Divergent Roles of Autophagy in Virus Infection

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
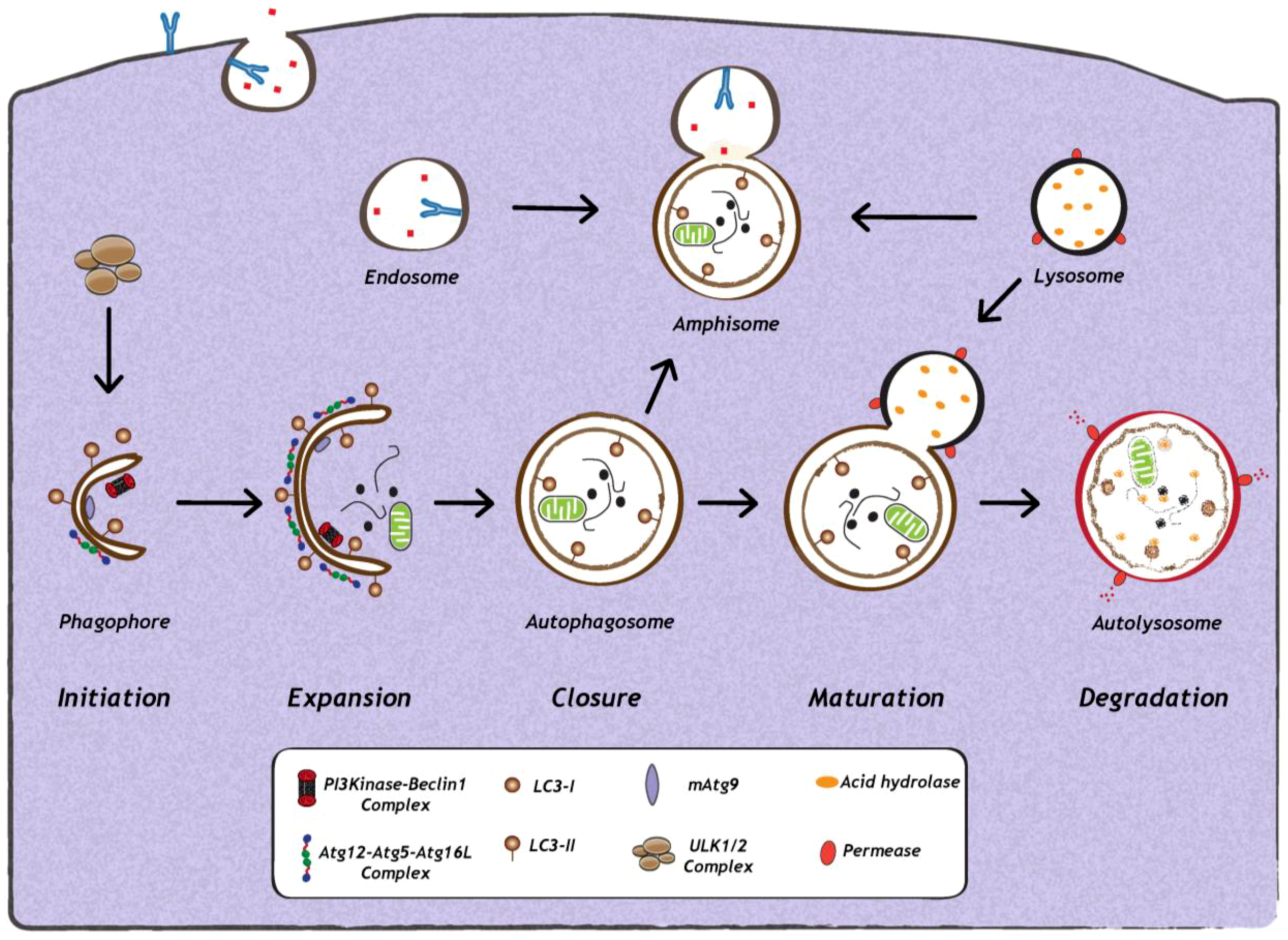
1. Introduction
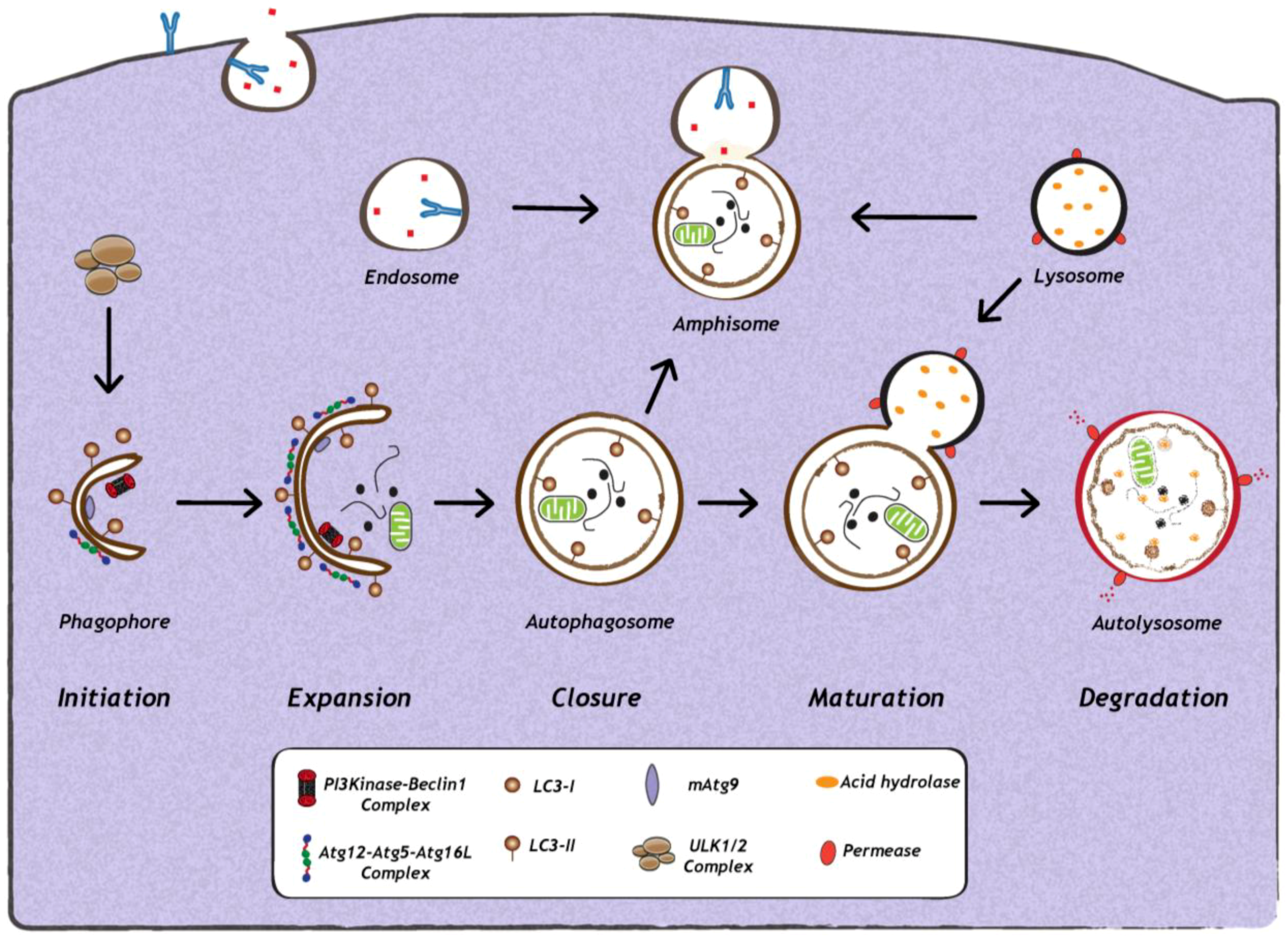
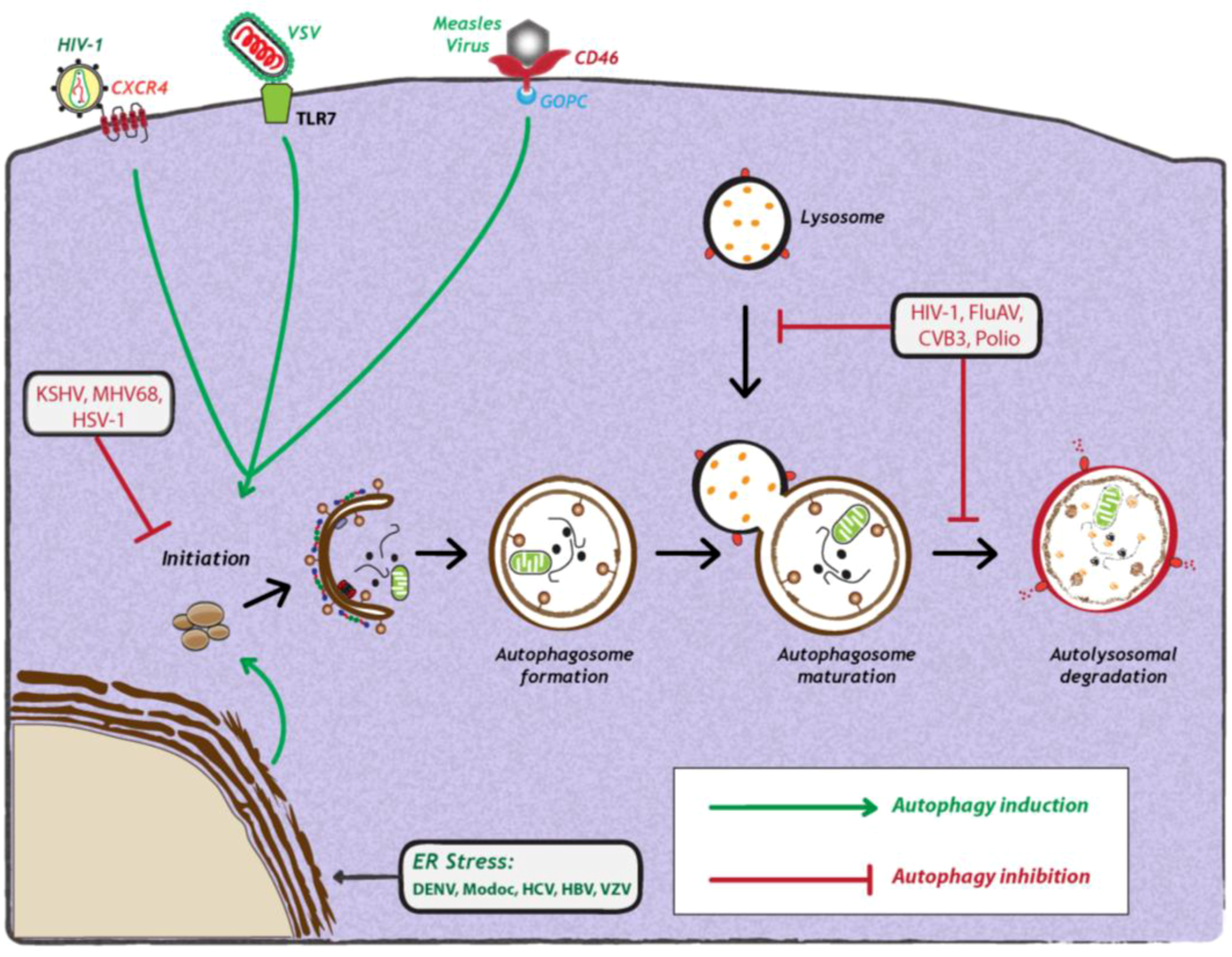
2. Impact of Viral Infection on Autophagy
2.1. Induction and Inhibition of Autophagy by Viruses
2.2. Cellular Signaling Events Influencing Autophagy During Viral Infection
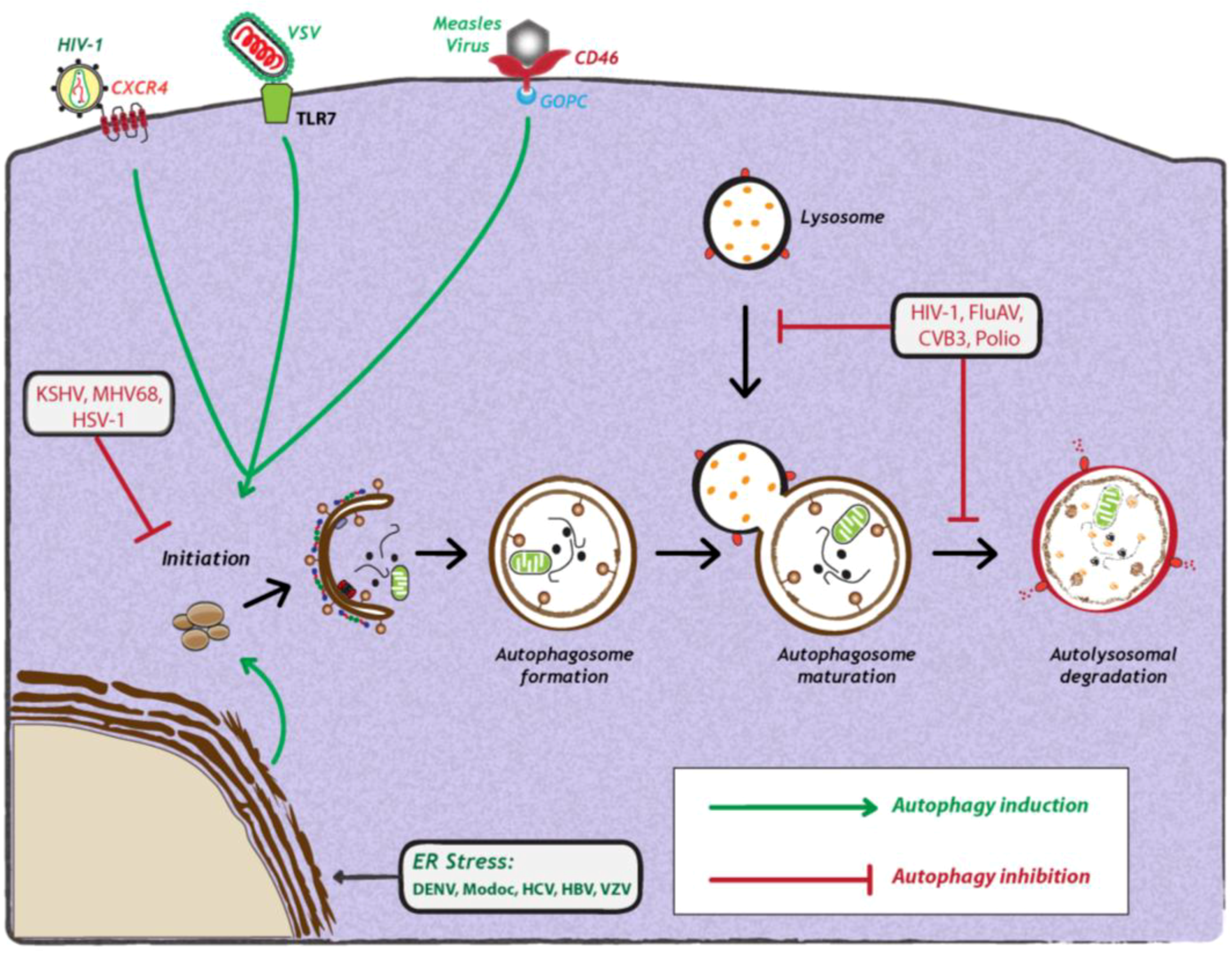
3. Relationship Between Autophagy and Viruses
3.1. Antiviral Functions of Autophagy
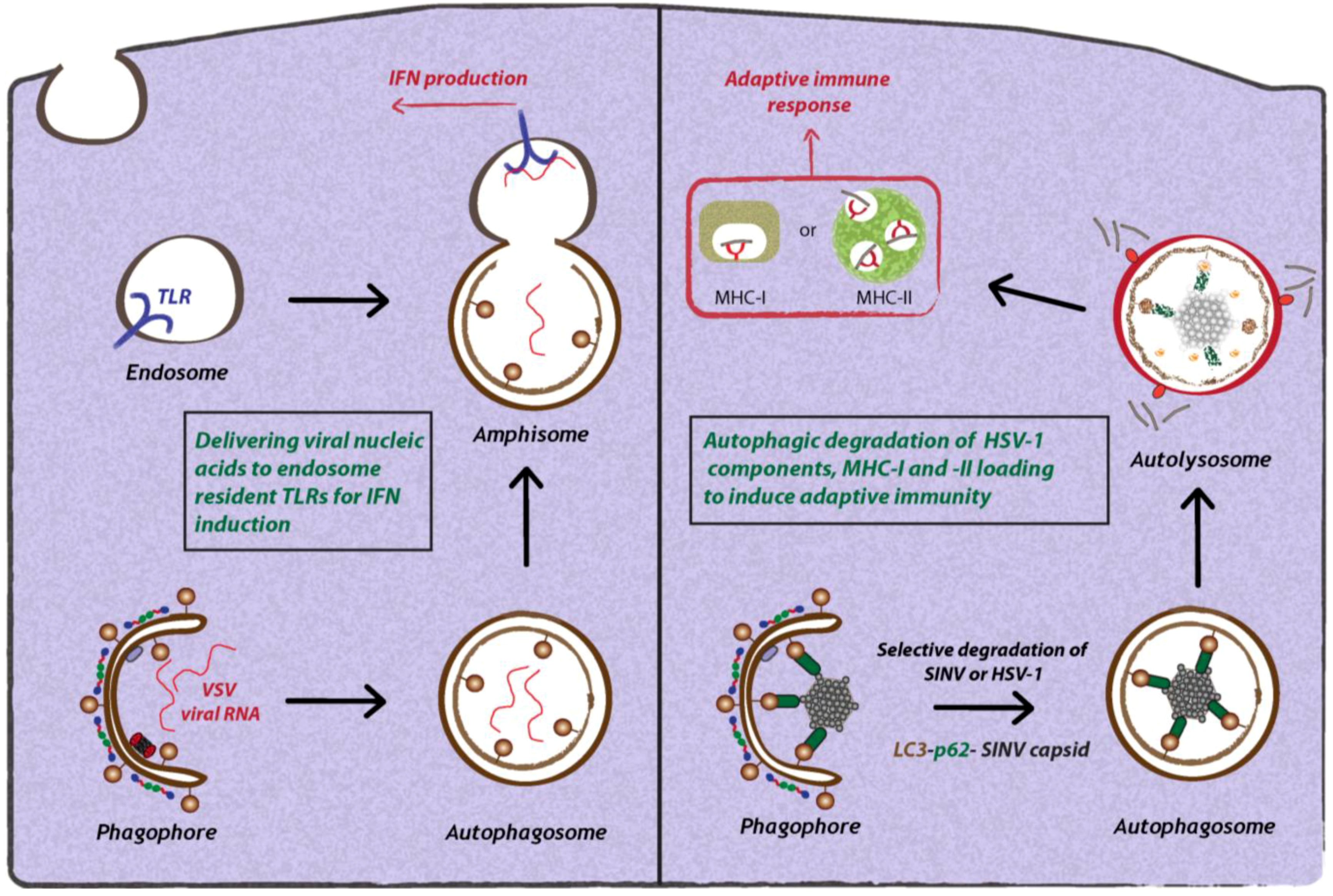
3.1.1. Interplay Between Toll-like Receptors (TLRs) and Autophagy During Viral Infection
3.1.2. Autophagic Degradation of Viruses by SLRs (Sequestosome 1/p62-like Receptors)
3.1.3. Role of Autophagy in Antigen Presentation
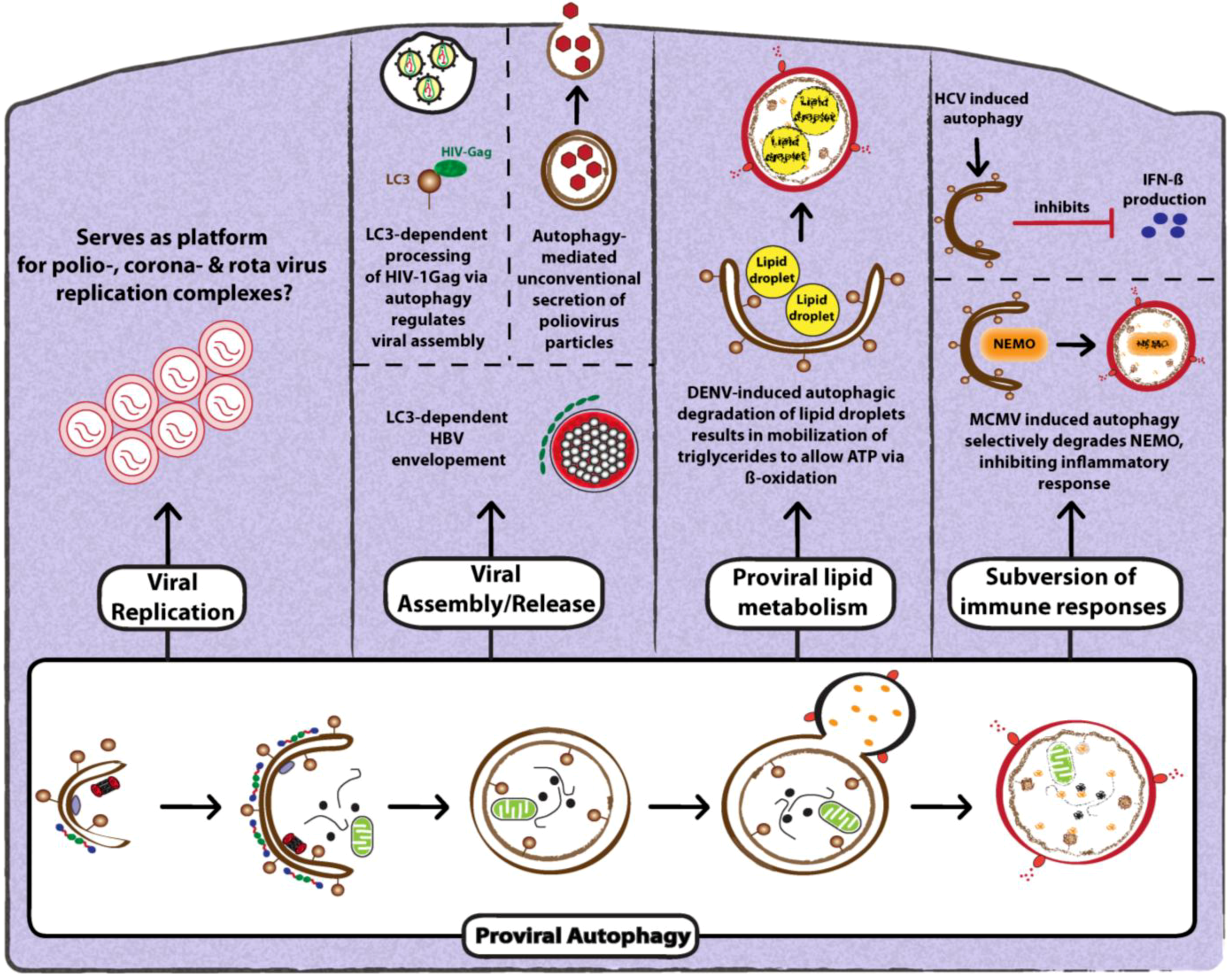
3.2. Proviral Functions of Autophagy
3.2.1. Role of Autophagy in Promoting Viral Replication
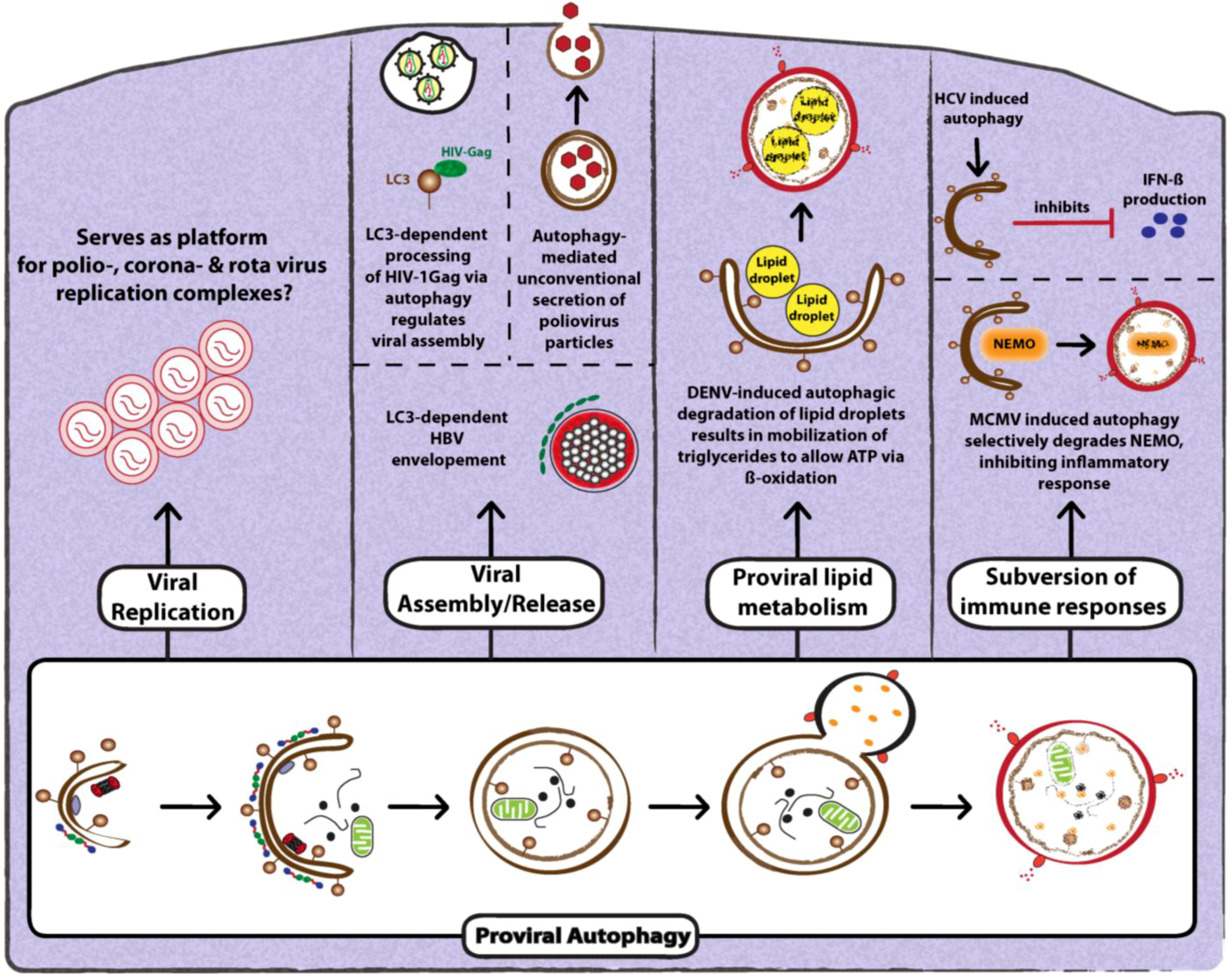
3.2.2. Role of Autophagy in Promoting Viral Assembly and Release
3.2.3. Role of Autophagy in Suppression of Anti-Viral Innate Immunity
4. Future Directions
Conflict of Interest
References
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef]
- Dunn, W.A., Jr. Studies on the mechanisms of autophagy: Formation of the autophagic vacuole. J. Cell Biol. 1990, 110, 1923–1933. [Google Scholar] [CrossRef]
- Fengsrud, M.; Erichsen, E.S.; Berg, T.O.; Raiborg, C.; Seglen, P.O. Ultrastructural characterization of the delimiting membranes of isolated autophagosomes and amphisomes by freeze-fracture electron microscopy. Eur. J. Cell Biol. 2000, 79, 871–882. [Google Scholar] [CrossRef]
- Eskelinen, E.L. Maturation of autophagic vacuoles in Mammalian cells. Autophagy 2005, 1, 1–10. [Google Scholar] [CrossRef]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nature Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef]
- Inoue, Y.; Klionsky, D.J. Regulation of macroautophagy in Saccharomyces cerevisiae. Semin. Cell Dev. Biol. 2010, 21, 664–670. [Google Scholar] [CrossRef]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef]
- Behrends, C.; Fulda, S. Receptor proteins in selective autophagy. Int. J. Cell Biol. 2012, 2012, 673290. [Google Scholar]
- Novak, I.; Dikic, I. Autophagy receptors in developmental clearance of mitochondria. Autophagy 2011, 7, 301–303. [Google Scholar] [CrossRef]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar]
- McEwan, D.G.; Dikic, I. The Three Musketeers of Autophagy: phosphorylation, ubiquitylation and acetylation. Trends Cell Biol. 2011, 21, 195–201. [Google Scholar] [CrossRef]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network organization of the human autophagy system. Nature 2010, 466, 68–76. [Google Scholar]
- Galluzzi, L.; Aaronson, S.A.; Abrams, J.; Alnemri, E.S.; Andrews, D.W.; Baehrecke, E.H.; Bazan, N.G.; Blagosklonny, M.V.; Blomgren, K.; Borner, C.; Bredesen, D.E.; Brenner, C.; Castedo, M.; Cidlowski, J.A.; Ciechanover, A.; Cohen, G.M.; De Laurenzi, V.; De Maria, R.; Deshmukh, M.; Dynlacht, B.D.; El-Deiry, W.S.; Flavell, R.A.; Fulda, S.; Garrido, C.; Golstein, P.; Gougeon, M.L.; Green, D.R.; Gronemeyer, H.; Hajnoczky, G.; Hardwick, J.M.; Hengartner, M.O.; Ichijo, H.; Jaattela, M.; Kepp, O.; Kimchi, A.; Klionsky, D.J.; Knight, R.A.; Kornbluth, S.; Kumar, S.; Levine, B.; Lipton, S.A.; Lugli, E.; Madeo, F.; Malomi, W.; Marine, J.C.; Martin, S.J.; Medema, J.P.; Mehlen, P.; Melino, G.; Moll, U.M.; Morselli, E.; Nagata, S.; Nicholson, D.W.; Nicotera, P.; Nunez, G.; Oren, M.; Penninger, J.; Pervaiz, S.; Peter, M.E.; Piacentini, M.; Prehn, J.H.; Puthalakath, H.; Rabinovich, G.A.; Rizzuto, R.; Rodrigues, C.M.; Rubinsztein, D.C.; Rudel, T.; Scorrano, L.; Simon, H.U.; Steller, H.; Tschopp, J.; Tsujimoto, Y.; Vandenabeele, P.; Vitale, I.; Vousden, K.H.; Youle, R.J.; Yuan, J.; Zhivotovsky, B.; Kroemer, G. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009, 16, 1093–1107. [Google Scholar]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. Plos Biol. 2005, 3, e156. [Google Scholar] [CrossRef]
- Berkova, Z.; Crawford, S.E.; Trugnan, G.; Yoshimori, T.; Morris, A.P.; Estes, M.K. Rotavirus NSP4 induces a novel vesicular compartment regulated by calcium and associated with viroplasms. JVI 2006, 80, 6061–6071. [Google Scholar] [CrossRef]
- Nakashima, A.; Tanaka, N.; Tamai, K.; Kyuuma, M.; Ishikawa, Y.; Sato, H.; Yoshimori, T.; Saito, S.; Sugamura, K. Survival of parvovirus B19-infected cells by cellular autophagy. Virology 2006, 349, 254–263. [Google Scholar] [CrossRef]
- Taylor, M.P.; Kirkegaard, K. Modification of cellular autophagy protein LC3 by poliovirus. JVI 2007, 81, 12543–12553. [Google Scholar] [CrossRef]
- Ait-Goughoulte, M.; Kanda, T.; Meyer, K.; Ryerse, J.S.; Ray, R.B.; Ray, R. Hepatitis C virus genotype 1a growth and induction of autophagy. JVI 2008, 82, 2241–2249. [Google Scholar] [CrossRef]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef]
- Taylor, M.P.; Kirkegaard, K. Potential subversion of autophagosomal pathway by picornaviruses. Autophagy 2008, 4, 286–289. [Google Scholar]
- Wong, J.; Zhang, J.; Si, X.; Gao, G.; Mao, I.; McManus, B.M.; Luo, H. Autophagosome supports coxsackievirus B3 replication in host cells. JVI 2008, 82, 9143–9153. [Google Scholar] [CrossRef]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. PNAS 2009, 106, 14046–14051. [Google Scholar]
- Gannage, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Ramer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; Pypaert, M.; Andersen, J.; Garcia-Sastre, A.; Munz, C. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; Federico, M.; Panganiban, A.; Vergne, I.; Deretic, V. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef]
- Panyasrivanit, M.; Khakpoor, A.; Wikan, N.; Smith, D.R. Linking dengue virus entry and translation/replication through amphisomes. Autophagy 2009, 5, 434–435. [Google Scholar] [CrossRef]
- Panyasrivanit, M.; Khakpoor, A.; Wikan, N.; Smith, D.R. Co-localization of constituents of the dengue virus translation and replication machinery with amphisomes. J. Gen. Virol. 2009, 90, 448–456. [Google Scholar] [CrossRef]
- Shelly, S.; Lukinova, N.; Bambina, S.; Berman, A.; Cherry, S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 2009, 30, 588–598. [Google Scholar] [CrossRef]
- Takahashi, M.N.; Jackson, W.; Laird, D.T.; Culp, T.D.; Grose, C.; Haynes, J.I., 2nd; Benetti, L. Varicella-zoster virus infection induces autophagy in both cultured cells and human skin vesicles. JVI 2009, 83, 5466–5476. [Google Scholar] [CrossRef]
- Tang, H.; Da, L.; Mao, Y.; Li, Y.; Li, D.; Xu, Z.; Li, F.; Wang, Y.; Tiollais, P.; Li, T.; Zhao, M. Hepatitis B virus X protein sensitizes cells to starvation-induced autophagy via up-regulation of beclin 1 expression. Hepatology 2009, 49, 60–71. [Google Scholar] [CrossRef]
- Zhou, Z.; Jiang, X.; Liu, D.; Fan, Z.; Hu, X.; Yan, J.; Wang, M.; Gao, G.F. Autophagy is involved in influenza A virus replication. Autophagy 2009, 5, 321–328. [Google Scholar] [CrossRef]
- Khakpoor, A.; Panyasrivanit, M.; Wikan, N.; Smith, D.R. A role for autophagolysosomes in dengue virus 3 production in HepG2 cells. J. Gen. Virol. 2009, 90, 1093–1103. [Google Scholar] [CrossRef]
- Ke, P.Y.; Chen, S.S. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Invest. 2011, 121, 37–56. [Google Scholar] [CrossRef]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Talloczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef]
- Liang, C.; E, X.; Jung, J.U. Downregulation of autophagy by herpesvirus Bcl-2 homologs. Autophagy 2008, 4, 268–272. [Google Scholar]
- Ku, B.; Woo, J.S.; Liang, C.; Lee, K.H.; Hong, H.S.; E, X.; Kim, K.S.; Jung, J.U.; Oh, B.H. Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine gamma-herpesvirus 68. PLoS Pathog. 2008, 4, e25. [Google Scholar] [CrossRef]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef]
- Levine, B.; Deretic, V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 2007, 7, 767–777. [Google Scholar] [CrossRef]
- Kemball, C.C.; Alirezaei, M.; Flynn, C.T.; Wood, M.R.; Harkins, S.; Kiosses, W.B.; Whitton, J.L. Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. JVI 2010, 84, 12110–12124. [Google Scholar] [CrossRef]
- Joubert, P.E.; Meiffren, G.; Gregoire, I.P.; Pontini, G.; Richetta, C.; Flacher, M.; Azocar, O.; Vidalain, P.O.; Vidal, M.; Lotteau, V.; Codogno, P.; Rabourdin-Combe, C.; Faure, M. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 2009, 6, 354–366. [Google Scholar] [CrossRef]
- Denizot, M.; Varbanov, M.; Espert, L.; Robert-Hebmann, V.; Sagnier, S.; Garcia, E.; Curriu, M.; Mamoun, R.; Blanco, J.; Biard-Piechaczyk, M. HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy 2008, 4, 998–1008. [Google Scholar]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Invest. 2006, 116, 2161–2172. [Google Scholar] [CrossRef]
- Nakamoto, M.; Moy, R.H.; Xu, J.; Bambina, S.; Yasunaga, A.; Shelly, S.S.; Gold, B.; Cherry, S. Virus recognition by Toll-7 activates antiviral autophagy in Drosophila. Immunity 2012, 36, 658–667. [Google Scholar] [CrossRef]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Martinon, F. The endoplasmic reticulum: A sensor of cellular stress that modulates immune responses. Microbes Infect. 2012, 14, 1293–1300. [Google Scholar] [CrossRef]
- Mohl, B.P.; Tedbury, P.R.; Griffin, S.; Harris, M. Hepatitis C virus-induced autophagy is independent of the unfolded protein response. JVI 2012, 86, 10724–10732. [Google Scholar] [CrossRef]
- McLean, J.E.; Wudzinska, A.; Datan, E.; Quaglino, D.; Zakeri, Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J. Biol. Chem. 2011, 286, 22147–22159. [Google Scholar]
- Carpenter, J.E.; Jackson, W.; Benetti, L.; Grose, C. Autophagosome formation during varicella-zoster virus infection following endoplasmic reticulum stress and the unfolded protein response. JVI 2011, 85, 9414–9424. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Fitzgerald, K.A. Cytosolic surveillance and antiviral immunity. Curr. Opin. Virol. 2011, 1, 455–462. [Google Scholar] [CrossRef]
- Ramos, H.J.; Gale, M., Jr. RIG-I like receptors and their signaling crosstalk in the regulation of antiviral immunity. Curr. Opin. Virol. 2011, 1, 167–176. [Google Scholar] [CrossRef]
- Arpaia, N.; Barton, G.M. Toll-like receptors: key players in antiviral immunity. Curr. Opin. Virol. 2011, 1, 447–454. [Google Scholar] [CrossRef]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar]
- Gorbea, C.; Makar, K.A.; Pauschinger, M.; Pratt, G.; Bersola, J.L.; Varela, J.; David, R.M.; Banks, L.; Huang, C.H.; Li, H.; Schultheiss, H.P.; Towbin, J.A.; Vallejo, J.G.; Bowles, N.E. A role for Toll-like receptor 3 variants in host susceptibility to enteroviral myocarditis and dilated cardiomyopathy. J. Biol. Chem. 2010, 285, 23208–23223. [Google Scholar]
- Into, T.; Inomata, M.; Takayama, E.; Takigawa, T. Autophagy in regulation of Toll-like receptor signaling. Cell Signal. 2012, 24, 1150–1162. [Google Scholar] [CrossRef]
- Delgado, M.A.; Deretic, V. Toll-like receptors in control of immunological autophagy. Cell Death Differ. 2009, 16, 976–983. [Google Scholar] [CrossRef]
- Shi, C.S.; Kehrl, J.H. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 2008, 283, 33175–33182. [Google Scholar]
- Deretic, V. Autophagy: an emerging immunological paradigm. J. Immunol. 2012, 189, 15–20. [Google Scholar] [CrossRef]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; Dotsch, V.; Bumann, D.; Dikic, I. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar]
- Orvedahl, A.; Sumpter, R., Jr.; Xiao, G.; Ng, A.; Zou, Z.; Tang, Y.; Narimatsu, M.; Gilpin, C.; Sun, Q.; Roth, M.; Forst, C.V.; Wrana, J.L.; Zhang, Y.E.; Luby-Phelps, K.; Xavier, R.J.; Xie, Y.; Levine, B. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011, 480, 113–117. [Google Scholar]
- Munz, C. Antigen processing by macroautophagy for MHC presentation. FIMMU 2011, 2, 42. [Google Scholar]
- Martinez-Borra, J.; Lopez-Larrea, C. The emergence of the major histocompatilibility complex. Adv. Exp. Med. Biol. 2012, 738, 277–289. [Google Scholar] [CrossRef]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar]
- Gannage, M.; Munz, C. MHC presentation via autophagy and how viruses escape from it. Semin. Immunopathol. 2010, 32, 373–381. [Google Scholar] [CrossRef]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippe, R.; Desjardins, M. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef]
- Leung, C.S.; Taylor, G.S. Nuclear shelter: The influence of subcellular location on the processing of antigens by macroautophagy. Autophagy 2010, 6. [Google Scholar]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Munz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar]
- Schmid, D.; Pypaert, M.; Munz, C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef]
- Schlegel, A.; Giddings, T.H., Jr.; Ladinsky, M.S.; Kirkegaard, K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. JVI 1996, 70, 6576–6588. [Google Scholar]
- Huang, S.C.; Chang, C.L.; Wang, P.S.; Tsai, Y.; Liu, H.S. Enterovirus 71-induced autophagy detected in vitro and in vivo promotes viral replication. J. Med. Virol. 2009, 81, 1241–1252. [Google Scholar]
- O'Donnell, V.; Pacheco, J.M.; LaRocco, M.; Burrage, T.; Jackson, W.; Rodriguez, L.L.; Borca, M.V.; Baxt, B. Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology 2011, 410, 142–150. [Google Scholar] [CrossRef]
- Belov, G.A.; Altan-Bonnet, N.; Kovtunovych, G.; Jackson, C.L.; Lippincott-Schwartz, J.; Ehrenfeld, E. Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. JVI 2007, 81, 558–567. [Google Scholar] [CrossRef]
- Belov, G.A.; Feng, Q.; Nikovics, K.; Jackson, C.L.; Ehrenfeld, E. A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLoS Pathog. 2008, 4, e1000216. [Google Scholar]
- Hsu, N.Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; Cameron, C.E.; Ehrenfeld, E.; van Kuppeveld, F.J.; Altan-Bonnet, N. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef]
- Taylor, M.P.; Burgon, T.B.; Kirkegaard, K.; Jackson, W.T. Role of microtubules in extracellular release of poliovirus. JVI 2009, 83, 6599–6609. [Google Scholar] [CrossRef]
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.H. Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043. [Google Scholar]
- Tanida, I.; Fukasawa, M.; Ueno, T.; Kominami, E.; Wakita, T.; Hanada, K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009, 5, 937–945. [Google Scholar] [CrossRef]
- Taguwa, S.; Kambara, H.; Fujita, N.; Noda, T.; Yoshimori, T.; Koike, K.; Moriishi, K.; Matsuura, Y. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. JVI 2011, 85, 13185–13194. [Google Scholar] [CrossRef]
- Shrivastava, S.; Raychoudhuri, A.; Steele, R.; Ray, R.; Ray, R.B. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011, 53, 406–414. [Google Scholar] [CrossRef]
- Prentice, E.; Jerome, W.G.; Yoshimori, T.; Mizushima, N.; Denison, M.R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 2004, 279, 10136–10141. [Google Scholar]
- Zhao, Z.; Thackray, L.B.; Miller, B.C.; Lynn, T.M.; Becker, M.M.; Ward, E.; Mizushima, N.N.; Denison, M.R.; Virgin, H.W.T. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 2007, 3, 581–585. [Google Scholar]
- Knoops, K.; Kikkert, M.; Worm, S.H.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef]
- Reggiori, F.; Monastyrska, I.; Verheije, M.H.; Cali, T.; Ulasli, M.; Bianchi, S.; Bernasconi, R.; de Haan, C.A.; Molinari, M. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe 2010, 7, 500–508. [Google Scholar] [CrossRef]
- Heaton, N.S.; Randall, G. Dengue virus and autophagy. Viruses 2011, 3, 1332–1341. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Wang, Z.; Liu, K.; Wang, Y.; Liu, J.; Ding, H.; Yuan, Z. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. JVI 2011, 85, 6319–6333. [Google Scholar] [CrossRef]
- Deretic, V.; Jiang, S.; Dupont, N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol. 2012.
- Fliss, P.M.; Jowers, T.P.; Brinkmann, M.M.; Holstermann, B.; Mack, C.; Dickinson, P.; Hohenberg, H.; Ghazal, P.; Brune, W. Viral mediated redirection of NEMO/IKKgamma to autophagosomes curtails the inflammatory cascade. PLoS Pathog. 2012, 8, e1002517. [Google Scholar]
- Gregoire, I.P.; Richetta, C.; Meyniel-Schicklin, L.; Borel, S.; Pradezynski, F.; Diaz, O.; Deloire, A.; Azocar, O.; Baguet, J.; Le Breton, M.; Mangeot, P.E.; Navratil, V.; Joubert, P.E.; Flacher, M.; Vidalain, P.O.; Andre, P.; Lotteau, V.; Biard-Piechaczyk, M.; Rabourdin-Combe, C.; Faure, M. IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 2011, 7, e1002422. [Google Scholar]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; Nibert, M.L.; Superti-Furga, G.; Kagan, J.C. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar]
- Zemirli, N.; Arnoult, D. Mitochondrial anti-viral immunity. Int. J. Biochem. Cell Biol. 2012, 44, 1473–1476. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chiramel, A.I.; Brady, N.R.; Bartenschlager, R. Divergent Roles of Autophagy in Virus Infection. Cells 2013, 2, 83-104. https://doi.org/10.3390/cells2010083
Chiramel AI, Brady NR, Bartenschlager R. Divergent Roles of Autophagy in Virus Infection. Cells. 2013; 2(1):83-104. https://doi.org/10.3390/cells2010083
Chicago/Turabian StyleChiramel, Abhilash I., Nathan R. Brady, and Ralf Bartenschlager. 2013. "Divergent Roles of Autophagy in Virus Infection" Cells 2, no. 1: 83-104. https://doi.org/10.3390/cells2010083
APA StyleChiramel, A. I., Brady, N. R., & Bartenschlager, R. (2013). Divergent Roles of Autophagy in Virus Infection. Cells, 2(1), 83-104. https://doi.org/10.3390/cells2010083