
Comparative Study of Small-RNA and Degradome Sequencing Reveals Role of Novel stu-miR8006 in Regulating Root Development in Solanum tuberosum L.
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Small-RNA Isolation and Sequencing
2.3. Sequencing Data Analysis and Identification of Potato miRNAs
2.4. Differential Expression Analysis of Potato miRNAs
2.5. Degradome Library Construction and Sequencing
2.6. Identification of miRNA Targets
2.7. RLM-5′-RACE Assay
2.8. qRT-PCR Analysis
2.9. Identification and Expressional Analysis of stu-miR8006-p5-1ss9AT
2.10. Vector Construction and Potato Transformation
2.11. Root Architecture Analysis
2.12. Statistical Analysis
3. Results
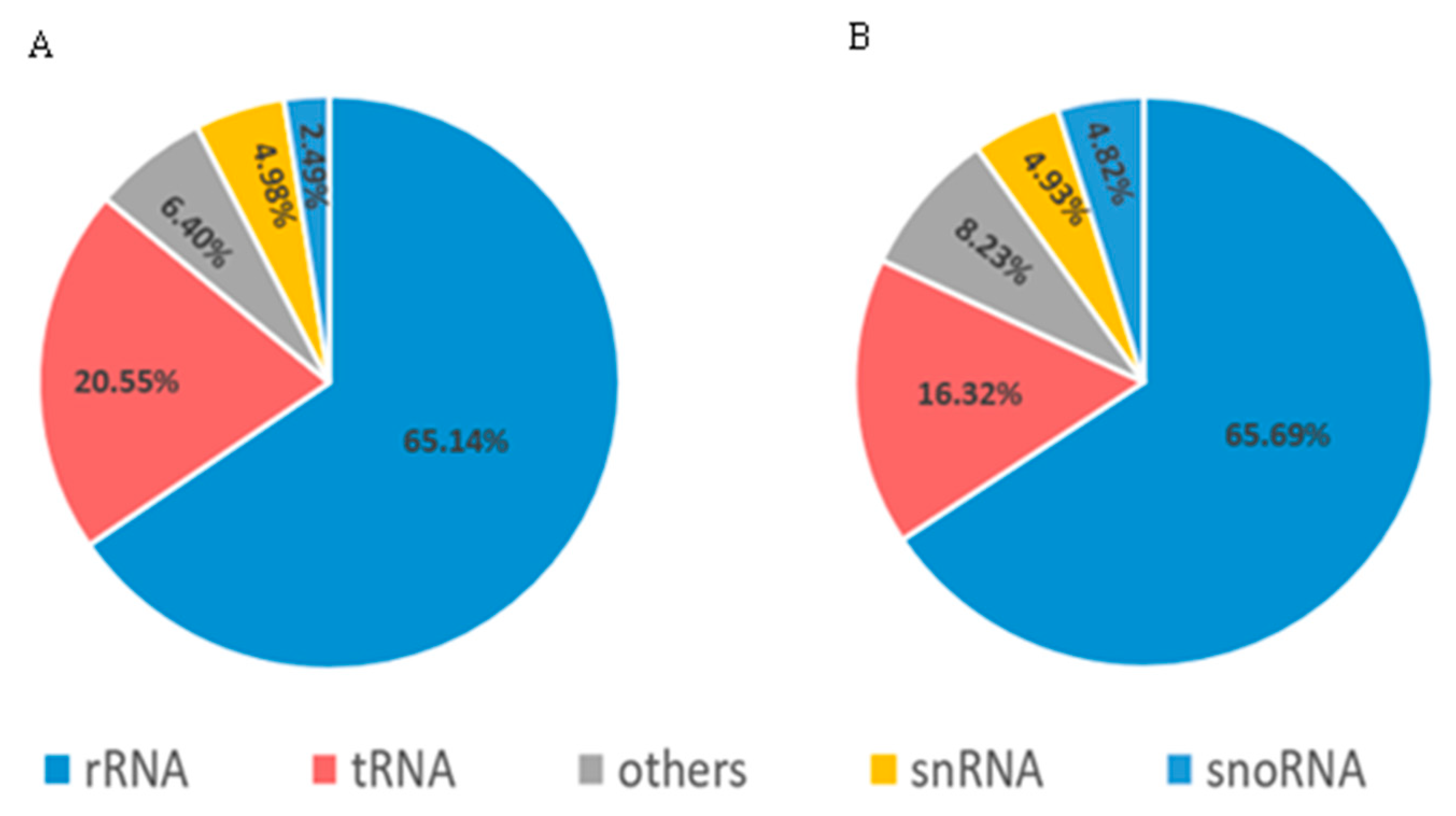
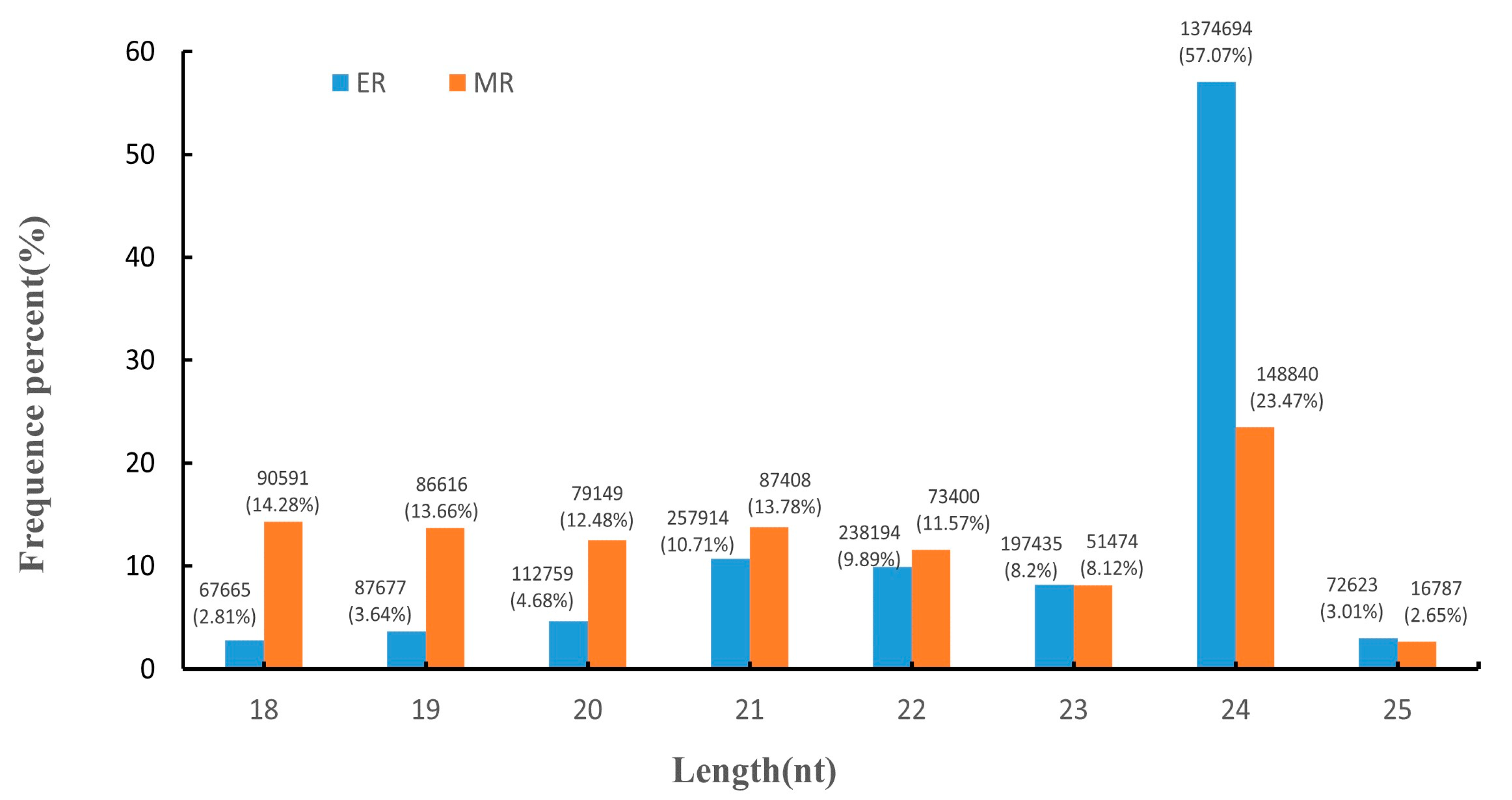
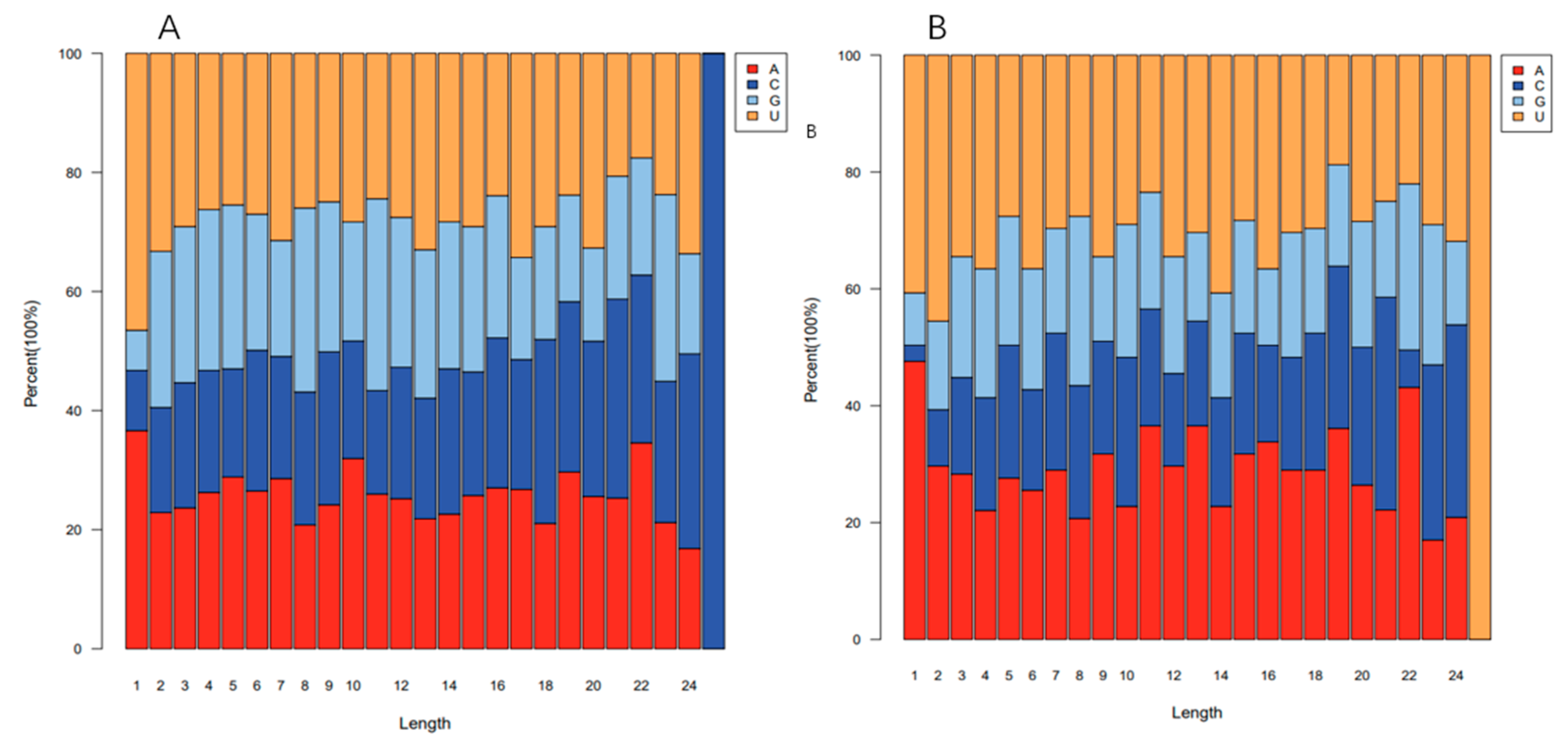
3.1. Overview of sRNA Sequencing
3.2. Identification of Known and Novel miRNAs in Potato
3.3. Differential Expression Analysis of miRNAs between ER and MR
3.4. Identification of miRNA Targets by Degradome Analysis
3.5. GO Enrichment and KEGG Pathway Analyses of Target Genes
3.6. Identification of miRNAs and Their Targets Involved in the Development of Potato Roots
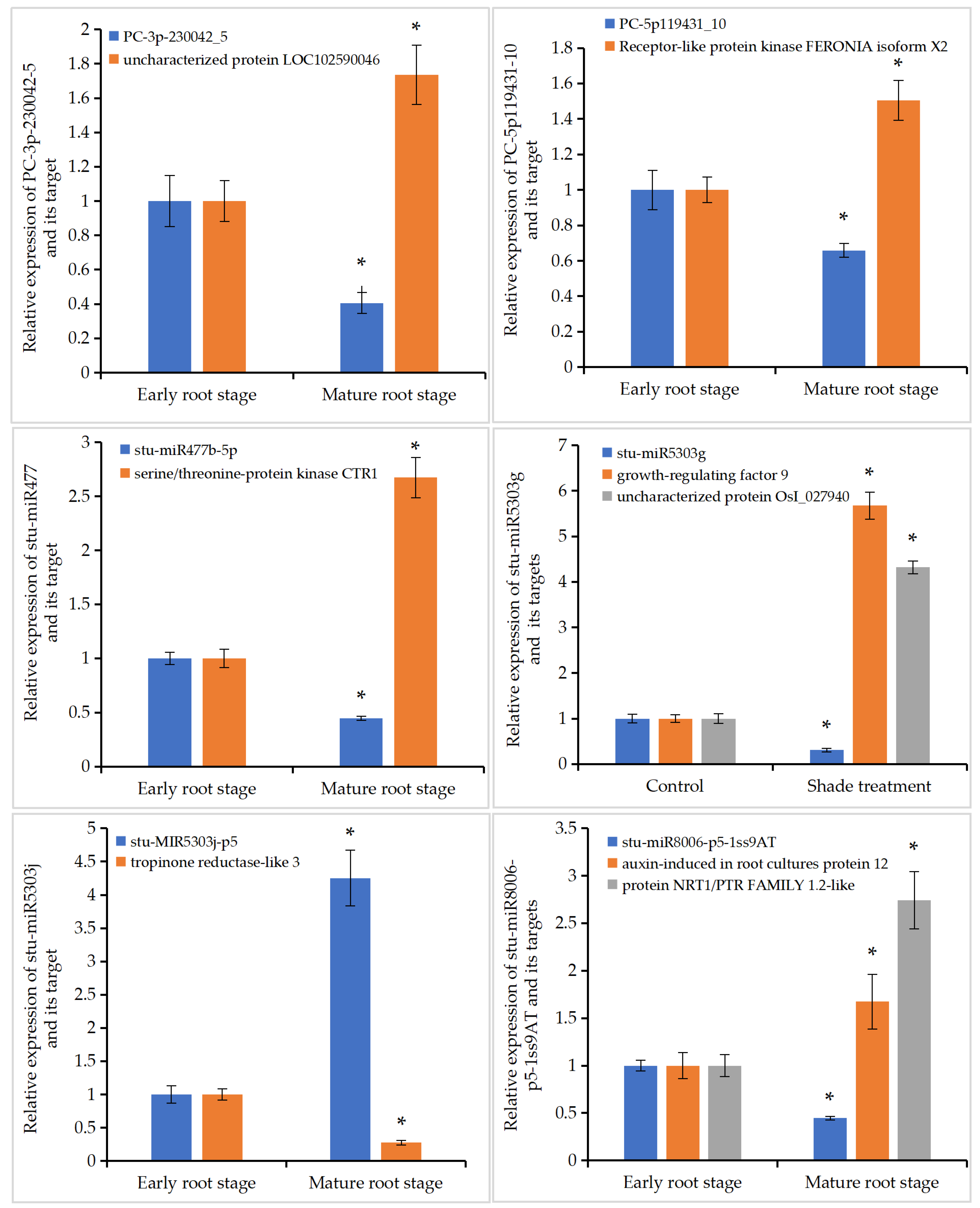
3.7. Validation of miRNAs’ Targets Involved in the Development of Potato Roots
3.8. Expression Analysis of Root-Development-Related miRNAs and Their Target Genes
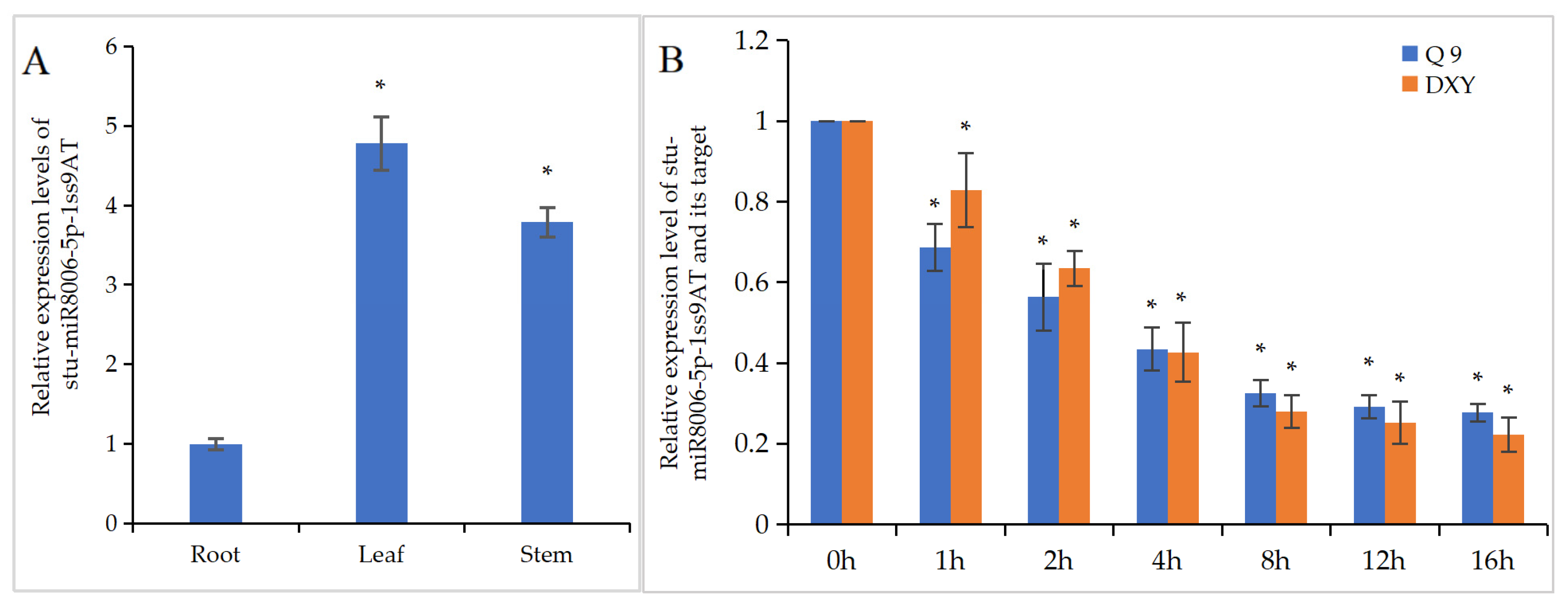
3.9. Clone, Characterization, and Expressional Analysis of stu-miR8006-p5-1ss9AT in Potato
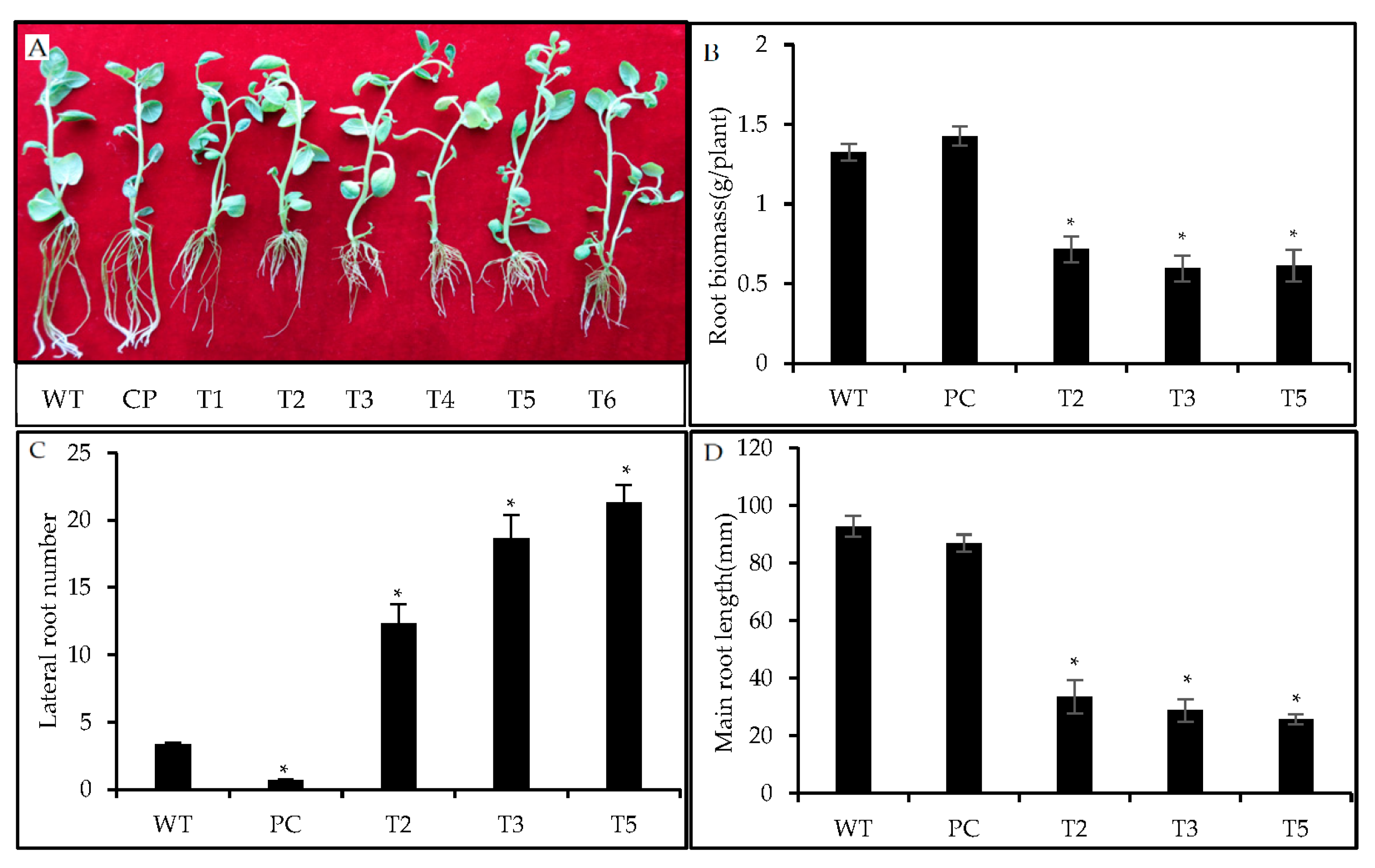
3.10. Suppression of stu-miR8006-p5-1ss9AT Resulted in Alteration in Potato Root Architecture
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bashir, K.; Matsui, A.; Rasheed, S.; Seki, M. Recent advances in the characterization of plant transcriptomes in response to drought, salinity, heat, and cold stress. F1000Research 2019, 8, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.; Sharma, I.; Kaur, N.; Pati, P.K. Auxin: A master regulator in plant root development. Plant Cell Rep. 2013, 32, 741–757. [Google Scholar] [CrossRef] [PubMed]
- Pélissier, P.M.; Motte, H.; Beeckman, T. Lateral root formation and nutrients: Nitrogen in the spotlight. Plant Physiol. 2021, 187, 1104–1116. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, A.; Roy, S.; Sarkar, A.K. SWP1 negatively regulates lateral root initiation and elongation in Arabidopsis. Plant Signal Behav. 2012, 7, 1522–1525. [Google Scholar] [CrossRef]
- Kang, Z.; Qin, T.; Zhao, Z. Overexpression of the zinc finger protein gene OsZFP350 improves root development by increasing resistance to abiotic stress in rice. Acta Biochim. Pol. 2019, 66, 183–190. [Google Scholar] [CrossRef]
- Yadav, P.K.; Gupta, N.; Verma, V.; Gupta, A.K. Overexpression of SlHSP90.2 leads to altered root biomass and architecture in tomato (Solanum lycopersicum). Physiol. Mol. Biol. Plants 2021, 27, 713–725. [Google Scholar] [CrossRef]
- Lu, G.; Coneva, V.; Casaretto, J.A.; Ying, S.; Mahmood, K.; Liu, F.; Nambara, E.; Bi, Y.M.; Rothstein, S.J. OsPIN5b modulates rice (Oryza sativa) plant architecture and yield by changing auxin homeostasis, transport and distribution. Plant J. 2015, 83, 913–925. [Google Scholar] [CrossRef]
- Prasad, M.E.; Schofield, A.; Lyzenga, W.; Liu, H.; Stone, S.L. Arabidopsis RING E3 ligase XBAT32 regulates lateral root production through its role in ethylene biosynthesis. Plant Physiol. 2010, 153, 1587–1596. [Google Scholar] [CrossRef]
- Lewis, D.R.; Negi, S.; Sukumar, P.; Muday, G.K. Ethylene inhibits lateral root development, increases IAA transport and expression of PIN3 and PIN7 auxin efflux carriers. Development 2011, 138, 3485–3495. [Google Scholar] [CrossRef]
- Mutum, R.D.; Kumar, S.; Balyan, S.; Kansal, S.; Mathur, S.; Raghuvanshi, S. Identification of novel miRNAs from drought tolerant rice variety Nagina 22. Sci. Rep. 2016, 6, 30786. [Google Scholar] [CrossRef]
- Natarajan, B.; Banerjee, A.K. MicroRNA160 regulates leaf curvature in potato (Solanum tuberosum L. cv. Désirée). Plant Signal Behav. 2020, 15, 1744373. [Google Scholar] [CrossRef]
- Navarro, C.; Abelenda, J.A.; Cruz-Oró, E.; Cuéllar, C.A.; Tamaki, S.; Silva, J.; Shimamoto, K.; Prat, S. Control of flowering and storage organ formation in potato by flowering locus T. Nature 2011, 478, 119–122. [Google Scholar] [CrossRef]
- Li, W.; Wang, B.; Wang, M.; Chen, M.; Yin, J.M.; Kaleri, G.M.; Zhang, R.J.; Zuo, T.N.; You, X.; Yang, Q. Cloning and characterization of a potato StAN11 gene involved in anthocyanin biosynthesis regulation. J. Integr. Plant Biol. 2014, 56, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, N.; Fu, X.; Zhang, H.; Liu, S.; Pu, X.; Wang, X.; Si, H. StTCP15 regulates potato tuber sprouting by modulating the dynamic balance between abscisic acid and gibberellic acid. Front. Plant Sci. 2022, 13, 1009552. [Google Scholar] [CrossRef] [PubMed]
- Hartje, S.; Zimmermann, S.; Klonus, D.; Mueller-Roeber, B. Functional characterisation of LKT1, a K+ uptake channel from tomato root hairs, and comparison with the closely related potato inwardly rectifying K+ channel SKT1 after expression in xenopus oocytes. Planta 2000, 210, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Ivanchenko, M.G.; Zhu, J.; Wang, B.; Medvecká, E.; Du, Y.; Azzarello, E.; Mancuso, S.; Megraw, M.; Filichkin, S.; Dubrovsky, J.G.; et al. The cyclophilin a diaceotropica gene affects auxin transport in both root and shoot to control lateral root formation. Development 2015, 142, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Tang, X.; Liu, W.; Fu, X.; Luo, H.; Ghimire, S.; Zhang, N.; Si, H. A potato RING-finger protein gene StRFP2 is involved in drought tolerance. Plant Physiol. Biochem. 2020, 146, 438–446. [Google Scholar] [CrossRef]
- Koroban, N.V.; Kudryavtseva, A.V.; Krasnov, G.S.; Sadritdinova, A.F.; Fedorova, M.S.; Snezhkina, A.V.; Bolsheva, N.L.; Muravenko, O.V.; Dmitriev, A.A.; Melnikova, N.V. The role of microRNA in abiotic stress response in plants. Mol. Biol. 2016, 50, 387–394. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef]
- Ahmad, H.M.; Wang, X.; Ijaz, M.; Rahman, M.U.; Oranab, S.; Ali, M.A.; Fiaz, S. Molecular aspects of microRNAs and phytohormonal signaling in response to drought stress: A review. Curr. Issues Mol. Biol. 2022, 44, 3695–3710. [Google Scholar] [CrossRef]
- Damodharan, S.; Corem, S.; Gupta, S.K.; Arazi, T. Tuning of SlARF10A dosage by sly-miR160a is critical for auxin-mediated compound leaf and flower development. Plant J. 2018, 96, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.C.; Dugas, D.V.; Bartel, D.P.; Bartel, B. MicroRNA regulation of NAC domaintargets is required for proper formation and separation of adjacent embryonic, vegetative, and floral organs. Curr. Biol. 2004, 14, 1035–1046. [Google Scholar] [CrossRef]
- Waheed, S.; Zeng, L. The critical role of miRNAs in regulation of flowering time and flower development. Genes 2020, 11, 319. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, F.T.; Sarkar, A.K.; Chitwood, D.H.; Timmermans, M.C. Organ polarity in plants is specified through the opposing activity of two distinct small regulatory RNAs. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Sanan-Mishra, N.; Varanasi, S.P.; Mukherjee, S.K. Micro-regulators of auxin action. Plant Cell Rep. 2013, 32, 733–740. [Google Scholar] [CrossRef]
- Ali, M.; Javaid, A.; Naqvi, S.H.; Batcho, A.; Kayani, W.K.; Lal, A.; Sajid, I.A.; Nwogwugwu, J.O. Biotic stress triggered small RNA and RNAi defense response in plants. Mol. Biol. Rep. 2020, 47, 5511–5522. [Google Scholar] [CrossRef] [PubMed]
- Mengistu, A.A.; Tenkegna, T.A. The role of miRNA in plant-virus interaction: A review. Mol. Biol. Rep. 2021, 48, 2853–2861. [Google Scholar] [CrossRef]
- Jeyaraj, A.; Elango, T.; Li, X.; Guo, G. Utilization of microRNAs and their regulatory functions for improving biotic stress tolerance in tea plant [Camellia sinensis (L.) O. Kuntze]. RNA Biol. 2020, 17, 1365–1382. [Google Scholar] [CrossRef]
- Megha, S.; Basu, U.; Kav, N.N.V. Regulation of low temperature stress in plants by microRNAs. Plant Cell Environ. 2018, 41, 1–15. [Google Scholar] [CrossRef]
- Singroha, G.; Sharma, P.; Sunkur, R. Current status of microRNA-mediated regulation of drought stress responses in cereals. Physiol. Plant 2021, 172, 1808–1821. [Google Scholar] [CrossRef]
- Um, T.; Choi, J.; Park, T.; Chung, P.J.; Jung, S.E.; Shim, J.S.; Kim, Y.S.; Choi, I.Y.; Park, S.C.; Oh, S.J.; et al. Rice microRNA171f/SCL6 module enhances drought tolerance by regulation of flavonoid biosynthesis genes. Plant Direct 2022, 6, e374. [Google Scholar] [CrossRef]
- Pandey, C.; Raghuram, B.; Sinha, A.K.; Gupta, M. miRNA plays a role in the antagonistic effect of selenium on arsenic stress in rice seedlings. Metallomics 2015, 7, 857–866. [Google Scholar] [CrossRef]
- Akdogan, G.; Tufekci, E.D.; Uranbey, S.; Unver, T. miRNA-based drought regulation in wheat. Funct. Integr. Genom. 2016, 16, 221–233. [Google Scholar] [CrossRef]
- Wu, H.J.; Wang, Z.M.; Wang, M.; Wang, X.J. Widespread long noncoding RNAs as endogenous target mimics for microRNAs in plants. Plant Physiol. 2013, 161, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Anna, W.; Marcin, P.; Zofifia, S.K. Construction of Artificial miRNAs to prevent drought stress in Solanum tuberosum. In Environmental Responses in Plants; Springer: New York, NY, USA, 2016; Volume 1398, pp. 271–290. [Google Scholar]
- Zhang, R.; Marshall, D.; Bryan, G.J.; Hornyik, C. Identification and characterization of miRNA transcriptome in potato by highthroughput sequencing. PLoS ONE 2013, 8, e57233. [Google Scholar]
- Shin, D.; Moon, S.J.; Han, S.; Kim, B.G.; Park, S.R.; Lee, S.K.; Yoon, H.J.; Lee, H.E.; Kwon, H.B.; Baek, D.; et al. Expression of StMYB1R-1, a novel potato single MYB-like domain transcription factor, increases drought tolerance. Plant Physiol. 2011, 155, 421–432. [Google Scholar] [CrossRef]
- Lovas, A.; Bimbó, A.; Szabó, L.; Bánfalvi, Z. Antisense repression of StubGAL83 affects root and tuber development in potato. Plant J. 2003, 33, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Islam, W.; Naveed, H.; Idress, A.; Ishaq, D.U.; Kurfi, B.G.; Zeng, F. Plant responses to metals stress: microRNAs in focus. Environ. Sci. Pollut. Res. Int. 2022, 29, 69197–69212. [Google Scholar] [CrossRef]
- Zhu, Q.H.; Upadhyaya, N.M.; Gubler, F.; Helliwell, C.A. Over-expression of miR172 causes loss of spikelet determinacy and floral organ abnormalities in rice (Oryza sativa). BMC Plant Biol. 2009, 9, 149. [Google Scholar] [CrossRef]
- Mecchia, M.A.; Debernardi, J.M.; Rodriguez, R.E.; Schommer, C.; Palatnik, J.F. MicroRNA miR396 and RDR6 synergistically regulate leaf development. Mech. Dev. 2013, 130, 2–13. [Google Scholar] [CrossRef]
- Ferdous, J.; Hussain, S.S.; Shi, B.J. Role of microRNAs in plant drought tolerance. Plant Biotechnol. J. 2015, 13, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Saminathan, T.; Bodunrin, A.; Singh, N.V.; Devarajan, R.; Nimmakayala, P.; Jeff, M.; Aradhya, M.; Reddy, U.K. Genome-wide identification of microRNAs in pomegranate (Punica granatum L.) by high-throughput sequencing. BMC Plant Biol. 2016, 16, 122. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Iwasaki, S.; Watanabe, T.; Utsumi, M.; Watanabe, Y. The mechanism selecting the guide strand from small RNA duplexes is different among argonaute proteins. Plant Cell Physiol. 2008, 49, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Olina, A.V.; Kulbachinskiy, A.V.; Aravin, A.A.; Esyunina, D.M. Argonaute proteins and mechanisms of RNA interference in eukaryotes and prokaryotes. Biochemistry 2018, 83, 483–497. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, S.; Du, S.; Jiang, J.; Wang, G. Transcriptome and metabonomic analysis of tamarix ramosissima Potassium (K+) channels and transporters in response to NaCl stress. Genes 2022, 13, 1313. [Google Scholar] [CrossRef] [PubMed]
- Back, K. Melatonin metabolism, signaling and possible roles in plants. Plant J. 2021, 105, 376–391. [Google Scholar] [CrossRef]
- Shen, W.; Cao, S.; Liu, J.; Zhang, W.; Chen, J.; Li, J.F. Overexpression of an osa-miR162a derivative in rice confers cross-kingdom RNA interference-mediated brown planthopper resistance without perturbing host development. Int. J. Mol. Sci. 2021, 22, 12652. [Google Scholar] [CrossRef]
- Gupta, O.P.; Dahuja, A.; Sachdev, A.; Kumari, S.; Jain, P.K.; Vinutha, T.; Praveen, S. Conserved miRNAs modulate the expression of potential transcription factors of isoflavonoid biosynthetic pathway in soybean seeds. Mol. Biol. Rep. 2019, 46, 3713–3730. [Google Scholar] [CrossRef]
- De Paolo, S.; Gaudio, L.; Aceto, S. Analysis of the TCP genes expressed in the inflorescence of the orchid orchis italica. Sci. Rep. 2015, 5, 16265. [Google Scholar] [CrossRef]
- Baulies, J.L.; Bresso, E.G.; Goldy, C.; Palatnik, J.F.; Schommer, C. Potent inhibition of TCP transcription factors by miR319 ensures proper root growth in Arabidopsis. Plant Mol. Biol. 2022, 108, 93–103. [Google Scholar] [CrossRef]
- Liu, X.; Huang, S.; Xie, H. Advances in the regulation of plant development and stress response by miR167. Front. Biosci. (Landmark Ed.) 2021, 26, 655–665. [Google Scholar] [CrossRef]
- Fei, Y.; Luo, C.; Tang, W. Differential expression of microRNAs during root formation in Taxus Chinensis Var. mairei cultivars. Open Life Sci. 2019, 14, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Deng, Y.; Wu, T.; Subramanian, S.; Yu, O. Misexpression of miR482, miR1512, and miR1515 increases soybean nodulation. Plant Physiol. 2010, 153, 1759–1770. [Google Scholar] [CrossRef]
- Arraes, F.B.M.; Beneventi, M.A.; De Sa, M.E.L.; Paixao, J.F.R.; Albuquerque, E.V.S.; Marin, S.R.R.; Purgatto, E.; Nepomuceno, A.L.; Grossi-De-Sa, M.F. Implications of ethylene biosynthesis and signaling in soybean drought stress tolerance. BMC Plant Biol. 2015, 15, 213. [Google Scholar] [CrossRef]
- Kim, S.; Nie, H.; Jun, B.; Kim, J.; Lee, J.; Kim, S.; Kim, E.; Kim, S. Functional genomics by integrated analysis of transcriptome of sweet potato (Ipomoea batatas (L.) Lam.) during root formation. Genes Genom. 2020, 42, 581–596. [Google Scholar] [CrossRef] [PubMed]
- Hewezi, T.; Maier, T.R.; Nettleton, D.; Baum, T.J. The Arabidopsis microRNA396-GRF1/GRF3 regulatory module acts as a developmental regulator in the reprogramming of root cells during cyst nematode infection. Plant Physiol. 2012, 159, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Mills, A.M.; Rasmussen, C.G. Defects in division plane positioning in the root meristematic zone affect cell organization in the differentiation zone. J. Cell Sci. 2022, 135, jcs260127. [Google Scholar] [CrossRef] [PubMed]
- IrinaI, V.; Kiril, M.; Thomas, D.; Valya, V.; Dominique, V.D.S. The Diverse Salt-Stress Response of Arabidopsis ctr1-1 and ein2-1 Ethylene Signaling Mutants Is Linked to Altered Root Auxin Homeostasis. Plants. 2021, 10, 452. [Google Scholar] [CrossRef]
- Abdelkawy, A.M.; Alshammari, S.O.; Hussein, H.A.; Abou El-Enain, I.M.M.; Abdelkhalek, E.S.; Radwan, A.M.; Kenawy, S.K.M.; Maaty, D.A.M.; Abed, N.N.; Sabry, S.; et al. Effect of silver nanoparticles on tropane alkaloid production of transgenic hairy root cultures of Hyoscyamus muticus L. and their antimicrobial activity. Sci. Rep. 2023, 13, 10397. [Google Scholar] [CrossRef]
- Ou, Y.; Kui, H.; Li, J. Receptor-like Kinases in Root Development: Current Progress and Future Directions. Mol. Plant 2021, 14, 166–185. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MiRNA | MiRNA Sequences | Target Gene | Transcript Annotation | GO_Term |
|---|---|---|---|---|
| PC-3p-230042_5 | CAAGGCTAGGACAGATGGAAAGAA | PGSC0003DMT400074930 | uncharacterized protein LOC102590046 isoform X1 | GO:0048364 (root development); GO: 0071433 (cell wall repair) |
| PC-5p119431_10 | TCTTAGGCCTAATTCACACCC | PGSC0003DMT400003464 | receptor-like protein kinase FERONIA isoform X2 | GO:0030308 (negative regulation of cell growth); GO: 0048364 (root development) |
| stu-miR477b-5p | ACTCTCCCTCAAAGGCTTCTG | PGSC0003DMT400021612 | serine/threonine-protein kinase CTR1-like isoform X1 | GO:2000069 (regulation of post-embryonic root development) |
| stu-miR5303g | TTTTTGAAGAGTCTGGGCAAC | PGSC0003DMT400009997 | growth-regulating factor 9-like | GO:0008285 (negative regulation of cell proliferation); GO: 0048364 (root development) |
| PGSC0003DMT400079970 | uncharacterized protein OsI_027940-like | GO:0010449 (root meristem growth); GO:0080037 (negative regulation of cytokinin-activated signaling pathway); GO:2000012 (regulation of auxin polar transport) | ||
| stu-miR5303j-p5 | ATTCAAATTTCTGCCCTA | PGSC0003DMT400034518 | tropinone reductase-like 3 | GO:0048767 (root hair elongation) |
| stu-miR8006-p5-1ss9AT | CATACTTTTGGGACATTG | PGSC0003DMT400013837 | auxin-induced in root cultures protein 12 isoform X1 | Function unknown |
| PGSC0003DMT400040282 | protein NRT1/PTR FAMILY 1.2-like isoform X2 | GO:0010015 (root morphogenesis); |
| Gene | Sequence of Oligonucleotides Forward (F) and Reverse (R) | Length (bp) |
|---|---|---|
| stu-miR8020 | R 5’-GGAATTTCATTGAGTATGTTGTTGTT-3’ | - |
| stu-miR167d-3p | R 5’-CAGATCATGTGGTTGCTTCAC-3’ | - |
| stu-miR477a-5p | R 5’-CCTCTCCCTCAAGGGCTTCT-3’ | - |
| stu-miR408a-3p | R 5’-TGCACAGCCTCTTCCCTG-3’ | - |
| stu-miR398a-3p | R 5’-TATGTTCTCAGGTCGCCCCT-3’ | - |
| stu-MIR319-p5 | R 5’-TGCTGCTGAATCATTGGTTC-3’ | - |
| PC-3p-28523_59 | R 5’-GCGTTATTTTCACCTGACAAGACT-3’ | - |
| PC-5p-82220_15 | R 5’-GCGTTCTAGGATATAAATTGCACTACT-3’ | - |
| PC-5p-138631_8 | R 5’-TCAACTATCCAGTCTTTCCTCAGA-3’ | - |
| PC-5p-107179_11 | R 5’-CGAGTTTCCTACCTGAACTATCACC-3’ | - |
| PC-5p-656396_2 | R 5’-ACTTCCTGATCACATGGCGA-3’ | - |
| PC-3p-291253_4 | R 5’-TAAGGTAGGGCGCTCTTCG-3’ | - |
| PC-3p-230042_5 | R 5’-CAAGGCTAGGACAGATGGAAAG-3’ | - |
| PC-5p119431_10 | R 5’-GTCTTAGGCCTAATTCACACCC-3’ | - |
| stu-miR477b-5p | R 5’-ACTCTCCCTCAAAGGCTTCTG-3’ | - |
| stu-miR5303g | R 5’-GTTTTTGAAGAGTCTGGGCAAC-3’ | - |
| stu-MIR5303j-p5 | R 5’-GCGATTCAAATTTCTGCCCTA-3’ | - |
| stu-miR8006-p5-1ss9AT | R 5’-CGCATACTTTTGGGACATTG-3’ | - |
| St18sRNA | R 5’-TTAGAGGAAGGAGAAGTCGTAACAA-3’ | - |
| uncharacterized protein LOC102590046 | F 5’-CAAGCATACCCACTTCCTCT-3’ | 182 |
| R 5’-GTCCTAACTCCTGAGACAACG-3’ | ||
| receptor-like protein kinase FERONIA | F 5’-CGCCGAGGATAGTTTACACAA-3’ | 150 |
| R 5’-ACCCTGTCGTTCACTTCCAG-3’ | ||
| serine/threonine-protein ki’-se CTR1-like | F 5’-TTGTGTTACCGACGCAGT-3’ | 187 |
| R 5’-GCAGGTTGCTTCAGTAGAGAT-3’ | ||
| growth-regulating factor 9-like | F 5’-CAGAGCCAAGAAGATGTAGGAG-3’ | 168 |
| R 5’-TTTGAAGAGTCCGAACAACG-3’ | ||
| uncharacterized protein OsI_027940 | F 5’-TGACTTCTCGGACTGGCTGT-3’ | 182 |
| R 5’-GCAGGATGTCTTCAACTCTGG-3’ | ||
| tropinone reductase-like 3 | F 5’-CTGAAAGGCTTGGTTTAGAAGG-3’ | 225 |
| R 5’-GCCACGGTAGGATTTACAGC-3’ | ||
| auxin-induced in root cultures protein | F 5’-CGTATTCCCTTCCATCCTATC-3’ | 254 |
| R 5’-GGCTGAGAAAGTTTGAGAGG-3’ | ||
| protein NRT1/PTR FAMILY 1.2 | F 5’- CTGATTCTTACTGTGGTCGCT -3’ | 118 |
| R 5’-CCTCTTGTTCTCGGGTTTC-3’ | ||
| Stef1α | F 5’- ATTGGAAACGGATATGCTCCA-3’ | 101 |
| R 5’- TCCTTACCTGAACGCCTGTCA-3’ |
| Term | ER | MR |
|---|---|---|
| Raw reads | 15,443,787 | 12,099,703 |
| 3ADT and length filter | 2,202,430 (54.26%) | 6,595,079 (54.51%) |
| Junk reads | 56,274 (0.36%) | 25,332 (0.21%) |
| Rfam | 1,693,395 (10.96%) | 1,189,792 (9.83%) |
| Repeats | 24,663 (0.16%) | 24,286 (0.20%) |
| Valid reads | 8,985,433 (5%) | 3,166,534 (26.17%) |
| Mapped reads | 46,382,473 (88.29%) | 44,040,313 (88.39%) |
| Unique Mapped reads | 30,578,981 (58.21%) | 28,408,840 (57.02%) |
| Q20 (%) | 99.73 | 99.72 |
| Q30 (%) | 98.10 | 98.08 |
| GC content (%) | 43 | 43.5 |
| Type | Number of Reads | Percentage (%) |
|---|---|---|
| Raw reads | 26,207,517 | 100 |
| Reads < 15 nt after removing 3 adapters | 229,844 | 0.88 |
| Mappable reads | 25,977,673 | 99.12 |
| Unique raw reads | 9,690,091 | 100 |
| Unique reads < 15 nt after removing 3 adapters | 78,098 | 0.81 |
| Unique mappable reads | 9,611,993 | 99.19 |
| Transcript mapped reads | 18,191,670 | 69.41 |
| Unique transcript mapped reads | 5,637,124 | 58.17 |
| Number of input transcript | 56,218 | 100 |
| Number of covered transcript | 43,889 | 78.07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, X.; Yang, J.; Zhang, F.; Han, Y.; Gong, Y.; Liu, M.; Zhang, N.; Si, H. Comparative Study of Small-RNA and Degradome Sequencing Reveals Role of Novel stu-miR8006 in Regulating Root Development in Solanum tuberosum L. Agronomy 2023, 13, 2942. https://doi.org/10.3390/agronomy13122942
Duan X, Yang J, Zhang F, Han Y, Gong Y, Liu M, Zhang N, Si H. Comparative Study of Small-RNA and Degradome Sequencing Reveals Role of Novel stu-miR8006 in Regulating Root Development in Solanum tuberosum L. Agronomy. 2023; 13(12):2942. https://doi.org/10.3390/agronomy13122942
Chicago/Turabian StyleDuan, Xiaoqin, Jiangwei Yang, Feiyan Zhang, Yuwen Han, Yating Gong, Mei Liu, Ning Zhang, and Huaijun Si. 2023. "Comparative Study of Small-RNA and Degradome Sequencing Reveals Role of Novel stu-miR8006 in Regulating Root Development in Solanum tuberosum L." Agronomy 13, no. 12: 2942. https://doi.org/10.3390/agronomy13122942
APA StyleDuan, X., Yang, J., Zhang, F., Han, Y., Gong, Y., Liu, M., Zhang, N., & Si, H. (2023). Comparative Study of Small-RNA and Degradome Sequencing Reveals Role of Novel stu-miR8006 in Regulating Root Development in Solanum tuberosum L. Agronomy, 13(12), 2942. https://doi.org/10.3390/agronomy13122942