HSV-1 ICP0: An E3 Ubiquitin Ligase That Counteracts Host Intrinsic and Innate Immunity


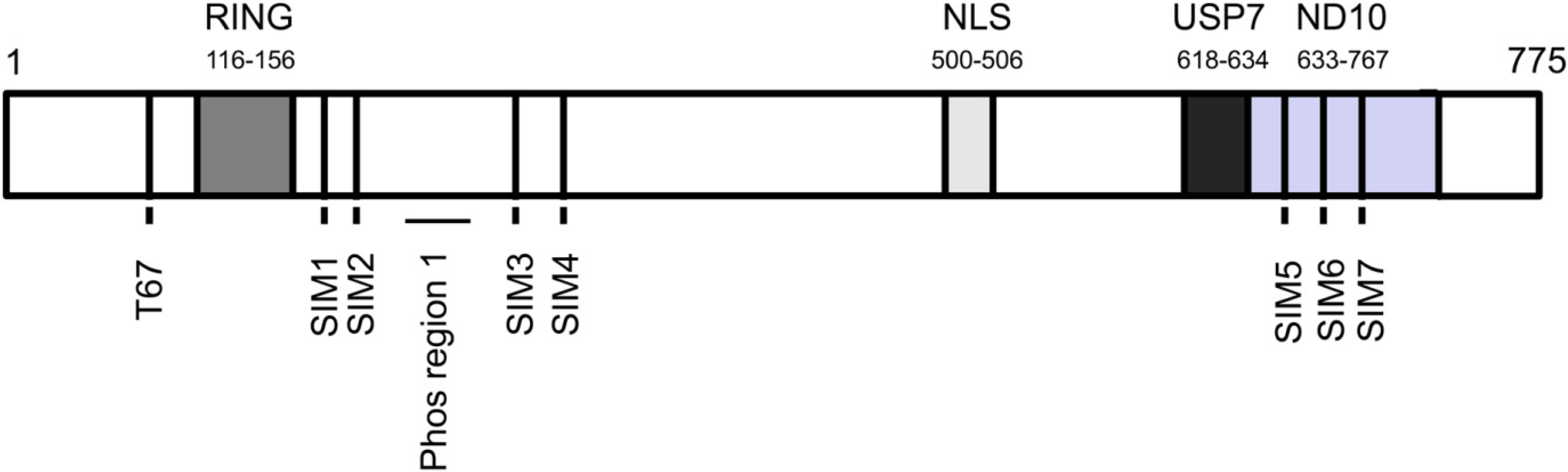{kind=link}
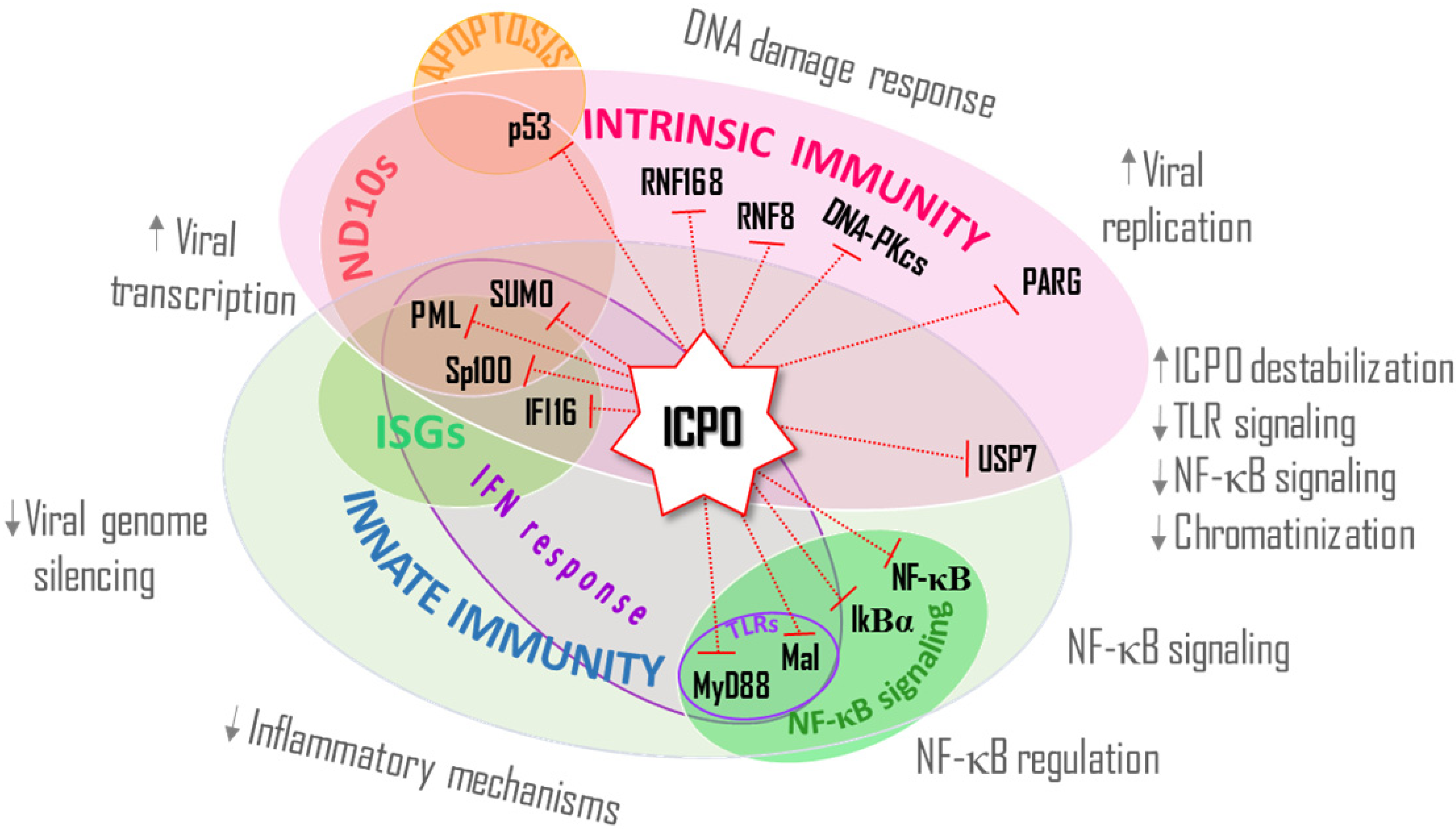
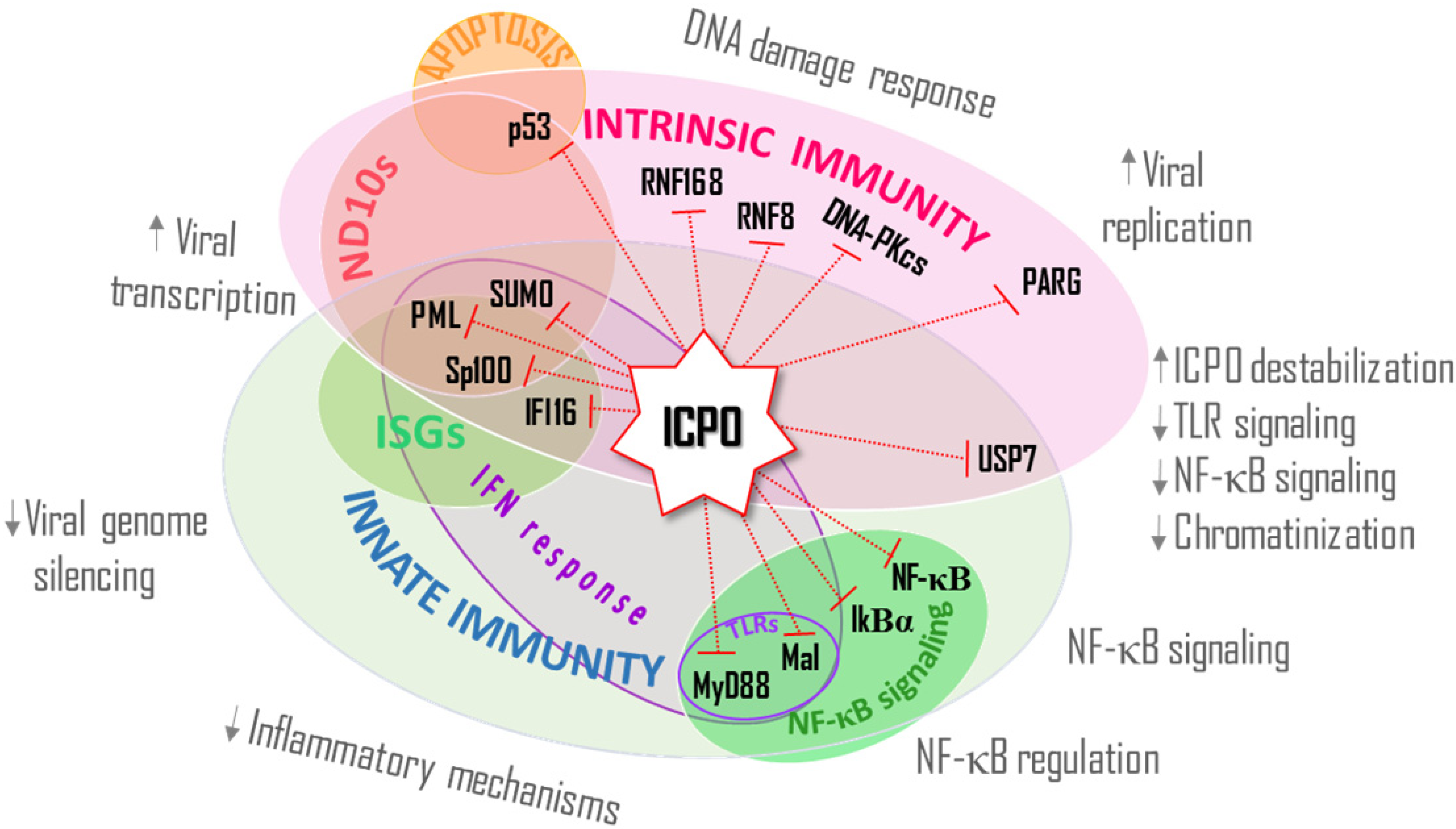{kind=link}
Abstract
:1. Introduction
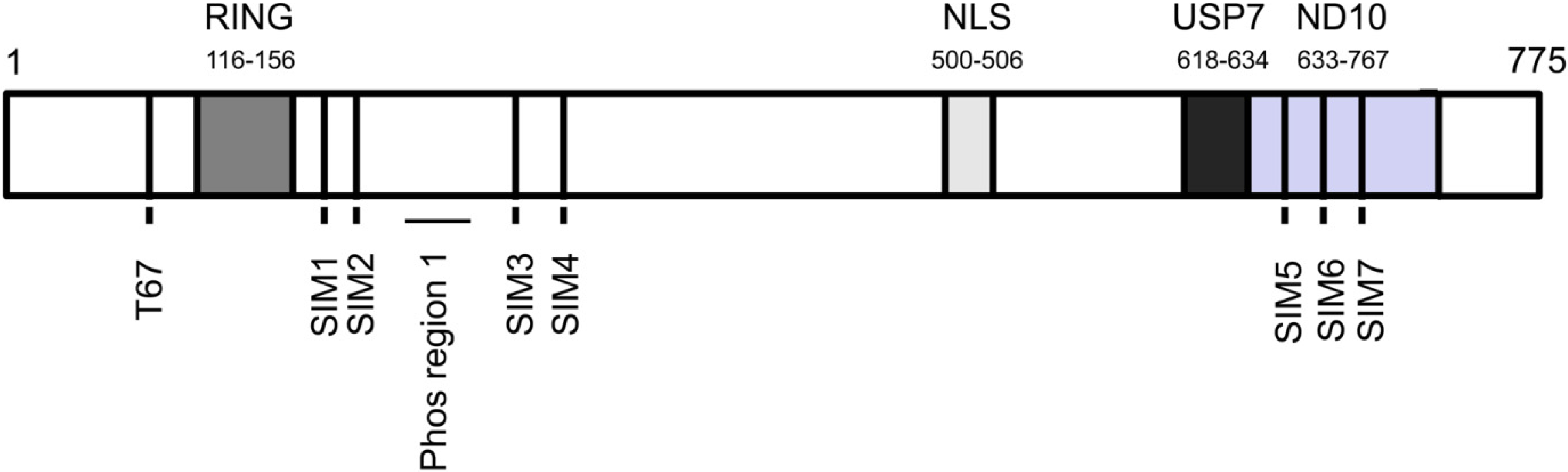
3. ICP0 Interferes With the DNA Damage Response, an Intrinsic Defense Against HSV-1
4. ICP0’s E3 Ub Ligase Activity Modulates the Activation and Establishment of the IFN Response, an Innate Host Defense
4.1. IFI16
4.2. MyD88 and Mal
4.3. NF-κB Signaling
5. ICP0 Directs the Degradation of USP7, a Mediator of Intrinsic and Innate Immunity
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interests
References
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Shabek, N.; Ciechanover, A. Degradation of ubiquitin: The fate of the cellular reaper. Cell Cycle 2010, 9, 523–530. [Google Scholar]
- Van Wijk, S.J.; Timmers, H.T. The family of ubiquitin-conjugating enzymes (E2s): Deciding between life and death of proteins. FASEB J. 2010, 24, 981–993. [Google Scholar] [CrossRef]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar]
- Welchman, R.L.; Gordon, C.; Mayer, R.J. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat. Rev. Mol. Cell Biol. 2005, 6, 599–609. [Google Scholar] [CrossRef]
- Jacobson, A.D.; Zhang, N.Y.; Xu, P.; Han, K.J.; Noone, S.; Peng, J.; Liu, C.W. The lysine 48 and lysine 63 ubiquitin conjugates are processed differently by the 26 s proteasome. J. Biol. Chem. 2009, 184, 35485–35494. [Google Scholar]
- Tokunaga, F.; Sakata, S.; Saeki, Y.; Satomi, Y.; Kirisako, T.; Kamei, K.; Nakagawa, T.; Kato, M.; Murata, S.; Yamaoka, S.; et al. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat. Cell Biol. 2009, 11, 123–132. [Google Scholar] [CrossRef]
- Roizman, R.; Whitley, R.J. Herpes Simplex Viruses; Knipe, D.M., Ed.; Lippincott Williams & Wilkins: New York, NY, USA, 2007; pp. 2501–2601. [Google Scholar]
- Yan, N.; Chen, Z.J. Intrinsic antiviral immunity. Nat. Immunol. 2012, 13, 214–222. [Google Scholar] [CrossRef]
- Bieniasz, P.D. Intrinsic immunity: A front-line defense against viral attack. Nat. Immunol. 2004, 5, 1109–1115. [Google Scholar] [CrossRef]
- Regad, T.; Saib, A.; Lallemand-Breitenbach, V.; Pandolfi, P.P.; de The, H.; Chelbi‐Alix, M.K. PML mediates the interferon-induced antiviral state against a complex retrovirus via its association with the viral transactivator. EMBO J. 2001, 20, 3495–3505. [Google Scholar] [CrossRef]
- McNally, B.A.; Trgovcich, J.; Maul, G.G.; Liu, Y.; Zheng, P. A role for cytoplasmic PML in cellular resistance to viral infection. PLoS One 2008, 3, e2277. [Google Scholar] [CrossRef]
- Tavalai, N.; Stamminger, T. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim. Biophys. Acta 2008, 1783, 2207–2221. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Coscoy, L.; Ganem, D. Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl. Acad. Sci. USA 2000, 97, 8051–8056. [Google Scholar] [CrossRef]
- Ishido, S.; Choi, J.K.; Lee, B.S.; Wang, C.; DeMaria, M.; Johnson, R.P.; Cohen, G.B.; Jung, J.U. Inhibition of natural killer cell-mediated cytotoxicity by Kaposi’s sarcoma-associated herpesvirus K5 protein. Immunity 2000, 13, 365–374. [Google Scholar] [CrossRef]
- Zhu, H.; Zheng, C.; Xing, J.; Wang, S.; Li, S.; Lin, R.; Mossman, K.L. Varicella-zoster virus immediate-early protein ORF61 abrogates the IRF3-mediated innate immune response through degradation of activated IRF3. J. Virol. 2011, 85, 11079–11089. [Google Scholar] [CrossRef]
- Wang, L.; Oliver, S.L.; Sommer, M.; Rajamani, J.; Reichelt, M.; Arvin, A.M. Disruption of PML nuclear bodies is mediated by ORF61 SUMO-interacting motifs and required for varicella-zoster virus pathogenesis in skin. PLoS Pathog. 2011, 7, e1002157. [Google Scholar] [CrossRef]
- De Bie, P.; Ciechanover, A. Ubiquitination of E3 ligases: Self-regulation of the ubiquitin system via proteolytic and non-proteolytic mechanisms. Cell Death Differ. 2011, 18, 1393–1402. [Google Scholar] [CrossRef]
- Leib, D.A.; Coen, D.M.; Bogard, C.L.; Hicks, K.A.; Yager, D.R.; Knipe, D.M.; Tyler, K.L.; Schaffer, P.A. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J. Virol. 1989, 63, 759–768. [Google Scholar]
- Cai, W.Z.; Schaffer, P.A. Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J. Virol. 1989, 63, 4579–4589. [Google Scholar]
- Boutell, C.; Everett, R.D. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J. Gen. Virol. 2013, 94, 465–481. [Google Scholar] [CrossRef]
- Sacks, W.R.; Schaffer, P.A. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. J. Virol. 1987, 61, 829–839. [Google Scholar]
- Cai, W.; Schaffer, P.A. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J. Virol. 1992, 66, 2904–2915. [Google Scholar]
- Everett, R.D. ICP0 induces the accumulation of colocalizing conjugated ubiquitin. J. Virol. 2000, 74, 9994–10005. [Google Scholar] [CrossRef]
- Boutell, C.; Canning, M.; Orr, A.; Everett, R.D. Reciprocal activities between herpes simplex virus type 1 regulatory protein ICP0, a ubiquitin E3 ligase, and ubiquitin-specific protease USP7. J. Virol. 2005, 79, 12342–12354. [Google Scholar] [CrossRef]
- Boutell, C.; Everett, R.D. The herpes simplex virus type 1 (HSV-1) regulatory protein ICP0 interacts with and Ubiquitinates p53. J. Biol. Chem. 2003, 178, 36596–36602. [Google Scholar] [CrossRef]
- Boutell, C.; Sadis, S.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 2002, 76, 841–850. [Google Scholar] [CrossRef]
- Hagglund, R.; Van Sant, C.; Lopez, P.; Roizman, B. Herpes simplex virus 1-infected cell protein 0 contains two E3 ubiquitin ligase sites specific for different E2 ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. USA 2002, 99, 631–636. [Google Scholar] [CrossRef]
- Gu, H.; Roizman, B. The degradation of promyelocytic leukemia and Sp100 proteins by herpes simplex virus 1 is mediated by the ubiquitin-conjugating enzyme UbcH5a. Proc. Natl. Acad. Sci. USA 2003, 100, 8963–8968. [Google Scholar] [CrossRef]
- Vanni, E.; Gatherer, D.; Tong, L.; Everett, R.D.; Boutell, C. Functional characterization of residues required for the herpes simplex virus 1 E3 ubiquitin ligase ICP0 to interact with the cellular E2 ubiquitin-conjugating enzyme UBE2D1 (UbcH5a). J. Virol. 2012, 86, 6323–6333. [Google Scholar] [CrossRef]
- Hagglund, R.; Roizman, B. Characterization of the novel E3 ubiquitin ligase encoded in exon 3 of herpes simplex virus-1-infected cell protein 0. Proc. Natl. Acad. Sci. USA 2002, 99, 7889–7894. [Google Scholar] [CrossRef]
- Everett, R.D. Herpes simplex virus type 1 regulatory protein ICP0 does not protect cyclins D1 and D3 from degradation during infection. J. Virol. 2004, 78, 9599–9604. [Google Scholar] [CrossRef]
- Everett, R.D.; Earnshaw, W.C.; Findlay, J.; Lomonte, P. Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. EMBO J. 1999, 18, 1526–1538. [Google Scholar] [CrossRef]
- Lomonte, P.; Sullivan, K.F.; Everett, R.D. Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J. Biol. Chem. 2001, 176, 5829–5835. [Google Scholar] [CrossRef]
- Lomonte, P.; Morency, E. Centromeric protein CENP-B proteasomal degradation induced by the viral protein ICP0. FEBS Lett. 2007, 181, 658–662. [Google Scholar] [CrossRef]
- Everett, R.D.; Murray, J.; Orr, A.; Preston, C.M. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J. Virol. 2007, 81, 10991–11004. [Google Scholar] [CrossRef]
- Everett, R.D.; Chelbi-Alix, M.K. PML and PML nuclear bodies: Implications in antiviral defence. Biochimie 2007, 89, 819–830. [Google Scholar] [CrossRef]
- Regad, T.; Chelbi-Alix, M.K. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene 2001, 20, 7274–7286. [Google Scholar] [CrossRef]
- Bernardi, R.; Papa, A.; Pandolfi, P.P. Regulation of apoptosis by PML and the PML-NBs. Oncogene 2008, 27, 6299–6312. [Google Scholar] [CrossRef]
- Everett, R.D.; Parada, C.; Gripon, P.; Sirma, H.; Orr, A. Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J. Virol. 2008, 82, 2661–2672. [Google Scholar] [CrossRef]
- Everett, R.D.; Rechter, S.; Papior, P.; Tavalai, N.; Stamminger, T.; Orr, A. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 2006, 80, 7995–8005. [Google Scholar] [CrossRef]
- Negorev, D.G.; Vladimirova, O.V.; Ivanov, A.; Rauscher, F. Maul GG Differential role of Sp100 isoforms in interferon-mediated repression of herpes simplex virus type 1 immediate-early protein expression. J. Virol. 2006, 80, 8019–8029. [Google Scholar] [CrossRef]
- Muller, S.; Dejean, A. Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J. Virol. 1999, 73, 5137–5143. [Google Scholar]
- Chelbi-Alix, M.K.; de, Thé, H. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 1999, 18, 935–941. [Google Scholar] [CrossRef]
- Everett, R.D.; Freemont, P.; Saitoh, H.; Dasso, M.; Orr, A.; Kathoria, M.; Parkinson, J. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 1998, 72, 6581–6591. [Google Scholar]
- Maul, G.G.; Everett, R.D. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J. Gen. Virol. 1994, 75, 1223–1233. [Google Scholar] [CrossRef]
- Everett, R.D.; Maul, G.G. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 1994, 13, 5062–5069. [Google Scholar]
- Everett, R.D.; Freemont, P.; Saitoh, H.; Dasso, M.; Orr, A.; Kathoria, M.; Parkinson, J. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 1998, 72, 6581–6591. [Google Scholar]
- Parkinson, J.; Everett, R.D. Alphaherpesvirus proteins related to herpes simplex virus type 1 ICP0 affect cellular structures and proteins. J. Virol. 2000, 74, 10006–10017. [Google Scholar] [CrossRef]
- Walters, M.S.; Kyratsous, C.A.; Silverstein, S.J. The RING finger domain of Varicella-Zoster virus ORF61p has E3 ubiquitin ligase activity that is essential for efficient autoubiquitination and dispersion of Sp100-containing nuclear bodies. J. Virol. 2010, 84, 6861–6865. [Google Scholar] [CrossRef]
- Lanfranca, M.P.; Mostafa, H.H.; Davido, D.J. Two overlapping regions within the N-terminal half of the herpes simplex virus 1 E3 ubiquitin ligase ICP0 facilitate the degradation and dissociation of PML and dissociation of Sp100 from ND10. J. Virol. 2013, 87, 13287–13296. [Google Scholar] [CrossRef]
- Lukashchuk, V.; Everett, R.D. Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J. Virol. 2010, 84, 4026–4040. [Google Scholar] [CrossRef]
- Glass, M.; Everett, R.D. Components of promyelocytic leukemia nuclear bodies (ND10) act cooperatively to repress herpesvirus infection. J. Virol. 2013, 87, 2174–2185. [Google Scholar] [CrossRef]
- Boutell, C.; Cuchet-Lourenco, D.; Vanni, E.; Orr, A.; Glass, M.; McFarlane, S.; Everett, R.D. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog. 2011, 7, e1002245. [Google Scholar] [CrossRef]
- Grotzinger, T.; Sternsdorf, T.; Jensen, K.; Will, H. Interferon-modulated expression of genes encoding the nuclear-dot-associated proteins Sp100 and promyelocytic leukemia protein. Eur. J. Biochem. 1996, 138, 554–560. [Google Scholar]
- Guldner, H.H.; Szostecki, C.; Grotzinger, T.; Will, H. IFN enhance expression of Sp100, an autoantigen in primary biliary cirrhosis. J. Immunol. 1992, 149, 4067–4073. [Google Scholar]
- Cuchet-Lourenco, D.; Boutell, C.; Lukashchuk, V.; Grant, K.; Sykes, A.; Murray, J.; Orr, A.; Everett, R.D. SUMO pathway dependent recruitment of cellular repressors to herpes simplex virus type 1 genomes. PLoS Pathog. 2011, 7, e1002123. [Google Scholar] [CrossRef]
- Hollenbach, A.D.; McPherson, C.J.; Mientjes, E.J.; Iyengar, R. Grosveld G Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J. Cell Sci. 2002, 115, 3319–3330. [Google Scholar]
- Li, R.; Pei, H.; Watson, D.K.; Papas, T.S. EAP1/Daxx interacts with ETS1 and represses transcriptional activation of ETS1 target genes. Oncogene 2000, 19, 745–753. [Google Scholar] [CrossRef]
- Drane, P.; Ouararhni, K.; Depaux, A.; Shuaib, M.; Hamiche, A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010, 24, 1253–1265. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.Y.; Li, X.; et al. Dstinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.M.; Stadler, S.C.; Allis, C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar]
- Newhart, A.; Rafalska-Metcalf, I.U.; Yang, T.; Negorev, D.G. Janicki SM Single-cell analysis of Daxx and ATRX-dependent transcriptional repression. J. Cell Sci. 2012, 125, 5489–5501. [Google Scholar] [CrossRef]
- Dantzer, F.; Ame, J.C.; Schreiber, V.; Nakamura, J.; Menissier-de, Murcia J; Murcia, G. Poly(ADP-ribose) polymerase-1 activation during DNA damage and repair. Methods Enzymol. 2006, 109, 493–510. [Google Scholar]
- Abraham, R.T. PI 3-kinase related kinases: “Big” players in stress-induced signaling pathways. DNA Repair (Amst) 2004, 3, 883–887. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Dery, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 183, 1197–1208. [Google Scholar]
- Turnell, A.S.; Grand, R.J. DNA viruses and the cellular DNA-damage response. J. Gen. Virol. 2012, 93, 2076–2097. [Google Scholar] [CrossRef]
- Shirata, N.; Kudoh, A.; Daikoku, T.; Tatsumi, Y.; Fujita, M.; Kiyono, T.; Sugaya, Y.; Isomura, H.; Ishizaki, K.; Tsurumi, T. Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elicited by herpes simplex virus infection. J. Biol. Chem. 2005, 180, 30336–30341. [Google Scholar]
- Lilley, C.E.; Carson, C.T.; Muotri, A.R.; Gage, F.H.; Weitzman, M.D. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 2005, 102, 5844–5849. [Google Scholar]
- Wilkinson, D.E.; Weller, S.K. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J. Virol. 2004, 78, 4783–4796. [Google Scholar] [CrossRef]
- Li, H.; Baskaran, R.; Krisky, D.M.; Bein, K.; Grandi, P.; Cohen, J.B.; Glorioso, J.C. Chk2 is required for HSV-1 ICP0-mediated G2/M arrest and enhancement of virus growth. Virology 2008, 175, 13–23. [Google Scholar]
- Lees-Miller, S.P.; Long, M.C.; Kilvert, M.A.; Lam, V.; Rice, S.A.; Spencer, C.A. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J. Virol. 1996, 70, 7471–7477. [Google Scholar]
- Parkinson, J.; Lees-Miller, S.P.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J. Virol. 1999, 73, 650–657. [Google Scholar]
- Ferguson, B.J.; Mansur, D.S.; Peters, N.E.; Ren, H.; Smith, G.L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 2012, 1, e00047. [Google Scholar] [CrossRef]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Everett, R.D. Weitzman MD The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog. 2011, 7, e1002084. [Google Scholar]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Landry, S.; Suh, J.; Panier, S.; Everett, R.D.; Stewart, S.G.; Durocher,, D.; et al. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 2010, 29, 943–955. [Google Scholar] [CrossRef]
- Bekker-Jensen, S.; Mailand, N. Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair (Amst) 2010, 9, 1219–1228. [Google Scholar] [CrossRef]
- Chaurushiya, M.S.; Lilley, C.E.; Aslanian, A.; Meisenhelder, J.; Scott, D.C.; Landry, S.; Ticau, S.; Boutell, C.; Yates, R.Y.; Schulman, A.B.; et al. Viral E3 ubiquitin ligase-mediated degradation of a cellular E3: Viral mimicry of a cellular phosphorylation mark targets the RNF8 FHA domain. Mol. Cell 2012, 46, 79–90. [Google Scholar] [CrossRef]
- Grady, S.L; Hwang, J.; Vastag, L.; Rabinowitz, J.D.; Shenk, T. Herpes simplex virus 1 infection activates poly(ADP-ribose) polymerase and triggers the degradation of poly(ADP-ribose) glycohydrolase. J. Virol. 2012, 86, 8259–8268. [Google Scholar] [CrossRef]
- Mossman, K. Analysis of anti-interferon properties of the herpes simplex virus type I ICP0 protein. Methods Mol. Med. 2005, 116, 195–205. [Google Scholar]
- Eidson, K.M.; Hobbs, W.E.; Manning, B.J.; Carlson, P.; DeLuca, N.A. Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral infection. J. Virol. 2002, 76, 2180–2191. [Google Scholar] [CrossRef]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. USA 2012, 109, E3008–E3017. [Google Scholar] [CrossRef]
- Soby, S.; Laursen, R.R.; Ostergaard, L.; Melchjorsen, J. HSV-1-induced chemokine expression via IFI16-dependent and IFI16-independent pathways in human monocyte-derived macrophages. Herpesviridae 2012, 3, 6. [Google Scholar] [CrossRef]
- Li, T.; Diner, B.A.; Chen, J.; Cristea, I.M. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc. Natl. Acad. Sci. USA 2012, 109, 10558–10563. [Google Scholar] [CrossRef]
- Cuchet-Lourenco, D.; Anderson, G.; Sloan, E.; Orr, A.; Everett, R.D. The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J. Virol. 2013, 87, 13422–13432. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Conwell, S.E.; Berrios, C.; DeCaprio, J.A.; Knipe, D.M. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc. Natl. Acad. Sci. USA 2013, 110, E4492–E4501. [Google Scholar]
- La Frazia, S.; Amici, C.; Santoro, M.G. Antiviral activity of proteasome inhibitors in herpes simplex virus-1 infection: Role of nuclear factor-kappaB. Antivir. Ther. 2006, 11, 995–1004. [Google Scholar]
- Lester, S.N.; Li, K. Toll-Like Receptors in Antiviral Innate Immunity. J. Mol. Biol. 2013, 426, 1246–1264. [Google Scholar] [CrossRef]
- Van Lint, A.L.; Murawski, M.R.; Goodbody, R.E.; Severa, M.; Fitzgerald, K.A.; Finberg, W.R.; Knipe, M.D.; Kurt-Jones, A.E. Herpes simplex virus immediate-early ICP0 protein inhibits Toll-like receptor 2-dependent inflammatory responses and NF-kappaB signaling. J. Virol. 2010, 84, 10802–10811. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Palsson-McDermott, E.M.; Bowie, A.G.; Jefferies, C.A.; Mansell, A.S.; Brady, G; Brint, E.; Dunne1, A.; Gray, P.; et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature 2001, 113, 78–83. [Google Scholar]
- Ea, C.K.; Deng, L.; Xia, Z.P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 2006, 22, 245–257. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef]
- Xing, J.; Ni, L.; Wang, S.; Wang, K.; Lin, R.; Zheng, C. Herpes simplex virus 1-encoded tegument protein VP16 abrogates the production of beta interferon (IFN) by inhibiting NF-kappaB activation and blocking IFN regulatory factor 3 to recruit its coactivator CBP. J. Virol. 2013, 87, 9788–9801. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, S.; Wang, K.; Zheng, C. Herpes simplex virus 1 DNA polymerase processivity factor UL42 inhibits TNF-alpha-induced NF-kappaB activation by interacting with p65/RelA and p50/NF-kappaB1. Med. MicroBiol. Immunol. 2013, 102, 313–325. [Google Scholar] [CrossRef]
- Jin, H.; Ma, Y.; Yan, Z.; Prabhakar, B.S.; He, B. Activation of NF-kappaB in CD8+ dendritic cells Ex Vivo by the gamma134.5 null mutant correlates with immunity against herpes simplex virus 1. J. Virol. 2012, 86, 1059–1068. [Google Scholar] [CrossRef]
- Cotter, C.R.; Kim, W.K.; Nguyen, M.L.; Yount, J.S.; Lopez, C.B.; Blaho, J.A.; Moran, T.M. The virion host shutoff protein of herpes simplex virus 1 blocks the replication-independent activation of NF-kappaB in dendritic cells in the absence of type I interferon signaling. J. Virol. 2011, 85, 12662–12672. [Google Scholar] [CrossRef]
- Kim, J.C.; Lee, S.Y.; Kim, S.Y.; Kim, J.K.; Kim, H.J.; Lee, HM; Choi, M.S.; Min, J.S.; Kim, M.J.; Choi, H.S.; et al. HSV-1 ICP27 suppresses NF-kappaB activity by stabilizing IkappaBalpha. FEBS Lett. 2008, 182, 2371–2376. [Google Scholar]
- Zhang, J.; Wang, K.; Wang, S.; Zheng, C. Herpes simplex virus 1 E3 ubiquitin ligase ICP0 protein inhibits tumor necrosis factor alpha-induced NF-kappaB activation by interacting with p65/RelA and p50/NF-kappaB1. J. Virol. 2013, 87, 12935–12948. [Google Scholar] [CrossRef]
- Diao, L.; Zhang, B.; Fan, J.; Gao, X.; Sun, S.; Yang, K.; Xin, D.; Jin, N.; Geng, Y.; Wang, C. Herpes virus proteins ICP0 and BICP0 can activate NF-kappaB by catalyzing IkappaBalpha ubiquitination. Cell Signal. 2005, 17, 217–229. [Google Scholar] [CrossRef]
- Meredith, M.; Orr, A.; Elliott, M.; Everett, R.D. Separation of sequence requirements for HSV-1 Vmw110 multimerisation and interaction with a 135-kDa cellular protein. Virology 1995, 109, 174–187. [Google Scholar]
- Meredith, M.; Orr, A.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein Vmw110 binds strongly and specifically to a 135-kDa cellular protein. Virology 1994, 100, 457–469. [Google Scholar] [CrossRef]
- Everett, R.D.; Meredith, M.; Orr, A.; Cross, A.; Kathoria, M.; Parkinson, J. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 1997, 16, 1519–1530. [Google Scholar] [CrossRef]
- Everett, R.D.; Meredith, M.; Orr, A. The ability of herpes simplex virus type 1 immediate-early protein Vmw110 to bind to a ubiquitin-specific protease contributes to its roles in the activation of gene expression and stimulation of virus replication. J. Virol. 1999, 73, 417–426. [Google Scholar]
- Canning, M.; Boutell, C.; Parkinson, J.; Everett, R.D. A RING finger ubiquitin ligase is protected from autocatalyzed ubiquitination and degradation by binding to ubiquitin-specific protease USP7. J. Biol. Chem. 2004, 179, 38160–38168. [Google Scholar]
- Mostafa, H.H.; Thompson, T.W.; Davido, D.J. N-terminal phosphorylation sites of herpes simplex virus type 1 ICP0 differentially regulate its activities and enhance viral replication. J. Virol. 2013, 87, 2109–2119. [Google Scholar] [CrossRef]
- Daubeuf, S.; Singh, D.; Tan, Y.; Liu, H.; Federoff, H.J.; Bowers, W.J.; Tolba, K. HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood 2009, 113, 3264–3275. [Google Scholar] [CrossRef]
- Sarkari, F.; Wheaton, K.; La Delfa, A.; Mohamed, M.; Shaikh, F.; Khatun, R.; Arrowsmith, C.H.; Frappier, L.; Saridakis, V.; Sheng, Y. Ubiquitin-specific protease 7 is a regulator of ubiquitin-conjugating enzyme UbE2E1. J. Biol. Chem. 2013, 188, 16975–16985. [Google Scholar]
- Boutell, C.; Orr, A.; Everett, R.D. PML residue lysine 160 is required for the degradation of PML induced by herpes simplex virus type 1 regulatory protein ICP0. J. Virol. 2003, 77, 8686–8694. [Google Scholar] [CrossRef]
- De Bie, P.; Zaaroor-Regev, D.; Ciechanover, A. Regulation of the Polycomb protein RING1B ubiquitination by USP7. Biochem. Biophys Res. Commun. 2010, 100, 389–395. [Google Scholar]
- Huang, Z.; Wu, Q.; Guryanova, O.A.; Cheng, L.; Shou, W.; Rich, J.N.; Bao, S. Deubiquitylase HAUSP stabilizes REST and promotes maintenance of neural progenitor cells. Nat. Cell Biol. 2011, 13, 142–152. [Google Scholar] [CrossRef]
- Kwiatkowski, D.L.; Thompson, H.W.; Bloom, D.C. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J. Virol. 2009, 83, 8173–8181. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Coen, D.M.; Knipe, D.M. Kinetics of facultative heterochromatin and polycomb group protein association with the herpes simplex viral genome during establishment of latent infection. MBio 2013, 4, e00590-12. [Google Scholar]
- Ferenczy, M.W.; DeLuca, N.A. Reversal of heterochromatic silencing of quiescent herpes simplex virus type 1 by ICP0. J. Virol. 2011, 85, 3424–3435. [Google Scholar] [CrossRef]
- Ferenczy, M.W.; Ranayhossaini, D.J.; Deluca, N.A. Activities of ICP0 involved in the reversal of silencing of quiescent herpes simplex virus 1. J. Virol. 2011, 85, 4993–5002. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Knipe, D.M. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol. 2008, 82, 12030–12038. [Google Scholar] [CrossRef]
- Coleman, H.M.; Connor, V.; Cheng, Z.S.; Grey, F.; Preston, C.M.; Efstathiou, S. Histone modifications associated with herpes simplex virus type 1 genomes during quiescence and following ICP0-mediated de-repression. J. Gen. Virol. 2008, 89, 68–77. [Google Scholar] [CrossRef]
- Lomonte, P.; Thomas, J.; Texier, P.; Caron, C.; Khochbin, S.; Epstein, A.L. Functional interaction between class II histone deacetylases and ICP0 of herpes simplex virus type 1. J. Virol. 2004, 78, 6744–6757. [Google Scholar] [CrossRef]
- Mostafa, H.H.; Thompson, T.W.; Kushnir, A.S.; Haenchen, S.D.; Bayless, A.M.; Hilliard, J.G.; Link, M.A.; Pitcher, L.A.; Loveday, E.; Schaffer, P.A.; et al. Herpes simplex virus 1 ICP0 phosphorylation site mutants are attenuated for viral replication and impaired for explant-induced reactivation. J. Virol. 2011, 85, 12631–12637. [Google Scholar] [CrossRef]
- Li, M.; Chen, D.; Shiloh, A.; Luo, J.; Nikolaev, A.Y.; Qin, J.; Gu, W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 2002, 116, 648–653. [Google Scholar]
- Boutell, C.; Everett, R.D. Herpes simplex virus type 1 infection induces the stabilization of p53 in a USP7- and ATM-independent manner. J. Virol. 2004, 78, 8068–8077. [Google Scholar]
- Marcos-Villar, L.; Perez-Giron, J.V.; Vilas, J.M.; Soto, A.; de la Cruz-Hererra, C.F.; Lang, V.; Collado, M.; Vidal, A.; Rodríguez, M.S.; Muñoz-Fontela, C.; et al. SUMOylation of p53 mediates interferon activities. Cell Cycle 2013, 12, 2809–2816. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Perusina Lanfranca, M.; Mostafa, H.H.; Davido, D.J. HSV-1 ICP0: An E3 Ubiquitin Ligase That Counteracts Host Intrinsic and Innate Immunity. Cells 2014, 3, 438-454. https://doi.org/10.3390/cells3020438
Perusina Lanfranca M, Mostafa HH, Davido DJ. HSV-1 ICP0: An E3 Ubiquitin Ligase That Counteracts Host Intrinsic and Innate Immunity. Cells. 2014; 3(2):438-454. https://doi.org/10.3390/cells3020438
Chicago/Turabian StylePerusina Lanfranca, Mirna, Heba H. Mostafa, and David J. Davido. 2014. "HSV-1 ICP0: An E3 Ubiquitin Ligase That Counteracts Host Intrinsic and Innate Immunity" Cells 3, no. 2: 438-454. https://doi.org/10.3390/cells3020438
APA StylePerusina Lanfranca, M., Mostafa, H. H., & Davido, D. J. (2014). HSV-1 ICP0: An E3 Ubiquitin Ligase That Counteracts Host Intrinsic and Innate Immunity. Cells, 3(2), 438-454. https://doi.org/10.3390/cells3020438