Recent Advances in Atmospheric Chemistry of Mercury



1
Department of Chemistry, Auburn University at Montgomery, 7400 East Dr., Montgomery, AL 36117, USA
2
Department of Chemistry & Department of Atmospheric and Oceanic Sciences, McGill University, 801 Sherbrooke St. W., Montreal, QC H3A 2K6, Canada
*
Authors to whom correspondence should be addressed.
Atmosphere 2018, 9(2), 76; https://doi.org/10.3390/atmos9020076
Submission received: 20 January 2018
/
Revised: 13 February 2018
/
Accepted: 15 February 2018
/
Published: 21 February 2018
(This article belongs to the Special Issue Atmospheric Metal Pollution)
Abstract
:Mercury is one of the most toxic metals and has global importance due to the biomagnification and bioaccumulation of organomercury via the aquatic food web. The physical and chemical transformations of various mercury species in the atmosphere strongly influence their composition, phase, transport characteristics and deposition rate to the ground. Modeling efforts to evaluate the mercury cycling in the environment require an accurate understanding of atmospheric mercury chemistry. We focus this article on recent studies (since 2015) on improving our understanding of the atmospheric chemistry of mercury. We discuss recent advances in (i) determining the dominant atmospheric oxidant of elemental mercury (Hg0); (ii) understanding the oxidation reactions of Hg0 by halogen atoms and by nitrate radical (NO3); (iii) the aqueous reduction of oxidized mercury compounds (HgII); and (iv) the heterogeneous reactions of Hg on atmospherically-relevant surfaces. The need for future research to improve understanding of the fate and transformation of mercury in the atmosphere is also discussed.
1. Introduction
Mercury (Hg) is one of the most toxic metals present globally in the environment [1]. Due to the rather long lifetime of atmospheric mercury, once mercury compounds are released into the atmosphere, they can be transported around the globe. As such, they not only have local impacts but also regional and global implications [2,3].
Hg is a Group IIB transition metal with an atomic number of 80 and a closed shell electronic configuration (5d10 6s2). Elemental mercury (Hg0) is the only liquid metal at room temperature and pressure. Mercury has been widely used in electrochemistry, in optical spectroscopy, in liquid mirror telescopes and also in medicine. However, tragic outbreaks of mercury-induced diseases have occurred in many areas of the world over the years, particularly in Japan and Iraq [4]. Mercury toxicity depends on its chemical species, with methylmercury being highly toxic. Humans and wildlife are exposed to mercury mainly through fish consumption. Exposure to mercury can damage the human nervous system, cause cardiovascular diseases in adults, impede cognitive development in children, as well as affect the reproduction of fish, mammals and birds ([5] and references therein).
Atmospheric deposition has been identified as a significant pathway for Hg transport into the aquatic and soil environment. It has been estimated that 5500–8900 tons of mercury are emitted per year into the atmosphere, with three major natural, anthropogenic and re-emitted sources [2]. Natural sources of Hg emission include volcanoes, soils, forests, natural waters, and the largest anthropogenic source of Hg emission is coal burning in coal fueled power plants [6]. Previously-deposited mercury can be re-emitted to the atmosphere through a series of physical, chemical and biological transformations in the environment [7,8]. Large uncertainties remain in existing emission inventories, particularly for natural and re-emission sources [9].
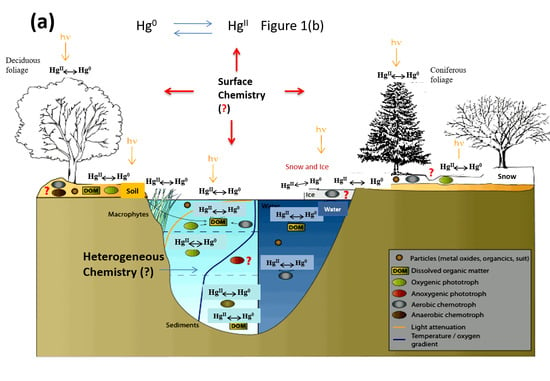
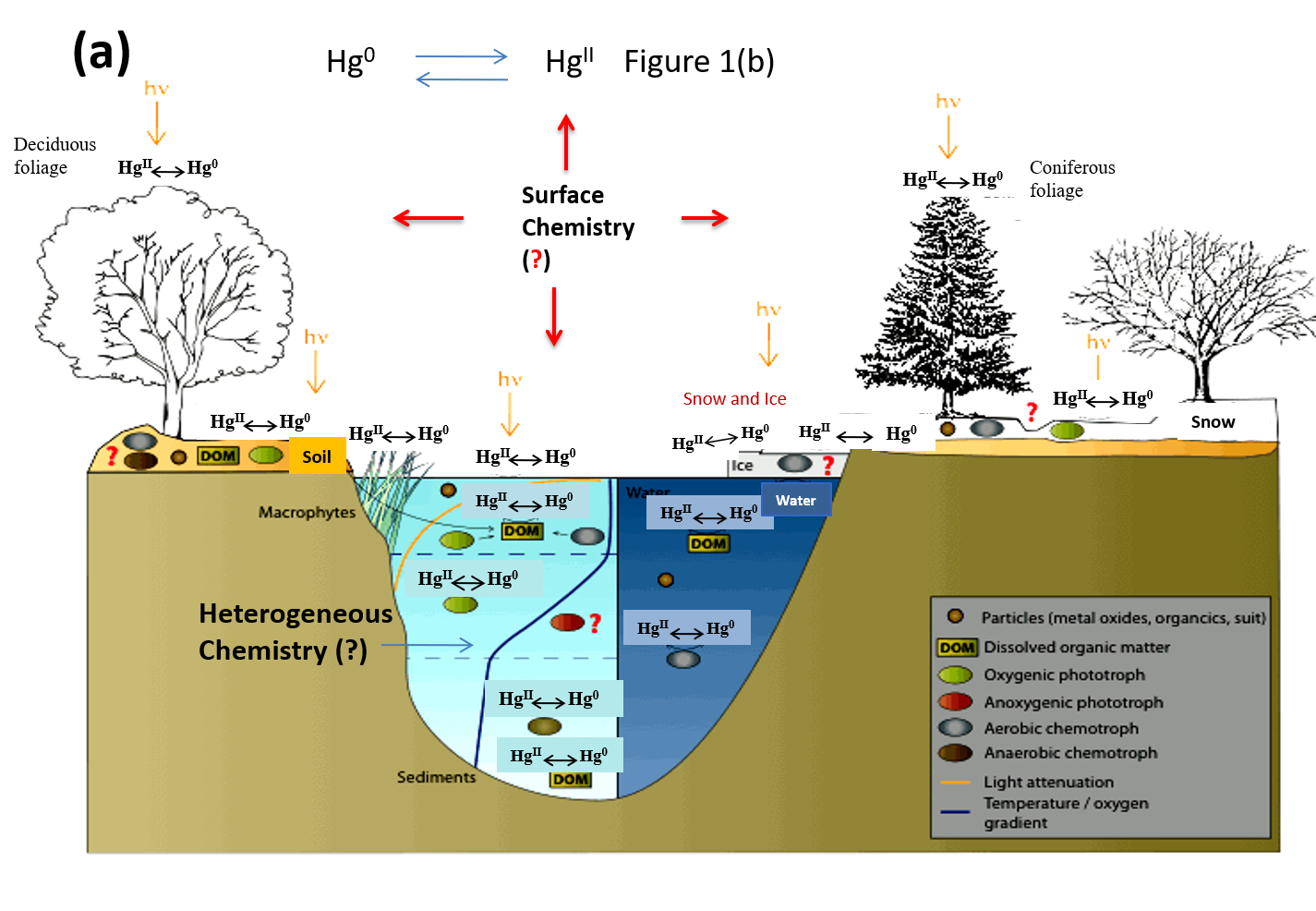
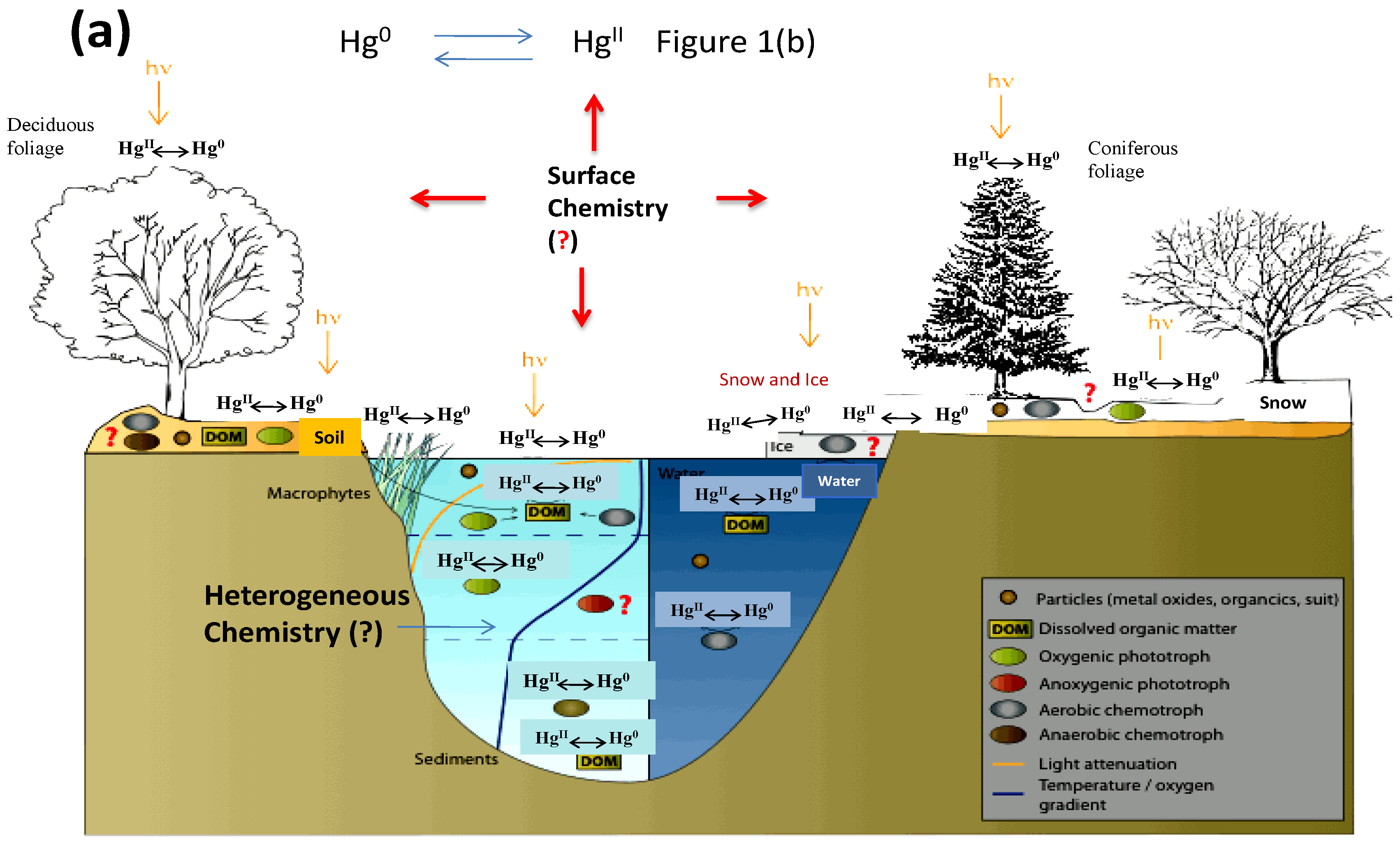
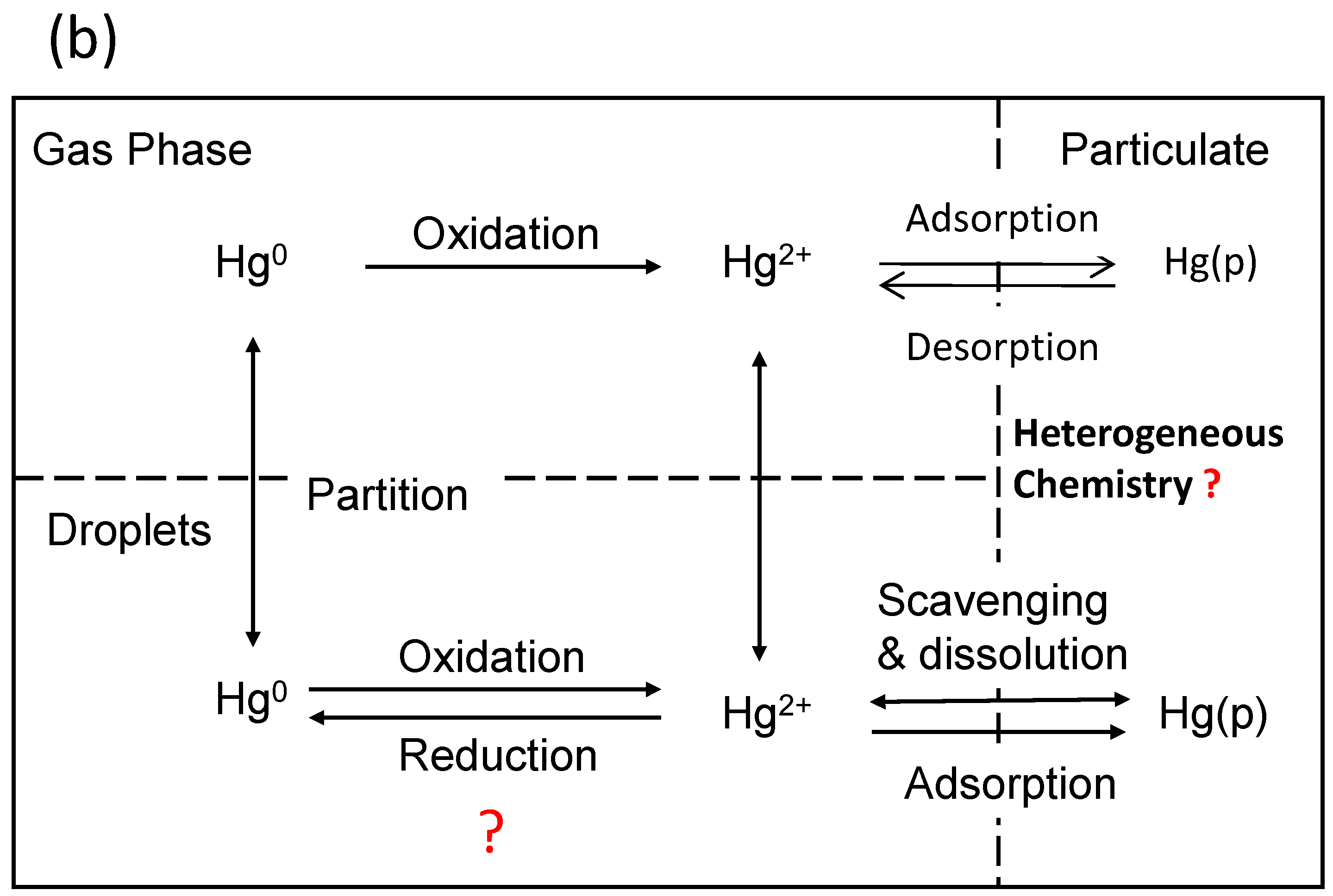
Figure 1a illustrates a simplified picture of mercury transformations in the environment, particularly on various environmental surfaces, and Figure 1b focuses on the atmospheric chemistry of mercury. In the atmospheric environment, mercury presents predominantly in oxidation states of 0 and +2 [10]. Atmospheric mercury exists in the gas phase, in atmospheric droplets, or in the particulate phase. Mercury speciation significantly affects the rates of dry and wet deposition and subsequently, the atmospheric lifetimes of different Hg species [11]. For example, gaseous elemental mercury (Hg0) has been estimated to have an atmospheric lifetime of about one year [4,12], which render sit subject to long-range transportation across the globe. However, atmospheric oxidation of Hg0 to HgBr2 will make mercury more subject to deposition, due to the low solubility of Hg0 and high solubility of HgBr2 in atmospheric droplets. The deposition of oxidized mercury can make Hg potentially become available to biota [4]. Field studies have demonstrated the fast oxidation of Hg0 to HgII in the Arctic and Antarctic regions after polar sunrise, a phenomenon known as Arctic mercury depletion events (AMDEs). The explanation of this fascinating phenomenon will rely on fully understanding the redox reactions of atmospheric Hg [13]. On the other hand, atmospheric Hg models are essential tools for interpreting field observations and evaluating pollution control policies [10]. The further advancement of atmospheric Hg modeling depends on accurate kinetic data on the oxidation and reduction reactions of various mercury species in the atmosphere. However, many important processes involved in the transformation and deposition of atmospheric Hg are yet to be identified or quantified [14]. Global modeling studies also suggest that large uncertainties exist in our current knowledge of the global cycling of mercury [15].
The chemistry of atmospheric mercury was previously reviewed in detail by Lin and Pehkonen [16], Ariya et al. [17], Subir et al. [18], Lin et al. [14] and Ariya et al. [4]. Several excellent contributions have been made to our understanding in this area since 2015. The objective of this article is to discuss the current understanding of atmospheric mercury chemistry, with more focus on recent laboratory, theoretical, field and model studies since 2015 on the interconverting reactions between Hg0 and HgII. Finally, future research is recommended in order to further advance our knowledge of the dynamic transformations of atmospheric mercury.
2. Chemical Redox Pathways in the Gas Phase
The oxidation of Hg0 is a very crucial step for the removal of Hg from the atmosphere. Understanding the oxidation reactions of Hg0 is important for estimating the atmospheric lifetime of mercury. To date, the proposed oxidation pathways of Hg0 in the gas phase include the oxidation of Hg0 by O3, H2O2, OH radical, NO3 radical and by various halogen species [14]. Table 1 lists the gas-phase oxidation reactions of Hg0 and the reported rate coefficients. As shown in Figure 2 in Subiret al. [18], large variations exist in the current reported rate constants of gas-phase oxidation reactions of Hg. Here we discuss recent advances in the understanding of the oxidation of Hg0 by halogen atoms and NO3 radicals, as well as determining the major oxidant(s) in the atmosphere.
2.1. Br-Initiated Oxidation of Hg0
The oxidation of Hg0 by Br occurs via a two-step process with HgBr as the intermediate. Previous quantum calculations by Dibble et al. [47] demonstrated that BrHg could react with the abundant radicals in the atmosphere to form stable BrHgY compounds where Y is NO2, HO2, ClO, or BrO. Previous laboratory studies reported apparent rate coefficients in the range of (3.6 ± 0.9) × 10−13 to (3.2 ± 0.3) × 10−12 cm3 molec−1 s−1, at 1 atm and 298 K [19,20,22]. Theoretical studies estimated the rate constants to be from 9.8 × 10−13 to 2.1 × 10−12 cm3 molec−1 s−1, at 1 atm and 298 K [3,21,23]. Theoretical and experimental rate constants for mercury oxidation by Br and Cl atoms at 298 K have previously been compared in detail in Table A.2 by Subir et al. [18].
The first kinetic study of the reactions of BrHg with NO2 and HO2 was recently reported by Jiao and Dibble [25], using computational chemistry. Quantum calculations were performed to obtain the rate constants and product yields for the oxidation reactions of BrHg with NO2 and HO2. The rate constant for the oxidation of HgBr by NO2 was larger than that for the oxidation of HgBr by HO2 (T = 200–320 K). The fate of HgBr replied more on the concentration ratio of [NO2]/[HO2] in the atmosphere than the ratio of their oxidation rate constants. While the addition reaction dominated the reaction of HgBy by HO2, the addition of HgBr by NO2 was competed by a-reduction reaction to form Hg + BrNO2 (up to 18% of the oxidation). The reaction product of the oxidation of HgBr by NO2 was computed to be syn-BrHgONO under atmospheric conditions. Because no experimental studies on the reaction of HgBr of NO2 and HO2 have been previously reported in the literature, the rate constants computed in this study are significant for future model and laboratory studies [25].
Sunet al. [24] used a relative rate technique with ethane and propene as references and determined the rate coefficients for Hg0 + Br• reactions to be (1.6 ± 0.8) × 10−12 cm3 molec−1 s−1, at 100 kPa and 298 ± 3 K. The determined rate coefficient was in the midst of previously reported rate constants. Measurements using a scanning mobility particle sizer by Sunet al. [24] also showed that Hg products largely existed as wall deposits in agreement with the previous product analysis by Ariyaet al. [19]. It has been suggested that the presence of aerosols in the Aitken mode during the oxidation reaction may be initiated by the vapor nucleation of mercury-containing products.
2.2. Cl-Initiated Oxidation of Hg0
Similar to Hg0-Br oxidation, the oxidation of Hg0 by atomic Cl was believed to occur via a two-step chlorination process with HgCl as the intermediate [19,24,28]. Theoretical calculations indicated that after the initializing step to form the HgCl intermediate, the secondary oxidation of HgCl could be carried out by NO2, HO2, ClO, or BrO [47]. Recently, Sunet al. [24] determined the rate coefficients using a relative rate technique with ethane and 2-chloro-propane as references. The rate coefficient for the Hg0 + Cl• reaction was reported to be (1.8 ± 0.5) × 10−11 cm3 molecule−1 s−1, at 100 kPa and 298 ± 3 K, which was in agreement with three earlier laboratory studies by Horne et al. [27], Spiceret al. [20] and Ariyaet al. [19] (1.0–6.4 × 10−11 cm3 molecule−1 s−1). However, the reported rate coefficient was about two orders of magnitude larger than that determined by Donohoueet al. [28] (5.4 × 10−13 cm3 molecule−1 s−1) and one order of magnitude smaller than that determined by Byunet al. [29] (1.2 × 10−10 cm3 molecule−1 s−1). The determined rate constant was one order of magnitude higher than the only theoretical estimation by Khalizov et al. [2] (2.81 × 10−12 cm3 molecule−1 s−1).
More studies are needed to reduce the uncertainties in the kinetic estimate of the oxidation reactions of atmospheric Hg. To render it more complex, as the existing reactions show the importance of surfaces in catalysis and heterogeneous reactions [4,48], further studies on atmospherically-relevant surfaces are recommended.
2.3. Oxidation of Hg0 by NO3
The experimentally-determined rate constant for the oxidation of Hg0 by NO3 has been previously reported to be <4 × 10−15 cm3 molecule−1 s−1 by Sommaret al. [39] and <7 × 10−15 cm3 molecule−1 s−1 by Sumner et al. [36] at 1 atm and 298 K. However, based on the new HgO thermochemistry, Hynes et al. estimated this reaction to be highly endothermic and, thus, suggested that this oxidation pathway is not viable in the atmosphere [14,49]. Furthermore, theoretical calculations by Dibble et al. [47] suggest that NO3 do not form strong bonds with Hg0 and therefore is unlikely to initiate gas-phase Hg0 oxidation reactions.
A recent study by Peleg et al. [50] provided field evidence of the possible participation of nitrate radicals (NO3) in the nighttime oxidation of Hg0 in the atmosphere. In their study, the role of NO3 inthe nighttime oxidation of Hg0 in the atmosphere was evaluated by measuring the concentrations of NO3, Hg0 and HgII continuously during a six-week period in the urban air shed of Jerusalem, Israel during the summer of 2012. The R2 average of 0.47 indicated a strong correlation between nighttime [HgII] and [NO3], while correlations of nighttime [HgII] with other environmental variables were either weak or absent. Detailed analyses implied that NO3 radicals may be involved in the Hg0 oxidation reaction in the atmosphere. Previous theoretical calculations suggested that NO3 may be unlikely to initiate Hg0 oxidation [47,49]; therefore, NO3 may be involved in the secondary oxidation reaction of unstable HgI species [50]. More laboratory and theoretical studies are required to assess the role of NO3 in secondary reactions of Hg0 oxidation in the real atmosphere, including on atmospherically-relevant surfaces.
2.4. Dominant Gaseous Oxidant for Hg0: O3/OH, Br or Others?
Previous studies have assumed O3 and OH to be the major Hg0 oxidants in the atmosphere [51,52,53,54,55,56]. Despite theoretical doubts concerning the thermal stability of gaseous HgOH and HgO products, many modeling results using O3/OH as the main atmospheric Hg oxidants showed good agreement with the observed Hg0 concentration and wet deposition flux. However, more complex atmospheric Hg oxidation reactions, particularly heterogeneous reactions involving O3 and OH, have been implied in recent studies ([57] and references therin).
Atomic bromine (Br) has been suggested to be the dominant oxidant of Hg0 in the marine boundary layer and in the Arctic [58,59,60]. A previous model study by Holmeset al. [61] implied that Br could be the major oxidant of Hg0 in the global atmosphere. The existence of BrO radical has been reported in the upper troposphere [62,63] and over the southeastern US [5,64]. Due to the rapid exchange between Br and BrO radicals [65] and the much slower oxidation rate of Hg0 by BrO [18] the field observations of BrO seem to support the dominant oxidation of Hg0 by Br in the atmosphere. Shah et al. [5] found that sensitivity simulations using the GEOS-Chem chemical transport model by either increasing Br concentrations, or using a faster rate constant for the oxidation reaction of Hg0 byBr, resulted in a better agreement between the modeled results and the aircraft observations. Yet, consistent BrO measurements do not exist in the lower troposphere where humans and biota exist. Moreover, note that the majority of chemical compounds are more concentrated and diversified in the boundary layer, which leads to much more complexity in chemistry, physics and biology due to various interfacial processes. The field studies by Gust in and co-workers during the last decades also suggested that Hg0 may be oxidized not just by Br or BrO, but also by various oxidants in the atmosphere. The composition of oxidized mercury showed that Hg0 is subject to oxidation processes with various oxidants and not necessarily Br. The assumption of using Br as a universal global oxidant for atmospheric Hg may need further evaluation [66].
The most recent modeling studies indicated more complex Hg chemistry in the atmosphere, and multiple oxidants may be significant under various atmospheric conditions. Ye et al. [67] developed a box model including up-to-date atmospheric Hg chemistry and used it to study the oxidation of Hg0 at three different locations in the northeastern United States. The simulated diurnal cycles of HgII agreed well with the observations. Model results showed that Hg0 oxidation was dominated by O3 and OH at the coastal and inland sites during the day and Hg0 oxidation initiated by H2O2 was significant at the inland site during the night. In the marine boundary layer (MBL), the model simulations indicated that Br/BrO were the major oxidants of Hg0 at midday while O3 became the dominant oxidant for the remainder of the day.
Travnikovet al. [57] compared simulation results from four state-of-the-art chemical transport models with field data from various global and regional monitoring networks. It was found that models using the Br oxidation mechanism correctly simulated the observed seasonal variation of the HgII/Hg0 ratio in the near-surface layer but failed to predict the observed wet deposition maximum in summer at monitoring sites in North America and Europe. Models applying OH chemistry successfully predicted both the observed amplitude of seasonal changes and the periods of maximum and minimum values, but did not catch the maximum HgII/Hg0 ratios observed in spring. Models using O3 chemistry could not predict the observed large seasonal variation of either Hg oxidation or wet deposition flux.
Gencarelliet al. [68] simulated the deposition, transport and chemical interactions of atmospheric Hg over Europe for the year of 2013. The outputs of 14 model sensitivity tests were compared with field data from 28 monitoring sites. In general, good agreement was achieved between the model results and the observations. However, the observed deposition in precipitation was significantly underestimated when employing either the O3 or OH reaction mechanism alone. Using the Br oxidation mechanism overestimated HgII at the ground level and produced a lower overall Hg wet deposition than the simulations using both O3 and OH as atmospheric oxidants for Hg0. These model results revealed that the filed data could not be reproduced using the oxidation of Hg0 by O3, OH or Br alone, which indicated a more complicated oxidation mechanism of atmospheric Hg.
Bieseret al. [69] performed a model comparison study evaluating the impact of oxidation schemes and emissions on atmospheric mercury. The models under study successfully simulated the concentration distribution of total Hg and Hg0 in the troposphere. It was found that the agreement between the observed HgII patterns and the model results employing different chemistry schemes seemed to depend on altitude. Although models using the Br oxidation scheme well simulated high concentrations of HgII in the upper troposphere, models applying O3 and OH chemistry better estimated elevated concentrations in the lower troposphere. However, more studies are needed to confirm this conclusion due to the possible significant influence of model results by the physical and chemical parameters used in these models.
Recent model studies seem to suggest that multiple oxidants are likely involved in the oxidation of atmospheric Hg0 dependent on seasons and locations. However, whether Br, O3/OH or multiple oxidants are the major oxidants of Hg0 in the global atmosphere is unclear. To address this important question, more studies on reducing the large uncertainties in rate constants, understanding the heterogeneous oxidation reactions of Hg0, as well as improving the treatment of chemical mechanisms in atmospheric Hg models and the accuracy of mercury emission inventories are needed [8,18,48].
3. Chemical Redox Reactions of Hg in the Aqueous Phase
To date, the proposed chemical oxidation pathways in atmospheric droplets include the aqueous-phase oxidation of Hg0 by O3, OH, chlorine (HOCl/OCl−) and bromine (Br2/HOBr/BrO−). The proposed chemical reduction pathways of mercury in the aqueous phase relevant to environmental conditions include the reduction of HgII by sulfite, photo-reduction of Hg(OH)2, photo-reduction of HgII by HO2 and photo-reduction of HgII-dicarboxylic acid complexes. The obtained rate constants and proposed mechanisms for these reactions were summarized in Table 2. Recent advances include the aqueous photoreduction of HgII-organic complexes and the effects of environmental variables on the aqueous reduction of HgII by sulfite.
3.1. Field Evidence for the Reduction of HgII
The first field observation of the sunlight-induced reduction of HgII in the atmosphere was recently reported by Foyet al. [82]. In their study, concentrations of Hg0, HgII and Hg(p) were monitored hourly over four winter months in a remote, high-altitude location. This study site is absent of local anthropogenic sources, which is ideal for studying atmospheric Hg chemical transformations. The parameters of a chemical box model required to reproduce the observations were determined using an optimization algorithm. It was found that the presence of a photolytic reduction reaction previously observed in laboratory studies was needed in order to match the model results with the field observations. The results suggested that the reduction reaction needs to be included in atmospheric Hg models in order to improve simulations of mercury deposition in the atmosphere.
3.2. Photoreduction of HgII-Organic Complexes
HgII could form strong complexes with organic ligands such as reduced sulfur groups [74,83,84,85]. The photoreduction of HgII in the presence of dissolved organic matter (DOM) has been widely documented in various natural water systems [86,87,88,89,90] and may also occur in atmospheric droplets and organic aerosols (OA) [91]. Recent model studies by Horowitz et al. supported the occurrence of the reduction of HgII-organic complexes in the atmosphere. Horowitz et al. [91] incorporated the updated chemical mechanism for atmospheric Hg into the GEOS-Chem global model and found that the inclusion of aqueous-phase photoreduction of HgII-organic complexes reported by Si and Ariya [76] in GEOS-Chem models, was critical for reproducing the observations. The major HgII deposition to the global oceans and the relatively low observed wet deposition of Hg over rural China may be due to different reduction rates of HgII with organic aerosols at various geographic locations.
3.3. Direct Reduction of HgII by Sulfite
The aqueous-phase reduction of Hg2+ with sulfite is believed to be a process relevant to atmospheric droplets. The first-order rate constant determined by van Loon and her co-workers [71] has been widely used in current atmospheric Hg models. The effects of pH (1–7), temperature (274–318 K) and HgII sources (Hg(NO3)2 or HgO) on the aqueous-phase reduction rate were recently examined by Feinberget al. [72] to better understand this reduction pathway. The results showed that the reduction could occur in the pH range of 1–7. The activation parameters of the aqueous HgO reduction by sulfite at pH 1 and 3 with T = 274–318 K, was in good agreement with the previous study [71]. The reduction rate at pH = 7 decreased with increasing ionic strength, especially with Hg(NO3)2. No statistical difference was found between the reduction rate constants of Hg(NO3)2 and HgO, which suggested that the reduction of HgII by sulfite may be independent of the HgII species in the aqueous phase. The results indicated the possible occurrence of this reduction reaction under various environmental conditions and, thus, the necessity of its universal inclusion in atmospheric Hg models.
4. Heterogeneous Redox Reactions of Hg
As shown in Figure 1b, gaseous mercury species can adsorb on atmospheric surfaces and then undergo desorption, dissolution to atmospheric droplets or surface-enhanced (photo)chemical reactions [48]. Despite the potential significant role of heterogeneous Hg reactions on atmospheric Hg chemistry and model simulations [37,92], scare data is available on mercury reactions and equilibrium processes on atmospheric surfaces such as aerosols [48]. A systematic understanding of the surface chemistry of Hg is extremely difficult [48,93] due to the varying concentration, size distribution and composition of aerosols at different locations, times and meteorological conditions [94,95]. Nevertheless, several recent laboratory studies focus on understanding the complex heterogeneous reactions between atmospheric mercury and various aerosols. The major findings in these studies are summarized in Table 3.
Recently, Feinberget al. [72] performed the first investigation of the kinetics of heterogeneous HgII reduction by sulfite (Na2SO3) in the presence of fly ash using UV absorption spectroscopy. Compared with the corresponding homogeneous reduction rates of HgII by sulfite, the addition of fly ash samples from Cumberland Power Plant (Tennessee, USA) and Shawnee Fossil Plant (Kentucky, USA) reduced the reduction rates by c.a.45% and 95%, respectively. The observation of the HgII reduction in presence of the fly ash samples from Cumberland Power Plant (Tennessee) without added Na2SO3, may be due to the richness of sulfite in these fly ash samples. The existence of a large proportion of nanoparticles in the fly ash samples suggested that there were adequate surfaces for heterogeneous chemical reactions to occur. These results indicated the need to incorporate heterogeneous Hg reduction reactions into various environmental models of mercury.
Previous laboratory studies showed that the heterogeneous reduction reaction of HgII could occur on iron and sodium chloride aerosol surfaces, which are important components of atmospheric aerosols [97]. Recently, theoretical calculations by Taceyet al. [99] supported the experimental results. In this study, theoretical calculations were performed for the heterogeneous reduction reactions of HgCl2, HgBr2, Hg(NO3)2 and HgSO4 on clean Fe(110), NaCl(100) and NaCl(111)Na surfaces. Here, Fe(110) was the most thermodynamically stable and, thus, the most abundant surface on metallic iron aerosols. The NaCl(100) facet is composed of neutrally charged layers with both Na and Cl atoms exposed on the surface, while NaCl(111)Na surfaces are charged layers exposing only Na atoms. The results indicated that the heterogeneous reduction reactionthatgeneratesHg0 is highly favorable on Fe(110) and NaCl(111)Na surfaces. The desorption of reduced Hg required either no energy input on the NaCl(111)Na surfaces or ~0.5 eV of external energy on the Fe(110) surfaces. The results suggested that many oxidized mercury species can be heterogeneously reduced on metallic Fe and NaCl surfaces and the photochemical reaction on the aerosol surfaces may be a necessary step to catalyze the reaction.
Kurienet al. [100] used various iron(oxyhydr)oxide (γ-Fe2O3, α-FeOOH, α-Fe2O3 and Fe3O4) nanoparticles as proxies for reactive components of mineral dust and determined the uptake coefficients for the heterogeneous reaction of Hg0(g) on these nanoparticles. Upon irradiation (λ = 290–700 nm), the Hg0(g) uptake kinetics significantly increased on γ-Fe2O3, α-FeOOH, α-Fe2O3 nanoparticles, but not on the Fe3O4 surface at P = 760 ± 5 Torr and T = 295 ± 2 K. The effect of radiation on the uptake of Hg0(g) by α-Fe2O3was retarded by relative humidity. The variation in the uptake behavior of the iron(oxyhydr) oxides nanoparticles was due to their different band gaps. More studies are needed to improve our understanding of such reactions.
Such research presents the need for further studies of the heterogeneous chemistry of mercury, because elemental mercury and many types of oxidized mercury are likely adsorbed and undergo (photo)chemical reactions in the presence of abundant atmospheric surfaces, such as particles and clouds.
5. Future Research Directions
Despite research progress in the understanding of the atmospheric processes of Hg, there are still major knowledge gaps in laboratory, theoretical and modeling studies, as well as field measurement. These gaps include but are not limited to mercury chemical speciation in the field, better kinetic and laboratory studies under various environmental conditions, and more consistent and sophisticated theoretical and modeling integration. We herein focus on the future research needs implied in recent studies since 2015.
Despite the potential importance of the oxidation of Hg0 by O3/OH in the atmosphere, large uncertainties exist in the current gas-phase reaction rate constants and more studies need to be carried out to reduce these uncertainties in the first place. More studies are needed to evaluate the contribution of heterogeneous processes to the obtained rate coefficients for the oxidation of Hg0 by O3 and/or OH, in order to fully understand the discrepancy between the consistent experimental values and theoretical studies. Secondly, recent theoretical studies indicated that the oxidation of Hg0 could be initiated by Br- or Cl-atoms and then the secondary oxidation of the HgX (X= Br or Cl) intermediate could be carried out by NO2, HO2, ClO, or BrO. More laboratory studies are welcome to confirm this oxidation mechanism. Future field work and model sensitivity studies will also provide valuable insights into the viability of these reactions in the atmosphere. Since several studies supported the possible significant role of Br in the oxidation of Hg0 in the atmosphere, more kinetic and mechanistic studies are required to reduce the discrepancy in the reported rate constants for the oxidation of Hg0 by halogen atoms.
Recent model studies seemed to suggest that multiple oxidants are likely involved in the oxidation of atmospheric Hg0 dependent on seasons and locations. However, whether Br, O3/OH or multiple oxidants are the major oxidants of Hg0 in the global atmosphere is unclear. Accurate measurements of vertical tropospheric concentration profiles of the species involved, such as various atmospheric oxidants, Hg0(g) as well as detailed chemical compositions of oxidized mercury using diverse techniques, are critical for verifying the significance of various oxidants in Hg removal in the atmosphere globally.
Recent field observations and model studies supported the occurrence of reduction reactions in the atmosphere. Furthermore, their results supported the hypothesis that the reduction of HgII–organic complexes may play an important role in atmospheric Hg cycling besides sulfite-mediated reduction and photo-reduction of Hg(OH)2. More kinetic and mechanistic studies of HgII reduction pathways under environmentally-relevant conditions are needed. A better understanding of the reduction of HgII by organic compounds will require studies on the possible reduction pathways as well as on the quantification of various organic compounds in atmospheric droplets and aerosols.
Another important knowledge gap is the understanding of the redox reactions of Hg on various environmental surfaces such as aerosol, water, ice, snow, soil, and vegetative surfaces [48]. Despite the experimental difficulties caused by the variability in the size and composition of aerosols, several recent studies using fly ash or model aerosols have provided valuable information on understanding heterogeneous reactions of Hg on aerosols. The measured reaction rates are likely important in the chemical transformation of mercury in the atmosphere and the incorporation of these recent laboratory data in future model studies is essential to reduce uncertainties in current atmospheric Hg models. Such endeavors will benefit from the identification and quantification of oxidized mercury compounds, which has been a major challenge in atmospheric Hg research. There are novel instruments including mercury mass spectrometry [101], which can be used to provide such information. Further complementary analytical innovations to accurately quantify mercury-containing compounds in the atmosphere and atmospheric interfaces are needed.
In the light of the Minamata convention, we encourage a more integrated multi-disciplinary approach to comprehend mercury transformation, dynamics, speciation and remediation. Such integration is required to translate sound science to sound policy and regulations.
Author Contributions
Both authors made significant contributions. In particular, L.S. wrote the manuscript and P.A.A. revised, commented, and edited it.
Acknowledgments
We would like to thank the editor and two anonymous reviewers for their comments. We would like to thank the Natural Sciences and Engineering Research Council of Canada (NSERC), Environment Canada [Clean Air Regulatory Agenda (CARA)] and Auburn University at Montgomery for financial support. Lin Si would like to thank Emma for help with references.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Miretzky, P.; Cirelli, A.F. Hg(II) removal from water by chitosan and chitosan derivatives: A review. J. Hazard. Mater. 2009, 167, 10–23. [Google Scholar] [CrossRef] [PubMed]
- UNEP. Global Mercury Assessment 2013: Sources, Emissions, Releases, and Environmental Transport; UNEP Chemicals Branch: Geneva, Switzerland, 2013. [Google Scholar]
- Khalizov, A.F.; Viswanathan, B.; Larregaray, P.; Ariya, P.A. Theoretical Study on the Reactions of Hg with Halogens: Atmospheric Implications. J. Phys. Chem. A 2003, 107, 6360–6365. [Google Scholar] [CrossRef]
- Ariya, P.A.; Amyot, M.; Dastoor, A.; Deeds, D.; Feinberg, A.; Kos, G.; Poulain, A.; Ryjkov, A.; Semeniuk, K.; Subir, M.; et al. Mercury Physicochemical and Biogeochemical Transformation in the Atmosphere and at Atmospheric Interfaces: A Review and Future Directions. Chem. Rev. 2015, 115, 3760–3802. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.; Jaeglé, L.; Gratz, L.E.; Ambrose, J.L.; Jaffe, D.A.; Selin, N.E.; Song, S.; Campos, T.L.; Flocke, F.M.; Reeves, M.; et al. Origin of oxidized mercury in the summertime free troposphere over the southeastern US. Atmos. Chem. Phys. 2016, 16, 1511–1530. [Google Scholar] [CrossRef]
- Driscoll, C.T.; Mason, R.P.; Chan, H.M.; Jacob, D.J.; Pirrone, N. Mercury as a Global Pollutant: Sources, Pathways, and Effects. Environ. Sci. Technol. 2013, 47, 4967–4983. [Google Scholar] [CrossRef] [PubMed]
- Bergan, T.; Gallardo, L.; Rodhe, H. Mercury in the global troposphere: A three dimensional model study. Atmos. Environ. 1999, 33, 1575–1585. [Google Scholar] [CrossRef]
- Zhang, L.; Lyman, S.; Mao, H.; Lin, C.-J.; Gay, D.A.; Wang, S.; Gustin, M.S.; Feng, X.; Wania, F. A synthesis of research needs for improving the understanding of atmospheric mercury cycling. Atmos. Chem. Phys. 2017, 17, 9133–9144. [Google Scholar] [CrossRef]
- Pacyna, J.M.; Travnikov, O.; Simone, F.D.; Hedgecock, I.M.; Sundseth, K.; Pacyna, E.G.; Steenhuisen, F.; Pirrone, N.; Munthe, J.; Kindbom, K. Current and future levels of mercury atmospheric pollution on a global scale. Atmos. Chem. Phys. 2016, 16, 12495–12511. [Google Scholar] [CrossRef]
- Schroeder, W.; Munthe, J. Atmospheric mercury—An overview. Atmos. Environ. 1998, 5, 809–822. [Google Scholar] [CrossRef]
- Ryaboshapko, A.; Bullock, R.; Ebinghaus, R.; Ilyin, I.; Lohman, K.; Munthe, J.; Peterson, G.; Seigneur, C.; Wangberg, I. Comparison of mercury chemistry models. Atmos. Environ. 2002, 36, 3881–3898. [Google Scholar] [CrossRef]
- Fitzgerald, W.F.; Mason, R.P. Biogeochemical cycling of mercury in the marine environment. Metal Ions Biol. Syst. 1997, 34, 53–111. [Google Scholar]
- Ariya, P.A.; Skov, H.; Grage, M.M.L.; Goodsite, E.M. Gaseous elemental mercury in the ambient atmosphere: Review of the application of theoretical calculations and experimental studies for determination of reaction coefficients and mechanisms with halogens and other reactants. Adv. Quantum Chem. 2008, 55, 43–55. [Google Scholar]
- Lin, C.-J.; Singhasuk, P.; Pehkonen, S.O. Atmospheric Chemistry of Mercury. In Environmental Chemistry and Toxicology of Mercury; Liu, G., Cai, Y., O’Driscoll, N., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 113–153. [Google Scholar]
- Jaffe, D.; Strode, S. Sources, fate and transport of atmospheric mercury from Asia. Environ. Chem. 2008, 5, 121–126. [Google Scholar] [CrossRef]
- Lin, C.-J.; Pehkonen, S.O. The chemistry of atmospheric mercury: A review. Atmos. Environ. 1999, 33, 2067–2079. [Google Scholar] [CrossRef]
- Ariya, P.A.; Peterson, K.; Snider, G.; Amyot, M. Mercury Chemical Transformations in the Gas, Aqueous and Heterogeneous Phases: State-of-the-Art Science and Uncertainties. In Mercury Fate and Transport in the Global Atmosphere; Springer: Dordrecht, The Netherlands, 2009; pp. 459–501. [Google Scholar]
- Subir, M.; Ariya, P.A.; Dastoor, A.P. A review of uncertainties in atmospheric modeling of mercury chemistry I. Uncertainties in existing kinetic parameters—Fundamental limitations and the importance of heterogeneous chemistry. Atmos. Environ. 2011, 45, 5664–5676. [Google Scholar] [CrossRef]
- Ariya, P.A.; Khalizov, A.F.; Gidas, A. Reaction of Gaseous Mercury with Atomic and Molecular Halogens: Kinetics, Product Studies, and Atmospheric Implications. J. Phys. Chem. A 2002, 106, 7310–7320. [Google Scholar] [CrossRef]
- Spicer, C.W.; Satola, J.; Abbgy, A.A.; Plastridge, R.A.; Cowen, K.A. Kinetics of Gas-Phase Elemental Mercury Reaction with Halogen Species, Ozone, and Nitrate Radical Under Atmospheric Conditions. In Final Report to Florida Department of Environmental Protection; Battelle: Columbus, OH, USA, 2002. [Google Scholar]
- Goodsite, M.E.; Plane, J.M.C.; Skov, H. A theoretical study of the oxidation of Hg0 to HgBr2 in the troposphere. Environ. Sci. Technol. 2004, 38, 1772–1776. [Google Scholar] [CrossRef] [PubMed]
- Donohoue, D.L.; Bauer, D.; Cossairt, B.; Hynes, A.J. Temperature and pressure dependent rate coefficients for the reaction of Hg with Br and the reaction of Br with Br: A pulsed laser photolysis-pulsed laser induced fluorescence study. J. Phys. Chem. A 2006, 110, 6623–6632. [Google Scholar] [CrossRef] [PubMed]
- Shepler, B.C.; Balabanov, N.B.; Peterson, K.A. Hg+Br–>HgBr recombination and collision induced dissociation dynamics. J. Chem. Phys. 2007, 127, 164–304. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Sommar, J.; Feng, X.; Lin, C.-J.; Ge, M.; Wang, W.; Yin, R.; Fu, X.; Shang, L. Mass-dependent and -independent fractionation of mercury isotope during gas-phase oxidation of elemental mercury vapor by atomic Cl and Br. Environ. Sci. Technol. 2016, 50, 9232–9241. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Dibble, T.S. First kinetic study of the atmospherically important reactions BrHg + NO2 and BrHg + HOO. Phys. Chem. Chem. Phys. 2017, 19, 1826–1838. [Google Scholar] [CrossRef] [PubMed]
- Greig, G.; Gunning, H.E.; Strausz, O.P. Reactions of metal atoms. II. The combination of mercury and bromine atoms and the dimerization of HgBr. J. Chem. Phys. 1970, 52, 3684–3690. [Google Scholar] [CrossRef]
- Horne, D.G.; Gosavi, R.; Strausz, O.P. Reactions of metal atoms: Combination of mercury and chlorine atoms and the dimerization of HgCl. J. Chem. Phys. 1968, 48, 4758–4764. [Google Scholar] [CrossRef]
- Donohoue, D.L.; Bauer, D.; Hynes, A.J. Temperature and pressure dependent rate coefficients for the reaction of Hg with Cl and the reaction of Cl with Cl: A pulsed laser photolysispulsed laser induced fluorescence study. J. Phys. Chem. A 2005, 109, 7732–7741. [Google Scholar] [CrossRef] [PubMed]
- Byun, Y.; Cho, M.; Namkung, W.; Lee, K.; Koh, D.J.; Shin, D.N. Insight into the unique oxidation chemistry of elemental mercury by chlorine-containing species: Experiment and simulation. Environ. Sci. Technol. 2010, 44, 1624–1629. [Google Scholar] [CrossRef] [PubMed]
- Slemr, F.; Schuster, G.; Seiler, W. Distribution, speciation, and budget of atmospheric mercury. J. Atmos. Chem. 1985, 3, 407–434. [Google Scholar] [CrossRef]
- P’yankov, V.A. O kinetike Reaktsii Parov Rtuti s Ozonom (Kinetics of the Reaction of Mercury Vapour with Ozone). Zhurmal Obscej. Chem. Akatemijaneuk SSSR 1949, 19, 224–229. [Google Scholar]
- Schroeder, W.; Yarwood, G.; Niki, H. Transformation processes involving mercury species in the atmosphere—Results from a literature survey. Water Air Soil Pollut. 1991, 56, 653–666. [Google Scholar] [CrossRef]
- Iverfeldt, A.A.; Lindqvist, O. Atmospheric oxidation of elemental mercury by ozone in the aqueous phase. Atmos. Environ. 1986, 20, 1567–1573. [Google Scholar] [CrossRef]
- Hall, B. The gas-phase oxidation of elemental mercury by ozone. Water Air Soil Pollut. 1995, 80, 301–315. [Google Scholar] [CrossRef]
- Pal, B.; Ariya, P.A. Studies of ozone initiated reactions of gaseous mercury: Kinetics, product studies, and atmospheric implications. Phys. Chem. Chem. Phys. 2004, 6, 572–579. [Google Scholar] [CrossRef]
- Sumner, A.; Spicer, C.; Satola, J.; Mangaraj, R.; Cowen, K.; Landis, M.; Stevens, R.; Atkeson, T. Environmental chamber studies of mercury reactions in the atmosphere. In Dynamics of Mercury Pollution on Regional and Global Scales; Springer: New York, NY, USA, 2005; pp. 193–212. [Google Scholar]
- Snider, G.; Raofie, F.; Ariya, P.A. Effects of relative humidity and CO(g) on the O3-initiated oxidation reaction of Hg0(g): Kinetic & product studies. Phys. Chem. Chem. Phys. 2008, 10, 5616–5623. [Google Scholar] [PubMed]
- Rutter, A.P.; Shakya, K.M.; Lehr, R.; Schauer, J.J.; Griffin, R.J. Oxidation of gaseous elemental mercury in the presence of secondary organic aerosols. Atmos. Environ. 2012, 59, 86–92. [Google Scholar] [CrossRef]
- Sommar, J.; Hallquist, M.; Ljungstrom, E.; Lindqvist, O. On the gas phase reactions between volatile biogenic mercury species and the nitrate radical. J. Atmos. Chem. 1997, 27, 233–247. [Google Scholar] [CrossRef]
- Miller, G.C.; Quashnick, J.; Hebert, V. Reaction rate of metallic mercury with hydroxyl radical in the gas phase. Abstr. Paper Am. Chem. Soc. 2001, 221, 16-AGRO. [Google Scholar]
- Bauer, D.; D’Ottone, L.; Campuzano-Jost, P.; Hynes, A.J. Gas phase elemental mercury: Acomparison of LIF detection techniques and study of the kinetics of reaction with thehydroxyl radical. J. Photochem. Photobiol. A 2003, 157, 247–256. [Google Scholar] [CrossRef]
- Pal, B.; Ariya, P.A. Gas-phase HO center dot-initiated reactions of elemental mercury: Kinetics, product studies, and atmospheric implications. Environ. Sci. Technol. 2004, 38, 5555–5566. [Google Scholar] [CrossRef] [PubMed]
- Raofie, F.; Snider, G.; Ariya, P.A. Reaction of gaseous mercury with molecular iodine, atomic iodine, and iodine oxide radicals—Kinetics, product studies, and atmospheric implications. Can. J. Chem. 2008, 86, 811–820. [Google Scholar] [CrossRef]
- Raofie, F.; Ariya, P.A. Product Study of the Gas-Phase BrO-Initiated Oxidation of Hg0: Evidence for Stable Hg1+ Compounds. Environ. Sci. Technol. 2004, 38, 4319–4326. [Google Scholar] [CrossRef] [PubMed]
- Seigneur, C.; Wrobel, J.; Constantinou, E. A chemical kinetic mechanism for atmospheric inorganic mercury. Environ. Sci. Technol. 1994, 28, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Tokos, J.J.S.; Hall, B.; Calhoun, J.A.; Prestbo, E.M. Homogeneous gas-phase reaction of Hg◦ with H2O2, O3, CH3I, and (CH3)2S: Implications for atmospheric Hg cycling. Atmos. Environ. 1998, 32, 823–827. [Google Scholar] [CrossRef]
- Dibble, T.S.; Zelie, M.J.; Mao, H. Thermodynamics of reactions of ClHg and BrHg radicals with atmospherically abundant free radicals. Atmos. Chem. Phys. 2012, 12, 10271–10279. [Google Scholar] [CrossRef]
- Subir, M.; Ariya, P.A.; Dastoor, A.P. A review of the sources of uncertainties in atmospheric mercury modeling II. Mercury surface and heterogeneous chemistry—A missing link. Atmos. Environ. 2012, 46, 1–10. [Google Scholar] [CrossRef]
- Hynes, A.J.; Donohoue, D.L.; Goodsite, M.E.; Hedgecock, I.M. Our current understanding of major chemical and physical processes affecting mercury dynamics in the atmosphere and at the air-water/terrestrial interfaces. In Mercury Fate and Transport in the Global Atmosphere; Springer: New York, NY, USA, 2009; pp. 427–457. [Google Scholar]
- Peleg, M.; Tas, E.; Obrist, D.; Matveev, V.; Moore, C.; Gabay, M.; Luria, M. Observational Evidence for Involvement of Nitrate Radicals in Nighttime Oxidation of Mercury. Environ. Sci. Technol. 2015, 49, 14008–14018. [Google Scholar] [CrossRef] [PubMed]
- Bergan, T.; Rodhe, H. Oxidation of elemental mercury in the atmosphere; constraints imposed by global scale modelling. J. Atmos. Chem. 2001, 40, 191–212. [Google Scholar] [CrossRef]
- Dastoor, A.P.; Larcoque, Y. Global circulation of atmospheric mercury: A modelling study. Atmos. Environ. 2004, 38, 147–161. [Google Scholar] [CrossRef]
- Selin, N.E.; Javob, D.J.; Park, R.J.; Yantosca, R.M.; Strode, S.; Jaeglé, L.; Jaffe, D. Chemical cycling and deposition of atmospheric mercury: Global constraints from observations. J. Geophys. Res. Atmos. 2007, 112, D02308. [Google Scholar] [CrossRef]
- Travnikov, O.; Ilyin, I. The EMEP/MSC-E Mercury Modeling System. In Mercury Fate and Transport in the Global Atmosphere: Emissions, Measurements, and Models; Pirrone, N., Mason, R.P., Eds.; Springer: New York, NY, USA, 2009; pp. 571–587. [Google Scholar]
- De Simone, F.; Gencarelli, C.N.; Hedgecock, I.M.; Pirrone, N. Global atmospheric cycle of mercury: A model study on the impact of oxidation mechanisms. Environ. Sci. Pollut. Res. 2014, 21, 4110–4123. [Google Scholar] [CrossRef] [PubMed]
- Gencarelli, C.N.; de Simone, F.; Hedgecock, I.M.; Sprovieri, F.; Pirrone, N. Development and Application of a Regional-Scale Atmospheric Mercury Model Based on WRF/Chem: A Mediterranean Area Investigation. Environ. Sci. Pollut. Res. 2014, 21, 4095–4109. [Google Scholar] [CrossRef] [PubMed]
- Travnikov, O.; Angot, H.; Artaxo, P.; Bencardino, M.; Bieser, J.; D’Amore, F.; Dastoor, A.; Simone, F.D.; Diéguez, M.d.C.; Dommergue, A.; et al. Multi-model study of mercury dispersion in the atmosphere: Atmospheric processes and model evaluation. Atmos. Chem. Phys. 2017, 17, 5271–5295. [Google Scholar] [CrossRef]
- Mason, R.P.; Sheu, G.R. Role of the ocean in the global mercury cycle. Glob. Biogeochem. Cycle 2002, 16, 1093. [Google Scholar] [CrossRef]
- Hedgecock, I.M.; Pirrone, N. Chasing quicksilver: Modeling the atmospheric lifetime of Hg-(g)(0) in the marine boundary layer at various latitudes. Environ. Sci. Technol. 2004, 38, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Dastoor, A.P.; Davignon, D.; Theys, N.; van Roozendael, M.; Steffen, A.; Ariya, P.A. Modeling dynamic exchange of gaseous elemental mercury at polar sunrise. Environ. Sci. Technol. 2008, 42, 5183–5188. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.D.; Jacob, D.J.; Yang, X. Global lifetime of elemental mercury against oxidation by atomic bromine in the free troposphere. Geophys. Res. Lett. 2006, 33, L20808. [Google Scholar] [CrossRef]
- Theys, N.; van Roozendael, M.; Hendrick, F.; Yang, X.; de Smedt, I.; Richter, A.; Begoin, M.; Errera, Q.; Johnston, P.V.; Kreher, K.; et al. Global observations of tropospheric BrO columns using GOME-2 satellite data. Atmos. Chem. Phys. 2011, 11, 1791–1811. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.Y.; Schmidt, J.A.; Baidar, S.; Coburn, S.; Dix, B.; Koenig, T.K.; Apel, E.; Bowdalo, D.; Campos, T.L.; Eloranta, E.; et al. Active and widespread halogen chemistry in the tropical and subtropical free troposphere. Proc. Natl. Acad. Sci. USA 2015, 112, 9281–9286. [Google Scholar] [CrossRef] [PubMed]
- Gratz, L.E.; Ambrose, J.L.; Jaffe, D.A.; Shah, V.; Jaegle, L.; Stutz, J.; Festa, J.; Spolaor, M.; Tsai, C.; Selin, N.E.; et al. Oxidation of mercury by bromine in the subtropical Pacific free troposphere. Geophys. Res. Lett. 2015, 42, 10494–10502. [Google Scholar] [CrossRef]
- Finlayson-Pitts, B.J.; Pitts, J.N.J. Chemistry of the Upper and Lower Atmosphere, Theory, Experiments, and Applications; Academic Press: San Diego, CA, USA, 2000. [Google Scholar]
- Gustin, M.S.; Amos, H.M.; Huang, J.; Miller, M.B.; Heidecorn, K. Measuring and modeling mercury in the atmosphere: A critical review. Atmos. Chem. Phys. 2015, 15, 5697–5713. [Google Scholar] [CrossRef]
- Ye, Z.; Mao, H.; Lin, C.-J.; Kim, S.Y. Investigation of processes controlling summertime gaseous elemental mercury oxidation at midlatitudinal marine, coastal, and inland sites. Atmos. Chem. Phys. 2016, 16, 8461–8478. [Google Scholar] [CrossRef]
- Gencarelli, C.N.; Bieser, J.; Carbone, F.; Simone, F.D.; Hedgecock, I.M.; Matthias, V.; Travnikov, O.; Yang, X.; Pirrone, N. Sensitivity model study of regional mercury dispersion in the atmosphere. Atmos. Chem. Phys. 2017, 17, 627–643. [Google Scholar] [CrossRef]
- Bieser, J.; Slemr, F.; Ambrose, J.; Brenninkmeijer, C.; Brooks, S.; Dastoor, A.; Simone, F.D.; Ebinghaus, R.; Gencarelli, C.N.; Geyer, B.; et al. Multi-model study of mercury dispersion in the atmosphere: Vertical and interhemispheric distribution of mercury species. Atmos. Chem. Phys. 2017, 17, 6925–6955. [Google Scholar] [CrossRef]
- Munthe, J.; Xiao, Z.F.; Lindqvist, O. The aqueous reduction of divalent mercury by sulfite. Water Air Soil Pollut. 1991, 56, 621–630. [Google Scholar] [CrossRef]
- Van Loon, L.; Mader, E.; Scott, S.L. Reduction of the aqueous mercuric ion by sulfite: UV Spectrum of HgSO3 and Its Intramolecular Redox Reaction. J.Phys.Chem. A 2000, 104, 1621–1626. [Google Scholar] [CrossRef]
- Feinberg, A.I.; Kurien, U.; Ariya, P.A. The Kinetics of Aqueous Mercury(II) Reduction by Sulfite Over an Array of Environmental Conditions. Water Air Soil Pollut. 2015, 226, 1–12. [Google Scholar] [CrossRef]
- Xiao, Z.F.; Munthe, J.; Stromberg, D.; Lindqvist, O. Photochemical behavior of inorganic mercury compounds in aqueous solution. In Mercury as a Global Pollutant-Integration and Synthesis; Watras, C.J., Huckabee, J.W., Eds.; Lewis Publishers: Boca Raton, USA, 1994; pp. 581–592. [Google Scholar]
- Pehkonen, S.O.; Lin, C.-J. Aqueous photochemistry of divalent mercury with organic acids. J. Air Waste Manag. Assoc. 1998, 48, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Gardfeldt, K.; Jonsson, M. Is bimolecular reduction of Hg(II) complexes possible in aqueous systems of environmental importance. J. Phys. Chem. A 2003, 107, 4478–4482. [Google Scholar] [CrossRef]
- Si, L.; Ariya, P.A. Reduction of oxidized mercury species by dicarboxylic acids (C2-C4): Kinetic and product studies. Environ. Sci. Technol. 2008, 42, 5150–5155. [Google Scholar] [CrossRef] [PubMed]
- Munthe, J. Aqueous oxidation of elemental Hg by O3. Atmos. Environ. 1992, 26, 1461–1468. [Google Scholar] [CrossRef]
- Gardfeldt, K.; Sommar, J.; Stromberg, D.; Feng, X. Oxidation of atomic mercury by hydroxyl radicals and photoinduced decomposition of methylmercury in the aqueous phase. Atmos. Environ. 2001, 35, 3039–3047. [Google Scholar] [CrossRef]
- Hines, N.A.; Brezonik, P.L. Mercury dynamics in a small Northern Minnesota lake: Water to air exchange and photoreactions of mercury. Mar. Chem. 2004, 90, 137–149. [Google Scholar] [CrossRef]
- Wang, Z.; Pehkonen, S.O. Oxidation of elemental mercury by aqueous bromine: Atmospheric implications. Atmos. Environ. 2004, 38, 3675–3688. [Google Scholar] [CrossRef]
- Lin, C.-J.; Pehkonen, S.O. Oxidation of elemental mercury by aqueous chlorine (HOCl−/OCl−): Implication for tropospheric mercury chemistry. J. Geophys. Res. 1998, 103, 28093–28102. [Google Scholar] [CrossRef]
- Foy, B.D.; Tong, Y.; Yin, X.; Zhang, W.; Kang, S.; Zhang, Q.; Zhang, G.; Wang, X.; Schauer, J.J. First field-based atmospheric observation of the reduction of reactive mercury driven by sunlight. Atmos. Environ. 2016, 134, 27–39. [Google Scholar] [CrossRef]
- Haitzer, M.; Aiken, G.R.; Ryan, J.N. Binding of mercury(II) to dissolved organic matter: The role of the mercuryto-DOM concentration ratio. Environ. Sci. Technol. 2002, 36, 3564–3570. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, M. Interactions between mercury and dissolved organic matter—A review. Chemosphere 2004, 55, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Hintelmann, H. Mercury isotope fractionation during photoreduction in natural water is controlled by its Hg-DOC ratio. Geochim. Cosmochim. Acta 2009, 73, 6704–6715. [Google Scholar] [CrossRef]
- Amyot, M.; Mierle, G.; Lean, D.R.S.; McQueen, D.J. Sunlight-induced formation of dissovled gaseous mercury in lake waters. Environ. Sci. Technol. 1994, 28, 2366–2371. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.F.; Stromberg, D.; Lindqvist, O. Influence of humic substances on photolysis of divalent mercury in aqueous solution. Water Air Soil Pollut. 1995, 80, 789–798. [Google Scholar] [CrossRef]
- O’Driscoll, N.J.; Siciliano, S.D.; Lean, D.R.S.; Amyot, M. Gross photoreduction kinetics of mercury in temperate freshwater lakes and rivers: Application to a general model of DGM dynamics. Environ. Sci. Technol. 2006, 40, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Whalin, L.; Mason, R. A new method for the investigation of mercury redox chemistry in natural waters utilizing deflatable Teflon (R) bags and additions of isotopically labeled mercury. Anal. Chim. Acta 2006, 558, 211–221. [Google Scholar] [CrossRef]
- Si, L.; Ariya, P.A. Aqueous photoreduction of oxidized mercury species in presence of selected alkanethiols. Chemosphere 2011, 84, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, H.M.; Jacob, D.J.; Zhang, Y.; Dibble, T.S.; Slemr, F.; Amos, H.M.; Schmidt, J.A.; Corbitt, E.S.; Marais, E.A.; Sunderland, E.M. A new mechanism for atmospheric mercury redox chemistry: Implications for the global mercury budget. Atmos. Chem. Phys. 2017, 17, 6353–6371. [Google Scholar] [CrossRef]
- Si, L.; Ariya, P.A. Photochemical Reactions of Divalent Mercury with Thioglycolic Acid: Formation of Mercuric Sulfide Particles. Chemosphere 2015, 119, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Prather, K.A.; Hatch, C.D.; Grassian, V.H. Analysis of atmospheric aerosols. Ann. Rev. Anal. Chem. 2008, 1, 485–514. [Google Scholar] [CrossRef] [PubMed]
- Finlayson-Pitts, B.J. Reactions at surfaces in the atmosphere: Integration of experiments and theory as necessary (but not necessarily sufficient) for prediction the physical chemistry of aerosols. Phys. Chem. Chem. Phys. 2009, 11, 7760–7779. [Google Scholar] [CrossRef] [PubMed]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006. [Google Scholar]
- Lin, C.-J.; Pehkonen, S.O. Aqueous free radical chemistry of mercury in the presence of iron oxides and ambient aerosol. Atmos. Environ. 1997, 31, 4125–4137. [Google Scholar] [CrossRef]
- Tong, Y.; Eichhorst, T.; Olson, M.R.; McGinnis, J.E.; Turner, I.; Rutter, A.P.; Shafer, M.M.; Wanga, X.; Schauer, J.J. Atmospheric photolytic reduction of Hg(II) in dry aerosols. Environ.Sci. Processes Impacts 2013, 15, 1883–1888. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Eichhorst, T.; Olson, M.R.; Rutter, A.P.; Shafer, M.M.; Wang, X.; Schauer, J.J. Comparison of heterogeneous photolytic reduction of Hg(II) in the coal fly ashes and synthetic aerosols. Atmos. Res. 2014, 138, 324–329. [Google Scholar] [CrossRef]
- Tacey, S.A.; Xu, L.; Mavrikakis, M.; Schauer, J.J. Heterogeneous Reduction Pathways for Hg(II) Species on Dry Aerosols: A First-Principles Computational Study. J. Phys. Chem. A 2016, 120, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Kurien, U.; Hu, Z.; Lee, H.; Dastoor, A.P.; Ariya, P.A. Radiation Enhanced Uptake of Hg0(g) on Iron (Oxyhydr)Oxide Nanoparticles. RSC Adv. 2017, 7, 45010–45021. [Google Scholar] [CrossRef]
- Deeds, D.; Ghoshdastidar, A.; Raofie, F.; Guerette, E.-A.; Tessier, A.; Ariya, P. Development of a Particle-Trap Preconcentration-Soft Ionization Mass Spectrometric Technique for the Quantification of mercury halides in air. Anal. Chem. 2015, 87, 5109–5116. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(a) Mercury transformations in the environment; (b) Atmospheric chemistry of mercury. Adapted and modified from [3]. The question mark (?) indicates some major knowledge gaps in Hg cycling in the environment.
Figure 1.
(a) Mercury transformations in the environment; (b) Atmospheric chemistry of mercury. Adapted and modified from [3]. The question mark (?) indicates some major knowledge gaps in Hg cycling in the environment.



{kind=link}
{kind=link}
{kind=link}
Table 1.
Current accumulated knowledge on the chemical redox pathways of Hg in the gas phase.
| Gas Phase Reaction | Diluent gas a (T = 298 K) | Rate Coefficient b (cm3 molec−1 s−1) | References |
|---|---|---|---|
| Air, N2, 1 atm | (3.2 ± 0.3) × 10−12 | [19] | |
| Air, NO, 1 atm | 9 × 10−13 | [20] | |
| N/A, 1 atmc | 1.01 × 10−12exp(209.03/T) | [3] | |
| N/A, 1 atm | 2.07 × 10−12 | ||
| N/A (180–400 K) | 1.1 × 10−12(T/298K)−2.37 | [21] | |
| N/A, 1 atm | 1.1 × 10−12 | ||
| N2, 1atm (243–298 K) | (1.46 ± 0.34) × 10−32 × (T/298)−(1.86 ± 1.49) cm6/molec2/s | [22] | |
| N2, 1 atm | (3.6 ± 0.9) × 10−13 | ||
| Ar, 1 atm (260 K)c | 1.2 × 10−12 | [23] | |
| Air, 1 atm | (1.6 ± 0.8) × 10−12 | [24] | |
M = NO2 or HO2 | N/A (220–320 K) | k ([M], T)d | [25] |
| CF3Br, 0.26 atm (397 K) | 7 × 10−17 | [26] | |
| N/A, 1 atm (180–400 K) | 2.5 × 10−10(T/298K)−0.57 | [21] | |
| N/A, 1 atm | 2.5 × 10−10 | ||
| Ar, 0.93 atm (383–443 K) | (3.2 ± 1.7) × 10−11 | [27] | |
| Air, NO, 1 atm | 6.4 × 10−11 | [20] | |
| Air, N2, 1 atm | (1.0 ± 0.2) × 10−11 | [19] | |
| N/A, 1 atmc | 1.38 × 10−12exp(208.02/T) | [3] | |
| N/A, 1 atm | 2.81 × 10−12 | ||
| N2 (243–298 K) | (2.2 ± 0.5) × 10−32 × exp [(680 ± 400)(1/T − 1/298)] cm6/molec2/s | [28] | |
| N2, 1 atm | 5.4 × 10−13 | ||
| N2, 1 atm | 1.2 × 10−10 | [29] | |
| Air, 1 atm | (1.8 ± 0.5) × 10−11 | [24] | |
| N/A, 1 atm (293 K) | 4.2 × 10−19 | [30,31] | |
| N/A, 1 atm (293 K) | 4.9 × 10−18 | [31,32] | |
| Air, 1 atm (293 K) | 1.7 × 10−18 | [33] | |
| N2/O2, 1 atm (293 K) | (3 ± 2) × 10−20 | [34] | |
| N2, 1 atm | (7.5 ± 0.9) × 10−19 | [35] | |
| Air, 1 atm | (6.4 ± 2.3) × 10−19 | [36] | |
| N2, 1 atm | (6.2 ± 1.1) × 10−19 | [37] | |
| Air, 1 atm | (7.4 ± 0.5) × 10−19 | [38] | |
| Air, 1 atm | (8.7 ± 2.8) × 10−14 | [39] | |
| N/A, 1 atm (343 K) | (1.6 ± 0.2) × 10−11 | [40] | |
| Air, 1 atm | <1.2 × 10−13 | [41] | |
| N/A, 1 atm (180–400 K) | 3.2 × 10−13(T/298K)−3.06 | [21] | |
| N/A, 1 atm | 3.2 × 10−13 | ||
| Air/N2, 1 atm | (9.0 ± 1.3) × 10−14 | [42] | |
| N/A, 1 atmc | 9.2 × 10−13exp(206.81/T) | [3] | |
| N/A, 1 atm | 1.86 × 10−12 | ||
| N/A, 1 atm (180–400 K) | 4.0 × 10−13(T/298 K)−2.38 | [21] | |
| N/A, 1 atm | 4.0 × 10−13 | ||
| Air, 1 atm | (1.8 ± 0.4) × 10−15 | [36] | |
| N2, 1 atm | ≤(1.27 ± 0.58) × 10−19 | [43] | |
| Air, N2, 1 atm | (2.6 ± 0.2) × 10−18 | [19] | |
| Air, 1 atm | (2.5 ± 0.9) × 10−18 | [36] | |
| N2, 1 atm | 4.3 × 10−15 | [29] | |
| Air, N2, 1 atm | <(0.9 ± 0.2) × 10−16 | [19] | |
| N2, 1 atm | 1.1 × 10−11 | [29] | |
| Air, NO, 1 atm | (3.0–6.4) × 10−14 | [20] | |
| N2, 1 atm | (1–100) × 10−15 | [44] | |
| N2, (5–10) × 10−3atm(294 K) | <4 × 10−15 | [39] | |
| Air, 1 atm | <7 × 10−15 | [36] | |
| N/A, 1 atm | ≤4.1 × 10−16 | [45] | |
| N2, N/A (293 K) | <8.5 × 10−19 | [46] |
a T = 298 K, unless otherwise stated; b The unit is cm3 molec−1 s−1 unless otherwise stated; c Temperature range is unknown. d where k0(T) and k∞(T) values are tabulated in [25].
Table 2.
Current accumulated knowledge on chemical redox pathways of Hg in the aqueous phase.
| Reactant(s) | Rate Constants | T(K) | pH | Potential Mechanism | Reference |
|---|---|---|---|---|---|
| Identified Aqueous Reduction Pathways of Hg2+ | |||||
| Hg2+ + sulfite (aq) | 0.6 s−1 | 299 | 3.0–4.84 | [70] | |
| 0.0106 ± 0.0009 s−1 | 298 | 3 | [71] | ||
| 0.013 ± 0.007 s−1 | 298 | 7 | Same as above | [72] | |
| <10−4 s−1 | 299 | 3.0–4.84 | [70] | ||
| Hg(OH)2 | 3 × 10−7 s−1 | 293 | 7 | [73] | |
| HgS22− | ~10−7 s−1 | 298 | Not available | [73] | |
| Hg2+ + HO2 | 1.7 × 104 M−1 s−1 | 298 | [74] | ||
| Not available | Intramolecular 2e− transfer via Hg2+-oxalate complex | [75] | |||
| Hg2++Dicarboxylic acids (C2–C4) | (1.2 ± 0.2) × 104 M−1 s−1(Oxalic) (4.9 ± 0.8) × 103 M−1 s−1(Malonic); (2.8 ± 0.5) × 103 M−1 s−1(Succinic) | 296 | 3.0 | Mainly intramolecular 2e− transfer via Hg2+-dicarboxylate complexes | [76] |
| Identified Aqueous Oxidation Pathways of Hg0 | |||||
| Hg0 + O3 | (4.7 ± 2.2) × 107 M−1 s−1 | 298 | 4.5–9.5 | [77] | |
| Hg0 + •OH | 2.0 × 109 M−1 s−1 | 298 | [74] | ||
| (2.4 ± 0.3) × 109 M−1 s–1 | 298 | [78] | |||
| 5.5 × 109 M–1 s–1 | Not available | [79] | |||
| Hg0 + aqueous bromine | 0.28 ± 0.02 M–1 s–1 0.27 ± 0.04 M–1 s–1 0.2 ± 0.03 M–1 s–1 | 294–296 | 2, 6.8, 11.7 | [80] | |
| Hg0 + HOCl/OCl− | (2.09 ± 0.06) × 106 M–1 s–1 | Ambient | [81] | ||
| (1.99 ± 0.05) × 106 M–1 s–1 | |||||
Table 3.
Current accumulated knowledge on heterogeneous chemical reactions of Hg in the atmosphere.
| Reactants | Surfaces | Major Findings | References |
|---|---|---|---|
| Hg2+ + organic acids | 0.1 g/L iron oxides particles or 0.01 g/L ambient aerosols |
| [96] |
| HgCl2 | Synthetic NaCl aerosols |
| [97] |
| HgCl2 | Coal fly ash or synthetic aerosols |
| [98] |
| Hg2+ + sulfite | Fly ash |
| [72] |
| HgCl2, HgBr2, Hg(NO3)2, HgSO4 | Fe(110), NaCl(100) and NaCl(111)Na |
| [99] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Si, L.; Ariya, P.A. Recent Advances in Atmospheric Chemistry of Mercury. Atmosphere 2018, 9, 76. https://doi.org/10.3390/atmos9020076
AMA Style
Si L, Ariya PA. Recent Advances in Atmospheric Chemistry of Mercury. Atmosphere. 2018; 9(2):76. https://doi.org/10.3390/atmos9020076
Chicago/Turabian StyleSi, Lin, and Parisa A. Ariya. 2018. "Recent Advances in Atmospheric Chemistry of Mercury" Atmosphere 9, no. 2: 76. https://doi.org/10.3390/atmos9020076
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.