Conservation and Occurrence of Trans-Encoded sRNAs in the Rhizobiales

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
: Post-transcriptional regulation by trans-encoded sRNAs, for example via base-pairing with target mRNAs, is a common feature in bacteria and influences various cell processes, e.g., response to stress factors. Several studies based on computational and RNA-seq approaches identified approximately 180 trans-encoded sRNAs in Sinorhizobium meliloti. The initial point of this report is a set of 52 trans-encoded sRNAs derived from the former studies. Sequence homology combined with structural conservation analyses were applied to elucidate the occurrence and distribution of conserved trans-encoded sRNAs in the order of Rhizobiales. This approach resulted in 39 RNA family models (RFMs) which showed various taxonomic distribution patterns. Whereas the majority of RFMs was restricted to Sinorhizobium species or the Rhizobiaceae, members of a few RFMs were more widely distributed in the Rhizobiales. Access to this data is provided via the RhizoGATE portal [1,2].1. Introduction
In the past two decades the appreciation of small noncoding RNAs (sRNAs) and their importance rose from the status of an exceptional occurrence to that of a general and ubiquitous feature of gene regulation in prokaryotic and eukaryotic life. sRNAs were characterized to be involved in several cellular processes, e.g., response to a variety of cell stresses, regulation of quorum sensing, and toxin antitoxin systems [3–5]. Depending on their location and perfect or imperfect sequence complementarity to specific mRNA targets two major classes were determined: (i) cis-encoded sRNAs, located in antisense to their target mRNA and thus possessing perfect sequence complementarity, and (ii) trans-encoded sRNAs located independently from potential targets, commonly in intergenic regions (IGRs), where sequence complementarity to a possible target can be imperfect or disrupted [6]. Noncoding transcripts, generally 50–250 nt in length, act as (i) activator or repressor of translation (OxyS/fhlA; DsrA/rpoS), are involved in (ii) mRNA stabilization or degradation (GadY/GadX; RyhB/sodB), or act as (iii) target mimicry (6S RNA/CsrB and CsrC) [6–14].
1.1. In Silico Prediction of sRNAs
RNA functional analyses revealed the relevance of RNA secondary structure for their function. Including RNA secondary structure information, various bioinformatics approaches were developed to identify and analyze sRNAs. In silico screens were performed in several bacteria. In Escherichia coli, initially four comprehensive analyses of IGRs were conducted, based on comparative sequence- and secondary structure-analyses as well as promoter and terminator predictions. Several hundred sRNA candidates were identified and 36 validated experimentally [15–18]. Following these studies, dozens of sRNA candidates were predicted in other bacteria using similar approaches, e.g., in Helicobacter pylori [19], Pseudomonas aeruginosa [20], Nitrosomonas europaea [21]. Tools for de novo sRNA gene finding, such as RNAz [22] and EvoFold [23] use multiple genome alignments and focus on sequence and structure conservation. In contrast, the application of cmsearch [24] requires prior knowledge about a family of related sRNAs in different species. Scans with cmsearch base on a combination of HMMs and covariance models. Agreement between approaches is low, and a potentially large number of false positives is predicted [25]. As validation of candidates by experimental methods is usually required anyway, researchers have increasingly turned towards experimental screens.
1.2. Experimental Screens
High throughput studies based on the deep sequencing and tiling array technologies elevated the potential of sRNA identification enormously. Transcriptome studies of e.g., H. pylori, Caulobacter crescentus, and Synechocystis sp. PCC6803 revealed hundreds of new sRNAs [26–28]. In the order of Rhizobiales, transcriptome analyses, focused on sRNA identification, were reported for Rhizobium etli, Agrobacterium tumefaciens, and S. meliloti 1021. A tiling array study of the R. etli transcriptome resulted in identification of 17 putative trans-encoded sRNAs and 49 cis-encoded antisense sRNAs [29,30]. A deep sequencing approach in A. tumefaciens C58 identified 228 sRNA transcripts, 22 of which were experimentally confirmed via Northern blot experiments [31]. Beside individually detected and characterized sRNAs in S. meliloti, e.g., IncA, tmRNA, 4.5S RNA, and RNase P, bioinformatics based studies identified a set of sRNAs which were further validated by Northern blot experiments [32–37]. Recently, a comprehensive deep sequencing approach combined with microarray analyses extended the number of trans-encoded sRNAs to approximately 180 [38].
An experimental screen delivers bona fide sRNA transcripts, with no obvious hints towards a potential functional role. By necessity, it starts from a single species and does not by itself incorporate phylogenetic information. Hence, it calls for a subsequent in silico study where the transcripts obtained for one species are taken as pivot elements to study their conservation and distribution in larger phylogenetic units. One intrinsic limitation of this approach is clear: an sRNA widely distributed, e.g., in the Rhizobiales, but lacking in S. meliloti, cannot be found. Hence, a complete survey of the phylogenetic order or even class from a single pivot organism is not possible.
1.3. Overview of the Present Study
The present study starts from S. meliloti 1021 as the pivot organism and from 52 trans-encoded sRNA transcripts obtained in our aforementioned study [38]. For each transcript, we performed homology searches and constructed RNA family models (RFMs). Our goals are twofold:
We want to increase our knowledge about the distribution pattern of potential sRNAs conserved in the Rhizobiales;
We want to automate the bioinformatics steps that are necessary for RFM construction, as far as it is possible utilizing present-day bioinformatics tools.
The present article describes the RFM construction process, and discusses our observations made when applying these models to the Rhizobiales.
1.4. Our Pivot Organism and Its Kind Relation
The endosymbiont S. meliloti exists in two different life forms, either in a free-living state as a soil bacterium or in a symbiotic relationship with its leguminous host plants, e.g., Medicago sativa. In response to flavonoids secreted by the host plant S. meliloti induces the formation of root nodules. These are colonized by the bacteria which inside the nodules differentiate to endosymbiotic bacteroids that are capable of nitrogen fixation. Bacteroids support the plant with ammonia and in turn receive C4-metabolites, e.g., succinate, from the host [39].
The genome of S. meliloti consists of three replicons, a single chromosome (3.65 Mbp) and two megaplasmids pSymA (1.35 Mbp) and pSymB (1.68 Mbp). The chromosome encodes 3,351 genes predominantly involved in housekeeping functions. The 1,293 genes on megaplasmid pSymA encode, among other functions, the symbiotic apparatus. pSymB carries 1,583 genes mainly involved in exopolysaccharide synthesis and transporter functions [40–42].
Within the order of Rhizobiales, sequenced plant symbionts include Mesorhizobium loti, Sinorhizobium fredii, R. etli, and Sinorhizobium medicae. The order of Rhizobiales also comprises completely sequenced human-, animal- as well as plant-pathogens. The animal pathogen B. melitensis, for example, generally infects sheep and goats, but can act as a human pathogen as well [43]. Bartonella henselae is responsible for the cat-scratch disease of humans [44]. A well studied plant-pathogen is A. tumefaciens, which infects several dicotyledons and acts as powerful tool in plant genetics [45].
2. Results and Discussion
2.1. From sRNA Transcripts to Family Models
We define our notion of RNA family models and give an informal overview of how they are constructed, before we proceed to report on the findings obtained with these models. Details of family model construction are presented in the Methods section.
2.1.1. RNA Family Models: Terminology
The deep sequencing approach by Schlüter et al. [38] elucidated the existence of approximately 1,100 noncoding transcripts encoded on the S. meliloti genome, about 180 of which were trans-encoded. Due to the presumed function as regulatory sRNAs, a subset of 52 trans-encoded transcripts was chosen for a first comparative study (see Section 3.1). Our pivotal transcripts are named SmelXnnn, consistent with Schlüter et al. [38], where X ∈ {A, B, C} denotes the location on pSymA, pSymB, or chromosome, respectively. Potentially related sRNAs found by candidate search are simply named RNA1, RNA2, etc.
For all transcripts, we constructed RNA family models. Informally speaking, an RFM is a set of related sRNAs combined with a method to scan genomes to search for additional family members. Creating RNA family models is not a fully automated process, but requires both high computational effort and human curation.
We constructed two types of RFMs:
Covariance models (CMs) are stochastic models, capturing sequence and structure conservation in an alignment of family members. CMs can be automatically constructed by infernal [24], given such an alignment;
Thermodynamic matchers (TDMs) are RNA folding programs, based on the established thermodynamic model, but tailored to a specific structural motif [46]. Production of such matchers is supported by the graphical editor Locomotif [47].
Both approaches to RFM construction are complementary. When sequence conservation is high enough such that a trustworthy multiple sequence alignment and consensus structure can be established, CMs can be constructed automatically. TDMs are appropriate if sequence conservation is much weaker than structure conservation, such that no candidates are found by sequence similarity search, or they cannot be aligned well. TDMs focus on structure and folding energy; they can ignore sequence conservation in some parts, e.g., in helices, and yet insist on conserved sequence motifs elsewhere, e.g., in loops. Building such a matcher requires human design decisions and some experimentation, and hence, it is more laborious. In this study, we constructed CMs as a rule and TDMs for selected families of special interest to promote identification of further family members.
2.1.2. Overview of the Model Construction Process
Figure 1 gives an overview of our CM construction pipeline. Phase 1 identifies putative homologous RNAs by iterative searches focusing on sequence similarity. Phase 2 constructs an initial family model based on sequence and conserved structure, and uses this model to search all Rhizobiales for further homologs. After adding these to the family, Phase 2 is also iterated.

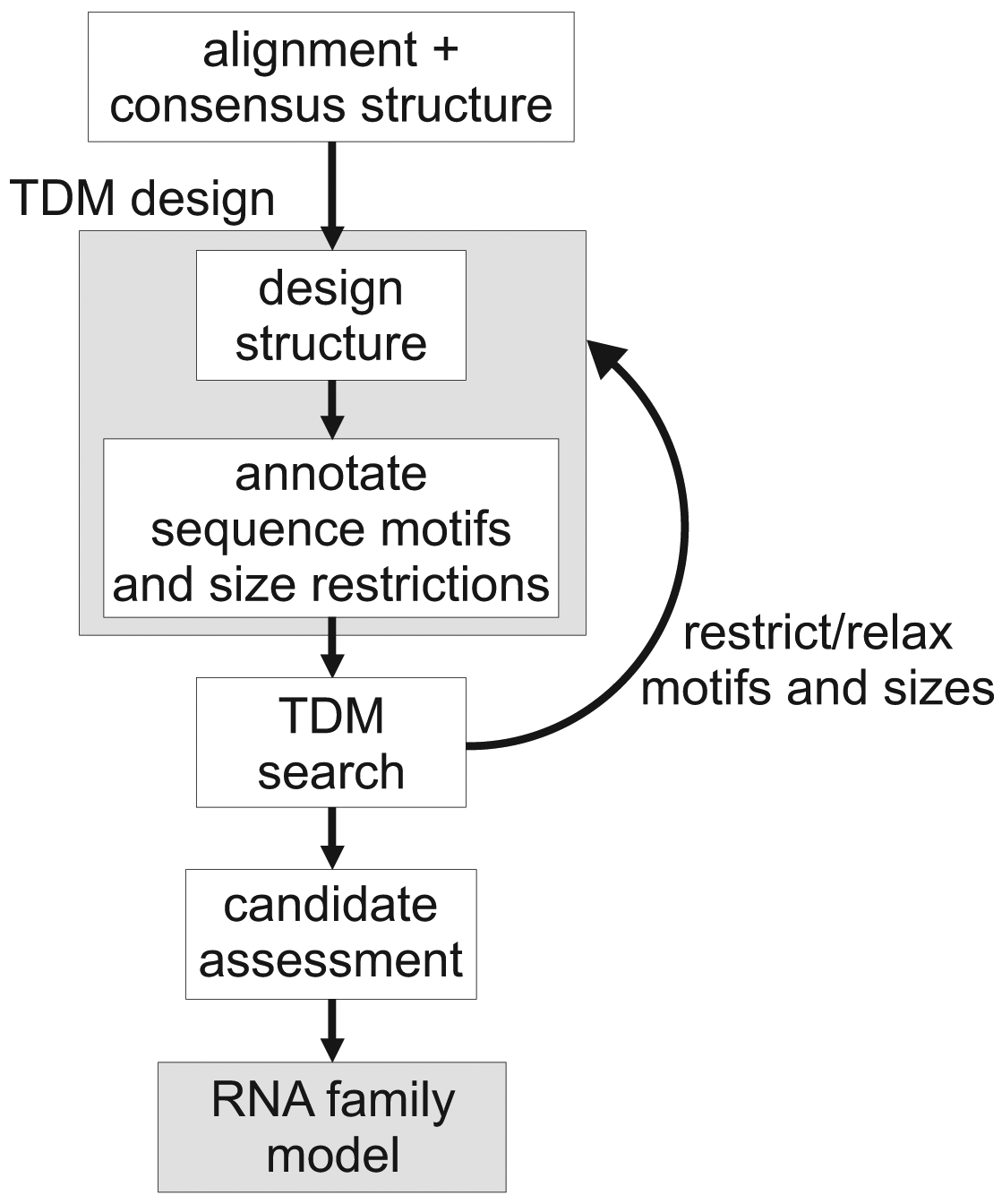
Figure 2 gives an overview of the TDM construction process. Here, we start from a transcript that has a well-defined secondary structure. First, we create a graphical description of this structure, using the Locomotif editor. The graphics can be annotated with size constraints for structural components, and with required sequence motifs. The graphics is then compiled by locomotif into a TDM. We use the TDM to scan other bacterial genomes in order to find subsequences that fold well into the described structure motif. The assessment of candidates is used to adapt the design of the TDM to be more restrictive or more relaxed.
Both methods of family model construction use the same assessment step, which checks for further evidence: Preservation of synteny, quality of alignment against the pivotal transcript, energy of a free folding. For the details, we refer the reader to the Methods section.
The family models are named after their pivotal elements, e.g., RFMSmelA001. These RFMs by themselves constitute an essential part of the results of this study, as they can be used (and extended further) to increase our knowledge about sRNAs in bacteria—beyond the findings that are reported here.

2.2. Distribution Pattern of Trans-Encoded sRNAs in the Rhizobiales
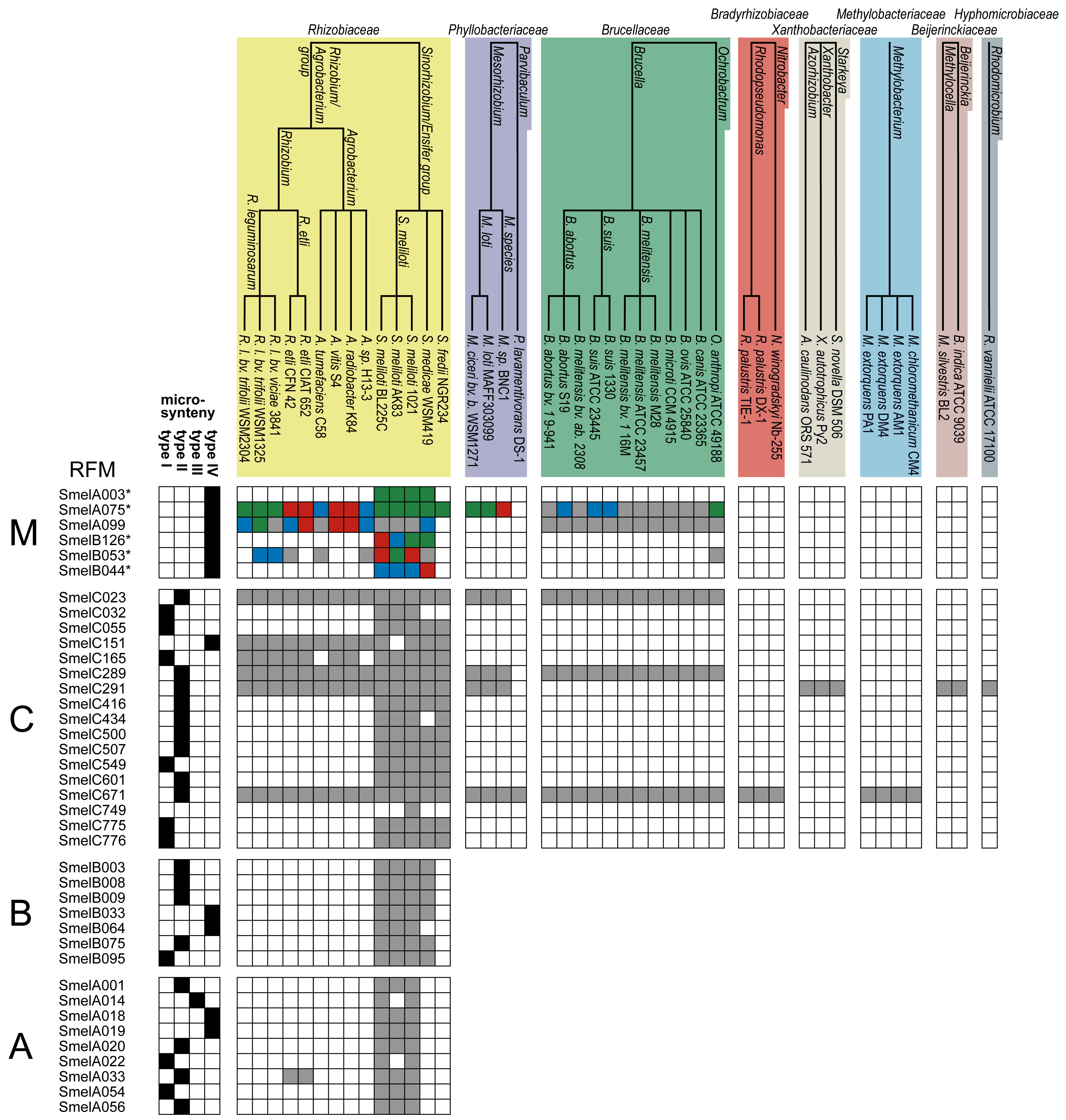
The 52 analyzed trans-encoded sRNAs from S. meliloti and their relatives are collected in 39 RFMs (Figure 3, Table S1). At the first glance, they show a distribution of sRNAs in good accordance with phylogeny. In this subsection, we study their distribution in detail, moving from S. meliloti species to higher taxonomic levels. We will refer to Figure 3 and Table S1 throughout this discussion.
Among our 52 transcripts, 34 map to a single origin in S. meliloti 1021 and give rise to 34 RFMs. These 34 transcripts reveal a relative distribution of 50% (17), 29.4% (10) and 20.6% (7) on the chromosome, pSymA and pSymB, respectively. The remaining 18 transcripts reveal strong sequence similarity to other transcripts, originate from multiple loci and replicons, and were summarized to five RFMs RFMSmelA003, RFMSmelA075, RFMSmelB044, RFMSmelB053, and RFMSmelB126.
2.2.1. Trans-Encoded sRNAs Delimited to the S. meliloti Strains 1021, BL225C, and AK83
Eleven of our transcripts appear to be restricted to S. meliloti strains, which share a core genome of approximately 5,100 genes dispersed on three replicons, a single chromosome, a second chromosome/megaplasmid and a symbiotic megaplasmid, respectively [40–42,49]. However, in AK83 two additional small plasmids were identified with a few genetic features corresponding to syntenic regions of the 1021 and BL225C symbiotic replicons [49]. RFMs of SmelA001, SmelA018, SmelA019, SmelA020, SmelA054, SmelA056, SmelB064, and SmelC032 reveal homologous sequences in the S. meliloti strains 1021, BL225C and AK83, while relatives of SmelA014 and SmelA022 are limited to BL225C, the most closely related strain of S. meliloti 1021 [49]. No homologous sequences were identified in case of SmelC749. Thus, it represents the only trans-encoded sRNA specific for S. meliloti 1021 identified in our study. RFMs deduced from pSymA-located sRNAs are composed of relatives located on the replicons psiNMEB01 and chromosome 3 of S. meliloti BL225C and AK83, respectively. Both, psiNMEB01 of S. meliloti BL225C and chromosome 3 of AK83 share functional similarities with the symbiotic megaplasmid pSymA. Along this line, the RFM members of SmelB064 (pSymB) are located on pSymB-like replicons (BL225C psiNMEB02 and AK83 chromosome 2), while SmelC032 relatives are located on chromosomal-like replicons (BL225C chromosome and AK83 chromosome 1) [40,41,49].
The RFMs of SmelA001, SmelA014, SmelA018, SmelA019, SmelA020, SmelA022, SmelA054, SmelA056, SmelB064, and SmelC032 contain relatives delimited to the three S. meliloti strains, which predominantly map to their particular symbiotic plasmids. Consequentially, on an evolutionary scale the emergence of these transcripts is a more recent S. meliloti-specific incident rather than a loss of the sRNA during evolution of all non-S. meliloti strains. This is in good agreement with Galibert et al. [40], who concluded that pSymA was acquired more recently in the evolutionary history of S. meliloti. Genomic analyses revealed a divergence of genome contents between pSymA and the two remaining replicons [40]. González et al. [50] hypothesized that emergence, remodeling, and annihilation of accessory plasmids in general, is highly variable within the Rhizobiales [50,51]. Our findings support the conclusion that emergence of trans-encoded sRNAs on accessory plasmids in general and on S. meliloti-specific symbiotic plasmids in particular is predominantly a recent evolutionary event. However, SmelB044 and SmelB064 have emerged on the pSymB-like replicon while SmelC032 and SmelC749 evolved on the ancestral chromosome. Thus, S. meliloti-specific sRNA emergence is not restricted to the symbiotic plasmids.
2.2.2. Trans-Encoded sRNAs in the Genus Sinorhizobium
18 RFMs show an extended set of relatives in the Sinorhizobium/Ensifer group. The genus, among others, is composed of the most closely related bacteria S. meliloti 1021, BL225C, and AK83, the next related S. medicae WSM419, and S. fredii NGR234 with the biggest phylogenetic gap to S. meliloti.
Trans-encoded sRNAs of RFMSmelA003, RFMSmelB003, RFMSmelB008, RFMSmelB009, RFMSmelB033, RFMSmelB044, RFMSmelB075, RFMSmelB095, and RFMSmelB126 were identified in S. medicae but not in S. fredii. The S. medicae WSM419 genome consists of four replicons, a circular chromosome and three plasmids, pSMED01 (1.5 Mbp), pSMED02 (1.2 Mbp), and pSMED03 (0.2 Mbp) [52]. The genomic distribution of these sRNAs reveals a strong overrepresentation (89%) on pSymB-like replicons (which are represented by pSMED01 in S. medicae).
In contrast, RFMSmelC055, RFMSmelC416, RFMSmelC434, RFMSmelC500, RFMSmelC507, RFMSmelC549, RFMSmelC601, RFMSmelC775, and RFMSmelC776 commonly include additional relatives in both S. medicae and S. fredii. S. fredii NGR234 has a single chromosome (3.93 Mbp) and two additional plasmids, pNGR234a (0.54 Mbp) and pNGR234b (2.43 Mbp), whereof the smaller plasmid encodes the symbiotic features [51]. RFMs derived from trans-encoding sRNAs occurring in all Sinorhizobium strains, including S. fredii, are composed of members that predominantly map to the chromosomal replicons. This is a notable difference to the 20 RFMs which are restricted to S. meliloti and S. medicae strains and whose members predominantly map to the megaplasmids. An exception is given by a single relative of SmelC434 that is located on megaplasmid pNGR234b in S. fredii. With exception of the multicopy sRNAs SmelA003 and SmelB126, not in a single case unique sRNAs were found on the symbiotic plasmids. This is in good agreement with the strong fluctuation of accessory plasmids [53]. Furthermore, sizes of the symbiotic plasmids in S. fredii (0.54 Mbp) and S. meliloti strains (1.3 Mbp) differ by approximately 0.8 Mbp [41,51] and thus indicate a broad remodeling pattern even in the closely related members of the Sinorhizobium/Ensifer group.

2.2.3. Trans-Encoded sRNAs in the Rhizobiaceae
Most of the analyzed sRNA families (32 out of 39) are restricted to the Rhizobiaceae. RFMs of SmelA033, SmelC151, and SmelC165 hold members with origins in the Sinorhizobium, Rhizobium, and Agrobacterium species. RFMSmelA033 shows an unusual distribution pattern with representatives in S. meliloti, S. medicae, and R. etli. RFMSmelC151 comprises members distributed in the whole Rhizobiaceae except for the Candidatus liberibacter genus and S. meliloti AK83. RFMSmelC165 displays a similar pattern but lacks relatives in A. tumefaciens and A. radiobacter.
2.2.4. Complex Distribution of Trans-Encoded sRNAs in the Order of Rhizobiales
Slater et al. [53] proposed that the ancestor of the Rhizobiales is an unichromosomal organism that acquired an additional, ancestral plasmid [53]. This was underlined by a high proportion of conserved, primary chromosomes in Rhizobiales genera, e.g., Rhizobium, Sinorhizobium, Brucella, Bradyrhizobium, and Mesorhizobium, respectively. The theory of an ancestral plasmid was supported by the existence of several gene clusters that are conserved on the second chromosomes/megaplasmids while generally missing on the primary chromosomes [53]. Further, due to intragenomic gene transfers essential genes, e.g., the tRNA-Arg encoding gene in S. meliloti, are sporadically rearranged to the second chromosomes/megaplasmids [40,53]. Further replicons, in addition to the ancestral chromosome and plasmid, were determined as accessory plasmids with beneficial but non-essential features [53]. All this is in good agreement with our findings about sRNAs in the Rhizobales.
The comprehensive RFMs of SmelA075, SmelA099, SmelB053, SmelC023, SmelC289, SmelC291, and SmelC671 comprise members in the Phyllobacteriaceae, Brucellaceae, Bartonellaceae, Bradyrhizobiaceae, Methylobacteriaceae, Beijerinckaceae, and Hyphomicrobiaceae. The occurrence of these transcripts is restricted to the chromosomal replicons with an exception of RFMSmelA075, RFMSmelA099, and RFMSmelB053, whose members occur several times in each genome with copies on each replicon. RFMs of SmelC023 and SmelC289 show an equal distribution pattern in the Rhizobiales. Both are represented by 29 sRNA relatives in the Rhizobiaceae, Brucellaceae, and Phyllobacteriaceae. SmelC671 has additional relatives in Bradyrhizobiaceae and Methylobacteriaceae. RFMSmelC291 exhibits a more fragmentary occurrence in the Rhizobiales with relatives in the Rhizobiaceae, Phyllobacteriaceae, Xanthobacteriaceae, Beijerinckaceae, and Hyphomicrobiaceae.
RFMs of SmelA075, SmelC023, SmelC289, SmelC291, and SmelC671 show a broad distribution pattern within the Rhizobiales. Each sRNA family has relatives on primary chromosomes in the Rhizobiales strains and thus an ancestral trans-encoded sRNA for each of these models presumably arose in the beginning of the Rhizobiales evolution. However, RFMSmelA075 consists of presumably paralogous copies on different replicons, e.g., each replicon in S. meliloti 1021 harbors at least a single copy. The strong conservation in the Rhizobiales and the occurrence of at least a subset of copies on primary chromosomes suggest that the duplication and transfer events have initially been emanated from origins on ancestral chromosomes. Members of RFMSmelC291 and RFMSmelC671 indeed are distributed to the whole Rhizobiales, but are differentially lacking in several taxonomy families, e.g., Bartonellaceae, Bradyrhizobiaceae and Xanthobacteriaceae.
Similar to that, the Rhizobiaceae-specific RFMSmelA033 and RFMSmelC165 show a dispersed occurrence pattern, the former with representatives only in the R. etli strains. The latter is widely distributed in the Rhizobiaceae but does not occur in A. tumefaciens C58 and A. sp. H13-3. Presumably, the functional relevance of these transcripts has been lost since the specific emergence of these taxonomy families in their ecological niche. Generally, trans-encoded sRNAs act via base pairing with their mRNA targets or interact with RNA binding proteins [6]. In a precedent evolutionary step the target mRNA was presumably removed from the genome or somehow disrupted. This event in turn left a redundant, non-functional sRNA that was removed in the course of time. RFMSmelC151 has relatives in the Rhizobium/Agrobacterium as well as the Sinorhizobium/Ensifer group and serves as a good example for a Rhizobia-specifc sRNA.
Our study reveals relatives of SmelA033, SmelA075, SmelA099, SmelB053, SmelC023, SmelC151, SmelC165, SmelC289, SmelC291, and SmelC671 in both Rhizobium etli species. A genome wide tiling array study for R. etli CFN42 [30] identified, among others, 17 noncoding RNAs. ReC06, ReC25, ReC26, and ReC71 were re-identified within this study as relatives of SmelC023, SmelC289, SmelC291, and SmelC671, respectively. Related transcripts of RFMSmelC291 were confirmed via Northern blot analyses in S. meliloti, S. fredii, R. etli and R. leguminosarum strains and thus underlines the informative value of the comparative approach applied in this study [54]. Recently, a deep sequencing study using the 454-pyrosequencing technology identified about 228 noncoding transcripts located on the three A. tumefaciens C58 replicons. A subset of 22 sRNAs were additionally confirmed via Northern blot analyses. Eight of the RFMs computed in our study comprise members in A. tumefaciens and five, namely C1 (RNA8 of RFMSmelA075), C2 (RNA12 of RFMSmelC023), C5 (RNA11 of RFMSmelC289), C6 (RNA12 of RFMSmelC291), and L5 (RNA25 of RFMSmelA099) were experimentally verified in A. tumefaciens by both deep sequencing and Northern blot experiments [31].
Remarkably, each RFM with relatives beyond the Sinorhizobium/Ensifer group has no corresponding sRNA in the genus Liberibacter. Phylogenetic analyses identified the Liberibacter species as the most divergent species within the Rhizobiaceae with the largest distance to the root of this family. Thus, it might explain the lack of homologs of Rhizobiaceae-specific sRNAs in this genus [55]. Even for the most conserved models, e.g., RFMSmelC671, relatives in the Liberibacter genus were not found. In this context, it has to be noted that the genome of the Liberibacter genus consists of a relatively small chromosome, only 1.2 Mbp in size. Compared to the remaining Rhizobiaceae, enormous genomic capacity, including open reading frames and sRNA genes, has been lost within the Liberibacter lineage.
2.3. Microsynteny
Microsynteny means the preservation of the adjacent protein-coding gene upstream or downstream of a putative sRNA locus. Gene function is usually not affected by its location in relation to its genomic neighborhood. Consequentially, the degree of synteny is lost much faster than sequence similarity and represents a sensitive indicator for genome evolution [56–58].
To classify the dimension of microsynteny for the 39 RFMs, four categories were specified.
Complete microsynteny (type I) is determined for relatives of both neighboring genes;
extensive microsynteny (type II) means the majority of genes shares homology but with a few exceptions;
partial microsynteny (type III) specifies the homology to a single adjacent gene and
fragmented microsynteny (type IV) is given by subsets of homologous genes within a RFM.
According to this definition, microsynteny of type I, II, III, IV was observed for 9, 17, 1, and 11 RFMs, respectively (Figure 3, Table S1). Microsynteny analyses for the stand-alone sRNA SmelC749 was not performed.
Complete microsynteny was observed for RFMs SmelA022, SmelA054, SmelB095, SmelC032, SmelC055, SmelC165, SmelC549, SmelC775, and SmelC776, which are predominantly (8 out of 9) restricted to the Sinorhizobium/Ensifer group. An exception is given by RFMSmelC165 with relatives in the Rhizobiaceae. Extensive microsynteny was observed for RFMs of SmelA001, SmelA020, SmelA056, SmelB003, SmelB008, SmelB009, SmelB075, SmelC289, SmelC416, SmelC500, SmelC507, SmelC601, SmelC671, SmelC023, SmelC434, SmelA033, and SmelC291 (Figure 3, Table S1). Similar to the aforementioned RFMs, with the exception of SmelA033, SmelC023, SmelC289, SmelC291, and SmelC671, this type of microsynteny was predominantly observed in the Sinorhizobium/Ensifer group as well. As expected, the degree of microsynteny is higher for RFMs that are restricted to closely related organisms and to RFMs with a predominant occurrence on descendants of the ancestral chromosome and megaplasmid [53,56].
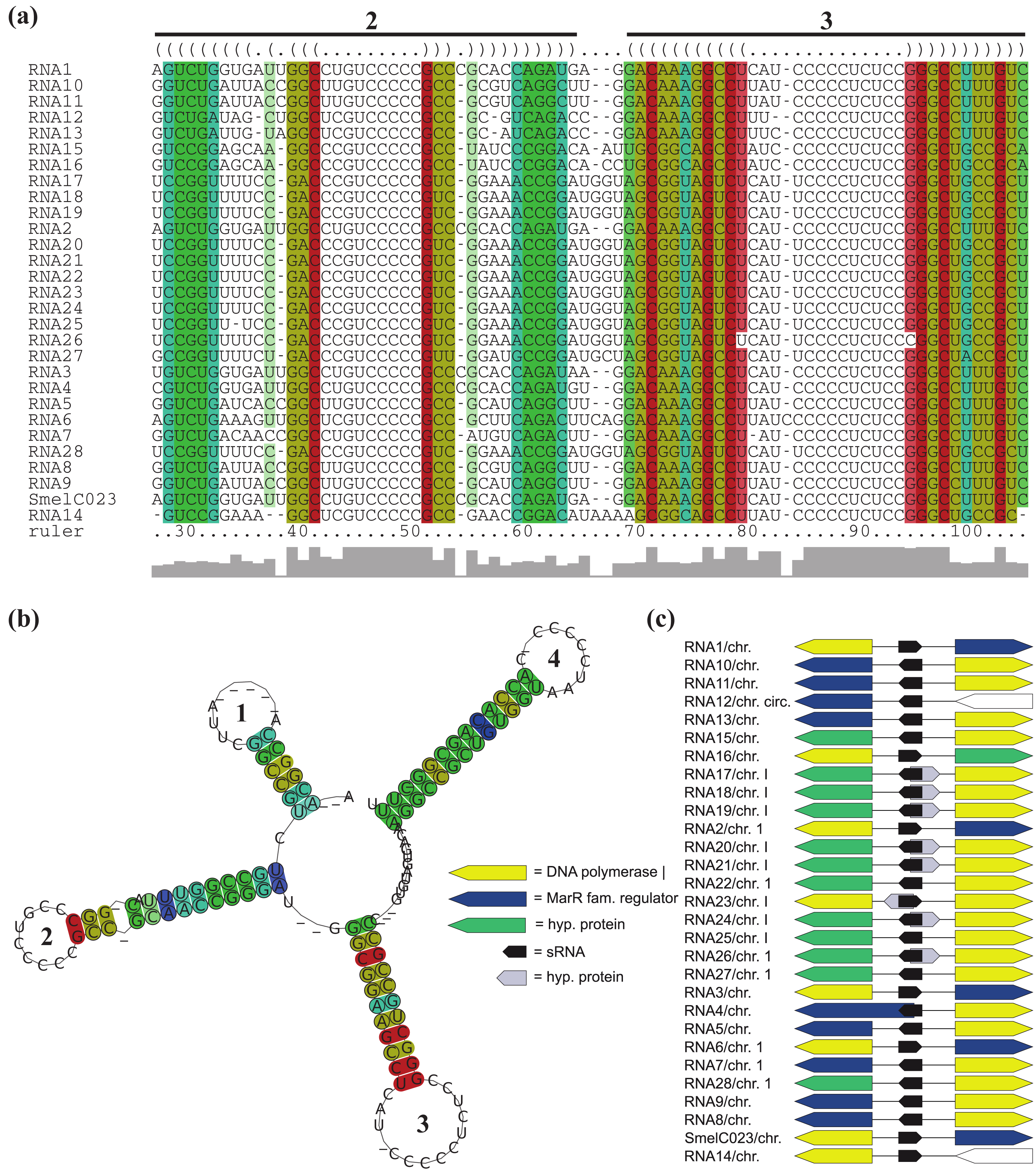
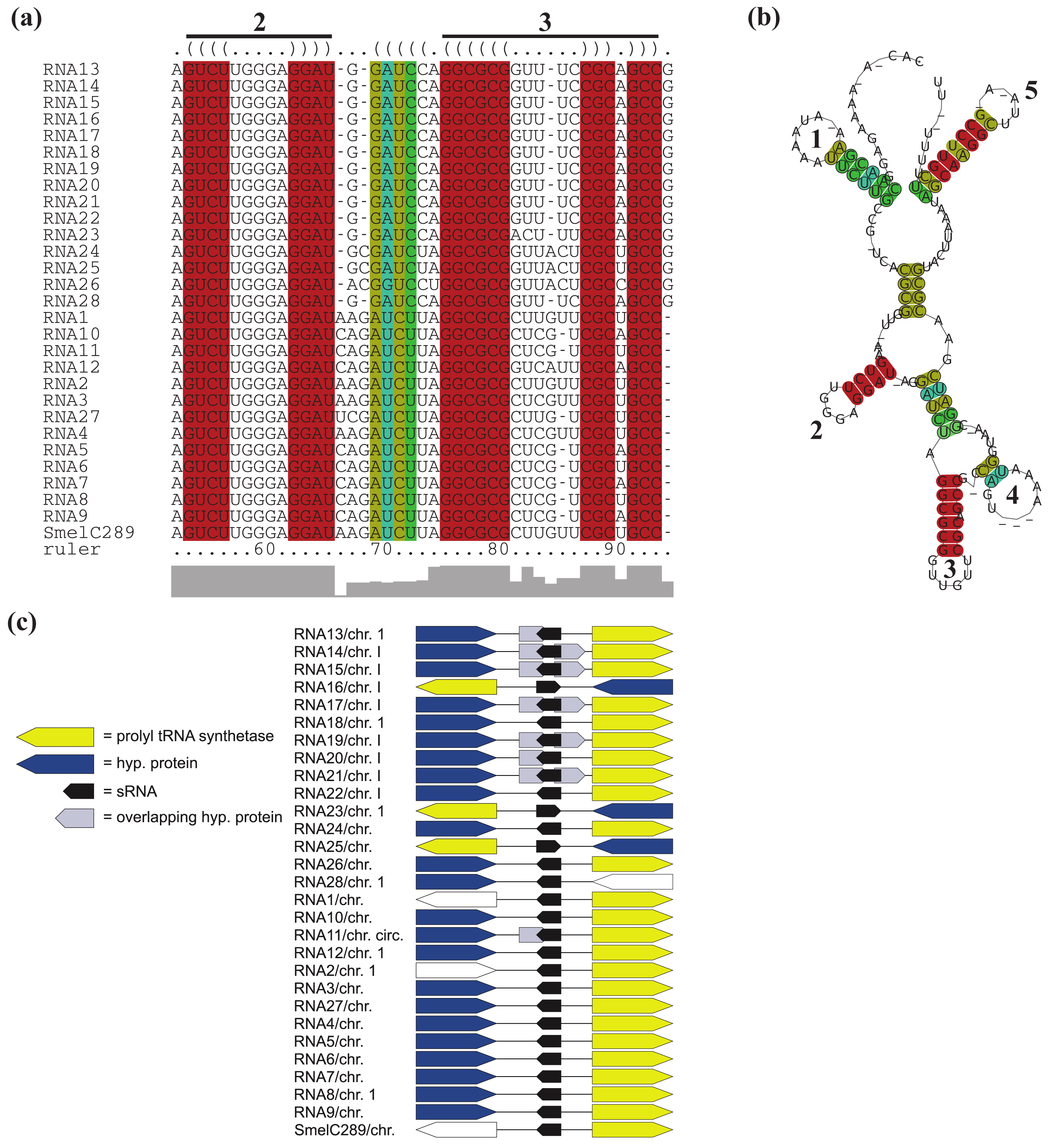
All RNAs of RFMSmelC023 are located adjacent to a DNA polymerase I encoding gene, except for RNA12 of A. tumefaciens str. C58. Furthermore, for the 14 RNAs identified in the Rhizobiaceae, a gene encoding a MarR-type transcriptional regulator is situated next to and in case of RNA4 of S. fredii overlaps the sRNA gene (Figure 5c, Table S1). Due to the aberrant length of the overlapping transcriptional regulator gene (compared to its homologous genes) and the presence of alternative start codons approximately 200 nt downstream of the predicted start we presume an annotation mistake. Except for RNA22, RNA25, and RNA28 of B. abortus S19, B. ovis ATCC25840 and B. melitensis M28, all RFMSmelC023 members that occur in the Brucellaceae are located antisense to a predicted small peptide encoding region. Due to the fact that the sequence of the predicted ORFs is also present in other Brucellaceae which lack this annotation, most likely this ORF was missed during gene prediction.
Except for RNA3 of S. medicae WSM419, all relatives of SmelC289 are located next to a prolyl-tRNA synthetase gene (Figure 6c, Table S1). In case of RNA11, RNA13, RNA14, RNA15, RNA17, RNA19, RNA20, and RNA21 of the Brucellaceae, the prolyl-tRNA synthetase gene is indeed located adjacent to the corresponding sRNA genes, but these sRNA genes are overlapped in antisense by one or two presumably misannotated, small hypothetical genes. For 18 RFMs, overlapping genes were predicted of which the majority is annotated as hypothetical and thus their function and existence remain in question.
Partial microsynteny was only observed for RFMSmelA014, while fragmented microsynteny is given for RFMs of SmelA003, SmelA018, SmelA019, SmelA075, SmelA099, SmelB033, SmelB044, SmelB053, SmelB064, SmelB126, and SmelC151 (Figure 3, Table S1).


In case of RFMs of SmelA003, SmelA075, SmelB053, SmelB044, and SmelB126, fragmented microsynteny is explained by their multiple copy numbers per genome. Members of these RFMs indicate a higher rate of intragenomic transfers. RFMSmelA075 and RFMSmelA099 occur with three and four hairpin loops, respectively, with similar loop motifs (see Section 2.7). Considering both RFMs, fragmented microsynteny is the dominant observation, but a closer look at specific taxonomy families, e.g., R. leguminosarum reveals “local” microsynteny. In detail, the R. leguminosarum relatives RNA9, RNA14, RNA16, RNA17, RNA18, RNA21, and RNA24 of RFMSmelA099 and RNA12, RNA17, RNA19, RNA26, RNA35, RNA42, and RNA54 of RFMSmelA075 occur in complete and extensive microsynteny, respectively RFMSmelB126 contains 4, 3, 2, and 5 copies in S. meliloti 1021, BL225c, Ak83, and S. medicae WSM419, respectively An association to a potassium transporter encoding gene was observed for at least a single sRNA copy in each genome. Thus it is tempting to speculate that the potassium transporter associated sRNAs represent the ancestral version of this RFM.
2.4. Copy Numbers and Association with Mobile Genetic Elements
Multiple copy numbers per genome as well as the scarce microsynteny of RFMs of SmelA003, SmelA075, SmelB044, SmelB053, and SmelB126 are in good agreement with the scattered occurrence of mobile genetic elements next to the sRNA loci. Mobile genetic elements probably contribute significantly to the genetic polymorphism in S. meliloti natural populations [59], since mobile genetic elements are able to copy and uncouple sRNA loci from their genomic context. Repeats and mobile genetic elements were also associated to members of RFMSmelA014, RFMSmelA054, RFMSmelB003, RFMSmelB008, RFMSmelB009, RFMSmelB064, RFMSmelB075, and RFMSmelC500.
2.5. Structural Features Conserved in RFMs
Generally, transcripts have a varying number of sub-structural RNA-domains determined as stacked base pairs, internal loops, bulges and hairpin loops [60]. A number of sRNAs, e.g., Yfr1 of several cyanobacteria, reveal typical Rho-independent terminator-like features with a 3′-located, GC rich hairpin followed by a poly-U-tail [61,62]. Additional examples for sRNAs with typical terminator features are represented by RprA and Qrr1 of E. coli and Vibrio cholera, respectively [63]. RFMs of SmelB126, SmelB053, SmelC434, SmelC507, SmelC151, SmelC023, SmelC289, and SmelC671 include typical terminator structures. On the contrary, RFMSmelB075, RFMSmelA014, RFMSmelA054, RFMSmelC416, RFMSmelC601, and RFMSmelC165 contain stems with atypical hairpin loops, e.g., disrupted with internal loops, followed by poly-U-tails (Figure 5b and Figure 6b, Supplement S1). Otaka et al. [64] reported that besides transcription termination, terminator poly-U-tails of the noncoding transcripts SgrS and RyhB in E. coli are essential for Hfq interaction and riboregulation [64]. A similar pattern could be presumed in case of the aforementioned trans-encoded sRNA models. However, the remaining models reveal no terminator-like features (Supplement S1).
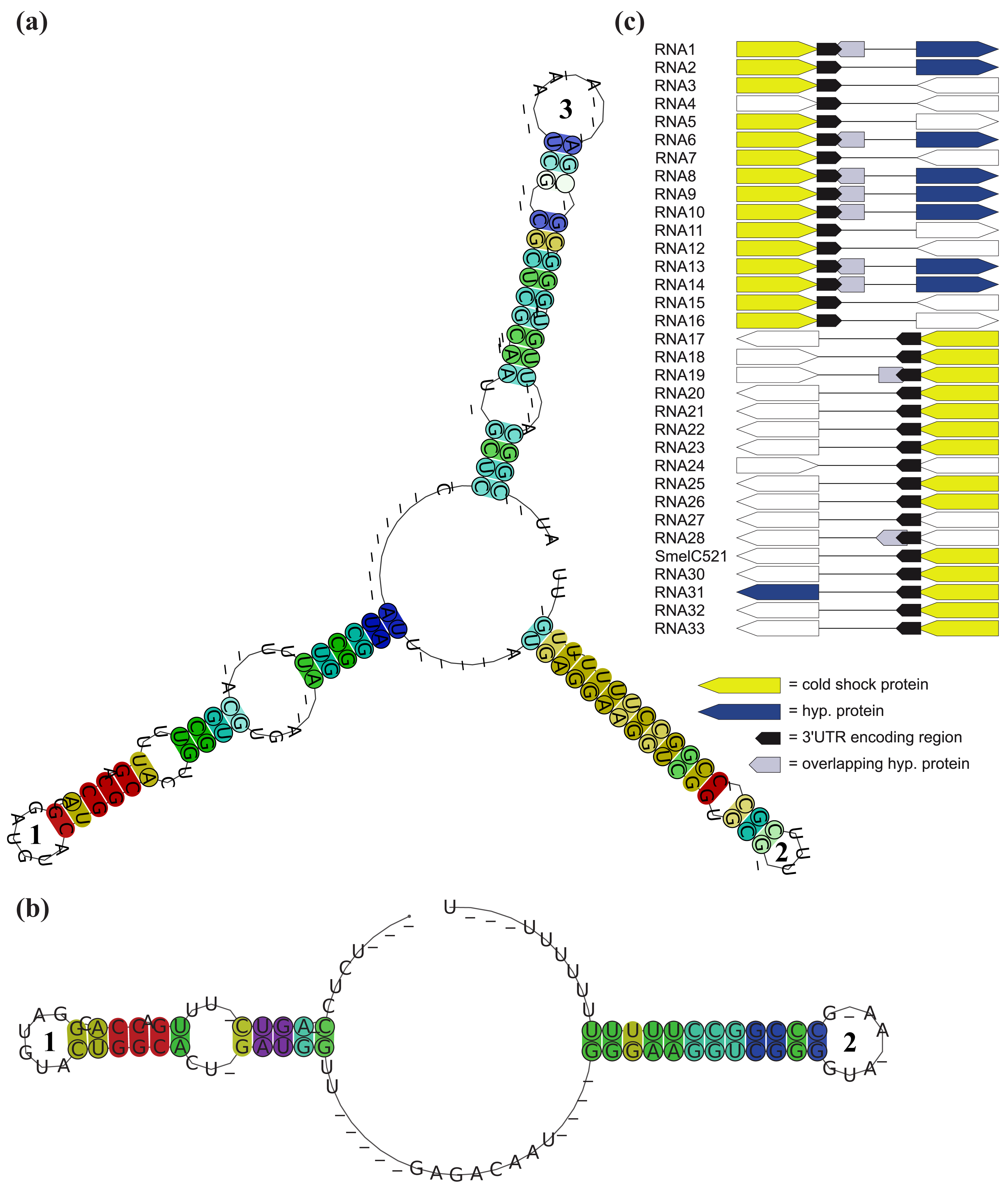
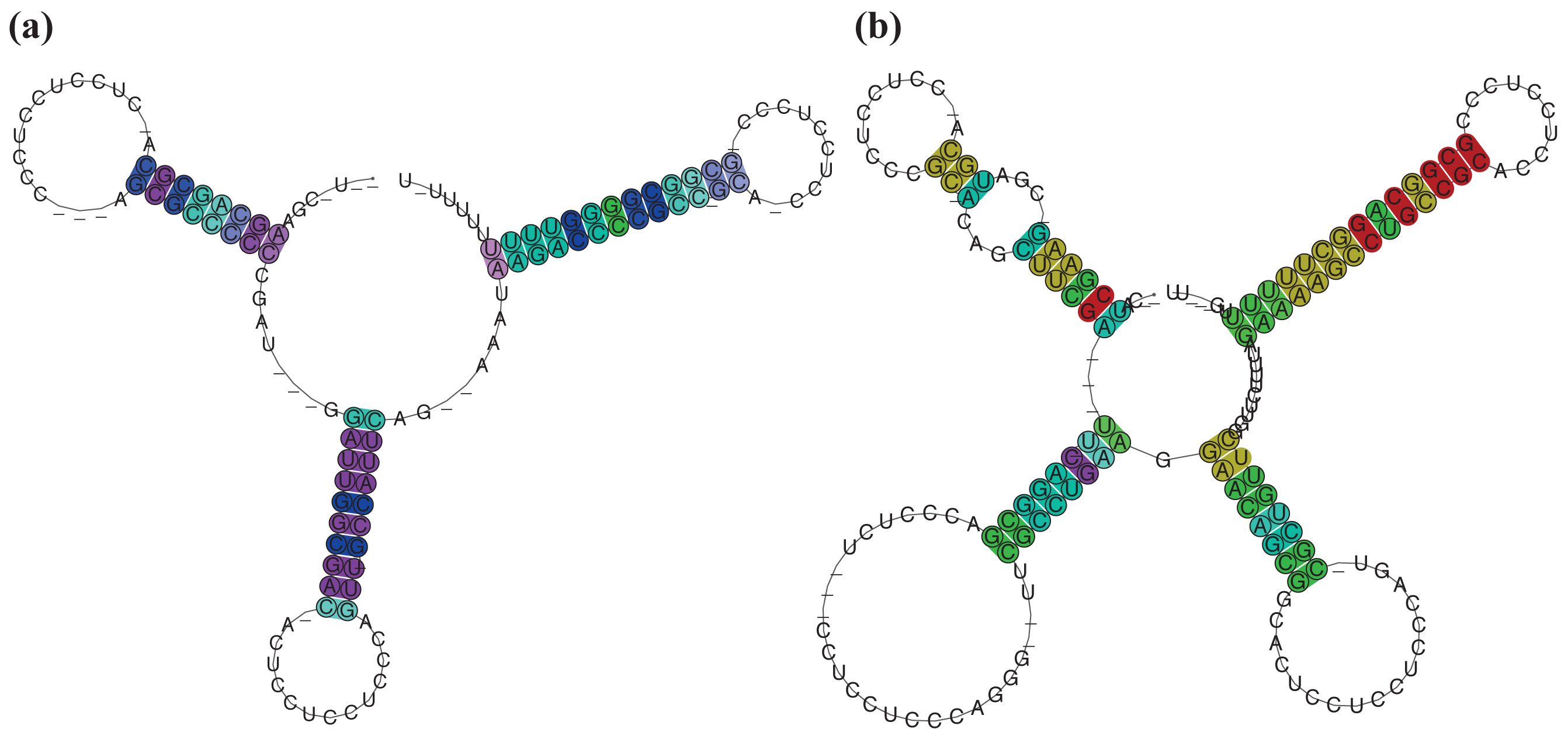
A complex situation is given for SmelB050, SmelB053, and SmelC691. The pSymB-located sRNA genes share typical trans-encoded sRNA gene features with a long distance to neighboring genes. Their transcripts have distinct 5′- and 3′-ends and form a triple stem loop structure [38]. SmelC691 has a similar pattern except for the first stem loop. The identified sRNA relatives, all collected in RFMSmelB053, are mainly found in the Rhizobiaceae; only a single member occurs in Ochrobactrum anthropii in the Brucellaceae (Figure 3, Table S1). Comparison of all homologous sequences indicates the first stem loop as the most variable, sometimes completely missing domain, while the second stem loop shows strong conservation, at least in the loop motifs (GGAUGUA). The third stem loop has typical Rho-independent terminator-like features (Figure 4a, Supplement S1). In addition to the identified sRNA relatives of RFMSmelB053, a number of putative 3′-UTRs were identified in the Rhizobiales, which occur in a sequence and structure pattern similar to the second and third stem loop of the SmelB053 relatives (Figure 4b). The majority of the identified 3′-UTRs (29 out of 33) are connected to genes coding for proteins involved in cold shock adaptation, e.g., SmelC521 in S. meliloti 1021 (Figure 4c, Table S2) [38]. Post-transcriptional regulation of cold shock genes via special 3′-UTR structures was reported in case of the 428 nt long cspA mRNA in E. coli. The mRNA has two stem loop structures at its 3′-end connected to regulation of degradation via binding of Hfq and Poly-(A)-polymerase I (PAP I) that prevents binding of polynucleotide phosphorylase and RNAseE [66,67]. Due to the structural similarity of the homologous sequences identified in our study compared to the cspA 3′-UTR of E. coli and the predominant connection of the homologous 3′-UTRs to cold shock genes in the Rhizobiales, we suggest similar functions of the 3′-UTRs in a Hfq dependent manner. The functional characteristics of the trans-encoded sRNA SmelB053 and its relatives remain unclear, but due to their conspicuous similarity to the conserved 3′-domains of cold shock genes, they might act as interceptor transcripts that sequester RNA degradation complexes and thus protect and stabilize mRNAs. Sequestration is a well characterized phenomenon in bacteria, e.g., exemplified by CsrB, 6S and GlmY RNA [14,63,68].
Strong sequence and thus structure conservation is a general feature between RFM members of S. meliloti-specific sRNAs (SmelA001, SmelA014, SmelA018, SmelA019, SmelA020, SmelA022, SmelA054, SmelA056, SmelB064, SmelC032). The lowest conservation is found in RFMSmelB064 with a structure conservation index (SCI, see Methods section) of 0.91, while the remaining transcripts of each model reveal strong sequence and thus structure conservation, with SCI values of approximately 1 (Supplement S1). Functional characteristics of trans-encoded sRNAs are commonly provided by sub-sequences and structures, e.g., in case of DsrA and RyhB in E.coli [63]. Consequently in sRNA relatives, these crucial domains of sRNAs should be conserved in a more stringent manner than in non-functional domains. However, due to the kinship between and the high sequence similarities of the S. meliloti sRNAs, conclusions about conspicuous functional sub-structures of these transcripts remain impractical. As a matter of course, we see more sequence divergence for a subset of 22 RFMs with additional relatives in the Rhizobiaceae. RFMSmelA003 and RFMSmelB126 have several genome internal copies in each strain (Figure 3, Table S1). Presumably due to the duplication events, transcripts of these RFMs show strong sequence divergence. However, the structure as well as a motif section of the second hairpin loop of RFMSmelA003 shows conservation (Supplement S1). RFMSmelB126 reveals piecewise sequence conservations at more or less conserved positions, as well as a conserved 3′-located terminator hairpin. The best structural conservation for the 22 RFMs is represented by RFMSmelC055 (SCI = 1.04), RFMSmelC151 (SCI = 1.02), RFMSmelB009 (SCI = 1.02), RFMSmelC500 (SCI = 1.01), RFMSmelC775 (SCI = 1.0), RFMSmelB075 (SCI = 1.0), and RFMSmelB008 (SCI = 1.0) (Supplement S1). The strongest nucleotide divergence is shown by RFMSmelC549 with relatives in S. meliloti and S. fredii (SCI = 0.69) (Supplement S1).
In general, RFMSmelC549 consists of four conserved stem loops but is disrupted by several internal loops and single stranded domains. These single stranded domains are the most deviating regions and thus they are responsible for the depressed SCI value. This high conservation implies a functional role of these four domains (Supplement S1).

The RFMs of SmelC023 (SCI = 1.11), SmelA075 (SCI = 0.99), SmelC289 (SCI = 0.9), SmelC291 (SCI = 0.55), and SmelC671 (SCI = 0.55) are composed of transcripts with variable sequence and structure conservations, presumably derived from a common ancestor (Supplement S1). In case of RFMSmelC023 and RFMSmelC289, four and five hairpin loops, respectively, are the main components of these transcripts. The 5′-located domain of RFMSmelC023 is highly variable both in length and nucleotide composition. The 3′-located stem loop of RFMSmelC023 is a Rho-independent terminator-like structure with high GC content (Supplement S1). This is supported by the poly-T-sequence following the annotated sRNA gene (data not shown). Presumably a degradation event of the initially annotated sRNA SmelC023 occurred and resulted in a processed 3′-end. The middle domain consists of two hairpin loop structures with highly conserved sequences within the loops, while the structural maintenance of these sub-domains is provided by stems with varying nucleotide compositions with evolutionary established base pair exchanges (Figure 5a,b, Supplement S1). This strongly suggests that the functional maintenance of this molecule is provided by both the hairpin structures and the loop sequences. sRNAs with several functional domains are a common feature in bacteria. Obvious examples are the trans-encoded sRNAs OxyS and DsrA in E. coli. The former binds to the fhlA mRNA in a Hfq-dependent manner. Two stem loops are presumed to be involved in mRNA binding, while the interaction site of Hfq is different to that of the mRNA binding site [3,69,70]. The latter exhibits different hairpins for different targets. In detail, DsrA consists of three stem loops of which the second is able to interact with hns mRNA, blocks the ribosome binding site (RBS) and thus inhibits mRNA translation. The third stem loop interacts with the rpoS mRNA and activates translation via remodeling of an inhibitory mRNA sub-structure [70]. Similar features could be presumed for RFMSmelC291 (sra33) [54]. The 5′-domain of RFMSmelC291 has a structurally conserved stem loop with a strongly conserved loop motif, UCCGCCGCAUCU, while the second stem loop shows extremely variable stem sequences but occurs with a dominant but different loop motif, UCCUCG as well (Supplement S1).
RFMSmelC289 shows a 5′-located stem loop characterized by an AU-rich loop, stabilized by a variable stem of organism-specific nucleotide contents. Similar to RFMSmelC023, the 3′-region contains a Rho-independent terminator-like structure and the middle domain consists of two conserved hairpin loops as well. The former hairpin shows complete conservation, while the latter reveals a highly conserved stem with a variable loop (Figure 6a,b, Supplement S1). However, the functional patterns of noncoding transcripts are not restricted to their presumed single stranded domains, e.g., the binding sites of RprA sRNA in E. coli are predominantly incorporated in stem structures [63]. Further hidden sRNA sequences that are essential for target interactions could be released due to a structural reformation of the sRNA, for example, the RNA binding protein Hfq is able to alter the secondary structure of the RydC sRNA in Enterobacteriaceae resulting in an active version of this transcript [71]. RFMSmelC671 represents the most variable model of related sRNAs in this study. The transcript has a long single-stranded domain with a conserved CUCCCUGU motif, enclosed by down- and upstream located, highly variable hairpin loops of which the 3′-located domain acts as terminator (Supplement S1). A similar pattern was observed for Qrr1 in Vibrio cholera. This transcript reveals a long single stranded domain between its first and second loop, which was verified as the mRNA interaction site. This motif is highly conserved within Qrr2-4, which are paralogs of Qrr1 [4,63].
2.6. sRNAs in Antisense
Large numbers of cis-encoded antisense RNAs were identified, e.g., via sequencing and tiling array studies in Synechocystis [28], H. pylori [26], S. meliloti [38], and R. etli [30]. Cis-encoded antisense sRNAs are located in antisense to their targets, act via perfect base pairing and mediate post-transcriptional regulation, e.g., stabilization or destabilization of target mRNAs [6]. Pairs of small noncoding transcripts that are located in antisense to each other remain rarely identified in bacteria. Georg and Hess [72] presumed that two small transcripts encoded by unlinked sRNA genes interact with each other due to a complementary section in both transcripts. However, the functional relevance of this feature needs to be further elucidated.
Here, we observe that SmelC776 relatives are located in antisense to RFMSmelC775 sRNAs and thus most likely allow a mutual interference of both transcripts. This is in good agreement with the strong sequence conservation of SmelC776 (SCI = 0.95) within the overlapping domain (Table S1, Supplement S1). Approximately 90% (17 out of 19) of the nucleotide exchanges of RFMSmelC776 in the S. fredii strain occur outside the overlapping part and thus suggest the antisense part as the functional transcript domain.
2.7. Focus on SmelA075 and SmelA099
A remarkable situation was found in case of RFMSmelA075 and RFMSmelA099, which exhibit three and four hairpin loops, respectively. Each hairpin loop carries similar loop motifs, CCUCCUCCC, representing an anti Shine-Dalgarno (aSD) sequence, while the stems show more variability in their nucleotide content (Figure 7, Supplement S1). In several sRNAs, e.g., RNA III in Staphylococcus aureus, CyaR in Enterobacteria and ABcR1 in A. tumefaciens C58, loop sequences with at least partial aSD motifs were observed. Functional analyses demonstrated that the aSD motif is indispensable for sRNA binding to the RBS of the particular mRNA targets, as well as the resulting translation inhibition [73–79]. The sequencing profile as well as Northern blot analyses of SmelA075, which is a member of RFMSmelA075, suggested SmelA075 as a stress-induced sRNA that occurs in several processed forms [38]. This is in good agreement with the trans-encoded sRNA RS0680a and its homologous transcripts identified in Rhodobacter sphaeroides. RS0680a represents a shorter version than the RFM members identified in this study. The transcript has two stem loops instead of three and four, and each comprises an aSD motif. It was suggested that RS0680a undergoes different maturation processes and is involved in the stress response in a more general pattern via binding to the RBS of several genes [80]. From a biological point of view, it was suggested to group all derivatives of RFMSmelA075 and RFMSmelA099 to a single sRNA family that consists of members with a varying number of hairpin modules. Implementing all facts, this RNA family consists of several copies per genome. It is somehow involved in stress adaption, presumably in post-transcriptional regulation via blocking the RBS of target mRNAs.

3. Experimental Section
In this section, we describe in detail the process of RNA family model construction from our S. meliloti transcripts which is not a fully automated method. We report on the automatic steps and on the points where human judgement or design decisions are involved. A genuine knowledge of common bioinformatics tools is assumed. All tools used are strictly concerned with secondary structure—non-standard base pairs, possible pseudoknots, or other tertiary interactions are not considered. Although including such features would be desirable, present day tools cannot achieve this.
3.1. Sequence Data and Databases
52 of approximately 180 trans-encoded sRNAs were selected and downloaded from GenDB [81], accessible via the RhizoGATE portal [1,2]. The choice of these 52 candidates was made in purely technical terms: Candidate transcripts should be well-covered by sequence reads, with clearly defined ends, and remote from any coding region. We plan to generate models for the remaining transcripts in the near future.
Complete genome sequences and annotations of all Rhizobiales available were obtained from the NCBI genomes FTP site [82]. For a complete list see Table S3. Additionally, whole genome and plasmid sequences were included that are not (yet) part of the above collection. Sequence data of A. sp. H13-3, B. melitensis M28, S. meliloti AK83, and S. meliloti BL225C, all members of the order of the Rhizobiales, were downloaded from the NCBI nucleotide database.
3.2. Construction of RNA Family Models
There is no standard and fully automated way to construct an RNA family model. The general difficulty of this process has been discussed, e.g., in [83]. Family model construction is supported by a variety of tools, but interspersed with modeling decisions and candidate screening by a human expert. In our case, we start with a trans-encoded sRNA from S. meliloti, say SmelXnnn, and construct an RNA family model RFMSmelXnnn, which (1) comprises a set of orthologous RNA sequences from related organisms; and provides (2) a search function to find further family members in the Rhizobiales and beyond. In a few cases, we find that several sRNAs should be collected into the same family model, which is then named arbitrarily after one of them.
We constructed types of RFMs, Covariance models and Thermodynamic matchers. The use of these two complementary methods has already been motivated above (Section 2.1). We now add some details about both methods, and about the assessment step used with both.
3.3. Automated Candidate Generation and CM Construction Steps
Recall Figure 1, which gives an overview of our RFM construction pipeline. Phase 1 identifies putative homologous RNAs by iterative searches focusing on sequence homology. Phase 2 constructs an initial family model based on sequence and conserved structure, and uses this model to search all Rhizobiales for further homologs.
3.3.1. Phase 1: Sequence Homology Search for SmelXnnn
In the first stage of the workflow, putative homologs are obtained by sequence homology searches using blastn [84] and GotohScan [85]. We initialize the search for homologous RNA sequences of a reference sequence SmelXnnn by employing blastn with E < 10−5 on the complete set of alphaproteobacterial genomes (For word-size and scoring function we set the parameters -W 7 -q -3 -r 2 -G 2 -E 2).
As fragmentation of conserved regions is a common characteristics of RNA families, candidate homologs often do not cover the complete reference sequence. Therefore, blastn matches must be postprocessed. First, the candidate sequence is extended on either side to cover the reference sRNA sequence plus an extra 10% of its length on either side. Next, the reference is semi-globally aligned to the candidate sequence and un-matched leading and trailing bases of the candidate are trimmed.
The detection of RNA homologs is complemented by GotohScan searches for SmelXnnn in three separated sequence databases representing the families of Rhizobiaceae, Brucellaceae, and Phyllobacteriaceae, using default parameters. The latter are the most closely related families of Rhizobiaceae, with S. meliloti as member, within the order of Rhizobiales.
Resulting candidates from both search methods are combined and undergo assessment in the same way.
3.3.2. Iteration of Homology Search
As is common practice in search of distant homologies [86], rather than searching with a relaxed threshold, we use a stringent threshold in each step. From the hits determined and assessed positively, a new search emerges, again with a stringent threshold. We use a maximum of three iterations.
3.3.3. Phase 2: CM Creation and Search
We enter Phase 2 with a first set of candidates for RFMSmelXnnn, given that at least two homologs of SmelXnnn were found. An initial covariance model CMSmelXnnn is to be built. This requires a multiple sequence alignment which supports a consensus structure. LocARNA [87] is used for creating the structural alignment, RNAalifold [88,89] for prediction of consensus structure from this alignment, and Infernal [24] for CM model construction and search. Figure 8 gives an example of the input for CM construction.

After constructing CMSmelXnnn by Infernal, a profile search is performed on all alphaproteobacterial genomes. The best 50 hits are analyzed and undergo assessment. The steps of model construction, search and hit assessment are repeated while new homologs are identified. A maximum of three cycles is allowed. The third iteration uses as a cut-off a CM-score of 25% of the highest CM-score of any present member of the model.
3.4. TDM Model Construction
For specific RNA families, we found that a CM was inadequate to express their peculiarities. For example, SmelA075 has three hairpins, all of which exhibit a perfect loop motif (CCUCCUCCC) (cf. Section 2.7). It is widely distributed among the Rhizobiales, with the stems sequences highly diverged. As a consequence, the discriminatory power of the CM decreases. A TDM can be designed such that it gives no weight to the stem sequences, but enforces the loop motif. SmelB053 is a similar case with a strongly conserved structure and low sequence similarity, except for a prominent loop motif (GAUGUA).
Figure 9 shows a snapshot of TDM construction, where we depict a structure graphics with the help of the Locomotif editor [47], annotate it with conserved sequence information and compiled it into a search program at the Bielefeld Bioinformatics Server [90].

TDMs tend to run a bit faster than CMs (when their HMM filter is turned off). We use them to scan all Rhizobiales genomes. Resulting candidates are assessed like other candidates.
Design of a TDM also requires some iteration, as the motif description can be made more or less specific. Generally, it is a good strategy to begin with a rather restrictive motif and check that the known sRNAs are actually found, which verifies that the design is correct. Then, the motif is gradually relaxed. Search results may suggest adjustment to the original TDM design, such as relaxing the number of paired bases in a stem, or increasing the allowed size for a loop. This interplay of human design, matcher compilation and search continues as long as candidates pass the assessment step.
3.5. Assessment: Taming the Flood of Candidates
When an RNA family model is available, its search procedure can be used with more or less stringent cut-offs. In this study, however, we need to construct such a model in the first place. We start from a single family member, which may not even be a typical one, but has the virtue of being based on an experimental screen rather than being an in silico prediction. We want to collect a large number of homologs, and use further evidence to weed out unplausible candidates.
Some sequences show strong sequence and structure conservation and thus can be unambiguously identified in an automatic fashion. No further effort, human or computational, is spent on them. Divergent sequences without global conservation have to be curated integrating sequence and structure conservation with additional sources of information, such as genomic context and phylogenetic distribution. Figure 10 gives an overview of this assessment, which is described further below.

3.5.1. Filtering of Sequence Homlogs Based on Pairwise LocARNA Scores
Let S = SmelXnnn for this section. Each putative homolog returned by sequence search is aligned (separately) to the S using LocARNA. For any further consideration, only candidates c are retained for which their LocARNA score loc(S, c) satisfies
This restriction is due to practical considerations, as a trade off between the number of candidates that we have to inspect against the possible loss of true homologs. It leaves us with a set of candidates that align well to S individually, yet the structures underlying their alignments may have little in common.
3.5.2. Analysis of Structure Conservation
We next promote candidates with strong structural conservation, which pass the selection process without further inspection: From the previous step, we have two criteria that measure individual structure conservation of each candidate c against S, the (pairwise) LocARNA score loc(S, c) and the (pairwise) structure conservation index sci(S, c), which is computed with the help of RNAfold and RNAalifold, both part of the Vienna RNA Package [91]. If an SCI value is low, the candidate rather folds into a native structure different from the consensus found by LocARNA. Now the pieces of information gained from pairwise considerations have to be related.
For the pairwise alignment of S and c, we map the consensus structure back to S and compute its abstract shape representation, computed with RNAshapes [92,93], extended by hairpin centers (akin to the “helix centers” of [94]). A hairpin center is calculated as (i + j)/2, where i and j are the positions of the hairpin closing base pair.
We refer to the combination of shape and hairpin center as a pointed shape. This yields (potentially different) pointed shapes pS,c for S, one for each candidate c. We also compute pS,S, which is the pointed shape of the minimum free energy structure for S. Candidate c qualifies as a family member if
The last equation borrows the idea of consensus shapes from [95] for a fast way to select candidates where structure conservation is obvious.
Candidates which do not pass the above test are not (yet) discarded, but subjected to the next step.
3.5.3. Synteny, Phylogeny, and Multiple Alignment
We perform BLAST comparisons of the protein-coding genes flanking SmelXnnn against the Rhizobiales database, with a maximum E-value of 10−6 to indicate gene synteny. If one or both flanking genes are conserved with respect to at least one of the family members as accepted at this point, the candidate is accepted.
Additionally, we examine the distribution of homologs related to phylogeny, replicon type, and copy number. Candidates are discarded, for example, when located on a different replicon in a closely related strain, or when there is only a single hit in a remote phylogenetic group.
Finally (this must be the last assessment step for computational reasons), candidates that passed the previous criteria are cast into a multiple structural alignment with LocARNA. Such an alignment is easily derailed by outliers that do not fit to a common structure. Obvious outliers, exhibiting a scattered alignment throughout the sequence, are removed by human inspection. As long as candidates are removed, the alignment is recalculated for the remaining family members.
3.6. Towards a More Automated RNA Family Model Construction Process
So far we completed 39 RFMs including 52 of 173 trans-encoded sRNAs published in [38]. There are 121 more to go. Further automation of the model construction process is highly desirable, but not easy to achieve. While some parts of our assessment step can be integrated into an automated workflow, there are also serious challenges arising from technical limitations of the available software. We discuss two such aspects in the sequel.
3.6.1. Modular Architecture of RNA Families
An important characteristic of RNA is its modular architecture. Similar substructures, shared among different RNA families, or multiple copies of modules within an RNA family may indicate related functionality. In RNA family reconstruction, shared modules complicate the identification of homologs by making it difficult to distinguish whether a candidate belongs to an already existing family or constitutes a completely new RNA family, merely sharing a similar sub-structure.
An example for different manifestations of a repetitive module, comprised of a single hairpin, are the trans-encoded sRNAs SmelC201 (not included in this study), SmelA075, and SmelA099. Their structures are composed of two, three, and four consecutive copies of similar hairpins. Neither CMs nor TDMs (at least not those created with Locomotif) can model a variable number of modules. In case of SmelA075 and SmelA099, our workaround was to build separate models for each module number. An extra classification step was required for matches obtained from the alternative models, because shorter models produced multiple overlapping hits to homologs that were members of RFMs with a higher number of module copies. From the algorithmic point of view, it should be simple to extend modeling techniques in this direction.
3.6.2. Conserved Terminators in Short sRNAs
Finding homologs for sRNAs with a length below 80 nucleotides is generally difficult if their sequence has diverged while retaining a conserved structure. In particular, when the structure is a GC-rich hairpin, it often matches to terminator hairpins in multiple locations. A source of confusion is for example RFMSmelB053, whose structure consists of three adjacent hairpins. The nuceotide sequence of the central hairpin is conserved, whereas the last one constitutes a bona fide terminator.
Situations like this ask for a generalization of models towards avoidance of specified motifs. This is probably more difficult than allowing for optional modules.
4. Conclusions
In this study, we aimed at the identification of homologous sRNAs in the Rhizobiales, starting with a set of well-defined trans-encoded sRNAs from S. meliloti 1021. This is the first comprehensive, comparative in silico approach in this group of bacteria. Definition of RNA family models and grouping of sRNAs into these families is complicated by the poor knowledge about relationships between sequence, structure, and functions of sRNA domains. Whereas strong sequence and structural conservation is a good indication for assignment to the same family, the process becomes more difficult if only short sequence motifs and some structural features show similarities. This also includes ambiguous situations of sRNAs showing limited similarities to different family models. Therefore, full automation of RFM construction has not yet been achieved in this study. It is also not clear how far up in the taxonomy an approach like ours can reach. Hence, numbers of false positives and negatives are likely to increase with the evolutionary distance of the organisms.
Several independent pieces of evidence suggest that the 39 family models delivered here are a trustworthy bases for further, experimental and bioinformatics analyses. Apart from the criteria of sequence, structure and synteny conservation, such independent evidence is the following:
Our initial, experimental screen [38] was able to recover transcripts of the majority of sRNAs known in S. meliloti at that time;
Generally, our family models exhibit a plausible distribution of their members with respect to phylogeny;
In particular, the specific distribution of observed transcripts on replicons is in agreement with the accepted view that the symbiotic plasmid is a late acquisition in Sinorhizobium;
Members of five of our family models, found in A. tumefaciens, were validated experimentally by deep sequencing and Northern blots in independent studies (cf. Section 2.2).
Our approach provides valuable insights into the distributions of conserved and the presence of species-, family-, and genus-specific sRNAs. Most of the RNA families are restricted to the Rhizobiaceae, but a few show a broader distribution, implying a more general conserved function. While functional studies of sRNAs may build on our predictions, the future bioinformatics tasks are to construct models for the remaining transcripts from S. meliloti, and the extension of the comparative analysis to the alphaproteobacteria, and possibly beyond.
Supplementary Files
References
- Becker, A.; Barnett, M.J.; Capela, D.; Dondrup, M.; Kamp, P.B.; Krol, E.; Linke, B.; Rüberg, S.; Runte, K.; Schroeder, B.K.; et al. A portal for rhizobial genomes: RhizoGATE integrates a Sinorhizobium meliloti genome annotation update with postgenome data. J. Biotechnol. 2009, 140, 45–50. [Google Scholar]
- RhizoGATE - The gateway to rhizobial genomes. Available online: http://www.rhizogate.de/ (accessed on 25 February 2011).
- Wassarman, K.M. Small RNAs in bacteria: Diverse regulators of gene expression in response to environmental changes. Cell 2002, 109, 141–144. [Google Scholar]
- Lenz, D.H.; Mok, K.C.; Lilley, B.N.; Kulkarni, R.V.; Wingreen, N.S.; Bassler, B.L. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell 2004, 118, 69–82. [Google Scholar]
- Fozo, E.M.; Hemm, M.R.; Storz, G. Small toxic proteins and the antisense RNAs that repress them. Microbiol. Mol. Biol. Rev. 2008, 72, 579–589. [Google Scholar]
- Storz, G.; Altuvia, S.; Wassarman, K.M. An abundance of RNA regulators. Annu. Rev. Biochem. 2005, 74, 199–217. [Google Scholar]
- Altuvia, S.; Zhang, A.; Argaman, L.; Tiwari, A.; Storz, G. The Escherichia coli OxyS regulatory RNA represses fhlA translation by blocking ribosome binding. EMBO J. 1998, 17, 6069–6075. [Google Scholar]
- Andersen, J.; Forst, S.A.; Zhao, K.; Inouye, M.; Delihas, N. The function of micF RNA. micF RNA is a major factor in the thermal regulation of OmpF protein in Escherichia coli. J. Biol. Chem. 1989, 264, 17961–17970. [Google Scholar]
- Chen, S.; Zhang, A.; Blyn, L.B.; Storz, G. MicC, a second small-RNA regulator of Omp protein expression in Escherichia coli. J. Bacteriol. 2004, 186, 6689–6697. [Google Scholar]
- Majdalani, N.; Cunning, C.; Sledjeski, D.; Elliott, T.; Gottesman, S. DsrA RNA regulates translation of RpoS message by an anti-antisense mechanism, independent of its action as an antisilencer of transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 12462–12467. [Google Scholar]
- Massé, E.; Escorcia, F.E.; Gottesman, S. Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli. Genes Dev. 2003, 17, 2374–2383. [Google Scholar]
- Massé, E.; Salvail, H.; Desnoyers, G.; Arguin, M. Small RNAs controlling iron metabolism. Curr. Opin. Microbiol. 2007, 10, 140–145. [Google Scholar]
- Opdyke, J.A.; Kang, J.G.; Storz, G. GadY, a small-RNA regulator of acid response genes in Escherichia coli. J. Bacteriol. 2004, 186, 6698–6705. [Google Scholar]
- Babitzke, P.; Romeo, T. CsrB sRNA family: Sequestration of RNA-binding regulatory proteins. Curr. Opin. Microbiol. 2007, 10, 156–163. [Google Scholar]
- Wassarman, K.M.; Repoila, F.; Rosenow, C.; Storz, G.; Gottesman, S. Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev. 2001, 15, 1637–1651. [Google Scholar]
- Argaman, L.; Hershberg, R.; Vogel, J.; Bejerano, G.; Wagner, E.G.; Margalit, H.; Altuvia, S. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr. Biol. 2001, 11, 941–950. [Google Scholar]
- Rivas, E.; Klein, R.J.; Jones, T.A.; Eddy, S.R. Computational identification of noncoding RNAs in E. coli by comparative genomics. Curr. Biol. 2001, 11, 1369–1373. [Google Scholar]
- Chen, S.; Lesnik, E.A.; Hall, T.A.; Sampath, R.; Griffey, R.H.; Ecker, D.J.; Blyn, L.B. A bioinformatics based approach to discover small RNA genes in the Escherichia coli genome. Biosystems 2002, 65, 157–177. [Google Scholar]
- Xiao, B.; Li, W.; Guo, G.; Li, B.; Liu, Z.; Jia, K.; Guo, Y.; Mao, X.; Zou, Q. Identification of small noncoding RNAs in Helicobacter pylori by a bioinformatics-based approach. Curr. Microbiol. 2009, 58, 258–263. [Google Scholar]
- Livny, J.; Brencic, A.; Lory, S.; Waldor, M.K. Identification of 17 Pseudomonas aeruginosa sRNAs and prediction of sRNA-encoding genes in 10 diverse pathogens using the bioinformatic tool sRNAPredict2. Nucleic Acids Res. 2006, 34, 3484–3493. [Google Scholar]
- Gvakharia, B.O.; Tjaden, B.; Vajrala, N.; Sayavedra-Soto, L.A.; Arp, D.J. Computational prediction and transcriptional analysis of sRNAs in Nitrosomonas europaea. FEMS Microbiol. Lett. 2010, 312, 46–54. [Google Scholar]
- Washietl, S.; Hofacker, I.L.; Stadler, P.F. Fast and reliable prediction of noncoding RNAs. Proc. Natl. Acad. Sci. USA 2005, 102, 2454–2459. [Google Scholar]
- Pedersen, J.S.; Bejerano, G.; Siepel, A.; Rosenbloom, K.; Lindblad-Toh, K.; Lander, E.S.; Kent, J.; Miller, W.; Haussler, D. Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput. Biol. 2006, 2, e33:0251–e33:0262. [Google Scholar]
- Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Infernal 1.0: Inference of RNA alignments. Bioinformatics 2009, 25, 1335–1337. [Google Scholar]
- Washietl, S.; Pedersen, J.S.; Korbel, J.O.; Stocsits, C.; Gruber, A.R.; Hackermüller, J.; Hertel, J.; Lindemeyer, M.; Reiche, K.; Tanzer, A.; et al. Structured RNAs in the ENCODE selected regions of the human genome. Genome Res. 2007, 17, 852–864. [Google Scholar]
- Sharma, C.M.; Hoffmann, S.; Darfeuille, F.; Reignier, J.; Findeiss, S.; Sittka, A.; Chabas, S.; Reiche, K.; Hackermüller, J.; Reinhardt, R.; et al. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 2010, 464, 250–255. [Google Scholar]
- Landt, S.G.; Abeliuk, E.; McGrath, P.T.; Lesley, J.A.; McAdams, H.H.; Shapiro, L. Small non-coding RNAs in Caulobacter crescentus. Mol. Microbiol. 2008, 68, 600–614. [Google Scholar]
- Mitschke, J.; Georg, J.; Scholz, I.; Sharma, C.M.; Dienst, D.; Bantscheff, J.; Voss, B.; Steglich, C.; Wilde, A.; Vogel, J.; et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp. PCC6803. Proc. Natl. Acad. Sci. USA 2011, 108, 2124–2129. [Google Scholar]
- Livny, J.; Teonadi, H.; Livny, M.; Waldor, M.K. High-throughput, kingdom-wide prediction and annotation of bacterial non-coding RNAs. PLoS One 2008, 3, e3197:1–e3197:12. [Google Scholar]
- Vercruysse, M.; Fauvart, M.; Cloots, L.; Engelen, K.; Thijs, I.M.; Marchal, K.; Michiels, J. Genome-wide detection of predicted non-coding RNAs in Rhizobium etli expressed during free-living and host-associated growth using a high-resolution tiling array. BMC Genomics 2010, 11, 53:1–54:12. [Google Scholar]
- Wilms, I.; Overlöper, A.; Nowrousian, M.; Sharma, C.M.; Narberhaus, F. Deep sequencing uncovers numerous small RNAs on all four replicons of the plant pathogen Agrobacterium tumefaciens. RNA Biol. 2012, 9. in presss. [Google Scholar]
- Venkova-Canova, T.; Soberón, N.E.; Ramírez-Romero, M.A.; Cevallos, M.A. Two discrete elements are required for the replication of a repABC plasmid: An antisense RNA and a stem-loop structure. Mol. Microbiol. 2004, 54, 1431–1444. [Google Scholar]
- MacLellan, S.R.; Smallbone, L.A.; Sibley, C.D.; Finan, T.M. The expression of a novel antisense gene mediates incompatibility within the large repABC family of alpha-proteobacterial plasmids. Mol. Microbiol. 2005, 55, 611–623. [Google Scholar]
- Ulvé, V.M.; Sevin, E.W.; Chéron, A.; Barloy-Hubler, F. Identification of chromosomal alpha-proteobacterial small RNAs by comparative genome analysis and detection in Sinorhizobium meliloti strain 1021. BMC Genomics 2007, 8, 467:1–467:16. [Google Scholar]
- Ulvé, V.M.; Chéron, A.; Trautwetter, A.; Fontenelle, C.; Barloy-Hubler, F. Characterization and expression patterns of Sinorhizobium meliloti tmRNA (ssrA). FEMS Microbiol. Lett. 2007, 269, 117–123. [Google Scholar]
- del Val, C.; Rivas, E.; Torres-Quesada, O.; Toro, N.; Jiménez-Zurdo, J.I. Identification of differentially expressed small non-coding RNAs in the legume endosymbiont Sinorhizobium meliloti by comparative genomics. Mol. Microbiol. 2007, 66, 1080–1091. [Google Scholar]
- Valverde, C.; Livny, J.; Schlüter, J.P.; Reinkensmeier, J.; Becker, A.; Parisi, G. Prediction of Sinorhizobium meliloti sRNA genes and experimental detection in strain 2011. BMC Genomics 2008, 9, 416:1–416:24. [Google Scholar]
- Schlüter, J.P.; Reinkensmeier, J.; Daschkey, S.; Evguenieva-Hackenberg, E.; Janssen, S.; Jänicke, S.; Becker, J.D.; Giegerich, R.; Becker, A. A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genomics 2010, 11, 245:1–245:35. [Google Scholar]
- Jones, K.M.; Kobayashi, H.; Davies, B.W.; Taga, M.E.; Walker, G.C. How rhizobial symbionts invade plants: The Sinorhizobium-Medicago model. Nat. Rev. Microbiol. 2007, 5, 619–633. [Google Scholar]
- Galibert, F.; Finan, T.M.; Long, S.R.; Puhler, A.; Abola, P.; Ampe, F.; Barloy-Hubler, F.; Barnett, M.J.; Becker, A.; Boistard, P.; et al. The composite genome of the legume symbiont Sinorhizobium meliloti. Science 2001, 293, 668–672. [Google Scholar]
- Barnett, M.J.; Fisher, R.F.; Jones, T.; Komp, C.; Abola, A.P.; Barloy-Hubler, F.; Bowser, L.; Capela, D.; Galibert, F.; Gouzy, J.; et al. Nucleotide sequence and predicted functions of the entire Sinorhizobium meliloti pSymA megaplasmid. Proc. Natl. Acad. Sci. USA 2001, 98, 9883–9888. [Google Scholar]
- Finan, T.M.; Weidner, S.; Wong, K.; Buhrmester, J.; Chain, P.; Vorhölter, F.J.; Hernandez-Lucas, I.; Becker, A.; Cowie, A.; Gouzy, J.; et al. The complete sequence of the 1,683-kb pSymB megaplasmid from the N2-fixing endosymbiont Sinorhizobium meliloti. Proc. Natl. Acad. Sci. USA 2001, 98, 9889–9894. [Google Scholar]
- Pappas, G.; Akritidis, N.; Bosilkovski, M.; Tsianos, E. Brucellosis. N. Engl. J. Med. 2005, 352, 2325–2336. [Google Scholar]
- Florin, T.A.; Zaoutis, T.E.; Zaoutis, L.B. Beyond cat scratch disease: Widening spectrum of Bartonella henselae infection. Pediatrics 2008, 121, e1413–e1425. [Google Scholar]
- McCullen, C.A.; Binns, A.N. Agrobacterium tumefaciens and plant cell interactions and activities required for interkingdom macromolecular transfer. Annu. Rev. Cell. Dev. Biol. 2006, 22, 101–127. [Google Scholar]
- Höchsmann, T.; Höchsmann, M.; Giegerich, R. Thermodynamic Matchers: Strengthening the Significance of RNA Folding Energies. Proceedings of the Computational Systems Bioinformatics Conference, Houston, TX, USA, August 2006; pp. 111–121.
- Reeder, J.; Reeder, J.; Giegerich, R. Locomotif: From graphical motif description to RNA motif search. Bioinformatics 2007, 23, i392–i400. [Google Scholar]
- Gillespie, J.J.; Wattam, A.R.; Cammer, S.A.; Gabbard, J.; Shukla, M.P.; Dalay, O.; Driscoll, T.; Hix, D.; Mane, S.P.; Mao, C.; et al. PATRIC: The comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect. Immun. 2011, 79, 4286–4298. [Google Scholar]
- Galardini, M.; Mengoni, A.; Brilli, M.; Pini, F.; Fioravanti, A.; Lucas, S.; Lapidus, A.; Cheng, J.F.; Goodwin, L.; Pitluck, S.; et al. Exploring the symbiotic pangenome of the nitrogen-fixing bacterium Sinorhizobium meliloti. BMC Genomics 2011, 12, 235:1–235:15. [Google Scholar]
- González, V.; Acosta, J.L.; Santamaría, R.I.; Bustos, P.; Fernández, J.L.; González, I.L.H.; Díaz, R.; Flores, M.; Palacios, R.; Mora, J.; et al. Conserved symbiotic plasmid DNA sequences in the multireplicon pangenomic structure of Rhizobium etli. Appl. Environ. Microbiol. 2010, 76, 1604–1614. [Google Scholar]
- Schmeisser, C.; Liesegang, H.; Krysciak, D.; Bakkou, N.; Quéré, A.L.; Wollherr, A.; Heinemeyer, I.; Morgenstern, B.; Pommerening-Röser, A.; Flores, M.; et al. Rhizobium sp. strain NGR234 possesses a remarkable number of secretion systems. Appl. Environ. Microbiol. 2009, 75, 4035–4045. [Google Scholar]
- Reeve, W.; Chain, P.; O'Hara, G.; Ardley, J.; Nandesena, K.; Bräu, L.; Tiwari, R.; Malfatti, S.; Kiss, H.; Lapidus, A.; et al. Complete genome sequence of the Medicago microsymbiont Ensifer (Sinorhizobium) medicae strain WSM419. Stand. Genomic Sci. 2010, 2, 77–86. [Google Scholar]
- Slater, S.C.; Goldman, B.S.; Goodner, B.; Setubal, J.C.; Farrand, S.K.; Nester, E.W.; Burr, T.J.; Banta, L.; Dickerman, A.W.; Paulsen, I.; et al. Genome sequences of three agrobacterium biovars help elucidate the evolution of multichromosome genomes in bacteria. J. Bacteriol. 2009, 191, 2501–2511. [Google Scholar]
- Voss, B.; Georg, J.; Schön, V.; Ude, S.; Hess, W.R. Biocomputational prediction of non-coding RNAs in model cyanobacteria. BMC Genomics 2009, 10, 123:1–123:15. [Google Scholar]
- Duan, Y.; Zhou, L.; Hall, D.G.; Li, W.; Doddapaneni, H.; Lin, H.; Liu, L.; Vahling, C.M.; Gabriel, D.W.; Williams, K.P.; et al. Complete genome sequence of citrus huanglongbing bacterium, ‘Candidatus Liberibacter asiaticus’ obtained through metagenomics. Mol. Plant Microbe Interact. 2009, 22, 1011–1020. [Google Scholar]
- Bentley, S.D.; Parkhill, J. Comparative genomic structure of prokaryotes. Annu. Rev. Genet. 2004, 38, 771–792. [Google Scholar]
- Mushegian, A.R.; Koonin, E.V. Gene order is not conserved in bacterial evolution. Trends Genet. 1996, 12, 289–290. [Google Scholar]
- Suyama, M.; Bork, P. Evolution of prokaryotic gene order: Genome rearrangements in closely related species. Trends Genet. 2001, 17, 10–13. [Google Scholar]
- Biondi, E.G.; Femia, A.P.; Favilli, F.; Bazzicalupo, M. IS Rm31, a new insertion sequence of the IS 66 family in Sinorhizobium meliloti. Arch. Microbiol. 2003, 180, 118–126. [Google Scholar]
- Chen, Q.; Chen, Y.P.P. Modeling conserved structure patterns for functional noncoding RNA. IEEE Trans. Biomed. Eng. 2011, 58, 1528–1533. [Google Scholar]
- Voss, B.; Gierga, G.; Axmann, I.M.; Hess, W.R. A motif-based search in bacterial genomes identifies the ortholog of the small RNA Yfr1 in all lineages of cyanobacteria. BMC Genomics 2007, 8, 375:1–375:11. [Google Scholar]
- Kingsford, C.L.; Ayanbule, K.; Salzberg, S.L. Rapid, accurate, computational discovery of Rho-independent transcription terminators illuminates their relationship to DNA uptake. Genome Biol. 2007, 8, R22:1–R22:12. [Google Scholar]
- Fröhlich, K.S.; Vogel, J. Activation of gene expression by small RNA. Curr. Opin. Microbiol. 2009, 12, 674–682. [Google Scholar]
- Otaka, H.; Ishikawa, H.; Morita, T.; Aiba, H. PolyU tail of rho-independent terminator of bacterial small RNAs is essential for Hfq action. Proc. Natl. Acad. Sci. USA 2011, 108, 13059–13064. [Google Scholar]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The Vienna RNA websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar]
- Hankins, J.S.; Denroche, H.; Mackie, G.A. Interactions of the RNA-binding protein Hfq with cspA mRNA, encoding the major cold shock protein. J. Bacteriol. 2010, 192, 2482–2490. [Google Scholar]
- Brennan, R.G.; Link, T.M. Hfq structure, function and ligand binding. Curr. Opin. Microbiol. 2007, 10, 125–133. [Google Scholar]
- Göpel, Y.; Lüttmann, D.; Heroven, A.K.; Reichenbach, B.; Dersch, P.; Görke, B. Common and divergent features in transcriptional control of the homologous small RNAs GlmY and GlmZ in Enterobacteriaceae. Nucleic Acids Res. 2011, 39, 1294–1309. [Google Scholar]
- Salim, N.N.; Feig, A.L. An upstream Hfq binding site in the fhlA mRNA leader region facilitates the OxyS-fhlA interaction. PLoS One 2010, 5, e13028:1–e13028:11. [Google Scholar]
- Brantl, S. Antisense-RNA regulation and RNA interference. Biochim. Biophys. Acta 2002, 1575, 15–25. [Google Scholar]
- Antal, M.; Bordeau, V.; Douchin, V.; Felden, B. A small bacterial RNA regulates a putative ABC transporter. J. Biol. Chem. 2005, 280, 7901–7908. [Google Scholar]
- Georg, J.; Hess, W.R. Regulatory RNAs in cyanobacteria: Developmental decisions, stress responses and a plethora of chromosomally encoded cis-antisense RNAs. Biol. Chem. 2011, 392, 291–297. [Google Scholar]
- Wilms, I.; Voss, B.; Hess, W.R.; Leichert, L.I.; Narberhaus, F. Small RNA-mediated control of the Agrobacterium tumefaciens GABA binding protein. Mol. Microbiol. 2011, 80, 492–506. [Google Scholar]
- Benito, Y.; Kolb, F.A.; Romby, P.; Lina, G.; Etienne, J.; Vandenesch, F. Probing the structure of RNAIII, the Staphylococcus aureus agr regulatory RNA, and identification of the RNA domain involved in repression of protein A expression. RNA 2000, 6, 668–679. [Google Scholar]
- Huntzinger, E.; Boisset, S.; Saveanu, C.; Benito, Y.; Geissmann, T.; Namane, A.; Lina, G.; Etienne, J.; Ehresmann, B.; Ehresmann, C.; et al. Staphylococcus aureus RNAIII and the endoribonuclease III coordinately regulate spa gene expression. EMBO J. 2005, 24, 824–835. [Google Scholar]
- Boisset, S.; Geissmann, T.; Huntzinger, E.; Fechter, P.; Bendridi, N.; Possedko, M.; Chevalier, C.; Helfer, A.C.; Benito, Y.; Jacquier, A.; et al. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev. 2007, 21, 1353–1366. [Google Scholar]
- Johansen, J.; Eriksen, M.; Kallipolitis, B.; Valentin-Hansen, P. Down-regulation of outer membrane proteins by noncoding RNAs: Unraveling the cAMP-CRP- and sigmaE-dependent CyaR-ompX regulatory case. J. Mol. Biol. 2008, 383, 1–9. [Google Scholar]
- Papenfort, K.; Pfeiffer, V.; Lucchini, S.; Sonawane, A.; Hinton, J.C.D.; Vogel, J. Systematic deletion of Salmonella small RNA genes identifies CyaR, a conserved CRP-dependent riboregulator of OmpX synthesis. Mol. Microbiol. 2008, 68, 890–906. [Google Scholar]
- Lay, N.D.; Gottesman, S. The Crp-activated small noncoding regulatory RNA CyaR (RyeE) links nutritional status to group behavior. J. Bacteriol. 2009, 191, 461–476. [Google Scholar]
- Berghoff, B.A.; Glaeser, J.; Sharma, C.M.; Vogel, J.; Klug, G. Photooxidative stress-induced and abundant small RNAs in Rhodobacter sphaeroides. Mol. Microbiol. 2009, 74, 1497–1512. [Google Scholar]
- Meyer, F.; Goesmann, A.; McHardy, A.C.; Bartels, D.; Bekel, T.; Clausen, J.; Kalinowski, J.; Linke, B.; Rupp, O.; Giegerich, R.; et al. GenDB—An open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 2003, 31, 2187–2195. [Google Scholar]
- NCBI genomes FTP site. Available online: ftp://ftp.ncbi.nlm.nih.gov/genbank/genomes/Bacteria/ (accessed on 25 February 2011).
- Menzel, P.; Gorodkin, J.; Stadler, P.F. The tedious task of finding homologous noncoding RNA genes. RNA 2009, 15, 2075–2082. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar]
- Hertel, J.; de Jong, D.; Marz, M.; Rose, D.; Tafer, H.; Tanzer, A.; Schierwater, B.; Stadler, P.F. Non-coding RNA annotation of the genome of Trichoplax adhaerens. Nucleic Acids Res. 2009, 37, 1602–1615. [Google Scholar]
- Krause, A.; Stoye, J.; Vingron, M. Large scale hierarchical clustering of protein sequences. BMC Bioinform. 2005, 6, 15:1–15:12. [Google Scholar]
- Will, S.; Reiche, K.; Hofacker, I.L.; Stadler, P.F.; Backofen, R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 2007, 3, e65:1–e65:15. [Google Scholar]
- Hofacker, I.L.; Fekete, M.; Stadler, P.F. Secondary structure prediction for aligned RNA sequences. J. Mol. Biol. 2002, 319, 1059–1066. [Google Scholar]
- Bernhart, S.H.; Hofacker, I.L.; Will, S.; Gruber, A.R.; Stadler, P.F. RNAalifold: Improved consensus structure prediction for RNA alignments. BMC Bioinform. 2008, 9, 474:1–474:13. [Google Scholar]
- The Bielefeld Bioinformatics Server. Available online: http://bibiserv.techfak.uni-bielefeld.de/ (accessed on 10 May 2011).
- Hofacker, I.L.; Fontana, W.; Stadler, P.F.; Bonhoeffer, L.S.; Tacker, M.; Schuster, P. Fast folding and comparison of RNA secondary structures. Monatsh. Chem. 1994, 125, 167–188. [Google Scholar]
- Giegerich, R.; Voss, B.; Rehmsmeier, M. Abstract shapes of RNA. Nucleic Acids Res. 2004, 32, 4843–4851. [Google Scholar]
- Steffen, P.; Voss, B.; Rehmsmeier, M.; Reeder, J.; Giegerich, R. RNAshapes: An integrated RNA analysis package based on abstract shapes. Bioinformatics 2006, 22, 500–503. [Google Scholar]
- Huang, J.; Voss, B. RNAHeliCes—Folding Space Analysis Based on Position Aware Structure Abstraction. German Conference on Bioinformatics, Weihenstephan, Germany, 7–9 September 2011.
- Reeder, J.; Giegerich, R. Consensus shapes: An alternative to the Sankoff algorithm for RNA consensus structure prediction. Bioinformatics 2005, 21, 3516–3523. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/.)
Share and Cite
Reinkensmeier, J.; Schlüter, J.-P.; Giegerich, R.; Becker, A. Conservation and Occurrence of Trans-Encoded sRNAs in the Rhizobiales. Genes 2011, 2, 925-956. https://doi.org/10.3390/genes2040925
Reinkensmeier J, Schlüter J-P, Giegerich R, Becker A. Conservation and Occurrence of Trans-Encoded sRNAs in the Rhizobiales. Genes. 2011; 2(4):925-956. https://doi.org/10.3390/genes2040925
Chicago/Turabian StyleReinkensmeier, Jan, Jan-Philip Schlüter, Robert Giegerich, and Anke Becker. 2011. "Conservation and Occurrence of Trans-Encoded sRNAs in the Rhizobiales" Genes 2, no. 4: 925-956. https://doi.org/10.3390/genes2040925