Gene Conversion in Human Genetic Disease




1
Institut National de la Santé et de la Recherche Médicale (INSERM), U613, Brest, France
2
Etablissement Français du Sang (EFS)-Bretagne, Brest, France
3
Faculté de Médecine et des Sciences de la Santé, Université de Bretagne Occidentale (UBO), Brest, France
4
Laboratoire de Génétique Moléculaire et d’Histocompatibilité, Centre Hospitalier Universitaire (CHU) de Brest, Hôpital Morvan, Brest, France
5
Institute of Medical Genetics, School of Medicine, Cardiff University, Heath Park, Cardiff CF14 4XN, UK
*
Author to whom correspondence should be addressed.
Genes 2010, 1(3), 550-563; https://doi.org/10.3390/genes1030550
Submission received: 15 October 2010
/
Revised: 12 November 2010
/
Accepted: 17 November 2010
/
Published: 22 December 2010
(This article belongs to the Special Issue Gene Conversion in Duplicated Genes)
Abstract
:Gene conversion is a specific type of homologous recombination that involves the unidirectional transfer of genetic material from a ‘donor’ sequence to a highly homologous ‘acceptor’. We have recently reviewed the molecular mechanisms underlying gene conversion, explored the key part that this process has played in fashioning extant human genes, and performed a meta-analysis of gene-conversion events known to have caused human genetic disease. Here we shall briefly summarize some of the latest developments in the study of pathogenic gene conversion events, including (i) the emerging idea of minimal efficient sequence homology (MESH) for homologous recombination, (ii) the local DNA sequence features that appear to predispose to gene conversion, (iii) a mechanistic comparison of gene conversion and transient hypermutability, and (iv) recently reported examples of pathogenic gene conversion events.
1. Introduction
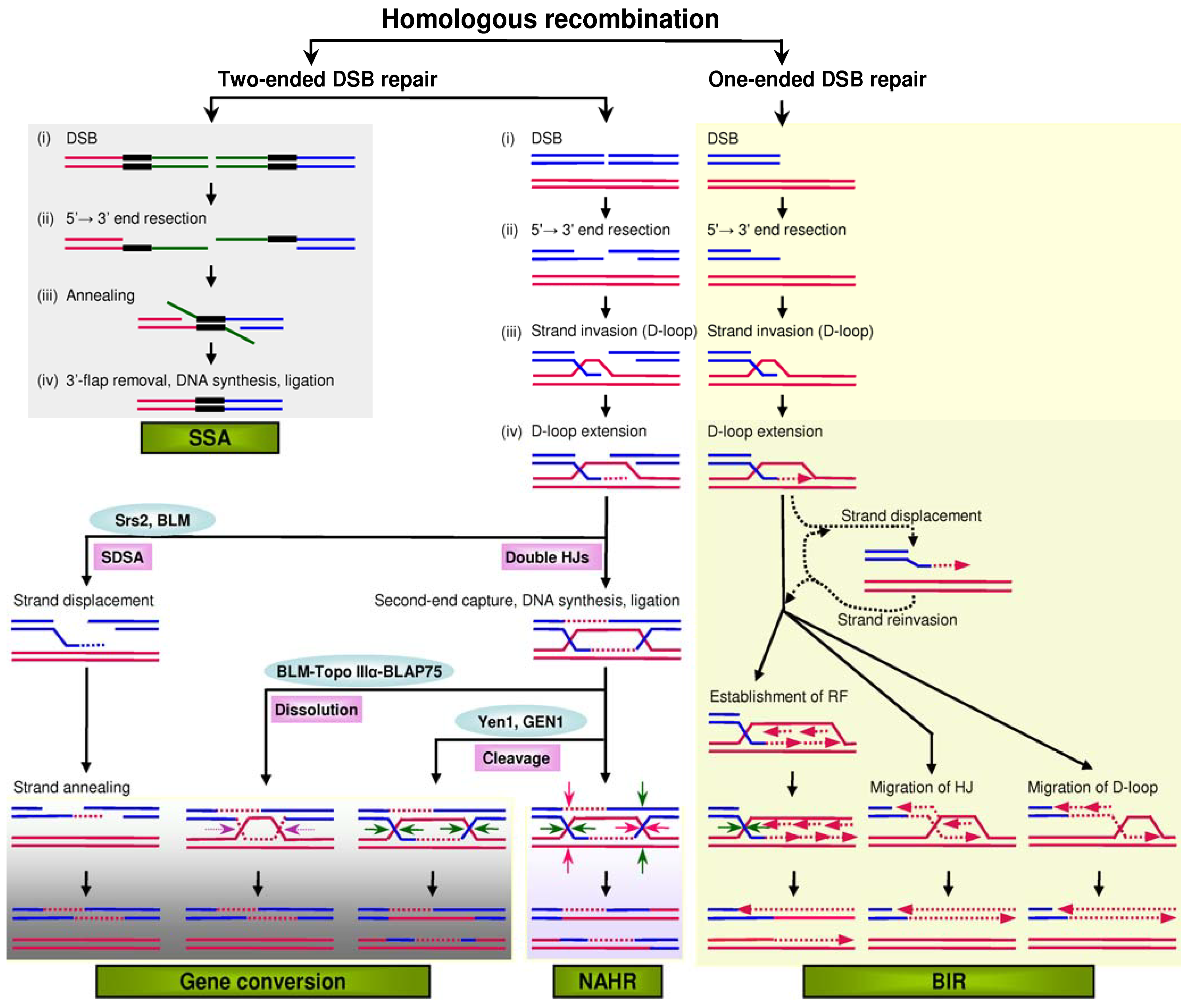
Gene conversion refers to the unidirectional transfer of genetic material from a ‘donor’ sequence to a highly homologous ‘acceptor’. It is one of four pathways of homologous recombination, the other three being non-allelic homologous recombination (NAHR), break-induced replication (BIR) and single-strand annealing (SSA) (Figure 1). All pathways share a similar initiating event: The double-strand break (DSB) generated within one of the duplicated (or repeated) sequences undergoes extensive 5'-end resection to form 3' single-stranded DNA tails. Gene conversion, NAHR and SSA all serve to repair DSBs with two ends, whereas BIR repairs DSBs with only one end. Gene conversion and NAHR may be considered to represent alternative outcomes of a common two-ended DSB repair process (for detailed description, see [1]).
In a recent review article, we assessed the current thinking in relation to the molecular mechanisms underlying gene conversion, surveyed the impact of gene conversion on human genome evolution, and performed a meta-analysis of pathogenic gene conversion events [2]. In this article, we shall briefly summarize some of the latest advances in the study of pathogenic gene conversion events.
2. The Emerging Idea of Minimal Efficient Sequence Homology for Homologous Recombination
Homologous recombination is one of the major mechanisms for the repair of DSBs (the other is non-homologous end joining [1]). As the term implies, homologous recombination is mediated through sequences which exhibit considerable similarity that presumably serves to stabilize chromosomal mispairing. In this regard, it is pertinent to note that the rate of gene conversion is directly proportional to the length of the uninterrupted sequence tract in the putatively converted region: in mouse cells, the minimal efficient processing segment (MEPS) for efficient meiotic homologous recombination is >200 bp [3,4] while in humans, it is estimated to be in the range of 337–456 bp [5].
How extensive does the global sequence similarity need to be for efficient homologous recombination to occur between two interacting sequences? Our analysis of 44 interlocus pathogenic gene conversion events revealed that the similarity between the interacting sequences is almost invariably >92% [2]. This finding has recently received strong support from the results of a study that approached this issue from an evolutionary perspective [6]. Both the human and chimpanzee growth hormone gene (GH1 in humans and GHN in chimpanzee) promoters are highly polymorphic, and all 14 human GH1 promoter SNPs and five of the nine chimpanzee GHN promoter SNPs could potentially have resulted from interlocus gene conversion (i.e., the minor allele occurs in at least one of the cis-linked paralogous genes); by contrast, no polymorphism was evident in the macaque GH1 gene promoter. Remarkably, the mean degree of pair-wise similarity between the GH1 promoter and its paralogs in macaque is 92.0%, significantly lower than in either chimpanzee (93.5%) or human (94.0%). Thus, it appears that if the degree of similarity between related gene sequences falls below a certain threshold (perhaps around 92%), then gene conversion may be significantly reduced or even abolished, with attendant consequences for the genetic variability manifested by the sequences in question [6].
Figure 1.
Mutational models of homologous recombination. In the models of gene conversion, NAHR (non-allelic homologous recombination) and BIR (break-induced replication), the invading strand invariably binds to a homologous sequence. In the model of SSA (single-strand annealing), the black bars indicate the direct repeats that flank a DSB (double-strand break). In the dissolution model of gene conversion, the two facing horizontal purple arrows indicate convergent branch migration. In the double HJs (Holliday junctions) cleavage model of gene conversion, the four horizontal green arrows indicate the orientation of resolution. In the double HJ cleavage model of NAHR, the dHJs can be cleaved as indicated by the green arrows or by the red arrows. In the model of BIR, the invading strand may undergo multiple rounds of displacement and annealing (indicated by dotted arrows) before a stable replication structure is established; this probably reflects repeated attempts to find the other end of the DSB. D-loop, displacement loop; RF, replication fork; SDSA, synthesis-dependent strand annealing. Reprinted from [1].
Figure 1.
Mutational models of homologous recombination. In the models of gene conversion, NAHR (non-allelic homologous recombination) and BIR (break-induced replication), the invading strand invariably binds to a homologous sequence. In the model of SSA (single-strand annealing), the black bars indicate the direct repeats that flank a DSB (double-strand break). In the dissolution model of gene conversion, the two facing horizontal purple arrows indicate convergent branch migration. In the double HJs (Holliday junctions) cleavage model of gene conversion, the four horizontal green arrows indicate the orientation of resolution. In the double HJ cleavage model of NAHR, the dHJs can be cleaved as indicated by the green arrows or by the red arrows. In the model of BIR, the invading strand may undergo multiple rounds of displacement and annealing (indicated by dotted arrows) before a stable replication structure is established; this probably reflects repeated attempts to find the other end of the DSB. D-loop, displacement loop; RF, replication fork; SDSA, synthesis-dependent strand annealing. Reprinted from [1].

By analogy to the concept of MEPS, we have proposed that efficient homologous recombination (including gene conversion) may also require a minimal efficient sequence homology (MESH; approximately 92%) between the interacting sequences [7]. Further supporting evidence for this idea has come from the accurate estimation of copy number variation and multicopy gene number in 159 human genomes; signatures consistent with gene conversion were virtually exclusive to high-identity duplications (>95% sequence similarity) and tended to occur in association with tandem duplications (≤1 Mbp) [8].
In summary, whereas the concept of MEPS defines a local sequence property of homologous recombination, MESH defines a global sequence property of homologous recombination.
3. Local Sequence Features Predisposing to Gene Conversion
A variety of DNA sequences, including direct repeats, inverted repeats (sometimes incorrectly termed palindromes), minisatellite repeats, the χ recombination hotspot, and alternating purine–pyrimidine tracts with Z-DNA-forming potential, have frequently been noted in association with gene conversion in human genes (see references in [9]). In addition, the convergence of biochemical, genetic, and genomic studies in the context of gross genomic deletions, inversions, duplications, and translocations has suggested that the ability of a given DNA sequence to adopt a non-B DNA conformation (e.g., slipped structures, triplexes and tetraplexes), rather than the DNA sequence per se (in the orthodox right-handed Watson-Crick B-form), could induce chromosomal DSBs (reviewed in [10]). However, no methodical statistically based analysis had been performed to formalize these observations until recently, when a series of well-characterized human gene conversion mutations were employed as a test system. The advantage of this novel approach lay in the fact that the extents of the maximal converted tracts (MaxCTs) and minimal converted tracts (MinCTs) associated with such pathological events could usually be fairly accurately determined and annotated [9]. In silico analysis of the DNA sequence tracts involved in 27 nonoverlapping pathogenic gene conversion events in 19 different genes yielded several novel findings [9]. First, gene conversion events tend to occur preferentially within (C + G)- and CpG-rich regions. Second, sequences with the potential to form non-B DNA structures occur disproportionately within MaxCTs and/or short flanking regions. Third, MaxCTs are enriched (P < 0.01) in a truncated version of the χ element (a TGGTGG motif), immunoglobulin heavy chain class switch repeats, translin target sites and several novel motifs including (or overlapping) the classical meiotic recombination hotspot, CCTCCCCT. Finally, gene conversions tend to occur in genomic regions that have the potential to fold into stable hairpin conformations [9]. These findings therefore provide support for the concept that recombination-inducing motifs, in association with alternative DNA conformations, can promote recombination in the human genome.
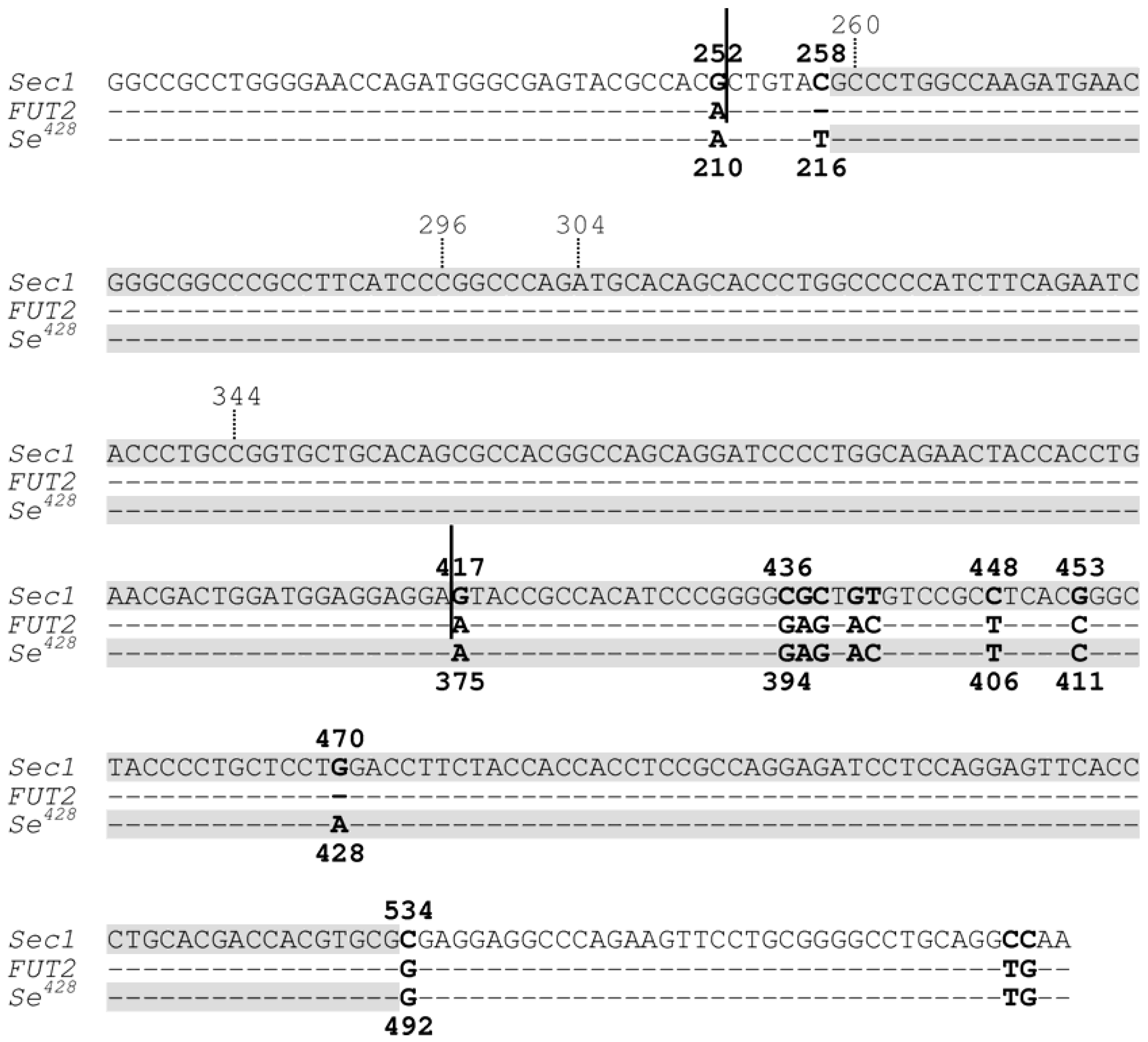
The importance of non-B DNA conformations in predisposing genomic rearrangements is perhaps best exemplified by a rather unique case of gene conversion. Soejima and colleagues reported a Sec1-FUT2-Sec1 hybrid allele that apparently resulted from a gene conversion event [11]. As pointed out by these authors, this allele is more appropriately termed Sec1-Se428-Sec1 because it is the Se428 mutant allele of the FUT2 gene that acts as the donor sequence (Figure 2). Interestingly, the 5' half of the MaxCT of this interlocus gene conversion event overlaps with the crossover region of the previously reported Sefus mutant allele (Figure 2). The Sefus allele was generated by NAHR via a process through which the 3'-part of the FUT2 gene was fused to the 5'-part of the Sec1 gene [12]. Based on our current understanding of mutational mechanisms, we reasoned that the initiating DSBs leading to the Sec1-Se428-Sec1 and Sefus alleles might have occurred within the aforementioned overlapping sequence tract.
Figure 2.
Partial sequence alignment of the Sec1 gene, the FUT2 gene and the Se428 mutant allele of the FUT2 gene. Dashes indicate identity with Sec1 DNA sequence. Nucleotides are numbered in accordance with previous publications (e.g., [11]) for easy comparison; numbers above the aligned sequences refer to Sec1 sequence whereas those below the aligned sequences refer to both FUT2 and Se428 sequences. As compared with the wild-type FUT2 gene, the Se428 mutant allele contains the 428G > A nonsense mutation and the 216C > T polymorphism. Sequence between the two vertical continued bars indicates the crossover region of the non-allelic homologous recombination-derived Sefus allele, in which the 3'-part of the FUT2 gene was fused to the 5'-part of the Sec1 gene. Shaded sequences indicated the maximal converted tract (MaxCT) of the gene conversion-derived Sec1-Se428-Sec1 allele (Sec1 is the acceptor gene whilst Se428 is the donor gene). The overlapping sequence tract between the MaxCT of the Sec1-Se428-Sec1 allele and the crossover region of the Sefus allele spans positions 259 to 416 in the context of the Sec1 sequence. Reprinted from [13].
Figure 2.
Partial sequence alignment of the Sec1 gene, the FUT2 gene and the Se428 mutant allele of the FUT2 gene. Dashes indicate identity with Sec1 DNA sequence. Nucleotides are numbered in accordance with previous publications (e.g., [11]) for easy comparison; numbers above the aligned sequences refer to Sec1 sequence whereas those below the aligned sequences refer to both FUT2 and Se428 sequences. As compared with the wild-type FUT2 gene, the Se428 mutant allele contains the 428G > A nonsense mutation and the 216C > T polymorphism. Sequence between the two vertical continued bars indicates the crossover region of the non-allelic homologous recombination-derived Sefus allele, in which the 3'-part of the FUT2 gene was fused to the 5'-part of the Sec1 gene. Shaded sequences indicated the maximal converted tract (MaxCT) of the gene conversion-derived Sec1-Se428-Sec1 allele (Sec1 is the acceptor gene whilst Se428 is the donor gene). The overlapping sequence tract between the MaxCT of the Sec1-Se428-Sec1 allele and the crossover region of the Sefus allele spans positions 259 to 416 in the context of the Sec1 sequence. Reprinted from [13].

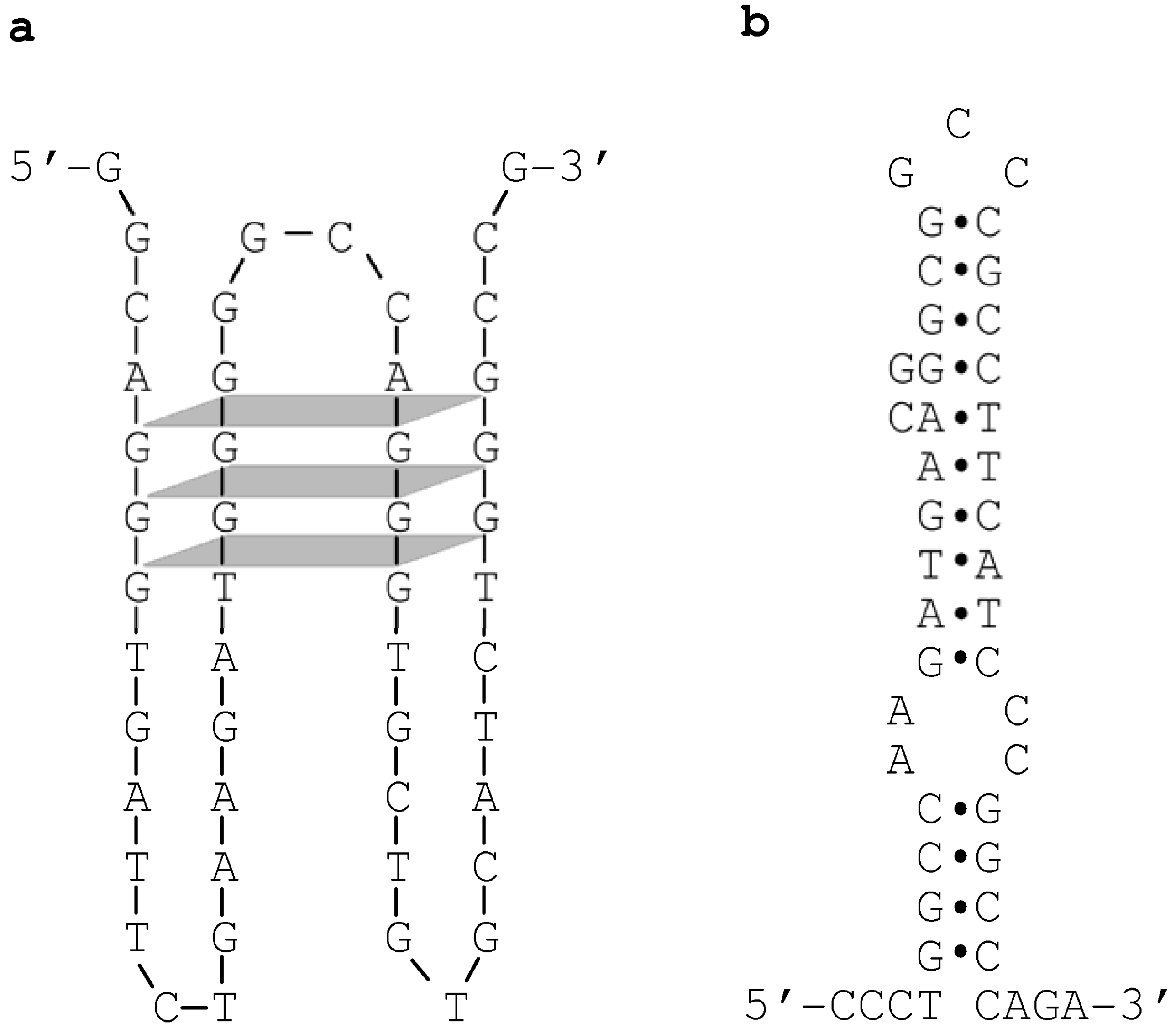
We further reasoned that, were this to be the case, the overlapping sequence tract might be capable of adopting non-B conformation(s) [13]. Indeed, upon inspection, four GGG repeats within the overlapping sequence tract were identified that would appear to have the potential to fold into a tetraplex structure (Figure 3a). In addition, by means of a previously established method for predicting the ‘local’ secondary structure of nucleotide sequences [14], we identified a hairpin structure that can potentially be formed by a specific pair of imperfect inverted repeats (Figure 3b). We speculate that these non-B DNA structures may have acted either individually or synergistically to promote the formation of DSBs, which in turn could have initiated the process of homologous recombination [13].
Figure 3.
Non-B conformations identified within the overlapping sequence tract between the maximal converted tract (MaxCT) of the Sec1-Se428-Sec1 allele and the crossover region of the Sefus allele (refer to Figure 2). (a) A tetraplex structure formed by four GGG repeats. The sequence illustrated corresponds to the reverse complement of Sec1 spanning positions 296 to 344 (see Figure 2); (b) A hairpin structure formed by a pair of imperfect inverted repeats. The sequence illustrated corresponds to Sec1 spanning positions 260 to 304 (see Figure 2). Reprinted from [13].
Figure 3.
Non-B conformations identified within the overlapping sequence tract between the maximal converted tract (MaxCT) of the Sec1-Se428-Sec1 allele and the crossover region of the Sefus allele (refer to Figure 2). (a) A tetraplex structure formed by four GGG repeats. The sequence illustrated corresponds to the reverse complement of Sec1 spanning positions 296 to 344 (see Figure 2); (b) A hairpin structure formed by a pair of imperfect inverted repeats. The sequence illustrated corresponds to Sec1 spanning positions 260 to 304 (see Figure 2). Reprinted from [13].

4. Gene Conversion vs Transient Hypermutability: A Mechanistic Comparison
Data from a wide variety of organisms (including viruses, prokaryotes and yeast, as well as cell lines and tissues from higher eukaryotes) have clearly demonstrated that the number of instances of multiple mutation is significantly higher than would be predicted simply from the mutation frequency and a random distribution of mutations [15]. For example, some of the multiple somatic HPRT mutations detected in a human epithelial cell line were closely spaced, with 4/12 mutation pairs being separated by only 6 bp on average, a much higher proportion than would be expected by chance alone [16]. The most robust data came from studies employing the Big Blue® transgenic mouse system [17,18]; complete sequencing of the 1.4 kb lacI transgene in thousands of mouse mutants from normal tissues and spontaneous tumors demonstrated that the distribution of the spacing between component mutations in doublets (two spatially separated mutations identified in cis) was highly non-random, with half the doublets being separated by <120 bp [18].
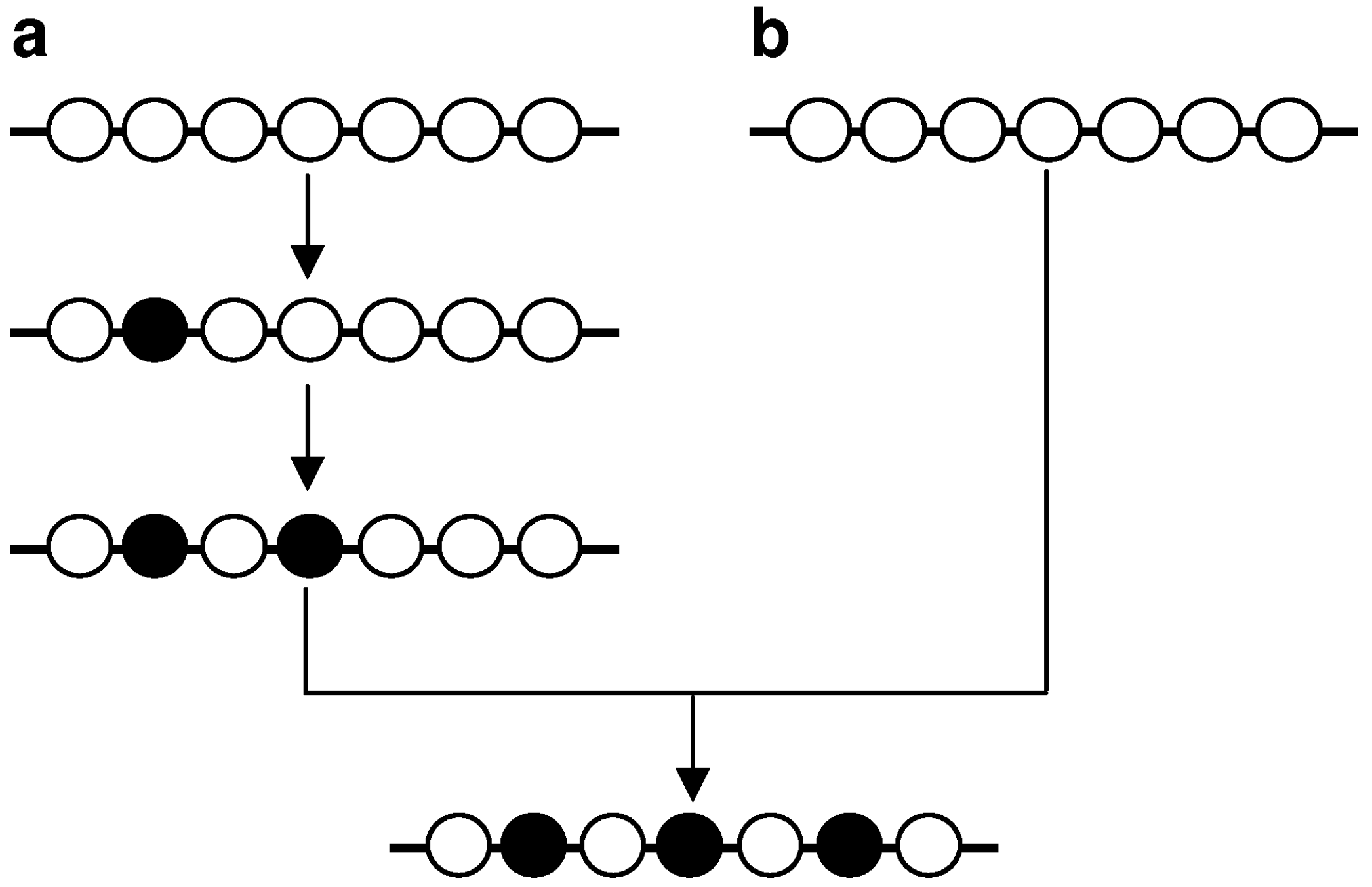
‘Multiple mutations’ can in principle be the observable net result of the sequential accumulation of single mutations independently generated during multiple cell replications (Figure 4a). However, known examples of such mutations [19,20] exhibit an essentially random inter-component spacing distribution, as would be expected for mutations of independent origin [15,21]. Consequently, the multiple mutations that exhibit non-random proximal spacing in higher eukaryotes [16,18]—termed ‘closely spaced multiple mutations’ (CSMMs; [22])—are most compatible with a model in which they are generated simultaneously or quasi-simultaneously in the same cell cycle (Figure 4b). Multiple synchronous mutations have been postulated to arise via transient hypermutability resulting from (i) the deregulated expression of, or conformational change in, either a replicative DNA polymerase or another protein involved in the maintenance of replication fidelity, (ii) the disruption of the balance of the nucleotide pool, or (iii) the recruitment of error-prone DNA polymerases in DNA replication or repair [15,23,24].
Figure 4.
Two concepts for generating multiple mutations. Multiple mutations can accumulate during multiple cell cycles (a) or can be generated in the same cell cycle and in rapid succession (b). Adapted from [22].
Figure 4.
Two concepts for generating multiple mutations. Multiple mutations can accumulate during multiple cell cycles (a) or can be generated in the same cell cycle and in rapid succession (b). Adapted from [22].

Recently, we have extended the concept of transient hypermutability from somatic cells to the germline, using human inherited disease-causing multiple mutations as a model system. Employing stringent criteria for data inclusion, we retrospectively identified 151 potential examples of pathogenic CSMMs [22]. Taken at face value, these examples possessed at least three features which were consistent with those noted with the Big Blue® transgenic mouse system [17,18]. First, a large fraction of the multiple mutations were closely spaced. Second, some single nucleotide substitutions (SNS) were found to coexist with other types of mutation. Finally, a small fraction of the collected multiple mutations comprised three or more distinct components. In particular, eight multiple mutations comprised three or more components within a sequence tract of <100 bp. The majority of these mutations may reasonably be assumed to have occurred as simultaneous or quasi-simultaneous events, thereby providing the first evidence to support the contention that the human germline can also experience transient hypermutability [22].
We then sought to procure evidence to support the postulate that the closely spaced double mutations causing human inherited disease arose predominantly through transient hypermutability. For reasons of simplicity, we focused our attention upon the 102 disease-causing double mutations that comprise exclusively SNS mutations. Transient hypermutability has been postulated to result from three different mechanisms (see before), all of which imply new DNA synthesis. One mutational mechanism which does not involve new DNA synthesis is methylation-mediated deamination of 5-methylcytosine, which gives rise to C > T transitions (or G > A transitions on the complementary strand). Since 5-methylcytosine in the human genome is almost exclusively confined to the CpG dinucleotide, this mechanism accounts for the CpG dinucleotide being a mutation hotspot. We therefore surmised that the proportion of CpG substitution, manifested by the component mutations from a given set of multiple mutations, could be used as a crude indicator of the relative likelihood of transient hypermutability: the lower the proportion of CpG substitution, the higher the likelihood that the multiple mutations would have arisen via transient hypermutability [22].
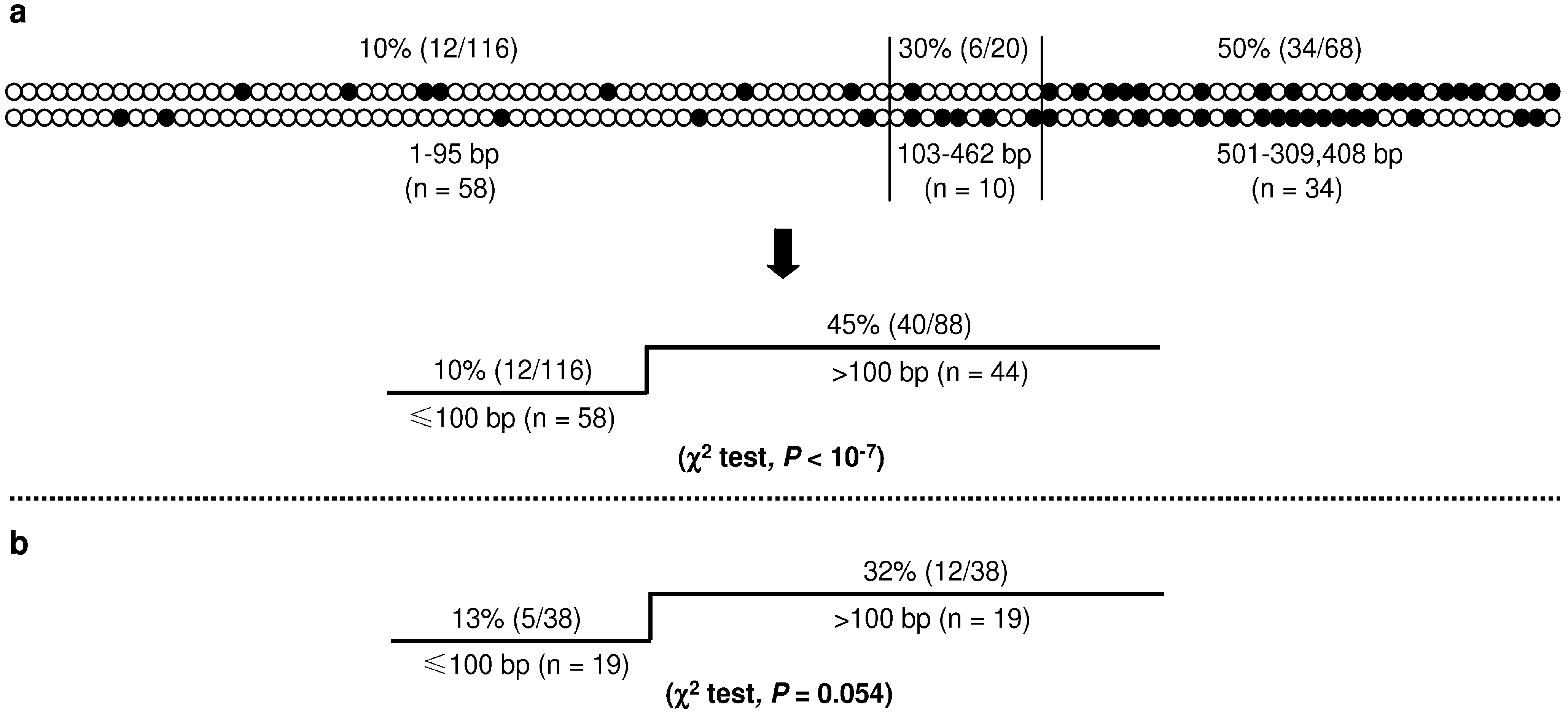
Figure 5.
CpG substitutions in double single nucleotide substitution (SNS) mutations. (a) Top panel illustrates the distribution of CpG substitutions in 102 pathogenic double SNS mutations. Each pair of vertical circles indicates a double SNS mutation. The upper and lower circles indicate the first and second components of a given double mutation, respectively. Solid circles indicate CpG substitutions. The lower panel compares the proportion of CpG substitutions in the group of ≤100 bp with that in the group of >100 bp; (b) Proportion of CpG substitutions in the group of ≤100 bp with that in the group of >100 bp; data were derived from a re-analysis of all double SNS mutations obtained from the Big Blue mice [17,18]. Reprinted from [22].
Figure 5.
CpG substitutions in double single nucleotide substitution (SNS) mutations. (a) Top panel illustrates the distribution of CpG substitutions in 102 pathogenic double SNS mutations. Each pair of vertical circles indicates a double SNS mutation. The upper and lower circles indicate the first and second components of a given double mutation, respectively. Solid circles indicate CpG substitutions. The lower panel compares the proportion of CpG substitutions in the group of ≤100 bp with that in the group of >100 bp; (b) Proportion of CpG substitutions in the group of ≤100 bp with that in the group of >100 bp; data were derived from a re-analysis of all double SNS mutations obtained from the Big Blue mice [17,18]. Reprinted from [22].

The 102 double SNS mutations could be roughly divided into three groups on the basis of the relative proportion of CpG substitution (top panel, Figure 5a). The first group comprised 58 events with an inter-component distance of 1–95 bp, and had the lowest proportion of CpG substitution (10%). The second group comprised 10 events with an inter-component distance of 103–462 bp, with an intermediate CpG substitution rate of 30.0%. The third group, comprising the remaining 34 events with an inter-component distance of 501–309,408 bp, exhibited the highest CpG substitution rate (50%). This distribution pattern coincided with two observations made in the Big Blue transgenic mice: first, nearly all the observed doublets were separated by an intervening sequence of <500 bp; second, it was those doublets with an inter-component distance of 1–100 bp that occurred more frequently than would be expected for independent mutations [18]. Adopting a fairly conservative strategy, we used the cutoff value of ≤100 bp to define CSMMs in the human context, manifesting a CpG substitution rate of 10%, significantly lower than that the 45% which characterized the remaining 44 double SNS events of >100 bp (lower panel, Figure 5a). Employing the same standard, we also revisited the double SNS mutations reported by Buettner et al. [17] and Hill et al. [18] and derived a P value of marginal significance: CpG substitution rates were 13% for the 19 events of ≤100 bp and 32% for the 19 events of >100 bp (Figure 5b).
The aforementioned findings strongly suggest that the two groups of double SNS mutations (i.e., ≤100 bp and >100 bp) arose via qualitatively quite different mutational mechanisms. This postulate was then given further support from the analysis of the highly informative homocoordinate mutations (multiple mutations in the same gene involving the same mutation type but occurring at different sites in cis [18]). Of the 102 double SNS mutations causing inherited disease, 17 were found to be homocoordinate mutations. Again, qualitative differences were apparent between the two groups of homocoordinate mutations. Only one of the six homocoordinate events in the ≤100 bp group involved a CpG substitution. By contrast, 10 of the 11 homocoordinate events in the >100 bp group were characterized by CpG substitutions [22].
Taking these data together, we proposed that CSMMs comprising at least one pair of mutations separated by ≤100 bp may constitute signatures of transient hypermutability in human genes [22]. Here it should however be emphasized that gene conversion events (with the exception of those that induce only a single nucleotide change) constitute in effect an important type of multiple mutation. They are also thought to be generated simultaneously or quasi-simultaneously in the same cell cycle; moreover, their MaxCTs are usually short, rarely exceeding 1 kb [2]. Mechanistically, there is a qualitative difference between these gene conversion events and multiple mutations originating via transient hypermutability. Whereas gene conversion constitutes a template-switching event through which a highly homologous template is faithfully copied by a normal replicative DNA polymerase, transient hypermutability-mediated multiple mutations are due to misincorporation of bases during DNA replication or repair.
5. New Examples of Pathogenic Gene Conversion Events
We initially collated 44 interlocus gene-conversion events (involving a total of 17 genes) that are known to have given rise to human inherited disease (see Table 1 in [2]). In the process of collating examples of multiple mutations that could have arisen through transient hypermutability [22], we identified eight further gene conversion mutations, all of which were included in our subsequent in silico analysis of the local sequence features that might predispose to gene conversion [9]. Here it is important to emphasize that all of these gene conversion events were collected in accordance with fairly stringent selection criteria. For example, all reported gene-conversion events comprising only a single nucleotide substitution were omitted from the analysis, since the possibility that these changes may have originated by simple point mutation can never be excluded. In addition, all gene conversion events that were not fully characterized at the nucleotide sequence level were also excluded [2]. Our collation therefore represents only a fraction of the actual number of pathogenic gene conversion events already described in one form or another in the literature. Hence, these collated data allow only a very conservative estimate to be made of the likely relative frequency of gene conversion as a cause of human inherited disease.
In a just published report, Boria and colleagues investigated the possible occurrence of pseudogene-mediated gene conversion in Diamond-Blackfan anemia (DBA) [25]. Mutations in nine ribosomal protein (RP) genes have so far been reported in ~50% of DBA patients. They aligned sequences of the most frequently mutated RP genes (i.e., RPS19, RPL5 and RPL11) with their respective pseudogene sequences and looked at 5 bp on each side of the mutation; coincidence was found in six mutations (Table 1). Evidently, they “could not exclude that the same changes arose independently in the gene and in the pseudogene” [25]. Here we would like to add that (i) both micro-deletions occurred within short direct repeats (three A and two AGAC, respectively) such that they are explicable by the classical model of replication slippage, a subclass of the recently coined “microhomology-mediated replication-dependent recombination (MMRDR)” mechanism [1], and (ii) three (i.e., c.403G > A, c.166C > T, and c.535C > T) of the four SNS mutations are CpG substitutions. By contrast, the c.191T > C mutation did not occur within a known mutational hotspot and thus may have a higher probability of being generated by gene conversion.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene (Chromosomal Localization) | Mutation | Wild-Type Sequence | Pseudogene Sequences | Pseudogene (Chromosomal Localization) |
|---|---|---|---|---|
| RPS19 (19q13.2) | c.384_385delAA | GGACA AAGAGAT | GGACA --GAGAT | RPS19P2 (1p13.2) |
| c.403G > A | GAATC GCCGGA | GAATC ACCGGA | RPS19P2 (1p13.2) | |
| c.191T > C | GCACC TGTACC | GCACC CGTACC | RPS19P4 (5q11.2) | |
| c.166C > T | ACACG CGAGCT | ACACG TGAGCT | RPS19P7 (10q11.21) | |
| RPL5 (1p22.1) | c.535C > T | CCAAA CGATTC | CCAAA TGATTC | RPL5P34 (22q13.2) |
| RPL11 (1p36.1-p35) | c.94_97delAGAC | GAGAC AGACTGAG | GAGAC ----TGACG | RPL11P5 (12q24.31) |
* From [25]
In certain cases, gene conversion is nevertheless more plausible than simple point mutation as an explanation for the observed single nucleotide changes. For example, Moradkhani et al. identified 14 different human hemoglobin (Hb) variants resulting from identical mutations on either one of the two human α-globin paralogous genes (HBA1 and HBA2) [26]. Interallelic gene conversion was regarded as the most plausible mechanism to account for “the same mutation being ‘transferred’ into different genomic contexts”. In support of this postulate is the fact that 13 out of the 14 Hb variants were located within exons 1 and 2; these two exons (but not exon 3) have been previously shown to be involved in gene conversion events [26]. Another example is provided by the identification of high frequency sequence exchange events between PMS2 and its pseudogene, PMS2CL, in which gene conversion has certainly played a key role [27].
Recently, Gardner et al. [28] reported a pathogenic gene conversion mutation that satisfies our previously established stringent selection criteria [2]. In a family with X-linked cone and cone-rod dystrophies, a missense mutation (c. 529T > C [p. W177R]) in exon 3 of both the long-wavelength-sensitive and medium-wavelength-sensitive cone opsin genes (OPN1LW and OPN1MW) was found to segregate with the disease. As opined by Gardner et al., the spontaneous occurrence of this point mutation in both genes is most unlikely. Much more likely was that the mutation first originated in one gene and was then transferred to the other by a gene conversion event. Indeed, the mutation in the OPN1LW gene was found to be embedded within a block of OPN1MW sequence [28] (a gene conversion mutation involving OPN1LW and OPN1MW was previously reported to cause blue cone monochromacy [29]). This new gene conversion event and the eight events described in Chuzhanova et al. [9] are summarized in Table 2. Additionally, a putative double gene conversion event causing spinal muscular atrophy has also been reported [30].
| Disease/Phenotype | Donor Gene | Acceptor Gene | Chromosomal Localization | Mutation | Ref. |
|---|---|---|---|---|---|
| Congenital adrenal hyperplasia | CYP21A1P | CYP21A2 | 6p21.3 | Intron 2 conversion | [31] |
| Increased CYP3A7 expression in adult liver and intestine | CYP3A4 | CYP3A7 | 7q21-q22.1 | Promoter conversion | [32] |
| Novel St glycophorin | GYPE | GYPA | 4q28-q31 | GPA-E-A hybrid gene | [33] |
| Microcytosis | HBA2 | HBA1 | 16p13.3 | α121 patchwork | [34] |
| Agammaglobulinemia | IGLL3 | IGLL1 | 22q11.23 | Conversion of exon 2 | [35] |
| Sec1–FUT2–Sec1 hybrid allele | FUT2 | Sec1 | 19q13.3 | Conversion involving exonic sequence | [11] |
| Atypical hemolytic uremic syndrome | CR1L | CD46 | 1q32 | D151N + Y155D | [36] |
| Pachyonychia congenita type 2 | KRT17P3 | KRT17 | 17q21.2 | 452G > A and 457T > C | [37] |
| X-linked cone and cone-rod dystrophies | OPN1MW | OPN1LW | Xq28 | c. 529T > C [p. W177R] | [28] |
6. Conclusions
Further gene conversion mutations causing human inherited disease will continue to be identified in the future and these should contribute to our emerging understanding of this important mutational mechanism. Indeed, the identification and analysis of such naturally occurring gene conversion events will serve as an invaluable source for refining the general characteristics of gene conversion in particular and homologous recombination in general. Although, to date, most relevant research has focused on the study of germline mutations, we speculate that somatic mosaicism resulting from interallelic gene conversion, which has until now largely escaped our attention, could turn out to be a potentially new and important modifier of human inherited disease [2]. Finally, it is pertinent to mention that an artificial form of gene conversion, brought about by means of homing endonucleases, holds great promise for targeted gene therapy in patients with monogenic diseases (reviewed in [38]).
References
- Chen, J.M.; Cooper, D.N.; Férec, C.; Kehrer-Sawatzki, H.; Patrinos, G.P. Genomic rearrangements in inherited disease and cancer. Semin. Cancer Biol. 2010, 20, 222–233. [Google Scholar] [CrossRef]
- Chen, J.M.; Cooper, D.N.; Chuzhanova, N.; Férec, C.; Patrinos, G.P. Gene conversion: mechanisms, evolution and human disease. Nat. Rev.Genet. 2007, 8, 762–775. [Google Scholar]
- Liskay, R.M.; Letsou, A.; Stachelek, J.L. Homology requirement for efficient gene conversion between duplicated chromosomal sequences in mammalian cells. Genetics 1987, 115, 161–167. [Google Scholar]
- Waldman, A.S.; Liskay, R.M. Dependence of intrachromosomal recombination in mammalian cells on uninterrupted homology. Mol. Cell Biol. 1988, 8, 5350–5357. [Google Scholar]
- Reiter, L.T.; Hastings, P.J.; Nelis, E.; de Jonghe, P.; van Broeckhoven, C.; Lupski, J.R. Human meiotic recombination products revealed by sequencing a hotspot for homologous strand exchange in multiple HNPP deletion patients. Am. J. Hum. Genet. 1998, 62, 1023–1033. [Google Scholar] [CrossRef]
- Wolf, A.; Millar, D.S.; Caliebe, A.; Horan, M.; Newsway, V.; Kumpf, D.; Steinmann, K.; Chee, I.S.; Lee, Y.H.; Mutirangura, A.; Pepe, G.; Rickards, O.; Schmidtke, J.; Schempp, W.; Chuzhanova, N.; Kehrer-Sawatzki, H.; Krawczak, M.; Cooper, D.N. A gene conversion hotspot in the human growth hormone (GH1) gene promoter. Hum. Mutat. 2009, 30, 239–247. [Google Scholar] [CrossRef]
- Chen, J.M.; Cooper, D.N.; Chuzhanova, N.; Férec, C.; Patrinos, G.P. Gene conversion in evolution and disease. In Encyclopedia of Life Sciences (ELS); John Wiley & Sons, Ltd.: Chichester, UK, 2009. [Google Scholar]
- Sudmant, P.H.; Kitzman, J.O.; Antonacci, F.; Alkan, C.; Malig, M.; Tsalenko, A.; Sampas, N.; Bruhn, L.; Shendure, J.; 1000 Genomes Project; Eichler, E.E. Diversity of human copy number variation and multicopy genes. Science 2010, 330, 641–646. [Google Scholar] [CrossRef]
- Chuzhanova, N.; Chen, J.M.; Bacolla, A.; Patrinos, G.P.; Férec, C.; Wells, R.D.; Cooper, D.N. Gene conversion causing human inherited disease: Evidence for involvement of non-B-DNA-forming sequences and recombination-promoting motifs in DNA breakage and repair. Hum. Mutat. 2009, 30, 1189–1198. [Google Scholar] [CrossRef]
- Wells, R.D. Non-B DNA conformations, mutagenesis and disease. Trends Biochem. Sci. 2007, 32, 271–278. [Google Scholar] [CrossRef]
- Soejima, M.; Fujihara, J.; Takeshita, H.; Koda, Y. Sec1-FUT2-Sec1 hybrid allele generated by interlocus gene conversion. Transfusion 2008, 48, 488–492. [Google Scholar] [CrossRef]
- Koda, Y.; Soejima, M.; Liu, Y.; Kimura, H. Molecular basis for secretor type alpha(1,2)-fucosyltransferase gene deficiency in a Japanese population: A fusion gene generated by unequal crossover responsible for the enzyme deficiency. Am. J. Hum. Genet. 1996, 59, 343–350. [Google Scholar]
- Chen, J.M.; Férec, C. Role of non-B DNA conformations in initiating the nonallelic homologous recombination-derived Se allele and the interlocus gene conversion-derived Sec1-FUT2-Sec1 hybrid allele. Transfusion 2008, 48, 1522–1523. [Google Scholar] [CrossRef]
- Chen, J.M.; Férec, C.; Cooper, D.N. A systematic analysis of disease-associated variants in the 3' regulatory regions of human protein-coding genes II: The importance of mRNA secondary structure in assessing the functionality of 3' UTR variants. Hum. Genet. 2006, 120, 301–333. [Google Scholar] [CrossRef]
- Drake, J.W. Too many mutants with multiple mutations. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 247–258. [Google Scholar] [CrossRef]
- Colgin, L.M.; Hackmann, A.F.; Emond, M.J.; Monnat, R.J., Jr. The unexpected landscape of in vivo somatic mutation in a human epithelial cell lineage. Proc. Natl. Acad. Sci. USA 2002, 99, 1437–1442. [Google Scholar] [CrossRef]
- Buettner, V.L.; Hill, K.A.; Scaringe, W.A.; Sommer, S.S. Evidence that proximal multiple mutations in Big Blue transgenic mice are dependent events. Mutat. Res. 2000, 452, 219–229. [Google Scholar] [CrossRef]
- Hill, K.A.; Wang, J.; Farwell, K.D.; Scaringe, W.A.; Sommer, S.S. Spontaneous multiple mutations show both proximal spacing consistent with chronocoordinate events and alterations with p53-deficiency. Mutat. Res. 2004, 554, 223–240. [Google Scholar] [CrossRef]
- Finette, B.A.; Homans, A.C.; Albertini, R.J. Emergence of genetic instability in children treated for leukemia. Science 2000, 288, 514–517. [Google Scholar]
- Barnett, L.B.; Tyl, R.W.; Shane, B.S.; Shelby, M.D.; Lewis, S.E. Transmission of mutations in the lacI transgene to the offspring of ENU-treated Big Blue male mice. Environ. Mol. Mutagen. 2002, 40, 251–257. [Google Scholar] [CrossRef]
- Crabbe, R.A.; Prtenjaca, A.; Tarnowski, H.E.; Hill, K.A. A novel germline mutation in Big Blue mice. Environ. Mol. Mutagen. 2009, 50, 114–120. [Google Scholar] [CrossRef]
- Chen, J.M.; Férec, C.; Cooper, D.N. Closely spaced multiple mutations as potential signatures of transient hypermutability in human genes. Hum. Mutat. 2009, 30, 1435–1448. [Google Scholar] [CrossRef]
- Drake, J.W.; Bebenek, A.; Kissling, G.E.; Peddada, S. Clusters of mutations from transient hypermutability. Proc. Natl. Acad. Sci. USA 2005, 102, 12849–12854. [Google Scholar] [CrossRef]
- Wang, J.; Gonzalez, K.D.; Scaringe, W.A.; Tsai, K.; Liu, N.; Gu, D.; Li, W.; Hill, K.A.; Sommer, S.S. Evidence for mutation showers. Proc. Natl. Acad. Sci. USA 2007, 104, 8403–8408. [Google Scholar] [CrossRef]
- Boria, I.; Garelli, E.; Gazda, H.T.; Aspesi, A.; Quarello, P.; Pavesi, E.; Ferrante, D.; Meerpohl, J.J.; Kartal, M.; Da Costa, L.; Proust, A.; Leblanc, T.; Simansour, M.; Dahl, N.; Frojmark, A.S.; Pospisilova, D.; Cmejla, R.; Beggs, A.H.; Sheen, M.R.; Landowski, M.; Buros, C.; Clinton, C.; Dobson, L.; Vlachos, A.; Atsidaftos, E.; Lipton, J.M.; Ellisq, S.R.; Ramenghi, U.; Dianzani, I. The ribosomal basis of diamond-blackfan anemia: Mutation and database update. Hum. Mutat. 2010, 31, 1269–1279. [Google Scholar] [CrossRef]
- Moradkhani, K.; Prehu, C.; Old, J.; Henderson, S.; Balamitsa, V.; Luo, H.Y.; Poon, M.C.; Chui, D.H.; Wajcman, H.; Patrinos, G.P. Mutations in the paralogous human alpha-globin genes yielding identical hemoglobin variants. Ann. Hematol. 2009, 88, 535–543. [Google Scholar] [CrossRef]
- van der Klift, H.M.; Tops, C.M.; Bik, E.C.; Boogaard, M.W.; Borgstein, A.M.; Hansson, K.B.; Ausems, M.G.; Gomez Garcia, E.; Green, A.; Hes, F.J.; Izatt, L.; van Hest, L.P.; Alonso, A.M.; Vriends, A.H.; Wagner, A.; van Zelst-Stams, W.A.; Vasen, H.F.; Morreau, H.; Devilee, P.; Wijnen, J.T. Quantification of sequence exchange events between PMS2 and PMS2CL provides a basis for improved mutation scanning of Lynch syndrome patients. Hum. Mutat. 2010, 31, 578–587. [Google Scholar]
- Gardner, J.C.; Webb, T.R.; Kanuga, N.; Robson, A.G.; Holder, G.E.; Stockman, A.; Ripamonti, C.; Ebenezer, N.D.; Ogun, O.; Devery, S.; Wright, G.A.; Maher, E.R.; Cheetham, M.E.; Moore, A.T.; Michaelides, M.; Hardcastle, A.J. X-linked cone dystrophy caused by mutation of the red and green cone opsins. Am. J. Hum. Genet. 2010, 87, 26–39. [Google Scholar] [CrossRef]
- Reyniers, E.; Van Thienen, M.N.; Meire, F.; De Boulle, K.; Devries, K.; Kestelijn, P.; Willems, P.J. Gene conversion between red and defective green opsin gene in blue cone monochromacy. Genomics 1995, 29, 323–328. [Google Scholar] [CrossRef]
- Maamouri, W.; Hammer, M.B.; Bouhlel, Y.; Souilem, S.; Khmiri, N.; Nehdi, H.; Hentati, F.; Amouri, R. Spinal muscular atrophy due to double gene conversion event. Int. J. Neurosci. [CrossRef]
- Globerman, H.; Amor, M.; Parker, K.L.; New, M.I.; White, P.C. Nonsense mutation causing steroid 21-hydroxylase deficiency. J. Clin. Invest. 1988, 82, 139–144. [Google Scholar] [CrossRef]
- Kuehl, P.; Zhang, J.; Lin, Y.; Lamba, J.; Assem, M.; Schuetz, J.; Watkins, P.B.; Daly, A.; Wrighton, S.A.; Hall, S.D.; Maurel, P.; Relling, M.; Brimer, C.; Yasuda, K.; Venkataramanan, R.; Strom, S.; Thummel, K.; Boguski, M.S.; Schuetz, E. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat. Genet. 2001, 27, 383–391. [Google Scholar] [CrossRef]
- Huang, C.H.; Chen, Y.; Blumenfeld, O.O. A novel St(a) glycophorin produced via gene conversion of pseudoexon III from glycophorin E to glycophorin A gene. Hum. Mutat. 2000, 15, 533–540. [Google Scholar] [CrossRef]
- Law, H.Y.; Luo, H.Y.; Wang, W.; Ho, J.F.; Najmabadi, H.; Ng, I.S.; Steinberg, M.H.; Chui, D. H.; Chong, S.S. Determining the cause of patchwork HBA1 and HBA2 genes: recurrent gene conversion or crossing over fixation events. Haematologica 2006, 91, 297–302. [Google Scholar]
- Conley, M.E.; Rapalus, L.; Boylin, E.C.; Rohrer, J.; Minegishi, Y. Gene conversion events contribute to the polymorphic variation of the surrogate light chain gene lambda 5/14.1. Clin. Immunol. 1999, 93, 162–167. [Google Scholar] [CrossRef]
- Fremeaux-Bacchi, V.; Moulton, E.A.; Kavanagh, D.; Dragon-Durey, M.A.; Blouin, J.; Caudy, A.; Arzouk, N.; Cleper, R.; Francois, M.; Guest, G.; Pourrat, J.; Seligman, R.; Fridman, W.H.; Loirat, C.; Atkinson, J.P. Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2006, 17, 2017–2025. [Google Scholar] [CrossRef]
- Hashiguchi, T.; Yotsumoto, S.; Shimada, H.; Terasaki, K.; Setoyama, M.; Kobayashi, K.; Saheki, T.; Kanzaki, T. A novel point mutation in the keratin 17 gene in a Japanese case of pachyonychia congenita type 2. J. Invest. Dermatol. 2002, 118, 545–547. [Google Scholar] [CrossRef]
- Marcaida, M.J.; Munoz, I.G.; Blanco, F.J.; Prieto, J.; Montoya, G. Homing endonucleases: From basics to therapeutic applications. Cell Mol. Life Sci. 2010, 67, 727–748. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Chen, J.-M.; Férec, C.; Cooper, D.N. Gene Conversion in Human Genetic Disease. Genes 2010, 1, 550-563. https://doi.org/10.3390/genes1030550
AMA Style
Chen J-M, Férec C, Cooper DN. Gene Conversion in Human Genetic Disease. Genes. 2010; 1(3):550-563. https://doi.org/10.3390/genes1030550
Chicago/Turabian StyleChen, Jian-Min, Claude Férec, and David N. Cooper. 2010. "Gene Conversion in Human Genetic Disease" Genes 1, no. 3: 550-563. https://doi.org/10.3390/genes1030550