The Chlamydiales Pangenome Revisited: Structural Stability and Functional Coherence


Abstract
:
1. Introduction
2. Experimental Section
2.1. Data Collection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ## | Species and Strain Name/Codes | Internal Identifier | Protein-Coding Genes |
|---|---|---|---|
| 01 | Candidatus Protochlamydia amoebophila UWE25 | CPRO-UWE-01 | 2,031 |
| 02 | Chlamydia muridarum Nigg | CMUR-NIG-01 | 911 |
| 03 | Chlamydia trachomatis 434/Bu | CTRA-434-01 | 874 |
| 04 | Chlamydia trachomatis A/HAR-13 | CTRA-AHA-01 | 919 |
| 05 | Chlamydia trachomatis B/Jali20/OT | CTRA-BJA-01 | 875 |
| 06 | Chlamydia trachomatis B/TZ1A828/OT | CTRA-BTZ-01 | 880 |
| 07 | Chlamydia trachomatis D/UW-3/CX | CTRA-DUW-01 | 895 |
| 08 | Chlamydia trachomatis L2b/UCH-1/proctitis | CTRA-L2B-01 | 874 |
| 09 | Chlamydophila abortus S26/3 | CABO-S26-01 | 932 |
| 10 | Chlamydophila caviae GPIC | CCAV-GPI-01 | 1,005 |
| 11 | Chlamydophila felis Fe/C-56 | CFEL-FEC-01 | 1,013 |
| 12 | Chlamydophila pneumoniae AR39 | CPNE-AR3-01 | 1,112 |
| 13 | Chlamydophila pneumoniae CWL029 | CPNE-CWL-01 | 1,052 |
| 14 | Chlamydophila pneumoniae J138 | CPNE-J13-01 | 1,069 |
| 15 | Chlamydophila pneumoniae TW-183 | CPNE-TW1-01 | 1,113 |
| 16 | Waddlia chondrophila WSU 86-1044 | WCHO-WSU-01 | 1,956 |
| 17 | Chlamydia trachomatis E/150 | CTRA-E15-01 | 927 |
| 18 | Chlamydophila pecorum E58 | CPEC-E58-01 | 988 |
| 19 | Chlamydophila psittaci 6BC | CPSI-6BC-01 | 975 |
| 20 | Chlamydophila abortus LLG | CABO-LLG-01 | 925 |
| 21 | Chlamydophila pneumoniae LPCoLN | CPNE-LPC-01 | 1,105 |
| 22 | Chlamydophila psittaci Cal10 | CPSI-CAL-01 | 1,005 |
| 23 | Parachlamydia acanthamoebae UV7 | PACA-UV7-01 | 2,788 |
| 24 | Parachlamydia acanthamoebae str. Hall’s coccus | PACA-HAL-01 | 2,809 |
| 25 | Simkania negevensis Z | SNEG-ZXX-01 | 2,518 |
| 26 | Waddlia chondrophila 2032/99 | WCHO-203-01 | 2,015 |
| 27 | Chlamydophila psittaci 01DC11 | CPSI-01D-01 | 975 |
| 28 | Chlamydophila psittaci 02DC15 | CPSI-02D-01 | 978 |
| 29 | Chlamydophila psittaci 08DC60 | CPSI-08D-01 | 973 |
| 30 | Chlamydia trachomatis D-EC | CTRA-DEC-01 | 878 |
| 31 | Chlamydia trachomatis D-LC | CTRA-DLC-01 | 878 |
| 32 | Chlamydia trachomatis E/11023 | CTRA-E11-01 | 926 |
| 33 | Chlamydia trachomatis G/11074 | CTRA-G74-01 | 919 |
| 34 | Chlamydia trachomatis G/11222 | CTRA-G22-01 | 927 |
| 35 | Chlamydia trachomatis G/9301 | CTRA-G93-01 | 921 |
| 36 | Chlamydia trachomatis G/9768 | CTRA-G97-01 | 920 |
| 37 | Chlamydia trachomatis Sweden2 | CTRA-SWE-01 | 875 |
| Total | 43,736 |
2.2. Sequence Comparison
2.3. Clustering and Annotation
2.4. Analysis of Unique Genes
2.5. Genome Trees
2.6. Sequence Alignments
2.7. Data Availability
3. Results
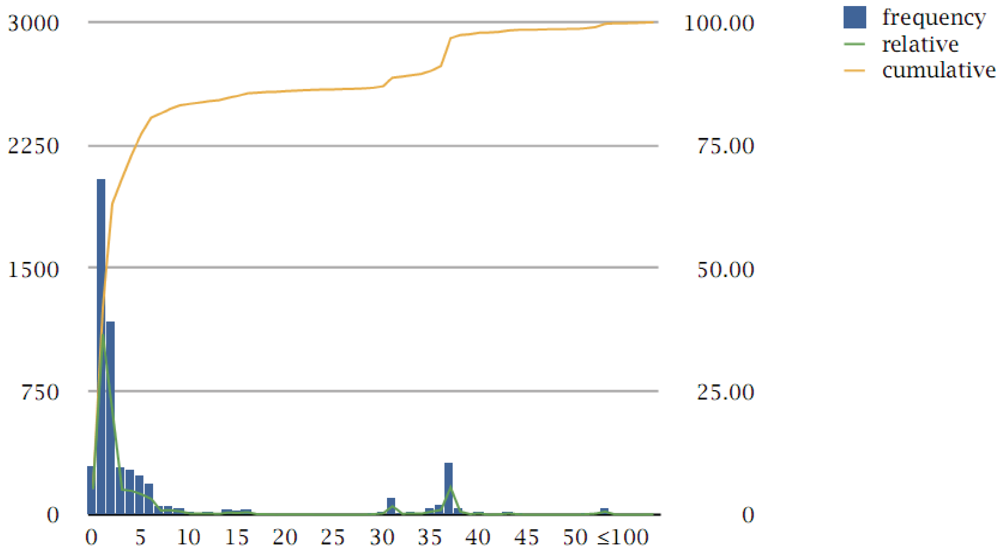
3.1. General Characteristics of the Chlamydiales Pangenome
3.2. Protein Families

3.3. Multi-Member Families

3.4. Core Genes
3.5. Peripheral Genes
3.6. Unique Genes

3.7. Properties of Unique Genes


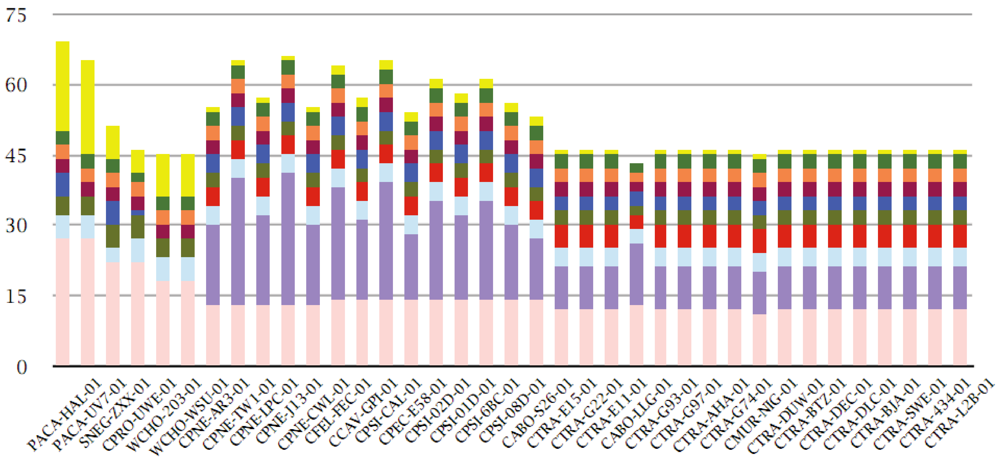
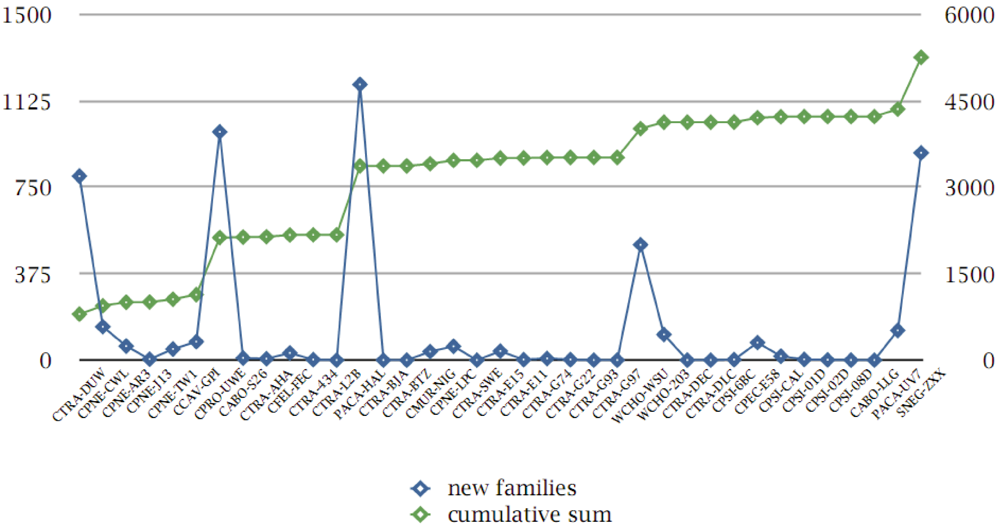
3.8. Protein Family Contributions from Genome Projects

3.9. Genome Phylogeny

4. Discussion
5. Conclusions
Acknowledgements
References
- Wyrick, P.B. Intracellular survival by Chlamydia. Cell. Microbiol. 2000, 2, 275–282. [Google Scholar]
- Corsaro, D.; Venditti, D. Emerging chlamydial infections. Crit. Rev. Microbiol. 2004, 30, 75–106. [Google Scholar]
- Horn, M. Chlamydiae as symbionts in eukaryotes. Annu. Rev. Microbiol. 2008, 62, 113–131. [Google Scholar]
- Everett, K.D.; Bush, R.M.; Andersen, A.A. Emended description of the order Chlamydiales, proposal of Parachlamydiaceae fam. nov. and Simkaniaceae fam. nov., each containing one monotypic genus, revised taxonomy of the family Chlamydiaceae, including a new genus and five new species, and standards for the identification of organisms. Int. J. Syst. Bacteriol. 1999, 49, 415–440. [Google Scholar]
- Ludwig, W.; Euzéby, J.; Whitman, W.B. Road map of the phyla Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes. In Bergey’s Manual of Systematic Bacteriology, 2nd; Krieg, N.R., Staley, J.T., Brown, D.R., Hedlund, B.P., Paster, B.J., Ward, N.L., Ludwig, W., Whitman, W.B., Eds.; Springer-Verlag: New York, NY, USA, 2010; Volume 4, pp. 1–19. [Google Scholar]
- Corsaro, D.; Valassina, M.; Venditti, D. Increasing diversity within Chlamydiae. Crit. Rev. Microbiol. 2003, 29, 37–78. [Google Scholar]
- Stephens, R.S.; Kalman, S.; Lammel, C.; Fan, J.; Marathe, R.; Aravind, L.; Mitchell, W.; Olinger, L.; Tatusov, R.L.; Zhao, Q.; et al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 1998, 282, 754–759. [Google Scholar]
- Stephens, R.S.; Myers, G.; Eppinger, M.; Bavoil, P.M. Divergence without difference: Phylogenetics and taxonomy of Chlamydia resolved. FEMS Immunol. Med. Microbiol. 2009, 55, 115–119. [Google Scholar]
- Wang, Y.; Kahane, S.; Cutcliffe, L.T.; Skilton, R.J.; Lambden, P.R.; Clarke, I.N. Development of a transformation system for Chlamydia trachomatis: Restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog. 2011, 7, e1002258. [Google Scholar]
- Horn, M.; Collingro, A.; Schmitz-Esser, S.; Beier, C.L.; Purkhold, U.; Fartmann, B.; Brandt, P.; Nyakatura, G.J.; Droege, M.; Frishman, D.; et al. Illuminating the evolutionary history of chlamydiae. Science 2004, 304, 728–730. [Google Scholar]
- Subtil, A.; Dautry-Varsat, A. Chlamydia: Five years A.G. (after genome). Curr. Opin. Microbiol. 2004, 7, 85–92. [Google Scholar]
- Vandahl, B.B.; Birkelund, S.; Christiansen, G. Genome and proteome analysis of Chlamydia. Proteomics 2004, 4, 2831–2842. [Google Scholar]
- Angiuoli, S.V.; Hotopp, J.C.; Salzberg, S.L.; Tettelin, H. Improving pan-genome annotation using whole genome multiple alignment. BMC Bioinformatics 2011, 12, 272. [Google Scholar]
- Donati, C.; Hiller, N.L.; Tettelin, H.; Muzzi, A.; Croucher, N.J.; Angiuoli, S.V.; Oggioni, M.; Dunning Hotopp, J.C.; Hu, F.Z.; Riley, D.R.; et al. Structure and dynamics of the pan-genome of Streptococcus pneumoniae and closely related species. Genome Biol. 2010, 11, R107. [Google Scholar] [CrossRef]
- Jacobsen, A.; Hendriksen, R.S.; Aaresturp, F.M.; Ussery, D.W.; Friis, C. The Salmonella enterica Pan-genome. Microb. Ecol. 2011, 62, 487–504. [Google Scholar]
- Iliopoulos, I.; Tsoka, S.; Andrade, M.A.; Enright, A.J.; Carroll, M.; Poullet, P.; Promponas, V.; Liakopoulos, T.; Palaios, G.; Pasquier, C.; et al. Evaluation of annotation strategies using an entire genome sequence. Bioinformatics 2003, 19, 717–726. [Google Scholar] [CrossRef]
- Ouzounis, C.A.; Karp, P.D. The past, present and future of genome-wide re-annotation. Genome Biol. 2002, 3, comment2001.1–comment2001.6. [Google Scholar]
- Collingro, A.; Tischler, P.; Weinmaier, T.; Penz, T.; Heinz, E.; Brunham, R.C.; Read, T.D.; Bavoil, P.M.; Sachse, K.; Kahane, S.; et al. Unity in variety—The pan-genome of the Chlamydiae. Mol. Biol. Evol. 2011, 28, 3253–3270. [Google Scholar] [CrossRef]
- Bertelli, C.; Collyn, F.; Croxatto, A.; Ruckert, C.; Polkinghorne, A.; Kebbi-Beghdadi, C.; Goesmann, A.; Vaughan, L.; Greub, G. The Waddlia genome: A window into chlamydial biology. PLoS One 2010, 5, e10890. [Google Scholar]
- Laing, C.; Buchanan, C.; Taboada, E.N.; Zhang, Y.; Kropinski, A.; Villegas, A.; Thomas, J.E.; Gannon, V.P. Pan-genome sequence analysis using Panseq: An online tool for the rapid analysis of core and accessory genomic regions. BMC Bioinformatics 2010, 11, 461. [Google Scholar]
- Lapierre, P.; Gogarten, J.P. Estimating the size of the bacterial pan-genome. Trends Genet. 2009, 25, 107–110. [Google Scholar]
- Janssen, P.; Enright, A.J.; Audit, B.; Cases, I.; Goldovsky, L.; Harte, N.; Kunin, V.; Ouzounis, C.A. COmplete GENome Tracking (COGENT): A flexible data environment for computational genomics. Bioinformatics 2003, 19, 1451–1452. [Google Scholar]
- Goldovsky, L.; Janssen, P.; Ahren, D.; Audit, B.; Cases, I.; Darzentas, N.; Enright, A.J.; Lopez-Bigas, N.; Peregrin-Alvarez, J.M.; Smith, M.; et al. CoGenT++: An extensive and extensible data environment for computational genomics. Bioinformatics 2005, 21, 3806–3810. [Google Scholar]
- Promponas, V.J.; Enright, A.J.; Tsoka, S.; Kreil, D.P.; Leroy, C.; Hamodrakas, S.; Sander, C.; Ouzounis, C.A. CAST: An iterative algorithm for the complexity analysis of sequence tracts. Complexity analysis of sequence tracts. Bioinformatics 2000, 16, 915–922. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- Enright, A.J.; van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar]
- Lefebure, T.; Bitar, P.D.; Suzuki, H.; Stanhope, M.J. Evolutionary dynamics of complete Campylobacter pan-genomes and the bacterial species concept. Genome Biol. Evol. 2010, 2, 646–655. [Google Scholar]
- Smith, M.; Kunin, V.; Goldovsky, L.; Enright, A.J.; Ouzounis, C.A. MagicMatch—Cross-referencing sequence identifiers across databases. Bioinformatics 2005, 21, 3429–3430. [Google Scholar]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bolton, E.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2011, 39, D38–D51. [Google Scholar]
- Pellegrini, M.; Marcotte, E.M.; Thompson, M.J.; Eisenberg, D.; Yeates, T.O. Assigning protein functions by comparative genome analysis: Protein phylogenetic profiles. Proc. Natl. Acad. Sci. USA 1999, 96, 4285–4288. [Google Scholar]
- Kunin, V.; Ahren, D.; Goldovsky, L.; Janssen, P.; Ouzounis, C.A. Measuring genome conservation across taxa: Divided strains and united kingdoms. Nucleic Acids Res. 2005, 33, 616–621. [Google Scholar]
- Snel, B.; Huynen, M.A.; Dutilh, B.E. Genome trees and the nature of genome evolution. Annu. Rev. Microbiol. 2005, 59, 191–209. [Google Scholar]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar]
- The 10 Supplements can be accessed at http://dx.doi.org/10.5061/dryad.rr064j8q/.
- Heinz, E.; Tischler, P.; Rattei, T.; Myers, G.; Wagner, M.; Horn, M. Comprehensive in silico prediction and analysis of chlamydial outer membrane proteins reflects evolution and life style of the Chlamydiae. BMC Genomics 2009, 10, 634. [Google Scholar]
- Stone, C.B.; Bulir, D.C.; Gilchrist, J.D.; Toor, R.K.; Mahony, J.B. Interactions between flagellar and type III secretion proteins in Chlamydia pneumoniae. BMC Microbiol. 2010, 10, 18. [Google Scholar]
- Muschiol, S.; Boncompain, G.; Vromman, F.; Dehoux, P.; Normark, S.; Henriques-Normark, B.; Subtil, A. Identification of a family of effectors secreted by the type III secretion system that are conserved in pathogenic Chlamydiae. Infect. Immun. 2011, 79, 571–580. [Google Scholar]
- Ouzounis, C.; Bork, P.; Sander, C. The modular structure of NifU proteins. Trends Biochem. Sci. 1994, 19, 199–200. [Google Scholar]
- Ouzounis, C.; Sander, C. Homology of the NifS family of proteins to a new class of pyridoxal phosphate-dependent enzymes. FEBS Lett. 1993, 322, 159–164. [Google Scholar]
- Devos, D.P.; Reynaud, E.G. Evolution. Intermediate steps. Science 2010, 330, 1187–1188. [Google Scholar] [CrossRef]
- McInerney, J.O.; Martin, W.F.; Koonin, E.V.; Allen, J.F.; Galperin, M.Y.; Lane, N.; Archibald, J.M.; Embley, T.M. Planctomycetes and eukaryotes: A case of analogy not homology. Bioessays 2011, 33, 810–817. [Google Scholar]
- Kunin, V.; Ouzounis, C.A. GeneTRACE-reconstruction of gene content of ancestral species. Bioinformatics 2003, 19, 1412–1416. [Google Scholar]
- Bush, R.M.; Everett, K.D. Molecular evolution of the Chlamydiaceae. Int. J. Syst. Evol. Microbiol. 2001, 51, 203–220. [Google Scholar]
- Janssen, P.J.; Audit, B.; Ouzounis, C.A. Strain-specific genes of Helicobacter pylori: Distribution, function and dynamics. Nucleic Acids Res. 2001, 29, 4395–4404. [Google Scholar]
- Brinkman, F.S.; Blanchard, J.L.; Cherkasov, A.; Av-Gay, Y.; Brunham, R.C.; Fernandez, R.C.; Finlay, B.B.; Otto, S.P.; Ouellette, B.F.; Keeling, P.J.; et al. Evidence that plant-like genes in Chlamydia species reflect an ancestral relationship between Chlamydiaceae, cyanobacteria, and the chloroplast. Genome Res. 2002, 12, 1159–1167. [Google Scholar] [CrossRef]
- Huang, J.; Gogarten, J.P. Did an ancient chlamydial endosymbiosis facilitate the establishment of primary plastids? Genome Biol. 2007, 8, R99. [Google Scholar] [CrossRef]
- Becker, B.; Hoef-Emden, K.; Melkonian, M. Chlamydial genes shed light on the evolution of photoautotrophic eukaryotes. BMC Evol. Biol. 2008, 8, 203. [Google Scholar]
- Moustafa, A.; Reyes-Prieto, A.; Bhattacharya, D. Chlamydiae has contributed at least 55 genes to Plantae with predominantly plastid functions. PLoS One 2008, 3, e2205. [Google Scholar]
- Kamneva, O.K.; Liberles, D.A.; Ward, N.L. Genome-wide influence of indel substitutions on evolution of bacteria of the PVC superphylum, revealed using a novel computational method. Genome Biol. Evol. 2010, 2, 870–886. [Google Scholar]
- Moran, N.A.; McCutcheon, J.P.; Nakabachi, A. Genomics and evolution of heritable bacterial symbionts. Annu. Rev. Genet. 2008, 42, 165–190. [Google Scholar]
- Merhej, V.; Royer-Carenzi, M.; Pontarotti, P.; Raoult, D. Massive comparative genomic analysis reveals convergent evolution of specialized bacteria. Biol. Direct 2009, 4, 13. [Google Scholar]
- Harris, S.R.; Clarke, I.N.; Seth-Smith, H.M.; Solomon, A.W.; Cutcliffe, L.T.; Marsh, P.; Skilton, R.J.; Holland, M.J.; Mabey, D.; Peeling, R.W.; et al. Whole-genome analysis of diverse Chlamydia trachomatis strains identifies phylogenetic relationships masked by current clinical typing. Nat. Genet. 2012, 44, 413–419. [Google Scholar]
Appendix
| Cluster ID | Lead Sequence | Master Sequence | Function Annotation from Master Sequence |
|---|---|---|---|
| 181 | CABO-LLG-01-000000 | CTRA-DUW-01-000647 | NA |
| 182 | CABO-LLG-01-000003 | CTRA-DUW-01-000644 | ENZYME UDP-N-acetylglucosamine pyrophosphorylase [EC] 2.7.7.23 |
| 183 | CABO-LLG-01-000004 | CTRA-DLC-01-000248 | FUNCTION PhoB-like protein |
| 184 | CABO-LLG-01-000015 | CTRA-DLC-01-000228 | FUNCTION RecA protein |
| 185 | PACA-UV7-01-001616 | CTRA-DUW-01-000487 | NA |
| 186 | CABO-LLG-01-000023 | CTRA-DUW-01-000658 | NA |
| 187 | CABO-LLG-01-000024 | CTRA-DUW-01-000624 | FUNCTION RNA Polymerase Sigma-54 factor RpoN |
| 188 | CABO-LLG-01-000026 | CTRA-DUW-01-000622 | ENZYME Uracil DNA glycosylase [EC] 3.2.2.- |
| 189 | CABO-LLG-01-000028 | CTRA-DUW-01-000620 | SIMILAR-TO NTPase HAM1 homolog [EC] 3.6.1.15 |
| 190 | CABO-LLG-01-000029 | CTRA-DLC-01-000273 | NA |
| 191 | CABO-LLG-01-000032 | CTRA-DUW-01-000616 | NA |
| 192 | CABO-LLG-01-000034 | CTRA-DLC-01-000278 | FUNCTION Peptidoglycan-associated lipoprotein |
| 193 | CABO-LLG-01-000035 | CTRA-DLC-01-000279 | FUNCTION TolB macromolecule uptake homolog |
| 194 | CABO-LLG-01-000037 | CTRA-DUW-01-000611 | FUNCTION TolR/ExbD macromolecule uptake homolog |
| 195 | CABO-LLG-01-000040 | CTRA-DLC-01-000284 | FUNCTION protein translocase TatD/MttC homolog |
| 196 | CABO-LLG-01-000047 | CTRA-DUW-01-000600 | ENZYME enolase [EC] 4.2.1.11 |
| 197 | CABO-LLG-01-000048 | CTRA-DUW-01-000599 | FUNCTION Excinuclease ABC subunit B |
| 198 | CABO-LLG-01-000049 | CTRA-DUW-01-000598 | ENZYME Tryptophanyl-tRNA Synthetase [EC] 6.1.1.2 |
| 199 | CTRA-G22-01-000161 | CTRA-DUW-01-000746 | ENZYME Seryl-tRNA Synthetase [EC] 6.1.1.11 |
| 200 | CABO-LLG-01-000054 | CTRA-DUW-01-000593 | FUNCTION Nickel transporter CnrT homolog |
| 201 | CABO-LLG-01-000061 | CTRA-DUW-01-000586 | NA |
| 202 | CABO-LLG-01-000062 | CTRA-DUW-01-000585 | FUNCTION type II secretion system protein D homolog |
| 203 | CABO-LLG-01-000063 | CTRA-DUW-01-000584 | FUNCTION type II secretion system protein E homolog |
| 204 | CABO-LLG-01-000064 | CTRA-DLC-01-000308 | FUNCTION type II secretion system protein F homolog |
| 205 | CABO-LLG-01-000065 | CTRA-DLC-01-000309 | NA |
| 206 | CABO-LLG-01-000070 | CTRA-DUW-01-000577 | FUNCTION protein secretion system YscT homolog |
| 207 | CABO-LLG-01-000072 | CTRA-DLC-01-000316 | FUNCTION protein secretion system YscR homolog |
| 208 | CABO-LLG-01-000073 | CTRA-DEC-01-000317 | FUNCTION protein secretion system YscL homolog |
| 209 | CABO-LLG-01-000074 | CTRA-DUW-01-000573 | NA |
| 210 | CABO-LLG-01-000076 | CTRA-DUW-01-000571 | ENZYME lipoate synthase [EC] 2.8.1.- |
| 211 | CABO-LLG-01-000081 | CTRA-DUW-01-000714 | ENZYME Endonuclease III [EC] 4.2.99.18 |
| 212 | CABO-LLG-01-000083 | CTRA-DUW-01-000716 | ENZYME Phosphatidylserine decarboxylase [EC] 4.1.1.65 |
| 213 | CABO-LLG-01-000085 | CTRA-DUW-01-000718 | FUNCTION preprotein translocase subunit SecA |
| 214 | CABO-LLG-01-000089 | CTRA-DUW-01-000722 | ENZYME ATP-dependent Clp protease ATP-binding subunit ClpX [EC] |
| 215 | CABO-LLG-01-000091 | CTRA-DUW-01-000724 | FUNCTION Trigger factor |
| 216 | CABO-LLG-01-000093 | CTRA-DUW-01-000726 | FUNCTION Rod shape-determining protein MreB |
| 217 | CABO-LLG-01-000094 | CTRA-DUW-01-000727 | ENZYME Phosphoenolpyruvate carboxykinase (GTP) [EC] 4.1.1.32 |
| 218 | CABO-LLG-01-000098 | CTRA-DUW-01-000731 | ENZYME Glycerol-3-phosphate dehydrogenase [NAD+] [EC] 1.1.1.8 |
| 219 | CABO-LLG-01-000099 | CTRA-DUW-01-000732 | ENZYME UDP-N-acetylhexosamine pyrophosphorylase [EC] 2.7.7.- |
| 220 | CCAV-GPI-01-000128 | CTRA-DUW-01-000503 | FUNCTION Transcription termination factor Rho |
| 221 | CABO-LLG-01-000104 | CTRA-DUW-01-000737 | DOMAIN NifU |
| 222 | PACA-HAL-01-002518 | CTRA-DUW-01-000261 | ENZYME NifS aminotransferase [EC] -.-.-.- |
| 223 | CABO-LLG-01-000109 | CTRA-DUW-01-000742 | ENZYME Biotin-[acetyl-CoA-carboxylase] synthetase [EC] 6.3.4.15 |
| 224 * | CABO-LLG-01-000121 | CTRA-DUW-01-000754 | DOMAIN SET |
| 225 | CABO-LLG-01-000122 | CTRA-DUW-01-000755 | SIMILAR-TO metallo-beta-lactamase [EC] 3.5.-.- |
| 226 | CABO-LLG-01-000123 | CTRA-DUW-01-000756 | FUNCTION Cell division protein FtsK C-terminus |
| 227 | CABO-LLG-01-000125 | CTRA-DUW-01-000757 | NA |
| 228 | CABO-LLG-01-000126 | CTRA-DUW-01-000758 | FUNCTION preprotein translocase complex subunit YajC |
| 229 | CABO-LLG-01-000130 | CTRA-DUW-01-000762 | ENZYME Protoporphyrinogen oxidase HemY [EC] 1.3.3.4 |
| 230 | CABO-LLG-01-000132 | CTRA-DUW-01-000764 | ENZYME Uroporphyrinogen decarboxylase HemE [EC] 4.1.1.37 |
| 231 | CABO-LLG-01-000134 | CTRA-DLC-01-000129 | ENZYME Alanyl-tRNA Synthetase [EC] 6.1.1.7 |
| 232 | CABO-LLG-01-000135 | CTRA-DUW-01-000767 | ENZYME Transketolase [EC] 2.2.1.1 |
| 233 | CABO-LLG-01-000136 | CTRA-DUW-01-000768 | SIMILAR-TO AMP nucleosidase [EC] 3.2.2.4 |
| 234 | CABO-LLG-01-000142 | CTRA-DUW-01-000774 | ENZYME Phospho-N-acetylmuramoyl-pentapeptide-transferase [EC] |
| 235 | CABO-LLG-01-000143 | CTRA-DUW-01-000775 | ENZYME UDP-N-acetylmuramoylalanine-D-glutamate ligase [EC] |
| 236 | CABO-LLG-01-000144 | CTRA-DUW-01-000776 | SIMILAR-TO N-acetylmuramoyl-L-alanine amidase C-terminus [EC] |
| 237 | CABO-LLG-01-000146 | CTRA-DLC-01-000117 | ENZYME UDP-N-acetylglucosamine-N-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine transferase |
| 238 | CABO-S26-01-000517 | CTRA-DUW-01-000125 | ENZYME Biotin carboxylase [EC] 6.3.4.14 |
| 239 | CABO-LLG-01-000150 | CTRA-DUW-01-000781 | NA |
| 240 | CABO-LLG-01-000155 | CTRA-DUW-01-000786 | NA |
| 241 | CABO-LLG-01-000157 | CTRA-DUW-01-000788 | ENZYME bis(5'-nucleosyl)-tetraphosphatase [EC] 3.6.1.17 |
| 242 | CABO-LLG-01-000168 | CTRA-DLC-01-000098 | ENZYME Cysteinyl-tRNA Synthetase [EC] 6.1.1.16 |
| 243 | CABO-LLG-01-000173 | CTRA-DUW-01-000804 | FUNCTION Ribosomal protein S14 |
| 244 | CABO-LLG-01-000174 | CTRA-DUW-01-000805 | NA |
| 245 | CABO-LLG-01-000176 | CTRA-DUW-01-000808 | ENZYME Excinuclease ABC subunit C [EC] -.-.-.- |
| 246 | CABO-LLG-01-000177 | CTRA-DUW-01-000809 | FUNCTION DNA mismatch repair protein MutS |
| 247 | CABO-LLG-01-000184 | CTRA-DUW-01-000815 | ENZYME CDP-diacylglycerol-glycerol-3-phosphate |
| 248 | CABO-LLG-01-000185 | CTRA-DUW-01-000816 | ENZYME Glycogen synthase [EC] 2.4.1.21 2 |
| 249 | CABO-LLG-01-000186 | CTRA-DUW-01-000817 | FUNCTION Ribosomal protein L25 |
| 250 | CABO-LLG-01-000187 | CTRA-DUW-01-000818 | ENZYME Peptidyl-tRNA hydrolase [EC] 3.1.1.29 |
| 251 | CABO-LLG-01-000188 | CTRA-DUW-01-000819 | FUNCTION Ribosomal protein S6 |
| 252 | CABO-LLG-01-000189 | CTRA-DUW-01-000820 | FUNCTION Ribosomal protein S18 |
| 253 | CABO-LLG-01-000190 | CTRA-DUW-01-000821 | FUNCTION Ribosomal protein L9 |
| 254 * | CABO-LLG-01-000193 | CTRA-DUW-01-000823 | NA |
| 255 * | CABO-LLG-01-000194 | CTRA-DUW-01-000824 | SIMILAR-TO Small-peptide endopeptidase [EC] 3.4.24.55 |
| 256 | CABO-LLG-01-000195 | CTRA-DLC-01-000073 | ENZYME Glycerol-3-phosphate acyltransferase [EC] 2.3.1.15 |
| 257 | CABO-LLG-01-000196 | CTRA-DLC-01-000072 | ENZYME Ribonuclease E [EC] 3.1.4.- |
| 258 | CABO-LLG-01-000197 | CTRA-DLC-01-000071 | NA |
| 259 | CABO-LLG-01-000214 | CTRA-DLC-01-000063 | ENZYME Glucosamine-fructose-6-phosphate aminotransferase [EC] |
| 260 | CABO-LLG-01-000218 | CTRA-DUW-01-000840 | ENZYME Succinyl-CoA synthetase beta chain [EC] 6.2.1.5 |
| 261 | CABO-LLG-01-000222 | CTRA-DUW-01-000843 | SIMILAR-TO Small-peptide endopeptidase [EC] 3.4.24.55 |
| 262 | CABO-LLG-01-000224 | CTRA-DUW-01-000845 | ENZYME CDP-diacylglycerol-serine O-phosphatidyltransferase [EC] |
| 263 | CABO-LLG-01-000229 | CTRA-DUW-01-000850 | ENZYME UDP-N-acetylenolpyruvoylglucosamine reductase [EC] |
| 264 | CABO-LLG-01-000230 | CTRA-DLC-01-000047 | FUNCTION Transcription termination protein NusB |
| 265 | CABO-LLG-01-000231 | CTRA-DLC-01-000046 | NA |
| 266 | CABO-LLG-01-000233 | CTRA-DUW-01-000854 | FUNCTION Ribosomal protein L20 |
| 267 | CABO-LLG-01-000234 | CTRA-DUW-01-000855 | ENZYME Phenylalanyl-tRNA Synthetase alpha chain [EC] 6.1.1.20 |
| 268 | CABO-LLG-01-000236 | CTRA-DUW-01-000857 | NA |
| 269 | CABO-LLG-01-000237 | CTRA-DUW-01-000858 | NA |
| 270 | CABO-LLG-01-000240 | CTRA-DUW-01-000861 | ENZYME Polynucleotide phosphorylase [EC] 2.7.7.8 |
| 271 | CABO-LLG-01-000241 | CTRA-DUW-01-000862 | NA |
| 272 * | CABO-LLG-01-000254 | CTRA-DUW-01-000874 | FUNCTION ABC transporter, ATP-binding protein N-terminus |
| 273 | CABO-LLG-01-000267 | CTRA-DUW-01-000385 | ENZYME Glucose-6-phosphate isomerase [EC] 5.3.1.9 |
| 274 | CABO-LLG-01-000269 | CTRA-DLC-01-000502 | ENZYME Malate dehydrogenase [EC] 1.1.1.82 |
| 275 | CABO-LLG-01-000271 | CTRA-DUW-01-000382 | SIMILAR-TO D-Amino Acid Dehydrogenase [EC] 1.-.-.- |
| 276 * | CABO-LLG-01-000276 | CTRA-DLC-01-000508 | ENZYME 3-dehydroquinate dehydratase [EC] 4.2.1.10 |
| 277 | CPRO-UWE-01-000881 | CTRA-DUW-01-000373 | ENZYME 3-phosphoshikimate 1-carboxyvinyltransferase [EC] |
| 278 | CABO-LLG-01-000277 | CTRA-DUW-01-000376 | ENZYME 3-dehydroquinate synthase [EC] 4.6.1.3 |
| 279 | CABO-LLG-01-000278 | CTRA-DUW-01-000375 | ENZYME Chorismate synthase [EC] 4.6.1.4 |
| 280 | CABO-LLG-01-000288 | CTRA-DUW-01-000371 | ENZYME Dihydrodipicolinate reductase [EC] 1.3.1.26 |
| 281 | CABO-LLG-01-000290 | CTRA-DUW-01-000369 | ENZYME Aspartokinase [EC] 2.7.2.4 |
| 282 | CABO-LLG-01-000298 | CTRA-DUW-01-000328 | NA |
| 283 | SNEG-ZXX-01-000625 | CTRA-DLC-01-000783 | FUNCTION Translation initiation factor IF-2 |
| 284 | CABO-LLG-01-000304 | CTRA-DUW-01-000322 | FUNCTION Ribosomal protein L11 |
| 285 | CABO-LLG-01-000305 | CTRA-DUW-01-000321 | FUNCTION Ribosomal protein L1 |
| 286 | CABO-LLG-01-000306 | CTRA-DUW-01-000320 | FUNCTION Ribosomal protein L10 |
| 287 | CABO-LLG-01-000308 | CTRA-DUW-01-000318 | ENZYME DNA-directed RNA polymerase beta subunit [EC] 2.7.7.6 |
| 288 | CABO-LLG-01-000309 | CTRA-DUW-01-000317 | ENZYME DNA-directed RNA polymerase beta prime subunit [EC] |
| 289 | CABO-LLG-01-000312 | CTRA-DUW-01-000314 | NA |
| 290 | CABO-LLG-01-000313 | CTRA-DLC-01-000569 | ENZYME vacuolar ATPase proteolipid subunit E [EC] 3.6.1.34 |
| 291 | CABO-LLG-01-000314 | CTRA-DUW-01-000312 | NA |
| 292 | CABO-LLG-01-000317 | CTRA-DUW-01-000309 | ENZYME vacuolar ATPase proteolipid subunit D [EC] 3.6.1.34 |
| 293 | CABO-LLG-01-000320 | CTRA-DLC-01-000576 | NA |
| 294 | CABO-LLG-01-000324 | CTRA-DUW-01-000337 | ENZYME Pyruvate kinase [EC] 2.7.1.40 |
| 295 | CPRO-UWE-01-001632 | CTRA-DUW-01-000012 | NA |
| 296 | CABO-LLG-01-000328 | CTRA-DUW-01-000013 | ENZYME Cytochrome Oxidase D subunit I [EC] 1.10.3.- |
| 297 | CABO-LLG-01-000329 | CTRA-DUW-01-000014 | ENZYME Cytochrome Oxidase D subunit II [EC] 1.10.3.- |
| 298 | CABO-LLG-01-000331 | CTRA-DLC-01-000860 | NA |
| 299 | CABO-LLG-01-000332 | CTRA-DLC-01-000861 | NA |
| 300 | CABO-LLG-01-000333 | CTRA-DUW-01-000015 | FUNCTION PhoH-like protein |
| 301 | CABO-LLG-01-000337 | CTRA-DLC-01-000856 | NA |
| 302 | CABO-LLG-01-000338 | CTRA-DUW-01-000022 | FUNCTION Ribosomal protein L31 |
| 303 | CABO-LLG-01-000342 | CTRA-DUW-01-000026 | FUNCTION Ribosomal protein S16 |
| 304 | CABO-LLG-01-000343 | CTRA-DUW-01-000027 | ENZYME tRNA (guanine N-1) methyltransferase [EC] 2.1.1.31 |
| 305 | CABO-LLG-01-000344 | CTRA-DUW-01-000028 | FUNCTION Ribosomal protein L19 |
| 306 | CABO-LLG-01-000345 | CTRA-DUW-01-000029 | ENZYME Ribonuclease HII [EC] 3.1.26.4 |
| 307 | CABO-LLG-01-000346 | CTRA-DUW-01-000030 | ENZYME Guanylate kinase [EC] 2.7.4.8 |
| 308 | CABO-LLG-01-000358 | CTRA-DUW-01-000215 | ENZYME Ribose 5-phosphate isomerase A [EC] 5.3.1.6 |
| 309 | CABO-LLG-01-000359 | CTRA-DLC-01-000666 | NA |
| 310 | CABO-LLG-01-000360 | CTRA-DUW-01-000213 | NA |
| 311 | CABO-LLG-01-000368 | CTRA-DLC-01-000765 | NA |
| 312 | CABO-LLG-01-000374 | CTRA-DUW-01-000147 | ENZYME DNA ligase (NAD+) [EC] 6.5.1.2 |
| 313 | CABO-LLG-01-000379 | CTRA-DUW-01-000210 | ENZYME 3-deoxy-d-manno-octulosonic-acid transferase [EC] 2.-.-.- |
| 314 | CABO-LLG-01-000392 | CTRA-DUW-01-000186 | NA |
| 315 | CABO-LLG-01-000393 | CTRA-DUW-01-000185 | ENZYME CTP synthetase [EC] 6.3.4.2 |
| 316 | CABO-LLG-01-000404 | CTRA-DUW-01-000195 | ENZYME Queuine tRNA-ribosyltransferase [EC] 2.4.2.29 |
| 317 | CABO-LLG-01-000420 | CTRA-DUW-01-000132 | NA |
| 318 | CABO-LLG-01-000423 | CTRA-DUW-01-000199 | ENZYME O-sialoglycoprotein endopeptidase [EC] 3.4.24.57 |
| 319 | CABO-LLG-01-000425 | CTRA-DUW-01-000187 | NA |
| 320 | CABO-LLG-01-000426 | CTRA-DUW-01-000188 | ENZYME Glucose-6-phosphate dehydrogenase [EC] -.-.-.- |
| 321 | CABO-LLG-01-000432 | CTRA-DLC-01-000753 | FUNCTION Ribosomal protein S9 |
| 322 | CABO-LLG-01-000433 | CTRA-DUW-01-000126 | FUNCTION Ribosomal protein L13 |
| 323 | CABO-LLG-01-000435 | CTRA-DUW-01-000152 | NA |
| 324 | CABO-LLG-01-000448 | CTRA-DUW-01-000138 | FUNCTION Sua5 homolog |
| 325 | CABO-LLG-01-000451 | CTRA-DUW-01-000190 | ENZYME Thymidylate kinase (dTMP kinase) [EC] 2.7.4.9 |
| 326 | CABO-LLG-01-000459 | CTRA-DUW-01-000217 | ENZYME Fructose-bisphosphate aldolase class I [EC] 4.1.2.13 |
| 327 | CABO-LLG-01-000471 | CTRA-DUW-01-000239 | FUNCTION acyl carrier protein ACP |
| 328 | PACA-UV7-01-000731 | CTRA-DUW-01-000105 | ENZYME Enoyl-[acyl-carrier protein] reductase (NADH) [EC] |
| 329 | CABO-LLG-01-000473 | CTRA-DLC-01-000640 | ENZYME Malonyl CoA-acyl carrier protein transacylase [EC] |
| 330 | CABO-LLG-01-000474 | CTRA-DLC-01-000639 | ENZYME 3-oxoacyl-[acyl-carrier-protein] synthase III [EC] |
| 331 | CABO-LLG-01-000475 | CTRA-DUW-01-000243 | FUNCTION Recombination protein RecR homolog |
| 332 | CABO-LLG-01-000477 | CTRA-DUW-01-000245 | NA |
| 333 | CPNE-TW1-01-000387 | CTRA-DUW-01-000055 | ENZYME 2-oxoglutarate dehydrogenase E1 component [EC] 1.2.4.2 |
| 334 | CABO-LLG-01-000486 | CTRA-DUW-01-000254 | FUNCTION Inner-membrane protein YidC |
| 335 | CABO-LLG-01-000489 | CTRA-DUW-01-000101 | ENZYME holo-[acyl-carrier protein] synthase [EC] 2.7.8.7 |
| 336 | CABO-LLG-01-000490 | CTRA-DUW-01-000100 | ENZYME Thioredoxin reductase (NADPH) [EC] 1.6.4.5 |
| 337 | CABO-LLG-01-000494 | CTRA-DUW-01-000096 | FUNCTION Ribosome-binding factor A RbfA |
| 338 | CABO-LLG-01-000496 | CTRA-DUW-01-000094 | ENZYME Riboflavin kinase [EC] 2.7.1.26 |
| 339 | CABO-LLG-01-000499 | CTRA-DUW-01-000090 | NA |
| 340 | CABO-LLG-01-000500 | CTRA-DUW-01-000089 | NA |
| 341 | CABO-LLG-01-000501 | CTRA-DLC-01-000793 | FUNCTION Ribosomal protein L28 |
| 342 | CABO-LLG-01-000508 | CTRA-DLC-01-000801 | ENZYME Methylenetetrahydrofolate dehydrogenase [EC] 1.5.1.15 |
| 343 | CABO-LLG-01-000509 | CTRA-DUW-01-000078 | FUNCTION Thiamine biosynthesis lipoprotein ApbE precursor |
| 344 | CABO-LLG-01-000510 | CTRA-DUW-01-000077 | FUNCTION Small protein B SmpB homolog |
| 345 | CABO-LLG-01-000511 | CTRA-DUW-01-000076 | ENZYME DNA polymerase III beta chain [EC] 2.7.7.7 |
| 346 | CABO-LLG-01-000514 | CTRA-DUW-01-000073 | SIMILAR-TO zinc protease [EC] -.-.-.- |
| 347 | CABO-LLG-01-000516 | CTRA-DUW-01-000071 | FUNCTION ABC transporter, permease protein TroD |
| 348 | CABO-LLG-01-000519 | CTRA-DUW-01-000068 | FUNCTION periplasmic substrate binding protein TroA |
| 349 | CPSI-CAL-01-000799 | CTRA-DUW-01-000423 | FUNCTION high-affinity ZnuA homolog |
| 350 | CABO-LLG-01-000520 | CTRA-DLC-01-000812 | NA |
| 351 | CABO-LLG-01-000523 | CTRA-DUW-01-000064 | ENZYME 6-phosphogluconate dehydrogenase [EC] 1.1.1.44 |
| 352 | CABO-LLG-01-000524 | CTRA-DUW-01-000063 | ENZYME Tyrosyl-tRNA Synthetase [EC] 6.1.1.1 |
| 353 | CABO-LLG-01-000535 | CTRA-DLC-01-000825 | NA |
| 354 | CABO-LLG-01-000541 | CTRA-DLC-01-000831 | NA |
| 355 | CABO-LLG-01-000544 | CTRA-DUW-01-000045 | FUNCTION single-stranded DNA-binding protein SSB |
| 356 | CABO-LLG-01-000545 | CTRA-DUW-01-000044 | NA |
| 357 | CABO-LLG-01-000547 | CTRA-DUW-01-000042 | NA |
| 358 | CABO-LLG-01-000554 | CTRA-DLC-01-000619 | ENZYME Protein phosphatase 2C [EC] 3.1.3.16 |
| 359 | CABO-LLG-01-000558 | CTRA-DUW-01-000257 | NA |
| 360 | CABO-LLG-01-000560 | CTRA-DUW-01-000108 | ENZYME A/G-specific adenine glycosylase [EC] 3.2.2.- |
| 361 | CABO-LLG-01-000564 | CTRA-DUW-01-000104 | NA |
| 362 | CABO-LLG-01-000571 | CTRA-DUW-01-000268 | ENZYME Acetyl-coenzyme A carboxylase carboxyl transferase |
| 363 | CABO-LLG-01-000574 | CTRA-DUW-01-000271 | ENZYME N-acetylmuramoyl-L-alanine amidase AmiB [EC] 3.5.1.28 |
| 364 | CABO-LLG-01-000577 | CTRA-DUW-01-000273 | FUNCTION Penicillin-binding protein 3 |
| 365 | CABO-LLG-01-000578 | CTRA-DUW-01-000274 | NA |
| 366 | CABO-LLG-01-000581 | CTRA-DUW-01-000277 | DOMAIN TPR |
| 367 | CABO-LLG-01-000585 | CTRA-DLC-01-000601 | NA |
| 368 | CABO-LLG-01-000586 | CTRA-DUW-01-000282 | NA |
| 369 | CABO-LLG-01-000587 | CTRA-DLC-01-000599 | NA |
| 370 | CPEC-E58-01-000614 | CTRA-DUW-01-000284 | NA |
| 371 | CABO-LLG-01-000591 | CTRA-DLC-01-000597 | FUNCTION Glycine cleavage system H protein |
| 372 | CABO-LLG-01-000594 | CTRA-DUW-01-000288 | SIMILAR-TO Lipoate-protein ligase A [EC] 6.3.4.- |
| 373 | CABO-LLG-01-000596 | CTRA-DUW-01-000290 | ENZYME tRNA (5-methylaminomethyl-2-thiouridylate)-methyltransferase |
| 374 | CABO-LLG-01-000601 | CTRA-DUW-01-000293 | ENZYME Nitrogen regulatory IIA protein A component [EC] 2.7.1.69 |
| 375 | WCHO-WSU-01-000243 | CTRA-DUW-01-000294 | ENZYME Nitrogen regulatory IIA protein A component [EC] 2.7.1.69 |
| 376 | CABO-LLG-01-000603 | CTRA-DUW-01-000295 | ENZYME dUTP pyrophosphatase [EC] 3.6.1.23 |
| 377 | CABO-LLG-01-000608 | CTRA-DUW-01-000300 | ENZYME Ribonuclease III [EC] 3.1.26.3 |
| 378 | CABO-LLG-01-000609 | CTRA-DLC-01-000581 | FUNCTION DNA repair protein RadA |
| 379 | CABO-LLG-01-000610 | CTRA-DUW-01-000302 | ENZYME Porphobilinogen deaminase [EC] 4.3.1.8 |
| 380 | CABO-LLG-01-000616 | CTRA-DUW-01-000340 | NA |
| 381 | CABO-LLG-01-000623 | CTRA-DUW-01-000346 | DOMAIN DnaJ |
| 382 | CABO-LLG-01-000624 | CTRA-DLC-01-000536 | FUNCTION Ribosomal protein S21 |
| 383 | CABO-LLG-01-000628 | CTRA-DUW-01-000351 | ENZYME Aryl-sulfate sulphohydrolase [EC] 3.1.6.1 |
| 384 | CABO-LLG-01-000631 | CTRA-DUW-01-000354 | FUNCTION Septum formation protein Maf homolog |
| 385 | CABO-LLG-01-000632 | CTRA-DUW-01-000355 | NA |
| 386 | WCHO-WSU-01-000567 | CTRA-DUW-01-000392 | NA |
| 387 | CABO-LLG-01-000633 | CTRA-DUW-01-000356 | NA |
| 388 | CABO-LLG-01-000636 | CTRA-DUW-01-000333 | ENZYME Triosephosphate isomerase [EC] 5.3.1.1 |
| 389 | CABO-LLG-01-000637 | CTRA-DUW-01-000334 | ENZYME Exonuclease VII large subunit [EC] 3.1.11.6 |
| 390 | CABO-LLG-01-000641 | CTRA-DUW-01-000360 | ENZYME Dimethyladenosine transferase [EC] 2.1.1.- |
| 391 | CABO-LLG-01-000642 | CTRA-DUW-01-000361 | NA |
| 392 | CABO-LLG-01-000643 | CTRA-DUW-01-000362 | DOMAIN Thioredoxin |
| 393 | CABO-LLG-01-000646 | CTRA-DLC-01-000868 | NA |
| 394 | CABO-LLG-01-000647 | CTRA-DLC-01-000869 | ENZYME Ribonuclease HII [EC] 3.1.26.4 |
| 395 | CABO-LLG-01-000651 | CTRA-DUW-01-000004 | ENZYME glutamyl-tRNA (Gln) amidotransferase, subunit B [EC] |
| 396 | CABO-LLG-01-000670 | CTRA-DUW-01-000386 | NA |
| 397 | CABO-LLG-01-000671 | CTRA-DUW-01-000387 | SIMILAR-TO metallo-beta-lactamase [EC] 3.5.-.- |
| 398 | CABO-LLG-01-000684 | CTRA-DLC-01-000488 | NA |
| 399 | CABO-LLG-01-000686 | CTRA-DUW-01-000396 | NA |
| 400 | CABO-LLG-01-000690 | CTRA-DLC-01-000484 | FUNCTION Heat-inducible transcription repressor HrcA |
| 401 | CABO-LLG-01-000691 | CTRA-DUW-01-000403 | FUNCTION GrpE protein |
| 402 | CABO-LLG-01-000698 | CTRA-DUW-01-000432 | NA |
| 403 | CABO-LLG-01-000701 | CTRA-DUW-01-000435 | NA |
| 404 | CABO-LLG-01-000705 | CTRA-DUW-01-000438 | ENZYME ubiquinone/menaquinone biosynthesis methlytransferase |
| 405 | CABO-LLG-01-000706 | CTRA-DLC-01-000449 | NA |
| 406 | CABO-LLG-01-000707 | CTRA-DUW-01-000440 | ENZYME Diaminopimelate epimerase [EC] 5.1.1.7 |
| 407 | CABO-LLG-01-000709 | CTRA-DLC-01-000446 | ENZYME Serine hydroxymethyltransferase [EC] 2.1.2.1 |
| 408 | CABO-LLG-01-000713 | CTRA-DUW-01-000406 | NA |
| 409 | CABO-LLG-01-000714 | CTRA-DLC-01-000479 | NA |
| 410 | CABO-LLG-01-000717 | CTRA-DUW-01-000410 | ENZYME Lipid A 4'-kinase [EC] 2.7.1.130 |
| 411 | CABO-LLG-01-000722 | CTRA-DLC-01-000471 | FUNCTION DnaK suppressor protein |
| 412 | CABO-LLG-01-000723 | CTRA-DUW-01-000416 | ENZYME Lipoprotein signal peptidase [EC] 3.4.23.36 |
| 413 | CABO-LLG-01-000735 | CTRA-DUW-01-000427 | FUNCTION Ribosomal protein L27 |
| 414 | CABO-LLG-01-000736 | CTRA-DLC-01-000459 | FUNCTION Ribosomal protein L21 |
| 415 | CABO-LLG-01-000738 | CTRA-DUW-01-000444 | NA |
| 416 | CABO-LLG-01-000739 | CTRA-DUW-01-000445 | ENZYME Sulfite reductase (NADPH) flavoprotein alpha-component |
| 417 | CABO-LLG-01-000740 | CTRA-DLC-01-000442 | FUNCTION Ribosomal protein S10 |
| 418 | CABO-LLG-01-000751 | CTRA-DUW-01-000456 | ENZYME Glutamyl-tRNA Synthetase [EC] 6.1.1.17 |
| 419 | CABO-LLG-01-000752 | CTRA-DLC-01-000431 | NA |
| 420 * | CABO-LLG-01-000753 | NA | |
| 421 | CABO-LLG-01-000754 | CTRA-DUW-01-000458 | ENZYME Single-stranded-DNA-specific exonuclease RecJ [EC] |
| 422 | CABO-LLG-01-000759 | CTRA-DUW-01-000463 | ENZYME Cytidylate kinase [EC] 2.7.4.14 |
| 423 | CABO-LLG-01-000761 | CTRA-DUW-01-000465 | ENZYME Arginyl-tRNA Synthetase [EC] 6.1.1.19 |
| 424 | CABO-LLG-01-000762 | CTRA-DUW-01-000466 | ENZYME UDP-N-acetylglucosamine 1-carboxyvinyltransferase [EC] |
| 425 | CABO-LLG-01-000764 | CTRA-DUW-01-000468 | NA |
| 426 | CABO-LLG-01-000778 | CTRA-DUW-01-000480 | NA |
| 427 | CABO-LLG-01-000779 | CTRA-DUW-01-000481 | NA |
| 428 | CABO-LLG-01-000784 | CTRA-DUW-01-000486 | ENZYME Phenylalanyl-tRNA Synthetase beta chain [EC] 6.1.1.20 |
| 429 * | CABO-LLG-01-000789 | CTRA-DUW-01-000491 | FUNCTION Dipeptide binding protein DppA |
| 430 | CABO-LLG-01-000792 | CTRA-DUW-01-000496 | NA |
| 431 | CABO-LLG-01-000793 | CTRA-DUW-01-000497 | ENZYME Protoheme ferro-lyase [EC] 4.99.1.1 |
| 432 | CABO-LLG-01-000794 | CTRA-DUW-01-000498 | FUNCTION Aminoacid-binding periplasmic protein precursor |
| 433 | CABO-LLG-01-000795 | CTRA-DUW-01-000499 | ENZYME HemK modification methylase homolog [EC] -.-.-.- |
| 434 | CABO-LLG-01-000796 | CTRA-DUW-01-000500 | NA |
| 435 | CABO-LLG-01-000801 | CTRA-DLC-01-000386 | DOMAIN ATP-binding |
| 436 | CABO-LLG-01-000802 | CTRA-DUW-01-000505 | ENZYME DNA polymerase I [EC] 2.7.7.7 |
| 437 | CABO-LLG-01-000803 | CTRA-DLC-01-000384 | NA |
| 438 | CABO-LLG-01-000805 | CTRA-DUW-01-000508 | ENZYME CDP-diacylglycerol-glycerol-3-phosphate |
| 439 | CABO-LLG-01-000807 | CTRA-DUW-01-000511 | FUNCTION Glucose inhibited division protein A GidA |
| 440 | CABO-LLG-01-000808 | CTRA-DUW-01-000512 | ENZYME Lipoate-protein ligase A [EC] 6.3.4.- |
| 441 | CABO-LLG-01-000810 | CTRA-DUW-01-000514 | ENZYME Holliday Junction DNA Helicase RuvA [EC] -.-.-.- |
| 442 | CABO-LLG-01-000811 | CTRA-DUW-01-000515 | ENZYME Holliday Junction DNA Helicase RuvC [EC] 3.1.22.4 |
| 443 | CABO-LLG-01-000813 | CTRA-DUW-01-000517 | NA |
| 444 | CABO-LLG-01-000814 | CTRA-DUW-01-000518 | ENZYME Glyceraldehyde 3-phosphate dehydrogenase [EC] 1.2.1.12 |
| 445 | CABO-LLG-01-000820 | CTRA-DUW-01-000524 | FUNCTION Ribosomal protein L15 |
| 446 | CABO-LLG-01-000821 | CTRA-DUW-01-000525 | FUNCTION Ribosomal protein S5 |
| 447 | CABO-LLG-01-000822 | CTRA-DUW-01-000526 | FUNCTION Ribosomal protein L18 |
| 448 | CABO-LLG-01-000824 | CTRA-DUW-01-000528 | FUNCTION Ribosomal protein S8 |
| 449 | CABO-LLG-01-000825 | CTRA-DUW-01-000529 | FUNCTION Ribosomal protein L5 |
| 450 | CABO-LLG-01-000826 | CTRA-DUW-01-000530 | FUNCTION Ribosomal protein L24 |
| 451 | CABO-LLG-01-000827 | CTRA-DUW-01-000531 | FUNCTION Ribosomal protein L14 |
| 452 | CABO-LLG-01-000828 | CTRA-DUW-01-000532 | FUNCTION Ribosomal protein S17 |
| 453 | CABO-LLG-01-000830 | CTRA-DUW-01-000534 | FUNCTION Ribosomal protein L16 |
| 454 | CABO-LLG-01-000831 | CTRA-DUW-01-000535 | FUNCTION Ribosomal protein S3 |
| 455 | CABO-LLG-01-000833 | CTRA-DUW-01-000537 | FUNCTION Ribosomal protein S19 |
| 456 | CABO-LLG-01-000834 | CTRA-DUW-01-000538 | FUNCTION Ribosomal protein L2 |
| 457 | CABO-LLG-01-000835 | CTRA-DUW-01-000539 | FUNCTION Ribosomal protein L23 |
| 458 | CABO-LLG-01-000836 | CTRA-DLC-01-000351 | FUNCTION Ribosomal protein L4 |
| 459 | CABO-LLG-01-000837 | CTRA-DUW-01-000541 | FUNCTION Ribosomal protein L3 |
| 460 * | CABO-LLG-01-000839 | CTRA-DUW-01-000543 | ENZYME Methionyl-tRNA formyltransferase [EC] 2.1.2.9 |
| 461 | CABO-LLG-01-000841 | CTRA-DUW-01-000545 | ENZYME (3R)-hydroxymyristoyl-[acyl carrier protein] dehydratase |
| 462 | CABO-LLG-01-000842 | CTRA-DLC-01-000345 | ENZYME UDP-3-O-[3-hydroxymyristoyl] N-acetylglucosamine |
| 463 | CABO-LLG-01-000843 | CTRA-DUW-01-000547 | ENZYME apolipoprotein N-acyltransferase [EC] 2.3.1. |
| 464 | CABO-LLG-01-000846 | CTRA-DUW-01-000550 | DOMAIN ATP-binding |
| 465 | CABO-LLG-01-000847 | CTRA-DUW-01-000551 | NA |
| 466 | CABO-LLG-01-000849 | CTRA-DUW-01-000553 | ENZYME rRNA methyltransferase SpoU homolog [EC] -.-.-.- |
| 467 | CABO-LLG-01-000852 | CTRA-DUW-01-000556 | ENZYME Histidyl-tRNA Synthetase [EC] 6.1.1.21 |
| 468 | CABO-LLG-01-000855 | CTRA-DUW-01-000558 | ENZYME DNA polymerase III alpha chain [EC] 2.7.7.7 |
| 469 | CABO-LLG-01-000856 | CTRA-DUW-01-000559 | NA |
| 470 | CABO-LLG-01-000857 | CTRA-DLC-01-000331 | NA |
| 471 | CABO-LLG-01-000858 | CTRA-DUW-01-000561 | NA |
| 472 | CABO-LLG-01-000860 | CTRA-DLC-01-000327 | ENZYME D-alanyl-D-alanine carboxypeptidase DacF [EC] 3.4.16.4 |
| 473 | CABO-LLG-01-000865 | CTRA-DUW-01-000710 | ENZYME Phosphoglycerate kinase [EC] 2.7.2.3 |
| 474 | CABO-LLG-01-000867 | CTRA-DUW-01-000708 | FUNCTION Phosphate transport system protein PhoU |
| 475 | CABO-LLG-01-000874 | CTRA-DUW-01-000703 | FUNCTION ABC transporter, ATP-binding protein |
| 476 | SNEG-ZXX-01-002117 | CTRA-DUW-01-000701 | FUNCTION ABC transporter, ATP-binding protein |
| 477 | CABO-LLG-01-000880 | CTRA-DUW-01-000697 | FUNCTION Ribosomal protein S2 |
| 478 | CABO-LLG-01-000881 | CTRA-DLC-01-000198 | FUNCTION Translation elongation factor EF-TS |
| 479 | CABO-LLG-01-000882 | CTRA-DUW-01-000695 | ENZYME Uridylate kinase [EC] 2.7.4. |
| 480 | CABO-LLG-01-000892 | CTRA-DUW-01-000684 | NA |
| 481 | CABO-LLG-01-000895 | CTRA-DUW-01-000681 | DOMAIN FHA |
| 482 | CABO-LLG-01-000897 | CTRA-DUW-01-000679 | ENZYME glutamyl-tRNA reductase [EC] 1.2.1. |
| 483 | CABO-LLG-01-000904 | CTRA-DUW-01-000672 | ENZYME KDO-8-phosphate synthetase [EC] 4.1.2.16 |
| 484 | CABO-LLG-01-000912 | CTRA-DLC-01-000254 | NA |
| 485 | CABO-LLG-01-000913 | CTRA-DUW-01-000640 | SIMILAR-TO Endonuclease IV [EC] 3.1.21.2 |
| 486 | CABO-LLG-01-000914 | CTRA-DUW-01-000641 | FUNCTION Ribosomal protein S4 |
| 487 | CABO-LLG-01-000916 | CTRA-DUW-01-000657 | FUNCTION Multidrug-efflux transporter |
| 488 | CABO-LLG-01-000917 | CTRA-DLC-01-000238 | ENZYME Exodeoxyribonuclease V gamma subunit [EC] 3.1.11.5 |
| 489 | CABO-LLG-01-000920 | CTRA-DUW-01-000652 | SIMILAR-TO Amino-acid aminotransferase class I [EC] 2.6.1. |
| 490 | CABO-LLG-01-000921 | CTRA-DUW-01-000651 | FUNCTION Transcription Elongation Factor GreA C-terminus |
| 491 | CABO-LLG-01-000923 | CTRA-DUW-01-000649 | NA |
| 492 | CABO-LLG-01-000924 | CTRA-DLC-01-000245 | ENZYME Porphobilinogen synthase [EC] 4.2.1.24 |
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Psomopoulos, F.E.; Siarkou, V.I.; Papanikolaou, N.; Iliopoulos, I.; Tsaftaris, A.S.; Promponas, V.J.; Ouzounis, C.A. The Chlamydiales Pangenome Revisited: Structural Stability and Functional Coherence. Genes 2012, 3, 291-319. https://doi.org/10.3390/genes3020291
Psomopoulos FE, Siarkou VI, Papanikolaou N, Iliopoulos I, Tsaftaris AS, Promponas VJ, Ouzounis CA. The Chlamydiales Pangenome Revisited: Structural Stability and Functional Coherence. Genes. 2012; 3(2):291-319. https://doi.org/10.3390/genes3020291
Chicago/Turabian StylePsomopoulos, Fotis E., Victoria I. Siarkou, Nikolas Papanikolaou, Ioannis Iliopoulos, Athanasios S. Tsaftaris, Vasilis J. Promponas, and Christos A. Ouzounis. 2012. "The Chlamydiales Pangenome Revisited: Structural Stability and Functional Coherence" Genes 3, no. 2: 291-319. https://doi.org/10.3390/genes3020291