Biodegradable Core–Multishell Nanocarriers: Influence of Inner Shell Structure on the Encapsulation Behavior of Dexamethasone and Tacrolimus


Abstract
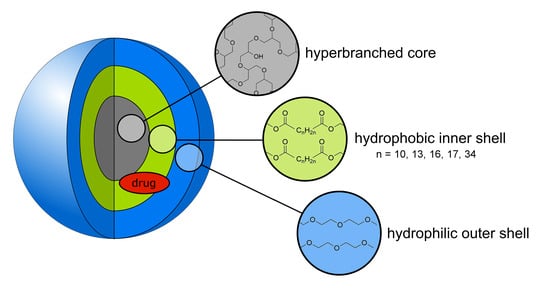
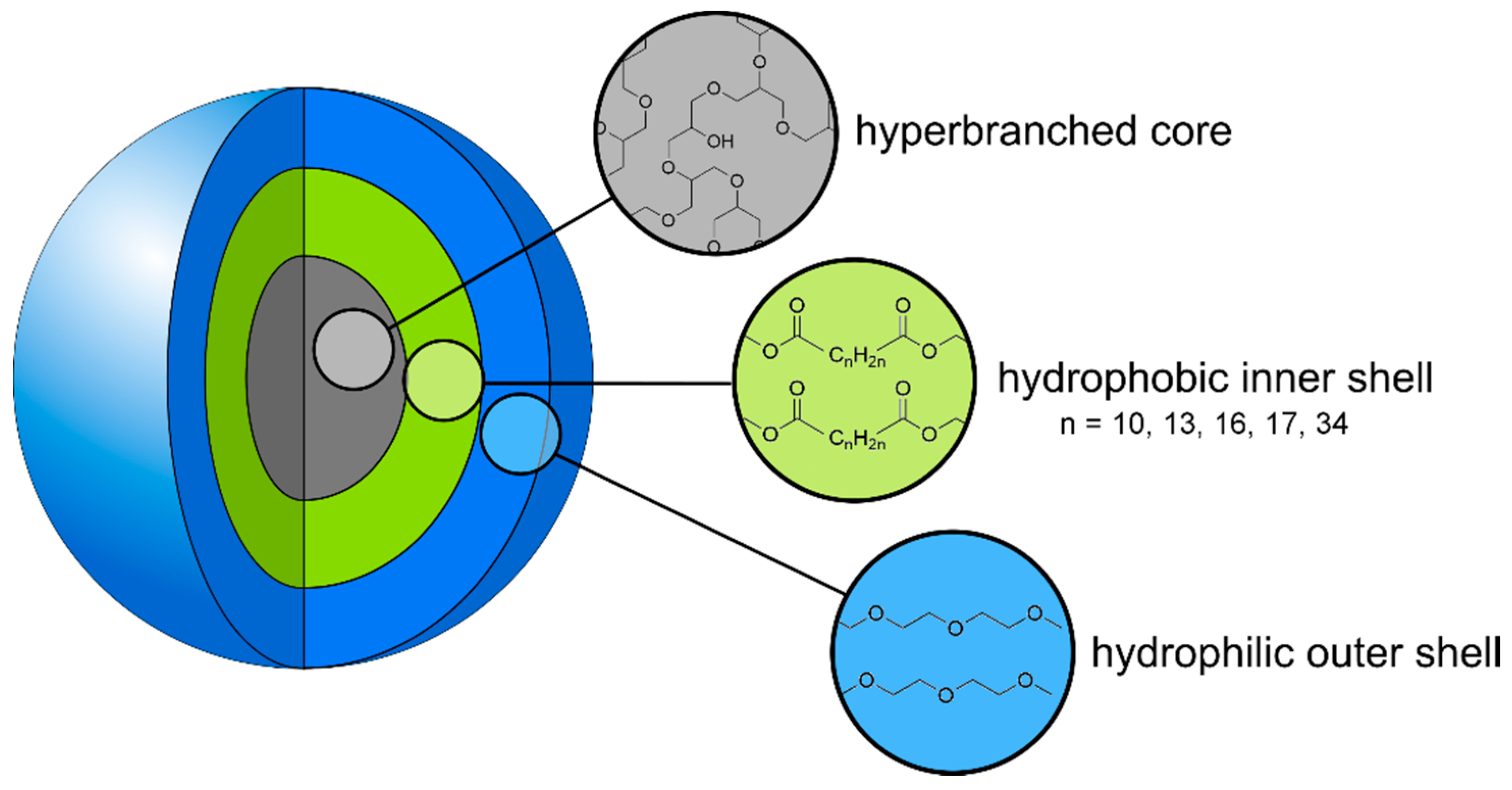
:
1. Introduction
2. Materials and Methods
2.1. Nuclear Magnetic Resonance (NMR)
2.2. Infrared (IR) Spectroscopy
2.3. Dynamic Light Scattering (DLS)
2.4. Gel Permeation Chromatography
2.5. Film Encapsulation Method
2.6. HPLC Analysis
2.7. Determination of Enzymatic Activity
2.8. Enzymatic Degradation
2.9. Release by Enzymatic Degradation
2.10. Synthesis
1,19-Nonadecandioic Acid
2.11. Double-Shell Building Blocks
2.11.1. C12-mPEG350
2.11.2. C15-mPEG350
2.11.3. C18-mPEG350
2.11.4. C19-mPEG350
2.11.5. C18b-mPEG750
2.12. Nanocarriers
2.12.1. CMS-E12
2.12.2. CMS-E15
2.12.3. CMS-E18
2.12.4. CMS-E19
2.12.5. CMS-E18b
2.12.6. CMS-A18
3. Results
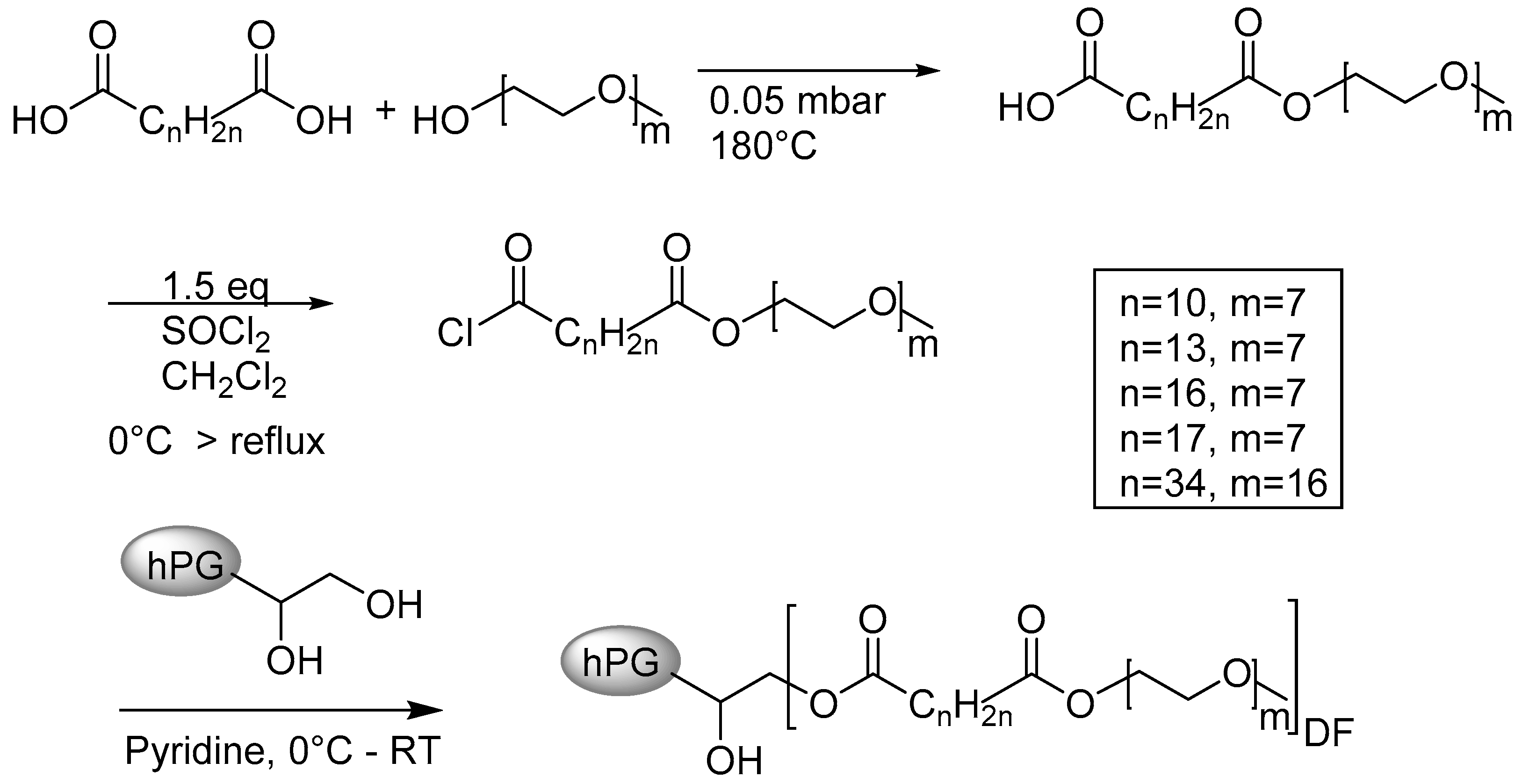
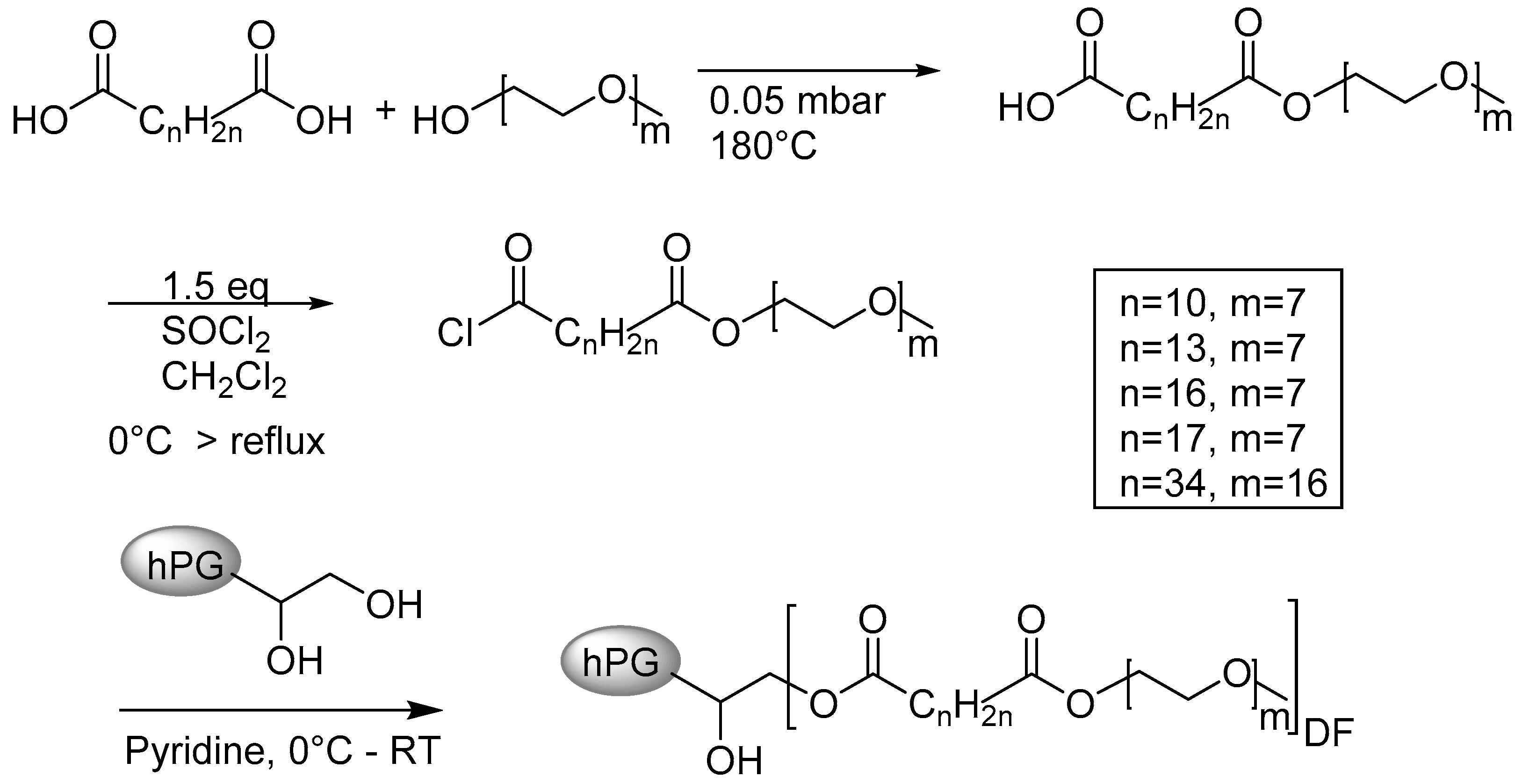
3.1. Synthesis of the Ester-Based Core–Multishell Nanocarriers
3.1.1. Synthesis of the Shell Molecule
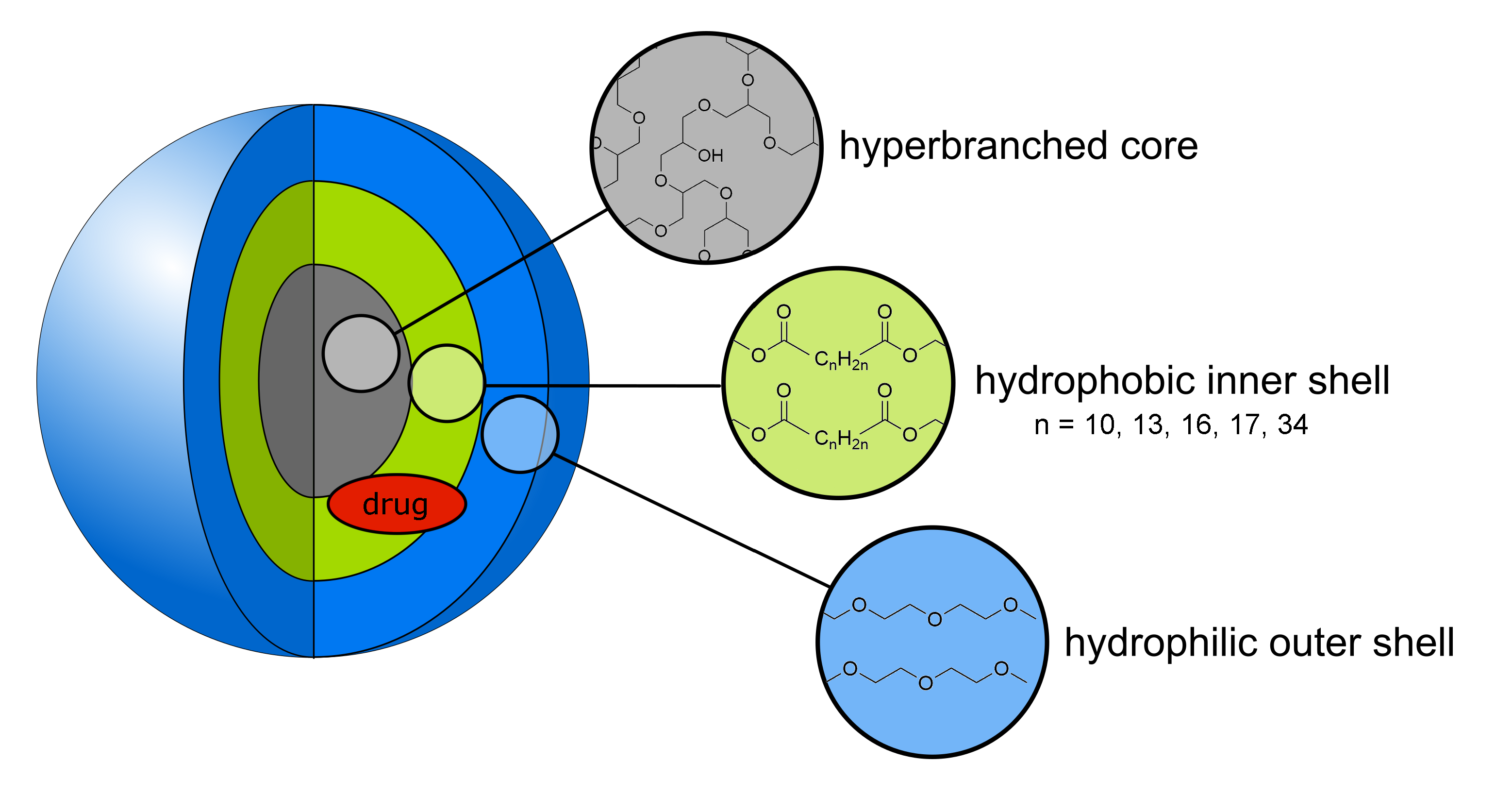
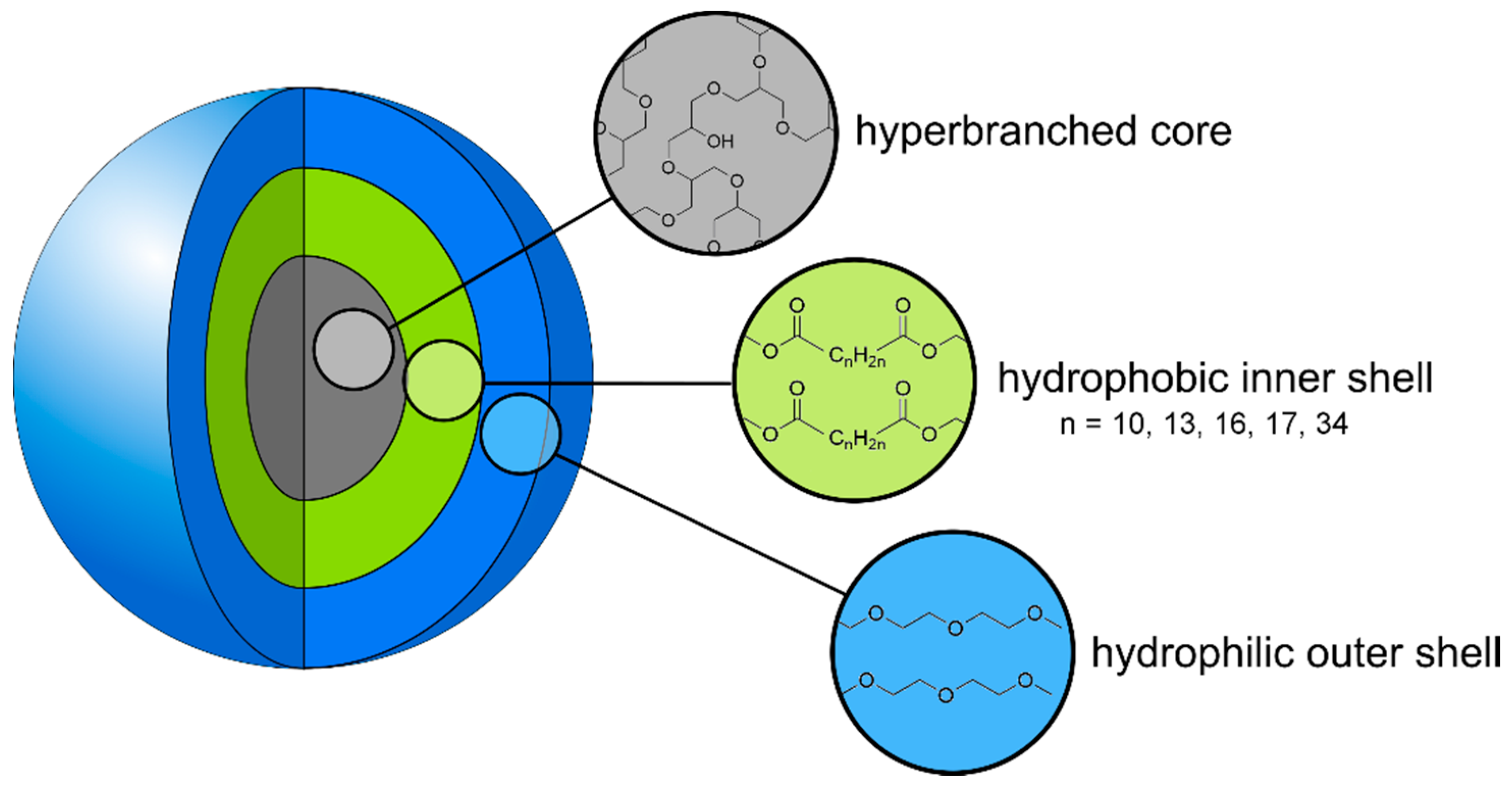
3.1.2. Synthesis of the CMS Architectures
3.2. Degree of Functionalisation
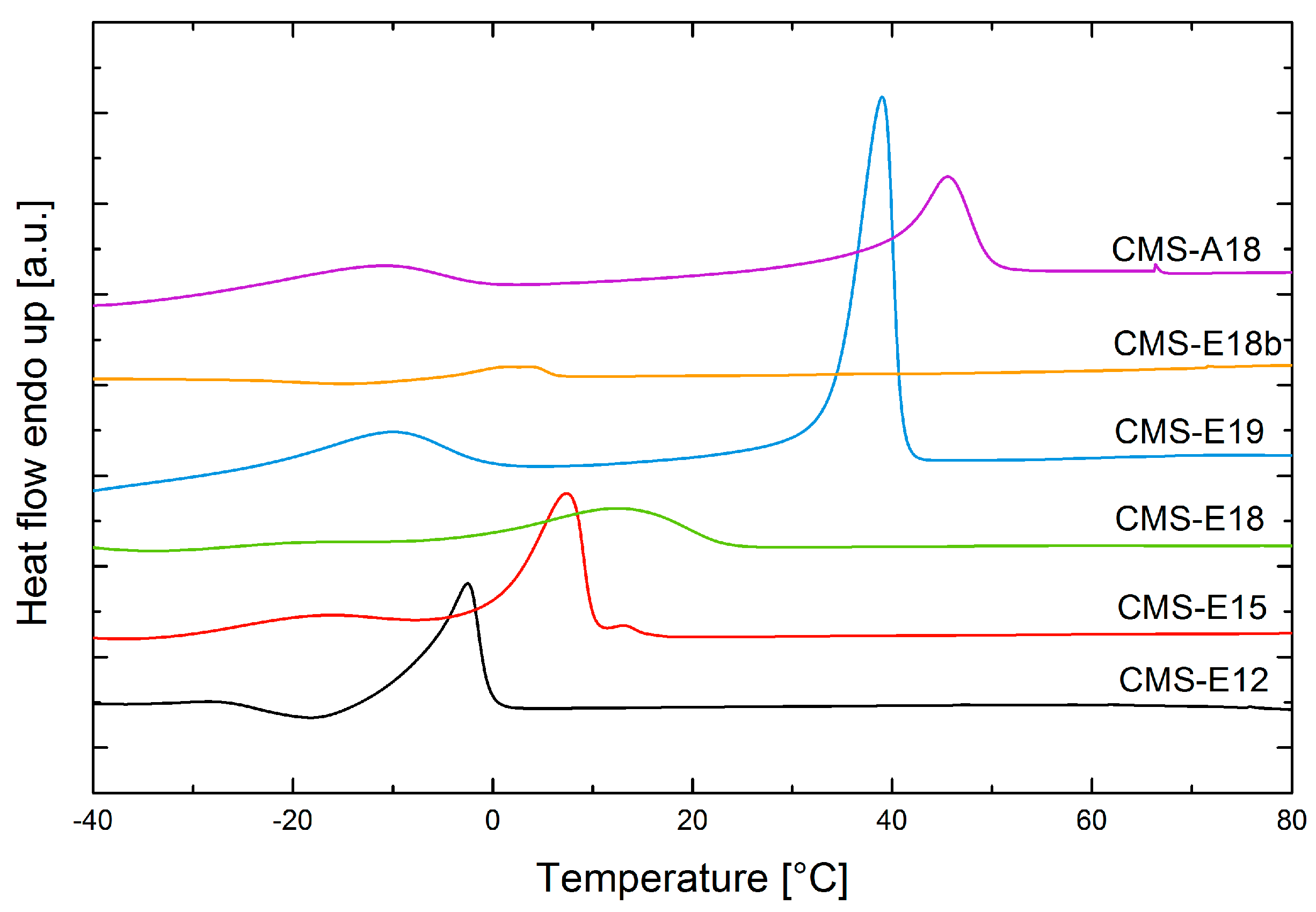
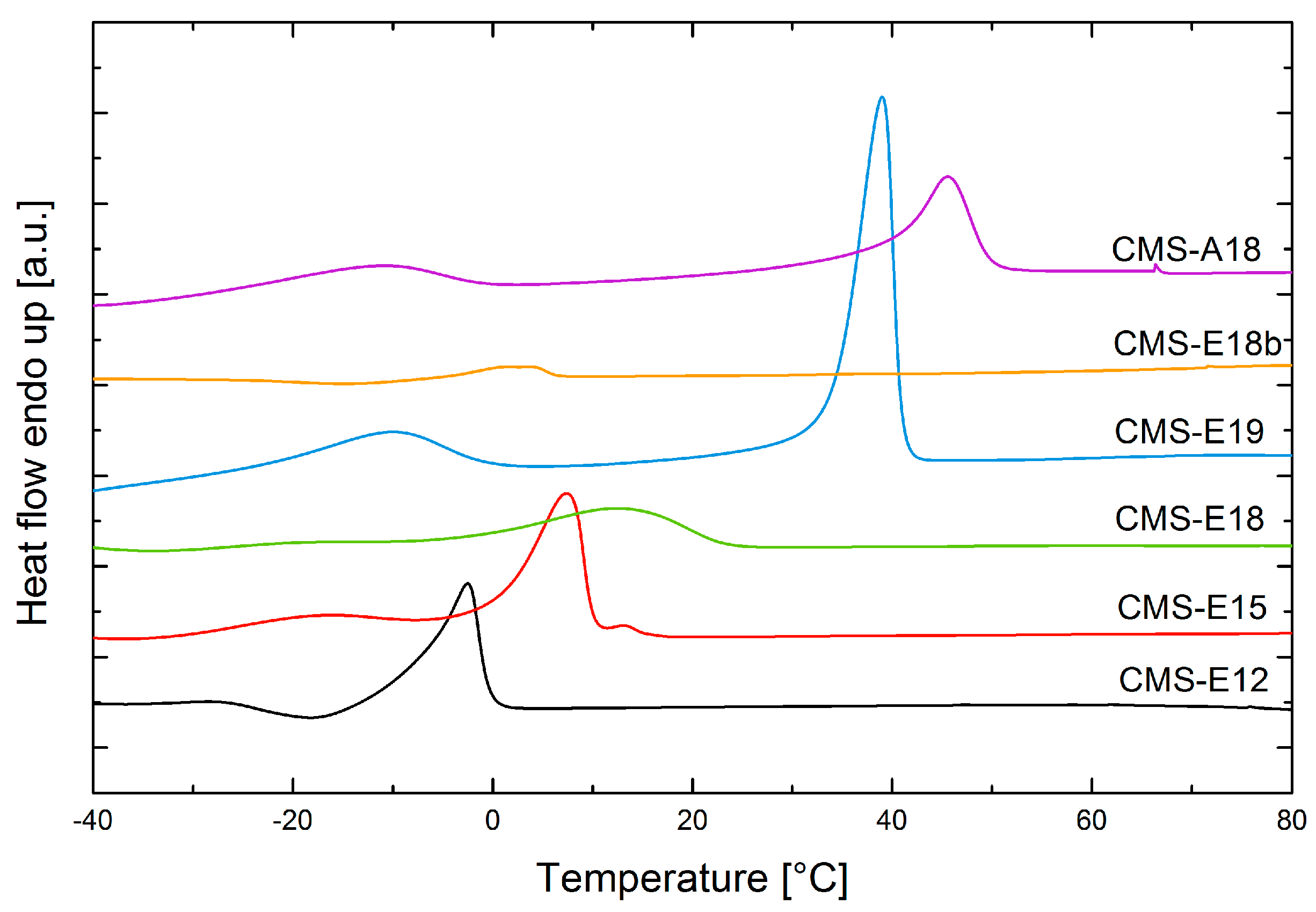
3.3. DSC Measurements
3.4. DLS Analysis of Loaded and Unloaded CMS Nanocarriers
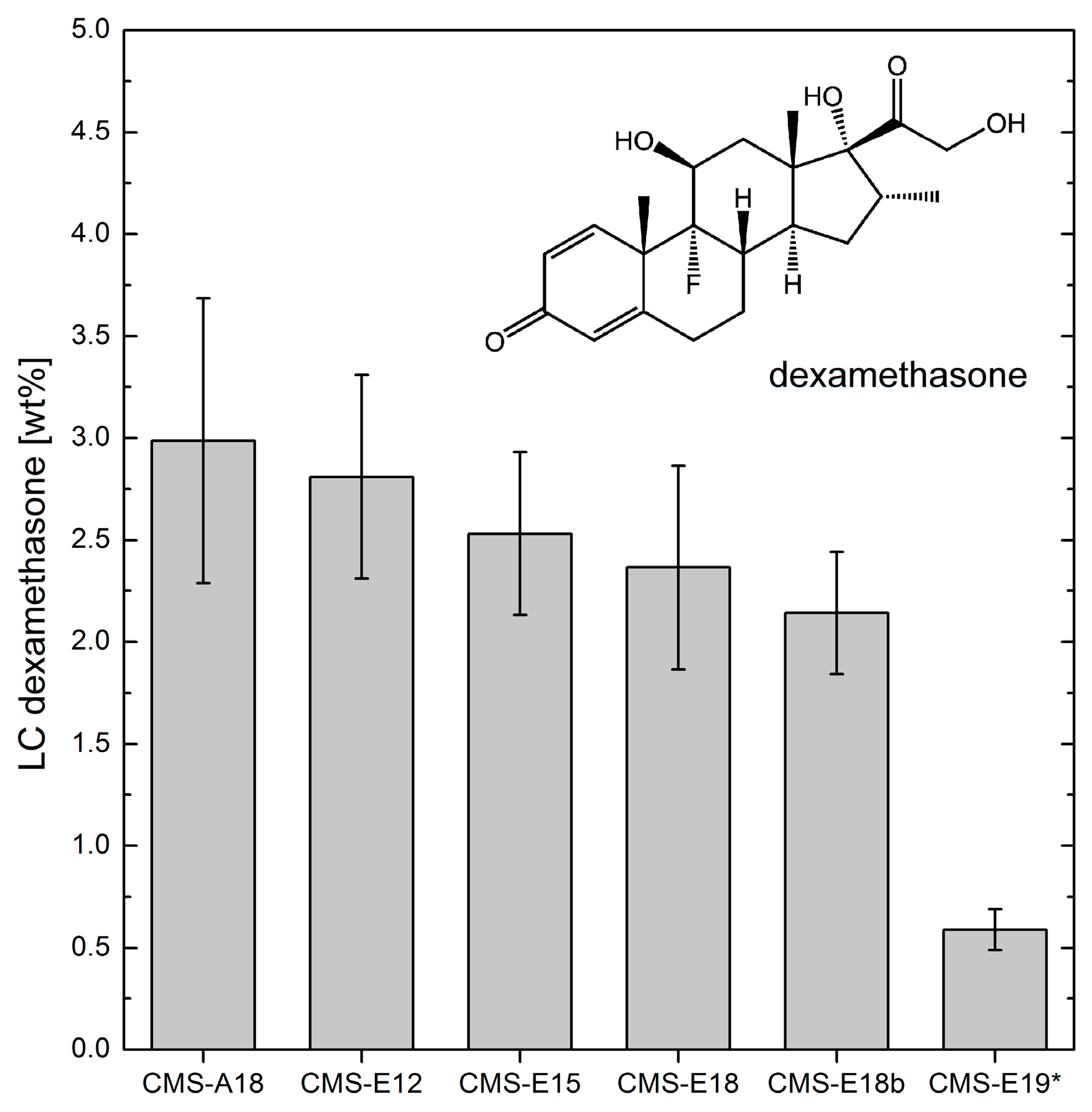
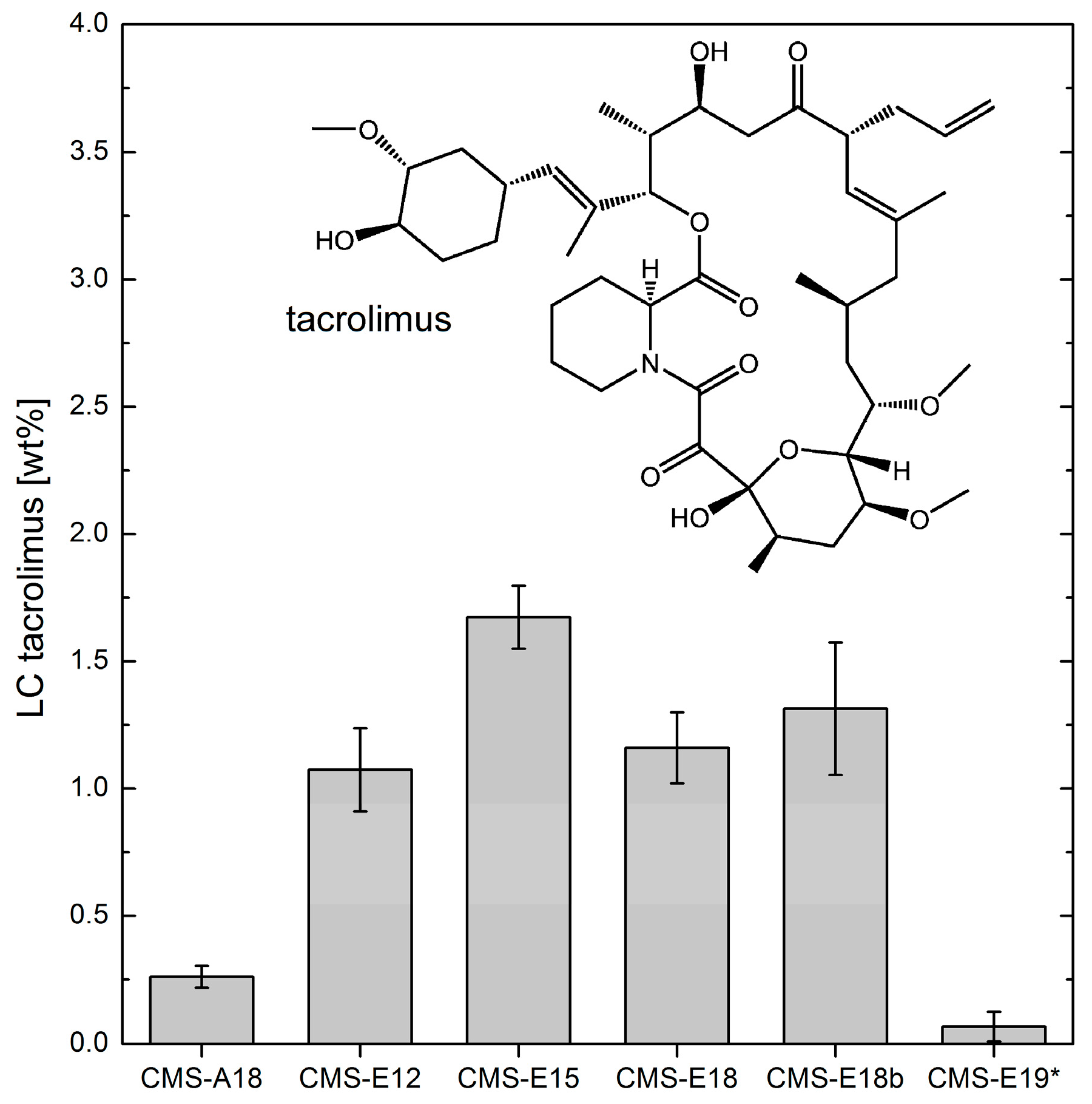
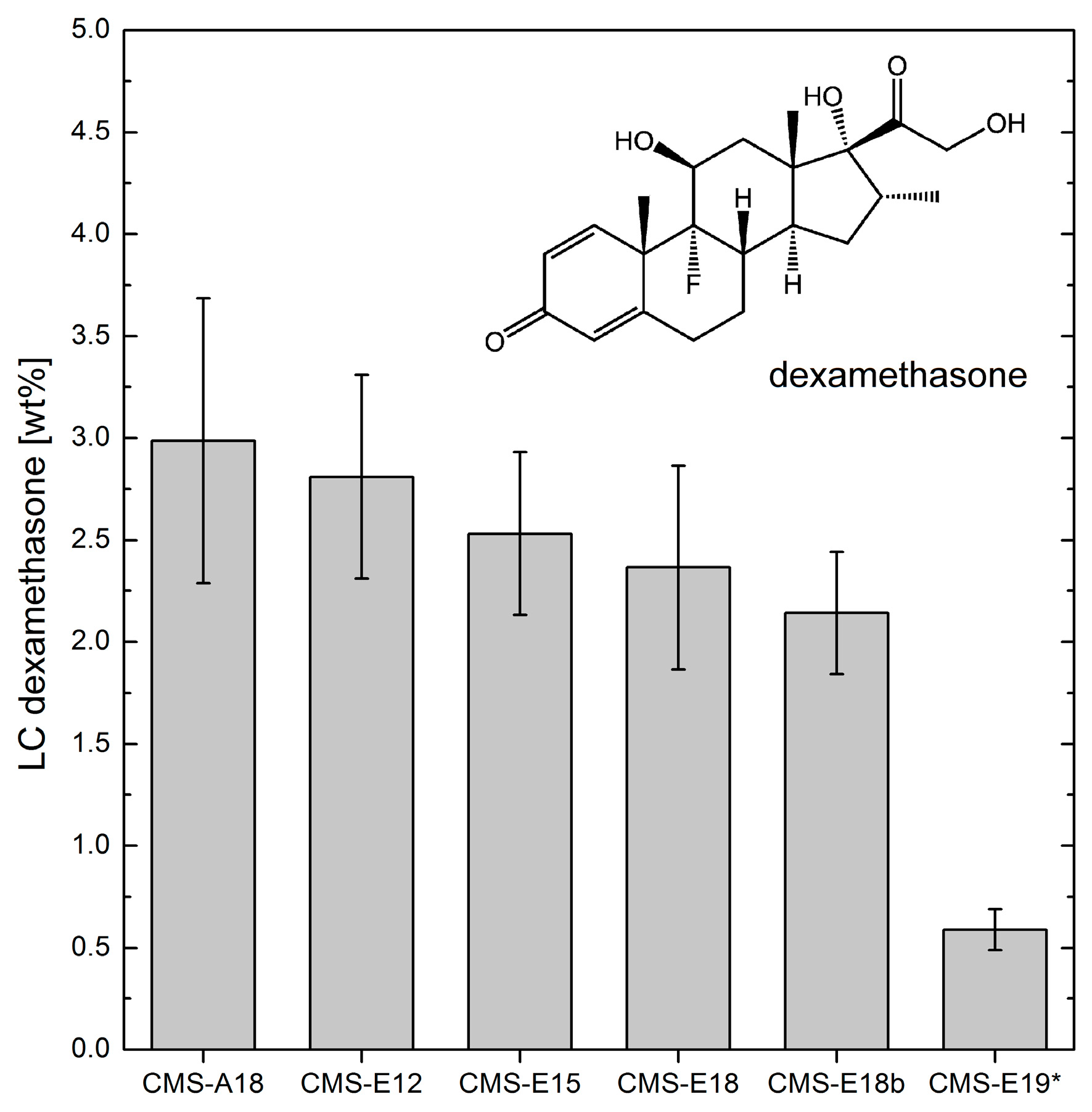
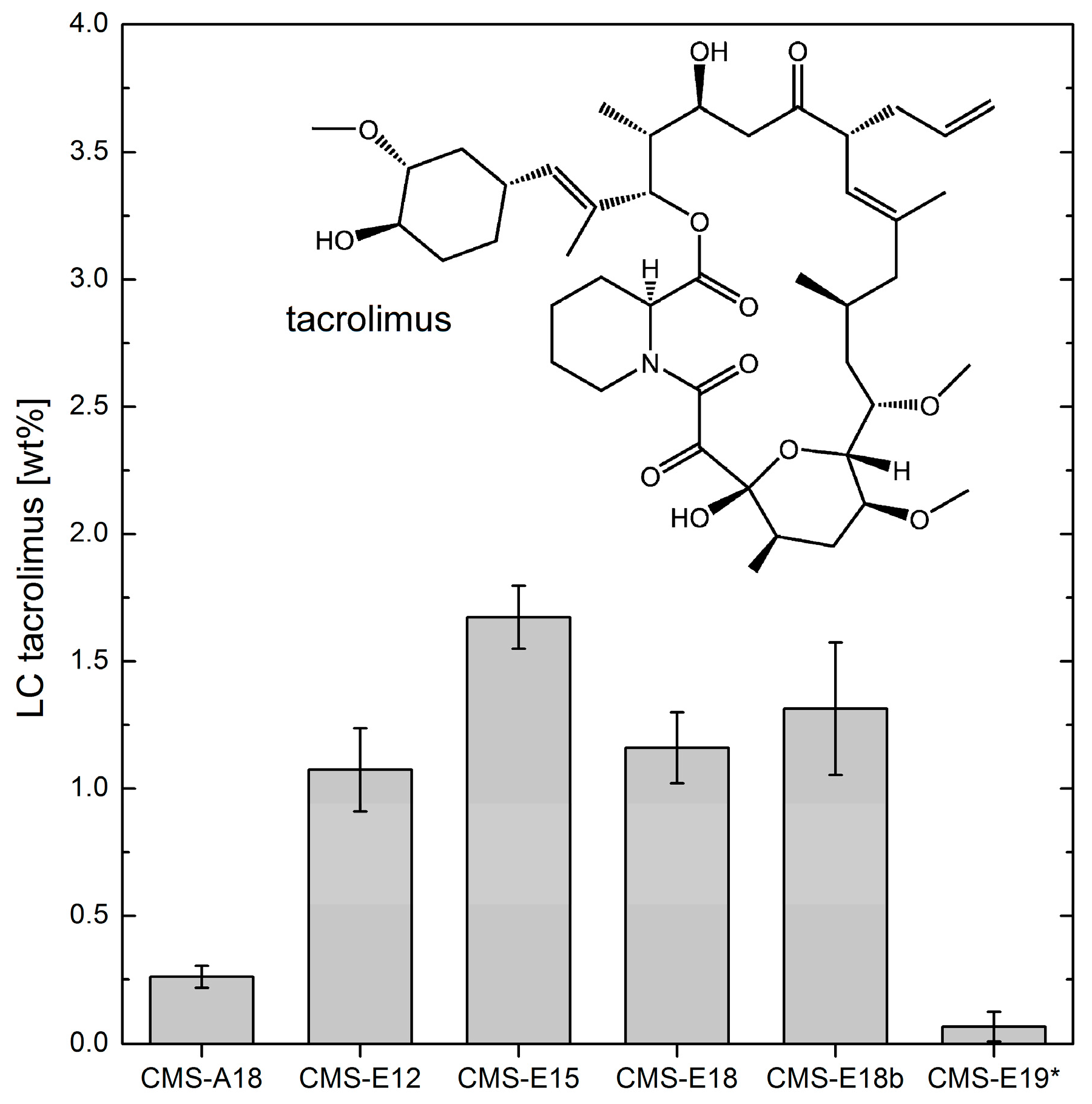
3.5. Loading Capacities of Dexamethasone and Tacrolimus
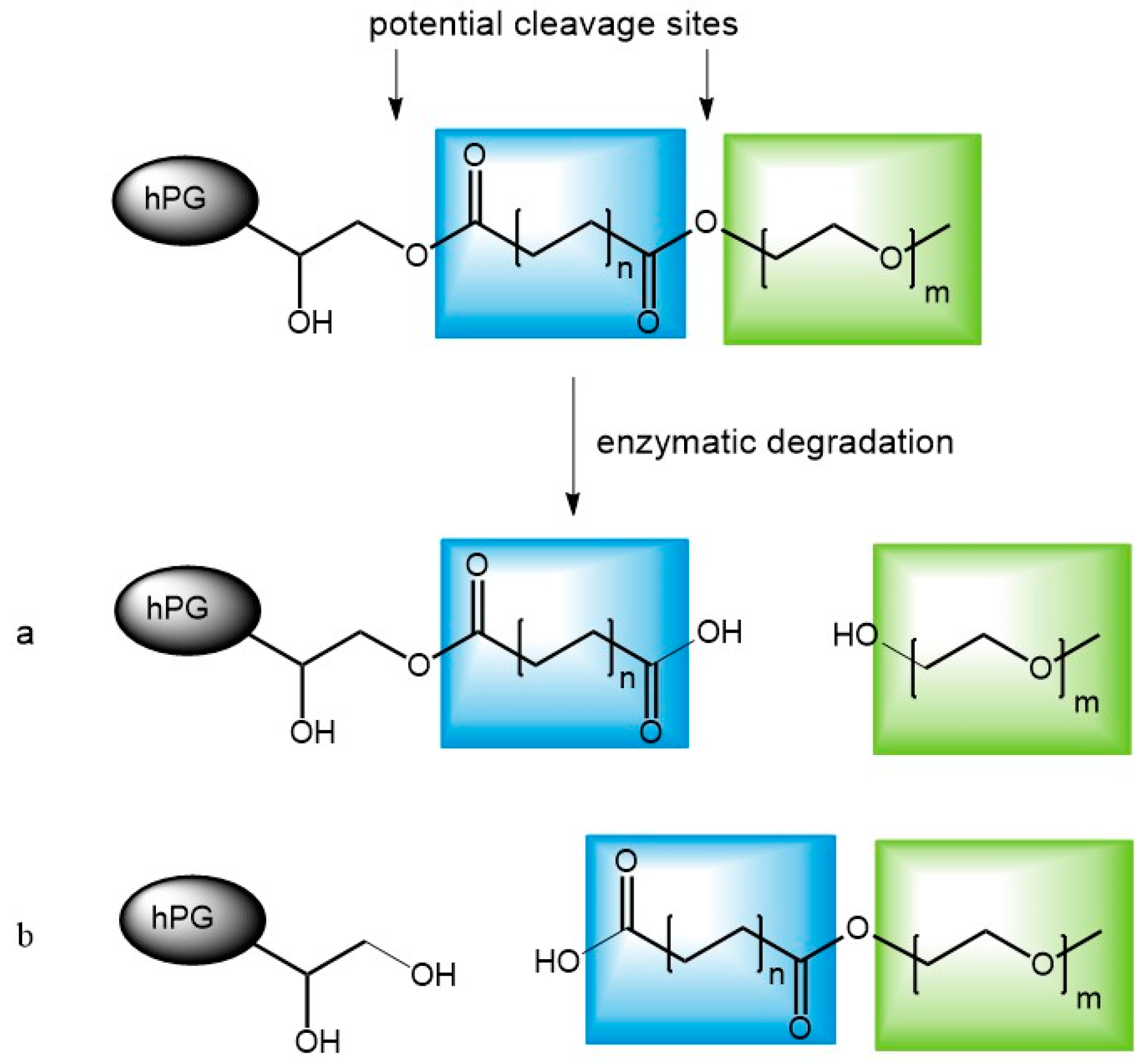
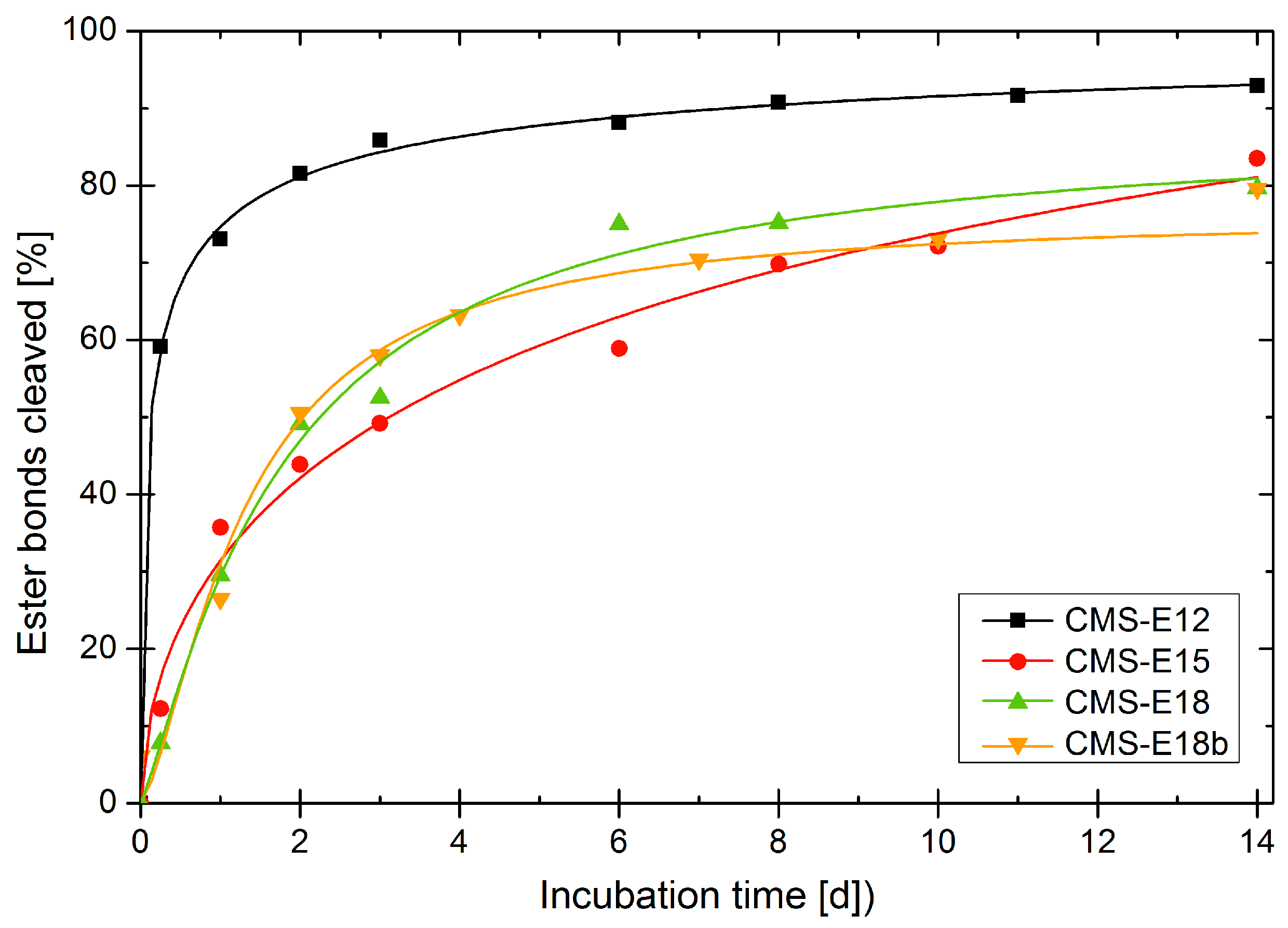
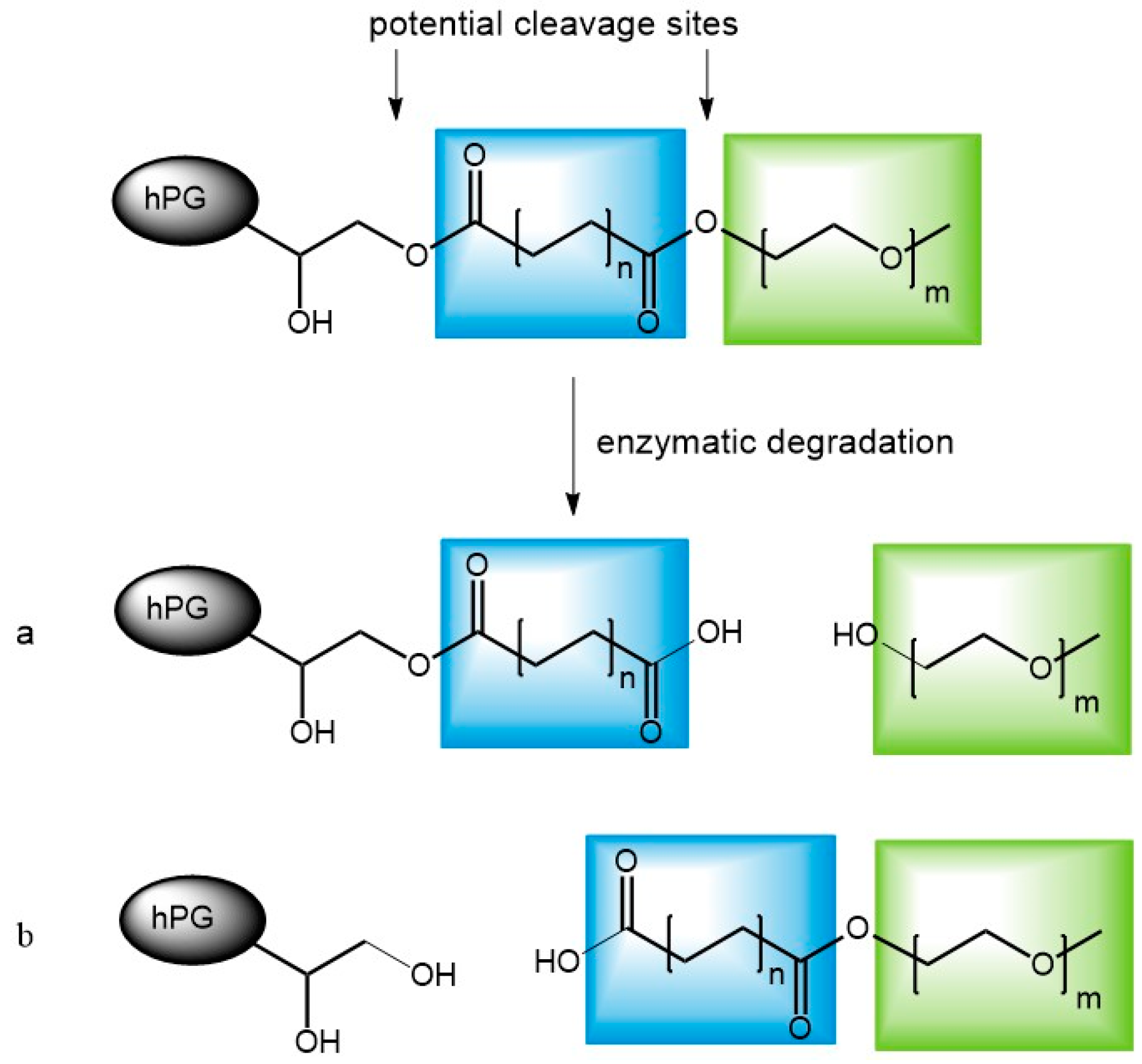
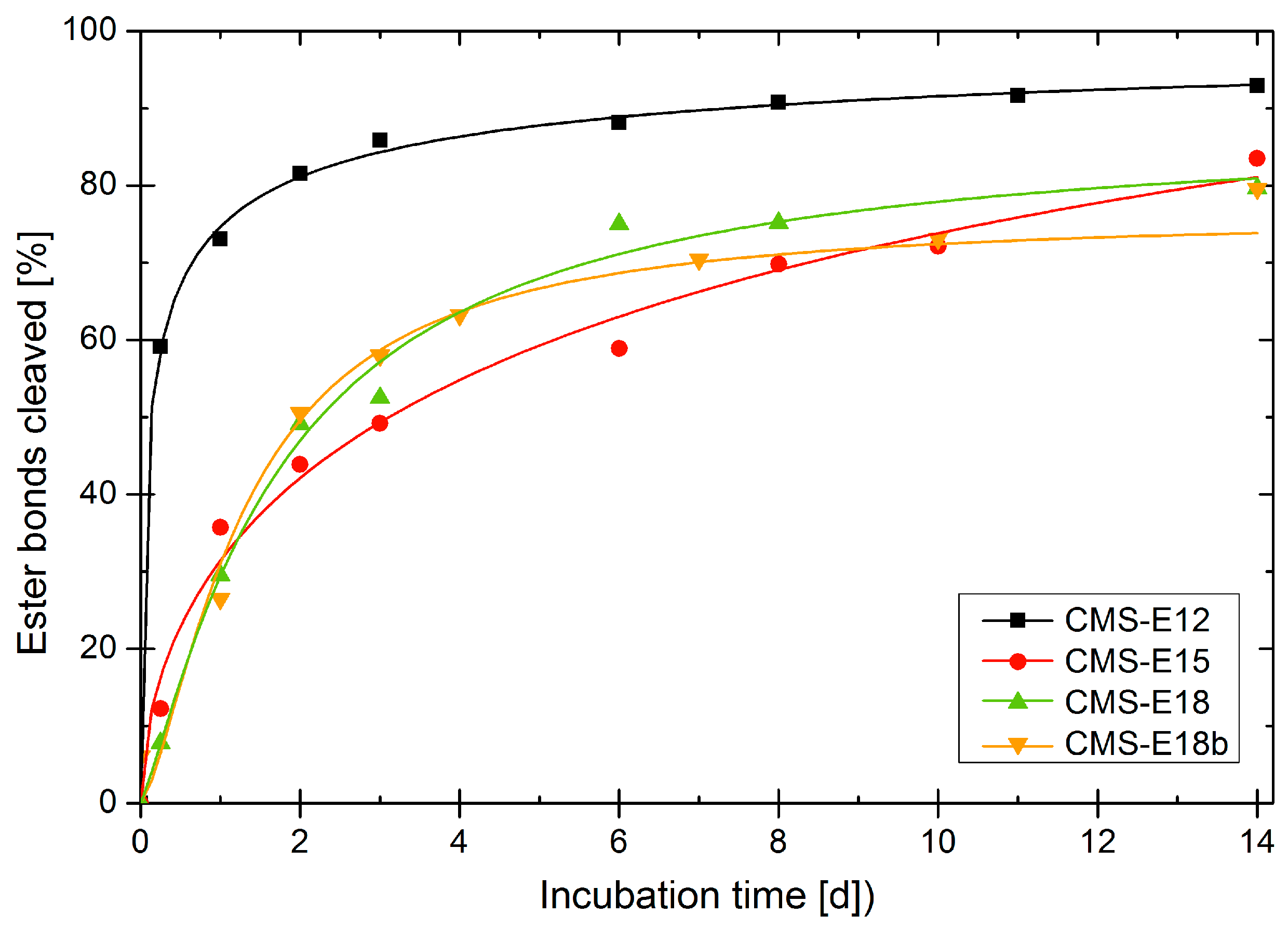
3.6. Enzymatic Degradation of Unloaded Carriers
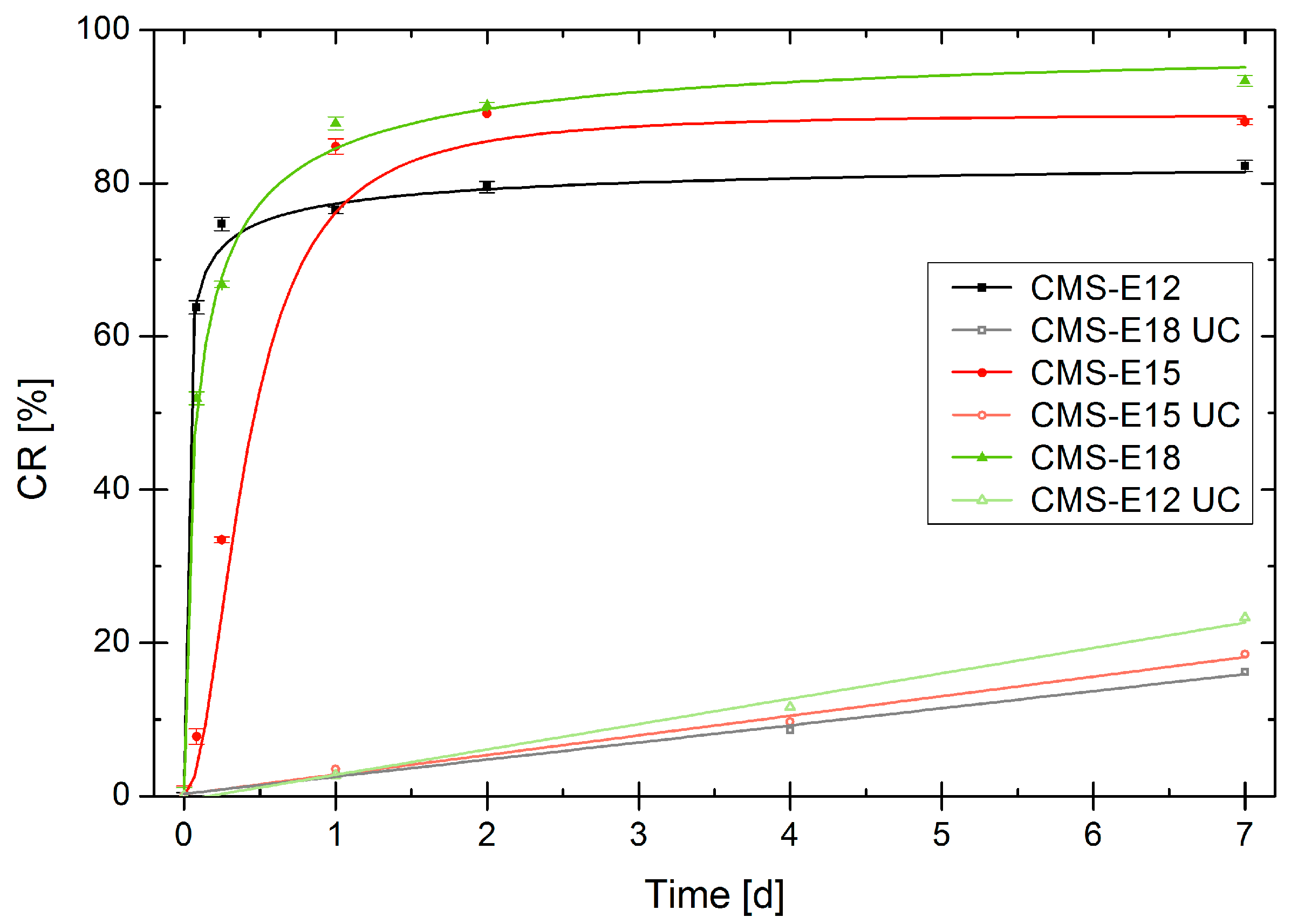
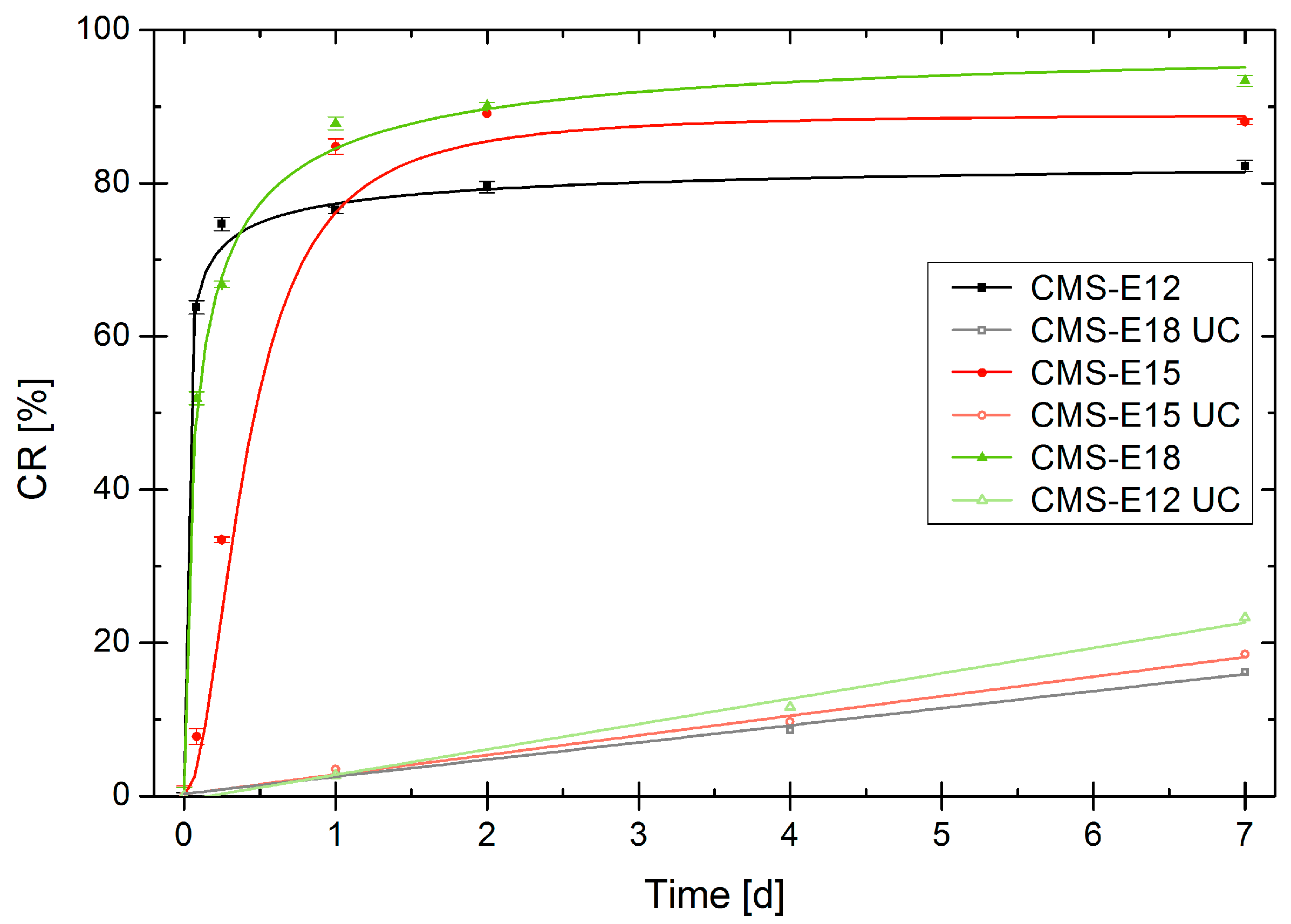
3.7. Release Mediated by Enzymatic Cleavage
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CMC | Critical micellar concentration |
| CMS | Core–multishell |
| CR | Cumulative release |
| DDS | Drug delivery system |
| DF | Degree of functionalization |
| hPG | Hyperbranched polyglycerol |
| hPG–NH2 | Hyperbranched polyglycerol amine |
| ITCC | Indotriscarbocyanine |
| PEI | Poly(ethyleneimine) |
References
- Williams, A.C.; Barry, B.W. Penetration enhancers. Adv. Drug Deliv. Rev. 2012, 64, 128–137. [Google Scholar] [CrossRef]
- Papakostas, D.; Rancan, F.; Sterry, W.; Blume-Peytavi, U.; Vogt, A. Nanoparticles in dermatology. Arch. Dermatol. Res. 2011, 303, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Bolzinger, M.-A.; Briançon, S.; Pelletier, J.; Chevalier, Y. Penetration of drugs through skin, a complex rate-controlling membrane. Curr. Opin. Colliod Interface Sci. 2012. [Google Scholar] [CrossRef]
- Alves, M.P.; Scarrone, A.L.; Santos, M.; Pohlmann, A.R.; Guterres, S.S. Human skin penetration and distribution of nimesulide from hydrophilic gels containing nanocarriers. Int. J. Pharm. 2007, 341, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Román, R.; Naik, A.; Kalia, Y.N.; Guy, R.H.; Fessi, H. Enhancement of Topical Delivery from Biodegradable Nanoparticles. Pharm. Res. 2004, 21, 1818–1825. [Google Scholar] [CrossRef] [PubMed]
- Rancan, F.; Papakostas, D.; Hadam, S.; Hackbarth, S.; Delair, T.; Primard, C.; Verrier, B.; Sterry, W.; Blume-Peytavi, U.; Vogt, A. Investigation of polylactic acid (PLA) nanoparticles as drug delivery systems for local dermatotherapy. Pharm. Res. 2009, 26, 2027–2036. [Google Scholar] [CrossRef] [PubMed]
- Patzelt, A.; Richter, H.; Knorr, F.; Schäfer, U.; Lehr, C.-M.; Dähne, L.; Sterry, W.; Lademann, J. Selective follicular targeting by modification of the particle sizes. J. Control. Release 2011, 150, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.; Seok Kang, H.; Park, W.-S.; Han, S.-H.; Kim, J.; Chang, I.-S. Transdermal delivery of mixnoxidil with block copolymer nanoparticles. J. Control. Release 2004, 97, 477–484. [Google Scholar] [CrossRef]
- Newkome, G.R.; Moorefield, C.N.; Baker, G.R.; Saunders, M.J.; Grossman, S.H. Unimolecular Micelles. Angew. Chem. Int. Ed. Engl. 1991, 30, 1178–1180. [Google Scholar] [CrossRef]
- Jansen, J.F.; de Brabander-van den Berg, E.M.; Meijer, E.W. Encapsulation of guest molecules into a dendritic box. Science 1994, 266, 1226–1229. [Google Scholar] [CrossRef] [PubMed]
- Kurniasih, I.N.; Keilitz, J.; Haag, R. Dendritic nanocarriers based on hyperbranched polymers. Chem. Soc. Rev. 2015, 44, 4145–4164. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Harada, A.; Nagasaki, Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv. Drug Deliv. Rev. 2001, 47, 113–131. [Google Scholar] [CrossRef]
- Prabaharan, M.; Grailer, J.J.; Pilla, S.; Steeber, D.A.; Gong, S. Amphiphilic Multi-Arm Block Copolymer Based on Hyperbranched Polyester, Poly(l-lactide) and Poly(ethylene glycol) as a Drug Delivery Carrier. Macromol. Biosci. 2009, 9, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qian, Y.; Liu, T.; Hu, X.; Zhang, G.; You, Y.; Liu, S. Amphiphilic multiarm star block copolymer-based multifunctional unimolecular micelles for cancer targeted drug delivery and MR imaging. Biomaterials 2011, 32, 6595–6605. [Google Scholar] [CrossRef] [PubMed]
- Radowski, M.R.; Shukla, A.; von Berlepsch, H.; Böttcher, C.; Pickaert, G.; Rehage, H.; Haag, R. Supramolecular aggregates of dendritic multishell architectures as universal nanocarriers. Angew. Chem. Int. Ed. Engl. 2007, 46, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Küchler, S.; Radowski, M.R.; Blaschke, T.; Dathe, M.; Plendl, J.; Haag, R.; Schäfer-Korting, M.; Kramer, K.D. Nanoparticles for skin penetration enhancement—A comparison of a dendritic core-multishell-nanotransporter and solid lipid nanoparticles. Eur. J. Pharm. Biopharm. 2009, 71, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Quadir, M.A.; Radowski, M.R.; Kratz, F.; Licha, K.; Hauff, P.; Haag, R. Dendritic multishell architectures for drug and dye transport. J. Control. Release 2008, 132, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Fleige, E.; Ziem, B.; Grabolle, M.; Haag, R.; Resch-Genger, U. Aggregation Phenomena of Host and Guest upon the Loading of Dendritic Core-Multishell Nanoparticles with Solvatochromic Dyes. Macromolecules 2012, 45, 9452–9459. [Google Scholar] [CrossRef]
- Weber, M.; Zoschke, C.; Sedighi, A.; Fleige, E.; Haag, R.; Schäfer-Korting, M. Free Energy Simulations of Cargo-Carrier Interactions for Core-Multishell Nanotransporters. J. Nanomed. Nanotechnol. 2014, 5. [Google Scholar] [CrossRef]
- Zhu, Y.; Hazeldine, S.; Li, J.; Oupický, D. Dendritic polyglycerol with secondary amine shell as an efficient gene delivery vector with reduced toxicity. Polym. Adv. Technol. 2014, 25, 940–947. [Google Scholar] [CrossRef]
- Roller, S.; Zhou, H.; Haag, R. High-loading polyglycerol supported reagents for Mitsunobu- and acylation-reactions and other useful polyglycerol derivatives. Mol. Divers. 2005, 9, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Hönzke, S.; Gerecke, C.; Elpelt, A.; Zhang, N.; Unbehauen, M.; Kral, V.; Fleige, E.; Paulus, F.; Haag, R.; Schäfer-Korting, M.; et al. Tailored dendritic core-multishell nanocarriers for efficient dermal drug delivery: A systematic top-down approach from synthesis to preclinical testing. J. Control. Release 2016, 242, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Stemp, F.; Quinzler, D.; Heckler, I.; Mecking, S. Long-Chain Linear C 19 and C 23 Monomers and Polycondensates from Unsaturated Fatty Acid Esters. Macromolecules 2011, 4159–4166. [Google Scholar] [CrossRef]
- Sunder, A.; Hanselmann, R.; Frey, H.; Mülhaupt, R. Controlled synthesis of hyperbranched polyglycerols by ring-opening multibranching polymerization. Macromolecules 1999, 32, 4240–4246. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Brundiek, H.; Sa, S.; Evitt, A.; Kourist, R.; Bornscheuer, U.T. The short form of the recombinant CAL-A-type lipase UM03410 from the smut fungus Ustilago maydis exhibits an inherent trans-fatty acid selectivity. Appl. Microbiol. Biotechnol. 2012, 94, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, C.; Ollivrin, X.; Alloina, F.; Lenest, J.-F.; Sanchez, J.-Y. Lithium salts based on oligoether sulfate esters. Electrochim. Acta 2005, 50, 3843–3852. [Google Scholar] [CrossRef]
- Rabe, C.; Fleige, E.; Vogtt, K.; Szekely, N.; Lindner, P.; Burchard, W.; Haag, R.; Ballauff, M. The multi-domain nanoparticle structure of a universal core-multi-shell nanocarrier. Polymer 2014, 55, 6735–6742. [Google Scholar] [CrossRef]
- Kurniasih, I.N.; Liang, H.; Kumar, S.; Mohr, A.; Sharma, S.K.; Rabe, J.P.; Haag, R. A bifunctional nanocarrier based on amphiphilic hyperbranched polyglycerol derivatives. J. Mater. Chem. B 2013, 1, 3569. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Mn [kDa] | PDI | DF (NMR) |
|---|---|---|---|
| CMS-A18 | 41.8 | 1.85 | 65% |
| CMS-E12 | 32.2 | 1.31 | 88% |
| CMS-E15 | 43.1 | 1.79 | 90% |
| CMS-E18 | 34.9 | 1.41 | 88% |
| CMS-E19 | 32.3 | 1.31 | 75% |
| CMS-E18b | 33.3 | 1.68 | 69% |
| Compound | Unloaded | Tacrolimus-Loaded | Dexamethasone-Loaded | ζ Potential | |||
|---|---|---|---|---|---|---|---|
| d [nm] | d [nm] | d [nm] | [mV] | ||||
| CMS-A18 | 15 | (81%) | 19 | (5%) | 32 | 0.07 ± 0.09 | |
| 210 | (19%) | 204 | (95%) | ||||
| CMS-E12 | 14 | (48%) | 14 | (5%) | 63 | (10%) | −1.27 ± 1.04 |
| 134 | (52%) | 215 | (95%) | 361 | (90%) | ||
| CMS-E15 | 15 | (23%) | 16 | (10%) | 16 | (9%) | 0.01 ± 0.06 |
| 138 | (77%) | 208 | (90%) | 270 | (91%) | ||
| CMS-E18 | 37 | 18 | (15%) | 24 | (25%) | −5.9 ± 0.7 | |
| 224 | (85%) | 272 | (74%) | ||||
| CMS-E19 * | 216 | 364 | 47 | (9%) | 0.01 ± 0.03 | ||
| 329 | (91%) | ||||||
| CMS-E18b | 76 | 123 | 121 | −0.003 ± 0.06 | |||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Unbehauen, M.L.; Fleige, E.; Paulus, F.; Schemmer, B.; Mecking, S.; Moré, S.D.; Haag, R. Biodegradable Core–Multishell Nanocarriers: Influence of Inner Shell Structure on the Encapsulation Behavior of Dexamethasone and Tacrolimus. Polymers 2017, 9, 316. https://doi.org/10.3390/polym9080316
Unbehauen ML, Fleige E, Paulus F, Schemmer B, Mecking S, Moré SD, Haag R. Biodegradable Core–Multishell Nanocarriers: Influence of Inner Shell Structure on the Encapsulation Behavior of Dexamethasone and Tacrolimus. Polymers. 2017; 9(8):316. https://doi.org/10.3390/polym9080316
Chicago/Turabian StyleUnbehauen, Michael L., Emanuel Fleige, Florian Paulus, Brigitta Schemmer, Stefan Mecking, Sam Dylan Moré, and Rainer Haag. 2017. "Biodegradable Core–Multishell Nanocarriers: Influence of Inner Shell Structure on the Encapsulation Behavior of Dexamethasone and Tacrolimus" Polymers 9, no. 8: 316. https://doi.org/10.3390/polym9080316