
Contrasting Key Bacteria and Fungi Related to Sugar Beet (Beta vulgaris L.) with Different Resistances to Beet Rot under Two Farming Modes
Abstract
:1. Introduction
2. Materials and Methods
2.1. Field Experiment and Sampling
2.2. DNA Extraction and Amplicon Sequencing
2.3. Soil Chemical Properties
2.4. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Trivedi, P.; Delgado-Baquerizo, M.; Trivedi, C.; Hamonts, K.; Anderson, I.C.; Singh, B.K. Keystone microbial taxa regulate the invasion of a fungal pathogen in agro-ecosystems. Soil. Biol. Biochem. 2017, 111, 10–14. [Google Scholar] [CrossRef]
- van Bruggen, A.H.; Semenov, A.M.; Diepeningen, A.D.V.; Vos, O.J.D.; Blok, W.J. Relation between soil health, wave-like fluctuations in microbial populations, and soil-borne plant disease management. Eur. J. Plant Pathol. 2006, 115, 105–122. [Google Scholar] [CrossRef]
- Kwak, M.J.; Kong, H.G.; Choi, K.; Kwon, S.K.; Song, J.Y.; Lee, J.; Lee, P.A.; Choi, S.Y.; Seo, M.; Lee, H.J.; et al. Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat. Biotechnol. 2018, 36, 1100–1109. [Google Scholar] [CrossRef]
- Zheng, Y.; Han, X.; Zhao, D.; Wei, K.; Yuan, Y.; Li, Y.; Liu, M.; Zhang, C.S. Exploring biocontrol agents from microbial keystone taxa associated to suppressive soil: A new attempt for a biocontrol strategy. Front. Plant Sci. 2021, 12, 655673. [Google Scholar] [CrossRef]
- Wen, T.; Yuan, J.; He, X.; Lin, Y.; Huang, Q.; Shen, Q. Enrichment of beneficial cucumber rhizosphere microbes mediated by organic acid secretion. Hortic. Res. 2020, 7, 154. [Google Scholar] [CrossRef]
- Carrión, V.J.; Perez-Jaramillo, J.; Cordovez, V.; Tracanna, V.; De Hollander, M.; Ruiz-Buck, D.; Mendes, L.W.; van Ljcken, W.F.J.; Gomez-Exposito, R.; Elsayed, S.S.; et al. Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome. Science 2019, 366, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Fan, X.; Wang, Y.; Kusstatscher, P.; Duan, J.; Wu, S.; Chen, S.; Qiao, K.; Wang, Y.; Ma, B.; et al. Bacterial seed endophyte shapes disease resistance in rice. Nat. Plants 2021, 7, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Wei, Z.; Wang, X.; Friman, V.P.; Huang, J.; Wang, X.; Xu, Y.; Shen, Q.; Jousset, A. Pathogen invasion indirectly changes the composition of soil microbiome via shifts in root exudation profile. Biol. Fertil. Soils 2016, 52, 997–1005. [Google Scholar] [CrossRef] [Green Version]
- Dini-Andreote, F. Endophytes: The second layer of plant defense. Trends Plant Sci. 2020, 25, 319–322. [Google Scholar] [CrossRef]
- Eichmann, R.; Richards, L.; Schäfer, P. Hormones as go-betweens in plant microbiome assembly. Plant J. 2021, 105, 518–541. [Google Scholar] [CrossRef]
- Liu, H.; Li, J.; Carvalhais, L.C.; Percy, C.D.; Prakash Verma, J.; Schenk, P.M.; Singh, B.K. Evidence for the plant recruitment of beneficial microbes to suppress soil-borne pathogens. New Phytol. 2021, 229, 2873–2885. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wilson, A.J.; Zhang, X.C.; Thoms, D.; Sohrabi, R.; Song, S.; Geissmann, Q.; Liu, Y.; Walgren, L.; He, S.Y.; et al. Feronia restricts Pseudomonas in the rhizosphere microbiome via regulation of reactive oxygen species. Nat. Plants 2021, 7, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Lin, M.; Du, C.; Hao, Y.; Zhang, Y.; Wang, Z. The use of manure shifts the response of α-diversity and network while not β-diversity of soil microbes to altered irrigation regimes. Appl. Soil Ecol. 2022, 174, 104423. [Google Scholar] [CrossRef]
- Olsen, S.R.; Cole, C.V.; Watanabe, F.S.; Dean, L.A. Estimation of Available Phosphorus in Soils by Extraction with Sodium Bicarbonate; U.S. Department of Agriculture: Washington, DC, USA, 1954.
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, D.M.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored rare biosphere. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar] [CrossRef] [Green Version]
- Faust, K.; Sathirapongsasuti, J.F.; Izard, J.; Segata, N.; Gevers, D.; Raes, J.; Huttenhower, C. Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 2012, 8, e1002606. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, K.; Zhao, X.; Zhang, H.; Li, D.; Li, J.; Shen, R. Balanced fertilization over four decades has sustained soil microbial communities and improved soil fertility and rice productivity in red paddy soil. Sci. Total. Environ. 2021, 793, 148664. [Google Scholar] [CrossRef]
- Merloti, L.F.; Mendes, L.W.; Pedrinho, A.; de Souza, L.F.; Ferrari, B.M.; Tsai, S.M. Forest-to-agriculture conversion in Amazon drives soil microbial communities and N-cycle. Soil. Bio. Biochem. 2019, 137, 107567. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, J.; Wu, F. Soil microbial communities in cucumber monoculture and rotation systems and their feedback effects on cucumber seedling growth. Plant Soil. 2017, 415, 507–520. [Google Scholar] [CrossRef]
- Shen, Z.; Penton, C.R.; Lv, N.; Xue, C.; Yuan, X.; Ruan, Y.; Li, R.; Shen, Q. Banana Fusarium wilt disease incidence is influenced by shifts of soil microbial communities under different monoculture spans. Microb. Ecol. 2018, 75, 739–750. [Google Scholar] [CrossRef]
- Mendes, L.W.; Raaijmakers, J.M.; de Hollander, M.; Mendes, R.; Tsai, S.M. 2018 Influence of resistance breeding in common bean on rhizosphere microbiome composition and function. ISME J. 2021, 12, 212–224. [Google Scholar] [CrossRef] [Green Version]
- Vannier, N.; Agler, M.; Hacquard, S. Microbiota-mediated disease resistance in plants. PLoS Pathog. 2019, 15, e1007740. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Jian, Y.; Chen, Y.; Kistler, H.C.; He, P.; Ma, Z.; Yin, Y. A phosphorylated transcription factor regulates sterol biosynthesis in Fusarium graminearum. Nat. Commun. 2019, 10, 1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Feng, H.; Schuelke, T.; De Santiago, A.; Zhang, Q.; Zhang, J.; Luo, C.; Wei, L. Rhizosphere microbiomes from root knot nematode non-infested plants suppress nematode infection. Microb. Ecol. 2019, 78, 470–481. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Q.; Wang, P.; Wang, X.; Hu, B.; Tao, L. Phytoremediation of cadmium-contaminated sediment using Hydrilla verticillata and Elodea canadensis harbor two same keystone rhizobacteria Pedosphaeraceae and Parasegetibacter. Chemosphere 2022, 286, 131648. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Feng, Q.; Wan, J.W. Application of five kinds of plant growth promoting rhizobacteria in improving agronomic characters of plants. Patent CN201510541017.2, 8 March 2017. [Google Scholar]
- Fernández-González, A.J.; Cardoni, M.; Gómez-Lama, C.C.; Valverde-Corredor, A.; Villadas, P.J.; Fernández-López, M.; Mercado-Blanco, J. Linking belowground microbial network changes to different tolerance level towards Verticillium wilt of olive. Microbiome 2020, 8, 11. [Google Scholar] [CrossRef]
- Lee, S.M.; Kong, H.G.; Song, G.C.; Ryu, C.M. Disruption of Firmicutes and Actinobacteria abundance in tomato rhizosphere causes the incidence of bacterial wilt disease. ISME J. 2021, 15, 330–347. [Google Scholar] [CrossRef] [PubMed]
- Snelders, N.C.; Rovenich, H.; Petti, G.C.; Rocafort, M.; van den Berg, G.; Vorholt, J.A.; Mesters, J.R.; Seidl, M.F.; Nijland, R.; Thomma, B.P. Microbiome manipulation by a soil-borne fungal plant pathogen using effector proteins. Nat. Plants 2020, 6, 1365–1374. [Google Scholar] [CrossRef]
- Köhl, J.; de Goossen-van Geijn, H.; Groenenboom-de Haas, L.; Henken, B.; Hauschild, R.; Hilscher, U.; Lombaers-vander Plasa, C.; van den Bosch, T.; Wikströmd, M. Stepwise screening of candidate antagonists for biological control of Blumeria graminis f. sp. tritici. Biol. Control 2019, 136, 104008. [Google Scholar] [CrossRef]
- Richter, C.; Yurkov, A.M.; Boekhout, T.; Stadler, M. Diversity of Tilletiopsis-like fungi in Exobasidiomycetes (Ustilaginomycotina) and description of six novel species. Front. Microbiol. 2019, 10, 2544. [Google Scholar] [CrossRef]
- Begerow, D.; Schäfer, A.M.; Kellner, R.; Yurkov, A.; Kemler, M.; Oberwinkler, F. Ustilaginomycotina in Systematics and Evolution, 2nd ed.; McLaughlin, D., Spatafora, J., Eds.; Springer: Berlin, Germany, 2014; pp. 295–329. [Google Scholar]
- Baric, S.; Lindner, L.; Marschall, K.; Dalla, V.J. Haplotype diversity of Tilletiopsis spp. causing white haze in apple orchards in Northern Italy. Plant Pathol. 2010, 59, 535–541. [Google Scholar] [CrossRef]
- Carstens, M.; Vivier, M.A.; Van Rensburg, P.; Pretorius, I.S. Overexpression, secretion and antifungal activity of the Saccharomyces cerevisiae chitinase. Ann. Microbiol. 2003, 53, 15–28. [Google Scholar]
- Gruhlke, M.C.; Nwachwukwu, I.; Arbach, M.; Anwar, A.; Noll, U.; Slusarenko, A.J. Allicin from garlic, effective in controlling several plant diseases, is a reactive sulfur species (RSS) that pushes cells into apoptosis. In Proceedings of the 16th International Reinhardsbrunn Symposium, Friedrichroda, Germany, 25–29 April 2011; pp. 325–330. [Google Scholar]
- Moussa, Z.; El-Hersh, M.S.; El-Khateeb, A.Y. Induction of potato resistance against bacterial wilt disease using Saccharomyces cerevisiae. Biotechnology 2017, 16, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Schlaeppi, K.; van der Heijden Marcel, G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef]
- Jia, H.; Muhae-Ud-Din, G.; Zhang, H.; Zong, Q.; Zhao, S.; Guo, Q.; Chen, W.; Gao, L. Characterization of rhizosphere microbial communities for disease incidence and optimized concentration of difenoconazole fungicide for controlling of wheat dwarf bunt. Front. Microbiol. 2022, 13, 853176. [Google Scholar] [CrossRef]
- Bonanomi, G.; Zotti, M.; Idbella, M.; Cesarano, G.; Al-Rowaily, S.L.; Abd-ElGawad, A.M. Mixtures of organic amendments and biochar promote beneficial soil microbiota and affect Fusarium oxysporum f. sp. lactucae, Rhizoctonia solani and Sclerotinia minor disease suppression. Plant Pathol. 2022, 71, 818–829. [Google Scholar] [CrossRef]
- Puopolo, G.; Tomada, S.; Pertot, I. The impact of the omics era on the knowledge and use of Lysobacter species to control phytopathogenic microorganisms. J. Appl. Microbiol. 2018, 124, 15–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Shen, D.; Yang, N.; Chou, S.H.; Gomelsky, M.; Qian, G. Coordinated control of the type IV pili and c-di-GMP-dependent antifungal antibiotic production in Lysobacter by the response regulator PilR. Mol. Plant Pathol. 2021, 22, 602–617. [Google Scholar] [CrossRef]
- Goh, Y.K.; Zoqratt, M.Z.H.M.; Goh, Y.K.; Ayub, Q.; Ting, A.S.Y. Determining soil microbial communities and their influence on Ganoderma disease incidences in oil palm (Elaeis guineensis) via high-throughput sequencing. Biology 2020, 9, 424. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, J.; Liu, Q.; Zhou, Z.; Chen, F.; Xiang, D. Effects of consecutive monoculture of sweet potato on soil bacterial community as determined by pyrosequencing. J. Basic Microb. 2019, 59, 181–191. [Google Scholar] [CrossRef]
- Yin, C.; Hulbert, S.H.; Schroeder, K.L.; Mavrodi, O.; Mavrodi, D.; Dhingra, A.; Schillinger, W.F.; Paulitz, T.C. Role of bacterial communities in the natural suppression of Rhizoctonia solani bare patch disease of wheat (Triticum aestivum L.). Appl. Environ. Microb. 2013, 79, 7428–7438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yim, B.; Nitt, H.; Wrede, A.; Jacquiod, S.; Sørensen, S.J.; Winkelmann, T.; Smalla, K. Effects of soil pre-treatment with Basamid® granules, Brassica juncea, Raphanus sativus, and Tagetes patula on bacterial and fungal communities at two apple replant disease sites. Front. Microbiol. 2017, 8, 1604. [Google Scholar] [CrossRef] [PubMed]
- Rosales, S.M.; Miller, M.W.; Williams, D.E.; Traylor-Knowles, N.; Young, B.; Serrano, X.M. Microbiome differences in disease-resistant vs. susceptible Acropora corals subjected to disease challenge assays. Sci. Rep. 2019, 9, 18279. [Google Scholar] [CrossRef] [Green Version]
- Patrmanova, T.; Krizkova, I.; Rapoport, D.; Kopecky, J.; Hrychova, S.; Sagova-Mareckova, M. Inoculations of soil by antagonistic strains modify tuberosphere bacterial communities and suppress common scab of potatoes. Appl. Soil Ecol. 2022, 176, 104491. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Q.; Cui, H.; Li, Y.; Xu, N.; Lu, T.; Penuelas, J.; Hu, B.; Qian, H. Composition identification and functional verification of bacterial community in disease-suppressive soils by machine learning. Environ. Microbiol. 2022, 24, 3405–3419. [Google Scholar] [CrossRef] [PubMed]
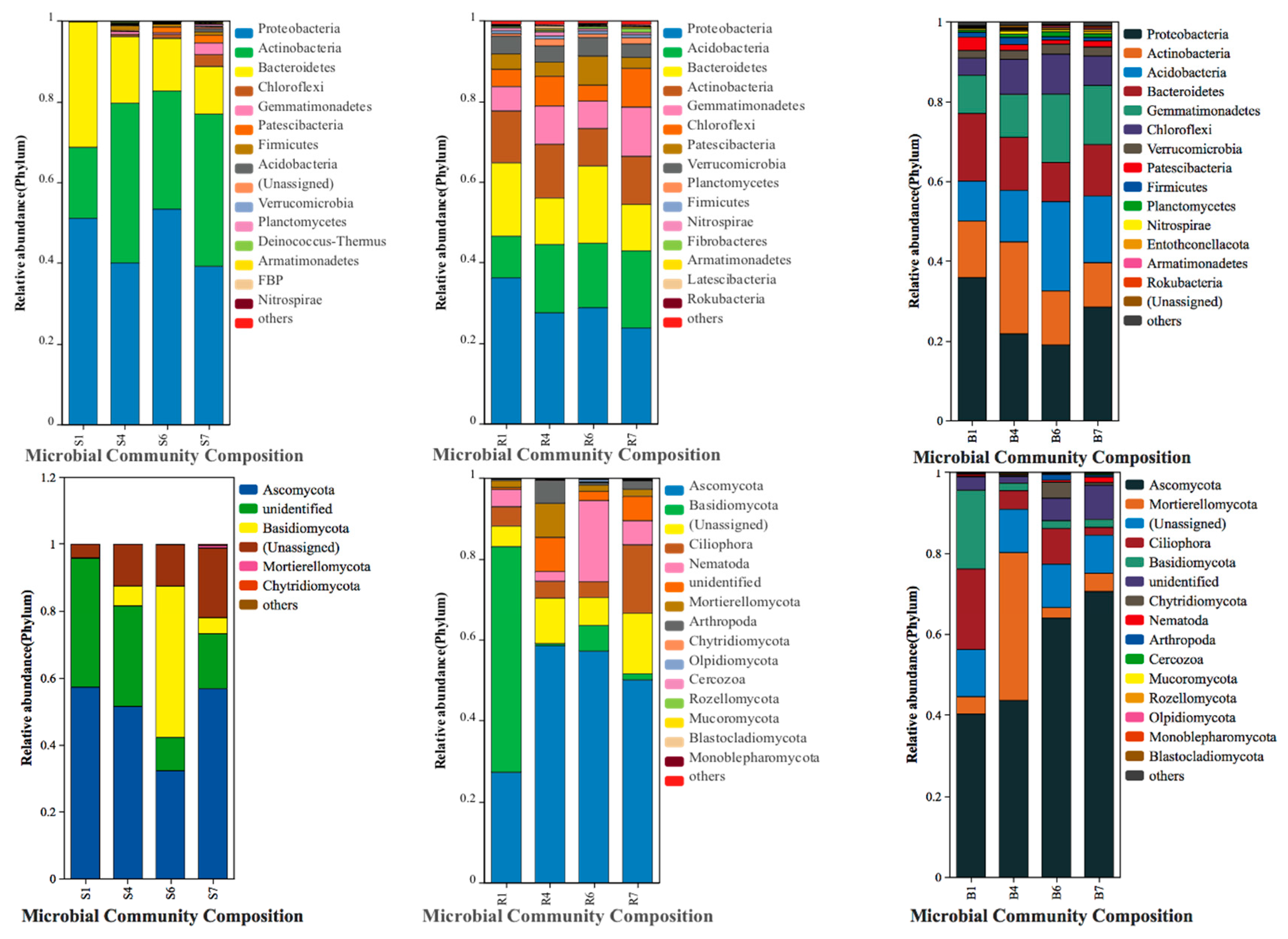

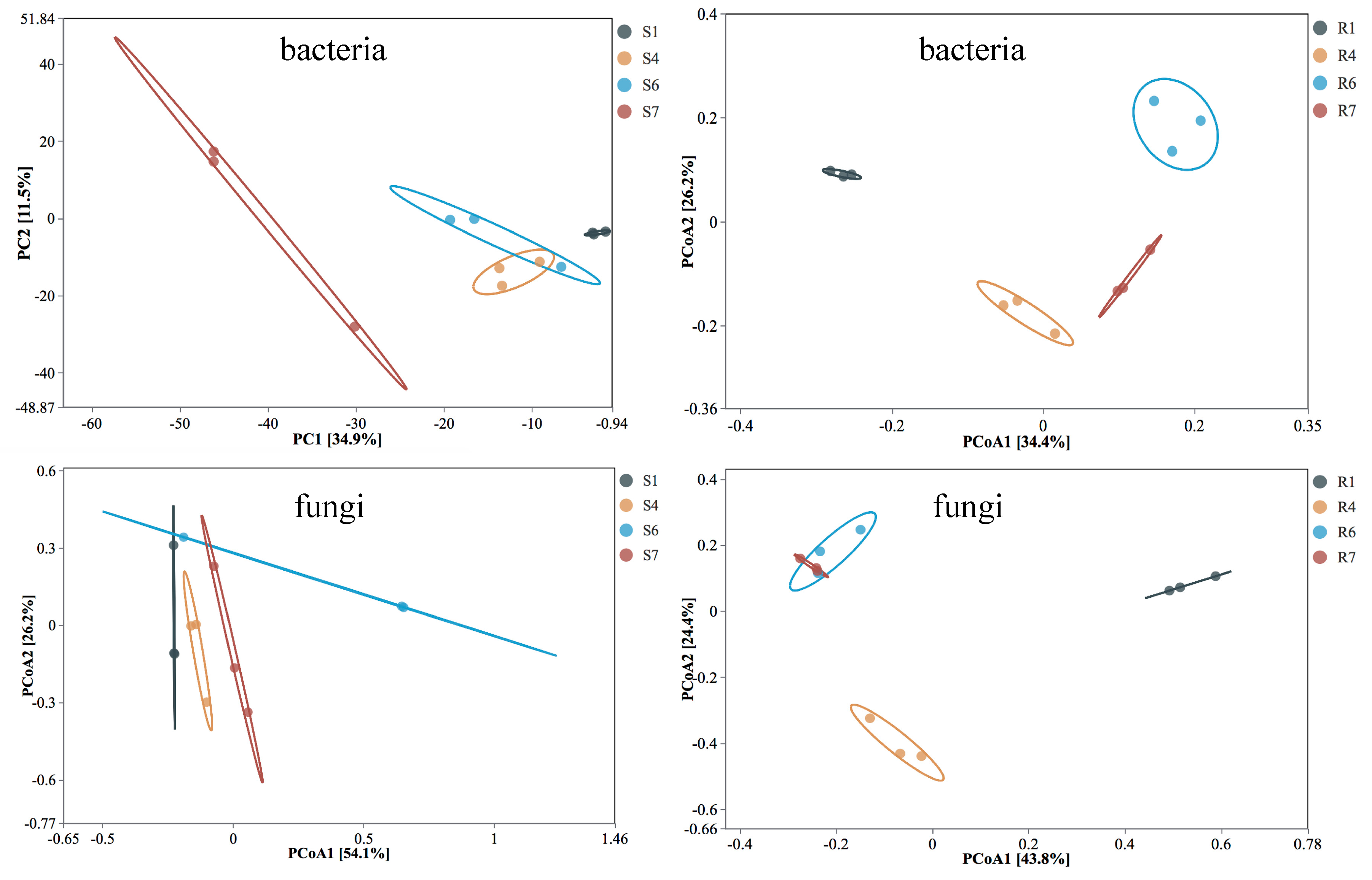

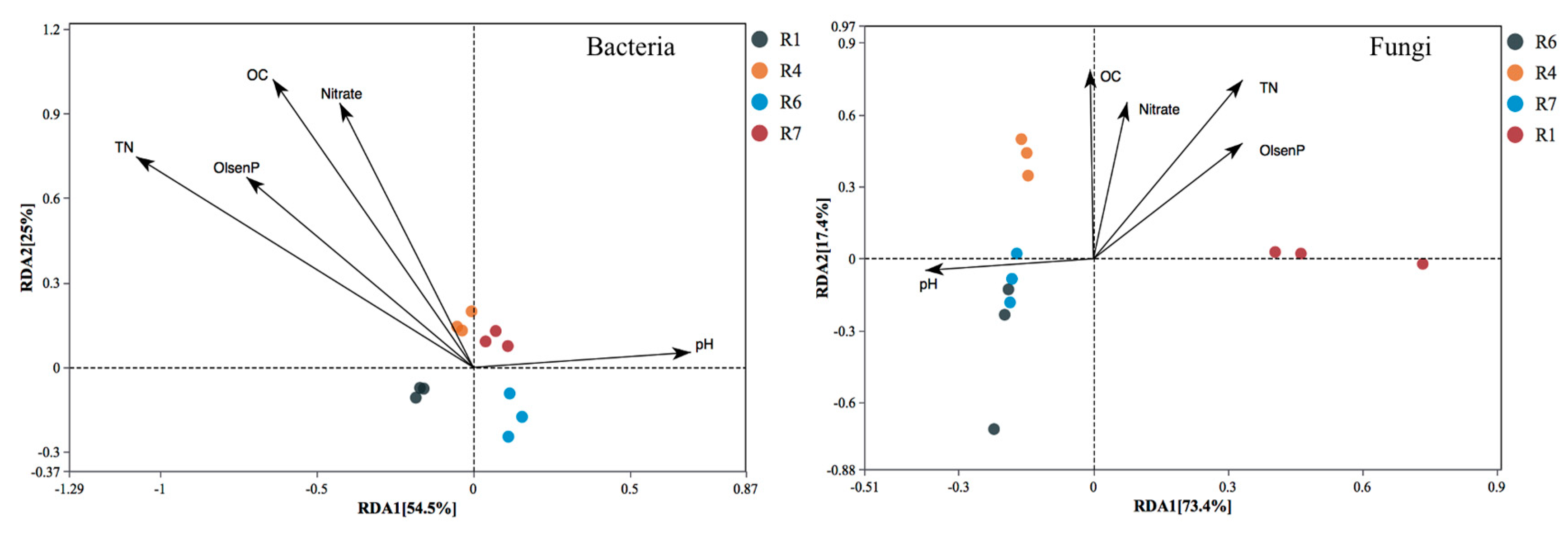
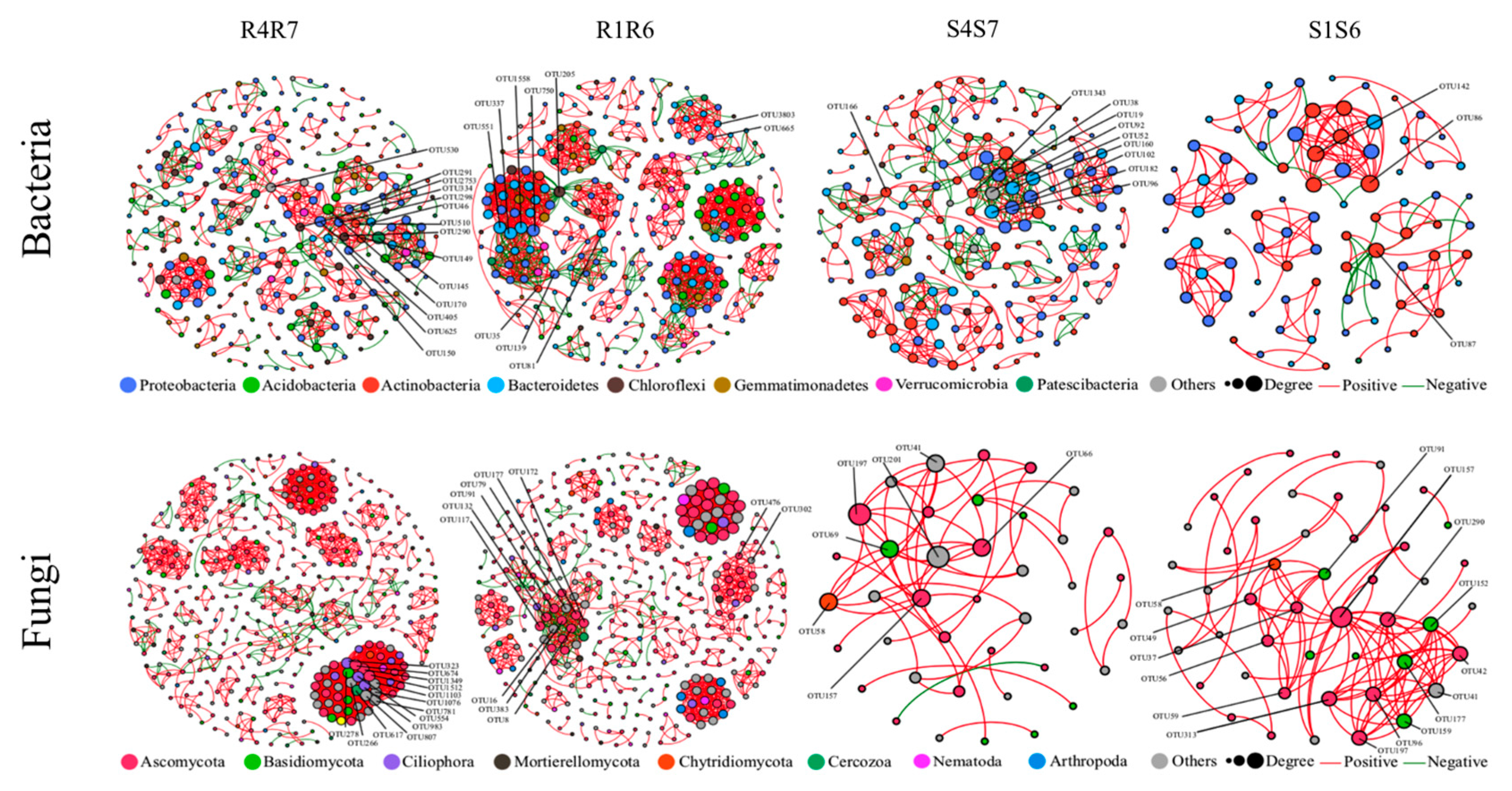

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Farming | Variety | α-Diversity of Bacteria | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Root Endosphere | Rhizosphere | ||||||||
| Richness | Chao1 | Shannon | Simpson | Richness | Chao1 | Shannon | Simpson | ||
| M ‡ | H ‡ | 319.3 c | 321.8 c | 4.64 c | 0.074 a | 2783.7 ab | 2785.2 ab | 9.10 ab | 0.006 a |
| M ‡ | C ‡ | 547.3 b | 548.4 b | 6.51 a | 0.035 a | 2874.0 a | 2875.3 a | 9.35 a | 0.004b |
| R ‡ | H ‡ | 646.3 ab | 647.3 ab | 5.45 b | 0.125 a | 2593.3 b | 2595.2 b | 8.66 b | 0.008 a |
| R ‡ | C ‡ | 896.0 a | 896.6 a | 7.77 a | 0.019 a | 2838.3 ab | 2839.8 ab | 9.29 a | 0.005 ab |
| Farming | *** | *** | * | ns | ns | ns | * | ns | |
| Variety | ** | ** | *** | ** | ns | ns | ** | * | |
| Farming × Variety | ns | ns | ns | ns | ns | ns | ns | ns | |
| Farming | Variety | α-Diversity of Fungi | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Root Endosphere | Rhizosphere | ||||||||
| Richness | Chao1 | Shannon | Simpson | Richness | Chao1 | Shannon | Simpson | ||
| M ‡ | H ‡ | 62.7 a | 68.4 a | 2.20 ab | 0.34 a | 463.3 a | 465.4 a | 514.1 b | 0.315 a |
| M ‡ | C ‡ | 68.7 a | 76.4 a | 3.56 a | 0.17 b | 386.7 a | 388.1 a | 403.0 a | 0.054 a |
| R ‡ | H ‡ | 67.0 a | 70.3 a | 2.18 b | 0.45 a | 354.3 a | 356.3 a | 426.5 ab | 0.116 a |
| R ‡ | C ‡ | 69.7 a | 82.0 a | 3.94 a | 0.15 b | 386.7 a | 388.4 a | 457.4 a | 0.057 a |
| Farming | ns | ns | ns | ns | ns | ns | ns | ns | |
| Variety | ns | ns | ** | ** | ns | ns | ** | * | |
| Farming × Variety | ns | ns | ns | ns | ns | ns | ns | ns | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, M.; Zhou, Y.; Xu, R.; Du, C.; Wang, R.; Lu, W.; Abudukadier, K.; Sun, Z. Contrasting Key Bacteria and Fungi Related to Sugar Beet (Beta vulgaris L.) with Different Resistances to Beet Rot under Two Farming Modes. Agronomy 2023, 13, 825. https://doi.org/10.3390/agronomy13030825
Lin M, Zhou Y, Xu R, Du C, Wang R, Lu W, Abudukadier K, Sun Z. Contrasting Key Bacteria and Fungi Related to Sugar Beet (Beta vulgaris L.) with Different Resistances to Beet Rot under Two Farming Modes. Agronomy. 2023; 13(3):825. https://doi.org/10.3390/agronomy13030825
Chicago/Turabian StyleLin, Ming, Yuanhang Zhou, Runlai Xu, Chenghang Du, Ronghua Wang, Weidan Lu, Kuerban Abudukadier, and Zhencai Sun. 2023. "Contrasting Key Bacteria and Fungi Related to Sugar Beet (Beta vulgaris L.) with Different Resistances to Beet Rot under Two Farming Modes" Agronomy 13, no. 3: 825. https://doi.org/10.3390/agronomy13030825