Modulating the Tg of Poly(alkylene succinate)s by Inserting Bio-Based Aromatic Units via Ring-Opening Copolymerization


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Measurements
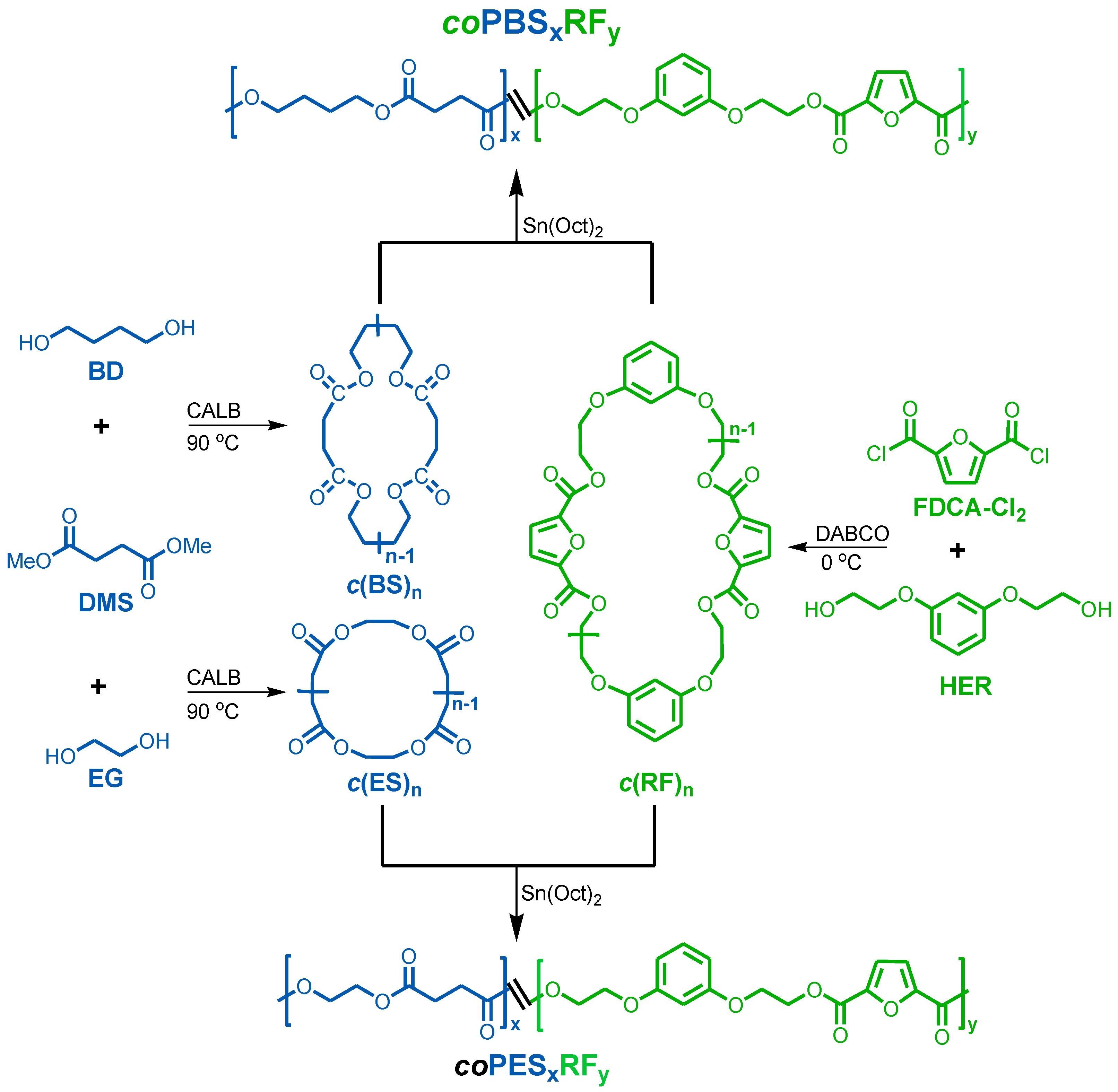
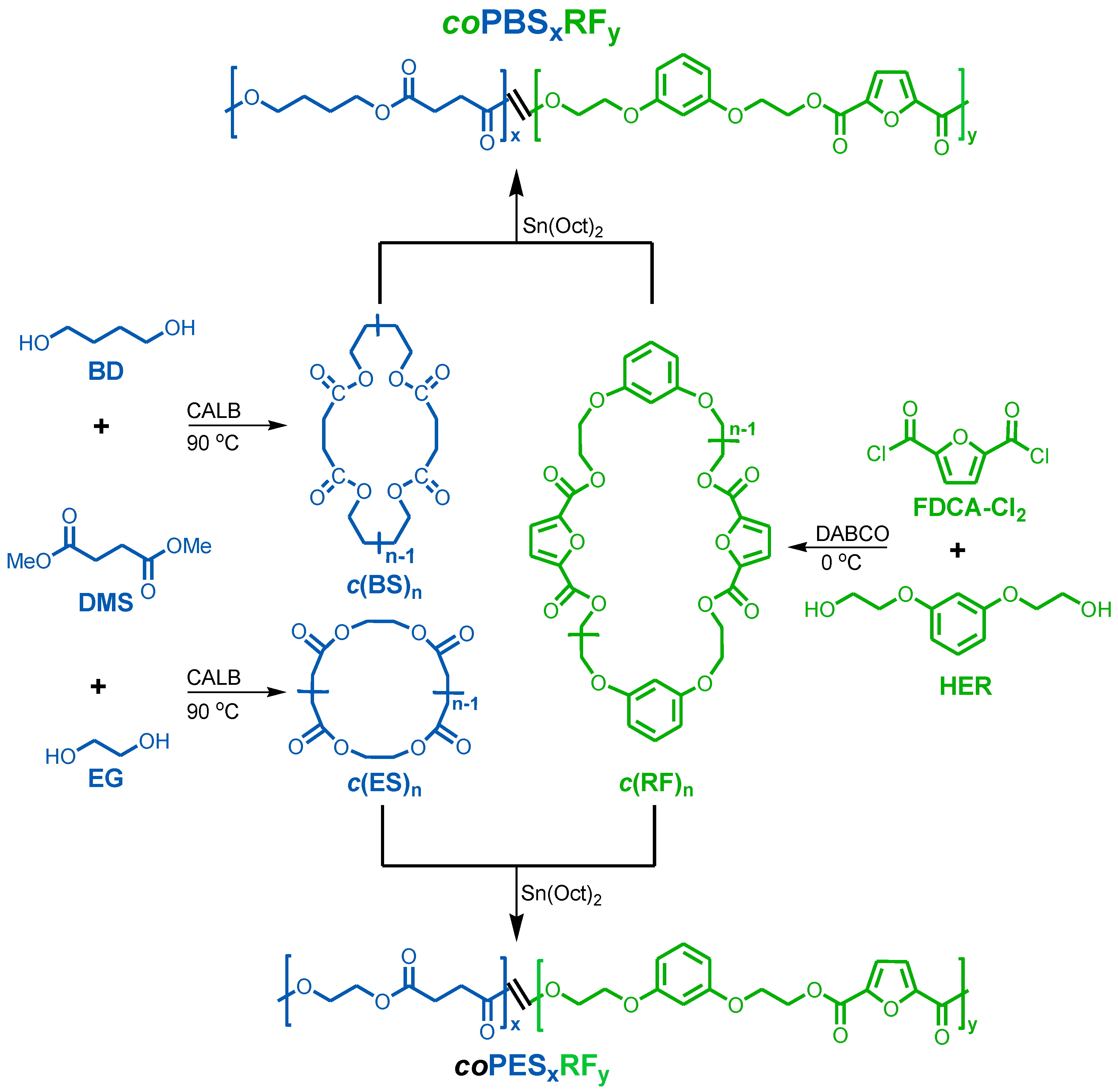
2.3. Synthesis of Cyclic Oligomers
2.4. Synthesis of Polyesters
2.5. Hydrolytic and Enzymatic Degradation
3. Results and Discussion
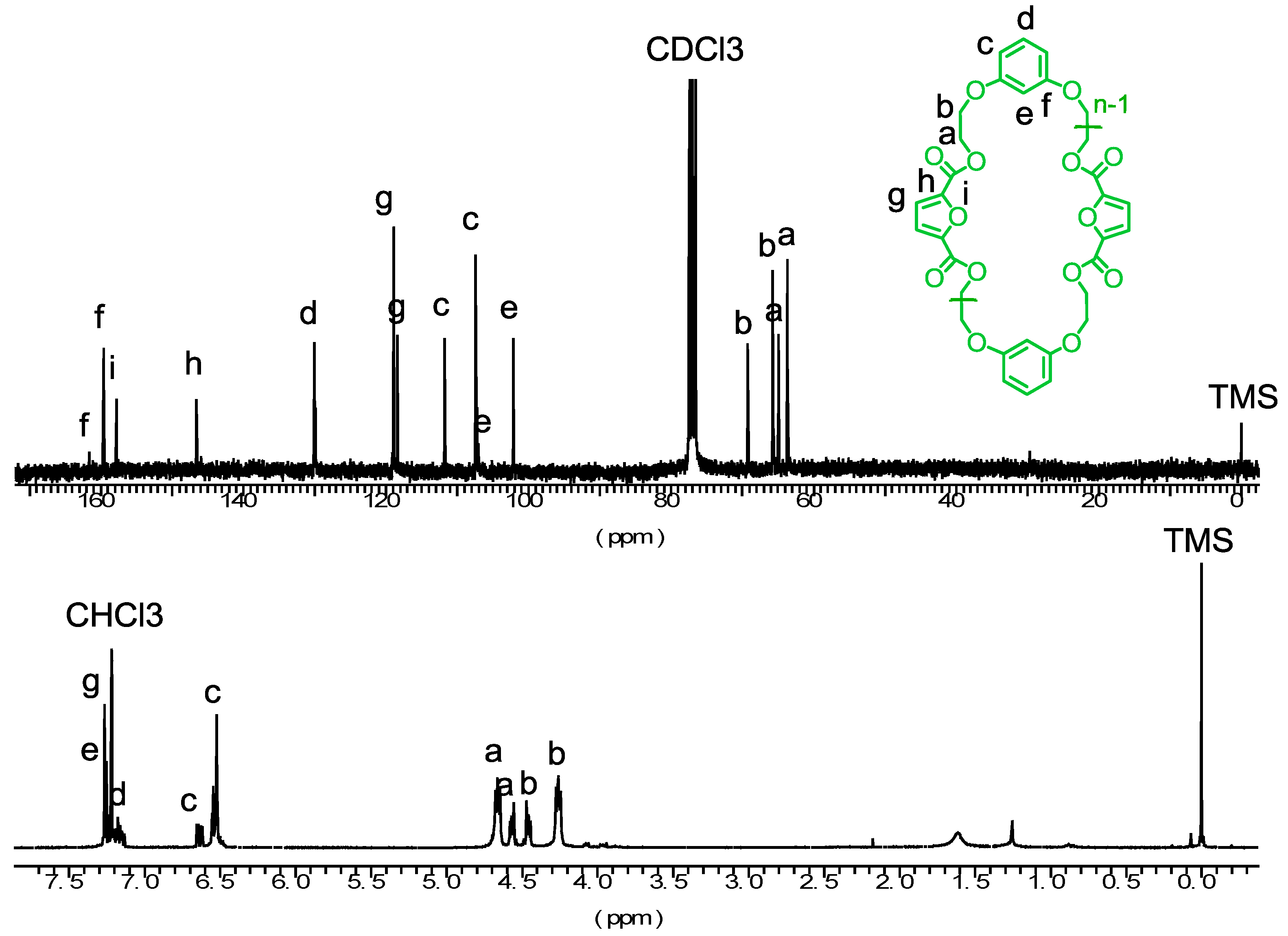
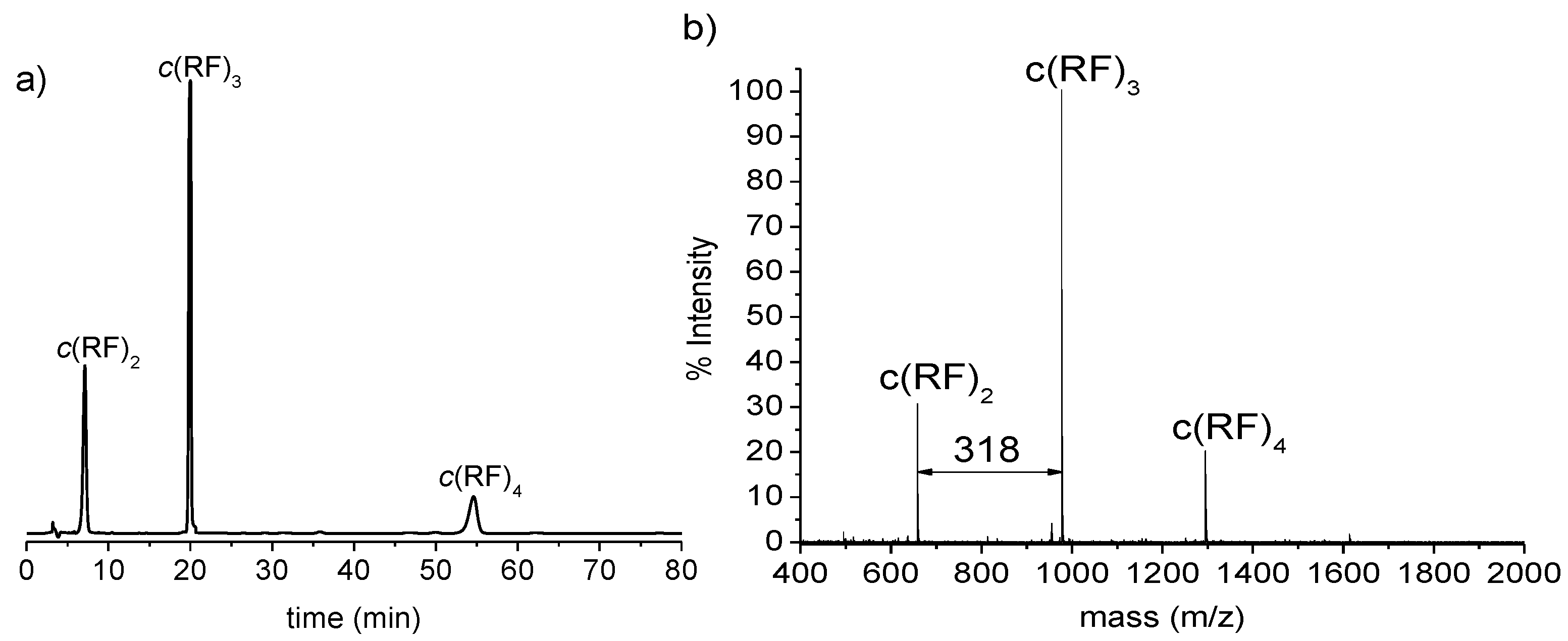
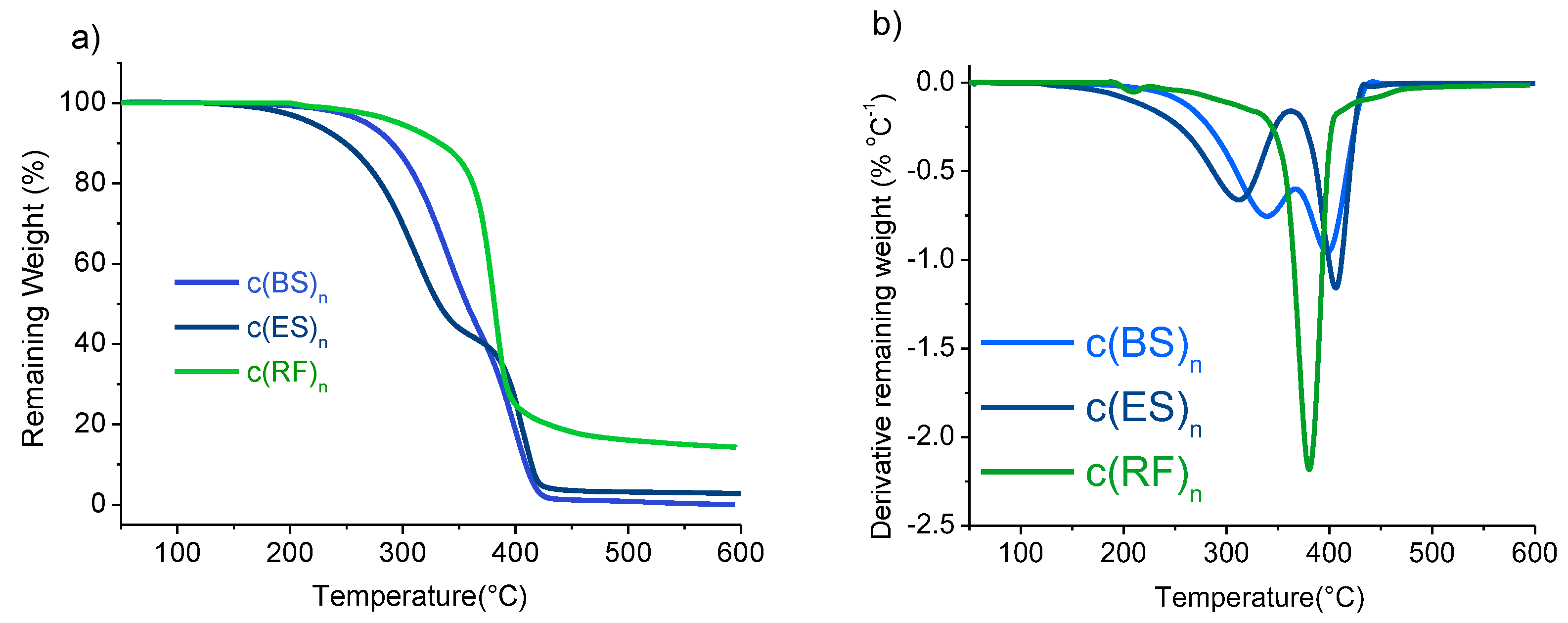
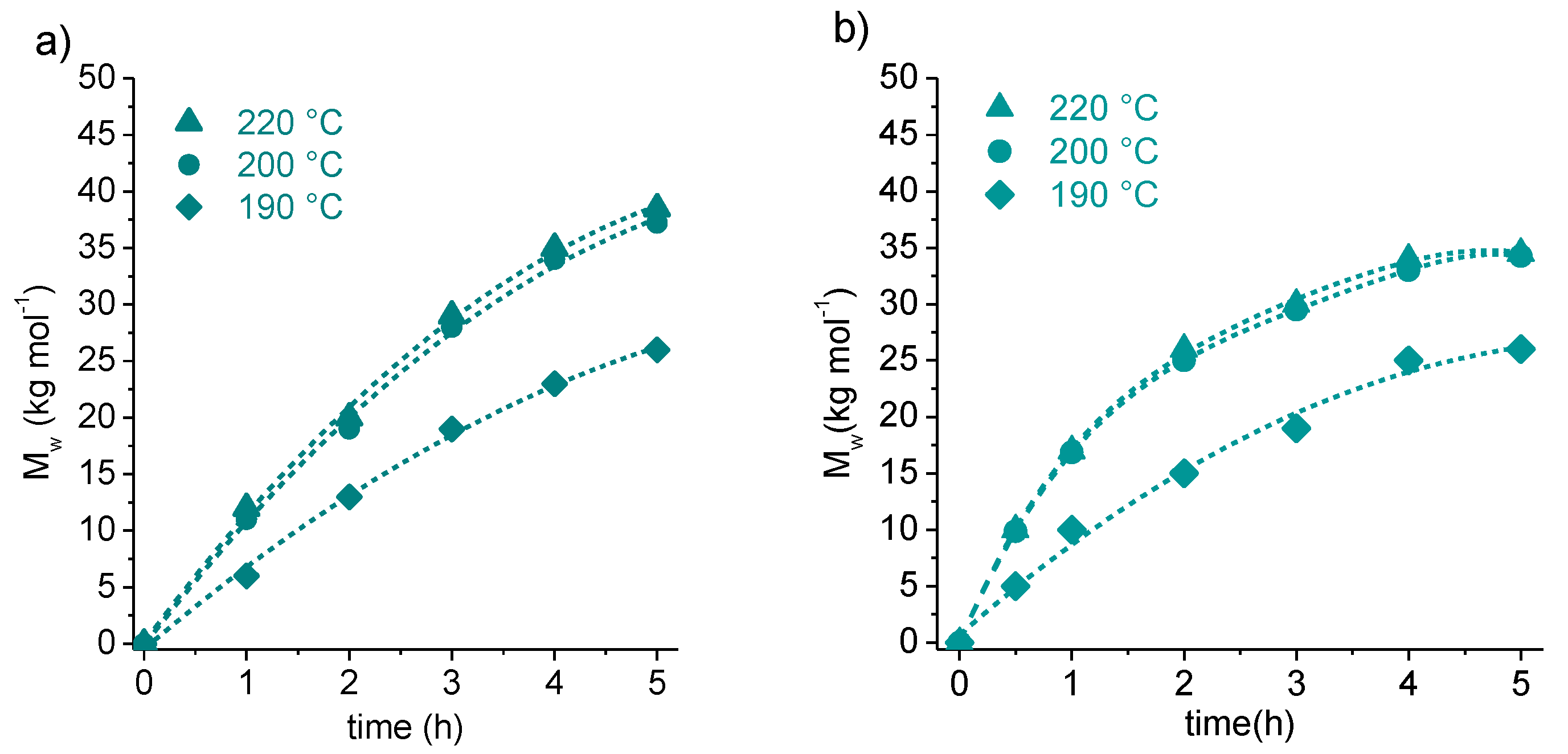
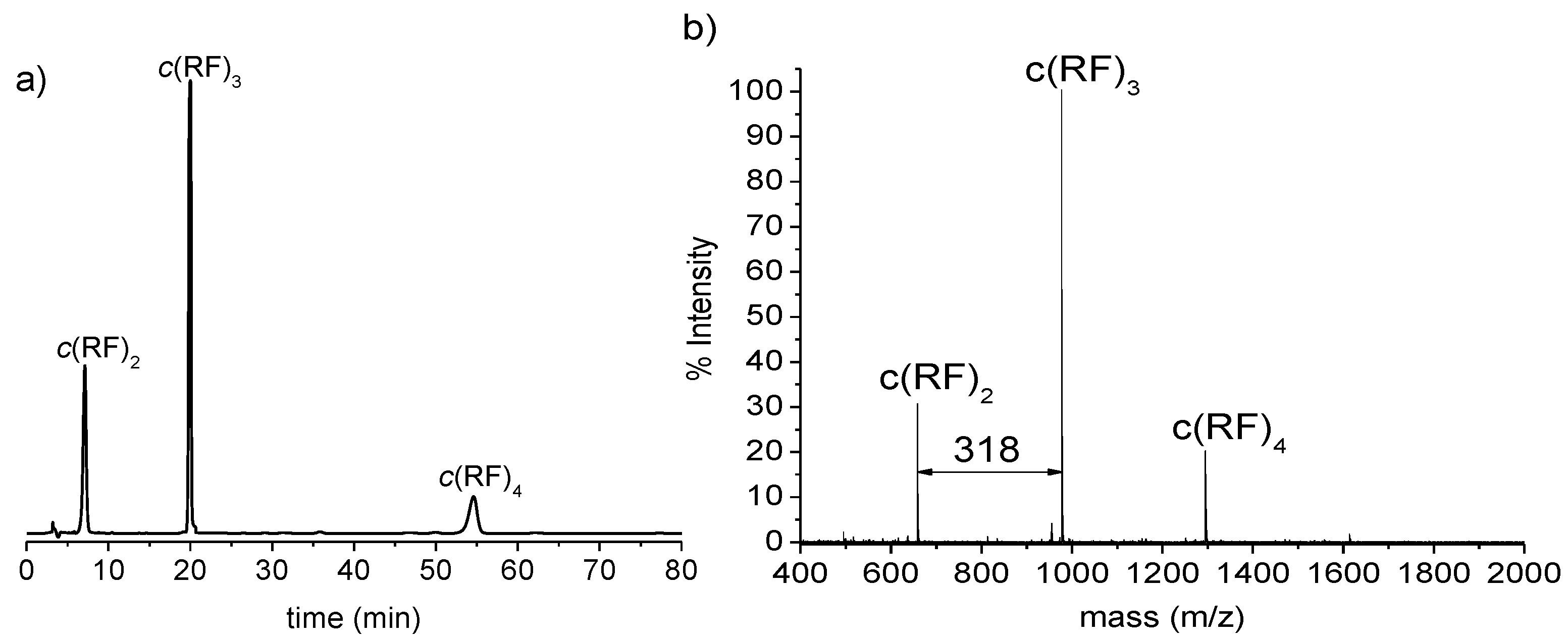
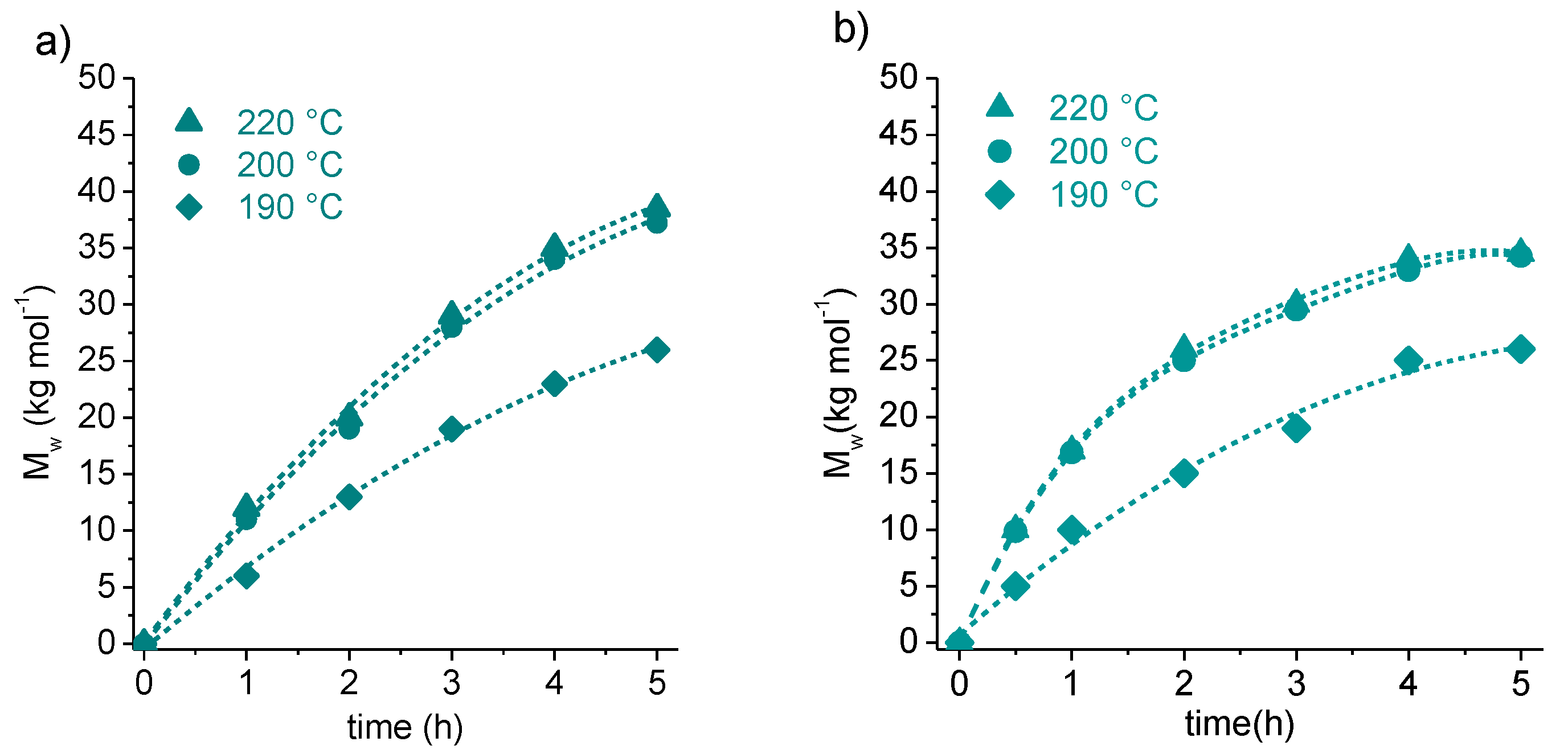
3.1. Synthesis of Cyclic Oligomers
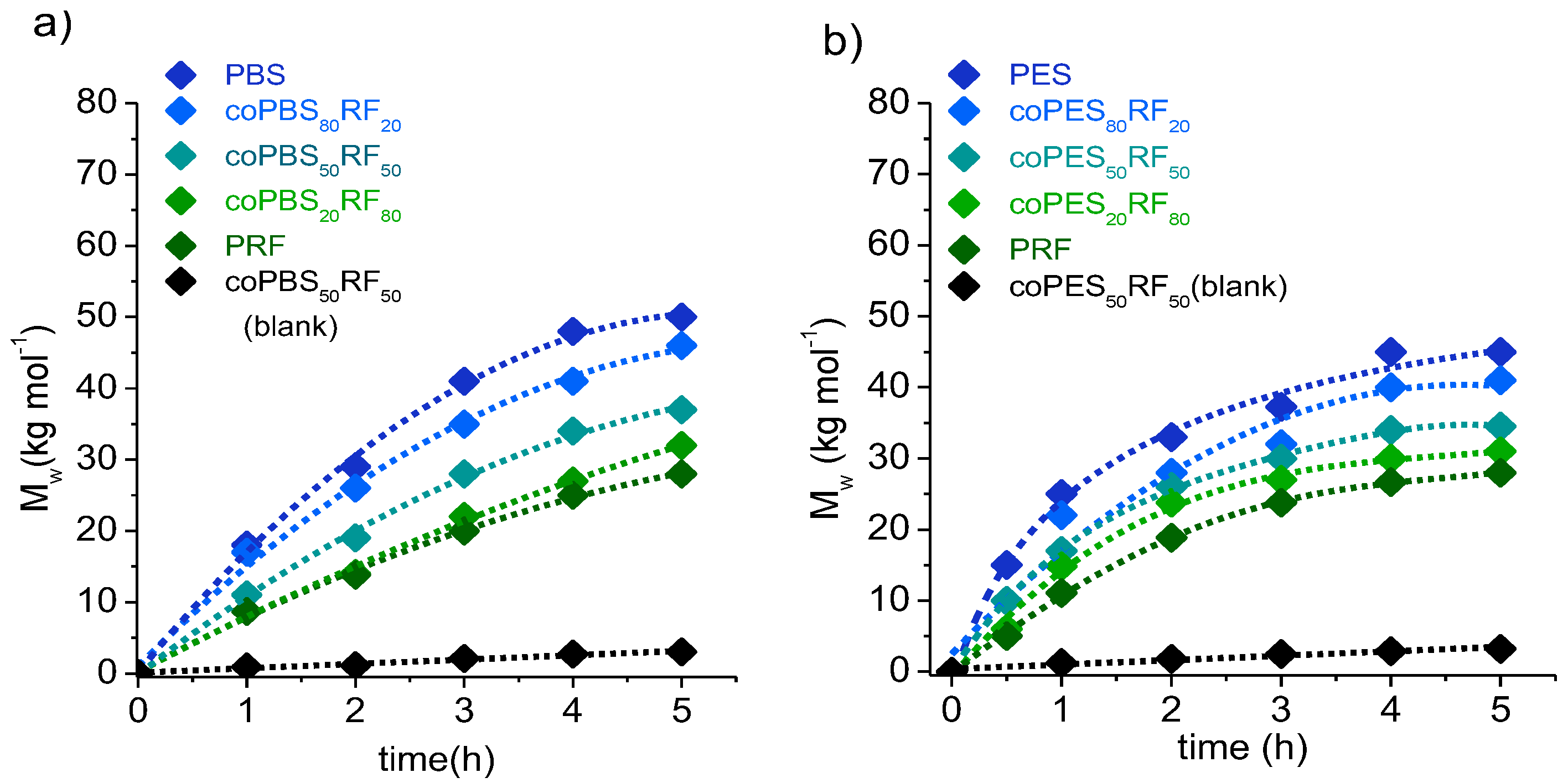
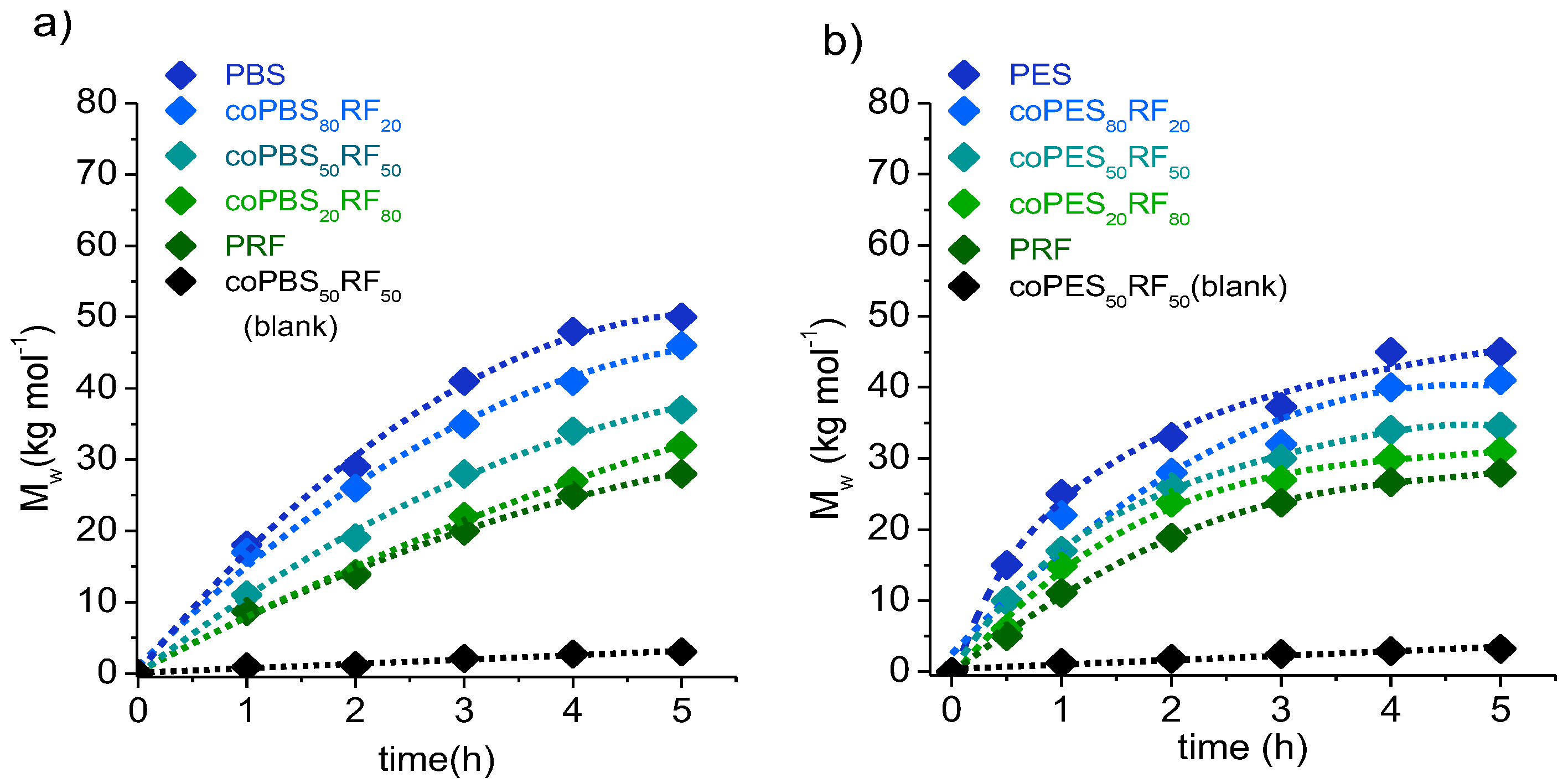
3.2. Synthesis of Copolyesters
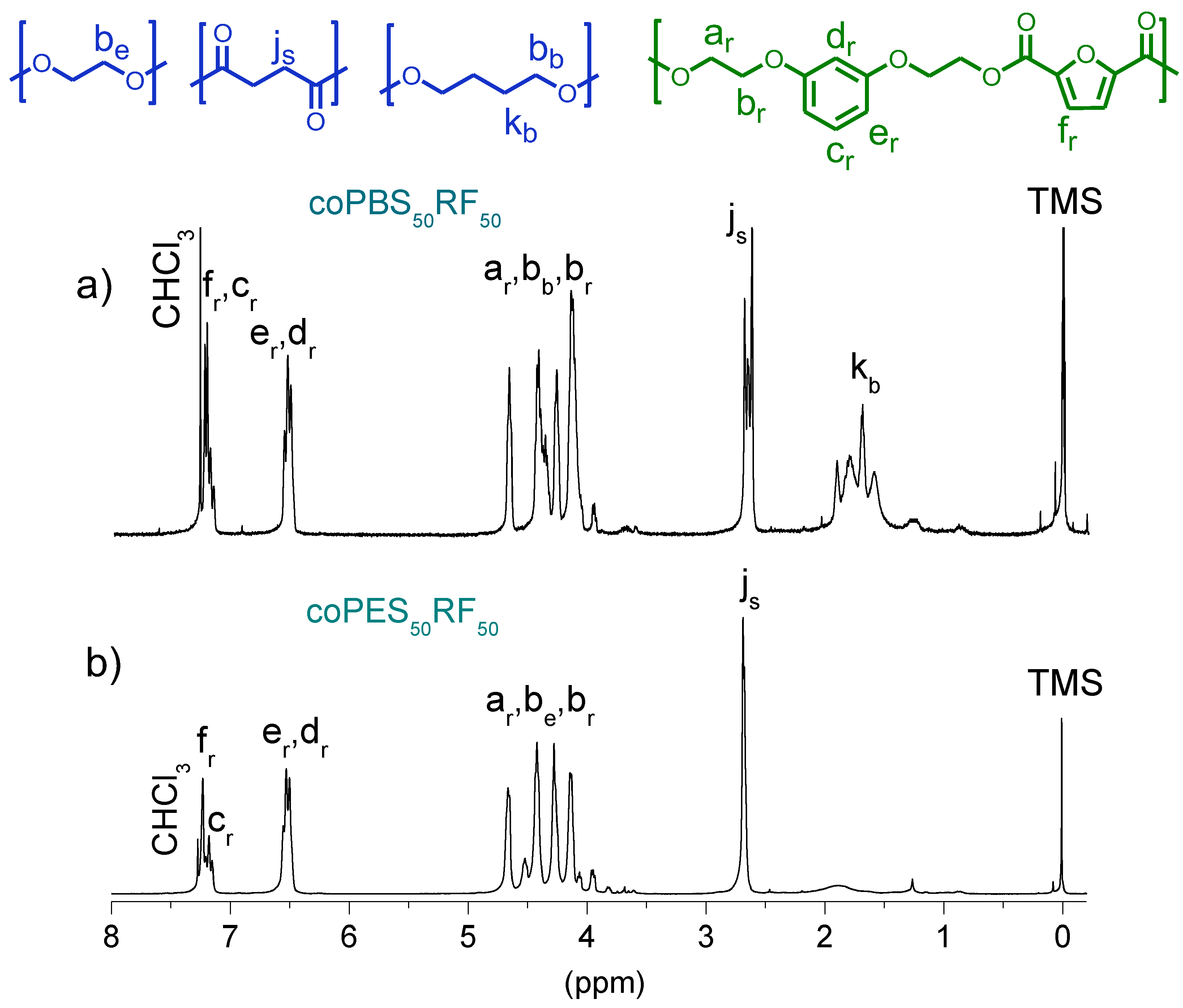
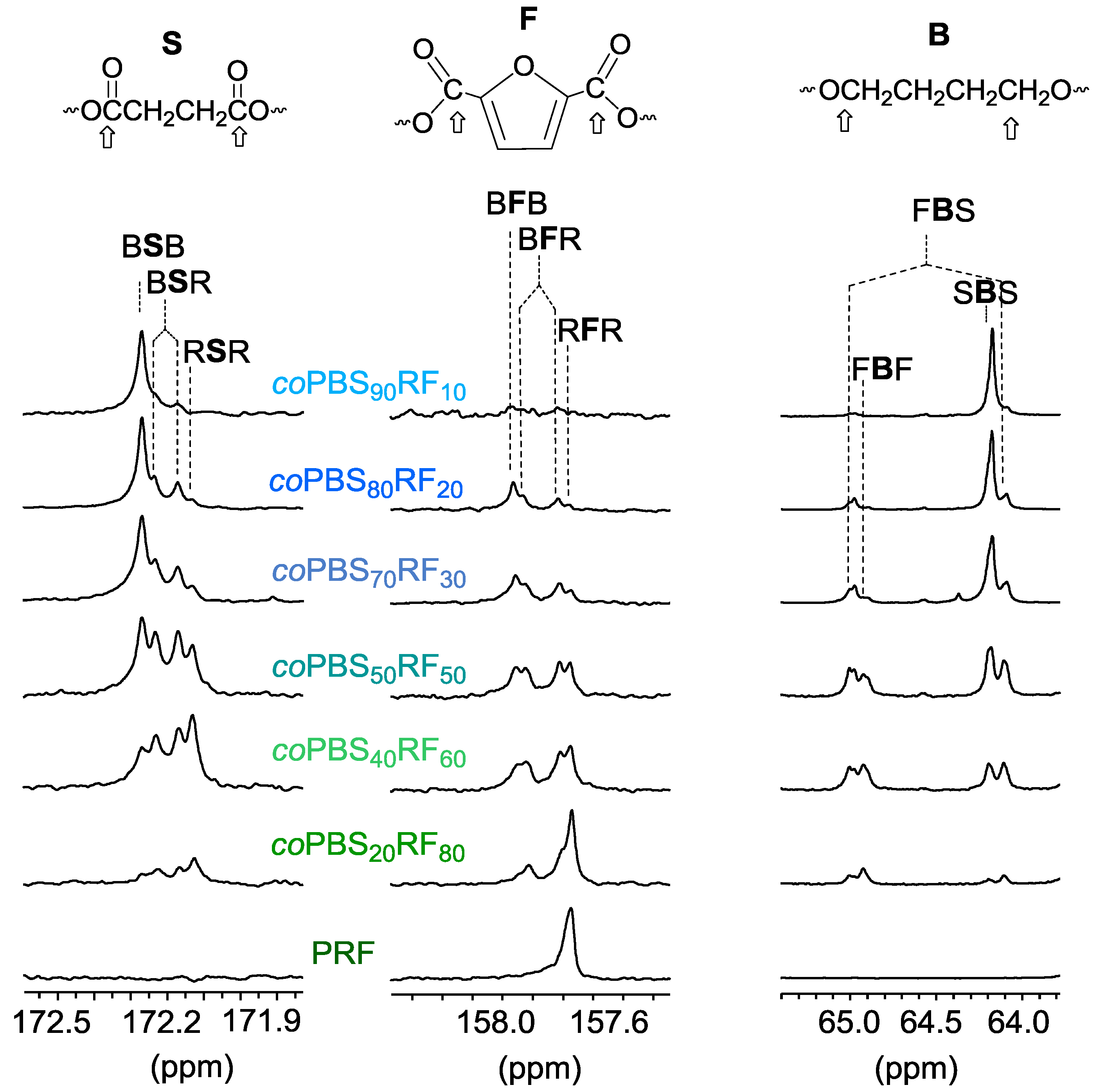
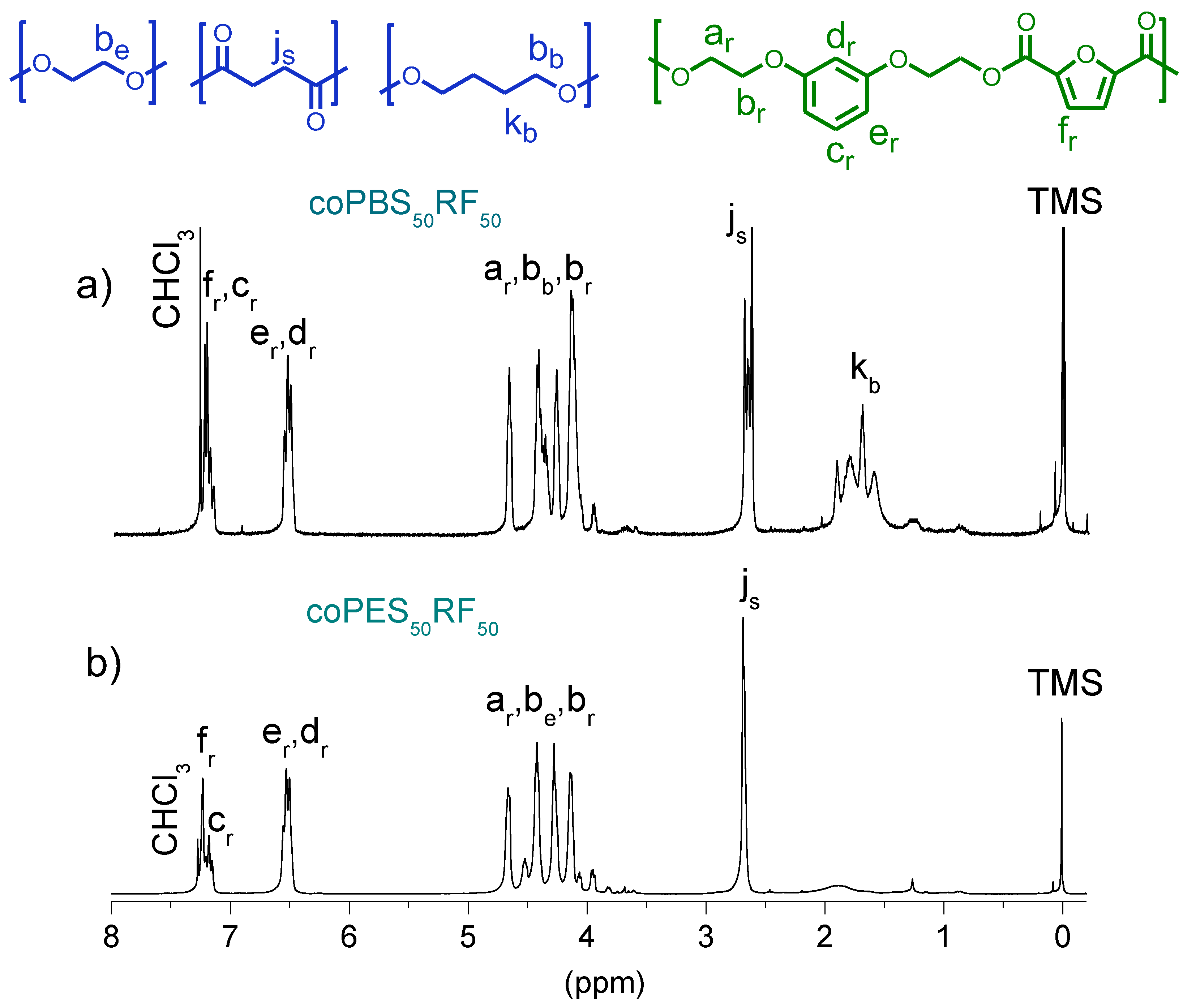
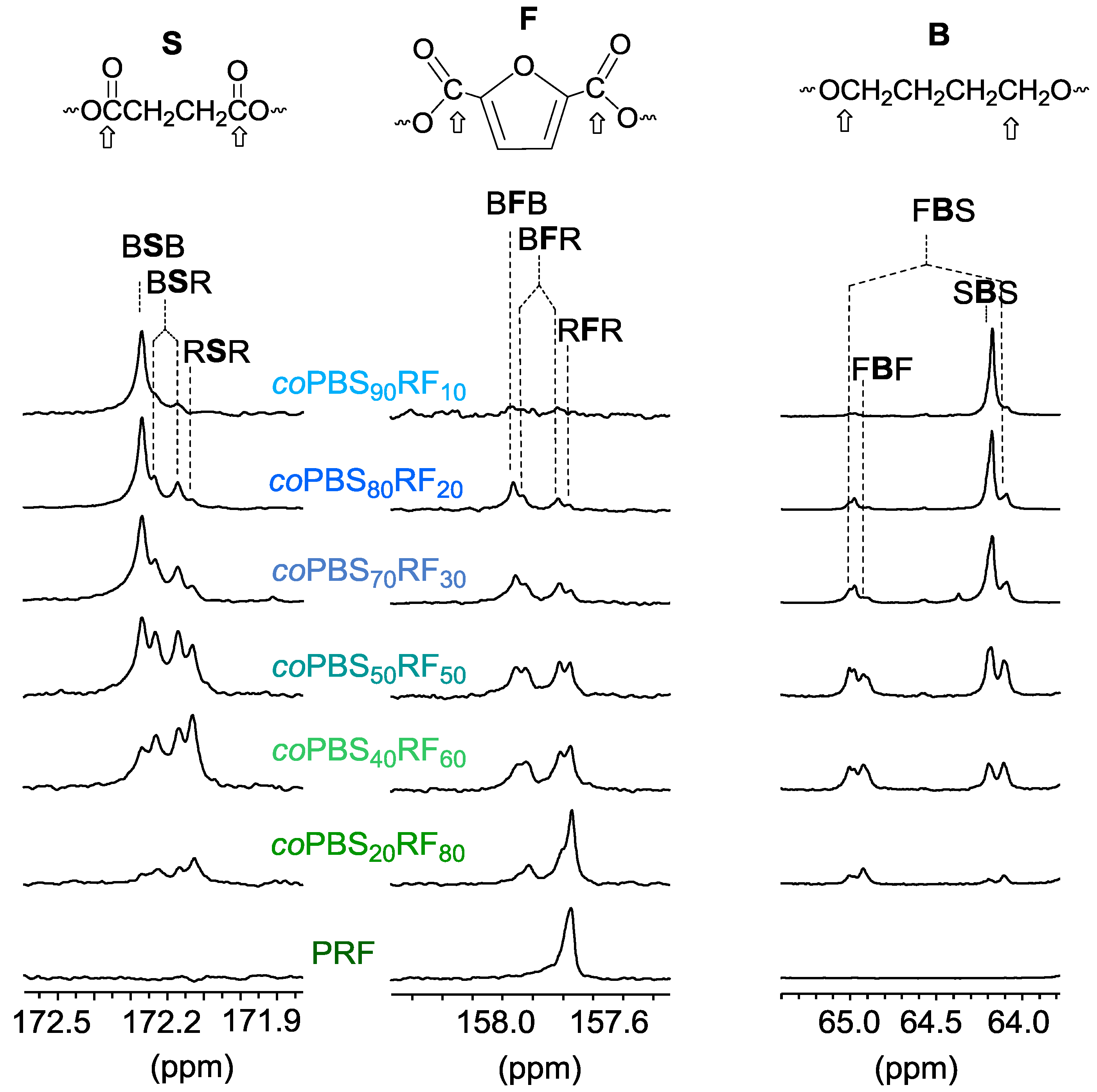
3.3. Microstructure of the Copolyesters
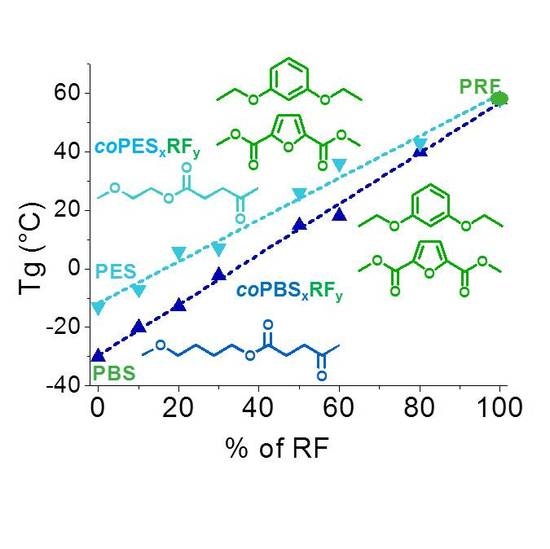
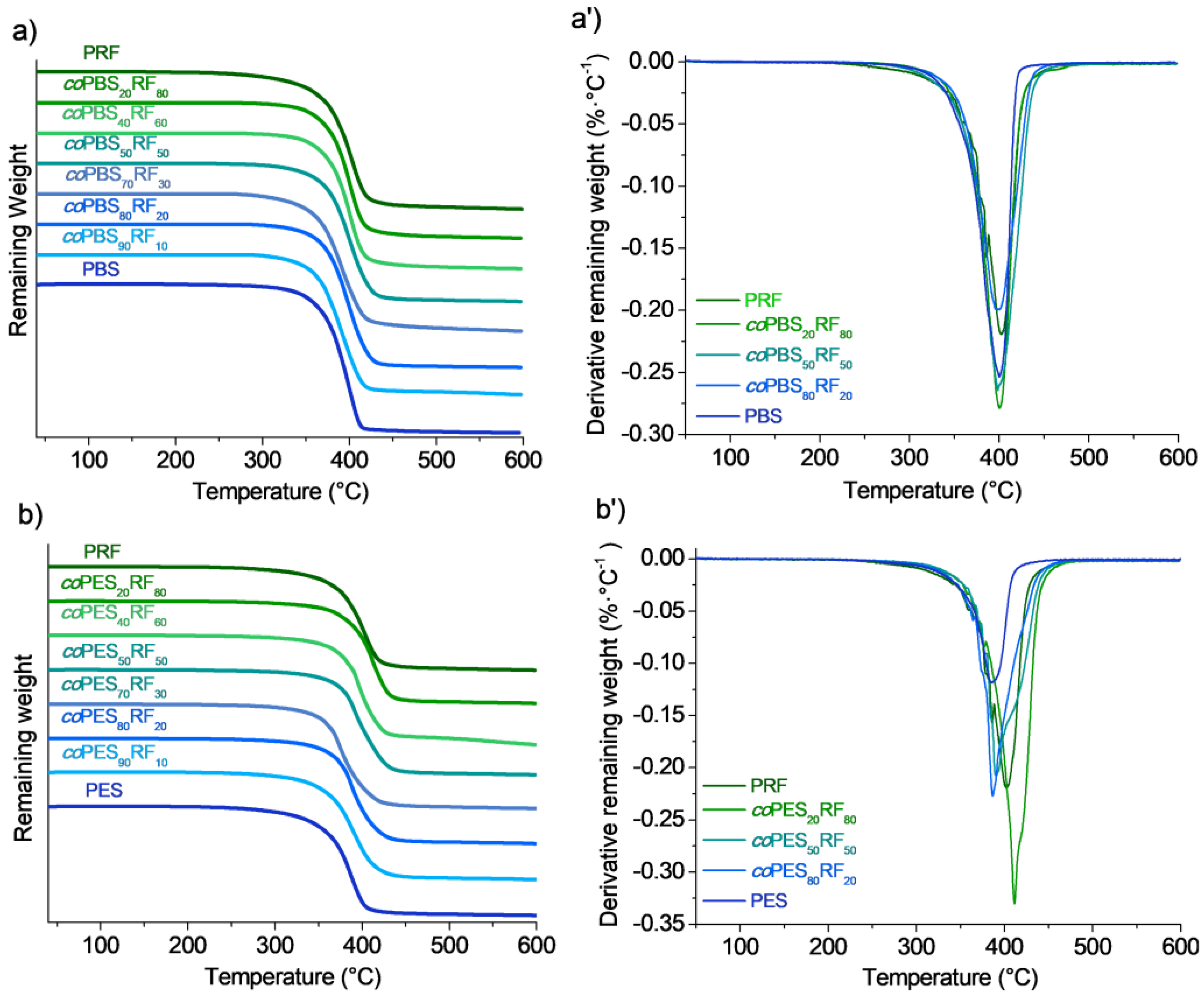
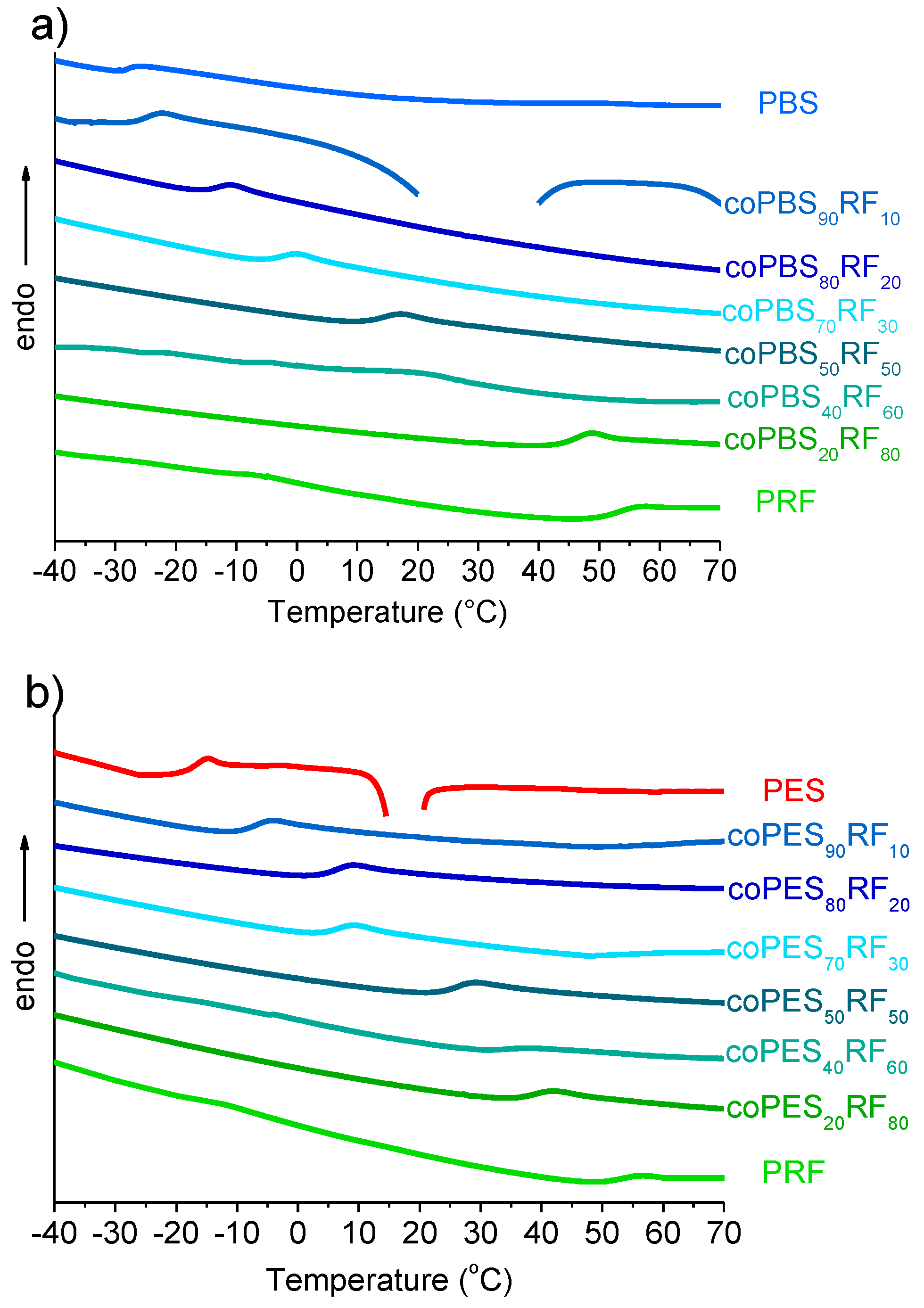
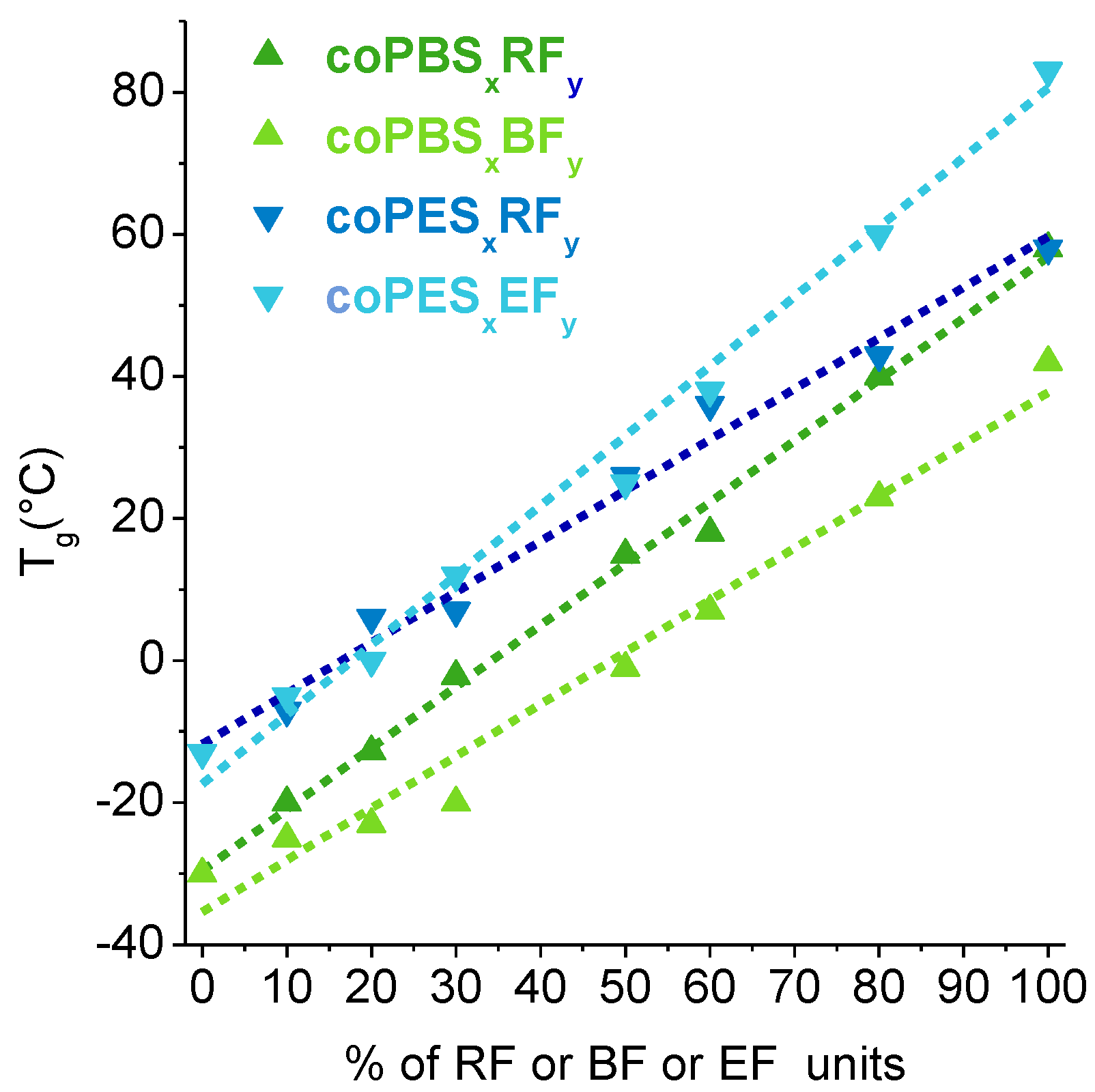
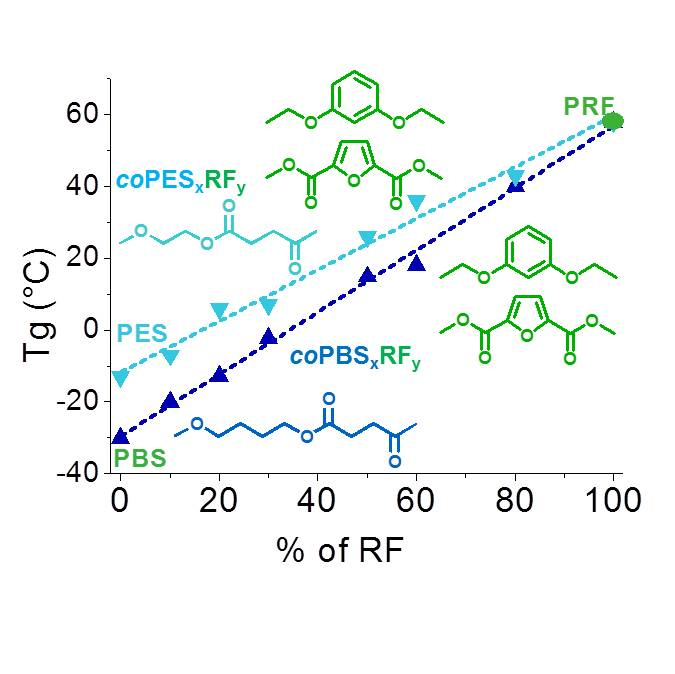
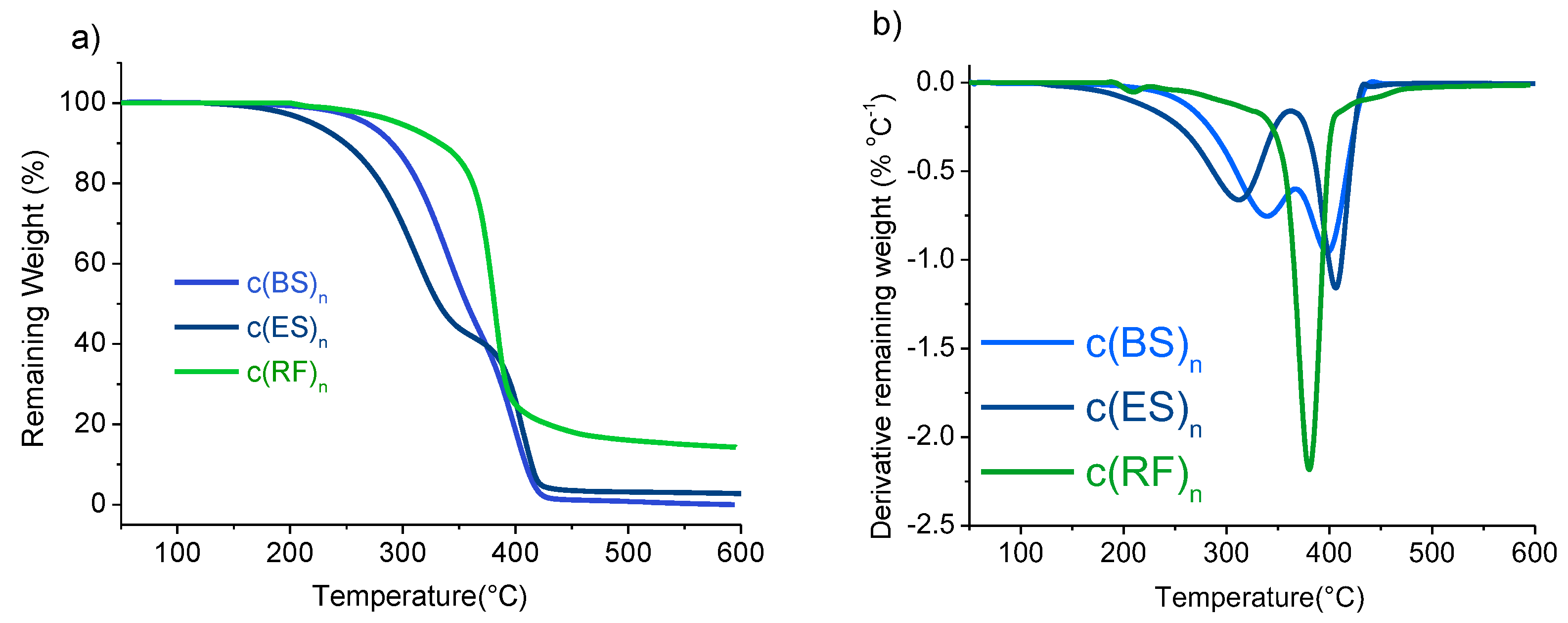
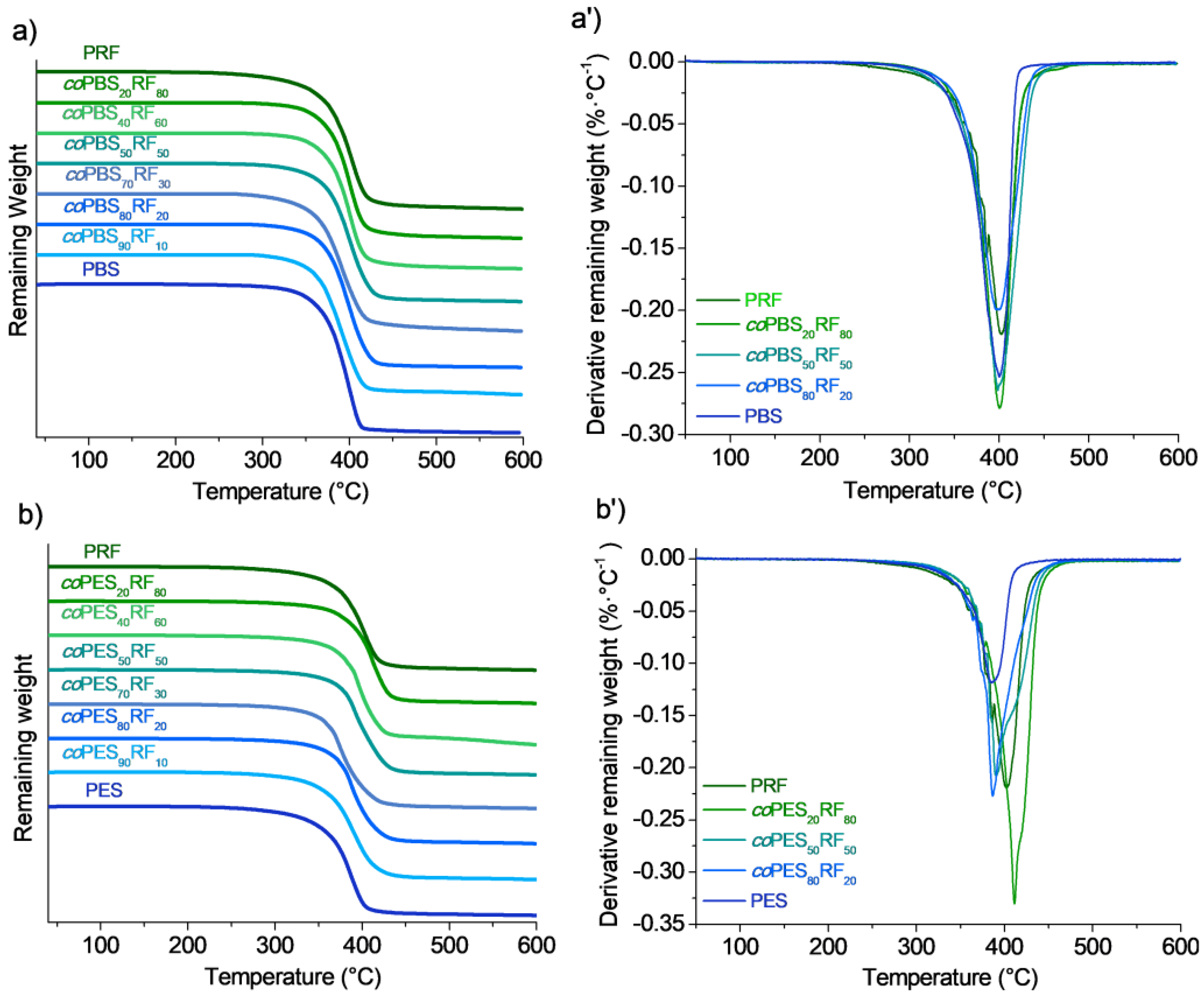
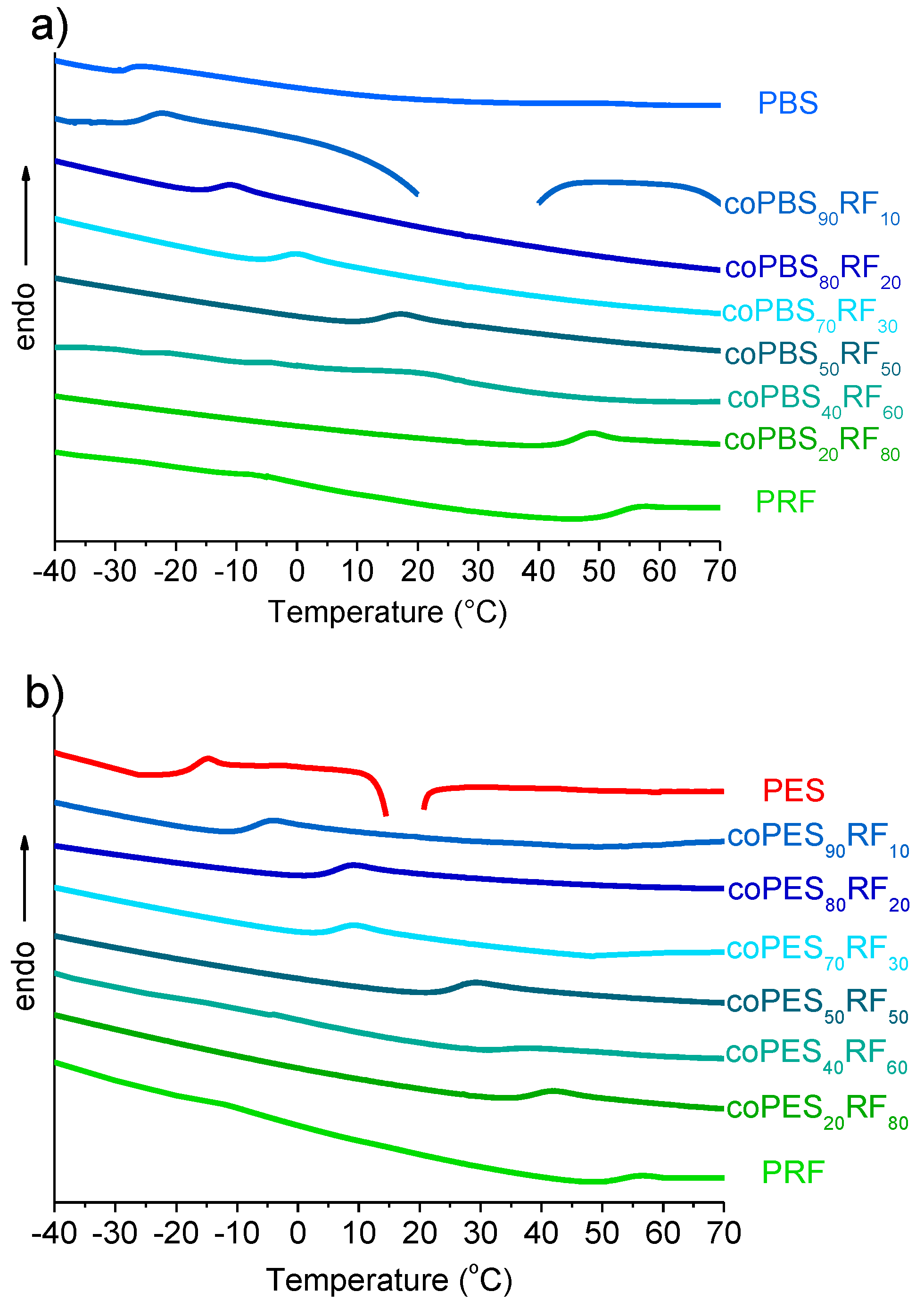
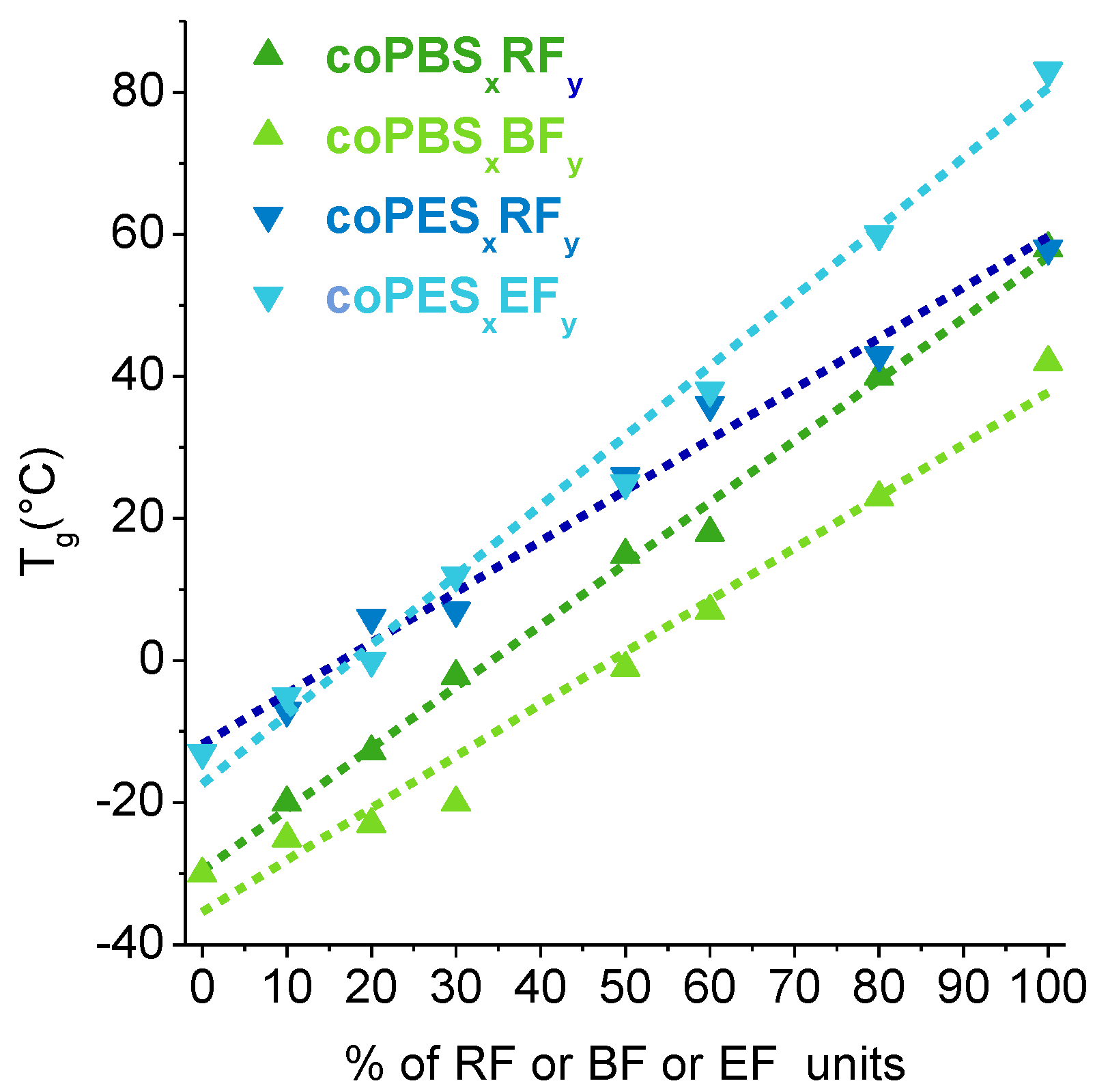
3.4. Thermal Properties
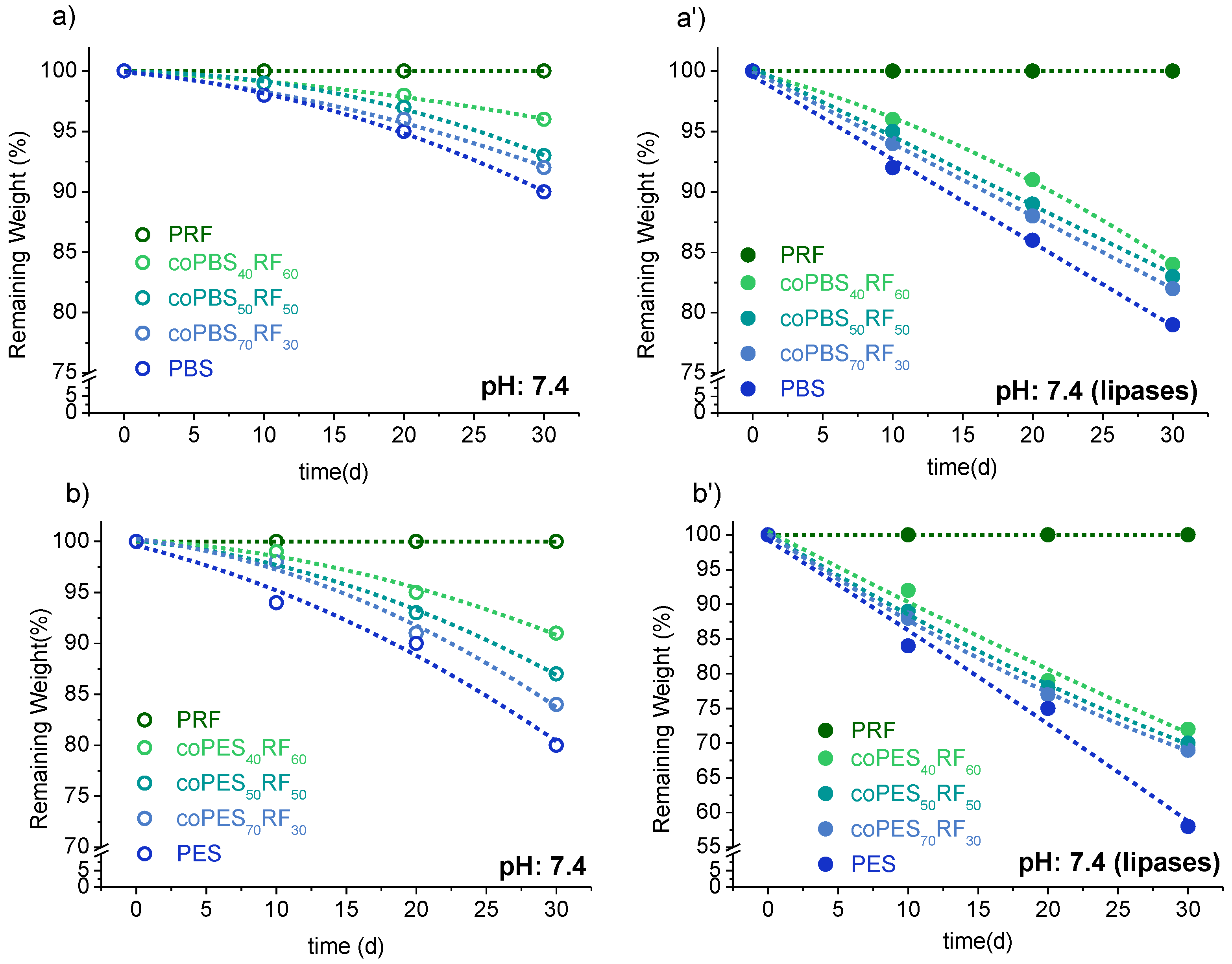
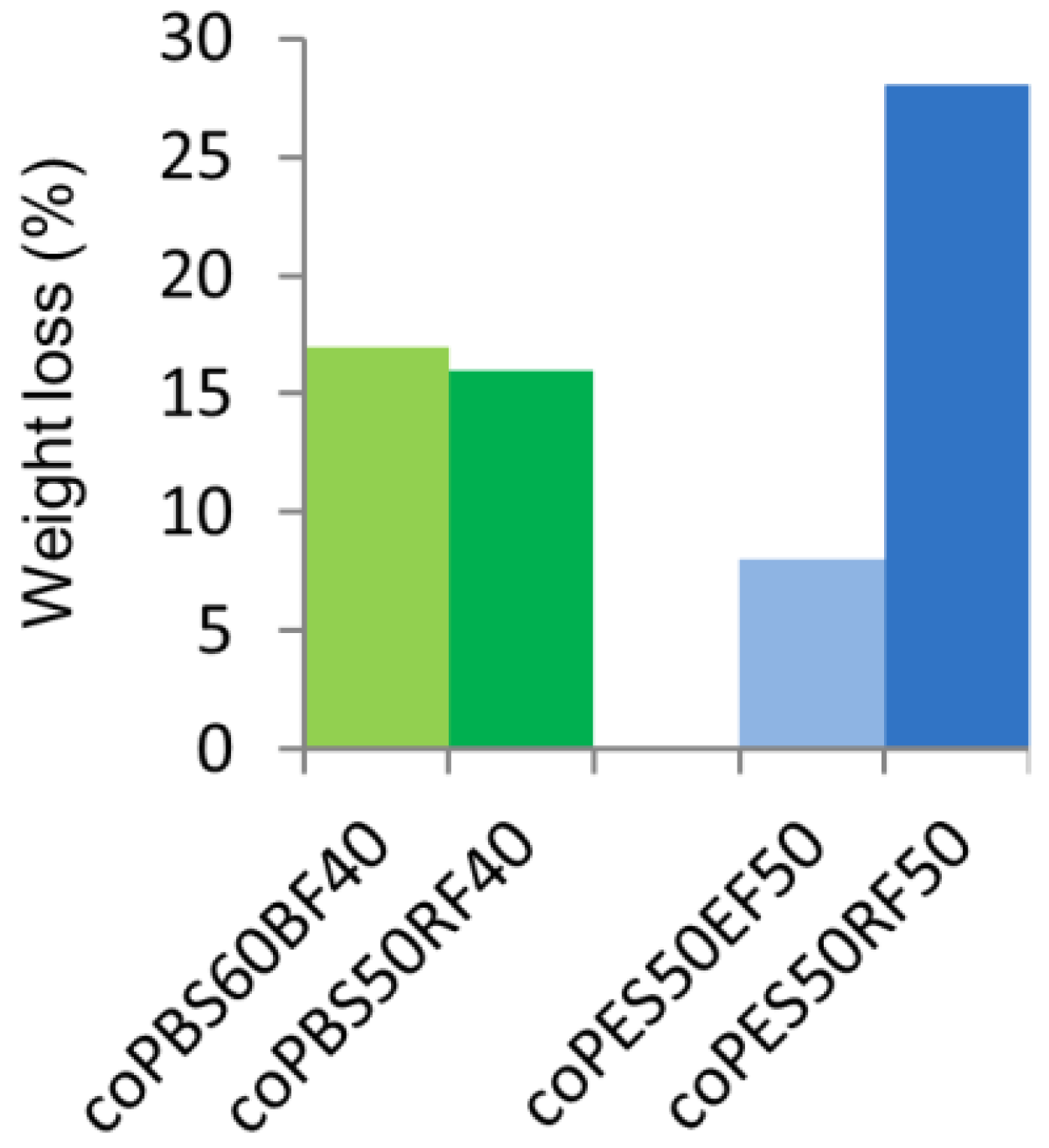
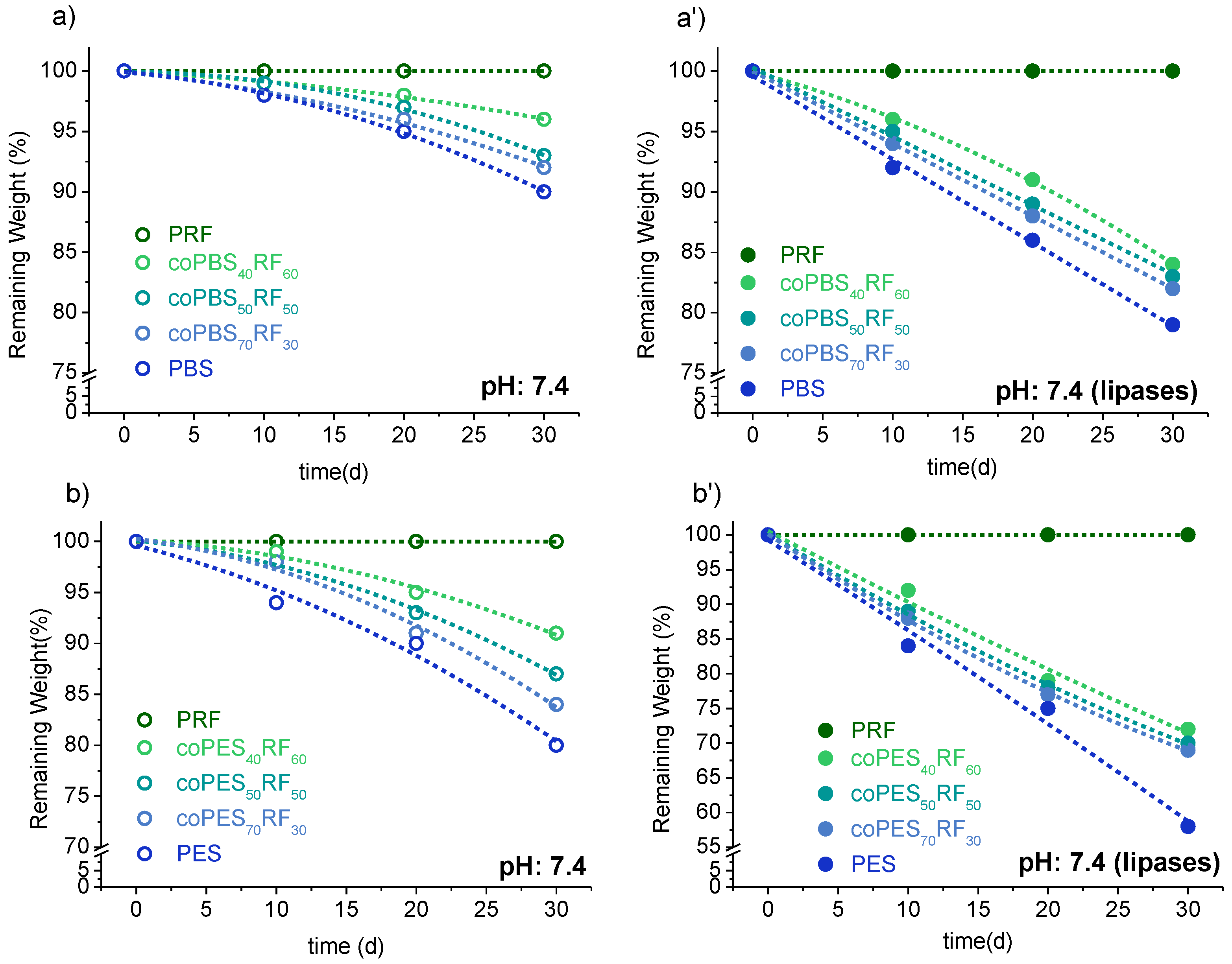
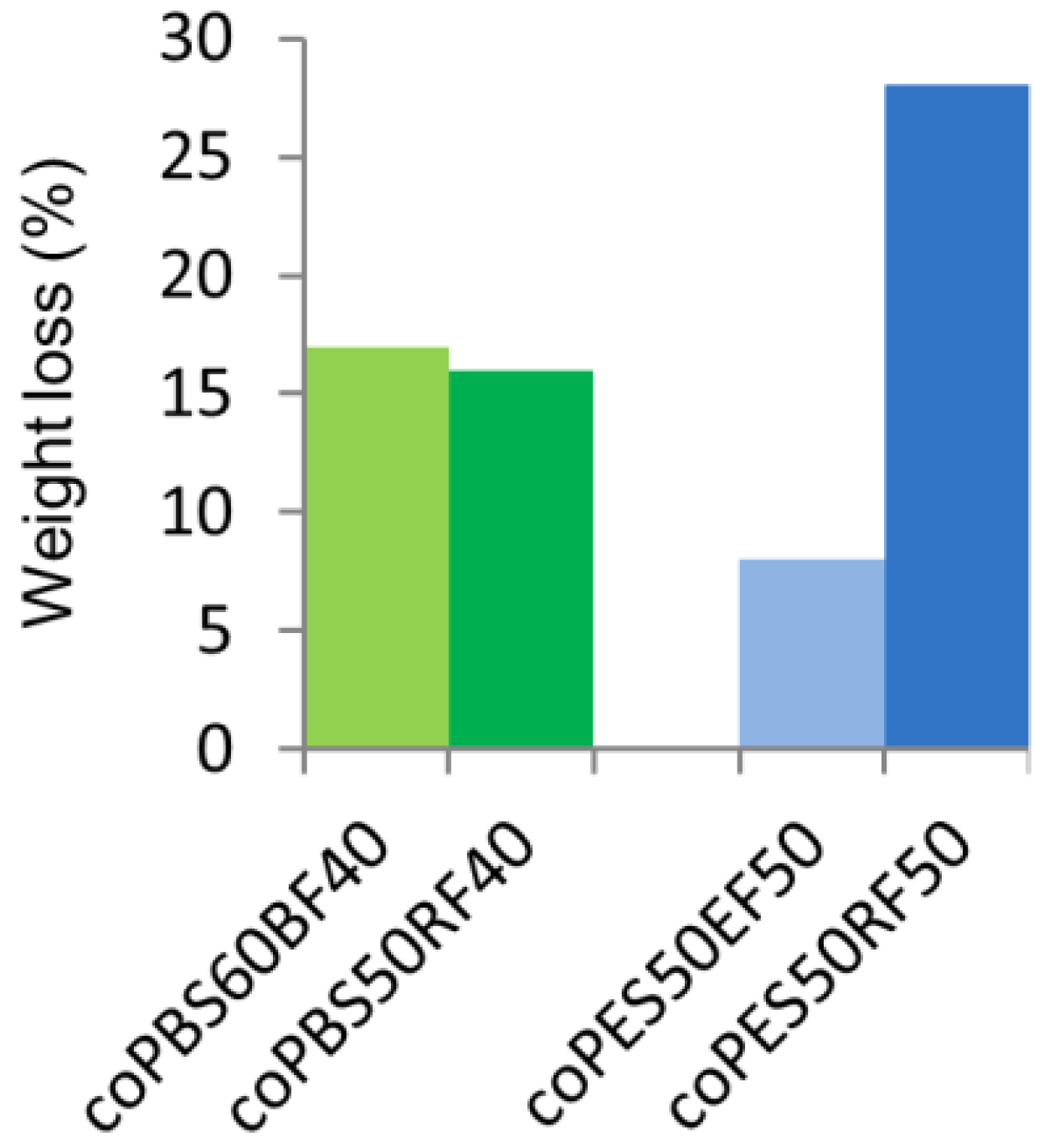
3.5. Biodegradability
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tsui, A.; Wright, Z.C.; Frank, C.W. Biodegradable polyesters from renewable resources. Annu. Rev. Chem. Biomol. 2013, 4, 143–170. [Google Scholar] [CrossRef] [PubMed]
- Zia, K.M.; Nooren, A.; Zuber, M.; Tabasum, S.; Mujahid, M. Recent developments and future prospects on bio-based polyesters derived from renewable resources: A review. Int. J. Biol. Macromol. 2015, 82, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Bechthold, I.; Bretz, K.; Kabasci, S.; Kopitzky, R.; Springer, A. Succinic acid: A new platform chemical for bio-based polymers from renewable resources. Chem. Eng. Technol. 2008, 31, 647–654. [Google Scholar] [CrossRef]
- Werpy, T.; Petersen, G. Top Value Added Chemicals from Biomass Volume I: Results of Screening for Potential Candidates from Sugars and Synthesis Gas; Pacific Northwest National Laboratory (PNNL): Richland, WA, USA; National Renewable Energy Laboratory (NREL): Golden, CO, USA, 2004. Available online: http://www.osti.gov/bridge (accessed on 12 October 2017).
- Cooper, J.S.; Vigon, B. Life Cycle Engineering Guidelines; Chapter 5: New Design; EPA/600/R-01/101; National Risk Management Research Laboratory, Office of Research and Development: Cincinnati, OH, USA, 2001; pp. 51–52.
- Wolf, O. (Ed.) Techno-Economic Feasibility of Large-Scale Production of Bio-Based Polymers in Europe; Technical Report EUR 22103 EN; Fraunhofer Institut: Munich, Germany, 2005. [Google Scholar]
- Papageorgiou, G.Z.; Bikiaris, D.N. Crystallization and melting behavior of three biodegradable poly (alkylene succinates). A comparative study. Polymer 2005, 46, 12081–12092. [Google Scholar] [CrossRef]
- Bikiaris, D.N.; Achilias, D.S. Synthesis of poly(alkylene succinate) biodegradable polyesters, Part II: Mathematical modelling of the polycondensation reaction. Polymer 2008, 49, 3677–3685. [Google Scholar] [CrossRef]
- Gigli, M.; Fabbri, M.; Lotti, N.; Gamberini, R.; Rimini, B.; Munari, A. Poly(butylene succinate)-based polyesters. Eur. Polym. J. 2016, 75, 431–460. [Google Scholar] [CrossRef]
- Wojtczak, M.; Dutkiewicz, S.; Galeski, A.; Gutowska, A. Classification of aliphatic-butylene terephthalate copolyesters in relation to aliphatic/aromatic ratio. Polymer 2017, 113, 119–134. [Google Scholar] [CrossRef]
- Witt, U.; Muller, R.-J.; Deckwer, W.-D. Biodegradation of polyester copolymers containing aromatic compounds. J. Macromol. Sci. Pure Appl. Chem. 1995, A32, 851–856. [Google Scholar] [CrossRef]
- Wu, L.; Mincheva, R.; Xu, Y.; Raquez, J.M.; Dubois, P. High Molecular Weight Poly(butylene succinate-co-butylene furandicarboxylate) Copolyesters: From Catalyzed Polycondensation Reaction to Thermomechanical Properties. Biomacromolecules 2012, 13, 2973–2981. [Google Scholar] [CrossRef] [PubMed]
- Jacquel, N.; Saint-Loup, R.; Pascault, J.P.; Rousseau, A.; Fenouillot, F. Synthesis and properties of poly(butylene succinate): Efficiency of different transesterification catalysts. Polymer 2015, 59, 234–242. [Google Scholar] [CrossRef]
- Morales-Huerta, J.C.; Ciulik, C.B.A.; Martínez de Ilarduya, A.; Muñoz-Guerra, S. Fully bio-based aromatic-aliphatic copolyesters: Poly(butylene furandicarboxylate-co-succinate)s obtained by ring opening polymerization. Polym. Chem. 2017, 8, 748–760. [Google Scholar] [CrossRef]
- Terzopoulou, Z.; Tsanaktsis, V.; Bikiaris, D.N.; Exarhopoulos, S.; Papageorgiou, D.G.; Papageorgiou, G.Z. Biobasedpoly(ethylene furanoate-co-ethylene succinate) copolyesters: Solid state structure, melting point depression and biodegradability. RSC Adv. 2016, 6, 84003–84015. [Google Scholar] [CrossRef]
- Gandini, A. Furans as offspring of sugars and polysaccharides and progenitors of a family of remarkable polymers: A review of recent progress. Polym. Chem. 2010, 1, 245–251. [Google Scholar] [CrossRef]
- Sousa, A.F.; Vilela, C.; Fonseca, A.C.; Matos, M.; Freire, C.S.; Gruter, G.J.M.; Coelho, J.F.J.; Silvestre, A.J.D. Biobased polyesters and other polymers from 2,5-furandicarboxylic acid: A tribute to furan excellency. Polym. Chem. 2015, 6, 5961–5983. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Papageorgiou, D.G.; Terzopoulou, Z.; Bikiaris, D.N. Production of bio-based 2,5-furan dicarboxylate polyesters: Recent progress and critical aspects in their synthesis and thermal properties. Eur. Polym. J. 2016, 83, 202–229. [Google Scholar] [CrossRef]
- Sanderson, K. US biofuels: A field in ferment. Nature 2006, 444, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Rocha, L.; Marston, A.; Potterat, O.; Kaplan, M.A.C.; Hostettmann, K. More phloroglucinols from Hypericumbrasiliense. Phytochemistry 1996, 42, 185–188. [Google Scholar] [CrossRef]
- Hansen, C.A.; Frost, J.W. Deoxygenation of polyhydroxybenzenes: An alternative strategy for the benzene-free synthesis of aromatic chemicals. J. Am. Chem. Soc. 2002, 124, 5926–5927. [Google Scholar] [CrossRef] [PubMed]
- Durairaj, R.B. Resorcinol: Chemistry, Technology and Applications; Springer: Berlin, Germany, 2005. [Google Scholar]
- Al-Muhtaseb, S.A.; Ritter, J.A. Preparation and properties of resorcinol-formaldehyde organic and carbon gels. Adv. Mat. 2003, 15, 101–114. [Google Scholar] [CrossRef]
- Vijayakumar, C.T.; Sivasamy, P.; Rajkumar, T. Synthesis and characterization of 1,3-bis (2-hydroxyethoxy) benzene based saturated and unsaturated polyesters. Eur. Polym. J. 2007, 43, 3028–3035. [Google Scholar] [CrossRef]
- Gioia, C.; Banella, M.B.; Vannini, M.; Celli, A.; Colonna, M.; Caretti, D. Resorcinol: A potentially bio-based building block for the preparation of sustainable polyesters. Eur. Polym. J. 2015, 73, 38–49. [Google Scholar] [CrossRef]
- Barbee, R.B.; Davis, B. Copolyesters from Ether-Containing Glycols and Acids. U.S. Patent 4518763, 21 May 1985. [Google Scholar]
- Takahashi, K.; Nakamachi, K.; Niimi, H.; Hiraoka, S.; Sakai, M.; Tsuboi, H. Polyester Pellets and Proccess for Preparing the Same. Patent Application No. EP 0939095A2, 1 September 1999. [Google Scholar]
- Brunelle, D.J.; Bradt, J.E.; Serth-Guzzo, J.; Takekoshi, T.; Evans, T.; Pearce, E.J.; Wilson, P. Semicrystalline polymers via ring-opening polymerization: Preparation and polymerization of alkylene phthalate cyclic oligomers. Macromolecules 1988, 31, 4782–4790. [Google Scholar] [CrossRef] [PubMed]
- Morales-Huerta, J.C.; Martínez de Ilarduya, A.; Muñoz-Guerra, S. A green strategy for the synthesis of poly(ethylene succinate) and its copolyesters via enzymatic ring opening polymerization. Eur. Polym. J. 2017, 95, 514–519. [Google Scholar] [CrossRef]
- Morales-Huerta, J.C.; Martínez de Ilarduya, A.; Muñoz-Guerra, S. Poly(alkylene 2,5-furandicarboxylate)s (PEF and PBF) by ring opening polymerization. Polymer 2016, 87, 148–158. [Google Scholar] [CrossRef]
- Devaux, J.; Godard, P.; Mercier, J.P. Bisphenol-A polycarbonate–poly(butylene terephthalate) transesterification. I. Theoretical study of the structure and of the degree of randomness in four-component copolycondensates. J. Polym. Sci. Polym. Phys. 1982, 20, 1875–1880. [Google Scholar] [CrossRef]
- Hodge, P. Entropically driven Ring-Opening Polymerization of strainless organic macrocycles. Chem. Rev. 2014, 114, 2278–2312. [Google Scholar] [CrossRef] [PubMed]
- Fox, T.G. Influence of diluent and of copolymer composition on the glass temperature of a polymer system. Bull. Am. Phys. Soc. 1956, 1, 123–135. [Google Scholar]
- Gordon, M.; Taylor, J.S. Ideal copolymers and the 2nd-ordertransitions of synthetic rubbers. 1. Non-crystalline copolymers. J. Appl. Chem. 1952, 2, 493–500. [Google Scholar] [CrossRef]
- Bikiaris, D.N.; Papageorgiou, G.Z.; Achilias, D.S. Synthesis and comparative biodegradability studies of three poly(alkylene succinate)s. Polym. Degrad. Stab. 2005, 91, 31–43. [Google Scholar] [CrossRef]
- Terzopoulou, Z.; Karakatsianopoulou, E.; Kasmi, N.; Tsanaktsis, V.; Nikolaidis, N.; Kostoglou, M.; Papageorgiou, G.Z.; Lambropoulou, D.A.; Bikiaris, D.N. Effect of catalyst type on molecular weight increase and coloration of poly(ethylene furanoate) biobased polyester during melt polycondensation. Polym. Chem. 2017, 8, 6895–6908. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fraction | Precursors | Yield (%) a | Composition (% wt) b | Thermal Properties | ||
|---|---|---|---|---|---|---|
| Tm (°C) c | °Td,5% (°C) d | maxTd (°C) d | ||||
| c(BS)n | DMS + BD | 70 | 50/40/10 | 105 | 266 | 340/400 |
| c(ES)n | DMS + EG | 70 | 0/45/55 | 100 | 200 | 300/400 |
| c(RF)n | FDCA-Cl2 + HER | 60 | 28/57/15 | 158/166/185 | 263 | 415 |
| Opolyester | Yield (%) | GPC a | Composition b x(BS or ES)/y(RF) | Chain Microstructure c | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mw (g·mol−1) | Đ | Feed | Copolyester | n(B/E)S | n(B/E)F | nRS | nRF | B | ||
| PBS | 90 | 50,000 | 1.9 | 100/- | 100/- | - | - | - | - | - |
| coPBS90RF10 | 96 | 48,000 | 2.0 | 90/10 | 86/14 | 17.3 | 1.1 | 2.8 | 1.5 | 0.72 |
| coPBS80RF20 | 95 | 46,000 | 1.8 | 80/20 | 78/22 | 7.2 | 1.2 | 3.8 | 1.4 | 0.89 |
| coPBS70RF30 | 90 | 42,500 | 1.6 | 70/30 | 64/36 | 4.2 | 1.3 | 2.8 | 1.6 | 0.90 |
| coPBS50RF50 | 80 | 37,200 | 2.0 | 50/50 | 49/51 | 2.3 | 1.9 | 2.1 | 1.9 | 0.96 |
| coPBS40RF60 | 80 | 36,000 | 1.7 | 40/60 | 37/63 | 1.8 | 2.2 | 1.7 | 2.5 | 0.96 |
| coPBS20RF80 | 85 | 32,200 | 1.8 | 20/80 | 21/79 | 1.5 | 3.2 | 1.3 | 3.9 | 0.92 |
| PES | 90 | 45,000 | 1.7 | 100/- | 100/- | - | - | - | - | - |
| coPES90RF10 | 95 | 42,500 | 1.9 | 90/10 | 86/14 | 4.6 | 1.3 | 7.3 | 1.2 | 1.07 |
| coPES80RF20 | 92 | 41,000 | 1.8 | 80/20 | 77/23 | 3.3 | 1.4 | 4.9 | 1.3 | 1.17 |
| coPES70RF30 | 80 | 39,200 | 1.5 | 70/30 | 76/24 | 3.0 | 1.5 | 4.2 | 1.3 | 1.08 |
| coPES50RF50 | 85 | 34,500 | 1.4 | 50/50 | 49/51 | 1.5 | 3.2 | 2.3 | 1.8 | 1.23 |
| coPES40RF60 | 90 | 34,000 | 1.6 | 40/60 | 36/64 | 1.7 | 2.5 | 1.6 | 2.7 | 0.98 |
| coPES20RF80 | 91 | 31,000 | 1.8 | 20/80 | 19/81 | 1.3 | 4.1 | 1.2 | 5.6 | 0.95 |
| PRF | 80 | 28,000 | 1.9 | -/100 | -/100 | - | - | - | - | - |
| Copolyester | TGA a | DSC b | ||||
|---|---|---|---|---|---|---|
| °Td (°C) | maxTd (°C) | Rw (%) | Tg (°C) | Tm (°C) | ∆H (J·mol−1) | |
| PBS | 350 | 399 | 3 | −30 | 114 | 82 |
| coPBS90RF10 | 336 | 391 | 8 | −20 | 100 | 74 |
| coPBS80RF20 | 344 | 398 | 6 | −13 | - | - |
| coPBS70RF30 | 326 | 391 | 9 | −2 | - | - |
| coPBS50RF50 | 336 | 399 | 10 | 15 | - | - |
| coPBS40RF60 | 331 | 391 | 11 | 18 | ||
| coPBS20RF80 | 345 | 400 | 9 | 40 | - | - |
| PES | 310 | 385 | 7 | −13 | 100 | 55 |
| coPES90RF10 | 323 | 389 | 7 | −7 | - | - |
| coPES80RF20 | 335 | 386 | 9 | 6 | - | - |
| coPES70RF30 | 325 | 374 | 9 | 8 | - | - |
| coPES50RF50 | 342 | 390 | 9 | 26 | - | - |
| coPES40RF60 | 340 | 396 | 5 | 36 | - | - |
| coPES20RF80 | 347 | 411 | 11 | 43 | - | - |
| PRF | 321 | 401 | 10 | 58 | - | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Huerta, J.C.; Martínez de Ilarduya, A.; Muñoz-Guerra, S. Modulating the Tg of Poly(alkylene succinate)s by Inserting Bio-Based Aromatic Units via Ring-Opening Copolymerization. Polymers 2017, 9, 701. https://doi.org/10.3390/polym9120701
Morales-Huerta JC, Martínez de Ilarduya A, Muñoz-Guerra S. Modulating the Tg of Poly(alkylene succinate)s by Inserting Bio-Based Aromatic Units via Ring-Opening Copolymerization. Polymers. 2017; 9(12):701. https://doi.org/10.3390/polym9120701
Chicago/Turabian StyleMorales-Huerta, Juan Carlos, Antxon Martínez de Ilarduya, and Sebastián Muñoz-Guerra. 2017. "Modulating the Tg of Poly(alkylene succinate)s by Inserting Bio-Based Aromatic Units via Ring-Opening Copolymerization" Polymers 9, no. 12: 701. https://doi.org/10.3390/polym9120701