14-3-3 Proteins are Regulators of Autophagy

Centro Andaluz de Biología Molecular y Medicina Regenerativa, Consejo Superior de Investigaciones Científicas. Av. Américo Vespucio s/n, Sevilla-41092, Spain
Cells 2012, 1(4), 754-773; https://doi.org/10.3390/cells1040754
Submission received: 30 June 2012
/
Revised: 3 August 2012
/
Accepted: 18 September 2012
/
Published: 15 October 2012
(This article belongs to the Special Issue Autophagy)
{kind=link}
Abstract
:14-3-3 proteins are implicated in the regulation of proteins involved in a variety of signaling pathways. 14-3-3-dependent protein regulation occurs through phosphorylation-dependent binding that results, in many cases, in the release of survival signals in cells. Autophagy is a cell digestion process that contributes to overcoming nutrient deprivation and is initiated under stress conditions. However, whether autophagy is a cell survival or cell death mechanism remains under discussion and may depend on context. Nevertheless, autophagy is a cellular process that determines cell fate and is tightly regulated by different signaling pathways, some of which, for example MAPK, PI3K and mTOR, are tightly regulated by 14-3-3 proteins. It is therefore important to understand the role of 14-3-3 protein in modulating the autophagic process. Within this context, direct binding of 14-3-3 to mTOR regulatory proteins, such as TSC2 and PRAS40, connects 14-3-3 with autophagy regulatory processes. In addition, 14-3-3 binding to human vacuolar protein sorting 34 (hVps34), a class III phosphatidylinositol-3-kinase (PI3KC3), indicates the involvement of 14-3-3 proteins in regulating autophagosome formation. hVps34 is involved in vesicle trafficking processes such as autophagy, and its activation is needed for initiation of autophagy. Chromatography and overlay techniques suggest that hVps34 directly interacts with 14-3-3 proteins under physiological conditions, thereby maintaining hVps34 in an inactive state. In contrast, nutrient starvation promotes dissociation of the 14-3-3–hVps34 complex, thereby enhancing hVps34 lipid kinase activity. Thus, 14-3-3 proteins are regulators of autophagy through regulating key components of the autophagic machinery. This review summarizes the role of 14-3-3 protein in the control of target proteins involved in regulating the master switches of autophagy.
1. Introduction
1.1. Overview of 14-3-3 Proteins
The cellular decision to live or die is finely controlled by tightly regulated signaling pathways. 14-3-3 proteins have a key role in this decision by controlling many of the signaling pathways that mediate this process.
14-3-3 proteins comprise a large family of acidic proteins that are expressed within all eukaryotic cells and function as homodimers and heterodimers [1,2]. They perform key regulatory roles by binding to and modulating the function of target proteins [3], principally through interacting with specific phosphoserine and phosphothreonine motifs [4]. Phosphorylation-dependent association with binding partners forms the mechanistic basis for the fundamental role for 14-3-3 in modulating kinase signaling pathways. A large number of 14-3-3 binding motifs have been established [5], and these have recently been reviewed [6]. Seven mammalian 14-3-3 isoforms have been reported (α/β, ε, γ, σ, ζ/δ, τ/θ and η) that are encoded by seven different genes and vary in their expression levels between tissues [7,8,9]. Moreover, highly redundant functions exist between the different 14-3-3 isoforms. Nevertheless, the 14–3-3σ isoform has distinct structural properties that principally promote the formation of homodimers, and this isoform is mainly associated with regulating cell proliferation. 14-3-3 binding can alter the enzymatic activity, subcellular localization, protein-protein interactions, phosphorylation status and proteolysis of target proteins [10]. Many 14-3-3 target proteins are deregulated in human diseases such as cancer, diabetes, Parkinson’s disease and other neurological diseases [11]. Furthermore, proteomics analysis suggests that 14-3-3 proteins are central regulators of different biological processes, including cell signaling, cell cycle progression, cytoskeletal dynamics, cell metabolism and making the decision between cell death and survival [12,13,14,15,16,17,18,19].
1.2. Role of 14-3-3 in Apoptosis
The role of 14-3-3 in apoptosis has been well documented and several reports indicate that 14-3-3 protein interaction with target binding partners initiates events that support cell survival, thus mediating an essential anti-apoptotic signal [20]. It has been reported that 14-3-3 proteins bind to members of the Bcl-2 family, Bcl-xL⁄Bcl-2-associated death promoter (BAD) and Bcl-2-associated X protein (BAX), thereby inhibiting their proapoptotic activities [21,22]. Additionally, 14-3-3 overexpression blocks cell death initiated by other death promoters, such as apoptosis signal-regulating kinase 1 (ASK1) [23]. Moreover, 14-3-3 protein interaction with a member of the forkhead family of transcription factors, forkhead box protein (Fox03a), blocks nuclear translocation and the transcription of death genes [24]. Thus, 14-3-3-dependent apoptosis suppression through association with ASK1, BAD and Fox03a suggests that 14-3-3 has a key anti-apoptotic function in cells. Furthermore, expression of a polypeptide that blocks 14-3-3 association with binding proteins promotes apoptosis and reduces viability in several cancer cell lines [25,26]. The use of 14-3-3 ζ antisense RNA molecules in cancer cell lines increases sensitivity to stress-induced apoptosis [27,28,29]. In addition, cell treatment with 2-methoxyestradiol results in reduced 14-3-3 expression that promotes apoptosis activation and inhibits cell growth [30]. A recent comprehensive proteomics analysis of 14-3-3-binding proteins in unchallenged cells, compared with those subject to an apoptosis stimulus, suggests new cell survival functions for 14-3-3 proteins [16]. Taking into account the finding that 14-3-3 protein association with binding partners induces a cell survival signal [20], it is of great interest to analyze the role of 14-3-3 protein in a mechanism that controls cell fate, such as autophagy.
1.3. Autophagy Process Summary
Macroautophagy (hereafter referred to as autophagy) is a self-digestion process that is enhanced by several cancer-related stimuli, as well as starvation, TNFα and ceramide. Autophagy may promote cell adaptation to starvation conditions produced by reduced extracellular or intracellular nutrients as a consequence of loss of growth factor signaling. Autophagy is characterized by the engulfment of the cell cytoplasm and organelles in double membrane vesicles, or autophagosomes, resulting in their degradation [31]. Autophagic degradation products are used as source materials for metabolic processes that enable the cell to obtain sufficient energy in order to survive under conditions of cellular stress and nutrient deprivation [32]. Under normal growth conditions, autophagy is a highly regulated process involved in the turnover of long-lived proteins or in eliminating damaged organelles [33,34]. It is notable that excessive autophagy may promote cell death [35].
The autophagic machinery includes groups of proteins that control autophagosome formation and thus the rate of degradation of sequestered material and cellular energy levels. Autophagosomes fuse with lysosomes to form autolysosomes, in which cellular components are degraded by lysosomal hydrolases [36] through a series of steps (reviewed in [37]). In yeast, the induction of autophagy involves inhibition of serine/threonine kinase TOR (target of rapamycin), which under normal growth condition blocks autophagy by constitutive phosphorylation of autophagy protein-13 (Atg13). Following TOR inhibition, Atg13 forms a protein complex containing Atg1 kinase and Atg17, which induces membrane isolation. The mammalian homologues of Atg1 are ULK1 and ULK2 (Unc-51-like kinase 1 and 2) and the Atg17 homologue is FIP200 (focal adhesion kinase family-interacting protein of 200 kDa) [38]. The ULKs and FIP200 form a complex with mammalian Atg13 that translocates to the phagophore under conditions of starvation and mediates autophagy initiation [39]. Under normal growth conditions, the mammalian homologous of Atg1 is inhibited by mammalian target of rapamycin signaling complex 1 (mTORC1) [40]. In the mammalian system, p62/SQSTM1 recognizes ubiquitinated proteins and binds to LC3 (microtubule-associated protein 1 light chain 3), leading to sequestration of polyubiquitinated proteins within the autophagosome and their subsequent degradation [41,42]. The subsequent activation of mammalian Vps34, a class III phosphatidylinositol 3-kinase (PI3KC3), generates phosphatidylinositol-3-phosphate (PtdIns3P) and initiates vesicle nucleation. Vps34 activation is dependent on the formation of a multiprotein complex involving Beclin-1, UVRAG (UV irradiation resistance-associated tumor suppressor gene), a myristylated kinase p150 and Atg14L. Vesicle elongation involves two ubiquitin-like conjugation systems. Thus, the conjugation of phosphatidylethanolamine to LC3 transforms the soluble form of LC3 (named LC3-I) to the autophagic vesicle-binding form (LC3-II). GFP-tagged LC3-II can be used as a marker of autophagy induction, as membrane association promotes a shift from a diffuse to a punctuate fluorescent signal. Autophagosomes mature by fusion with lysosomes to form autolysosomes that degrade the contents of the autophagic vacuoles using lysosomal enzymes [37].
Under different experimental conditions, autophagy has been reported to constitute a stress adaptation to avoid cell death, a failed attempt to rescue stressed cells from death or an alternative cell death pathway [36,43,44,45,46,47]. Whether autophagy is a survival strategy, a death mechanism or both is still being debated: autophagy is essential for maintaining cell survival under conditions of nutrient and growth factor deprivation, but C2-ceramide-induced autophagy may be a form of caspase-independent cell death [48]. Within this dichotomy, determining the role of 14-3-3 proteins in autophagy regulation is of key interest.
14-3-3 proteins form a ubiquitous eukaryotic adaptor protein family involved in numerous cell biology processes, including those related to cell survival or death decisions. This review summarizes the involvement of 14-3-3 family members in cell fate, and emphasizes their recently discovered regulatory roles in autophagy.
2. Role of 14-3-3 Proteins in Autophagy
2.1. Regulation of TCS2 by 14-3-3 Proteins
Autophagy is tightly controlled by signaling pathways that sense the energy and nutrient status of cells. The AMP-activated protein kinases (AMPKs) are important cellular sensors of energy levels that function by sensing the AMP:ATP ratio and regulating metabolic pathways. AMPKs are activated by AMP binding to their γ-subunit, leading to phosphorylation of Thr172 within the activation loop of the catalytic α-subunit. This phosphorylation is promoted by an upstream kinase, such as the tumor suppressor liver kinase B1 (LKB1/STK11) [49]. AMPKs respond to low energy stress by suppressing cell growth and biosynthesis, in part through inhibition of the TOR signaling complex 1 (TORC1), an established regulator of autophagy. TORC1 plays a critical role in sensing and responding to low energy conditions by linking nutrient levels with cell fate [50]. TORC1 controls biogenesis and protein translation through phosphorylation of S6 kinase 1 (S6K1) and eIF4E-binding protein 1 (4E-BP1). An important modulator of the mammalian TORC1 (mTORC1) is the GAP-containing protein complex of tuberous sclerosis proteins 1 and 2 (TSC1/hamartin and TSC2/tuberin). The TSC2 tumor suppressor is reported to be phosphorylated by AMPK at conserved serine sites [51,52,53,54]. Through this phosphorylation-dependent mechanism, TSC1/2 inhibit the Rheb GTPase (a Ras homologue enriched in brain), which is an inducer of mTORC1, thus suppressing mTORC1 activation and leading to autophagy initiation [55]. In addition, under normal growth conditions, stimulation of the insulin signaling pathway promotes Akt (protein kinase B) activation by phosphoinositide-dependent kinase-1(PDK1) and the rapamycin-insensitive companion of TOR (RICTOR)-containing TOR complex 2 (TORC2). Following IGF-1 or insulin stimulation, Akt phosphorylate TSC2 protein at Ser939 and Thr1462, leading to 14-3-3 protein association. Further analysis using point mutants indicates that TSC2 phospho-Ser939 is the primary recognition motif for 14-3-3 proteins [56]. However, other reports indicate that TSC2 Ser1210 phosphorylation by MAP-kinase activated protein kinase 2 (MK2) enhances the interaction between TSC2 and 14-3-3 proteins. Additional studies also established that TSC2 localization and its potential to regulate mTOR/S6K are controlled via 14-3-3 binding [57], which suggests an essential role for 14-3-3 in controlling the initial steps of autophagy through regulating the mTORC1 pathway. Thus, once TSC2 is inhibited by this 14-3-3 binding, GTP-bound Rheb can activate the mTORC1 complex, resulting in protein synthesis, cell growth and inhibition of autophagy [58] (Figure 1).
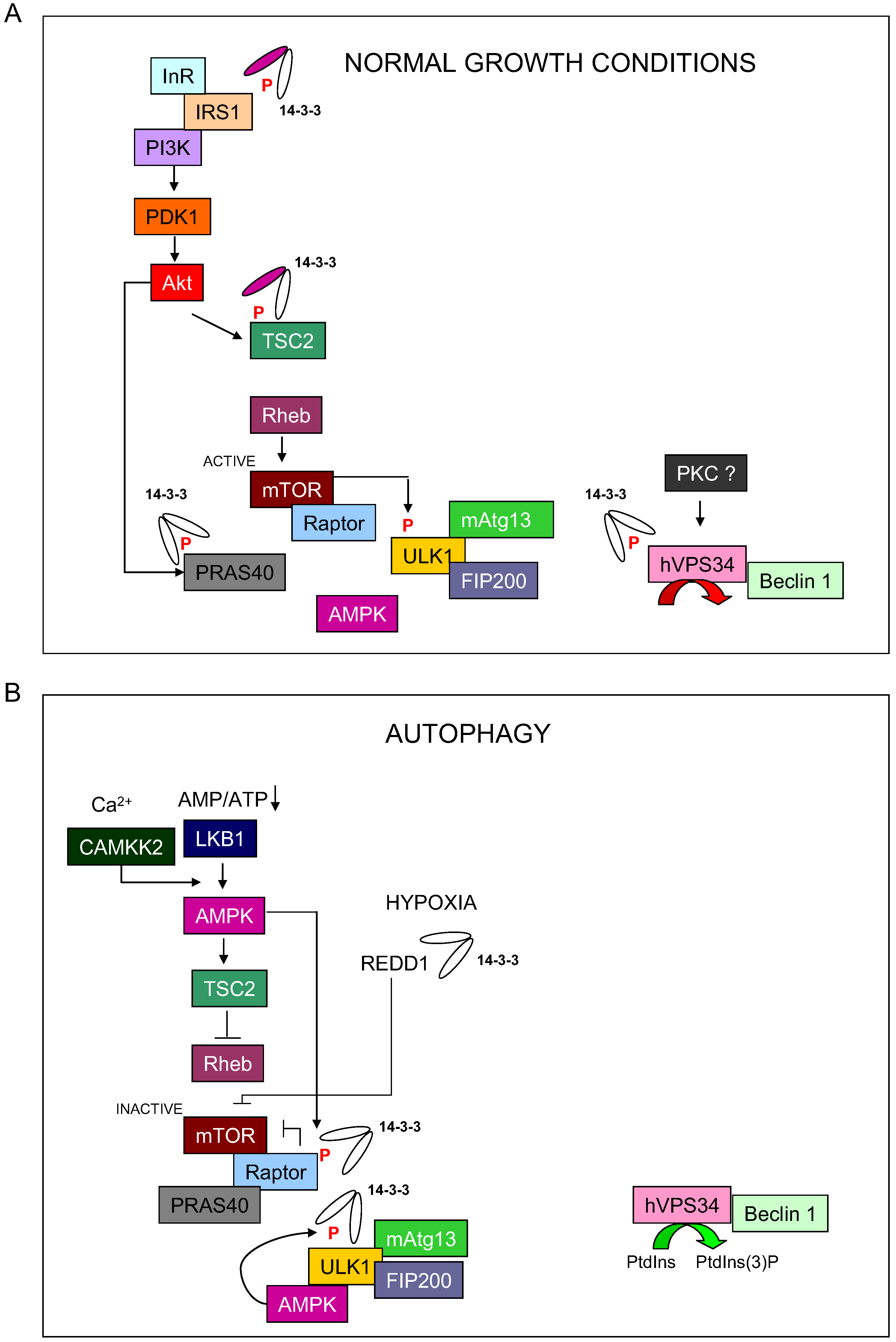
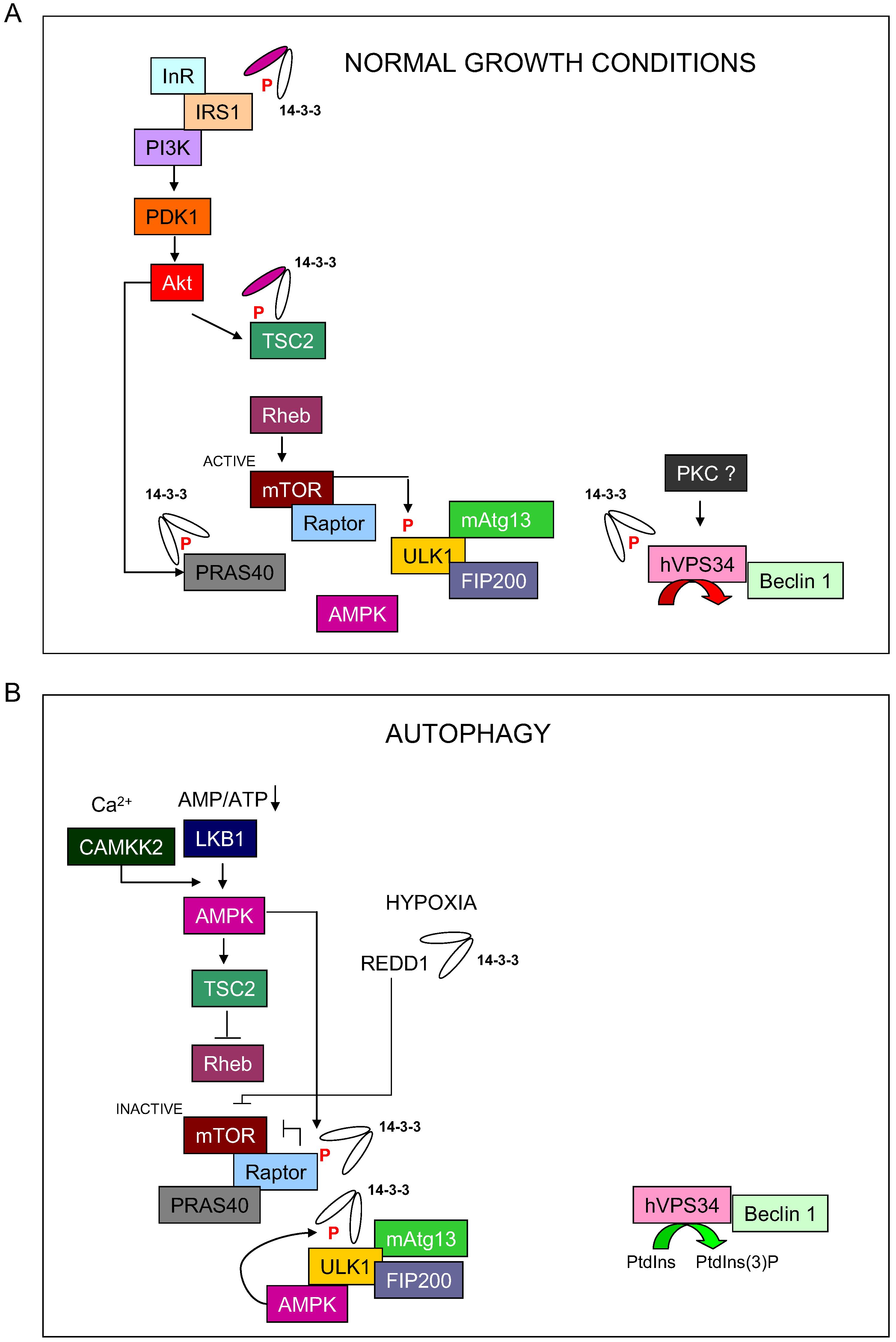
Figure 1.
Model of 14-3-3 regulation on autophagy initiation process. (A) During normal growth conditions, 14-3-3 proteins bind and regulate TCS2, PRAS40 and hVps34 to block autophagy. (B) During nutrient starvation, 14-3-3 proteins bind to Raptor and ULK1 to promote the initiation of autophagy.
Figure 1.
Model of 14-3-3 regulation on autophagy initiation process. (A) During normal growth conditions, 14-3-3 proteins bind and regulate TCS2, PRAS40 and hVps34 to block autophagy. (B) During nutrient starvation, 14-3-3 proteins bind to Raptor and ULK1 to promote the initiation of autophagy.
2.2. Regulation of PRAS40 by 14-3-3 Proteins
In addition to TSC2, the 14-3-3-binding partner proline-rich Akt substrate 40 (PRAS40) is reported to be essential for mediating mTORC1 signaling and triggering autophagy [59]. PRAS40 is a component of the mTORC1 complex that probably interacts with Raptor, which mediates PI3K pathway regulation of TORC1 signaling under nutrient or serum deprivation or following inhibition of mitochondrial metabolism [60]. Phosphorylation of PRAS40 Thr246 by Akt and PRAS40 Ser183 and Ser221 by mTORC1 promotes 14-3-3 binding to PRAS40 and its dissociation from mTORC1 [61]. Phosphorylation of PRAS40 Thr246 has been reported to facilitate the efficient phosphorylation of PRAS40 on its mTORC1-dependent sites [62]. In addition, Ser221 to Ala mutation reduces 14-3-3 interaction to the same extent as mutation of the Thr246 Akt site. Moreover, Ser221 mutation increases PRAS40 inhibition of mTORC1 [61]. PRAS40 binding to 14-3-3 proteins is blocked by TSC1/2 and stimulated by Rheb in a rapamycin-sensitive manner, suggesting an important role for PRAS40 in regulating mTORC1 [63]. Thus, PRAS40 functions as a negative regulator when bound to mTORC1, and dissociates from mTORC1 in response to insulin stimulation. PRAS40 phosphorylation and 14-3-3-binding therefore mediate the regulation of mTORC1 by nutrient and growth factor availability, leading to initiation of the autophagy process.
2.3. Regulation of Raptor by 14-3-3 Proteins
The mTOR binding partner Raptor has been reported to be a direct substrate of AMPK. Raptor phosphorylation by AMPK at two highly conserved serine residues, Ser722 and Ser792, promotes direct binding to 14-3-3, thus leading to inhibition of mTORC1 kinase activity [64]. AMPK phosphorylation of Raptor is thought to lead to changes in the availability of 14-3-3 for PRAS40 binding, which acts to suppress mTOR kinase activity, thus leading to autophagy initiation.
2.4. Regulation of REDD by 14-3-3 Proteins
In addition, hypoxic stress suppresses mTOR activity and induces autophagy. REDD1 (Regulated in development and DNA damages responses 1) expression is reported to be highly induced in response to hypoxia [65]. In mammalian cells, REDD1 overexpression inhibits mTORC1 activity, while suppression of REDD1 prevents mTORC1 downregulation in response to hypoxia [66,67]. Regulation of mTORC1 activity in response to hypoxia has been suggested to occur through REDD1-mediated dissociation of the TCS2–14-3-3 complex. 14-3-3 binding to REDD1 is necessary and sufficient to promote TSC2–14-3-3 dissociation and mTORC1 inhibition [68]. Thus, REDD1 specifically blocks PI3K/Akt-induced TSC2–14-3-3 association and promotes mTORC1 inactivation, leading to autophagy initiation under hypoxia conditions.
In addition, 14-3-3 proteins may control mTORC1 through PI3K pathway regulation in response to nutrient and growth factor depletion by direct binding to IRS-1 (insulin receptor substrate-1). 14-3-3 protein interaction with IRS-1 promotes dissociation of the IRS-1–PI3K class I complex during the insulin desensitization process [69].
2.5. 14-3-3 Proteins Regulate MAPK Pathway
Mitogen-activated protein kinases (MAPKs) are serine/threonine kinases that respond to signals such as growth factors and stress, leading to MAPK-dependent activation of a signaling cascade comprising of kinases and transcription factors. MAPK family members include ERK, which is activated by growth signals, and p38 and JNK, which are activated in response to various stresses. Extracellular signals promote activation of Ras, which then binds to and activates Raf, leading to phosphorylation and activation of the two ERK isoforms, ERK1 (p44) and ERK2 (p42) [70]. ERK has been proposed to control autophagy, as well as cell proliferation, migration, differentiation and death [71,72]. ERK induction of autophagy occurs in response to anti-tumor/cytotoxic stimuli through modulating Beclin-1 expression [73,74,75,76,77,78]. In contrast, ERK has also been reported to facilitate malignant growth by inhibiting autophagy-associated tumor suppression [79]. Within these contrasting processes, 14-3-3 proteins may function as adaptor proteins to hold Raf-1 in a partially active conformation prior to full activation [80]. For instance, when the N-terminal Raf-1 14-3-3-binding site is mutated, Raf-1 can activate the MAPK pathway, but is unable to induce cell transformation and differentiation [81]. In addition, 14-3-3 binding to PKA-phosphorylated Raf-1 inhibits Raf-1 translocation to the plasma membrane for interaction with its upstream activator Ras [82].
On the other hand, p38 MAPK has been implicated in cell cycle inhibition, the induction of apoptosis and terminal differentiation [83]. However, p38 has also been implicated in tumor progression, with reported roles in inducing cell invasion, angiogenesis and inflammation [83]. Despite this controversy, p38 is also known to regulate autophagy [84]. Induction of autophagy by p38 is reported to involve increased expression of Atg proteins, such as Beclin-1 and Atg5 [85,86,87,88]. However, other studies have linked p38 inhibition to increased Beclin-1 expression and the induction of autophagy [89,90]. It was recently suggested that p38 inhibits autophagy by competing with the transmembrane protein mAtg9 for binding to p38 interacting protein (p38IP), which is required for mAtg19 cycling during autophagy [84,91].
In this context, MAPK/ERK kinase kinase 3 (MEKK3) is activated by autophosphorylation of Ser526. Association between MEKK3 and 14-3-3 seems to be dependent on Ser526 phosphorylation and this interaction prevents dephosphorylation of Ser526 by PP2A [92]. Thus, transfection of cultured fibroblasts with a double mutant form of 14-3-3 ζ (DN-14-3-3-ζ) inhibited serum-stimulated ERK/MAPK activation, but increased the basal activation of JNK1 and p38 MAPK [93].
2.6. 14-3-3 Proteins Regulate Protein Kinase C
Protein kinase C (PKC) comprises a family of phospholipid-dependent serine/threonine kinases that regulate diverse cellular functions. On the basis of their requirement for Ca2+ and diacylglycerol and their structural characteristics, PKC isoforms involved in distinct roles in signal transduction pathways are classified as conventional (PKCα, βI, βII and γ), novel (PKCδ, ε, η and θ) and atypical (PKCζ and ɩ) [94]. The hypothesis that PKCs may have a role in autophagy originated with the identification of PKCδ as a suppressor of autophagy in pancreatic ductal carcinoma cells [95,96]. In contrast, PKCδ is thought to promote autophagy in rat parotid epithelial cells in response to hypoxic stress [97]. Thus, a controversy exists regarding the role of PKCδ role in determining cell fate, and PKCδ activation is thought to induce or inhibit autophagy depending on cellular context and the potency of the stimulus. PKCθ is also reported to be involved in endoplasmic reticulum (ER) stress-induced autophagy but has no involvement in amino acid starvation-induced autophagy [98]. ER stress inducers lead to calcium-dependent phosphorylation of PKCθ and translocation of PKCθ to LC3-II containing vesicles in the cytoplasm. In contrast, other conventional PKCs isoforms have been proposed to inhibit autophagy induced by starvation or rapamycin in a PI3K-independent manner [99]. Despite this controversy, a regulatory role for PKC isoforms in autophagy has been reported by several groups. On the other hand, 14-3-3 proteins have been reported to regulate PKC activity, although the nature of this effect varies between PKC isoforms: classical PKC isozymes show an approximate twofold activation, PKCδ shows no significant increase in activity, while PKCε is strongly activated by 14-3-3 proteins [100]. In addition, 14-3-3ε has been reported to modulate PKCα activity [101]. Moreover, several other studies have established that an interaction exists between 14-3-3 ζ and PKCs in rat retina, rodent brain and PC12 cells [102,103]. As mentioned above, there are contradictory reports regarding modulation of PKC activity by 14-3-3 proteins, with either inhibition [104,105,106] or activation [107,108] of PKCs, depending on the 14-3-3 and PKC isoforms involved and the cellular conditions.
2.7. 14-3-3 Proteins Regulate CaMKKβ
It is interesting to note that under ER stress, induction of autophagy facilitates the removal of unfolded proteins [109]. Among the ER stress-activated kinases, Ca2+/calmodulin-dependent kinase kinase-beta (CaMKKβ) binds 14-3-3, which may have important consequences for the regulation of Akt, CaMKI, CaMKIV and ERK signaling pathways [110]. Under ER stress conditions, the combined effect of CaMKKβ activation and the activation of death associated protein kinase (DAPK) and PKR-like ER-regulated kinase (PERK) can lead to upregulation of autophagy-related genes and downregulation of autophagy inhibitors.
3. Role of 14-3-3 Proteins during Autophagy Initiation Process
3.1. 14-3-3 Proteins Regulate ULK1
Autophagy is a highly regulated degradation process due to the energy cost involved and its important role in determining cell fate, and many different pathways cooperate to regulate this degradation process. In addition, genetic screens in yeast have identified a number of autophagy-related genes, including the serine/threonine protein kinase Atg1 that forms an active complex with Atg13 and Atg17 to regulate the autophagy initiation process [111,112,113]. Under normal physiological conditions, Atg13 is phosphorylated by TOR leading to Atg1–Atg13–Atg17 complex dissociation and inhibition of autophagy [114]. In mammals, the Atg1 kinase homologues ULK1 and ULK2 [115] are involved in starvation- and rapamycin-induced autophagy [39,115,116,117]. AMPK is reported to induce autophagy by directly activating ULK1 through phosphorylation during nutrient starvation. Two independent groups demonstrated that AMPK directly phosphorylates ULK1, although controversy exists regarding the ULK1 phosphorylation sites: Ser317 and Ser777 have been identified by one group and Ser722 and Ser792 by another [118,119]. In any case, phosphorylation at these sites seems to be required for full activation of ULK1 in response to glucose starvation. ULK1 has also been identified as an AMPK-binding protein during starvation, and the association is mediated by a proline–serine rich (PS) domain in ULK1. Further analysis revealed that AMPK-dependent ULK1 Ser555 phosphorylation promotes 14-3-3-binding by ULK1 in vivo and in vitro. Thus, Ser555 to Ala mutation blocks 14-3-3 binding to ULK1, and in vivo ULK1 phosphorylation is inhibited by expression of a dominant-negative AMPK mutant. Although further investigation is required, these data suggest a role for 14-3-3 in regulating a key protein during the autophagy initiation process. Moreover, mTOR has been reported to interact with the ULK1 kinase domain. In addition, formation of a complex between ULK1, mTORC1, and AMPK coincides with 14-3-3–Raptor binding and initiation of autophagy [119]. Under feeding conditions, increased mTOR activity blocks ULK1 activation through Ser757 phosphorylation, leading to ULK1–AMPK complex dissociation and inhibition of autophagy [118]. These results show ULK1 to be an important point of convergence between pathways that control the autophagy initiation process, which mediates feeding signals from mTOR and starvation signals from AMPK. While a role for 14-3-3 in this pathway has been suggested, further investigation is needed to establish the mechanism whereby 14-3-3 controls ULK1.
3.2. 14-3-3 Proteins Regulate hVps34 (PI3KC3)
14-3-3 proteins have also been reported to interact with proteins involved in the vesicle nucleation process during autophagy, such as hVps34 (human version of yeast Vps34), the class III phosphatidylinositol-3-kinase (PI3KC3). Phosphoinositide 3-kinases are enzymes that catalyze phosphorylation of the 3ʹ hydroxyl group of the inositol ring in phosphatidylinositol (PtdIns). PI3Ks are classified into three types based on their structure, substrate specificity and functionality [120]. Class I PI3Ks are usually activated by growth factors, leading the production of phosphatidylinositol-3-phosphate PtdIns(3)P, PtdIns(3,4)P2, and PtdIns(3,4,5)P3. These second messengers increase the membrane localization of pleckstrin-homology (PH) domain-containing proteins, such as PDK1 and its substrate Akt, leading to their activation [121]. Class II PI3Ks appear to be related to cell migration and the control of vascular smooth muscle contraction [122,123]. There is only one class III PI3K member, Vps34 (vacuolar protein sorting 34), which has been shown to mediate vesicular trafficking of proteins containing a PtdIns(3)P binding domain, leading to their localization to the endosomal and lysosomal membranes [124]. Vps34 plays an important role in mediating autophagosome formation by promoting the rapid recruitment of proteins with phospholipid-binding domain, such as the WD40-repeat proteins hWIPI-1alpha [125] and hWIPI2 [126], the human orthologues of the yeast autophagy gene Atg18, to the autophagosome structure during the vesicle nucleation process. WIPI-1alpha has been reported to colocalize with LC3 under conditions of nutrient deprivation [125]. Additional data suggests that the Atg18 phospholipid-binding domain binds PtdIns(3)P and is involved in the formation of the pre-autophagosome structure [127]. Furthermore, the presence of WD40 repeats in hWIPI may mediate multiprotein complex formation that may either play a scaffolding role during autophagosome generation or be involved in LC3 lipidation [125,126,128].
hVps34 forms a complex with Beclin-1 that participates in autophagosome formation. Regulatory mechanisms directing Beclin-1/hVps34 specificity were initially shown to involve complex formation with proteins such as Atg14L and Rubicon. Further analysis of these proteins suggested that Atg14L enhances hVps34 lipid kinase activity and upregulates autophagy, whereas Rubicon reduces hVps34 activity and downregulates autophagy [129]. Following initial reports that 14-3-3 proteins may also regulate hVps34 [130], 14-3-3-binding protein affinity purification and DIG-14-3-3 overlay assays revealed that hVps34 directly interacts with 14-3-3 under normal growth condition in a phosphorylation-dependent manner. Additionally, 14-3-3 was established to dissociate from hVps34 during C2-ceramide-induced autophagy, probably in response to protein phosphatase activation during C2-ceramide treatment. Thus, endogenous 14-3-3 and hVps34 proteins interact under normal growth conditions and this association is reversed during autophagy initiation by starvation or treatment with C2-ceramide, rapamycin or etoposide. Furthermore, hVps34 lipid kinase activity is increased following dissociation from 14-3-3 during starvation-induced autophagy or downregulation of 14-3-3 ζ. Moreover, 14-3-3 ζ overexpression decreases hVps34 activity under normal growth conditions. These data suggest that 14-3-3 proteins regulate hVps34 activity, leading to important effects on the autophagy initiation process. As mention above, 14-3-3 regulation of hVps34 seems to be dependent on phosphorylation. Stimulation of cells with the phorbol ester PMA promotes hVps34–14-3-3 binding and decreases hVps34 kinase activity. This effect is dependent on the PKC inhibitor H-7, suggesting that PKC isoforms, or a kinase activated downstream of PKC, induce phosphorylation-dependent hVps34 binding to 14-3-3. Moreover, in vitro phosphorylation by PKC promotes hVps34–14-3-3 binding. Additionally, experiments using deletion mutants and site-directed mutagenesis indicate that the N-terminal region of hVps34 mediates 14-3-3 binding and that phosphorylation at several sites may be required for hVps34–14-3-3 binding (Thr197 and Ser212). These data are consistent with the negative regulation of autophagy by PKC and the role of 14-3-3 in negatively regulating both hVps34 and the autophagy initiation process. Data also suggest that 14-3-3 ζ overexpression inhibits autophagy promoted by C2-ceramide or starvation in HeLa and HEK293T cells. Furthermore, depletion of 14-3-3 ζ promotes autophagy in cervix and breast cancer cell lines under normal culture conditions. A significant reduction in Beclin-1 protein expression correlating with positive regulation of autophagy has been observed in U20S cell lines stably expressing inducible or transient siRNA against 14-3-3 τ (θ) [131]. Nevertheless, siRNA-mediated downregulation of 14-3-3 ζ, σ or θ specifically depletes the respective 14-3-3 isoform and induces autophagy, but has no effect on the levels of expression of autophagy-related proteins, included Beclin-1. These data suggest a role for 14-3-3 proteins as negative regulators of autophagy and indicate that this function is mediated by inhibition of hVps34 kinase activity.
4. Conclusions
It is becoming clear that autophagy is a tightly regulated process and that this cellular degradation process must only be initiated under specific cellular conditions. 14-3-3 proteins provide an additional level of regulation to this process by binding and thereby controlling the function of key regulatory proteins involved in autophagy. 14-3-3 proteins can directly interact with IRS-1 and this interaction may regulate the ability of the IRS-1 to recruit and activate PI3K, leading to consequences for mTOR regulation and autophagy initiation [69]. Furthermore, 14-3-3 controls autophagy by binding to two regulators of mTOR activation, namely TSC2 and PRAS40 proteins, and probably by association with other autophagy-related proteins that remain to be identified. Under normal growth conditions, 14-3-3 protein binding to TSC2 and PRAS40 leads to mTOR activation. Nutrient or serum deprivation promotes mTOR inactivation through dissociation of the TSC2–14-3-3 and PRAS40–14-3-3 complexes. Moreover, 14-3-3 proteins may also control the autophagy process at a later stage by interacting with and regulating proteins involved in autophagosome formation, such as hVps34. This interaction blocks hVps34 activity under normal growth conditions, while nutrient deprivation promotes 14-3-3–hVps34 dissociation, leading to hVps34 activation. In addition, 14-3-3 proteins may associate with Raptor and ULK1 under conditions of nutrient deprivation and phosphorylation-dependent 14-3-3 association with Raptor may reduce the availability of 14-3-3 for PRAS40 binding, which may have the combined effect of suppressing mTOR kinase activity and inducing autophagy.
More research is required to fully define the role of 14-3-3 in autophagy. However, in addition to having an anti-apoptotic role, 14-3-3 proteins, along with other anti-apoptotic proteins such as Bcl-2 [132] during autophagosome generation and cFLIP during in autophagosome elongation [133], may also regulate cell fate by their anti-autophagic function. Further studies will increase our knowledge about the molecular processes involved in autophagy and may help to increase the efficacy of anti-tumor therapies that target cell fate control.
Acknowledgments
This study was supported by the ‘Ministerio de Educación y Ciencia’ grant BFU2006–01088/BMC and ‘Programa Ramón y Cajal’ contract (BOE 17/02/2004 ORDEN CTE/351/2004) given to Mercedes Pozuelo Rubio.
Conflict of Interest
The author declares no conflict of interest.
References
- Chaudhri, M.; Scarabel, M.; Aitken, A. Mammalian and yeast 14-3-3 isoforms form distinct patterns of dimers in vivo. Biochem. Biophys. Res. Commun. 2003, 300, 679–685. [Google Scholar] [CrossRef]
- Jones, D.H.; Ley, S.; Aitken, A. Isoforms of 14-3-3 protein can form homo- and heterodimers in vivo and in vitro: implications for function as adapter proteins. FEBS Lett. 1995, 368, 55–58. [Google Scholar] [CrossRef]
- Mackintosh, C. Dynamic interactions between 14-3-3 proteins and phosphoproteins regulate diverse cellular processes. Biochem J. 2004, 381, 329–342. [Google Scholar] [CrossRef]
- Muslin, A.J.; Tanner, J.W.; Allen, P.M.; Shaw, A.S. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 1996, 84, 889–897. [Google Scholar] [CrossRef]
- Yaffe, M.B.; Rittinger, K.; Volinia, S.; Caron, P.R.; Aitken, A.; Leffers, H.; Gamblin, S.J.; Smerdon, S.J.; Cantley, L.C. The structural basis for 14-3-3: phosphopeptide binding specificity. Cell 1997, 91, 961–971. [Google Scholar] [CrossRef]
- Johnson, C.; Crowther, S.; Stafford, M.J.; Campbell, D.G.; Toth, R.; MacKintosh, C. Bioinformatic and experimental survey of 14-3-3-binding sites. Biochem J. 2010, 427, 69–78. [Google Scholar] [CrossRef]
- Aitken, A.; Howell, S.; Jones, D.; Madrazo, J.; Patel, Y. 14-3-3 alpha and delta are the phosphorylated forms of raf-activating 14-3-3 beta and zeta. In vivo stoichiometric phosphorylation in brain at a Ser-Pro-Glu-Lys MOTIF. In J. Biol. Chem.; 1995; Volume 270, pp. 5706–5709. [Google Scholar]
- Moreira, J.M.; Shen, T.; Ohlsson, G.; Gromov, P.; Gromova, I.; Celis, J.E. A combined proteome and ultrastructural localization analysis of 14-3-3 proteins in transformed human amnion (AMA) cells: definition of a framework to study isoform-specific differences. Mol. Cell. Proteomics 2008, 7, 1225–1240. [Google Scholar] [CrossRef]
- Kilani, R.T.; Medina, A.; Aitken, A.; Jalili, R.B.; Carr, M.; Ghahary, A. Identification of different isoforms of 14-3-3 protein family in human dermal and epidermal layers. Mol. Cell. Biochem. 2008, 314, 161–169. [Google Scholar] [CrossRef]
- Yaffe, M.B. How do 14-3-3 proteins work? Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett. 2002, 513, 53–57. [Google Scholar] [CrossRef]
- Wilker, E.; Yaffe, M.B. 14-3-3 Proteins—a focus on cancer and human disease. J. Mol. Cell. Cardiol. 2004, 37, 633–642. [Google Scholar] [CrossRef]
- Meek, S.E.; Lane, W.S.; Piwnica-Worms, H. Comprehensive proteomic analysis of interphase and mitotic 14-3-3-binding proteins. J. Biol. Chem. 2004, 279, 32046–32054. [Google Scholar] [CrossRef]
- Pozuelo Rubio, M.; Geraghty, K.M.; Wong, B.H.; Wood, N.T.; Campbell, D.G.; Morrice, N.; Mackintosh, C. 14-3-3-affinity purification of over 200 human phosphoproteins reveals new links to regulation of cellular metabolism, proliferation and trafficking. Biochem. J. 2004, 379, 395–408. [Google Scholar] [CrossRef]
- Jin, J.; Smith, F.D.; Stark, C.; Wells, C.D.; Fawcett, J.P.; Kulkarni, S.; Metalnikov, P.; O’Donnell, P.; Taylor, P.; Taylor, L.; Zougman, A.; Woodgett, J.R.; Langeberg, L.K.; Scott, J.D.; Pawson, T. Proteomic, functional, and domain-based analysis of in vivo 14-3-3 binding proteins involved in cytoskeletal regulation and cellular organization. Curr. Biol. 2004, 14, 1436–1450. [Google Scholar] [CrossRef]
- Kjarland, E.; Keen, T.J.; Kleppe, R. Does isoform diversity explain functional differences in the 14-3-3 protein family’? Curr. Pharm. Biotechnol. 2006, 7, 217–223. [Google Scholar] [CrossRef]
- Pozuelo-Rubio, M. Proteomic and biochemical analysis of 14-3-3-binding proteins during C2- ceramide-induced apoptosis. FEBS J. 2010, 277, 3321–3342. [Google Scholar] [CrossRef]
- Chang, I.F.; Curran, A.; Woolsey, R.; Quilici, D.; Cushman, J.C.; Mittler, R.; Harmon, A.; Harper, J.F. Proteomic profiling of tandem affinity purified 14-3-3 protein complexes in Arabidopsis thaliana. Proteomics 2009, 9, 2967–2985. [Google Scholar] [CrossRef]
- Puri, P.; Myers, K.; Kline, D.; Vijayaraghavan, S. Proteomic analysis of bovine sperm YWHA binding partners identify proteins involved in signaling and metabolism. Biol. Reprod. 2008, 79, 1183–1191. [Google Scholar] [CrossRef]
- Benzinger, A.; Muster, N.; Koch, H.B.; Yates, J.R., 3rd; Hermeking, H. Targeted proteomic analysis of 14-3-3 sigma, a p53 effector commonly silenced in cancer. Mol. Cell. Proteomics 2005, 4, 785–795. [Google Scholar] [CrossRef]
- Masters, S.C.; Subramanian, R.R.; Truong, A.; Yang, H.; Fujii, K.; Zhang, H.; Fu, H. Survival-promoting functions of 14-3-3 proteins. Biochem. Soc. Trans. 2002, 30, 360–365. [Google Scholar]
- Zha, J.; Harada, H.; Yang, E.; Jockel, J.; Korsmeyer, S.J. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 1996, 87, 619–628. [Google Scholar] [CrossRef]
- Nomura, M.; Shimizu, S.; Sugiyama, T.; Narita, M.; Ito, T.; Matsuda, H.; Tsujimoto, Y. 14-3-3 Interacts directly with and negatively regulates pro-apoptotic Bax. J. Biol. Chem. 2003, 278, 2058–2065. [Google Scholar]
- Zhang, L.; Chen, J.; Fu, H. Suppression of apoptosis signal-regulating kinase 1-induced cell death by 14-3-3 proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 8511–8515. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Masters, S.C.; Fu, H. 14-3-3 proteins mediate an essential anti-apoptotic signal. J. Biol. Chem. 2001, 276, 45193–45200. [Google Scholar] [CrossRef]
- Cao, W.; Yang, X.; Zhou, J.; Teng, Z.; Cao, L.; Zhang, X.; Fei, Z. Targeting 14-3-3 protein, difopein induces apoptosis of human glioma cells and suppresses tumour growth in mice. Apoptosis 2010, 15, 230–241. [Google Scholar] [CrossRef]
- Qi, W.; Martinez, J.D. Reduction of 14-3-3 proteins correlates with increased sensitivity to killing of human lung cancer cells by ionizing radiation. Radiat. Res. 2003, 160, 217–223. [Google Scholar] [CrossRef]
- Niemantsverdriet, M.; Wagner, K.; Visser, M.; Backendorf, C. Cellular functions of 14-3-3 zeta in apoptosis and cell adhesion emphasize its oncogenic character. Oncogene 2008, 27, 1315–1319. [Google Scholar] [CrossRef]
- Neal, C.L.; Yao, J.; Yang, W.; Zhou, X.; Nguyen, N.T.; Lu, J.; Danes, C.G.; Guo, H.; Lan, K.H.; Ensor, J.; Hittelman, W.; Hung, M.C.; Yu, D. 14-3-3zeta overexpression defines high risk for breast cancer recurrence and promotes cancer cell survival. Cancer Res. 2009, 69, 3425–3432. [Google Scholar] [CrossRef]
- Kumar, A.P.; Garcia, G.E.; Orsborn, J.; Levin, V.A.; Slaga, T.J. 2-Methoxyestradiol interferes with NF kappa B transcriptional activity in primitive neuroectodermal brain tumors: implications for management. Carcinogenesis 2003, 24, 209–216. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: a double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Gestwicki, J.E.; Murphy, L.O.; Klionsky, D.J. Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 2007, 6, 304–312. [Google Scholar] [CrossRef]
- Galluzzi, L.; Aaronson, S.A.; Abrams, J.; Alnemri, E.S.; Andrews, D.W.; Baehrecke, E.H.; Bazan, N.G.; Blagosklonny, M.V.; Blomgren, K.; Borner, C.; Bredesen, D.E.; Brenner, C.; Castedo, M.; Cidlowski, J.A.; Ciechanover, A.; Cohen, G.M.; De Laurenzi, V.; De Maria, R.; Deshmukh, M.; Dynlacht, B.D.; El-Deiry, W.S.; Flavell, R.A.; Fulda, S.; Garrido, C.; Golstein, P.; Gougeon, M.L.; Green, D.R.; Gronemeyer, H.; Hajnoczky, G.; Hardwick, J.M.; Hengartner, M.O.; Ichijo, H.; Jaattela, M.; Kepp, O.; Kimchi, A.; Klionsky, D.J.; Knight, R.A.; Kornbluth, S.; Kumar, S.; Levine, B.; Lipton, S.A.; Lugli, E.; Madeo, F.; Malomi, W.; Marine, J.C.; Martin, S.J.; Medema, J.P.; Mehlen, P.; Melino, G.; Moll, U.M.; Morselli, E.; Nagata, S.; Nicholson, D.W.; Nicotera, P.; Nunez, G.; Oren, M.; Penninger, J.; Pervaiz, S.; Peter, M.E.; Piacentini, M.; Prehn, J.H.; Puthalakath, H.; Rabinovich, G.A.; Rizzuto, R.; Rodrigues, C.M.; Rubinsztein, D.C.; Rudel, T.; Scorrano, L.; Simon, H.U.; Steller, H.; Tschopp, J.; Tsujimoto, Y.; Vandenabeele, P.; Vitale, I.; Vousden, K.H.; Youle, R.J.; Yuan, J.; Zhivotovsky, B.; Kroemer, G. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009, 16, 1093–1107. [Google Scholar]
- Kroemer, G.; Jaattela, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 2005, 5, 886–897. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Chang, Y.Y.; Juhasz, G.; Goraksha-Hicks, P.; Arsham, A.M.; Mallin, D.R.; Muller, L.K.; Neufeld, T.P. Nutrient-dependent regulation of autophagy through the target of rapamycin pathway. Biochem. Soc. Trans. 2009, 37, 232–236. [Google Scholar] [CrossRef]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar]
- Levine, B.; Yuan, J. Autophagy in cell death: an innocent convict? J. Clin. Invest. 2005, 115, 2679–2688. [Google Scholar] [CrossRef]
- Baehrecke, E.H. Autophagy: dual roles in life and death? Nat. Rev. Mol. Cell Biol. 2005, 6, 505–510. [Google Scholar] [CrossRef]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef]
- Gozuacik, D.; Kimchi, A. Autophagy and cell death. Curr. Top. Dev. Biol. 2007, 78, 217–245. [Google Scholar] [CrossRef]
- Lockshin, R.A.; Zakeri, Z. Programmed cell death and apoptosis: origins of the theory. Nat Rev. Mol. Cell Biol. 2001, 2, 545–550. [Google Scholar] [CrossRef]
- Daido, S.; Kanzawa, T.; Yamamoto, A.; Takeuchi, H.; Kondo, Y.; Kondo, S. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004, 64, 4286–4293. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Corradetti, M.N.; Inoki, K.; Bardeesy, N.; DePinho, R.A.; Guan, K.L. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004, 18, 1533–1538. [Google Scholar] [CrossRef]
- Shaw, R.J.; Bardeesy, N.; Manning, B.D.; Lopez, L.; Kosmatka, M.; DePinho, R.A.; Cantley, L.C. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 2004, 6, 91–99. [Google Scholar] [CrossRef]
- Liu, L.; Cash, T.P.; Jones, R.G.; Keith, B.; Thompson, C.B.; Simon, M.C. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol. Cell 2006, 21, 521–531. [Google Scholar] [CrossRef]
- Shaw, R.J. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 2009, 196, 65–80. [Google Scholar] [CrossRef]
- Miyazaki, M.; McCarthy, J.J.; Esser, K.A. Insulin like growth factor-1-induced phosphorylation and altered distribution of tuberous sclerosis complex (TSC)1/TSC2 in C2C12 myotubes. FEBS J. 2010, 277, 2180–2191. [Google Scholar] [CrossRef]
- Cai, S.L.; Tee, A.R.; Short, J.D.; Bergeron, J.M.; Kim, J.; Shen, J.; Guo, R.; Johnson, C.L.; Kiguchi, K.; Walker, C.L. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J. Cell Biol. 2006, 173, 279–289. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 2009, 37, 217–222. [Google Scholar] [CrossRef]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef]
- Vander Haar, E.; Lee, S.I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef]
- Wang, L.; Harris, T.E.; Lawrence, J.C., Jr. Regulation of proline-rich Akt substrate of 40 kDa (PRAS40) function by mammalian target of rapamycin complex 1 (mTORC1)-mediated phosphorylation. J. Biol. Chem. 2008, 283, 15619–15627. [Google Scholar]
- Nascimento, E.B.; Snel, M.; Guigas, B.; van der Zon, G.C.; Kriek, J.; Maassen, J.A.; Jazet, I.M.; Diamant, M.; Ouwens, D.M. Phosphorylation of PRAS40 on Thr246 by PKB/AKT facilitates efficient phosphorylation of Ser183 by mTORC1. Cell Signal. 2010, 22, 961–967. [Google Scholar] [CrossRef]
- Fonseca, B.D.; Smith, E.M.; Lee, V.H.; MacKintosh, C.; Proud, C.G. PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J. Biol. Chem. 2007, 282, 24514–24524. [Google Scholar]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Corradetti, M.N.; Inoki, K.; Guan, K.L. The stress-induced proteins RTP801 and RTP801L are negative regulators of the mammalian target of rapamycin pathway. J. Biol. Chem. 2005, 280, 9769–9772. [Google Scholar]
- Sofer, A.; Lei, K.; Johannessen, C.M.; Ellisen, L.W. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol. Cell Biol. 2005, 25, 5834–5845. [Google Scholar] [CrossRef]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2- mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef]
- Xiang, X.; Yuan, M.; Song, Y.; Ruderman, N.; Wen, R.; Luo, Z. 14-3-3 facilitates insulin-stimulated intracellular trafficking of insulin receptor substrate 1. Mol. Endocrinol. 2002, 16, 552–562. [Google Scholar] [CrossRef]
- McKay, M.M.; Morrison, D.K. Integrating signals from RTKs to ERK/MAPK. Oncogene 2007, 26, 3113–3121. [Google Scholar] [CrossRef]
- Murphy, L.O.; Blenis, J. MAPK signal specificity: the right place at the right time. Trends Biochem. Sci. 2006, 31, 268–275. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.C. ERK and cell death: mechanisms of ERK-induced cell death—apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Ellington, A.A.; Berhow, M.A.; Singletary, K.W. Inhibition of Akt signaling and enhanced ERK1/2 activity are involved in induction of macroautophagy by triterpenoid B-group soyasaponins in colon cancer cells. Carcinogenesis 2006, 27, 298–306. [Google Scholar] [CrossRef]
- Choi, C.H.; Jung, Y.K.; Oh, S.H. Autophagy induction by capsaicin in malignant human breast cells is modulated by p38 and extracellular signal-regulated mitogen-activated protein kinases and retards cell death by suppressing endoplasmic reticulum stress-mediated apoptosis. Mol. Pharmacol. 2010, 78, 114–125. [Google Scholar] [CrossRef]
- Wang, S.H.; Shih, Y.L.; Lee, C.C.; Chen, W.L.; Lin, C.J.; Lin, Y.S.; Wu, K.H.; Shih, C.M. The role of endoplasmic reticulum in cadmium-induced mesangial cell apoptosis. Chem. Biol. Interact. 2009, 181, 45–51. [Google Scholar] [CrossRef]
- Yang, L.Y.; Wu, K.H.; Chiu, W.T.; Wang, S.H.; Shih, C.M. The cadmium-induced death of mesangial cells results in nephrotoxicity. Autophagy 2009, 5, 571–572. [Google Scholar] [CrossRef]
- Cheng, Y.; Qiu, F.; Tashiro, S.; Onodera, S.; Ikejima, T. ERK and JNK mediate TNFalpha- induced p53 activation in apoptotic and autophagic L929 cell death. Biochem. Biophys. Res.Commun. 2008, 376, 483–488. [Google Scholar] [CrossRef]
- Wang, J.; Whiteman, M.W.; Lian, H.; Wang, G.; Singh, A.; Huang, D.; Denmark, T. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J. Biol.Chem. 2009, 284, 21412–21424. [Google Scholar] [CrossRef]
- Corcelle, E.; Nebout, M.; Bekri, S.; Gauthier, N.; Hofman, P.; Poujeol, P.; Fenichel, P.; Mograbi, B. Disruption of autophagy at the maturation step by the carcinogen lindane is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Cancer Res. 2006, 66, 6861–6870. [Google Scholar]
- Tzivion, G.; Luo, Z.; Avruch, J. A dimeric 14-3-3 protein is an essential cofactor for Raf kinase activity. Nature 1998, 394, 88–92. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Meikle, S.; Peyssonnaux, C.; Grindlay, J.; Kaiser, C.; Steen, H.; Shaw, P.E.; Mischak, H.; Eychene, A.; Kolch, W. A Raf-1 mutant that dissociates MEK/extracellular signal-regulated kinase activation from malignant transformation and differentiation but not proliferation. Mol. Cell Biol. 2003, 23, 1983–1993. [Google Scholar]
- Dumaz, N.; Marais, R. Protein kinase A blocks Raf-1 activity by stimulating 14-3-3 binding and blocking Raf-1 interaction with Ras. J. Biol. Chem. 2003, 278, 29819–29823. [Google Scholar] [CrossRef]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Webber, J.L.; Tooze, S.A. New insights into the function of Atg9. FEBS Lett. 2010, 584, 1319–1326. [Google Scholar] [CrossRef]
- Cui, Q.; Tashiro, S.; Onodera, S.; Minami, M.; Ikejima, T. Oridonin induced autophagy in human cervical carcinoma HeLa cells through Ras, JNK, and P38 regulatio. J. Pharmacol. Sci. 2007, 105, 317–325. [Google Scholar] [CrossRef]
- Liao, P.C.; Ng, L.T.; Lin, L.T.; Richardson, C.D.; Wang, G.H.; Lin, C.C. Resveratrolarrests cell cycle and induces apoptosis in human hepatocellular carcinoma Huh-7 cells. J. Med. Food 2010, 13, 1415–1423. [Google Scholar] [CrossRef]
- Kim, D.S.; Kim, J.H.; Lee, G.H.; Kim, H.T.; Lim, J.M.; Chae, S.W.; Chae, H.J.; Kim, H.R. p38 Mitogen-activated protein kinase is involved in endoplasmic reticulum stress-induced cell death and autophagy in human gingival fibroblasts. Biol. Pharm. Bull. 2010, 33, 545–549. [Google Scholar] [CrossRef]
- Lim, S.C.; Hahm, K.S.; Lee, S.H.; Oh, S.H. Autophagy involvement in cadmium resistance through induction of multidrug resistance-associated protein and counterbalance of endoplasmic reticulum stress WI38 lung epithelial fibroblast cells. Toxicology 2010, 276, 18–26. [Google Scholar] [CrossRef]
- Thyagarajan, A.; Jedinak, A.; Nguyen, H.; Terry, C.; Baldridge, L.A.; Jiang, J.; Sliva, D. Triterpenes from Ganoderma Lucidum induce autophagy in colon cancer through the inhibition of p38 mitogen-activated kinase (p38 MAPK). Nutr. Cancer 2010, 62, 630–640. [Google Scholar] [CrossRef]
- Colosetti, P.; Puissant, A.; Robert, G.; Luciano, F.; Jacquel, A.; Gounon, P.; Cassuto, J.P.; Auberger, P. Autophagy is an important event for megakaryocytic differentiation of the chronic myelogenous leukemia K562 cell line. Autophagy 2009, 5, 1092–1098. [Google Scholar] [CrossRef]
- Webber, J.L.; Tooze, S.A. Coordinated regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP. Embo J. 2010, 29, 27–40. [Google Scholar] [CrossRef]
- Fritz, A.; Brayer, K.J.; McCormick, N.; Adams, D.G.; Wadzinski, B.E.; Vaillancourt, R.R. Phosphorylation of serine 526 is required for MEKK3 activity, and association with 14-3-3 blocks dephosphorylation. J. Biol. Chem. 2006, 281, 6236–6245. [Google Scholar]
- Xing, H.; Zhang, S.; Weinheimer, C.; Kovacs, A.; Muslin, A.J. 14-3-3 proteins block apoptosis and differentially regulate MAPK cascades. Embo J. 2000, 19, 349–358. [Google Scholar] [CrossRef]
- Basu, A. The potential of protein kinase C as a target for anticancer treatment. Pharmacol.Ther. 1993, 59, 257–280. [Google Scholar] [CrossRef]
- Ozpolat, B.; Akar, U.; Mehta, K.; Lopez-Berestein, G. PKC delta and tissue transglutaminase are novel inhibitors of autophagy in pancreatic cancer cells. Autophagy 2007, 3, 480–483. [Google Scholar]
- Lorand, L.; Graham, R.M. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Biol. 2003, 4, 140–156. [Google Scholar] [CrossRef]
- Chen, J.L.; Lin, H.H.; Kim, K.J.; Lin, A.; Forman, H.J.; Ann, D.K. Novel roles for protein kinase Cdelta-dependent signaling pathways in acute hypoxic stress-induced autophagy. J. Biol. Chem. 2008, 283, 34432–34444. [Google Scholar]
- Sakaki, K.; Wu, J.; Kaufman, R.J. Protein kinase Ctheta is required for autophagy in response to stress in the endoplasmic reticulum. J. Biol. Chem. 2008, 283, 15370–15380. [Google Scholar]
- Jiang, H.; Cheng, D.; Liu, W.; Peng, J.; Feng, J. Protein kinase C inhibits autophagy andphosphorylates LC3. Biochem. Biophys. Res. Commun. 2010, 395, 471–476. [Google Scholar] [CrossRef]
- Acs, P.; Szallasi, Z.; Kazanietz, M.G.; Blumberg, P.M. Differential activation of PKC isozymes by 14-3-3 zeta protein. Biochem. Biophys. Res. Commun. 1995, 216, 103–109. [Google Scholar] [CrossRef]
- Oriente, F.; Andreozzi, F.; Romano, C.; Perruolo, G.; Perfetti, A.; Fiory, F.; Miele, C.; Beguinot, F.; Formisano, P. Protein kinase C-alpha regulates insulin action and degradation by interacting with insulin receptor substrate-1 and 14-3-3 epsilon. J. Biol. Chem. 2005, 280, 40642–40649. [Google Scholar]
- Kim, Y.H.; Kim, Y.S.; Kang, S.S.; Noh, H.S.; Kim, H.J.; Cho, G.J.; Choi, W.S. Expression of 14-3-3 zeta and interaction with protein kinase C in the rat retina in early diabetes. Diabetologia 2005, 48, 1411–1415. [Google Scholar] [CrossRef]
- Dai, J.G.; Murakami, K. Constitutively and autonomously active protein kinase C associated with 14-3-3 zeta in the rodent brain. J. Neurochem. 2003, 84, 23–34. [Google Scholar]
- Hausser, A.; Storz, P.; Link, G.; Stoll, H.; Liu, Y.C.; Altman, A.; Pfizenmaier, K.; Johannes, F.J. Protein kinase C mu is negatively regulated by 14-3-3 signal transduction proteins. J. Biol. Chem. 1999, 274, 9258–9264. [Google Scholar]
- Meller, N.; Liu, Y.C.; Collins, T.L.; Bonnefoy-Berard, N.; Baier, G.; Isakov, N.; Altman, A. Direct interaction between protein kinase C theta (PKC theta) and 14-3-3 tau in T cells: 14-3-3 overexpression results in inhibition of PKC theta translocation and function. Mol. Cell Biol. 1996, 16, 5782–5791. [Google Scholar]
- Robinson, K.; Jones, D.; Patel, Y.; Martin, H.; Madrazo, J.; Martin, S.; Howell, S.; Elmore, M.; Finnen, M.J.; Aitken, A. Mechanism of inhibition of protein kinase C by 14-3-3 isoforms. 14- 3–3 isoforms do not have phospholipase A2 activity. Biochem. J. 1994, 29, 853–861. [Google Scholar]
- Van Der Hoeven, P.C.; Van Der Wal, J.C.; Ruurs, P.; Van Blitterswijk, W.J. Protein kinase C activation by acidic proteins including 14-3-3. Biochem. J. 2000, 347, 781–785. [Google Scholar] [CrossRef]
- Tanji, M.; Horwitz, R.; Rosenfeld, G.; Waymire, J.C. Activation of protein kinase C by purified bovine brain 14-3-3: comparison with tyrosine hydroxylase activation. J. Neurochem. 1994, 63, 1908–1916. [Google Scholar]
- Hoyer-Hansen, M.; Jaattela, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007, 14, 1576–1582. [Google Scholar] [CrossRef]
- Davare, M.A.; Saneyoshi, T.; Guire, E.S.; Nygaard, S.C.; Soderling, T.R. Inhibition of calcium/calmodulin-dependent protein kinase kinase by protein 14-3-3. J. Biol. Chem. 2004, 279, 52191–52199. [Google Scholar]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 2000, 150, 1507–1513. [Google Scholar] [CrossRef]
- Kabeya, Y.; Kamada, Y.; Baba, M.; Takikawa, H.; Sasaki, M.; Ohsumi, Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol. Biol. Cell 2005, 16, 2544–2553. [Google Scholar] [CrossRef]
- Kamada, Y.; Yoshino, K.; Kondo, C.; Kawamata, T.; Oshiro, N.; Yonezawa, K.; Ohsumi, Y. Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol. Cell Biol. 2010, 30, 1049–1058. [Google Scholar] [CrossRef]
- Kuroyanagi, H.; Yan, J.; Seki, N.; Yamanouchi, Y.; Suzuki, Y.; Takano, T.; Muramatsu, M.; Shirasawa, T. Human ULK1, a novel serine/threonine kinase related to UNC-51 kinase of Caenorhabditis elegans: cDNA cloning, expression, and chromosomal assignmet. Genomics 1998, 51, 76–85. [Google Scholar] [CrossRef]
- Chan, E.Y.; Kir, S.; Tooze, S.A. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 2007, 282, 25464–25474. [Google Scholar] [CrossRef]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.; Natsume, T.; Guan, J.L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Lee, J.W.; Park, S.; Takahashi, Y.; Wang, H.G. The association of AMPK with ULK1 regulates autophagy. PLoS One 2010, e15394, 1–9. [Google Scholar]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Lawlor, M.A.; Alessi, D.R. PKB/Akt: A key mediator of cell proliferation, survival and insulin responses? J. Cell Sci. 2001, 114, 2903–2910. [Google Scholar]
- Wang, Y.; Yoshioka, K.; Azam, M.A.; Takuwa, N.; Sakurada, S.; Kayaba, Y.; Sugimoto, N.; Inoki, I.; Kimura, T.; Kuwaki, T.; Takuwa, Y. Class II phosphoinositide 3-kinase alpha-isoform regulates Rho, myosin phosphatase and contraction in vascular smooth muscle. Biochem. J. 2006, 394, 581–592. [Google Scholar] [CrossRef]
- Domin, J.; Harper, L.; Aubyn, D.; Wheeler, M.; Florey, O.; Haskard, D.; Yuan, M.; Zicha, D. The class II phosphoinositide 3-kinase PI3K-C2beta regulates cell migration by a PtdIns3P dependent mechanism. J. Cell Physiol. 2005, 205, 452–462. [Google Scholar] [CrossRef]
- Backer, J.M. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem. J. 2008, 410, 1–17. [Google Scholar] [CrossRef]
- Proikas-Cezanne, T.; Waddell, S.; Gaugel, A.; Frickey, T.; Lupas, A.; Nordheim, A. WIPI- 1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene 2004, 23, 9314–9325. [Google Scholar] [CrossRef]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6. [Google Scholar]
- Krick, R.; Tolstrup, J.; Appelles, A.; Henke, S.; Thumm, M. The relevance of the phosphatidylinositolphosphat-binding motif FRRGT of Atg18 and Atg21 for the Cvt pathway and autophagy. FEBS Lett. 2006, 580, 4632–4638. [Google Scholar] [CrossRef]
- Ghosh, P.; Wu, M.; Zhang, H.; Sun, H. mTORC1 signaling requires proteasomal function and the involvement of CUL4-DDB1 ubiquitin E3 ligase. Cell Cycle 2008, 7, 373–381. [Google Scholar] [CrossRef]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1- phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef]
- Pozuelo-Rubio, M. Regulation of autophagic activity by 14-3-3zeta proteins associated with class III phosphatidylinositol-3-kinase. Cell Death Differ. 2011, 18, 479–492. [Google Scholar] [CrossRef]
- Wang, B.; Lin, S.; Lin, W.-C. 14-3-3tau regulates Beclin 1 and is required for autophagy. 2010, 5, e10409, 1–10. [Google Scholar]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; Jung, J.U. FLIP-mediated autophagy regulation in cell death control. Nat. Cell. Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Pozuelo-Rubio, M. 14-3-3 Proteins are Regulators of Autophagy. Cells 2012, 1, 754-773. https://doi.org/10.3390/cells1040754
AMA Style
Pozuelo-Rubio M. 14-3-3 Proteins are Regulators of Autophagy. Cells. 2012; 1(4):754-773. https://doi.org/10.3390/cells1040754
Chicago/Turabian StylePozuelo-Rubio, Mercedes. 2012. "14-3-3 Proteins are Regulators of Autophagy" Cells 1, no. 4: 754-773. https://doi.org/10.3390/cells1040754