Long-Term N Fertilization Decreased Diversity and Altered the Composition of Soil Bacterial and Archaeal Communities

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Site Descriptions and Soil Sampling
2.2. Soil Characterization
2.3. Soil DNA Extraction, qPCR, and Sequencing
2.4. Bioinformatic Analysis
2.5. Statistical Analysis
3. Results
3.1. General Surface Soil Properties
3.2. Sequencing Summary
3.3. Soil Prokaryotic Community Diversity
3.4. Soil Prokaryotic Community Composition
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Robertson, G.; Vitousek, P.M. Nitrogen in Agriculture: Balancing the Cost of an Essential Resource. Annu. Rev. Environ. Resour. 2009, 34, 97–125. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.P.; Zhang, L.M.; Guo, J.F.; Ray, J.L.; He, J.Z. Impact of long-term fertilization practices on the abundance and composition of soil bacterial communities in Northeast China. Appl. Soil Ecol. 2010, 46, 119–124. [Google Scholar] [CrossRef]
- Figueiredo, M.; Seldin, L.; Araujo, F.; Mariano, R. Plant Growth Promoting Rhizobacteria: Fundamentals and Applications. In Plant Growth and Health Promoting Bacteria. Microbiology Monographs; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Gupta, V.V.S.R.; Germida, J.J. Soil aggregation: Influence on microbial biomass and implications for biological processes. Soil Biol. Biochem. 2015, 80, A3–A9. [Google Scholar] [CrossRef]
- LeBauer, D.S.; Treseder, K.K. Nitrogen limitation of net primary productivity in terrestrial ecosystems is globally distributed. Ecology 2008, 89, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.H.; Gu, T.; Wang, W.; Zhang, B.; Lin, X.G.; Huang, Q.R.; Shen, W.S. The effects of mineral fertilizer and organic manure on soil microbial community and diversity. Plant Soil 2010, 326, 511–522. [Google Scholar] [CrossRef]
- Daniel, R. The metagenomics of soil. Nat. Rev. Microbiol. 2005, 3, 470–478. [Google Scholar] [CrossRef]
- Myrold, D.D.; Zeglin, L.H.; Jansson, J.K. The Potential of Metagenomic Approaches for Understanding Soil Microbial Processes. Soil Sci. Soc. Am. J. 2014, 78, 3–10. [Google Scholar] [CrossRef]
- Paul Chowdhury, S.; Babin, D.; Sandmann, M.; Jacquiod, S.; Sommermann, L.; Sorensen, S.J.; Fliessbach, A.; Mader, P.; Geistlinger, J.; Smalla, K.; et al. Effect of long-term organic and mineral fertilization strategies on rhizosphere microbiota assemblage and performance of lettuce. Environ. Microbiol. 2019, 21, 2426–2439. [Google Scholar] [CrossRef]
- Francioli, D.; Schulz, E.; Lentendu, G.; Wubet, T.; Buscot, F.; Reitz, T. Mineral vs. Organic Amendments: Microbial Community Structure, Activity and Abundance of Agriculturally Relevant Microbes Are Driven by Long-Term Fertilization Strategies. Front. Microbiol. 2016, 7, 1446. [Google Scholar] [CrossRef] [Green Version]
- Babin, D.; Deubel, A.; Jacquiod, S.; Sørensen, S.J.; Geistlinger, J.; Grosch, R.; Smalla, K. Impact of long-term agricultural management practices on soil prokaryotic communities. Soil Biol. Biochem. 2019, 129, 17–28. [Google Scholar] [CrossRef]
- Hartmann, M.; Frey, B.; Mayer, J.; Mäder, P.; Widmer, F. Distinct soil microbial diversity under long-term organic and conventional farming. ISME J. 2015, 9, 1177–1194. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Riggins, C.; Rodríguez-Zas, S.; Zabaloy, M.; Villamil, M. Long-term N fertilization imbalances potential N acquisition and transformations by soil microbes. Sci. Total Environ. 2019, 691, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Lauber, C.L.; Ramirez, K.S.; Zaneveld, J.; Bradford, M.A.; Knight, R. Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 2012, 6, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Yevdokimov, I.; Gattinger, A.; Buegger, F.; Munch, J.C.; Schloter, M. Changes in microbial community structure in soil as a result of different amounts of nitrogen fertilization. Biol. Fertil. Soils 2008, 44, 1103–1106. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, H.; He, X.; Thomas, B.W.; Lupwayi, N.Z.; Hao, X.; Thomas, M.C.; Shi, X. Fertilization Shapes Bacterial Community Structure by Alteration of Soil pH. Front. Microbiol. 2017, 8, 1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Qiu, S.; Cao, C.; Zheng, C.; Zhou, W.; He, P. Responses of soil properties, microbial community and crop yields to various rates of nitrogen fertilization in a wheat–maize cropping system in north-central China. Agric. Ecosyst. Environ. 2014, 194, 29–37. [Google Scholar] [CrossRef]
- Ramirez, K.S.; Craine, J.M.; Fierer, N. Consistent effects of nitrogen amendments on soil microbial communities and processes across biomes. Glob. Chang. Biol. 2012, 18, 1918–1927. [Google Scholar] [CrossRef]
- Bender, S.F.; Wagg, C.; Van Der Heijden, M.G. An Underground Revolution: Biodiversity and Soil Ecological Engineering for Agricultural Sustainability. Trends Ecol. Evol. 2016, 31, 440–452. [Google Scholar] [CrossRef]
- Geisseler, D.; Scow, K.M. Long-term effects of mineral fertilizers on soil microorganisms—A review. Soil Biol. Biochem. 2014, 75, 54–63. [Google Scholar] [CrossRef]
- McDaniel, M.D.; Tiemann, L.K.; Grandy, A.S. Does agricultural crop diversity enhance soil microbial biomass and organic matter dynamics? A meta-analysis. Ecol. Appl. 2014, 24, 560–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suding, K.N.; Collins, S.L.; Gough, L.; Clark, C.; Cleland, E.E.; Gross, K.L.; Milchunas, D.G.; Pennings, S. Functional-and abundance-based mechanisms explain diversity loss due to N fertilization. Proc. Natl. Acad. Sci. USA 2005, 102, 4387–4392. [Google Scholar] [CrossRef] [PubMed]
- Powlson, D.S.; Stirling, C.M.; Jat, M.L.; Gerard, B.G.; Palm, C.A.; Sanchez, P.A.; Cassman, K.G. Limited potential of no-till agriculture for climate change mitigation. Nat. Clim. Chang. 2014, 4, 678–683. [Google Scholar] [CrossRef]
- Kramer, S.B.; Reganold, J.P.; Glover, J.D.; Bohannan, B.J.M.; Mooney, H.A. Reduced nitrate leaching and enhanced denitrifier activity and efficiency in organically fertilized soils. Proc. Natl. Acad. Sci. USA 2006, 103, 4522–4527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagadamma, S.; Lal, R.; Hoeft, R.G.; Nafziger, E.D.; Adee, E.A. Nitrogen fertilization and cropping systems effects on soil organic carbon and total nitrogen pools under chisel-plow tillage in Illinois. Soil Tillage Res. 2007, 95, 348–356. [Google Scholar] [CrossRef]
- Fierer, N.; Jackson, J.A.; Vilgalys, R.; Jackson, R.B. Assessment of soil microbial community structure by use of taxon-specific quantitative PCR assays. Appl. Environ. Microbiol. 2005, 71, 4117–4120. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Aronesty, E. Comparison of Sequencing Utility Programs. Open Bioinform. J. 2013, 7, 1–8. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Reddy, T.B.; Thomas, A.D.; Stamatis, D.; Bertsch, J.; Isbandi, M.; Jansson, J.; Mallajosyula, J.; Pagani, I.; Lobos, E.A.; Kyrpides, N.C. The Genomes OnLine Database (GOLD) v.5: A metadata management system based on a four level (meta) genome project classification. Nucleic Acids Res. 2015, 43, D1099–D1106. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Chao, A. Nonparametric-Estimation of the Number of Classes in a Population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Walther, B.A.; Moore, J.L. The concepts of bias, precision and accuracy, and their use in testing the performance of species richness estimators, with a literature review of estimator performance. Ecography 2005, 28, 815–829. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Warton, D.I.; Wright, S.T.; Wang, Y. Distance-based multivariate analyses confound location and dispersion effects. Methods Ecol. Evol. 2012, 3, 89–101. [Google Scholar] [CrossRef]
- Yeater, K.M.; Villamil, M.B. Chapter 14: Multivariate Methods for Agricultural Research, in Applied Statistics in Agricultural, Biological, and Environmental Sciences; American Society of Agronomy, Crop Science Society of America, and Soil Science Society of America, Inc.: Madison, WI, USA, 2018; pp. 371–400. [Google Scholar] [CrossRef]
- Cumming, G.; Fidler, F.; Vaux, D.L. Error bars in experimental biology. J. Cell Biol. 2007, 177, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Yang, Y.; Luo, Y.; Fang, C.; Zhou, X.; Chen, J.; Yang, X.; Li, B. Responses of ecosystem nitrogen cycle to nitrogen addition: A meta-analysis. New Phytol. 2011, 189, 1040–1050. [Google Scholar] [CrossRef]
- Leff, J.W.; Jones, S.E.; Prober, S.M.; Barberán, A.; Borer, E.T.; Firn, J.L.; Harpole, W.S.; Hobbie, S.E.; Hofmockel, K.S.; Knops, J.M.H.; et al. Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe. Proc. Natl. Acad. Sci. USA 2015, 112, 10967–10972. [Google Scholar] [CrossRef] [Green Version]
- Madigan, M.T.; Martinko, J.M.; Bender, K.S.; Buckley, D.H.; Stahl, D.A. Brock Biology of Microorganisms, 14th ed.; Pearson: Boston, MA, USA, 2015. [Google Scholar]
- Li, H.; Zhang, Y.; Yang, S.; Wang, Z.; Feng, X.; Liu, H.; Jiang, Y. Variations in soil bacterial taxonomic profiles and putative functions in response to straw incorporation combined with N fertilization during the maize growing season. Agric. Ecosyst. Environ. 2019, 283, 106578. [Google Scholar] [CrossRef]
- Ding, J.; Jiang, X.; Ma, M.; Zhou, B.; Guan, D.; Zhao, B.; Zhou, J.; Cao, F.; Li, L.; Li, J. Effect of 35 years inorganic fertilizer and manure amendment on structure of bacterial and archaeal communities in black soil of northeast China. Appl. Soil Ecol. 2016, 105, 187–195. [Google Scholar] [CrossRef]
- Guo, J.H.; Liu, X.J.; Zhang, Y.; Shen, J.L.; Han, W.X.; Zhang, W.F.; Christie, P.; Goulding, K.W.T.; Vitousek, P.M.; Zhang, F.S. Significant acidification in major Chinese croplands. Science 2010, 327, 1008–1010. [Google Scholar] [CrossRef] [PubMed]
- Barak, P.; Jobe, B.O.; Krueger, A.R.; Peterson, L.A.; Laird, D.A. Effects of long-term soil acidification due to nitrogen fertilizer inputs in Wisconsin. Plant Soil 1997, 197, 61–69. [Google Scholar] [CrossRef]
- Chávez-Romero, Y.; Navarro-Noya, Y.E.; Reynoso-Martínez, S.C.; Sarria-Guzmán, Y.; Govaerts, B.; Verhulst, N.; Dendooven, L.; Luna-Guido, M. 16S metagenomics reveals changes in the soil bacterial community driven by soil organic C, N-fertilizer and tillage-crop residue management. Soil Tillage Res. 2016, 159, 1–8. [Google Scholar] [CrossRef]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Debruyn, J.M.; Nixon, L.T.; Fawaz, M.N.; Johnson, A.M.; Radosevich, M. Global biogeography and quantitative seasonal dynamics of Gemmatimonadetes in soil. Appl. Environ. Microbiol. 2011, 77, 6295–6300. [Google Scholar] [CrossRef] [PubMed]
- Van Der Bom, F.; Nunes, I.; Raymond, N.S.; Hansen, V.; Bonnichsen, L.; Magid, J.; Nybroe, O.; Jensen, L.S. Long-term fertilisation form, level and duration affect the diversity, structure and functioning of soil microbial communities in the field. Soil Biol. Biochem. 2018, 122, 91–103. [Google Scholar] [CrossRef]
- Nemergut, D.R.; Townsend, A.R.; Sattin, S.R.; Freeman, K.R.; Fierer, N.; Neff, J.C.; Bowman, W.D.; Schadt, C.W.; Weintraub, M.N.; Schmidt, S.K. The effects of chronic nitrogen fertilization on alpine tundra soil microbial communities: Implications for carbon and nitrogen cycling. Environ. Microbiol. 2008, 10, 3093–3105. [Google Scholar] [CrossRef]
- Carbonetto, B.; Rascovan, N.; Álvarez, R.; Mentaberry, A.; Vazquez, M.P. Structure, composition and metagenomic profile of soil microbiomes associated to agricultural land use and tillage systems in Argentine Pampas. PLoS ONE 2014, 9, e99949. [Google Scholar] [CrossRef]
- Rinke, C.; Schwientek, P.; Sczyrba, A.; Ivanova, N.N.; Anderson, I.J.; Cheng, J.-F.; Darling, A.; Malfatti, S.; Swan, B.K.; Gies, E.A.; et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 2013, 499, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.X.; Méndez-García, C.; Dombrowski, N.; Servín-Garcidueñas, L.E.; Eloe-Fadrosh, E.A.; Fang, B.Z.; Luo, Z.H.; Tan, S.; Zhi, X.Y.; Hua, Z.S.; et al. Metabolic versatility of small archaea Micrarchaeota and Parvarchaeota. ISME J. 2018, 12, 756–775. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, P.H.; Abu Al-Soud, W.; Bergmark, L.; Magid, J.; Hansen, L.H.; Sørensen, S.J. Effects of fertilization with urban and agricultural organic wastes in a field trial—Prokaryotic diversity investigated by pyrosequencing. Soil Biol. Biochem. 2013, 57, 784–793. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
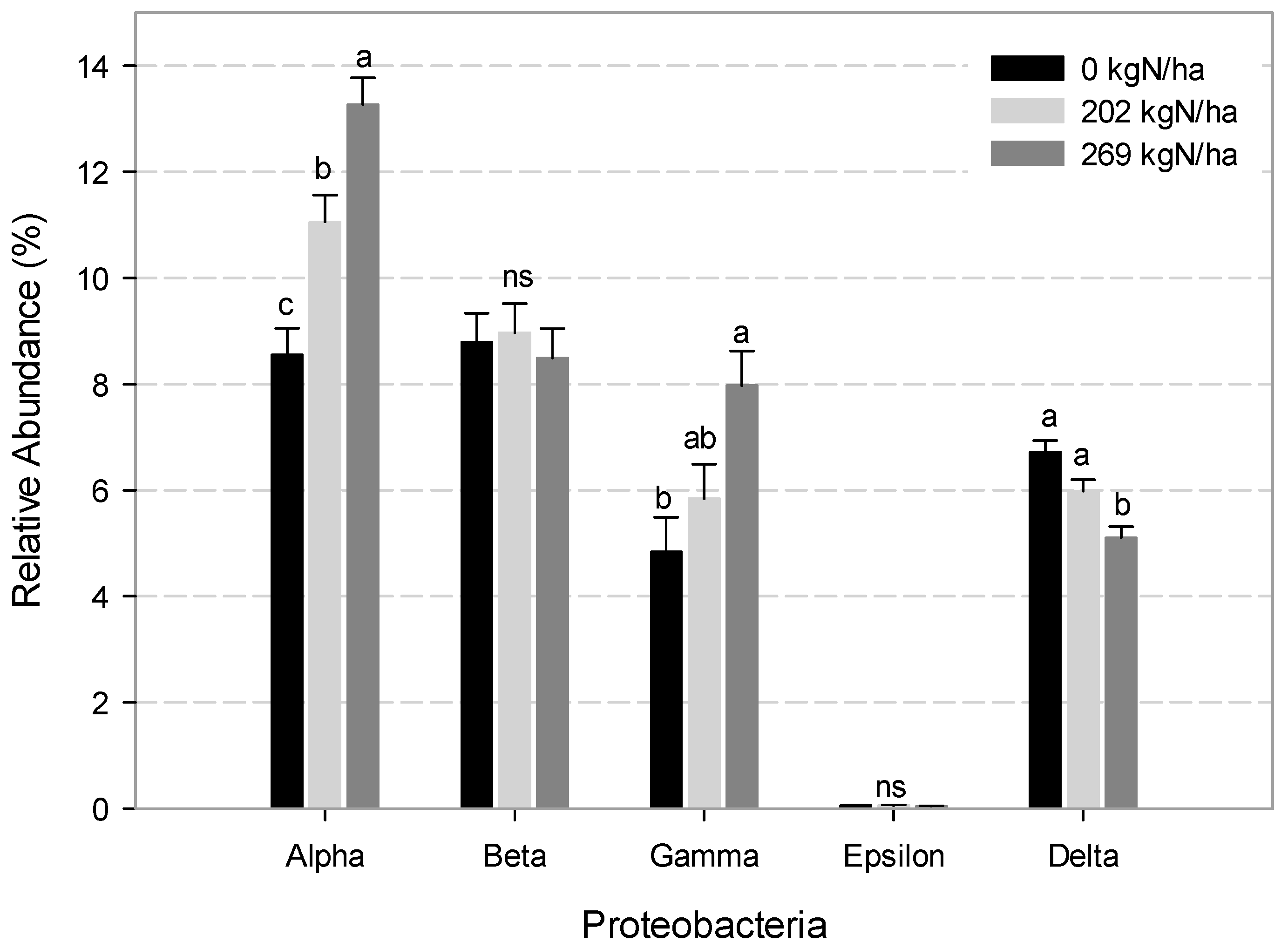{kind=link}
{kind=link}
| N Rate (kg N/ha) | pH | CEC (cmol/kg) | SOM (%) | NO3− mg/kg | NH4+ mg/kg |
|---|---|---|---|---|---|
| 0 | 6.33 a | 18.96 b | 3.62 a | 11.01 b | 2.71 b |
| 202 | 6.04 ab | 19.93 ab | 3.93 a | 12.63 b | 3.68 ab |
| 269 | 5.42 b | 22.51 a | 3.98 a | 24.47 a | 4.28 a |
| p-value | 0.0178 | 0.0490 | 0.2414 | 0.0045 | 0.0480 |
| N Rate (kg N/ha) | Observed OTUs | PD | Chao1 | Shannon’s | ACE | Fisher’s |
|---|---|---|---|---|---|---|
| 0 | 2258 a | 122.0 a | 2379 a | 10.00 a | 2340 a | 420.3 a |
| 202 | 2245 a | 120.8 a | 2367 a | 9.93 a | 2336 a | 414.0 a |
| 269 | 2055 b | 111.7 b | 2191 b | 9.62 b | 2151 b | 370.3 b |
| p-value | 0.0193 | 0.0034 | 0.0385 | 0.0007 | 0.0274 | 0.0102 |
| N Rate | 0 | 202 | 269 | |||
|---|---|---|---|---|---|---|
| Variable | Mean | SEM | Mean | SEM | Mean | SEM |
| Proteobacteria | 29.02 | 0.84 | 31.96 | 0.72 | 34.90 | 0.82 |
| Acidobacteria | 17.45 | 0.81 | 16.56 | 0.60 | 15.76 | 0.76 |
| Bacteroidetes | 11.83 | 0.67 | 12.53 | 0.96 | 10.22 | 0.53 |
| Actinobacteria | 9.22 | 0.87 | 7.59 | 0.49 | 8.24 | 0.72 |
| Chloroflexi | 5.85 | 0.43 | 4.59 | 0.30 | 4.07 | 0.20 |
| Verrucomicrobia | 5.57 | 0.20 | 5.99 | 0.18 | 5.09 | 0.23 |
| Gemmatimonadetes | 5.19 | 0.36 | 6.02 | 0.28 | 7.23 | 0.31 |
| Planctomycetes | 4.26 | 0.08 | 4.03 | 0.11 | 3.60 | 0.12 |
| Euryarchaeota | 1.87 | 0.07 | 2.25 | 0.06 | 2.39 | 0.13 |
| Crenarchaeota | 1.74 | 0.18 | 1.43 | 0.10 | 1.56 | 0.17 |
| Cyanobacteria | 1.45 | 0.33 | 0.50 | 0.14 | 0.23 | 0.03 |
| Firmicutes | 1.25 | 0.06 | 1.05 | 0.07 | 0.92 | 0.07 |
| Cumulative RA | 94.70 | 94.50 | 94.21 | |||
| N rate | 0 | 202 | 269 | |||
|---|---|---|---|---|---|---|
| Variable | Mean | SEM | Mean | SEM | Mean | SEM |
| WS3 | 0.980 | 0.110 | 0.640 | 0.050 | 0.270 | 0.050 |
| Nitrospirae | 0.850 | 0.080 | 0.450 | 0.050 | 0.330 | 0.040 |
| Armatimonadetes | 0.580 | 0.050 | 0.780 | 0.090 | 0.810 | 0.060 |
| Parvarchaeota | 0.340 | 0.040 | 0.860 | 0.130 | 1.070 | 0.160 |
| Aquificae | 0.180 | 0.040 | 0.460 | 0.090 | 0.700 | 0.110 |
| Tenericutes | 0.621 | 0.037 | 0.441 | 0.048 | 0.274 | 0.032 |
| Other | 0.568 | 0.063 | 0.415 | 0.034 | 0.427 | 0.062 |
| Elusimicrobia | 0.292 | 0.046 | 0.334 | 0.041 | 0.324 | 0.045 |
| Chlorobi | 0.181 | 0.024 | 0.174 | 0.024 | 0.222 | 0.055 |
| Spirochaetes | 0.143 | 0.033 | 0.149 | 0.030 | 0.119 | 0.018 |
| Fibrobacteres | 0.083 | 0.029 | 0.115 | 0.079 | 0.062 | 0.015 |
| OP3 | 0.075 | 0.019 | 0.034 | 0.010 | 0.026 | 0.006 |
| AD3 | 0.072 | 0.031 | 0.212 | 0.057 | 0.327 | 0.061 |
| Thermi | 0.070 | 0.015 | 0.040 | 0.011 | 0.047 | 0.011 |
| Lentisphaerae | 0.070 | 0.024 | 0.004 | 0.002 | 0.001 | 0.001 |
| Thermotogae | 0.051 | 0.013 | 0.036 | 0.013 | 0.009 | 0.004 |
| FCPU426 | 0.043 | 0.019 | 0.110 | 0.028 | 0.137 | 0.027 |
| Synergistetes | 0.033 | 0.009 | 0.011 | 0.003 | 0.004 | 0.003 |
| WS2 | 0.019 | 0.009 | 0.007 | 0.003 | 0.001 | 0.000 |
| BRC1 | 0.014 | 0.005 | 0.007 | 0.002 | 0.013 | 0.007 |
| EM19 | 0.014 | 0.006 | 0.009 | 0.003 | 0.011 | 0.008 |
| TM6 | 0.010 | 0.006 | 0.020 | 0.010 | 0.017 | 0.006 |
| TM7 | 0.008 | 0.006 | 0.053 | 0.018 | 0.240 | 0.089 |
| Deferribacteres | 0.007 | 0.004 | 0.004 | 0.002 | 0.007 | 0.003 |
| OD1 | 0.005 | 0.003 | 0.016 | 0.006 | 0.040 | 0.015 |
| Chlamydiae | 0.002 | 0.002 | 0.007 | 0.003 | 0.039 | 0.012 |
| WPS2 | 0.002 | 0.001 | 0.105 | 0.026 | 0.219 | 0.048 |
| OP11 | 0.001 | 0.001 | 0.007 | 0.004 | 0.019 | 0.012 |
| OP8 | 0.001 | 0.000 | 0.006 | 0.003 | 0.018 | 0.008 |
| Cumulative RA | 5.32 | 5.50 | 5.78 | |||
| PC1 | PC2 | PC3 | PC4 | PC5 | |
|---|---|---|---|---|---|
| Eigenvalue | 11.99 | 3.81 | 3.37 | 2.75 | 2.55 |
| Cum. Proportion | 0.29 | 0.39 | 0.47 | 0.53 | 0.60 |
| Microbial group | Eigenvalues | ||||
| Acidobacteria | 0.1 | 0.0 | 0.5 | 0.3 | 0.4 |
| Actinobacteria | 0.2 | −0.1 | −0.9 | 0.1 | −0.2 |
| AD3 | −0.7 | −0.5 | 0.0 | 0.0 | 0.0 |
| Aquificae | −0.8 | −0.5 | 0.0 | 0.1 | −0.1 |
| Armatimonadetes | −0.6 | −0.1 | −0.2 | 0.3 | −0.3 |
| Bacteroidetes | 0.4 | −0.1 | 0.3 | −0.6 | −0.3 |
| BR−C1 | 0.2 | 0.3 | 0.0 | 0.2 | −0.2 |
| Chlamydiae | −0.6 | 0.6 | −0.1 | 0.1 | 0.1 |
| Chlorobi | −0.2 | 0.7 | 0.1 | 0.0 | 0.1 |
| Chloroflexi | 0.7 | 0.1 | −0.6 | 0.1 | 0.3 |
| Crenarchaeota | 0.4 | 0.2 | 0.2 | 0.5 | −0.4 |
| Cyanobacteria | 0.7 | 0.0 | −0.3 | 0.0 | −0.1 |
| Deferribacteres | 0.1 | 0.4 | 0.2 | 0.3 | −0.2 |
| Elusimicrobia | −0.1 | −0.5 | 0.3 | 0.5 | 0.2 |
| EM19 | 0.1 | −0.4 | 0.3 | 0.1 | 0.6 |
| Euryarchaeota | −0.6 | 0.2 | 0.2 | 0.2 | 0.0 |
| FCPU426 | −0.7 | 0.0 | 0.0 | −0.2 | 0.3 |
| Fibrobacteres | 0.2 | 0.3 | −0.1 | 0.1 | 0.0 |
| Firmicutes | 0.7 | 0.2 | −0.1 | 0.2 | 0.0 |
| Gemmatimonadetes | −0.7 | −0.3 | −0.1 | 0.4 | 0.1 |
| Lentisphaerae | 0.6 | 0.2 | 0.4 | 0.3 | −0.2 |
| Nitrospirae | 0.8 | −0.1 | 0.2 | 0.4 | 0.2 |
| OD1 | −0.5 | 0.2 | 0.0 | −0.1 | 0.1 |
| OP11 | −0.3 | −0.2 | 0.5 | 0.0 | 0.4 |
| OP3 | 0.6 | 0.0 | −0.2 | 0.1 | 0.3 |
| OP8 | −0.4 | −0.3 | −0.1 | 0.0 | −0.3 |
| Other | 0.5 | 0.0 | −0.1 | 0.2 | −0.5 |
| Parvarchaeota | −0.7 | 0.3 | −0.1 | 0.2 | 0.0 |
| Planctomycetes | 0.7 | −0.4 | −0.1 | −0.1 | −0.1 |
| Proteobacteria | −0.7 | 0.3 | 0.2 | −0.3 | 0.2 |
| Spirochaetes | 0.2 | 0.4 | 0.5 | −0.3 | −0.1 |
| Synergistetes | 0.6 | −0.1 | −0.4 | 0.0 | 0.5 |
| Tenericutes | 0.8 | 0.0 | 0.0 | 0.2 | 0.2 |
| Thermi | 0.4 | 0.5 | 0.0 | −0.1 | 0.2 |
| Thermotogae | 0.6 | 0.1 | −0.1 | −0.4 | 0.1 |
| TM6 | −0.2 | 0.1 | 0.4 | 0.2 | −0.1 |
| TM7 | −0.6 | 0.6 | −0.2 | 0.0 | 0.2 |
| Verrucomicrobia | 0.4 | −0.3 | 0.4 | −0.3 | −0.5 |
| WPS2 | −0.8 | 0.0 | −0.2 | 0.3 | −0.2 |
| WS2 | 0.3 | −0.1 | 0.0 | −0.3 | 0.2 |
| WS3 | 0.8 | 0.0 | 0.4 | 0.3 | 0.0 |
| Factor | p-values | ||||
| N rate | <0.0001 | 0.6047 | 0.3156 | 0.3707 | 0.6335 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, R.; Zhang, P.; Riggins, C.W.; Zabaloy, M.C.; Rodríguez-Zas, S.; Villamil, M.B. Long-Term N Fertilization Decreased Diversity and Altered the Composition of Soil Bacterial and Archaeal Communities. Agronomy 2019, 9, 574. https://doi.org/10.3390/agronomy9100574
Sun R, Zhang P, Riggins CW, Zabaloy MC, Rodríguez-Zas S, Villamil MB. Long-Term N Fertilization Decreased Diversity and Altered the Composition of Soil Bacterial and Archaeal Communities. Agronomy. 2019; 9(10):574. https://doi.org/10.3390/agronomy9100574
Chicago/Turabian StyleSun, Renpeng, Pan Zhang, Chance W. Riggins, María C. Zabaloy, Sandra Rodríguez-Zas, and María B. Villamil. 2019. "Long-Term N Fertilization Decreased Diversity and Altered the Composition of Soil Bacterial and Archaeal Communities" Agronomy 9, no. 10: 574. https://doi.org/10.3390/agronomy9100574