
2′-O-Methyl RNA/Ethylene-Bridged Nucleic Acid Chimera Antisense Oligonucleotides to Induce Dystrophin Exon 45 Skipping

 ,
,
Abstract
:1. Introduction
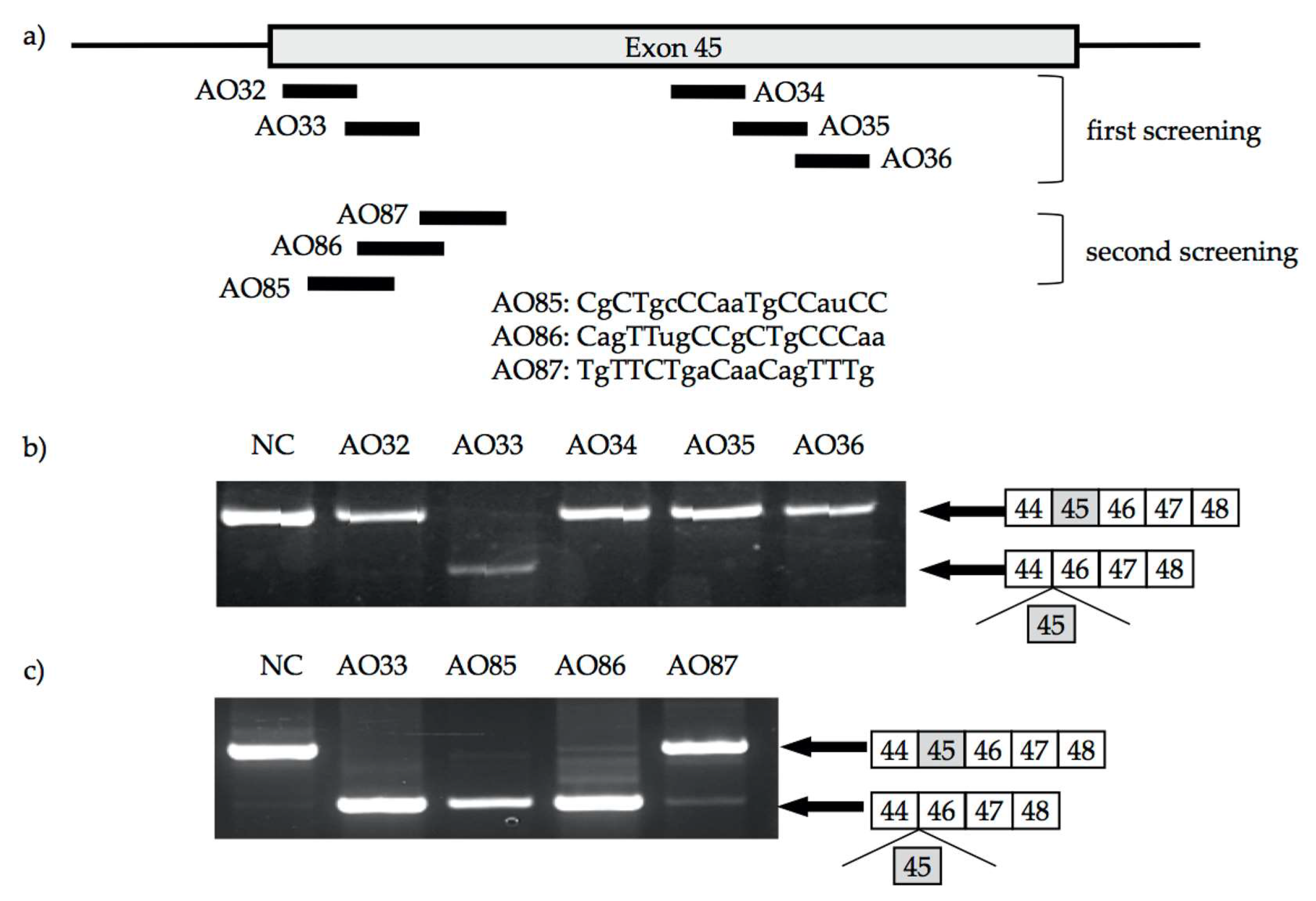
2. Identification of AO for Exon 45 Skipping
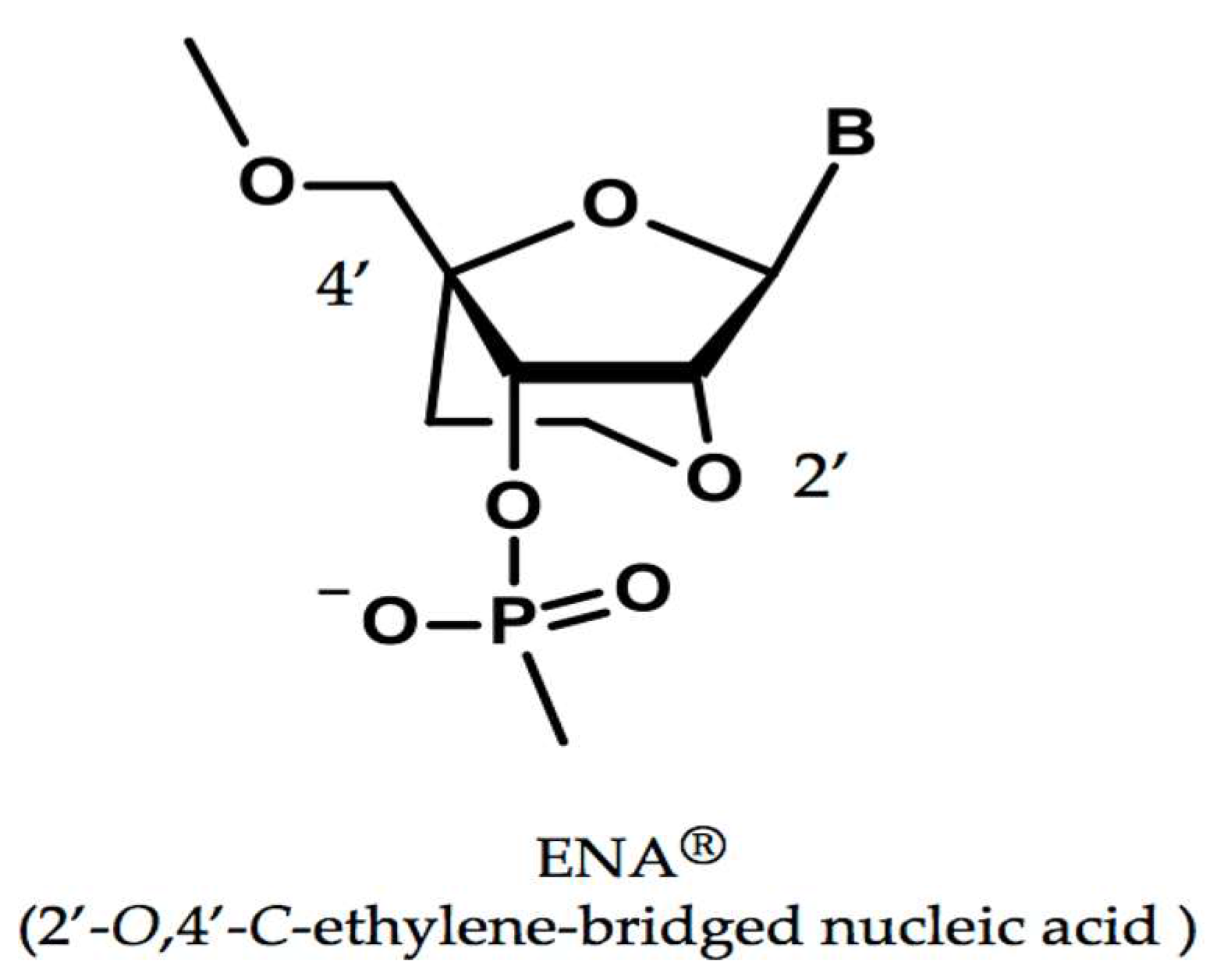
2.1. A Modified Nucleic Acid of ENA®
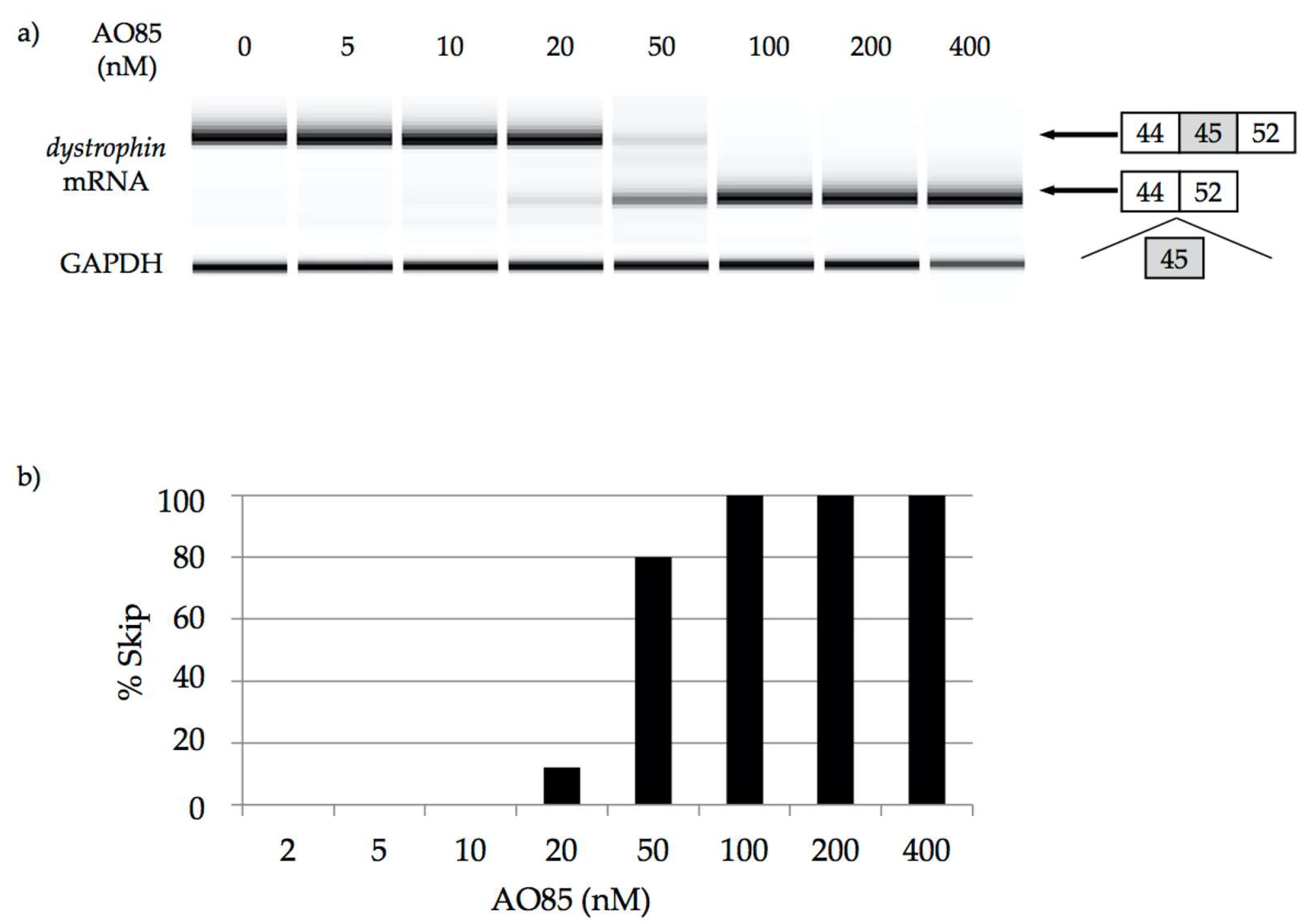
2.2. 2′-OMeRNA/ENA Chimera AO to Skip Exon 45
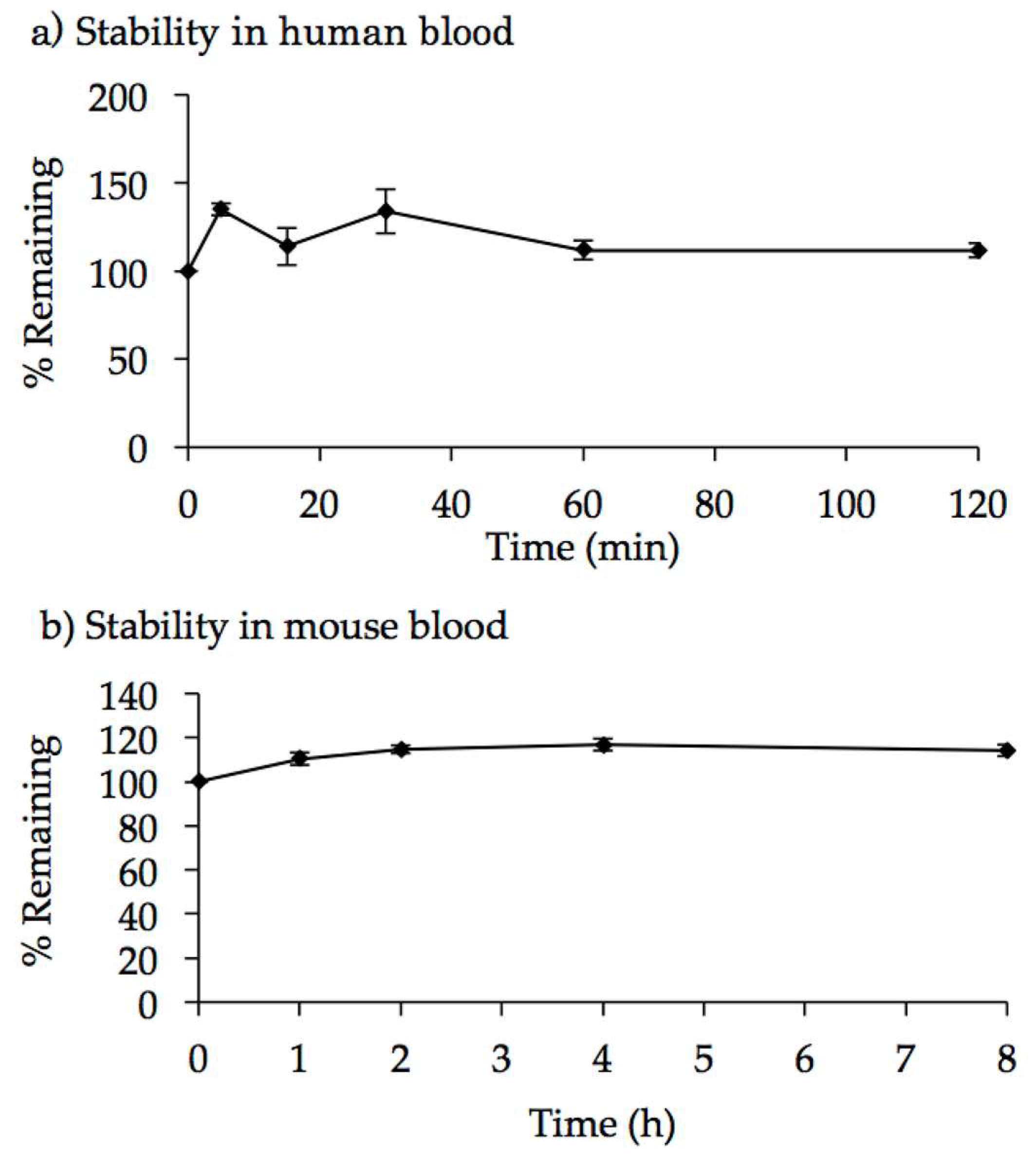
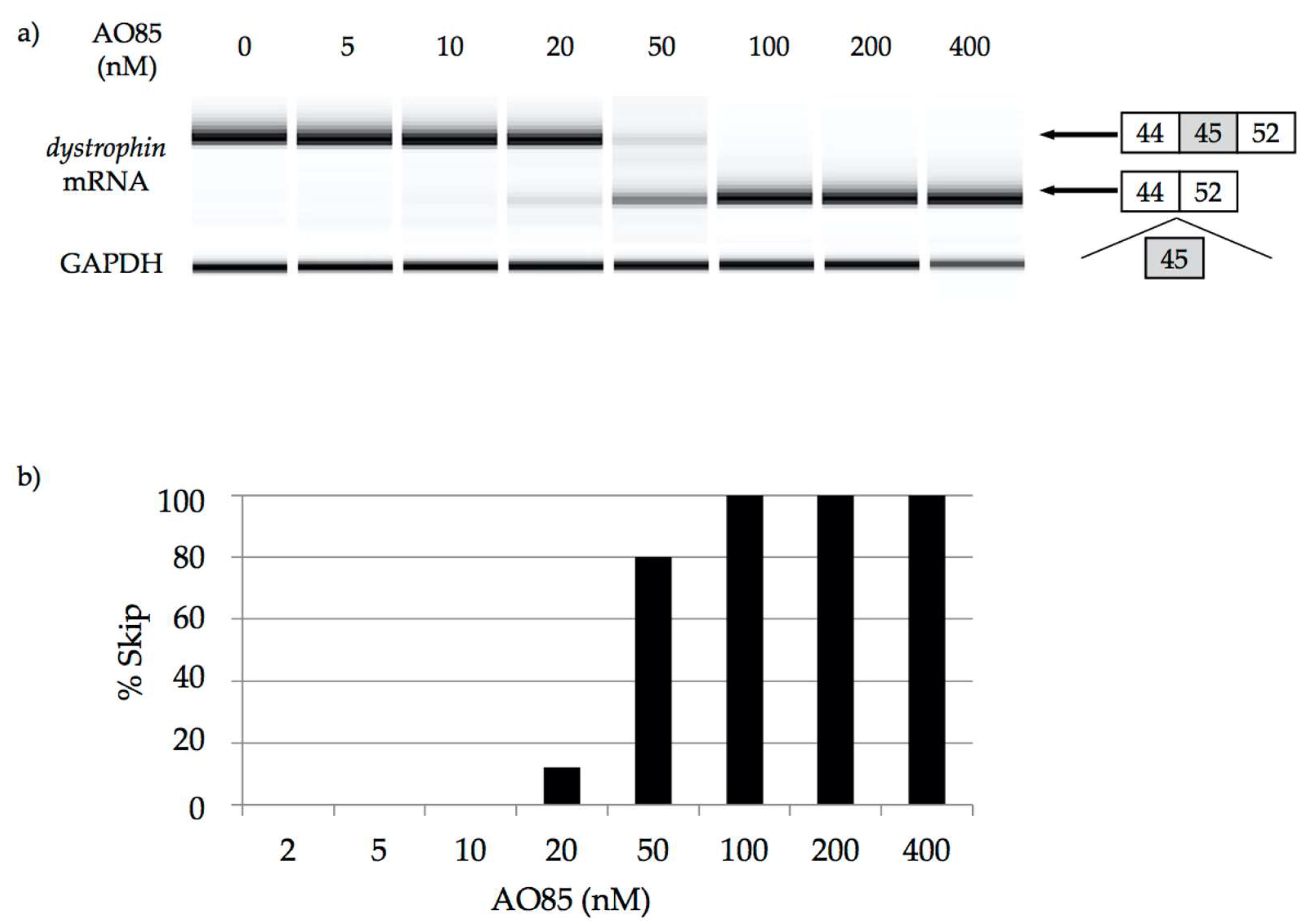
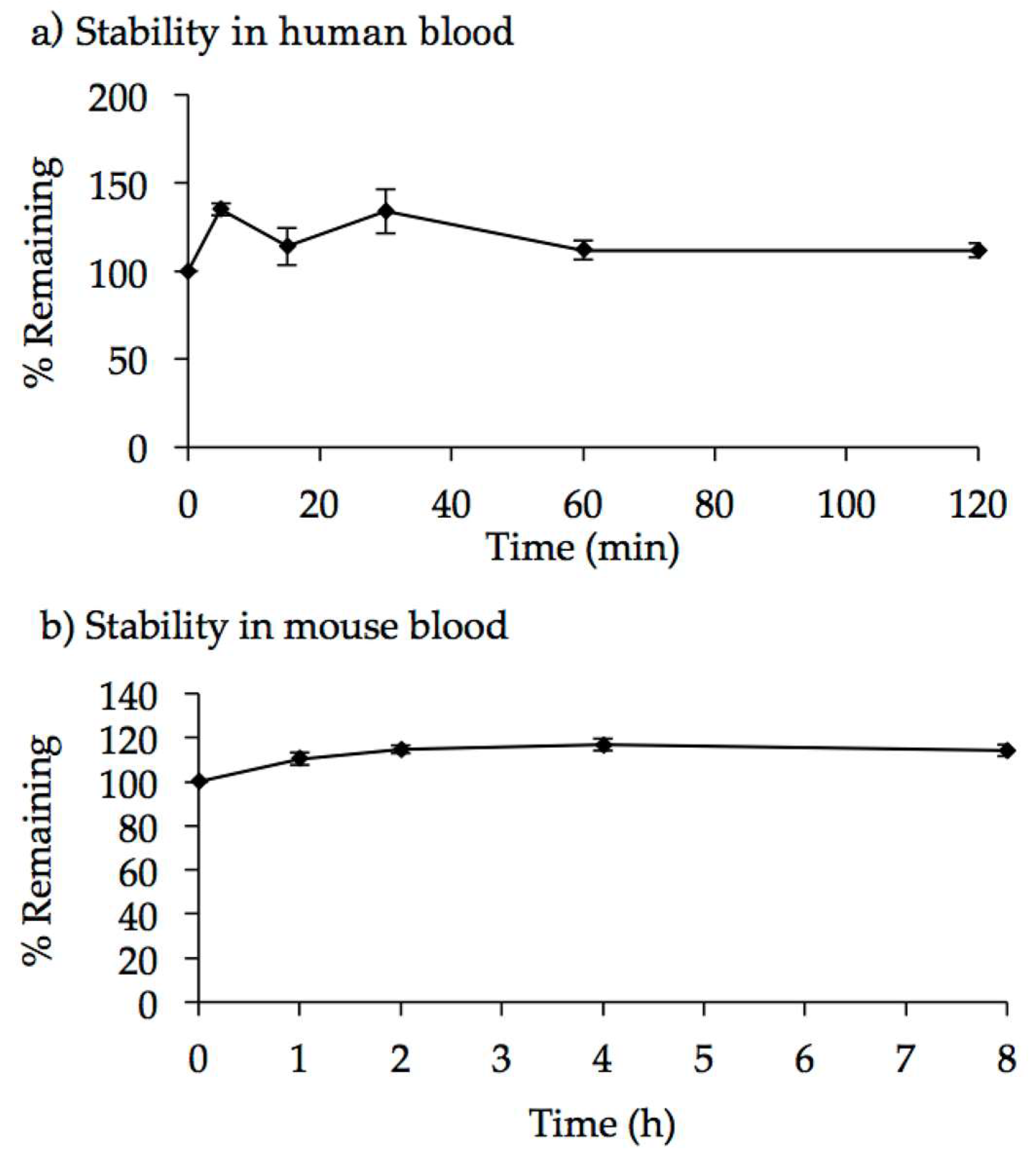
2.3. Characterization of AO85
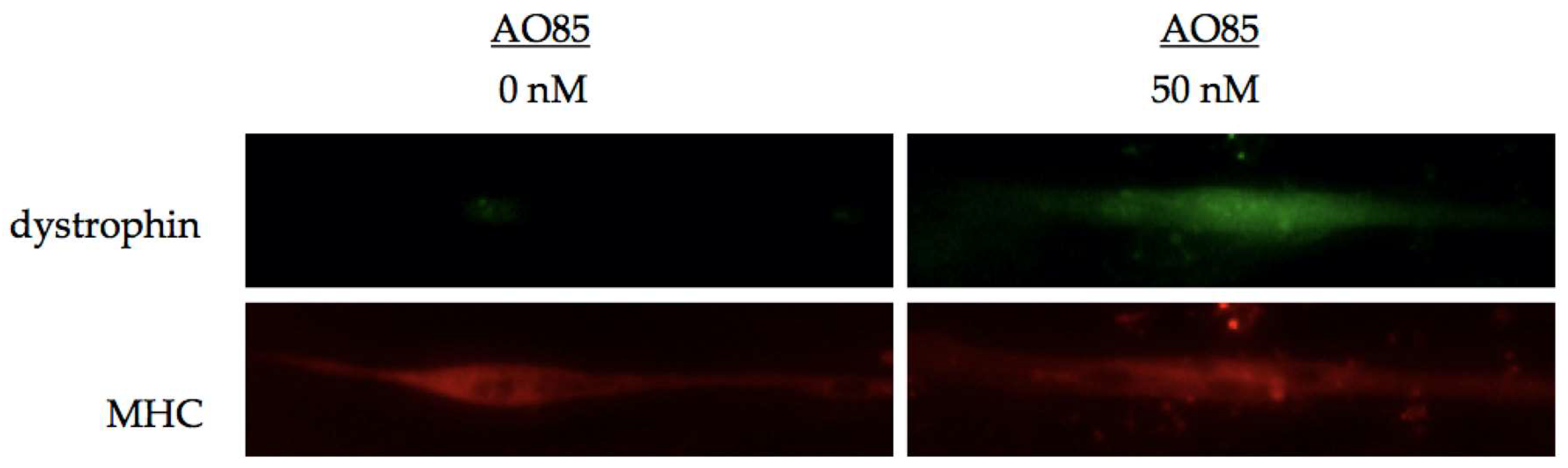
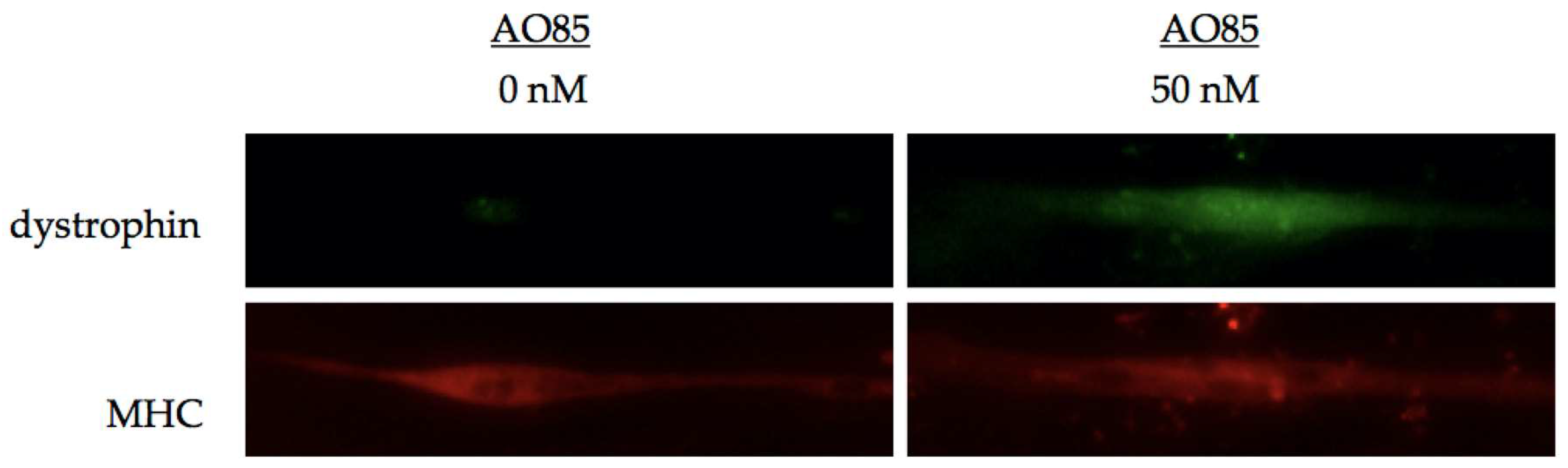
3. Efficacy of AO85 in Myotubes from DMD Patients
3.1. AO85 Efficacy in Myotubes from a DMD Patient with a Deletion Mutation of Exon 46–51
3.2. AO85 Efficacy in Myotubes from 13 DMD Patients
4. Discussion
5. Conclusions
Acknowledgements
Conflicts of Interest
References
- Eagle, M.; Baudouin, S.V.; Chandler, C.; Giddings, D.R.; Bullock, R.; Bushby, K. Survival in Duchenne muscular dystrophy: Improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord. 2002, 12, 926–929. [Google Scholar] [CrossRef]
- Passamano, L.; Taglia, A.; Palladino, A.; Viggiano, E.; D′Ambrosio, P.; Scutifero, M.; Rosaria Cecio, M.; Torre, V.; DE Luca, F.; Picillo, E.; et al. Improvement of survival in Duchenne muscular dystrophy: Retrospective analysis of 835 patients. Acta Myol. 2012, 31, 121–125. [Google Scholar] [PubMed]
- Van Deutekom, J.C.; van Ommen, G.J. Advances in Duchenne muscular dystrophy gene therapy. Nat. Rev. Genet. 2003, 4, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Robinson-Hamm, J.N.; Gersbach, C.A. Gene therapies that restore dystrophin expression for the treatment of Duchenne muscular dystrophy. Hum. Genet. 2016, 135, 1029–1040. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, Y.; Nishio, H.; Sakamoto, H.; Nakamura, H.; Matsuo, M. Modulation of in vitro splicing of the upstream intron by modifying an intra-exon sequence which is deleted from the dystrophin gene in dystrophin kobe. J. Clin. Investig. 1995, 95, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Matsuo, M. Duchenne/becker muscular dystrophy: From molecular diagnosis to gene therapy. Brain Dev. 1996, 18, 167–172. [Google Scholar] [CrossRef]
- Takeshima, Y.; Yagi, M.; Wada, H.; Ishibashi, K.; Nishiyama, A.; Kakumoto, M.; Sakaeda, T.; Saura, R.; Okumura, K.; Matsuo, M. Intravenous infusion of an antisense oligonucleotide results in exon skipping in muscle dystrophin mRNA of Duchenne muscular dystrophy. Pediatr. Res. 2006, 59, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, M.; Takeshima, Y.; Nishio, H. Contributions of Japanese patients to development of antisense therapy for DMD. Brain Dev. 2016, 38, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Wilton, S.D.; Veedu, R.N.; Fletcher, S. The emperor′s new dystrophin: Finding sense in the noise. Trends Mol. Med. 2015, 21, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Le, B.T.; Veedu, R.N.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide development for the treatment of muscular dystrophies. Expert Opin. Orph. Drugs 2016, 4, 139–152. [Google Scholar]
- Chen, S.; Le, B.T.; Rahimizadeh, K.; Shaikh, K.; Mohal, N.; Veedu, R.N. Synthesis of a morpholino nucleic acid(MNA)-uridine phosphoramidite, and exon skipping using MNA/20-O-Methyl mixmer antisense oligonucleotide. Molecules 2016. [Google Scholar] [CrossRef] [PubMed]
- Le, B.T.; Chen, S.; Abramov, M.; Herdewijn, P.; Veedu, R.N. Evaluation of anhydrohexitol nucleic acid, cyclohexenyl nucleic acid and d-altritol nucleic acid-modified 2′-O-methyl RNA mixmer antisense oligonucleotides for exon skipping in vitro. Chem. Commun. 2016, 52, 13467–13470. [Google Scholar] [CrossRef] [PubMed]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P.; et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918–928. [Google Scholar] [CrossRef]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic administration of PRO051 in Duchenne′s muscular dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Voit, T.; Topaloglu, H.; Straub, V.; Muntoni, F.; Deconinck, N.; Campion, G.; De Kimpe, S.J.; Eagle, M.; Guglieri, M.; Hood, S.; et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): An exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014, 13, 987–996. [Google Scholar] [CrossRef]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Goemans, N.M.; Tulinius, M.; van den Hauwe, M.; Kroksmark, A.K.; Buyse, G.; Wilson, R.J.; van Deutekom, J.C.; de Kimpe, S.J.; Lourbakos, A.; Campion, G. Long-term efficacy, safety, and pharmacokinetics of drisapersen in Duchenne muscular dystrophy: Results from an open-label extension study. PLoS ONE 2016, 11, e0161955. [Google Scholar]
- Aartsma-Rus, A.; Ferlini, A.; Goemans, N.; Pasmooij, A.M.; Wells, D.J.; Bushby, K.; Vroom, E.; Balabanov, P. Translational and regulatory challenges for exon skipping therapies. Hum. Gene Ther. 2014, 25, 885–892. [Google Scholar] [CrossRef] [PubMed]
- FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. Avaliable online: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm521263.htm (accessed on 19 September 2016).
- Aartsma-Rus, A.; Krieg, A.M. FDA approves eteplirsen for Duchenne muscular dystrophy: The next chapter in the eteplirsen saga. Nucleic Acid Ther. 2016. [Google Scholar] [CrossRef] [PubMed]
- Unger, E.F.; Califf, R.M. Regarding eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2016, 81, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R. Eteplirsen improves function and partially restores dystrophin. Ann. Neurol. 2016. [Google Scholar] [CrossRef]
- Stein, C.A. Eteplirsen approved for Duchenne muscular dystrophy: The FDA faces a difficult choice. Mol. Ther. 2016, 24, 1884–1885. [Google Scholar] [CrossRef] [PubMed]
- Toh, Z.Y.; Thandar Aung-Htut, M.; Pinniger, G.; Adams, A.M.; Krishnaswarmy, S.; Wong, B.L.; Fletcher, S.; Wilton, S.D. Deletion of dystrophin in-frame exon 5 leads to a severe phenotype: Guidance for exon skipping strategies. PLoS ONE 2016, 11, e0145620. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, M. ENA oligonucleotides as therapeutics. Curr. Opin. Mol. Ther. 2006, 8, 144–149. [Google Scholar] [PubMed]
- Morita, K.; Hasegawa, C.; Kaneko, M.; Tsutsumi, S.; Sone, J.; Ishikawa, T.; Imanishi, T.; Koizumi, M. 2′-O,4′-C-ethylene-bridged nucleic acids (ENA) with nuclease-resistance and high affinity for RNA. Nucleic Acids Res. Suppl. 2001, 241–242. [Google Scholar] [CrossRef]
- Morita, K.; Hasegawa, C.; Kaneko, M.; Tsutsumi, S.; Sone, J.; Ishikawa, T.; Imanishi, T.; Koizumi, M. 2′-O,4′-C-ethylene-bridged nucleic acids (ENA): Highly nuclease-resistant and thermodynamically stable oligonucleotides for antisense drug. Bioorg. Med. Chem. Lett. 2002, 12, 73–76. [Google Scholar] [CrossRef]
- Morita, K.; Takagi, M.; Hasegawa, C.; Kaneko, M.; Tsutsumi, S.; Sone, J.; Ishikawa, T.; Imanishi, T.; Koizumi, M. Synthesis and properties of 2′-O,4′-C-ethylene-bridged nucleic acids (ENA) as effective antisense oligonucleotides. Bioorg. Med. Chem. 2003, 11, 2211–2226. [Google Scholar] [CrossRef]
- Koizumi, M. 2′-O,4′-C-ethylene-bridged nucleic acids (ENA) as next-generation antisense and antigene agents. Biol. Pharm. Bull. 2004, 27, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Yagi, M.; Takeshima, Y.; Suruno, A.; Takagi, M.; Koizumi, M.; Matsuo, M. Chimeric RNA and 2′-O,4′-C-ethylene-bridged nucleic acids have stronger activity than phosphorothioate oligodeoxynucleotides in induction of exon-19 skipping in dystropin mRNA. Oligonucleotides 2004, 14, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Surono, A.; Tran, V.K.; Takshima, Y.; Wada, H.; Yagi, M.; Takagi, M.; Koizumi, M.; Matsuo, M. Chimeric RNA/ethylene bridged nucleic acids promote dystrophin expression in myocytes of Duchenne muscular dystrophy by inducing skipping of the nonsense-mutation-encoding exon. Hum. Gene Ther. 2004, 15, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, Y.; Yagi, M.; Okizuka, Y.; Awano, H.; Zhang, Z.; Yamauchi, Y.; Nishio, H.; Matsuo, M. Mutation spectrum of the dystrophin gene in 442 duchenne/becker muscular dystrophy cases from one Japanese referral center. J. Hum. Genet. 2010, 55, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, T.; Yagi, M.; Matsuo, M. Optimizing RNA/ENA chimeric antisense oligonucleotides using in vitro splicing. In Exon Skipping: Methods and Protocols; Aartsma-Rus, A., Ed.; Human Press: New York, NY, USA, 2012; Volume 867, pp. 131–141. [Google Scholar]
- Malueka, R.; Yagi, M.; Awano, H.; Lee, T.; Dwianingsih, E.K.; Nishida, A.; Takeshima, Y.; Matsuo, M. Antisense oligonucleotide induced dystrophin exon 45 skipping at a low half-maximal effective concentration in a cell-free splicing system. Nucleic Acid Ther. 2011, 21, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Malueka, R.G.; Dwianingsih, E.K.; Yagi, M.; Lee, T.; Nishida, A.; Iijima, K.; Takeshima, Y.; Matsuo, M. Phosphorothioate modification of chimeric 2′-O-methyl RNA/ethylene-bridged nucleic acid oligonucleotides increases dystrophin exon 45 skipping capability and reduces cytotoxicity. Kobe J. Med. Sci. 2015, 60, E86–E94. [Google Scholar] [PubMed]
- Takeshima, Y.; Lee, T.; Shimomura, H.; Tanaka, Y.; Awano, H.; Nishida, A.; Ojima, I.; Minami, S.; Nakagawa, A.; Iijima, K.; et al. A new antisense oligonucleotide composed of RNA/ENA chimera (AO85) against dystrophin exon 45 significantly increased six-minute walk distance in Duchenne muscular dystrophy. (Abstract/Program #P08-111). In Presented at the 5th International Congress of Myology, Lyon, France, 14–18 March 2016.
- Dwianingsih, E.K.; Malueka, R.G.; Nishida, A.; Itoh, K.; Lee, T.; Yagi, M.; Iijima, K.; Takeshima, Y.; Matsuo, M. A novel splicing silencer generated by dmd exon 45 deletion junction could explain upstream exon 44 skipping that modifies dystrophinopathy. J. Hum. Genet. 2014, 59, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Findlay, A.R.; Wein, N.; Kaminoh, Y.; Taylor, L.E.; Dunn, D.M.; Mendell, J.R.; King, W.M.; Pestronk, A.; Florence, J.M.; Mathews, K.D.; et al. Clinical phenotypes as predictors of the outcome of skipping around dmd exon 45. Ann. Neurol. 2015, 77, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Kendall, G.C.; Mokhonova, E.I.; Moran, M.; Sejbuk, N.E.; Wang, D.W.; Silva, O.; Wang, R.T.; Martinez, L.; Lu, Q.L.; Damoiseaux, R.; et al. Dantrolene enhances antisense-mediated exon skipping in human and mouse models of Duchenne muscular dystrophy. Sci. Transl. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Arechavala-Gomeza, V.; Anthony, K.; Morgan, J.; Muntoni, F. Antisense oligonucleotide-mediated exon skipping for Duchenne muscular dystrophy: Progress and challenges. Curr. Gene Ther. 2012, 12, 152–160. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AO85 (mg/kg) | Number of Deaths (Case) | General Condition | Pathological Findings (at Day 14) |
|---|---|---|---|
| 0 | 0 | Good | Normal |
| 5 | 0 | Good | Normal |
| 15 | 0 | Good | Normal |
| 50 | 0 | Good | Renal enlargement (4/5 cases) Cyst on renal surface (2/5 cases) |
| AO85 (mg/kg) | Deaths | General Condition | Laboratory Examination | Pathological Findings (at Day 28) |
|---|---|---|---|---|
| 0 | 1 | Good | Normal | Normal |
| 0.5 | 0 | Good | Normal | Normal |
| 5 | 0 | Good | Normal | Normal |
| No. | Age | Deleted Exon | Exon-45-Skipped mRNA (%) | Dystrophin Immunostaining |
|---|---|---|---|---|
| 1 | 5 | 44 | 65 | Positive |
| 2 | 5 | 70 | Positive | |
| 3 | 5 | 46–47 | 43 | Positive |
| 4 | 5 | 30 | Positive | |
| 5 | 10 | 40 | Positive | |
| 6 | 6 | 46–48 | 40 | Positive |
| 7 | 8 | 46–49 | 65 | Positive |
| 8 | 5 | 51 | Positive | |
| 9 | 5 | 46–51 | 100 | Positive |
| 10 | 5 | 100 | Positive | |
| 11 | 6 | 82 | Negative | |
| 12 | 6 | 46–53 | 65 | Positive |
| 13 | 3 | 45 | Negative |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, T.; Awano, H.; Yagi, M.; Matsumoto, M.; Watanabe, N.; Goda, R.; Koizumi, M.; Takeshima, Y.; Matsuo, M. 2′-O-Methyl RNA/Ethylene-Bridged Nucleic Acid Chimera Antisense Oligonucleotides to Induce Dystrophin Exon 45 Skipping. Genes 2017, 8, 67. https://doi.org/10.3390/genes8020067
Lee T, Awano H, Yagi M, Matsumoto M, Watanabe N, Goda R, Koizumi M, Takeshima Y, Matsuo M. 2′-O-Methyl RNA/Ethylene-Bridged Nucleic Acid Chimera Antisense Oligonucleotides to Induce Dystrophin Exon 45 Skipping. Genes. 2017; 8(2):67. https://doi.org/10.3390/genes8020067
Chicago/Turabian StyleLee, Tomoko, Hiroyuki Awano, Mariko Yagi, Masaaki Matsumoto, Nobuaki Watanabe, Ryoya Goda, Makoto Koizumi, Yasuhiro Takeshima, and Masafumi Matsuo. 2017. "2′-O-Methyl RNA/Ethylene-Bridged Nucleic Acid Chimera Antisense Oligonucleotides to Induce Dystrophin Exon 45 Skipping" Genes 8, no. 2: 67. https://doi.org/10.3390/genes8020067