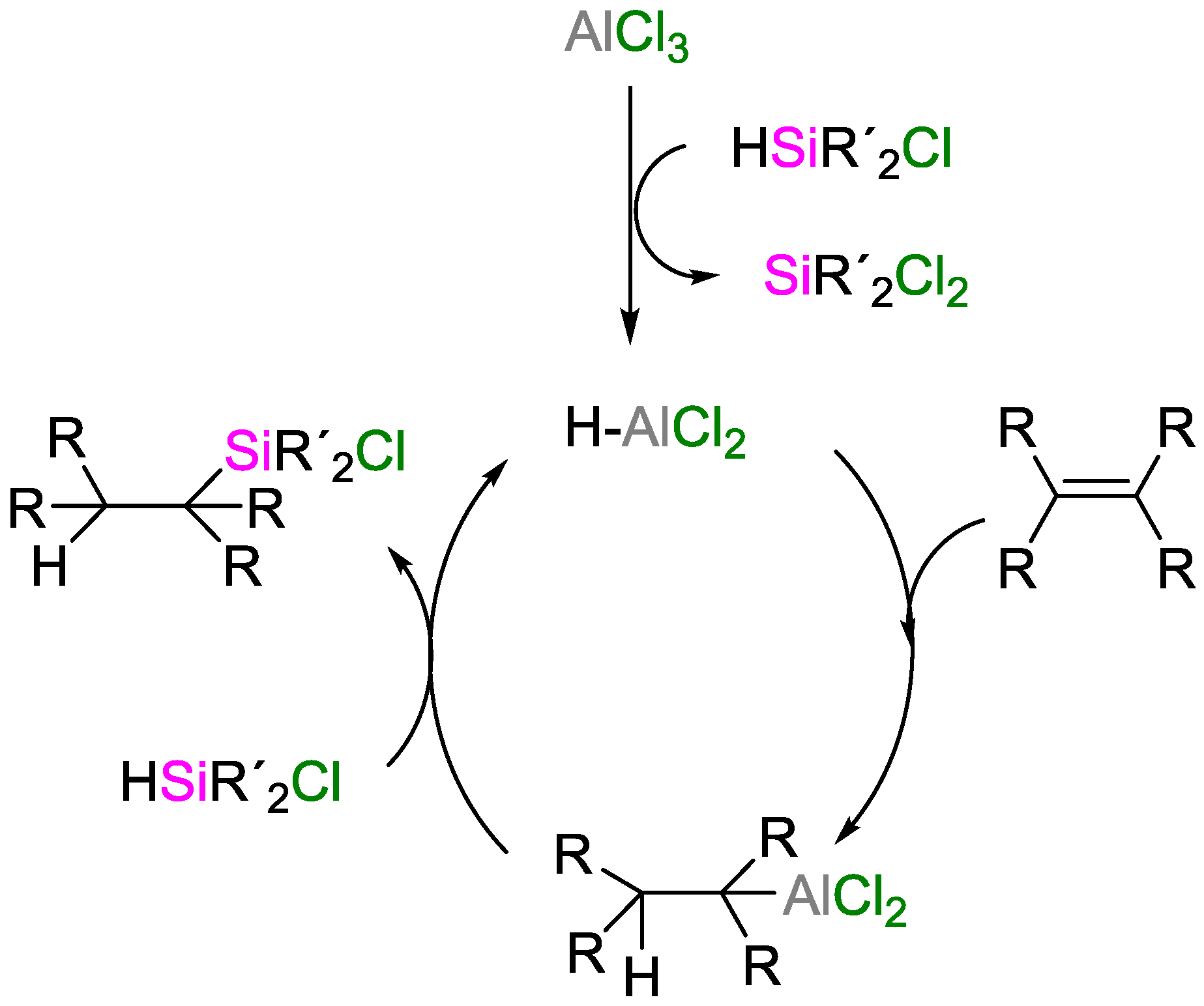
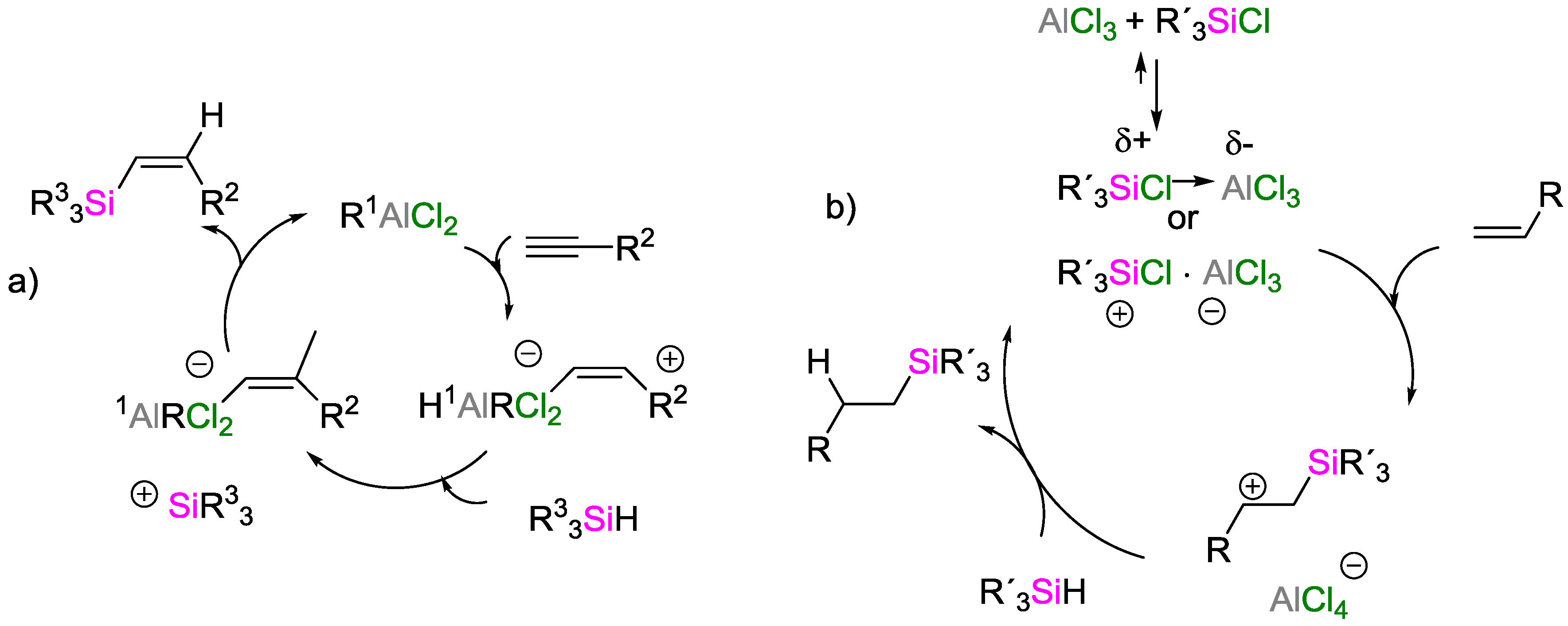
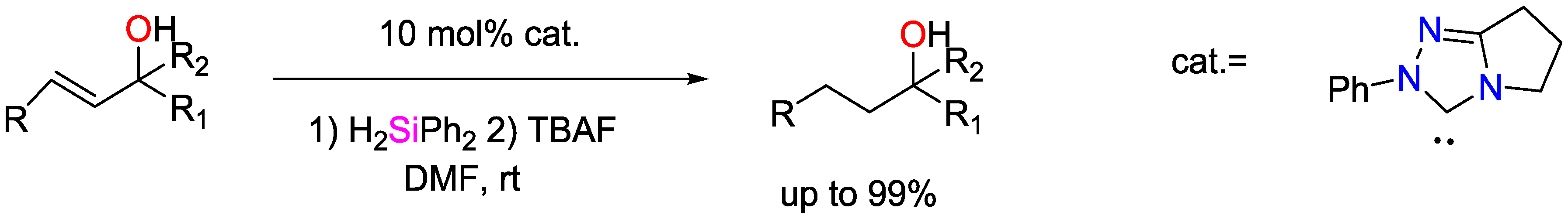
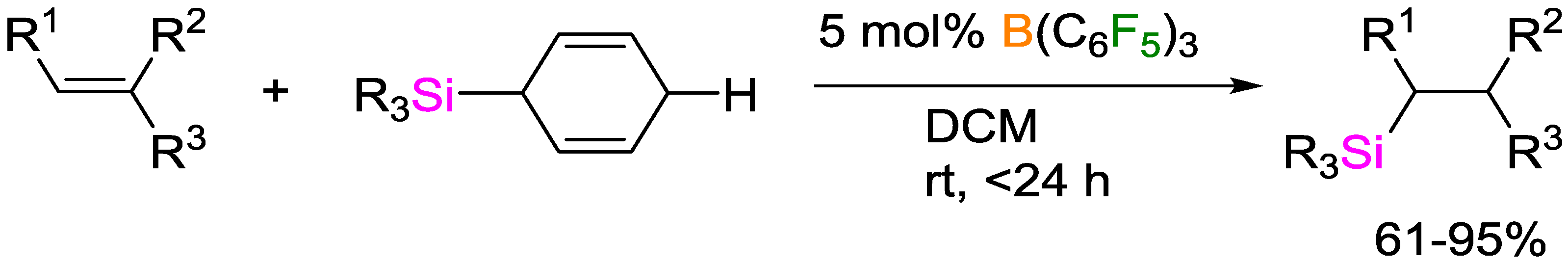
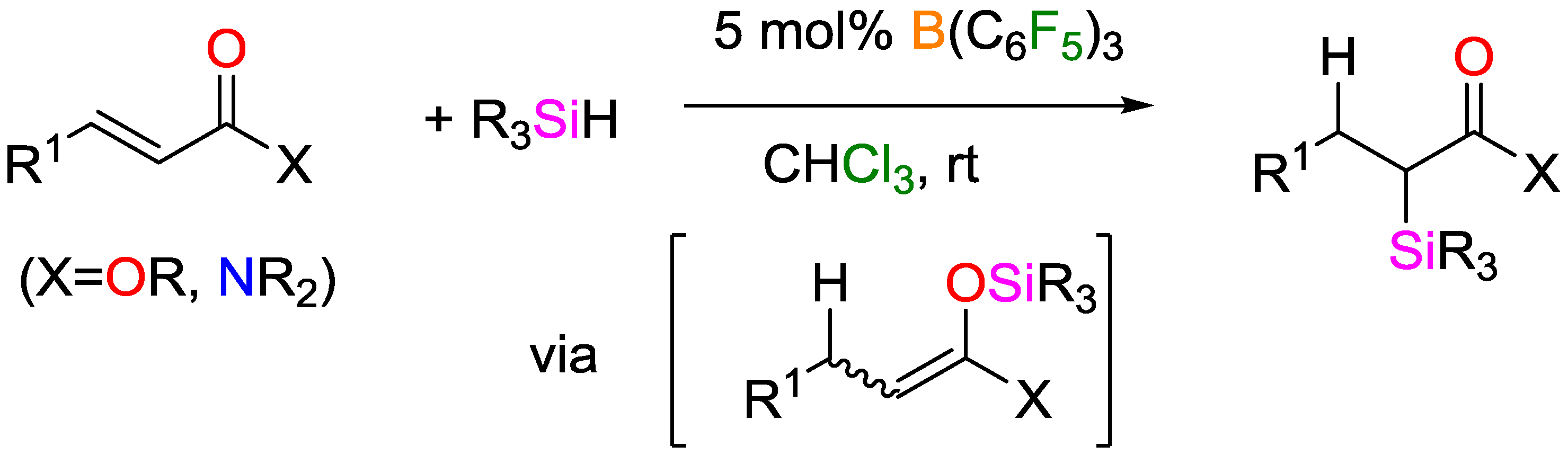
3. Non-Metal Catalysts for Hydrosilylation Reactions
4. Non-Thermal Stimuli for Hydrosilylation Reactions
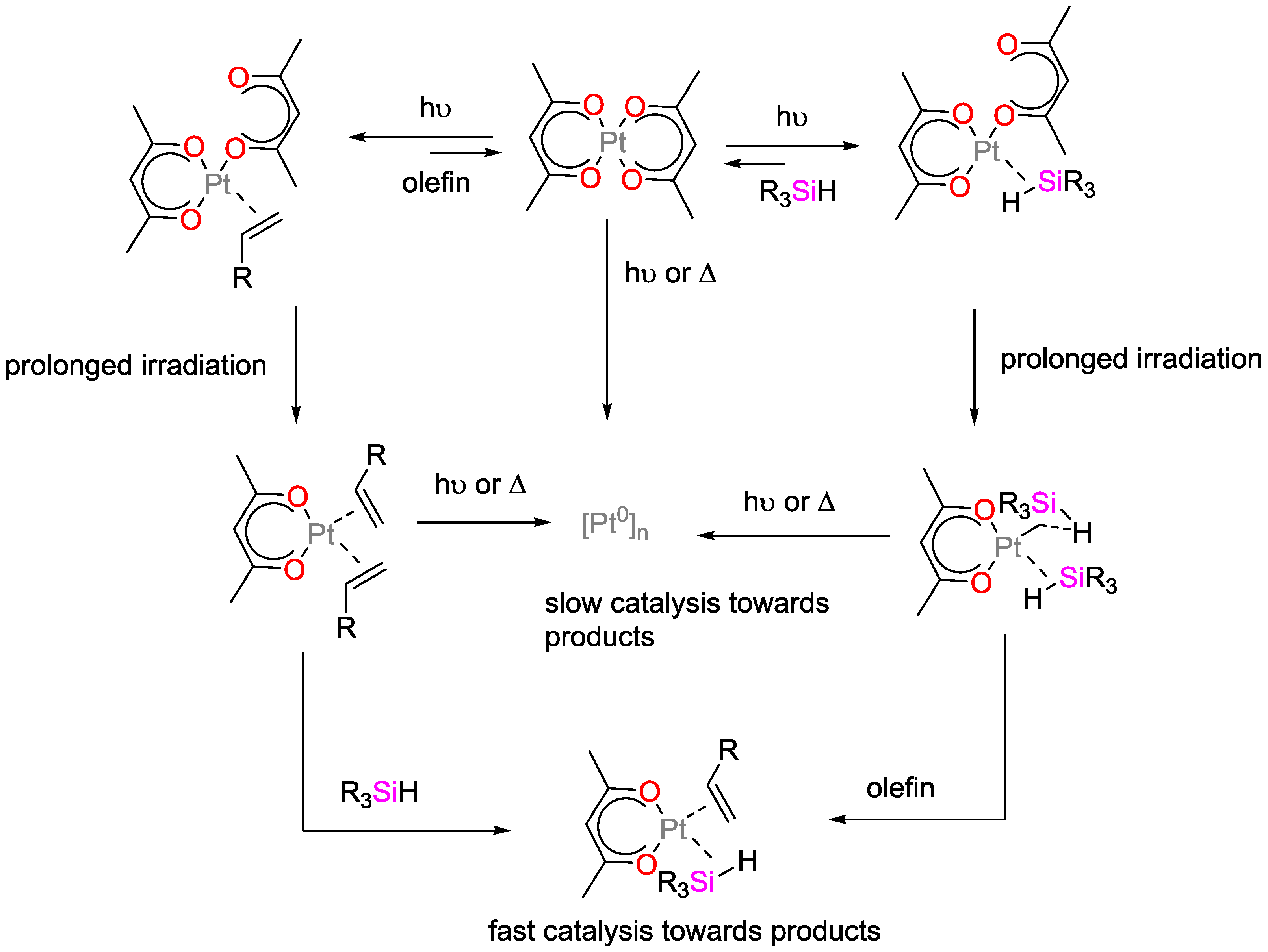
4.1. Light-Initiated Hydrosilylation Reactions
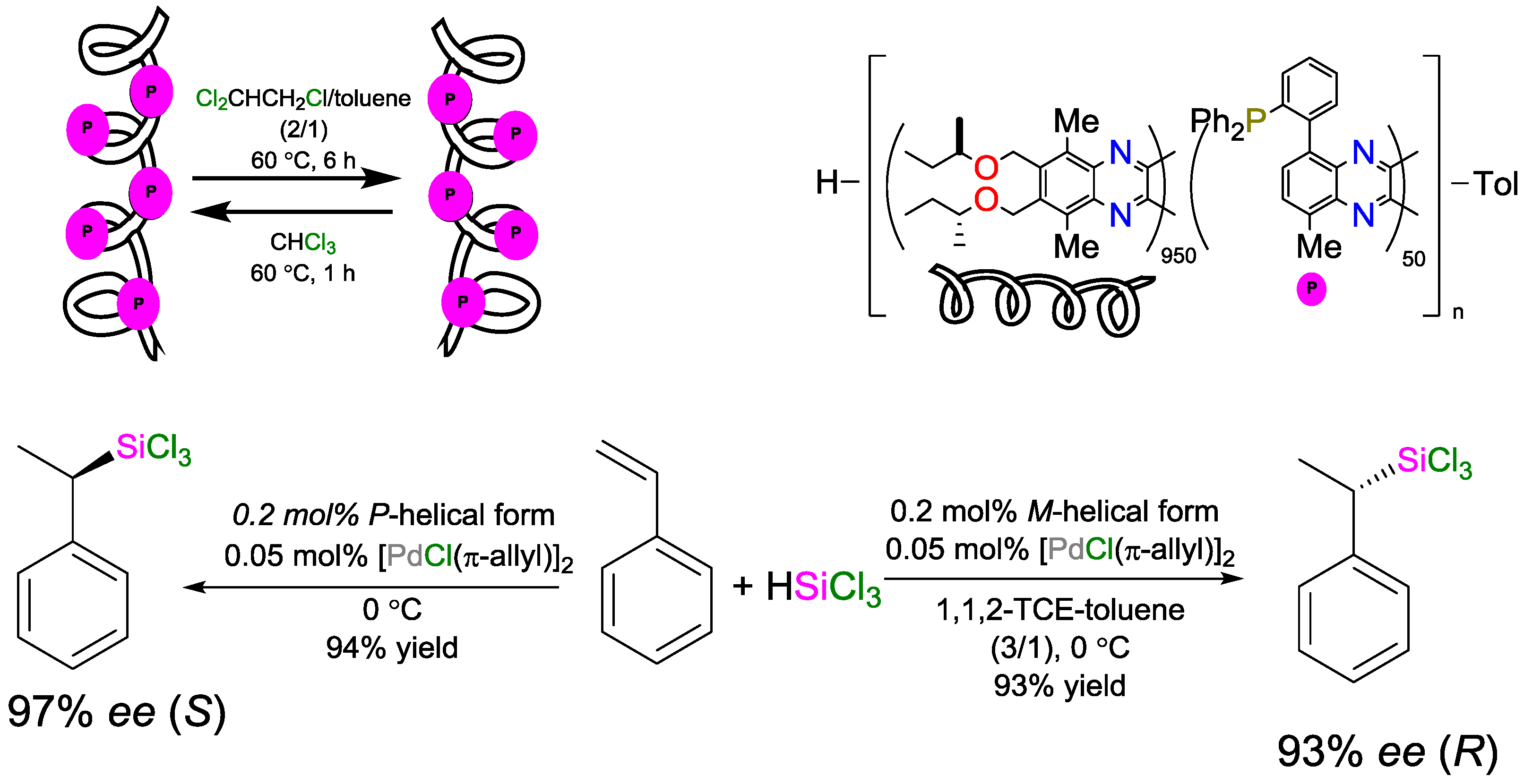
4.2. Solvent as a Trigger to Influence the Stereochemical Configuration of Hydrosilylated Products
4.3. Microwave-Initiated Hydrosilylation Reactions
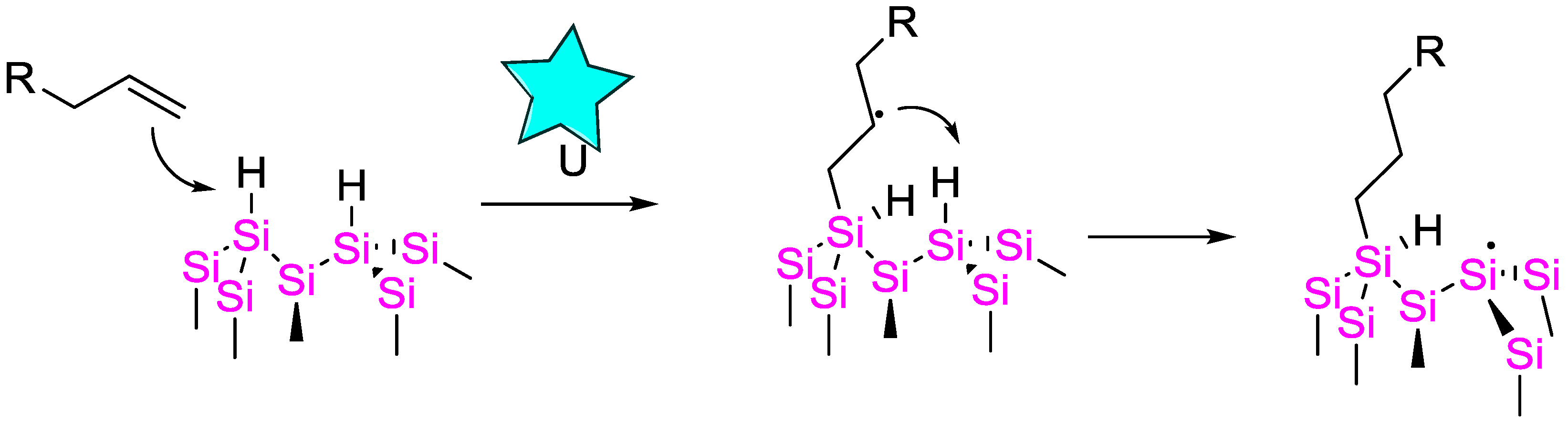
4.4. Sonication-Triggered Hydrosilylation Reactions
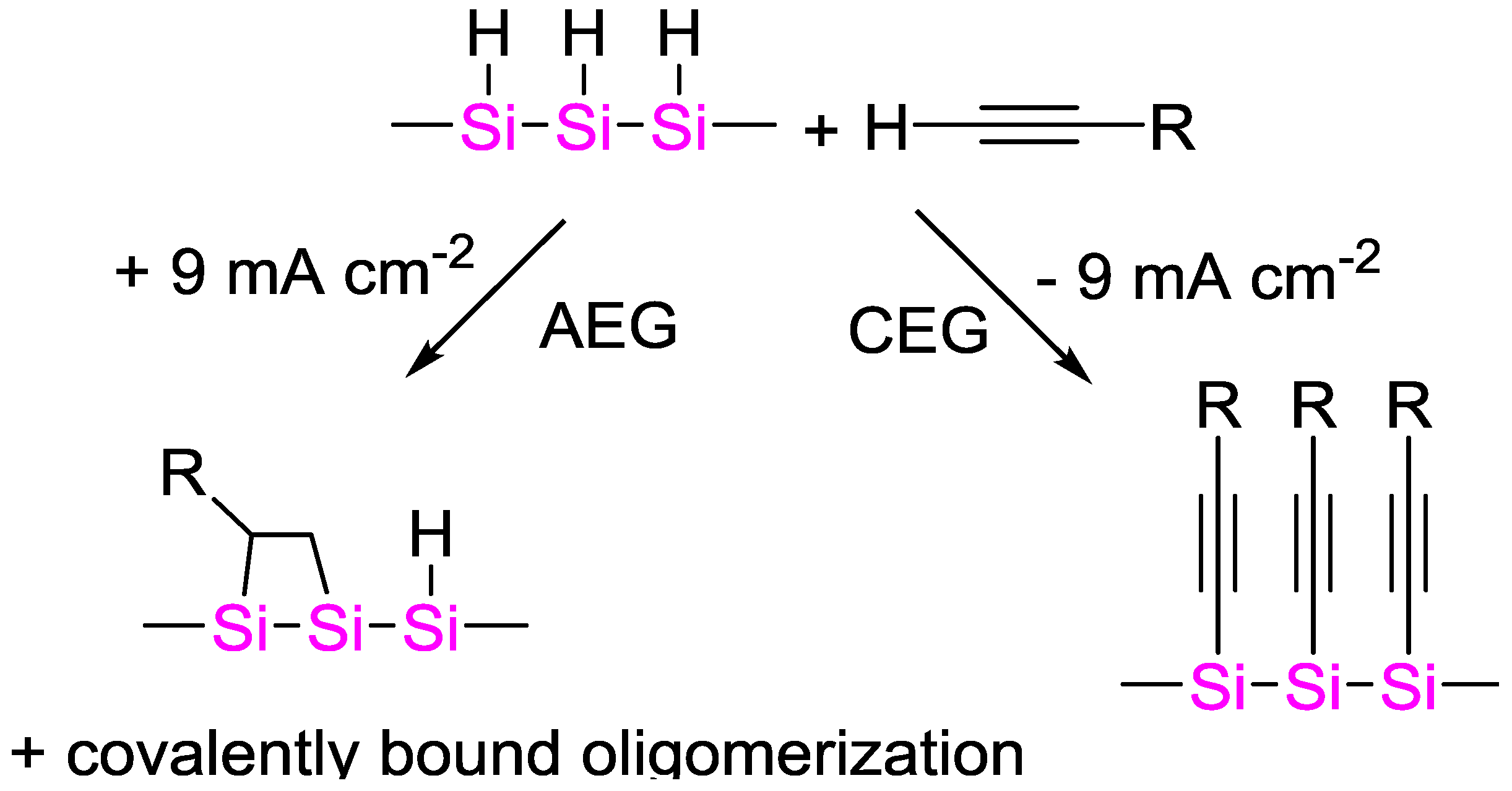
4.5. Electrochemically Initiated Hydrosilylation Reactions
5. Industrial Applications
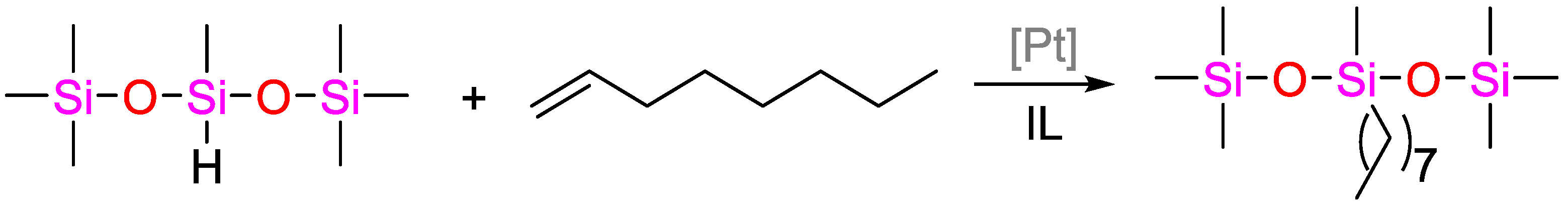
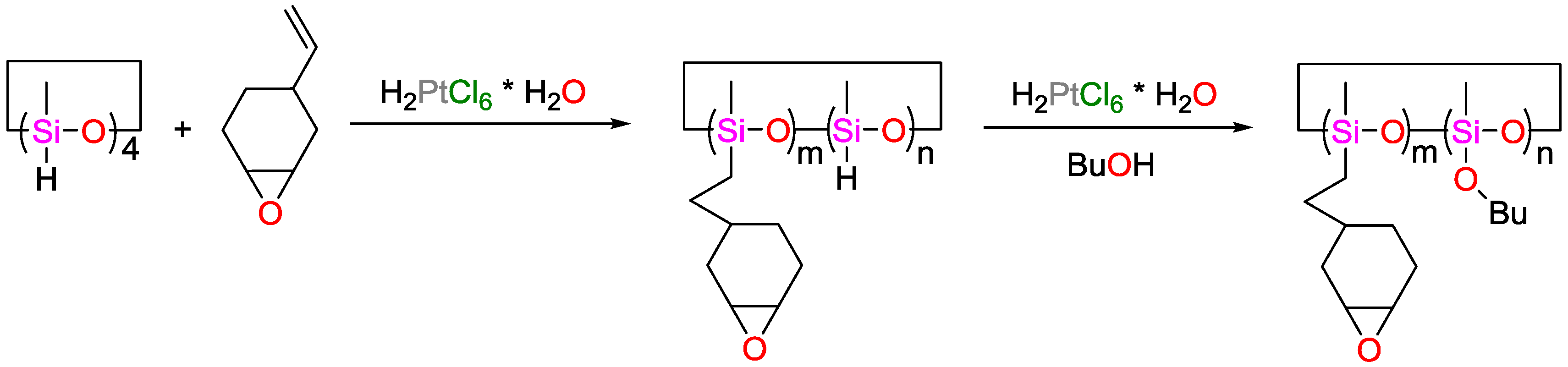
5.1. High Efficient/Low-Cost Hydrosilylation (Solvent-Free Conditions)
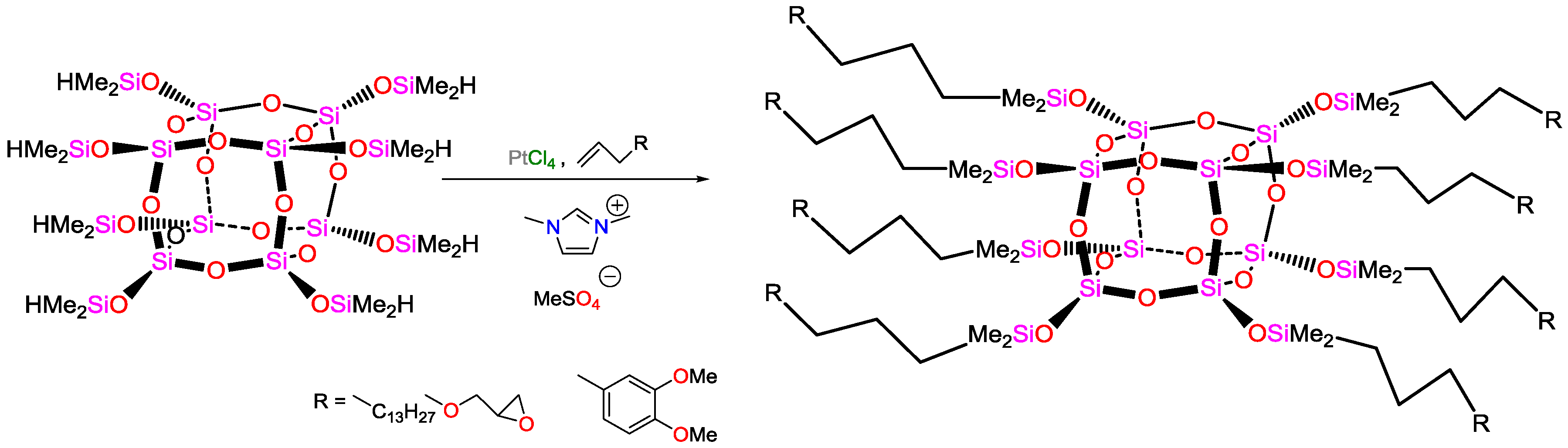
5.2. Recent Advances in Functional Materials Using Hydrosilylation
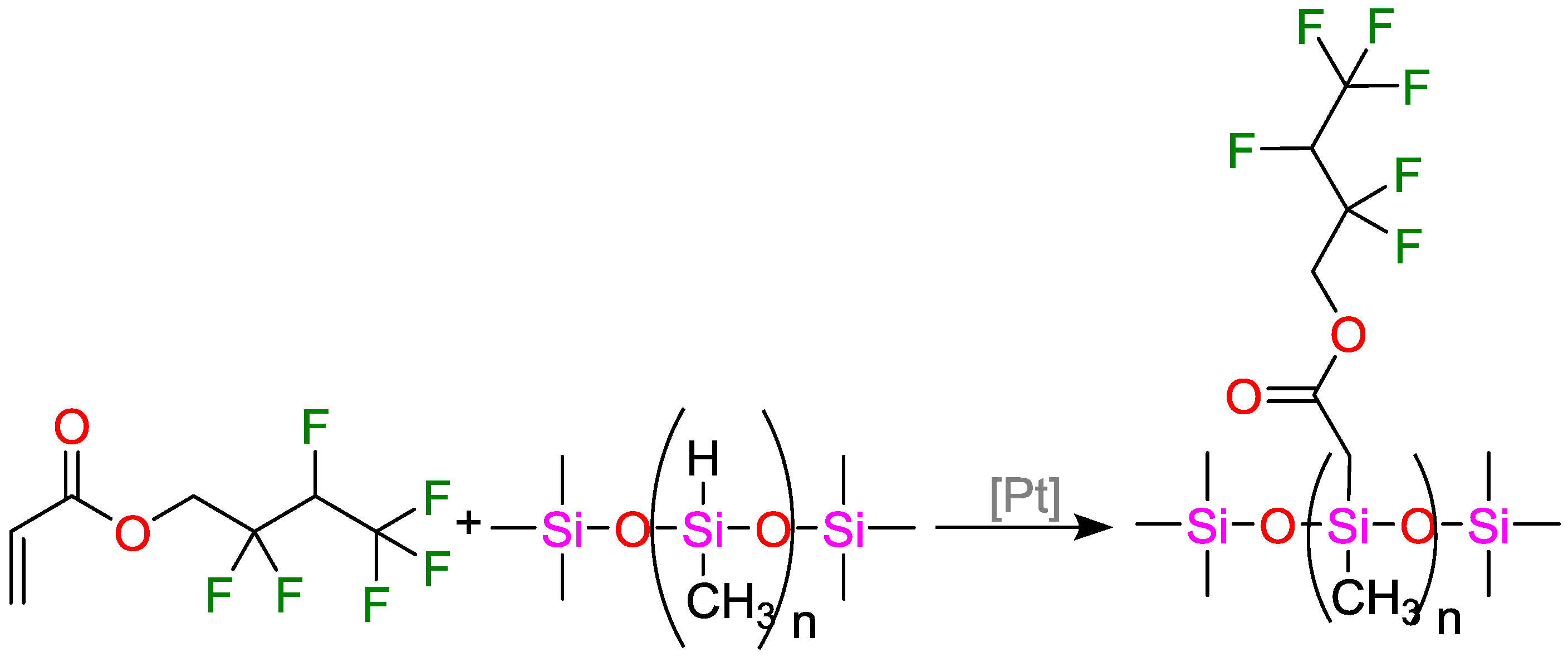
5.2.1. Coatings
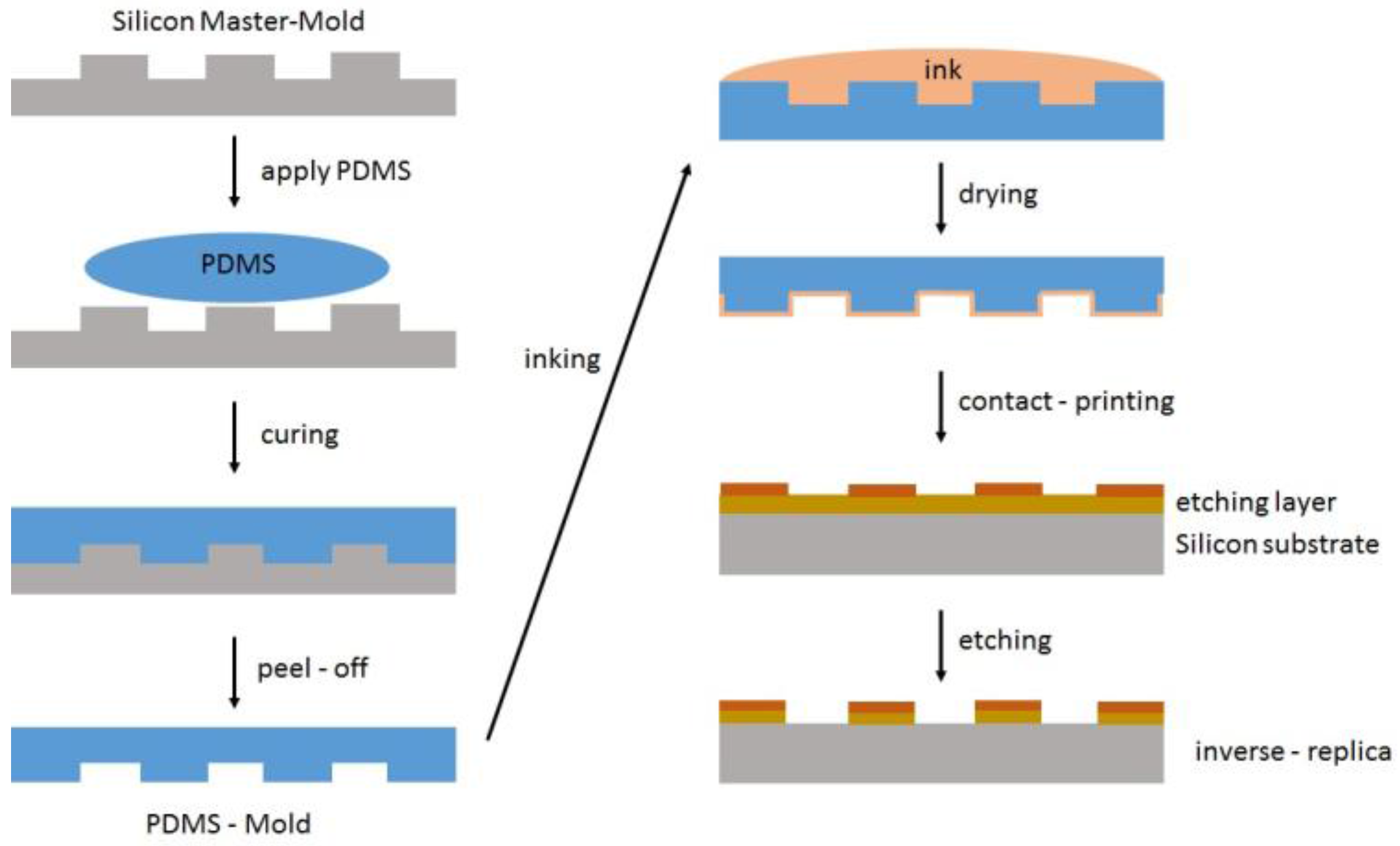
5.2.2. Printing and Inks
5.2.3. Microelectronic Applications
6. Conclusions
- Low-cost catalysts: Non-platinum transition metal catalysts have drastically lower the catalyst prices compared to platinum, and can be used in very low catalyst loadings.
- Non-metal catalysts: In addition to their cost efficiency, catalysts such as borane-based Lewis acids can be easily separated from the final (polymeric) products. They meet demands of the area of microelectronics, in which traces of metals may cause issues of performance.
- Triggered hydrosilylation: Stimuli other than heat give further flexibility to the processing schedule and eventually enhance the storage stability. Light is a favorable stimulus, as it allows for spatiotemporal control and specific activation.
- Solvent-free processes: These hydrosilylation reactions represent a big step towards green chemistry.
- Selectivity: Chemoselective hydrosilylation can pave the way to novel materials from a broad range of different substrates.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sommer, L.H.; Pietrusza, E.W.; Whitmore, F.C. Peroxide-catalyzed addition of trichlorosilane to 1-octene. J. Am. Chem. Soc. 1947, 69, 188. [Google Scholar] [CrossRef]
- Speier, J.L.; Webster, J.A.; Barnes, G.H. The addition of silicon hydrides to olefinic double bonds. Part II. The use of group VIII metal catalysts. J. Am. Chem. Soc. 1957, 79, 974–979. [Google Scholar] [CrossRef]
- Marciniec, B. Hydrosilylation: A Comprehensive Review on Recent Advances; Springer Science & Business Media: Berlin, Germany, 2008; ISBN 978-1-4020-8172-9. [Google Scholar]
- Zhang, M.; Zhang, A. Iron-catalyzed hydrosilylation reactions. Appl. Organomet. Chem. 2010, 24, 751–757. [Google Scholar] [CrossRef]
- Sun, J.; Deng, L. Cobalt Complex-Catalyzed Hydrosilylation of Alkenes and Alkynes. ACS Catal. 2016, 6, 290–300. [Google Scholar] [CrossRef]
- Troegel, D.; Stohrer, J. Recent advances and actual challenges in late transition metal catalyzed hydrosilylation of olefins from an industrial point of view. Coord. Chem. Rev. 2011, 255, 1440–1459. [Google Scholar] [CrossRef]
- Nakajima, Y.; Shimada, S. Hydrosilylation reaction of olefins: Recent advances and perspectives. RSC Adv. 2015, 5, 20603–20616. [Google Scholar] [CrossRef]
- Meister, T.K.; Riener, K.; Gigler, P.; Stohrer, J.; Herrmann, W.A.; Kühn, F.E. Platinum Catalysis Revisited- Unraveling Principles of Catalytic Olefin Hydrosilylation. ACS Catal. 2016, 6, 1274–1284. [Google Scholar] [CrossRef]
- Du, X.; Huang, Z. Advances in Base-Metal-Catalyzed Alkyne Hydrosilylation. ACS Catal. 2017, 7, 1227–1243. [Google Scholar] [CrossRef]
- NASDAQ. Available online: http://www.nasdaq.com/markets/platinum.aspx. (accessed on 23 June 2017).
- Tondreau, A.M.; Atienza, C.C.H.; Weller, K.J.; Nye, S.A.; Lewis, K.M.; Delis, J.G.; Chirik, P.J. Iron catalysts for selective anti-markovnikov alkene hydrosilylation using tertiary silanes. Science 2012, 335, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Sunada, Y.; Noda, D.; Soejima, H.; Tsutsumi, H.; Nagashima, H. Combinatorial Approach to the Catalytic Hydrosilylation of Styrene Derivatives: Catalyst Systems Composed of Organoiron (0) or (II) Precursors and Isocyanides. Organometallics 2015, 34, 2896–2906. [Google Scholar] [CrossRef]
- Marciniec, B.; Kownacka, A.; Kownacki, I.; Hoffmann, M.; Taylor, R. Hydrosilylation vs. dehydrogenative silylation of styrene catalysed by iron (0) carbonyl complexes with multivinylsilicon ligands–Mechanistic implications. J. Organomet. Chem. 2015, 791, 58–65. [Google Scholar] [CrossRef]
- Marciniec, B.; Kownacka, A.; Kownacki, I.; Taylor, R. Hydrosilylation cross-linking of silicon fluids by a novel class of iron (0) catalysts. Appl. Catal. A 2014, 486, 230–238. [Google Scholar] [CrossRef]
- Sunada, Y.; Tsutsumi, H.; Shigeta, K.; Yoshida, R.; Hashimoto, T.; Nagashima, H. Catalyst design for iron-promoted reductions: An iron disilyl-dicarbonyl complex bearing weakly coordinating η2-(H–Si) moieties. Dalton Trans. 2013, 42, 16687–16692. [Google Scholar] [CrossRef] [PubMed]
- Kamata, K.; Suzuki, A.; Nakai, Y.; Nakazawa, H. Catalytic hydrosilylation of alkenes by iron complexes containing terpyridine derivatives as ancillary ligands. Organometallics 2012, 31, 3825–3828. [Google Scholar] [CrossRef]
- Hayasaka, K.; Kamata, K.; Nakazawa, H. Highly Efficient Olefin Hydrosilylation Catalyzed by Iron Complexes with Iminobipyridine Ligand. Bull. Chem. Soc. Jpn. 2015, 89, 394–404. [Google Scholar] [CrossRef]
- Toya, Y.; Hayasaka, K.; Nakazawa, H. Hydrosilylation of Olefins Catalyzed by Iron Complexes Bearing Ketimine-Type Iminobipyridine Ligands. Organometallics 2017, 36, 1727–1735. [Google Scholar] [CrossRef]
- Chen, J.; Cheng, B.; Cao, M.; Lu, Z. Iron-Catalyzed Asymmetric Hydrosilylation of 1,1-Disubstituted Alkenes. Angew. Chem. 2015, 127, 4744–4747. [Google Scholar] [CrossRef]
- Zuo, Z.; Zhang, L.; Leng, X.; Huang, Z. Iron-catalyzed asymmetric hydrosilylation of ketones. Chem. Commun. 2015, 51, 5073–5076. [Google Scholar] [CrossRef] [PubMed]
- Ruddy, A.J.; Sydora, O.L.; Small, B.L.; Stradiotto, M.; Turculet, L. (N-Phosphinoamidinate) cobalt-Catalyzed Hydroboration: Alkene Isomerization Affords Terminal Selectivity. Chem. Eur. J. 2014, 20, 13918–13922. [Google Scholar] [CrossRef] [PubMed]
- Metsänen, T.T.; Gallego, D.; Szilvási, T.; Driess, M.; Oestreich, M. Peripheral mechanism of a carbonyl hydrosilylation catalysed by an SiNSi iron pincer complex. Chem. Sci. 2015, 6, 7143–7149. [Google Scholar] [CrossRef]
- Blom, B.; Enthaler, S.; Inoue, S.; Irran, E.; Driess, M. Electron-Rich N-Heterocyclic Silylene (NHSi)–Iron Complexes: Synthesis, Structures, and Catalytic Ability of an Isolable Hydridosilylene–Iron Complex. J. Am. Chem. Soc. 2013, 135, 6703–6713. [Google Scholar] [CrossRef] [PubMed]
- Gorczyński, A.; Zaranek, M.; Witomska, S.; Bocian, A.; Stefankiewicz, A.R.; Kubicki, M.; Patroniak, V.; Pawluć, P. The cobalt (II) complex of a new tridentate Schiff-base ligand as a catalyst for hydrosilylation of olefins. Catal. Commun. 2016, 78, 71–74. [Google Scholar] [CrossRef]
- Ibrahim, A.D.; Entsminger, S.W.; Zhu, L.; Fout, A.R. A Highly Chemoselective Cobalt Catalyst for the Hydrosilylation of Alkenes using Tertiary Silanes and Hydrosiloxanes. ACS Catal. 2016, 6, 3589–3593. [Google Scholar] [CrossRef]
- Noda, D.; Tahara, A.; Sunada, Y.; Nagashima, H. Non-Precious-Metal Catalytic Systems Involving Iron or Cobalt Carboxylates and Alkyl Isocyanides for Hydrosilylation of Alkenes with Hydrosiloxanes. J. Am. Chem. Soc. 2016, 138, 2480–2483. [Google Scholar] [CrossRef] [PubMed]
- Raya, B.; Biswas, S.; RajanBabu, T.V. Selective Cobalt-Catalyzed Reduction of Terminal Alkenes and Alkynes Using (EtO)2Si(Me)H as a Stoichiometric Reductant. ACS Catal. 2016, 6, 6318–6323. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Lu, Z. Highly Chemo-, Regio-, and Stereoselective Cobalt-Catalyzed Markovnikov Hydrosilylation of Alkynes. Angew. Chem. Int. Ed. 2016, 55, 10835–10838. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Shen, X.; Lu, Z. Regio- and Enantioselective Cobalt-Catalyzed Sequential Hydrosilylation/Hydrogenation of Terminal Alkynes. Angew. Chem. Int. Ed. 2017, 56, 615–618. [Google Scholar] [CrossRef]
- Mo, Z.; Xiao, J.; Gao, Y.; Deng, L. Regio- and stereoselective hydrosilylation of alkynes catalyzed by three-coordinate cobalt(I) alkyl and silyl complexes. J. Am. Chem. Soc. 2014, 136, 17414–17417. [Google Scholar] [CrossRef] [PubMed]
- Teo, W.J.; Wang, C.; Tan, Y.W.; Ge, S. Cobalt-Catalyzed Z-Selective Hydrosilylation of Terminal Alkynes. Angew. Chem. Int. Ed. 2017, 56, 4328–4332. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Hou, W.; Zhang, Y.; Huang, Z. Pincer cobalt complex-catalyzed Z-selective hydrosilylation of terminal alkynes. Org. Chem. Front. 2017, 4, 1517–1521. [Google Scholar] [CrossRef]
- Zuo, Z.; Yang, J.; Huang, Z. Cobalt-Catalyzed Alkyne Hydrosilylation and Sequential Vinylsilane Hydroboration with Markovnikov Selectivity. Angew. Chem. Int. Ed. 2016, 55, 10839–10843. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Peng, D.; Du, X.; Huang, Z.; Ma, S. Identifying a cobalt catalyst for highly selective hydrosilylation of allenes. Org. Chem. Front. 2017, 4, 1829–1832. [Google Scholar] [CrossRef]
- Xi, T.; Lu, Z. Cobalt-Catalyzed Hydrosilylation/Cyclization of 1,6-Enynes. J. Org. Chem. 2016, 81, 8858–8866. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, V.; Nakajima, Y.; Ando, W.; Sato, K.; Shimada, S. Bis(acetylacetonato)Ni(II)/NaBHEt3-catalyzed hydrosilylation of 1,3-dienes, alkenes and alkynes. J. Organomet. Chem. 2016, 809, 57–62. [Google Scholar] [CrossRef]
- Srinivas, V.; Nakajima, Y.; Ando, W.; Sato, K.; Shimada, S. (Salicylaldiminato)Ni(ii)-catalysts for hydrosilylation of olefins. Catal. Sci. Technol. 2015, 5, 2081–2084. [Google Scholar] [CrossRef]
- Buslov, I.; Becouse, J.; Mazza, S.; Montandon-Clerc, M.; Hu, X. Chemoselective Alkene Hydrosilylation Catalyzed by Nickel Pincer Complexes. Angew. Chem. 2015, 127, 14731–14734. [Google Scholar] [CrossRef]
- Chakraborty, S.; Bhattacharya, P.; Dai, H.; Guan, H. Nickel and iron pincer complexes as catalysts for the reduction of carbonyl compounds. Acc. Chem. Res. 2015, 48, 1995–2003. [Google Scholar] [CrossRef] [PubMed]
- Buslov, I.; Song, F.; Hu, X. An Easily Accessed Nickel Nanoparticle Catalyst for Alkene Hydrosilylation with Tertiary Silanes. Angew. Chem. 2016, 128, 12483–12487. [Google Scholar] [CrossRef]
- Pappas, I.; Treacy, S.; Chirik, P.J. Alkene Hydrosilylation Using Tertiary Silanes with α-Diimine Nickel Catalysts. Redox-Active Ligands Promote a Distinct Mechanistic Pathway from Platinum Catalysts. ACS Catal. 2016, 6, 4105–4109. [Google Scholar] [CrossRef]
- Wei, Y.; Liu, S.-X.; Mueller-Bunz, H.; Albrecht, M. Synthesis of Triazolylidene Nickel Complexes and Their Catalytic Application in Selective Aldehyde Hydrosilylation. ACS Catal. 2016, 6, 8192–8200. [Google Scholar] [CrossRef]
- Steiman, T.J.; Uyeda, C. Reversible substrate activation and catalysis at an intact metal-metal bond using a redox-active supporting ligand. J. Am. Chem. Soc. 2015, 137, 6104–6110. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sun, H.; Li, X. Imine Nitrogen Bridged Binuclear Nickel Complexes via N-H Bond Activation: Synthesis, Characterization, Unexpected C,N-Coupling Reaction, and Their Catalytic Application in Hydrosilylation of Aldehydes. Organometallics 2015, 34, 5175–5182. [Google Scholar] [CrossRef]
- Benkeser, R.A. Chemistry of trichlorosilane-tertiary amine combinations. Acc. Chem. Res. 2002, 4, 94–100. [Google Scholar] [CrossRef]
- Benkeser, R.A.; Gaul, J.M.; Smith, W.E. Silylation of organic halides. New method of forming the carbon-silicon bond. J. Am. Chem. Soc. 1969, 91, 3666–3667. [Google Scholar] [CrossRef]
- Hu, X.-Y.; Zhang, M.-M.; Shu, C.; Zhang, Y.-H.; Liao, L.-H.; Yuan, W.-C.; Zhang, X.-M. Enantioselective Lewis-Base-Catalyzed Asymmetric Hydrosilylation of Substituted Benzophenone N -Aryl Imines: Efficient Synthesis of Chiral (Diarylmethyl)amines. Adv. Synth. Catal. 2014, 356, 3539–3544. [Google Scholar] [CrossRef]
- Pike, R.A. Base-Catalyzed Additions of Trichlorosilane to Hydrocarbon Olefins. J. Org. Chem. 1962, 27, 2186–2190. [Google Scholar] [CrossRef]
- Cho, Y.S.; Kang, S.-H.; Han, J.S.; Yoo, B.R.; Jung, I.N. Novel Phosphonium Chloride-Catalyzed Dehydrohalogenative Si–C Coupling Reaction of Alkyl Halides with Trichlorosilane. J. Am. Chem. Soc. 2001, 123, 5584–5585. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-H.; Han, J.S.; Yoo, B.R.; Lee, M.E.; Jung, I.N. Phosphonium Chloride-Catalyzed Dehydrochlorinative Coupling Reactions of Alkyl Halides with Hydridochlorosilanes. Organometallics 2003, 22, 529–534. [Google Scholar] [CrossRef]
- Oertle, K.; Wetter, H. Hydrosilylation of tetrasubstituted olefins. Tetrahedron Lett. 1985, 26, 5511–5514. [Google Scholar] [CrossRef]
- Asao, N.; Sudo, T.; Yamamoto, Y. Lewis Acid-Catalyzed trans -Hydrosilylation of Alkynes. J. Org. Chem. 1996, 61, 7654–7655. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.-S.; Yoo, B.R.; Lee, G.-H.; Jung, I.N. Lewis Acid-Catalyzed Regio- and Stereoselective Hydrosilylation of Alkenes with Trialkylsilanes. Organometallics 1999, 18, 3109–3115. [Google Scholar] [CrossRef]
- Kim, D.W.; Joung, S.; Kim, J.G.; Chang, S. Metal-Free Hydrosilylation Polymerization by Borane Catalyst. Angew. Chem. 2015, 127, 15018–15022. [Google Scholar] [CrossRef]
- Kostjuk, S.V.; Ganachaud, F. Cationic Polymerization of Styrene in Solution and Aqueous Suspension Using B(C6F5)3 as a Water-Tolerant Lewis Acid. Macromolecules 2006, 39, 3110–3113. [Google Scholar] [CrossRef]
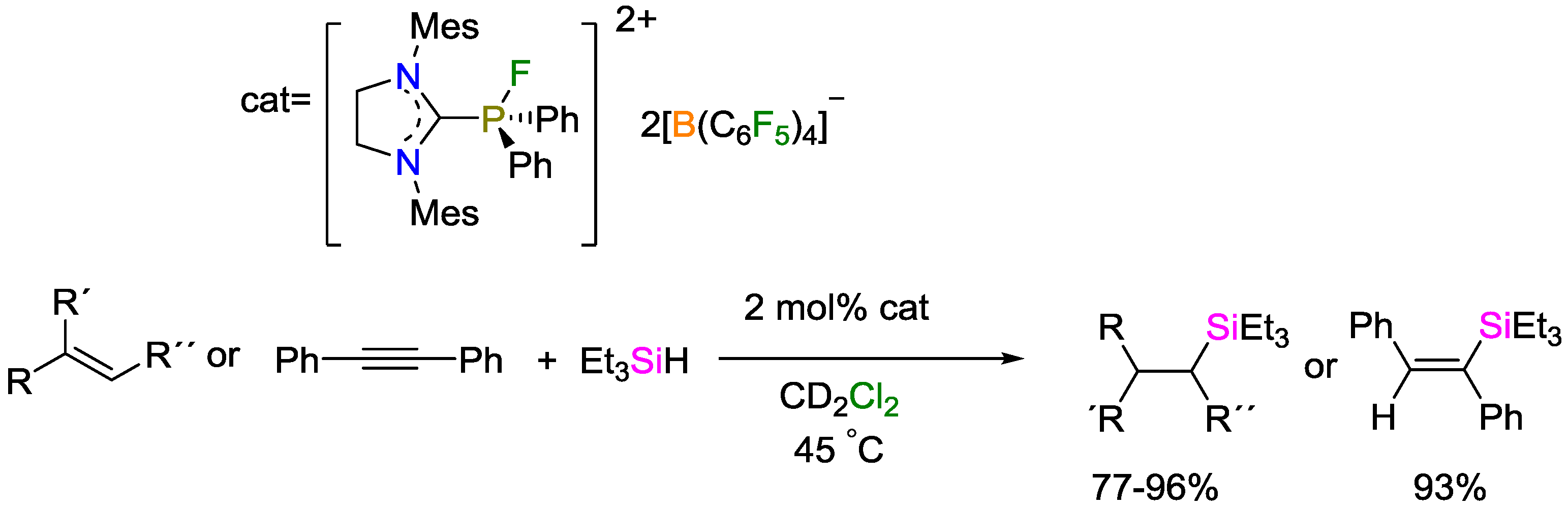
- Pérez, M.; Hounjet, L.J.; Caputo, C.B.; Dobrovetsky, R.; Stephan, D.W. Olefin isomerization and hydrosilylation catalysis by Lewis acidic organofluorophosphonium salts. J. Am. Chem. Soc. 2013, 135, 18308–18310. [Google Scholar] [CrossRef] [PubMed]
- Holthausen, M.H.; Mehta, M.; Stephan, D.W. The highly Lewis acidic dicationic phosphonium salt: (SIMes)PFPh₂B(C₆F₅)₄₂. Angew. Chem. Int. Ed. 2014, 53, 6538–6541. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, K.; Chu, T.; Nikonov, G.I. Hydrosilylation of Olefins Catalyzed by Well-Defined Cationic Aluminum Complexes: Lewis Acid vs. Insertion Mechanisms. ACS Catal. 2016, 6, 7350–7356. [Google Scholar] [CrossRef]
- Parks, D.J.; Piers, W.E. Tris(pentafluorophenyl)boron-Catalyzed Hydrosilation of Aromatic Aldehydes, Ketones, and Esters. J. Am. Chem. Soc. 1996, 118, 9440–9441. [Google Scholar] [CrossRef]
- Roesler, R.; Har, B.J.N.; Piers, W.E. Synthesis and Characterization of (Perfluoroaryl)borane-Functionalized Carbosilane Dendrimers and Their Use as Lewis Acid Catalysts for the Hydrosilation of Acetophenone. Organometallics 2002, 21, 4300–4302. [Google Scholar] [CrossRef]
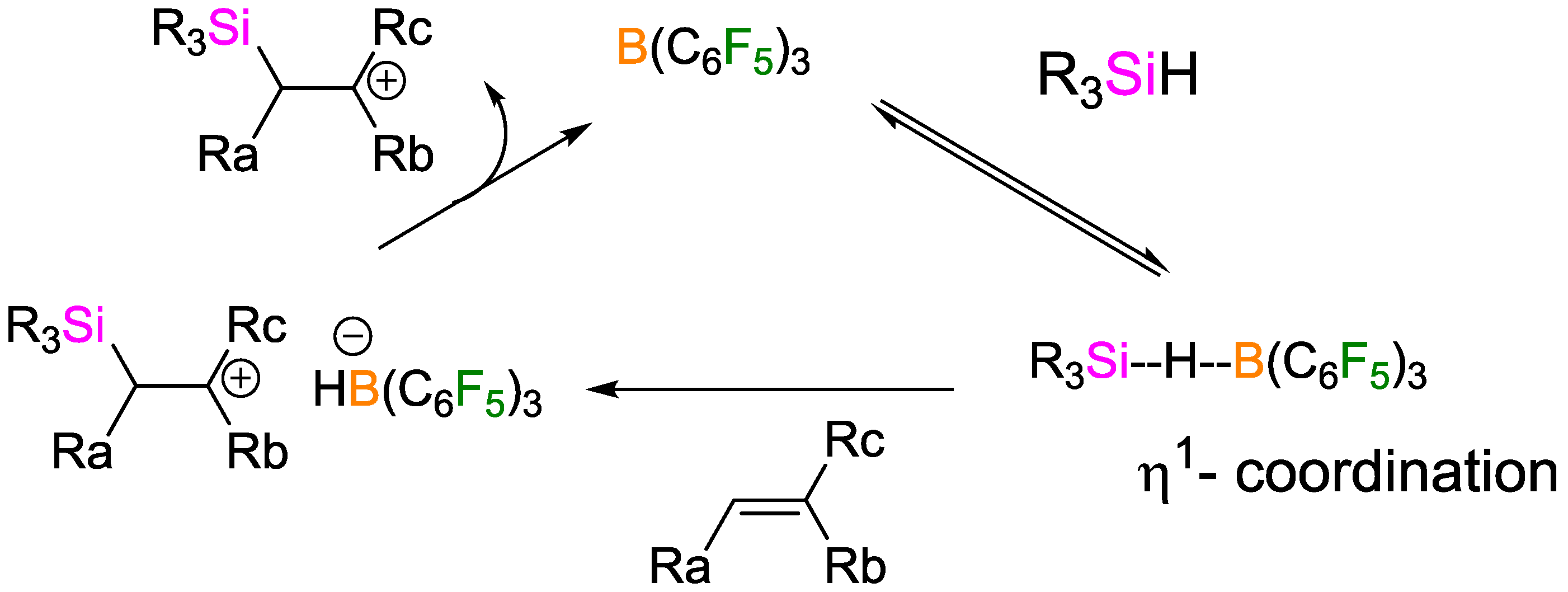
- Houghton, A.Y.; Hurmalainen, J.; Mansikkamäki, A.; Piers, W.E.; Tuononen, H.M. Direct observation of a borane-silane complex involved in frustrated Lewis-pair-mediated hydrosilylations. Nat. Chem. 2014, 6, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Rendler, S.; Oestreich, M. Conclusive evidence for an S(N)2–Si mechanism in the B(C6F5)3-catalyzed hydrosilylation of carbonyl compounds: Implications for the related hydrogenation. Angew. Chem. Int. Ed. 2008, 47, 5997–6000. [Google Scholar] [CrossRef] [PubMed]
- Mewald, M.; Oestreich, M. Illuminating the mechanism of the borane-catalyzed hydrosilylation of imines with both an axially chiral borane and silane. Chem. Eur. J. 2012, 18, 14079–14084. [Google Scholar] [CrossRef] [PubMed]
- Hermeke, J.; Mewald, M.; Oestreich, M. Experimental analysis of the catalytic cycle of the borane-promoted imine reduction with hydrosilanes: Spectroscopic detection of unexpected intermediates and a refined mechanism. J. Am. Chem. Soc. 2013, 135, 17537–17546. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, E.Y.-X. Elusive silane-alane complex Si–H⋅⋅⋅Al: Isolation, characterization, and multifaceted frustrated Lewis pair type catalysis. Angew. Chem. Int. Ed. 2015, 54, 6842–6846. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Curran, D.P.; Malacria, M.; Fensterbank, L.; Goddard, J.-P.; Lacôte, E. N-heterocyclic carbene-catalyzed hydrosilylation of styryl and propargylic alcohols with dihydrosilanes. Chem. Eur. J. 2011, 17, 9911–9914. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, A.; Oestreich, M. 3-Silylated cyclohexa-1,4-dienes as precursors for gaseous hydrosilanes: The B(C6F5)3-catalyzed transfer hydrosilylation of alkenes. Angew. Chem. Int. Ed. 2013, 52, 11905–11907. [Google Scholar] [CrossRef] [PubMed]
- Keess, S.; Simonneau, A.; Oestreich, M. Direct and Transfer Hydrosilylation Reactions Catalyzed by Fully or Partially Fluorinated Triarylboranes: A Systematic Study. Organometallics 2015, 34, 790–799. [Google Scholar] [CrossRef]
- Kim, Y.; Chang, S. Borane-Catalyzed Reductive α-Silylation of Conjugated Esters and Amides Leaving Carbonyl Groups Intact. Angew. Chem. 2016, 128, 226–230. [Google Scholar] [CrossRef]
- Park, S.; Brookhart, M. Development and mechanistic investigation of a highly efficient iridium(V) silyl complex for the reduction of tertiary amides to amines. J. Am. Chem. Soc. 2012, 134, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Müther, K.; Mohr, J.; Oestreich, M. Silylium Ion Promoted Reduction of Imines with Hydrosilanes. Organometallics 2013, 32, 6643–6646. [Google Scholar] [CrossRef]
- LeBlanc, F.A.; Piers, W.E.; Parvez, M. Selective hydrosilation of CO2 to a bis(silylacetal) using an anilido bipyridyl-ligated organoscandium catalyst. Angew. Chem. Int. Ed. 2014, 53, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Süsse, L.; Hermeke, J.; Oestreich, M. The Asymmetric Piers Hydrosilylation. J. Am. Chem. Soc. 2016, 138, 6940–6943. [Google Scholar] [CrossRef] [PubMed]
- Blanco, V.; Leigh, D.A.; Marcos, V. Artificial switchable catalysts. Chem. Soc. Rev. 2015, 44, 5341–5370. [Google Scholar] [CrossRef] [PubMed]
- Vlatković, M.; Collins, B.S.L.; Feringa, B.L. Dynamic Responsive Systems for Catalytic Function. Chem. Eur. J. 2016, 22, 17080–17111. [Google Scholar] [CrossRef] [PubMed]
- De Vekki, D.A. Hydrosilylation on photoactivated catalysts. Russ. J. Gen. Chem. 2011, 81, 1480–1492. [Google Scholar] [CrossRef]
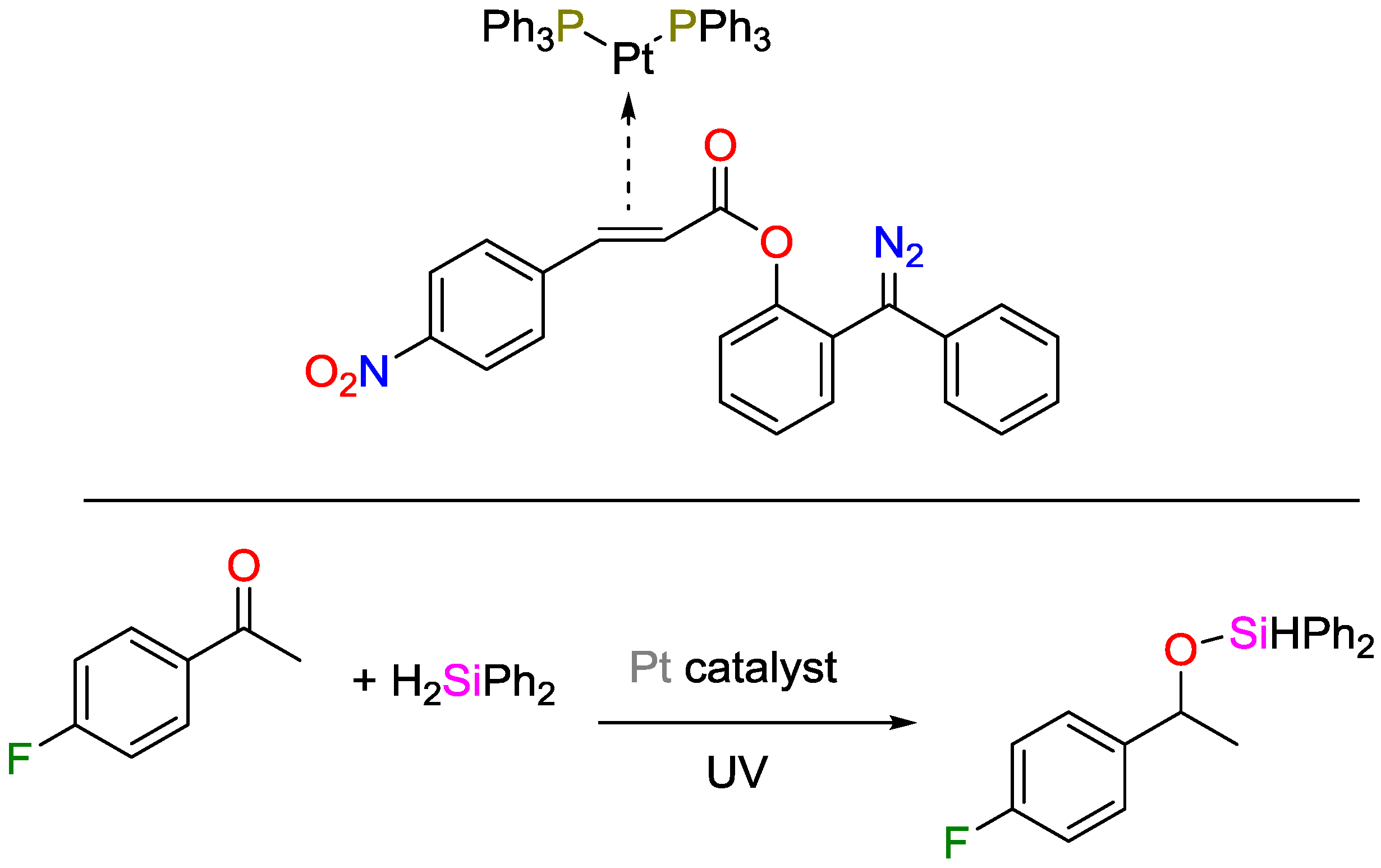
- Paonessa, R.S.; Prignano, A.L.; Trogler, W.C. Photochemical generation of bis(phosphine)palladium and bis(phosphine)platinum equivalents. Organometallics 1985, 4, 647–657. [Google Scholar] [CrossRef]
- Prignano, A.L.; Trogler, W.C. Silica-supported bis(trialkylphosphine)platinum oxalates. Photogenerated catalysts for hydrosilylation of olefins. J. Am. Chem. Soc. 1987, 109, 3586–3595. [Google Scholar] [CrossRef]
- Lippert, T.; Dauth, J.; Deubzer, B.; Weis, J.; Wokaun, A. Photolysis of an arylalkyl-triazenido-platinum-IV complex. Radiat. Phys. Chem. 1996, 47, 889–897. [Google Scholar] [CrossRef]
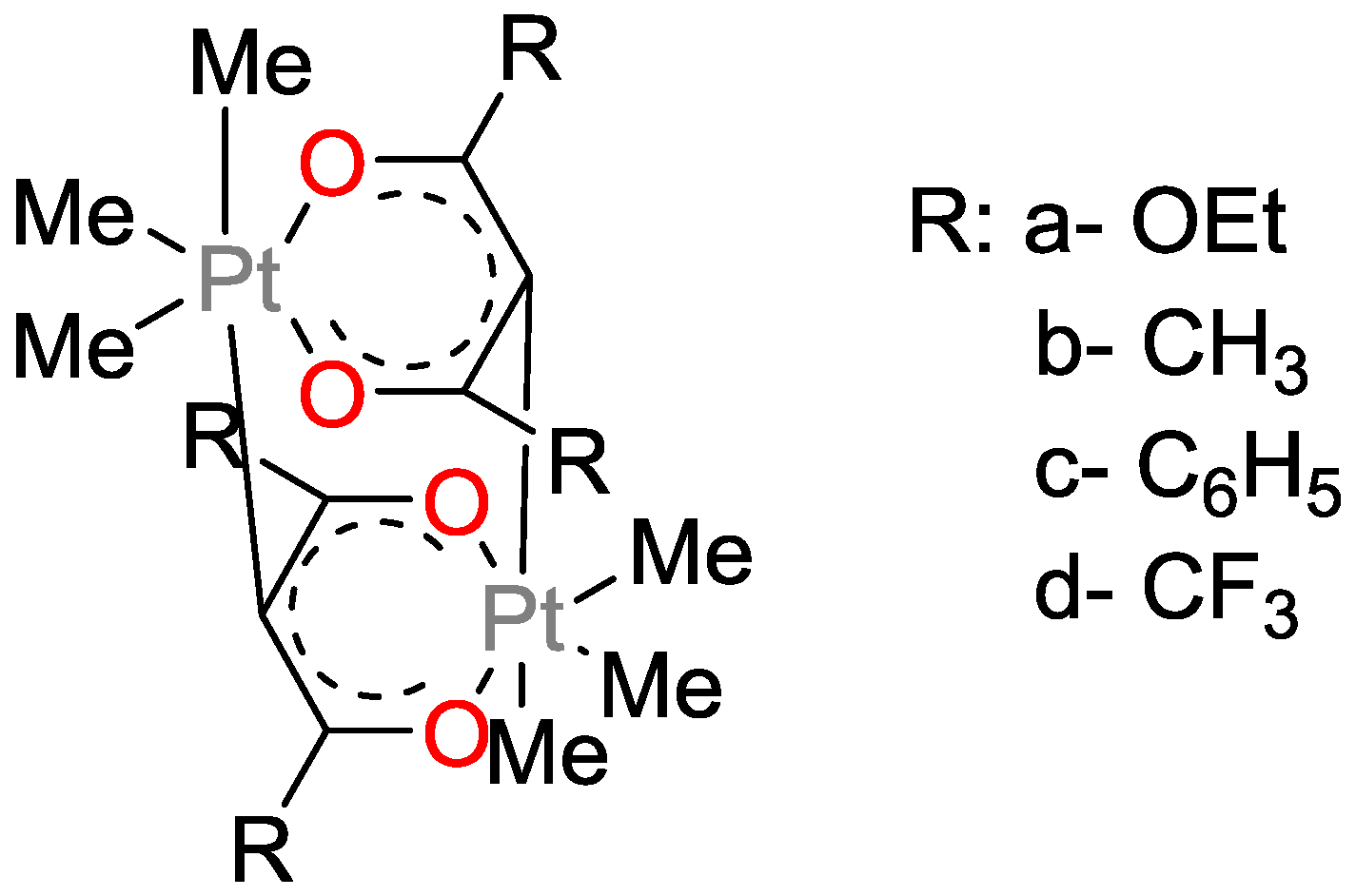
- Lewis, F.D.; Salvi, G.D. Platinum(II) Bis(beta-diketonates) as Photoactivated Hydrosilation Catalysts. Inorg. Chem. 1995, 34, 3182–3189. [Google Scholar] [CrossRef]
- Jakubek, V.; Lees, A.J. Quantitative photochemistry of Cp’Pt(CH3)3 (Cp’ = η5-C5H4CH3) in solution: A highly efficient organometallic photoinitiator for hydrosilylation. Inorg. Chem. 2004, 43, 6869–6871. [Google Scholar] [CrossRef] [PubMed]
- Boardman, L.D. (η5-Cyclopentadienyl)trialkylplatinum photohydrosilylation catalysts. Mechanism of active catalyst formation and preparation of a novel bis(silyl)platinum hydride. Organometallics 1992, 11, 4194–4201. [Google Scholar] [CrossRef]
- Mayer, T.; Burget, D.; Mignani, G.; Fouassier, J.P. Photohydrosilylation reaction of silicone polymers. Platinum-based photocatalysts: Trimethyl (β-dicarbonyl) platinum IV complexes. J. Polym. Sci. Pol. Chem. 1996, 34, 3141–3146. [Google Scholar] [CrossRef]
- Galeandro-Diamant, T.; Zanota, M.-L.; Sayah, R.; Veyre, L.; Nikitine, C.; Bellefon, C.; de Marrot, S.; Meille, V.; Thieuleux, C. Platinum nanoparticles in suspension are as efficient as Karstedt’s complex for alkene hydrosilylation. Chem. Commun. 2015, 51, 16194–16196. [Google Scholar] [CrossRef] [PubMed]
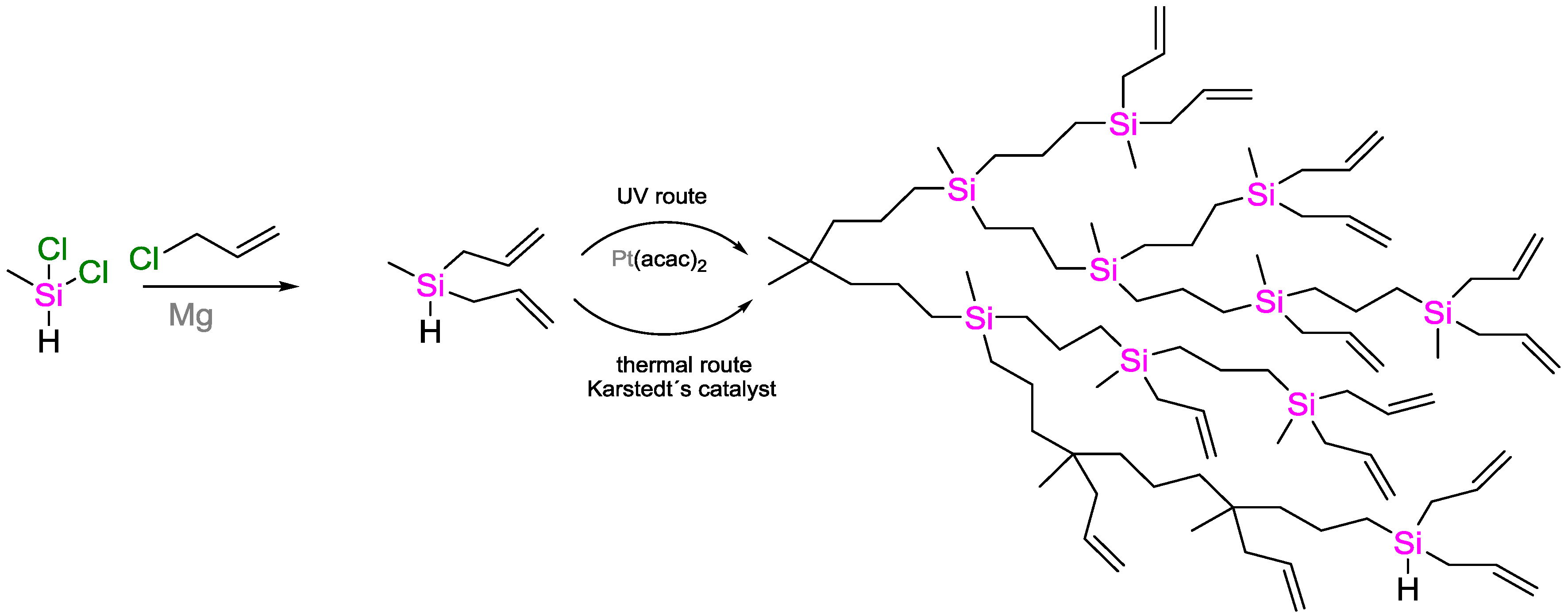
- Fry, B.E.; Neckers, D.C. Rapid Photoactivated Hydrosilation Polymerization of Vinyldimethylsilane. Macromolecules 1996, 29, 5306–5312. [Google Scholar] [CrossRef]
- Vekki, D.A.; de Skvortsov, N.K. Hydrosilylation of Vinylsiloxanes with Hydrosiloxanes in the Presence of Thermo- and Photoactivated Platinum(II) Phosphine Complexes. Russ. J. Gen. Chem. 2004, 74, 197–206. [Google Scholar] [CrossRef]
- Vekki, D.A.; de Skvortsov, N.K. Hydrosilylation of low-molecular siloxanes in the presence of photactivated alkene and sulfoxide platinum(II) complexes. Russ. J. Gen. Chem. 2006, 76, 116–121. [Google Scholar] [CrossRef]
- Fry, B.E.; Guo, A.; Neckers, D.C. Photoactivated hydrosilation curing of a ceramic precursor: Crosslinking and pyrolysis of branched oligo [(methylsilylene) methylene]. J. Organomet. Chem. 1997, 538, 151–161. [Google Scholar] [CrossRef]
- Marchi, S.; Sangermano, M.; Meier, P.; Kornmann, X. Visible light-activated hydrosilation reaction. J. Photochem. Photobiol. A 2015, 303–304, 86–90. [Google Scholar] [CrossRef]
- Zhang, G.B.; Kong, J.; Fan, X.D.; Li, X.G.; Tian, W.; Huang, M.R. UV-activated hydrosilylation: A facile approach for synthesis of hyperbranched polycarbosilanes. Appl. Organometal. Chem. 2009, 23, 277–282. [Google Scholar] [CrossRef]
- Marchi, S.; Sangermano, M.; Ligorio, D.; Meier, P.; Kornmann, X. Impressive Rate Raise of the Hydrosilation Reaction Through UV-Activation: Energy and Time Saving. Macromol. Mater. Eng. 2016, 301, 610–613. [Google Scholar] [CrossRef]
- Marchi, S.; Sangermano, M.; Meier, P.; Kornmann, X. Preparation and characterization of PDMS composites by UV-hydrosilation for outdoor polymeric insulators. Polym. Compos. 2014, 35, 1253–1262. [Google Scholar] [CrossRef]
- Sangermano, M.; Marchi, S.; Ligorio, D.; Meier, P.; Kornmann, X. UV-Induced Frontal Polymerization of a Pt-Catalyzed Hydrosilation Reaction. Macromol. Chem. Phys. 2013, 214, 943–947. [Google Scholar] [CrossRef]
- Marchi, S.; Sangermano, M.; Meier, P.; Kornmann, X. UV-cured silicone composites obtained via hydrosilation and in-situ generation of inorganic particles. Polym. Eng. Sci. 2016, 56, 3–8. [Google Scholar] [CrossRef]
- Sangermano, M.; Marchi, S.; Meier, P.; Kornmann, X. UV-activated hydrosilation reaction for silicone polymer crosslinking. J. Appl. Polym. Sci. 2012, 1, 1521–1526. [Google Scholar] [CrossRef]
- Marchi, S.; Sangermano, M.; Meier, P.; Kornmann, X. A Comparison of the Reactivity of Two Platinum Catalysts for Silicone Polymer Cross-Linking by UV-Activated Hydrosilation Reaction. Macromol. React. Eng. 2015, 9, 360–365. [Google Scholar] [CrossRef]
- Guo, A.; Fry, B.E.; Neckers, D.C. Highly Active Visible-Light Photocatalysts for Curing a Ceramic Precursor1. Chem. Mater. 1998, 10, 531–536. [Google Scholar] [CrossRef]
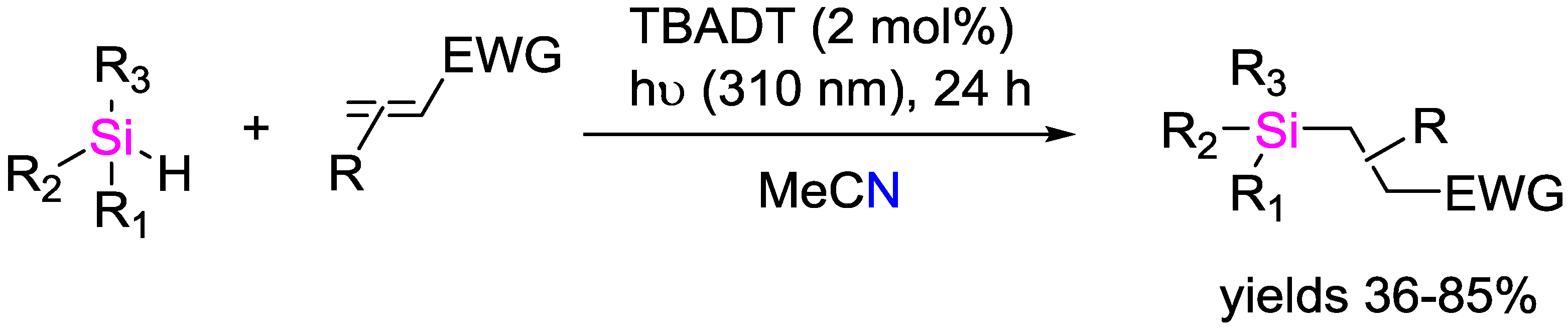
- Qrareya, H.; Dondi, D.; Ravelli, D.; Fagnoni, M. Decatungstate-Photocatalyzed Si–H/C–H Activation in Silyl Hydrides: Hydrosilylation of Electron-Poor Alkenes. ChemCatChem 2015, 7, 3350–3357. [Google Scholar] [CrossRef]
- Stoll, R.S.; Hecht, S. Artificial light-gated catalyst systems. Angew. Chem. Int. Ed. 2010, 49, 5054–5075. [Google Scholar] [CrossRef] [PubMed]
- Buchner, M.R.; Bechlars, B.; Ruhland, K. A new approach to light-gated Pt catalysts for the hydrosilylation. J. Organomet. Chem. 2013, 744, 60–67. [Google Scholar] [CrossRef]
- Misal Castro, L.C.; Li, H.; Sortais, J.-B.; Darcel, C. Selective switchable iron-catalyzed hydrosilylation of carboxylic acids. Chem. Commun. 2012, 48, 10514–10516. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Han, S.; Kim, J.; Soh, S.; Grzybowski, B.A. Photoswitchable catalysis mediated by dynamic aggregation of nanoparticles. J. Am. Chem. Soc. 2010, 132, 11018–11020. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Yamada, T.; Nagata, Y.; Suginome, M. High-molecular-weight polyquinoxaline-based helically chiral phosphine (PQXphos) as chirality-switchable, reusable, and highly enantioselective monodentate ligand in catalytic asymmetric hydrosilylation of styrenes. J. Am. Chem. Soc 2010, 132, 7899–7901. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.-Z.; Nagata, Y.; Yamada, T.; Suginome, M. Majority-Rules-Type Helical Poly(quinoxaline-2,3-diyl)s as Highly Efficient Chirality-Amplification Systems for Asymmetric Catalysis. Angew. Chem. Int. Ed. 2015, 54, 9333–9337. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Qian, C.; Mastronardi, M.L.; Wei, M.; Ozin, G.A. Hydrosilylation kinetics of silicon nanocrystals. Chem. Commun. 2013, 49, 11361–11363. [Google Scholar] [CrossRef] [PubMed]
- Boukherroub, R.; Petit, A.; Loupy, A.; Chazalviel, J.-N.; Ozanam, F. Microwave-Assisted Chemical Functionalization of Hydrogen-Terminated Porous Silicon Surfaces. J. Phys. Chem. B 2003, 107, 13459–13462. [Google Scholar] [CrossRef]
- Small, J.C.; Dam, H.M.; Siegel, J.L.; Crepinsek, A.J.; Neal, T.A.; Althoff, A.A.; Line, N.S.; Porter, L.A. Alkyl-functionalization of porous silicon via multimode microwave-assisted hydrosilylation. Polyhedron 2016, 114, 225–231. [Google Scholar] [CrossRef]
- Linford, M.R.; Fenter, P.; Eisenberger, P.M.; Chidsey, C.E.D. Alkyl Monolayers on Silicon Prepared from 1-Alkenes and Hydrogen-Terminated Silicon. J. Am. Chem. Soc. 1995, 117, 3145–3155. [Google Scholar] [CrossRef]
- Shiohara, A.; Hanada, S.; Prabakar, S.; Fujioka, K.; Lim, T.H.; Yamamoto, K.; Northcote, P.T.; Tilley, R.D. Chemical reactions on surface molecules attached to silicon quantum dots. J. Am. Chem. Soc. 2010, 132, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Robins, E.G.; Stewart, M.P.; Buriak, J.M. Anodic and cathodic electrografting of alkynes on porous silicon. Chem. Commun. 1999, 2479–2480. [Google Scholar] [CrossRef]
- Cicero, R.L.; Linford, M.R.; Chidsey, C.E.D. Photoreactivity of Unsaturated Compounds with Hydrogen-Terminated Silicon(111). Langmuir 2000, 16, 5688–5695. [Google Scholar] [CrossRef]
- Sieval, A.B.; Demirel, A.L.; Nissink, J.W.M.; Linford, M.R.; van der Maas, J.H.; de Jeu, W.H.; Zuilhof, H.; Sudhölter, E.J.R. Highly Stable Si-C Linked Functionalized Monolayers on the Silicon (100) Surface. Langmuir 1998, 14, 1759–1768. [Google Scholar] [CrossRef]
- Liu, W.; Sharp, I.D.; Tilley, T.D. Multifunctional silicon surfaces: Reaction of dichlorocarbene generated from Seyferth reagent with hydrogen-terminated silicon(111) surfaces. Langmuir 2014, 30, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.L.; Bernasek, S.L. Mild and efficient functionalization of hydrogen-terminated Si(111) via sonochemical activated hydrosilylation. J. Am. Chem. Soc. 2011, 133, 8118–8121. [Google Scholar] [CrossRef] [PubMed]
- Frederick, E.; Dickerson, P.N.; Zhong, Y.L.; Bernasek, S.L. Substituent effects on the kinetics of bifunctional styrene SAM formation on H-terminated Si. Langmuir 2014, 30, 7687–7694. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Hecht, M.B.; Kavara, A.; Brennessel, W.W.; Mercado, B.Q.; Weix, D.J.; Holland, P.L. Rapid, Regioconvergent, Solvent-Free Alkene Hydrosilylation with a Cobalt Catalyst. J. Am. Chem. Soc. 2015, 137, 13244–13247. [Google Scholar] [CrossRef] [PubMed]
- Andersson, P.G.; Munslow, I.J. Imine Hydrogenation. In Modern Reduction Methods; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2008; pp. 235–269. [Google Scholar]
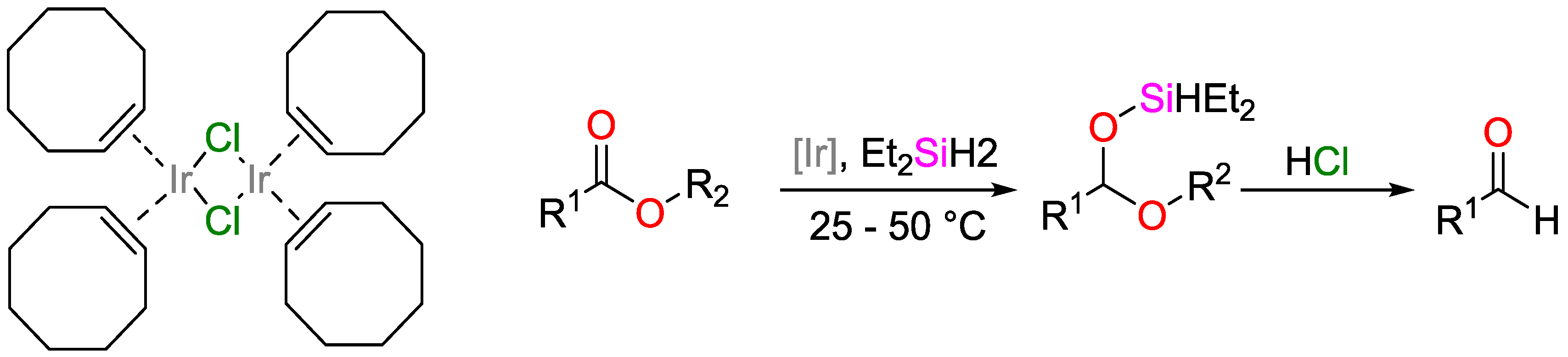
- Cheng, C.; Brookhart, M. Efficient reduction of esters to aldehydes through iridium-catalyzed hydrosilylation. Angew. Chem. Int. Ed. 2012, 51, 9422–9424. [Google Scholar] [CrossRef] [PubMed]
- Wekesa, F.S.; Arias-Ugarte, R.; Kong, L.; Sumner, Z.; McGovern, G.P.; Findlater, M. Iron-Catalyzed Hydrosilylation of Aldehydes and Ketones under Solvent-Free Conditions. Organometallics 2015, 34, 5051–5056. [Google Scholar] [CrossRef]
- Motokura, K.; Kashiwame, D.; Miyaji, A.; Baba, T. Copper-catalyzed formic acid synthesis from CO2 with hydrosilanes and H2O. Org. Lett. 2012, 14, 2642–2645. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cheng, J.; Hou, Z. Highly efficient catalytic hydrosilylation of carbon dioxide by an N-heterocyclic carbene copper catalyst. Chem. Commun. 2013, 49, 4782–4784. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, M.; Ciriminna, R.; Wong Chi Man, M.; Campestrini, S. Better chemistry through ceramics: The physical bases of the outstanding chemistry of ORMOSIL. J. Phys. Chem. B 2006, 110, 1976–1988. [Google Scholar] [CrossRef] [PubMed]
- Ciriminna, R.; Pandarus, V.; Gingras, G.; Béland, F.; Pagliaro, M. Closing the Organosilicon Synthetic Cycle: Efficient Heterogeneous Hydrosilylation of Alkenes over Silia Cat Pt(0). ACS Sustain. Chem. Eng. 2013, 1, 249–253. [Google Scholar] [CrossRef]
- Mason, B.P.; Price, K.E.; Steinbacher, J.L.; Bogdan, A.R.; McQuade, D.T. Greener approaches to organic synthesis using microreactor technology. Chem. Rev. 2007, 107, 2300–2318. [Google Scholar] [CrossRef] [PubMed]
- Kukawka, R.; Pawlowska-Zygarowicz, A.; Dutkiewicz, M.; Maciejewski, H.; Smiglak, M. New approach to hydrosilylation reaction in ionic liquids as solvent in microreactor system. RSC Adv. 2016, 6, 61860–61868. [Google Scholar] [CrossRef]
- Maciejewski, H.; Szubert, K.; Marciniec, B. New approach to synthesis of functionalised silsesquioxanes via hydrosilylation. Catal. Commun. 2012, 24, 1–4. [Google Scholar] [CrossRef]
- Barnes, T.J.; Jarvis, K.L.; Prestidge, C.A. Recent advances in porous silicon technology for drug delivery. Ther. Deliv. 2013, 4, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Bujak, P.; Kulszewicz-Bajer, I.; Zagorska, M.; Maurel, V.; Wielgus, I.; Pron, A. Polymers for electronics and spintronics. Chem. Soc. Rev. 2013, 42, 8895–8999. [Google Scholar] [CrossRef] [PubMed]
- Teplyakov, A.V.; Bent, S.F. Semiconductor surface functionalization for advances in electronics, energy conversion, and dynamic systems. J. Vac. Sci. Technol. 2013, 31, 50810. [Google Scholar] [CrossRef]
- Chatterjee, S.; Carter, R.; Oakes, L.; Erwin, W.R.; Bardhan, R.; Pint, C.L. Electrochemical and Corrosion Stability of Nanostructured Silicon by Graphene Coatings: Toward High Power Porous Silicon Supercapacitors. J. Phys. Chem. C 2014, 118, 10893–10902. [Google Scholar] [CrossRef]
- DeBenedetti, W.J.I.; Chabal, Y.J. Functionalization of oxide-free silicon surfaces. J. Vac. Sci. Technol. 2013, 31, 50826. [Google Scholar] [CrossRef]
- Kamino, B.A.; Bender, T.P. The use of siloxanes, silsesquioxanes, and silicones in organic semiconducting materials. Chem. Soc. Rev. 2013, 42, 5119–5130. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Fu, B.; Li, Y.; Shang, Q.; Xiao, G. Antigraffiti polyurethane coating containing fluorocarbon side chains grafted polymethylsiloxane. J. Coat. Technol. Res. 2013, 10, 361–369. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Z.; Li, Y.; Xu, C. Highly transparent and durable superhydrophobic hybrid nanoporous coatings fabricated from polysiloxane. ACS Appl. Mater. Interfaces 2014, 6, 10014–10021. [Google Scholar] [CrossRef] [PubMed]
- Gunkel, G.; Huck, W.T.S. Cooperative adsorption of lipoprotein phospholipids, triglycerides, and cholesteryl esters are a key factor in nonspecific adsorption from blood plasma to antifouling polymer surfaces. J. Am. Chem. Soc. 2013, 135, 7047–7052. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Niu, M.; Yuan, S.; Teng, H. Durable antimicrobial finishing of cellulose with QSA silicone by supercritical adsorption. Appl. Surf. Sci. 2013, 264, 171–175. [Google Scholar] [CrossRef]
- Cheng, L.; Liu, Q.; Lei, Y.; Lin, Y.; Zhang, A. The synthesis and characterization of carboxybetaine functionalized polysiloxanes for the preparation of anti-fouling surfaces. RSC Adv. 2014, 4, 54372–54381. [Google Scholar] [CrossRef]
- Yeh, S.-B.; Chen, C.-S.; Chen, W.-Y.; Huang, C.-J. Modification of silicone elastomer with zwitterionic silane for durable antifouling properties. Langmuir 2014, 30, 11386–11393. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.D.H.; Perrin, F.-X.; Nguyen, D.L. New hybrid materials based on poly(ethyleneoxide)-grafted polysilazane by hydrosilylation and their anti-fouling activities. Beilstein J. Nanotechnol. 2013, 4, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Cheng, L.; Hong, S.; Yang, C.; Lin, Y. Preparation of anti-fouling silicone elastomers by covalent immobilization of carboxybetaine. RSC Adv. 2015, 5, 88456–88463. [Google Scholar] [CrossRef]
- Kelly, A.M.; Kaltenhauser, V.; Mühlbacher, I.; Rametsteiner, K.; Kren, H.; Slugovc, C.; Stelzer, F.; Wiesbrock, F. Poly(2-oxazoline)-derived Contact Biocides: Contributions to the Understanding of Antimicrobial Activity. Macromol. Biosci. 2013, 13, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.; Reinhoudt, D.N.; Huskens, J. Microcontact Printing: Limitations and Achievements. Adv. Mater. 2009, 21, 2257–2268. [Google Scholar] [CrossRef]
- Wisser, F.M.; Schumm, B.; Mondin, G.; Grothe, J.; Kaskel, S. Precursor strategies for metallic nano- and micropatterns using soft lithography. J. Mater. Chem. C 2015, 3, 2717–2731. [Google Scholar] [CrossRef]
- Kastner, J.; Lorret, O.; Rank, A.; Schwarzinger, C.; Dittert, B.; Mühlberger, M. Nanocontact printing stamp material via bi-functionalization of polyhedral oligomeric silsesquioxanes. Eur. Polym. J. 2015, 65, 221–231. [Google Scholar] [CrossRef]
- Masuda, T.; Shen, Z.; Takagishi, H.; Ohdaira, K.; Shimoda, T. Amorphous silicon carbide films prepared using vaporized silicon ink. Jpn. J. Appl. Phys. 2014, 53, 31304. [Google Scholar] [CrossRef]
- Pi, X.; Zhang, L.; Yang, D. Enhancing the Efficiency of Multicrystalline Silicon Solar Cells by the Inkjet Printing of Silicon-Quantum-Dot Ink. J. Phys. Chem. C 2012, 116, 21240–21243. [Google Scholar] [CrossRef]
- Gao, N.; Liu, W.; Yan, Z.; Wang, Z. Synthesis and properties of transparent cycloaliphatic epoxy–silicone resins for opto-electronic devices packaging. Opt. Mater. 2013, 35, 567–575. [Google Scholar] [CrossRef]
- Zong, Y.; Gui, D.; Yu, S.; Liu, C.; Chen, W.; Qi, Z. Preparation and properties of silicone-cycloaliphatic epoxy resins for LED packaging. In Proceedings of the 17th International Conference on Electronic Packaging Technology (ICEPT), Wuhan, China, 16–19 August 2016; pp. 244–248. [Google Scholar]
- Li, D.D.; Li, S.; Zhang, S.; Liu, X.W.; Wong, C.P. Thermo and Dynamic Mechanical Properties of the High Refractive Index Silicone Resin for Light Emitting Diode Packaging. IEEE Trans. Compon. Packag. Manufact. Technol. 2014, 4, 190–197. [Google Scholar] [CrossRef]
- Cordelair, J.; Greil, P. Electrical Characterization of Polymethylsiloxane/MoSi2-Derived Composite Ceramics. J. Am. Ceram. Soc. 2001, 84, 2256–2259. [Google Scholar] [CrossRef]
- Haluschka, C.; Engel, C.; Riedel, R. Silicon carbonitride ceramics derived from polysilazanes Part II. Investigation of electrical properties. J. Eur. Ceram. Soc. 2000, 20, 1365–1374. [Google Scholar] [CrossRef]
- Dalcanale, F.; Grossenbacher, J.; Blugan, G.; Gullo, M.R.; Lauria, A.; Brugger, J.; Tevaearai, H.; Graule, T.; Niederberger, M.; Kuebler, J. Influence of carbon enrichment on electrical conductivity and processing of polycarbosilane derived ceramic for MEMS applications. J. Eur. Ceram. Soc. 2014, 34, 3559–3570. [Google Scholar] [CrossRef]
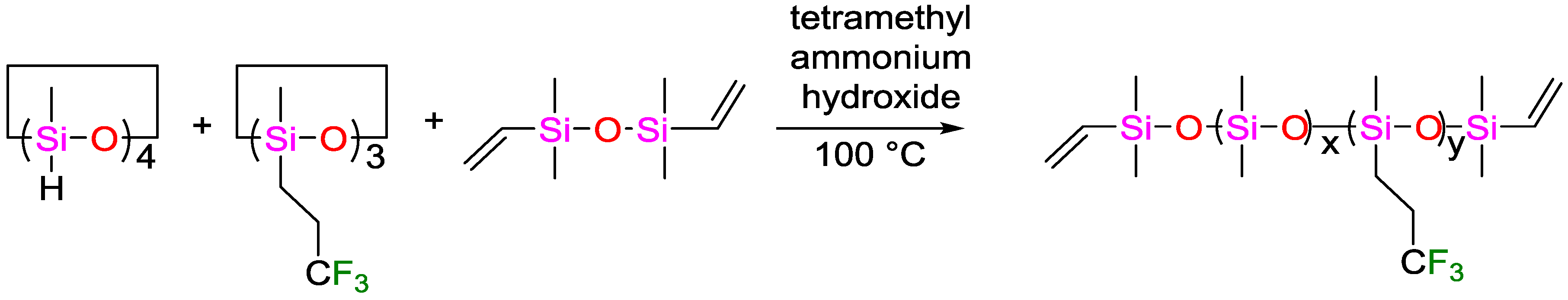
- Dascalu, M.; Dünki, S.J.; Quinsaat, J.-E.Q.; Ko, Y.S.; Opris, D.M. Synthesis of silicone elastomers containing trifluoropropyl groups and their use in dielectric elastomer transducers. RSC Adv. 2015, 5, 104516–104523. [Google Scholar] [CrossRef]
- Madsen, F.B.; Yu, L.; Daugaard, A.E.; Hvilsted, S.; Skov, A.L. Silicone elastomers with high dielectric permittivity and high dielectric breakdown strength based on dipolar copolymers. Polymer 2014, 55, 6212–6219. [Google Scholar] [CrossRef]











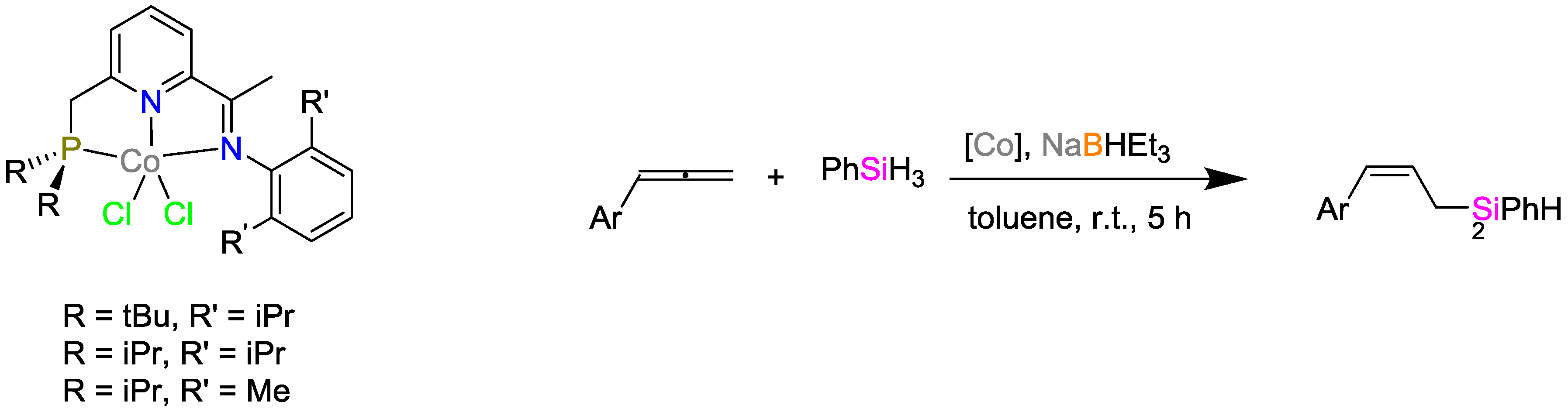













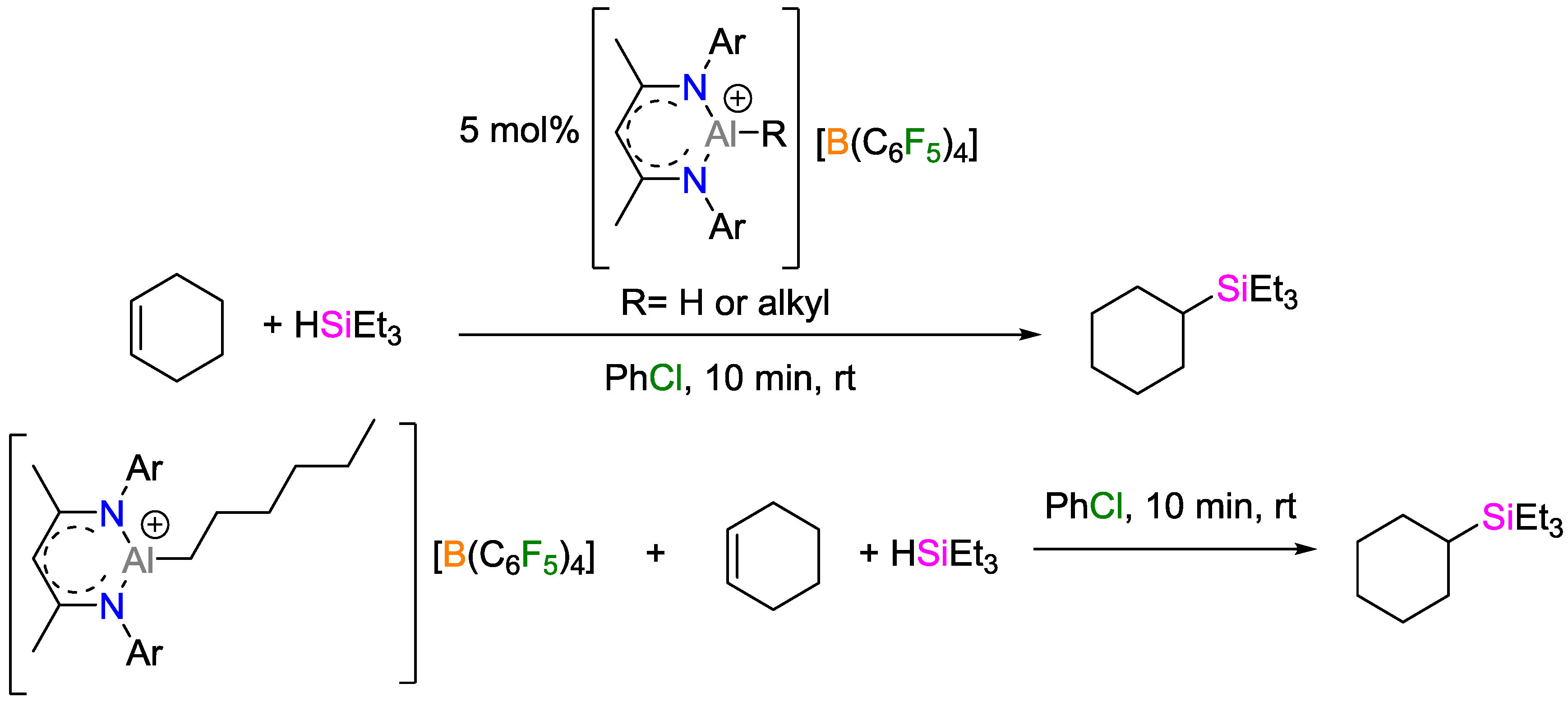























© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hofmann, R.J.; Vlatković, M.; Wiesbrock, F. Fifty Years of Hydrosilylation in Polymer Science: A Review of Current Trends of Low-Cost Transition-Metal and Metal-Free Catalysts, Non-Thermally Triggered Hydrosilylation Reactions, and Industrial Applications. Polymers 2017, 9, 534. https://doi.org/10.3390/polym9100534
Hofmann RJ, Vlatković M, Wiesbrock F. Fifty Years of Hydrosilylation in Polymer Science: A Review of Current Trends of Low-Cost Transition-Metal and Metal-Free Catalysts, Non-Thermally Triggered Hydrosilylation Reactions, and Industrial Applications. Polymers. 2017; 9(10):534. https://doi.org/10.3390/polym9100534
Chicago/Turabian StyleHofmann, Robin J., Matea Vlatković, and Frank Wiesbrock. 2017. "Fifty Years of Hydrosilylation in Polymer Science: A Review of Current Trends of Low-Cost Transition-Metal and Metal-Free Catalysts, Non-Thermally Triggered Hydrosilylation Reactions, and Industrial Applications" Polymers 9, no. 10: 534. https://doi.org/10.3390/polym9100534