Current and Potential Uses of Immunocytokines as Cancer Immunotherapy

Abstract
:1. Cancer Immunotherapy: Broad Application
2. ADCC and FcRs
3. Augmenting ADCC with Effector Activation, Ch 14.18 + Cytokines
4. Immunocytokines: Linking IL2 to Anti-GD2 mAb; Preclinical Development
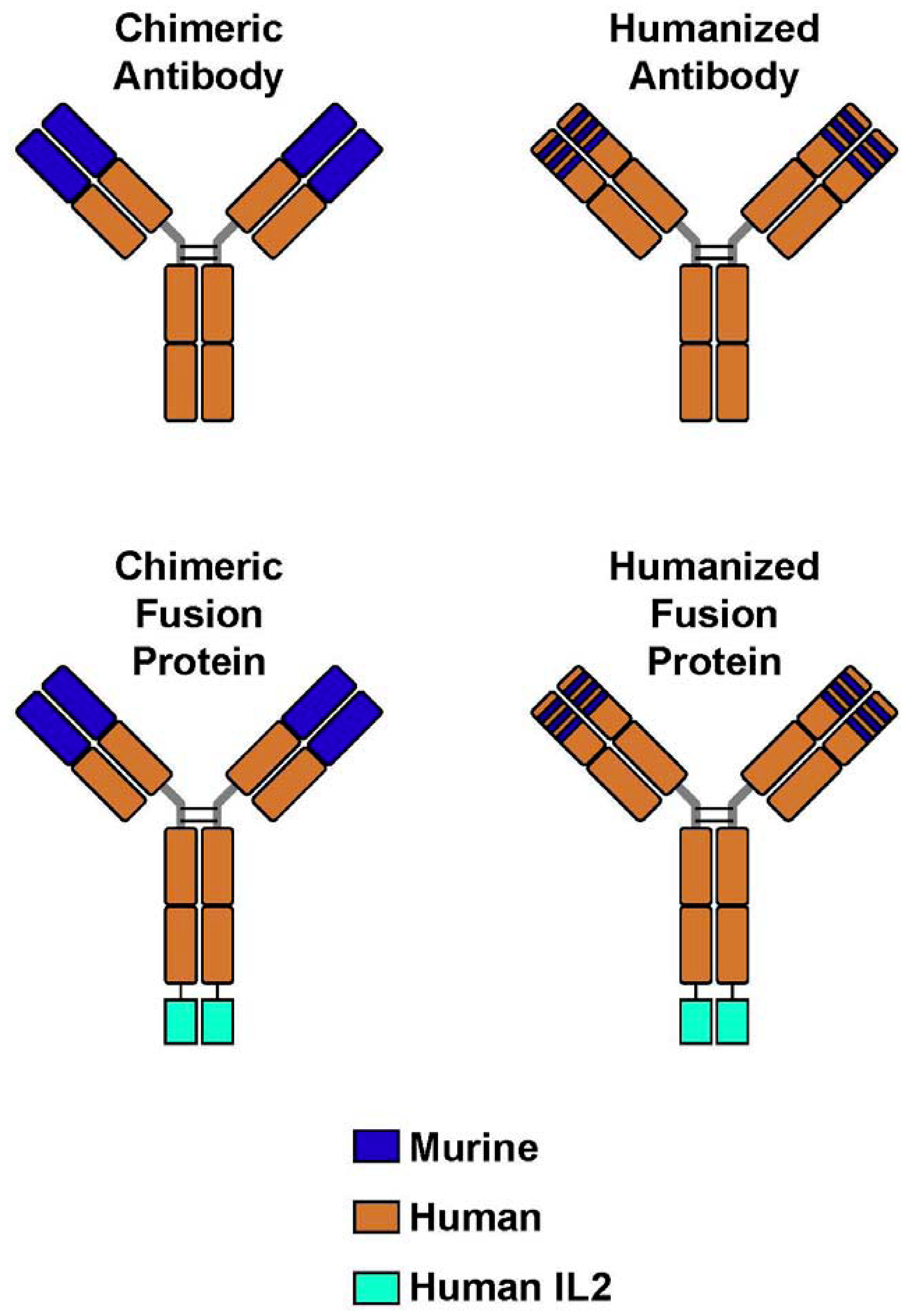

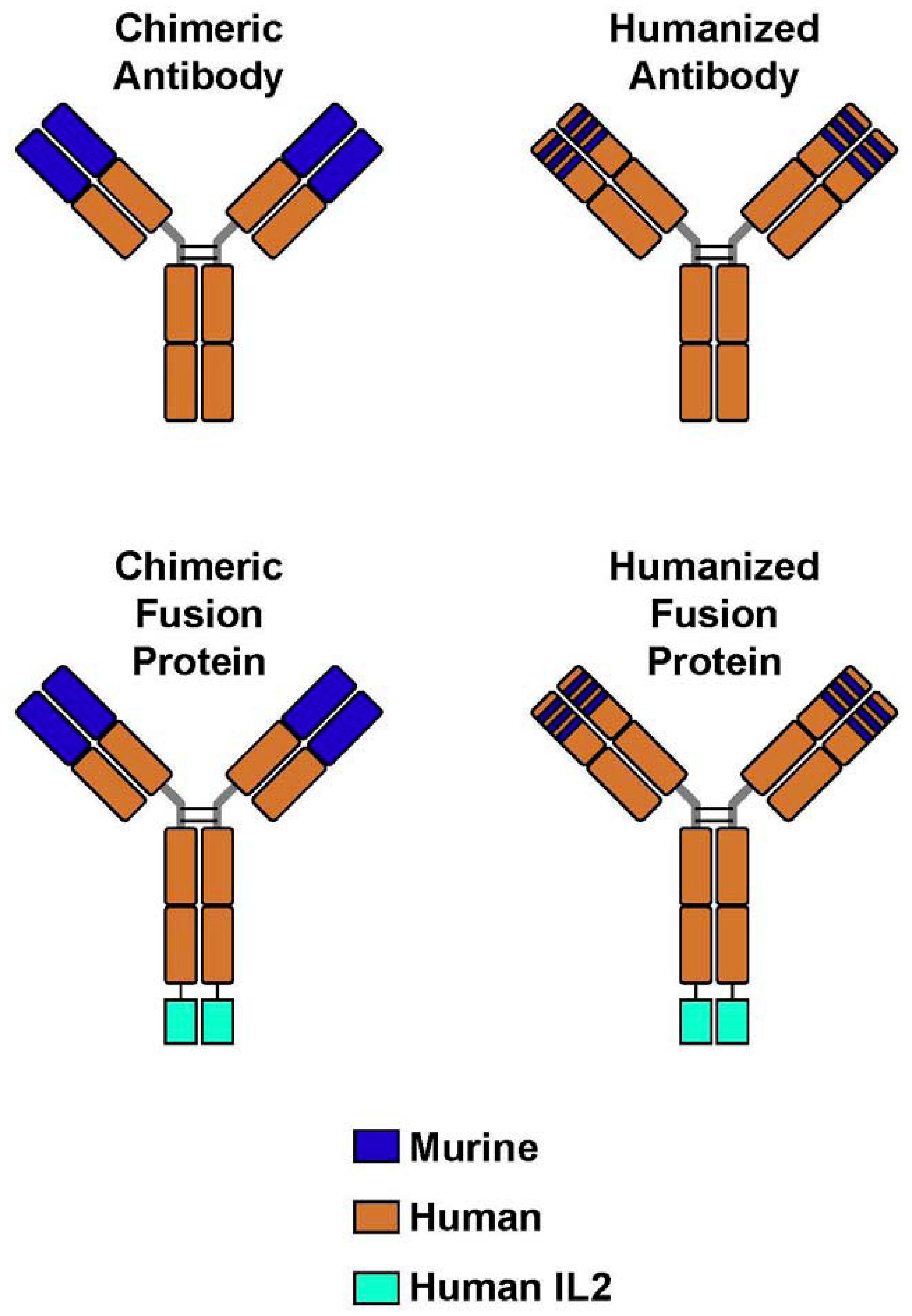{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Tumor | Number of Tumor Foci |
|---|---|---|
| PBS | *NXS2 | >250, >250, >250, >250, 240, 115 |
| IL2+ch14.18 | *NXS2 | 174, 134, 105, 102, 91, 83 |
| **ch14.18-IL2 | *NXS2 | 0, 0, 0, 0, 0, 0, |
| ch14.18-IL2 | #B16 | >500, >500, >500, >500, >500, 138, 97 |
| PBS | #B78 +B16 | >500, >500, >500, >500, >500, >500, >500,>500 |
| IL2+ch14.18 | #B78 + B16 | >500, >500, >500, >500, 189, 179, 104 |
| ch14.18-IL2 | #B78 + B16 | ##0, 0, 2, 7, 9, 12, 21, 43 |
| Receptor | Ligand |
|---|---|
| KIR2DL1 (CD158a) | HLA-C2 (Lys80) |
| KIR2DL2/KIR2DL3 (CD158b) | HLA-C1 (Asp80) |
| KIR3DL1 (CD158e) | HLA-Bw4, HLA-ABw4 |
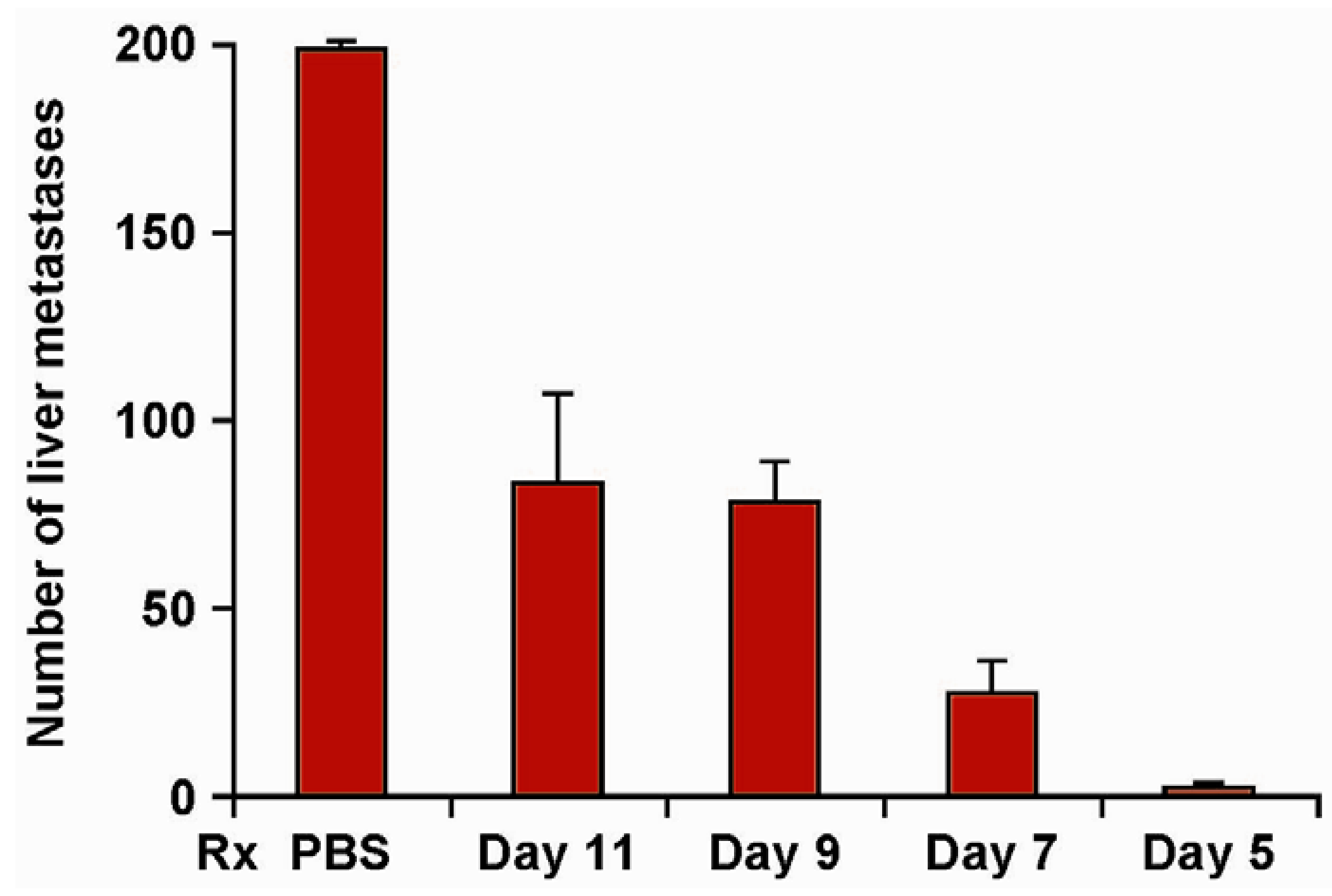
4.1. Phase I Clinical Testing of hu14.18-IL2
4.2. Phase II Clinical Testing of hu14.18-IL2
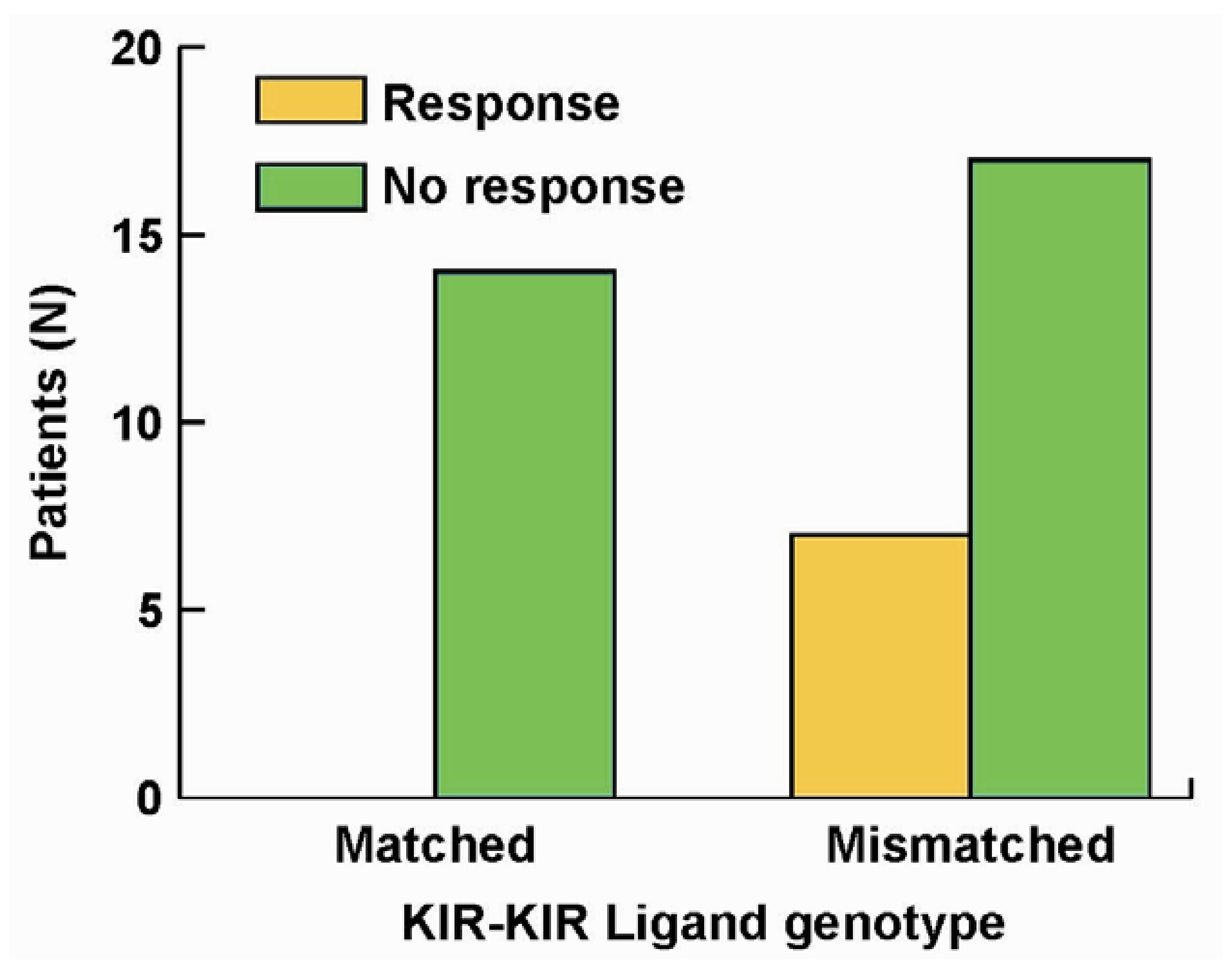
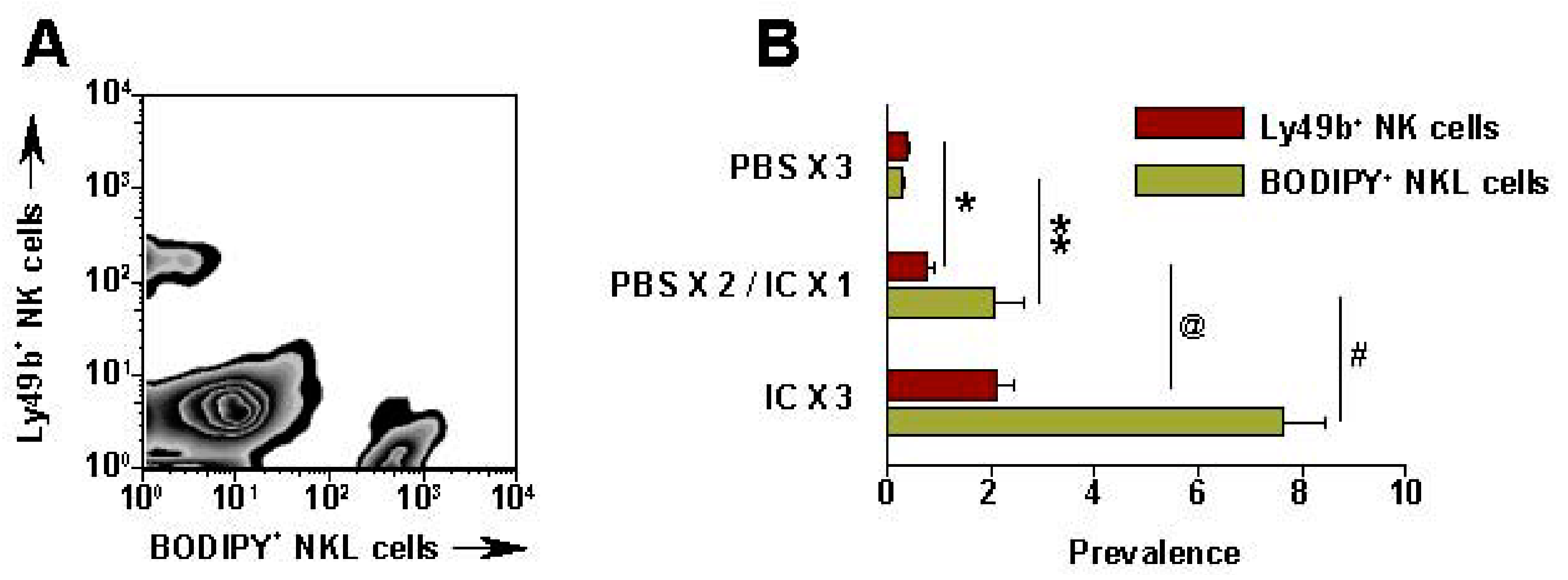
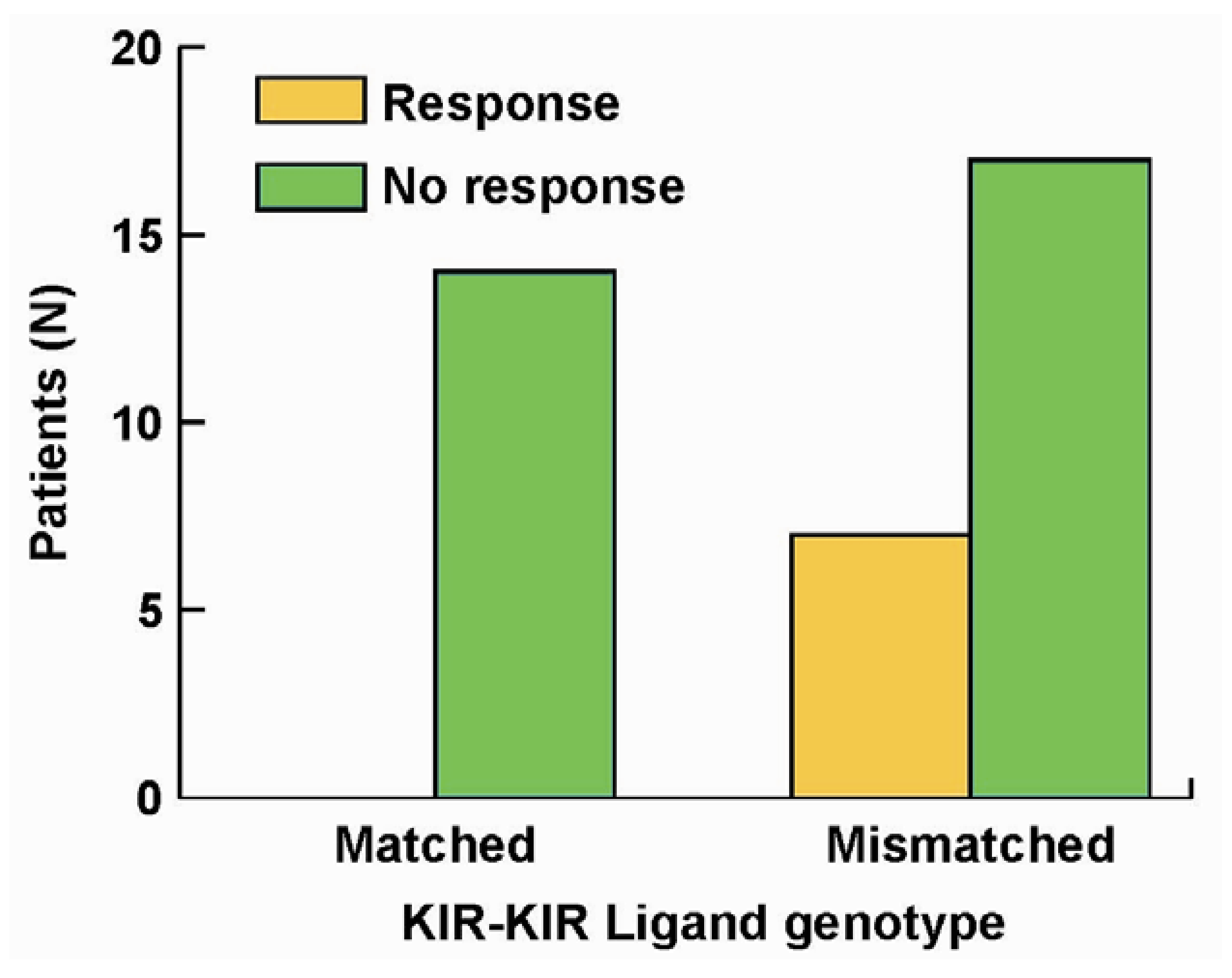
5. The Roles of KIR/KIR-Ligand (KIR-L) and FcR Genotypes in the Responses Induced by hu14.18-IL2
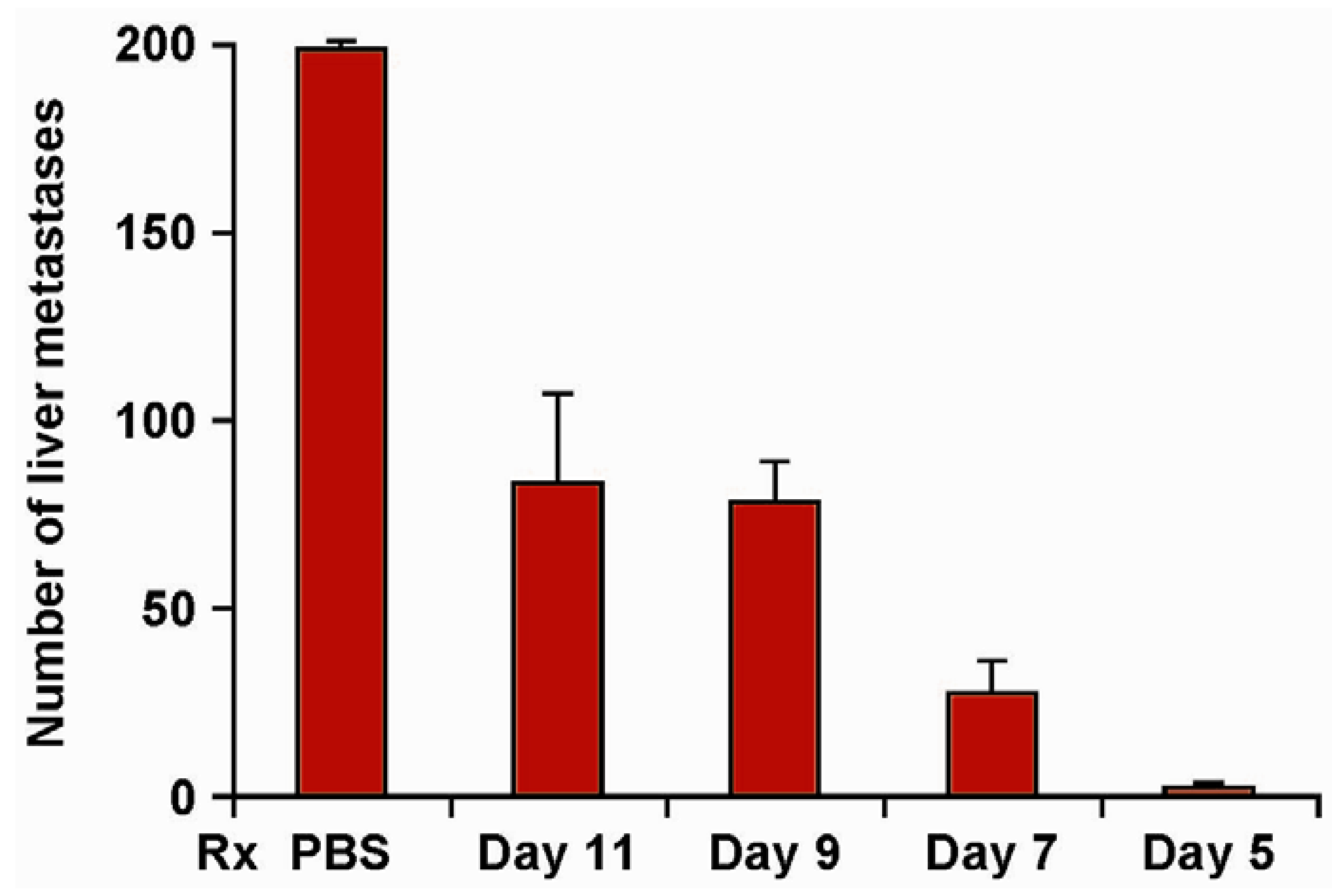
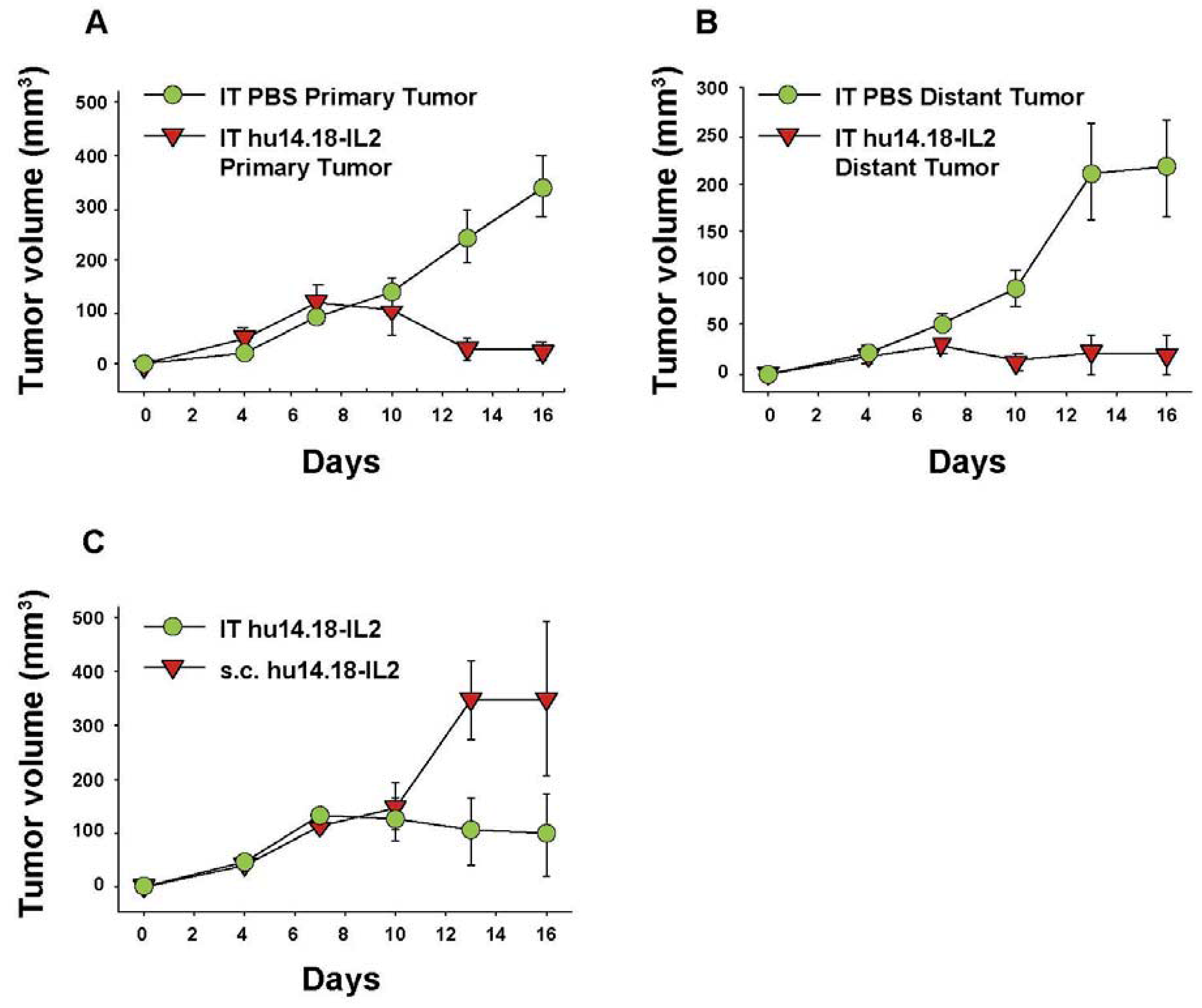
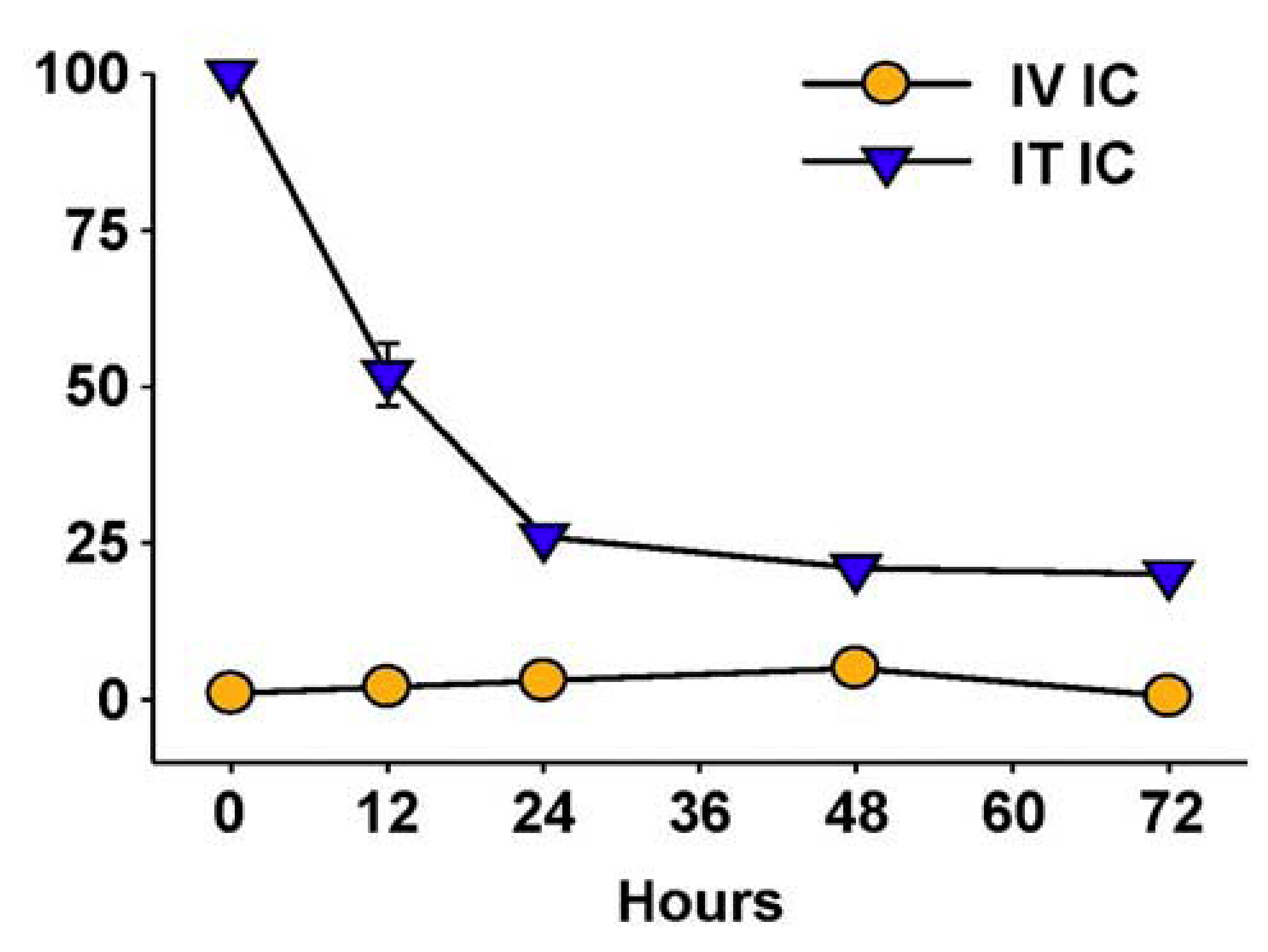
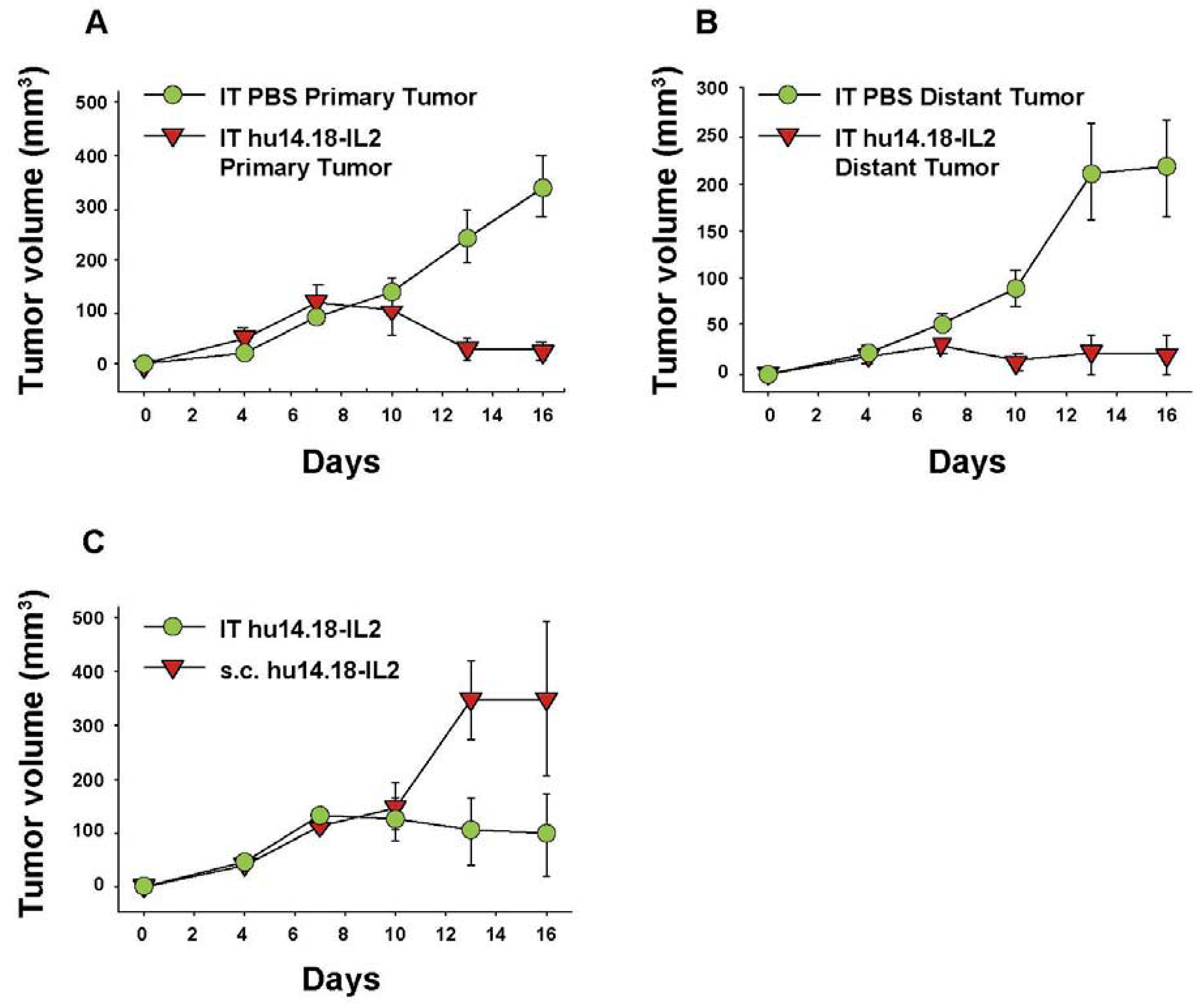
6. Augmenting Local Antitumor Activity by IT Injection of IC
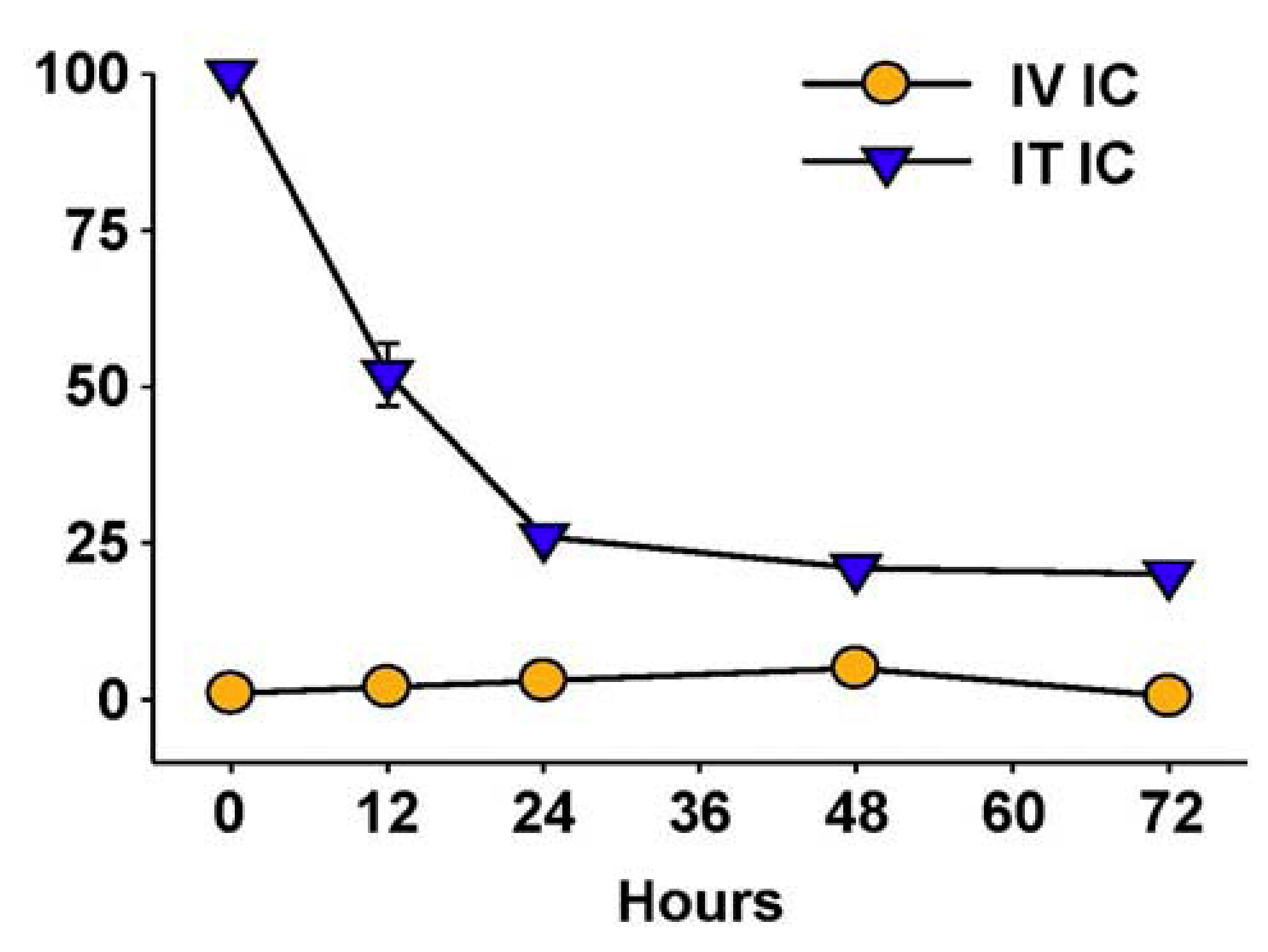
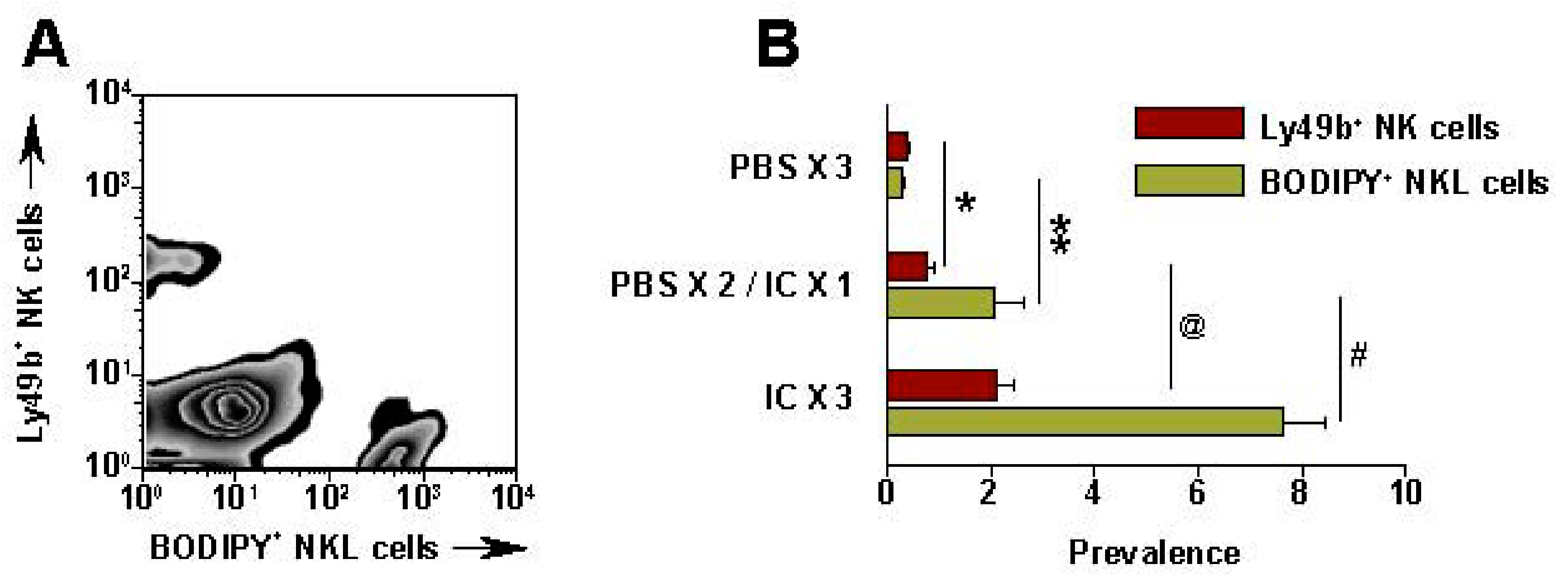
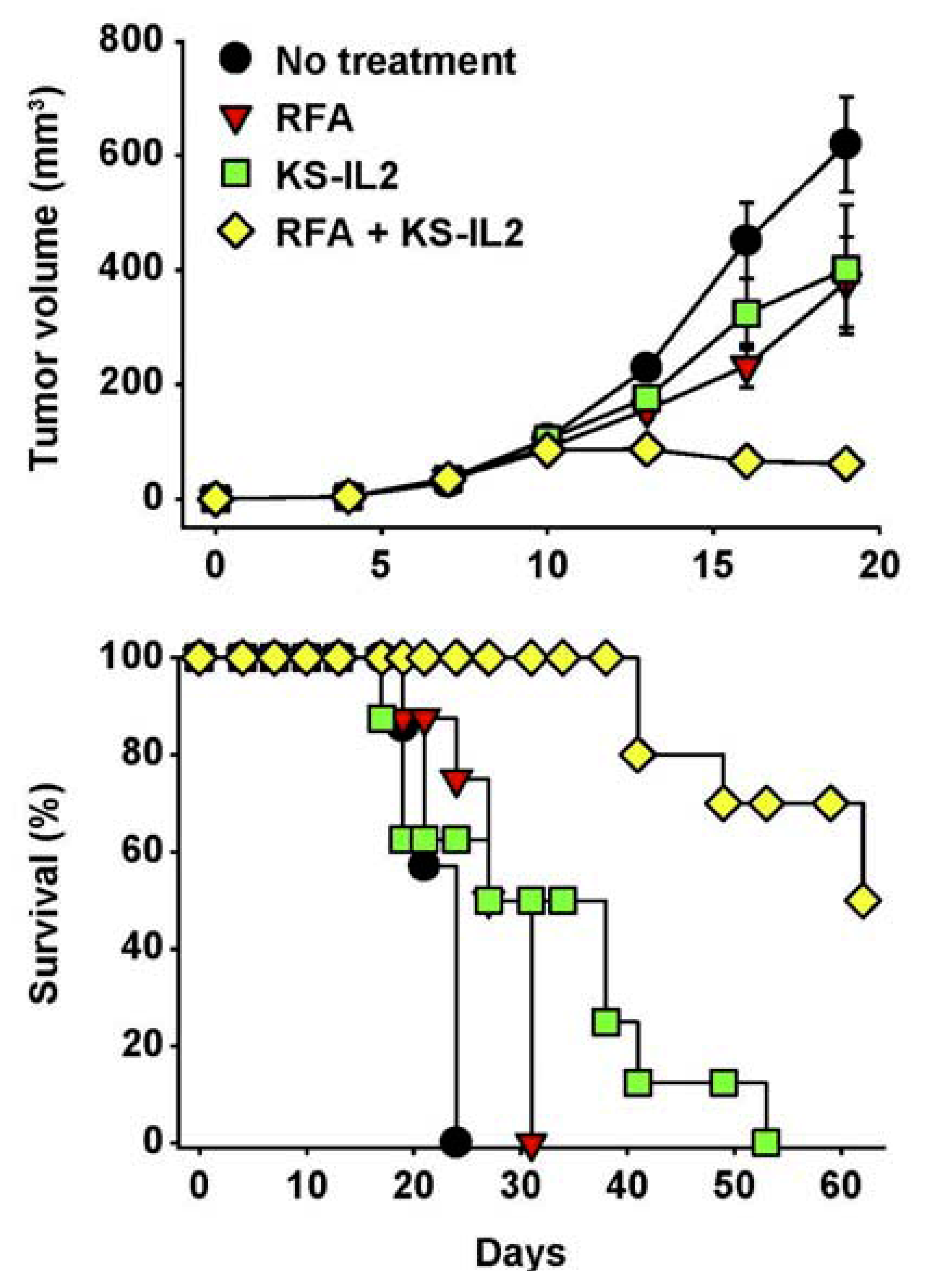
7. Future Directions for IC Development and Treatment
Acknowledgements
References
- Horowitz, M.M.; Gale, R.P.; Sondel, P.M.; Goldman, J.M.; Kerse, J.; Kolb, H.J.; Rimm, A.A.; Ringden, O.; Rozman, C.; Speck, B. Graft-versus-leukemia reactions after bone marrow transplantation. Blood 1990, 75, 555–562. [Google Scholar]
- Ferris, R.L.; Jaffee, E.M.; Ferrone, S. Tumor antigen-targeted, monoclonal antibody-based immunotherapy: Clinical response, cellular immunity, and immunoescape. J. Clin. Oncol. 2010, 28, 4390–4399. [Google Scholar] [CrossRef]
- Krege, S.; Giani, G.; Meyer, R.; Otto, T.; Rubben, H. A randomized multicenter trial of adjuvant therapy in superficial bladder cancer: Transurethral resection only versus transurethral resection plus mitomycin C versus transurethral resection plus bacillus Calmette-Guerin. Participating Clinics. J. Urol. 1996, 156, 962–966. [Google Scholar]
- Rosenberg, S.A.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA 1994, 271, 907–913. [Google Scholar]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar]
- Hodi, F.S.; O'Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar]
- Fong, L.; Small, E.J. Anti-cytotoxic T-lymphocyte antigen-4 antibody: The first in an emerging class of immunomodulatory antibodies for cancer treatment. J. Clin. Oncol. 2008, 265, 275–283. [Google Scholar]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.; Chen, H.; Smith, M.; Anderson, B.; Villablanca, J.; Matthay, K.K.; et al. Chimeric Anti-GD2 Antibody with GM-CSF, IL2 and 13-cis Retinoic Acid for High-risk Neuroblastoma: A Children’s Oncology Group (COG) Phase 3 Study. N. Engl. J. Med. 2010, 335, 1324–1334. [Google Scholar]
- Basham, T.Y.; Race, E.R.; Campbell, M.J.; Reid, T.R.; Levy, R.; Merigan, T.C. Synergistic antitumor activity with IFN and monoclonal anti-idiotype for murine B cell lymphoma. Mechanism of action. J. Immunol. 1988, 141, 2855–2860. [Google Scholar]
- Primus, F.J.; Finch, M.D.; Wetzel, S.A.; Masci, A.M.; Schlom, J.; Kashmiri, S.V. Monoclonal antibody gene transfer. Implications for tumor-specific cell-mediated cytotoxicity. Ann. NY Acad. Sci. 1994, 716, 165–166. [Google Scholar]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Jefferis, R. Antibody therapeutics: Isotype and glycoform selection. Expert Opin. Biol. Th. 2007, 7, 1401–1413. [Google Scholar] [CrossRef]
- Farag, S.S.; VanDeusen, J.B.; Fehniger, T.A.; Caligiuri, M.A. Biology and clinical impact of human natural killer cells. Int. J. Hematol. 2003, 78, 7–17. [Google Scholar] [CrossRef]
- Roda, J.M.; Parihar, R.; Magro, C.; Nuovo, G.J.; Tridandapani, S.; Carson, W.E., 3rd. Natural killer cells produce T cell-recruiting chemokines in response to antibody-coated tumor cells. Cancer Res. 2006, 66, 517–526. [Google Scholar]
- Orange, J.S. Formation and function of the lytic NK-cell immunological synapse. Nat. Rev. Immunol. 2008, 8, 713–725. [Google Scholar] [CrossRef]
- Carlsten, M.; Malmberg, K.J.; Ljunggren, H.G. Natural killer cell-mediated lysis of freshly isolated human tumor cells. Int. J. Cancer 2009, 124, 757–762. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Analyzing antibody-Fc-receptor interactions. Methods Mol. Biol. 2008, 415, 151–162. [Google Scholar] [CrossRef]
- Cartron, G.; Dacheux, L.; Salles, G.; Solal-Celigny, P.; Bardos, P.; Colombat, P.; Watier, H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 2002, 99, 754–758. [Google Scholar]
- Weng, W.K.; Levy, R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J. Clin. Oncol. 2003, 21, 3940–3947. [Google Scholar] [CrossRef]
- Treon, S.P.; Mitsiades, C.; Mitsiades, N.; Young, G.; Doss, D.; Schlossman, R.; Anderson, K.C. Tumor Cell Expression of CD59 Is Associated With Resistance to CD20 Serotherapy in Patients With B-Cell Malignancies. J. Immunother. 2001, 24, 263–271. [Google Scholar] [CrossRef]
- Dzietczenia, J.; Wróbel, T.; Mazur, G.; Poreba, R.; Jaźwiec, B.; Kuliczkowski, K. Expression of complement regulatory proteins: CD46, CD55, and CD59 and response to rituximab in patients with CD20+ non-Hodgkin's lymphoma. Med. Oncol. 2010, 27, 743–746. [Google Scholar] [CrossRef]
- Binyamin, L.; Alpaugh, R.K.; Hughes, T.L.; Lutz, C.T.; Campbell, K.S.; Weiner, L.M. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J. Immunol. 2008, 180, 6392–6401. [Google Scholar]
- Wang, S.Y.; Veeramani, S.; Racila, E.; Cagley, J.; Fritzinger, D.C.; Vogel, C.W.; St John, W.; Weiner, G.J. Depletion of the C3 component of complement enhances the ability of rituximab-coated target cells to activate human NK cells and improves the efficacy of monoclonal antibody therapy in an in vivo model. Blood 2009, 114, 5322–5330. [Google Scholar]
- Shawver, L.K.; Slamon, D.; Ullrich, A. Smart drugs: Tyrosine kinase inhibitors in cancer therapy. Cancer Cell 2002, 1, 117–123. [Google Scholar] [CrossRef]
- Bibeau, F.; Lopez-Crapez, E.; Di Fiore, F.; Thezenas, S.; Ychou, M.; Blanchard, F.; Lamy, A.; Penault-Llorca, F.; Frébourg, T.; Michel, P.; et al. Impact of FcgRIIa-FcgRIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J. Clin. Oncol. 2009, 27, 1122–1129. [Google Scholar]
- Musolino, A.; Naldi, N.; Bortesi, B.; Pezzuolo, D.; Capelletti, M.; Missale, G.; Laccabue, D.; Zerbini, A.; Camisa, R.; Bisagni, G.; et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J. Clin. Oncol. 2008, 26, 1789–1796. [Google Scholar]
- Shan, D.; Ledbetter, J.A.; Press, O.W. Apoptosis of malignant human B cells by ligation of CD20 with monoclonal antibodies. Blood 1998, 91, 1644–1652. [Google Scholar]
- Cheung, N.K.; Sowers, R.; Vickers, A.J.; Cheung, I.Y.; Kushner, B.H.; Gorlick, R. FCGR2A polymorphism is correlated with clinical outcome after immunotherapy of neuroblastoma with anti-GD2 antibody and granulocyte macrophage colony-stimulating factor. J. Clin. Oncol. 2006, 24, 2885–2890. [Google Scholar]
- Weng, W.K.; Czerwinski, D.; Levy, R. Humoral immune response and immunoglobulin G Fc receptor genotype are associated with better clinical outcome following idiotype vaccination in follicular lymphoma patients regardless of their response to induction chemotherapy. Blood 2007, 109, 951–953. [Google Scholar]
- Wang, B.; Kokhaei, P.; Mellstedt, H.; Liljefors, M. FcγR polymorphisms and clinical outcome in colorectal cancer patients receiving passive or active antibody treatment. Int. J. Oncol. 2010, 37, 1599–1606. [Google Scholar]
- Roda, J.M.; Joshi, T.; Butchar, J.P.; McAlees, J.W.; Lehman, A.; Tridandapani, S.; Carson, W.E., 3rd. The activation of natural killer cell effector functions by cetuximab-coated, epidermal growth factor receptor positive tumor cells is enhanced by cytokines. Clin. Cancer Res. 2007, 13, 6419–6428. [Google Scholar]
- Kloess, S.; Huenecke, S.; Piechulek, D.; Esser, R.; Koch, J.; Brehm, C.; Soerensen, J.; Gardlowski, T.; Brinkmann, A.; Bader, P.; et al. IL-2-activated haploidentical NK cells restore NKG2D-mediated NK-cell cytotoxicity in neuroblastoma patients by scavenging of plasma MICA. Eur. J. Immunol. 2010, 40, 3255–3267. [Google Scholar]
- Hank, J.A.; Kohler, P.C.; Weil-Hillman, G.; Rosenthal, N.; Moore, K.H.; Storer, B.; Minkoff, D.; Bradshaw, J.; Bechhofer, R.; Sondel, P.M. In vivo induction of the lymphokine-activated killer phenomenon: Interleukin 2-dependent human non-major histocompatibility complex-restricted cytotoxicity generated in vivo during administration of human recombinant Interleukin 2. Cancer Res. 1988, 48, 1965–1971. [Google Scholar]
- Hank, J.A.; Robinson, R.R.; Surfus, J.; Mueller, B.M.; Reisfeld, R.A.; Cheung, N.-K.; Sondel, P.M. Augmentation of antibody dependent cell mediated cytotoxicity following in vivo therapy with recombinant Interleukin-2. Cancer Res. 1990, 50, 5234–5239. [Google Scholar]
- Sosman, J.A.; Hank, J.A.; Sondel, P.M. In vivo activation of lymphokine-activated killer activity with Interleukin-2: Prospects for combination therapies. Semin. Oncol. 1990, 17, 22–30. [Google Scholar]
- Hank, J.A.; Albertini, M.R.; Schiller, J.; Sondel, P.M. Activation of multiple effector mechanisms to enhance tumor immunotherapy. J. Immunother. 1993, 14, 329–335. [Google Scholar] [CrossRef]
- Mujoo, K.; Kipps, T.J.; Yang, H.M.; Cheresh, D.A.; Wargalla, U.; Sander, D.J.; Reisfeld, R.A. Functional properties and effect on growth suppression of human neuroblastoma tumors by isotype switch variants of monoclonal antiganglioside GD2 antibody 14.18. Cancer Res. 1989, 49, 2857–2861. [Google Scholar]
- Kendra, K.; Malkovska, V.; Allen, M.; Guzman, J.; Albertini, M. In vivo binding and antitumor activity of Ch14.18. J. Immunother. 1999, 5, 23–430. [Google Scholar]
- Neal, Z.C.; Yang, J.C.; Rakhmilevich, A.L.; Buhtoiarov, I.; Lum, H.E.; Imboden, M.; Hank, J.A.; Lode, H.N.; Reisfeld, R.A.; Gillies, S.D.; et al. Enhanced activity of hu14.18-IL2 immunocytokine against the murine NXS2 neuroblastoma when combined with IL2 therapy. Clin. Cancer Res. 2004, 10, 4839–4847. [Google Scholar]
- Hank, J.; Surfus, J.; Gan, J.; Chew, T.-L.; Hong, R.; Tans, K.; Reisfeld, R.; Seeger, R.; Reynolds, C.P.; Bauer, M.; et al. Treatment of neuroblastoma patients with antiganglioside GD2 antibody plus Interleukin-2 induces antibody dependent cellular cytotoxicity against neuroblastoma detected in vitro. J. Immunother. 1994, 15, 29–37. [Google Scholar] [CrossRef]
- Frost, J.D.; Ettinger, L.J.; Hank, J.A.; Cairo, M.S.; Reaman, G.H.; Blazar, B.R.; Frierdich, S.; Krailo, M.; Seeger, R.C.; Matthay, K.; et al. Phase I/IB trial of murine monoclonal anti-GD2 antibody 14.G2a plus IL-2 in children with refractory neuroblastoma: A report of the Children’s Cancer Group. Cancer 1997, 80, 317–333. [Google Scholar] [CrossRef]
- Albertini, M.R.; Hank, J.A.; Schiller, J.H.; Khorsand, M.; Borchert, A.A.; Gan, J.; Bechhofer, R.; Storer, B.; Reisfeld, R.A.; Sondel, P.M. Phase IB trial of chimeric anti-GD2 antibody plus interleukin-2 for melanoma patients. Clin. Cancer Res. 1997, 3, 1277–1288. [Google Scholar]
- Gilman, A.L.; Ozkaynak, F.; Matthay, K.; Krailo, M.; Yu, A.; Gan, J.; Sternberg, A.; Hank, J.; Seeger, R.; Reaman, G.; et al. Phase I Study of ch14.18 with GM-CSF and IL-2 in Children with Neuroblastoma Following Autologous Bone Marrow Transplant or Stem Cell Rescue: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2009, 27, 85–91. [Google Scholar]
- Yu, A.L.; Batova, A.; Alvarado, C.; Rao, V.J.; Castleberry, R.P. Usefulness of a chimeric anti-GD2 (ch14.18) and GM-CSF for refractory neuroblastoma. Proc. Am. Assoc. Clin. Oncol. 1997, 6, 1846. [Google Scholar]
- Kushner, B.H.; Kramer, K.; Cheung, N.K. Phase II trial of the anti-G(D2) monoclonal antibody 3F8 and granulocyte-macrophage colony-stimulating factor for neuroblastoma. J. Clin. Oncol. 2001, 19, 4189–4194. [Google Scholar]
- Arndt, C.A.; Koshkina, N.V.; Inwards, C.Y.; Hawkins, D.S.; Krailo, M.D.; Villaluna, D.; Anderson, P.M.; Goorin, A.M.; Blakely, M.L.; Bernstein, M.; et al. Inhaled granulocyte-macrophage colony stimulating factor for first pulmonary recurrence of osteosarcoma: Effects on disease-free survival and immunomodulation. A report from the Children's Oncology Group. Clin. Cancer Res. 2010, 16, 4024–4030. [Google Scholar]
- Simon, T.; Hero, B.; Faldum, A.; Handgretinger, R.; Schrappe, M.; Niethammer, D.; Berthold, F. Consolidation treatment with chimeric anti-GD2-antibody ch14.18 in children older than 1 year with metastatic neuroblastoma. J. Clin. Oncol. 2004, 22, 3549–3557. [Google Scholar] [CrossRef]
- Weil-Hillman, G.; Fisch, P.; Prieves, A.F.; Sosman, J.A.; Hank, J.A.; Sondel, P.M. Lymphokine-activated killer activity induced by in vivo interleukin-2 therapy: Predominant role for lymphocytes with increased expression of CD2 and Leul9 antigens but negative expression of CD16 antigens. Cancer Res. 1989, 49, 3680–3688. [Google Scholar]
- Voss, S.D.; Robb, R.J.; Weil-Hillman, G.; Hank, J.A.; Sugamum, K.; Tsudo, M.; Sondel, P.M. Increased expression of the interleukin-2 (IL2) receptor beta chain (p70) on CD56+ natural killer cells after in vivo IL2 therapy: p70 expression does not alone predict the level of intermediate affinity IL2 binding. J. Exp. Med. 1990, 172, 1101–1114. [Google Scholar]
- Sondel, P.M.; Kohler, P.C.; Hank, J.A.; Moore, K.H.; Rosenthal, N.; Sosman, J.; Bechhofer, R.; Storer, B. Clinical and immunological effects of recombinant interleukin-2 given by repetitive weekly cycles to subjects with cancer. Cancer Res. 1988, 48, 2561–2567. [Google Scholar]
- Gillies, S.D.; Reilly, E.B.; Lo, K.-M.; Reisfeld, R.A. Antibody-targeted interleukin 2 stimulates the T-cell killing of autologous tumor cells. Proc. Natl. Acad. Sci. USA 1992, 89, 1428. [Google Scholar]
- Hank, J.A.; Surfus, J.E.; Gan, J.; Jaeger, P.; Gillies, S.; Reisfeld, R.A.; Sondel, P.M. Activation of human effector cells by a tumor reactive recombinant anti-ganglioside GD2/interleukin-2 immunocytokine (ch14.18-IL2). Clin. Cancer Res. 1996, 2, 1951–1959. [Google Scholar]
- Sabzevari, H.; Gillies, S.D.; Mueller, B.M.; Pancook, J.D.; Reisfeld, R.A. A recombinant antibody-interleukin 2 immunocytokine suppresses growth of hepatic human neuroblastoma metastases in severe combined immunodeficiency mice. Proc. Natl. Acad. Sci. USA 1994, 91, 9626. [Google Scholar]
- Becker, J.C.; Pancook, J.D.; Gillies, S.D.; Furukawa, K.; Reisfeld, R.A. T cell mediated eradiation of murine metastatic melanoma induced by targeted interleukin-2 therapy. J. Exp. Med. 1996, 183, 2361. [Google Scholar] [CrossRef]
- Becker, J.C.; Varki, N.; Gillies, S.D.; Furukawa, K.; Reisfeld, R.A. An antibody-interleukin-2 fusion protein overcomes tumor heterogeneity by induction of a cellular immune response. Proc. Natl. Acad. Sci. USA 1996, 93, 7826–7831. [Google Scholar]
- Lode, H.N.; Xiang, R.; Varki, N.M.; Dolman, C.S.; Gillies, S.D.; Reisfeld, R.A. Targeted interleukin-2 therapy of spontaneous neuroblastoma to bone marrow. J. Natl. Cancer Inst. 1997, 89, 1586–1591. [Google Scholar] [CrossRef]
- Lode, H.N.; Xiang, R.; Drier, T.; Varki, N.M.; Gillies, S.D.; Reisfeld, R.A. Natural killer cell mediated eradication of neuroblastoma metastases to bone marrow by targeted IL2 therapy. Blood 1998, 91, 1706–1715. [Google Scholar]
- Neal, Z.C.; Imboden, M.; Rakhmilevich, A.L.; Kim, K.M.; Hank, J.A.; Surfus, J.; Dixon, J.R.; Lode, H.N.; Reisfeld, R.A.; Gillies, S.D. NXS2 murine neuroblastomas express increased levels of MHC class I antigens upon recurrence following NK-dependent immunotherapy. Cancer Immunol Immun. 2003, 53, 41–52. [Google Scholar]
- King, D.M.; Albertini, M.R.; Schalch, H.; Hank, J.A.; Gan, J.; Surfus, J.; Mahvi, D.; Schiller, J.H.; Warner, T.; Kim, K.M.; et al. A Phase I Clinical Trial of the Immunocytokine EMD 273063 (hu14.18-IL2) in Patients with melanoma. J. Clin. Oncol. 2004, 22, 4463–4473. [Google Scholar]
- Osenga, K.L.; Hank, J.A.; Albertini, M.R.; Gan, J.; Sternberg, A.G.; Eickhoff, J.; Seeger, R.C.; Matthay, K.K.; Reynolds, C.P.; Twist, C.; et al. A Phase I Clinical Trial of Hu14.18-IL2 (EMD 273063) as a Treatment for Children with Refractory or Recurrent Neuroblastoma and Melanoma: a Study of the Children’s Oncology Group. Clin. Cancer Res. 2005, 12, 1750–1759. [Google Scholar]
- Shusterman, S.; London, W.B.; Gillies, S.D.; Hank, J.A.; Voss, S.; Seeger, R.C.; Reynolds, C.P.; Kimball, J.; Albertini, M.A.; Wagner, B.; Gan, J.; et al. Anti-tumor activity of hu14.18-IL2 in relapsed/refractory neuroblastoma patients: A Children’s Oncology Group (COG) phase II study. J. Clin. Oncol. 2010, 28, 4969–4975. [Google Scholar]
- Joncker, N.T.; Shifrin, N.; Delebecque, F.; Raulet, D.H. Mature natural killer cells reset their responsiveness when exposed to an altered MHC environment. J. Exp. Med. 2010, 207, 2065–2072. [Google Scholar] [CrossRef]
- Elliott, J.M.; Wahle, J.A.; Yokoyama, W.M. MHC class I-deficient natural killer cells acquire a licensed phenotype after transfer into an MHC class I-sufficient environment. J. Exp. Med. 2010, 207, 2073–2079. [Google Scholar] [CrossRef]
- Sun, J.C. Re-educating natural killer cells. J. Exp. Med. 2010, 207, 2049–2052. [Google Scholar] [CrossRef]
- Orr, M.T.; Lanier, L.L. Natural killer cell education and tolerance. Cell 2010, 142, 847–856. [Google Scholar] [CrossRef]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar]
- Anfossi, N.; André, P.; Guia, S.; Falk, C.S.; Roetynck, S.; Stewart, C.A.; Breso, V.; Frassati, C.; Reviron, D.; Middleton, D.; et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity 2006, 25, 331–342. [Google Scholar] [CrossRef]
- Yawata, M.; Yawata, N.; Draghi, M.; Partheniou, F.; Little, A.M.; Parham, P. MHC class I-specific inhibitory receptors and their ligands structure diverse human NK-cell repertoires toward a balance of missing self-response. Blood 2008, 112, 2369–2380. [Google Scholar] [CrossRef]
- Sola, C.; André, P.; Lemmers, C.; Fuseri, N.; Bonnafous, C.; Bléry, M.; Wagtmann, N.R.; Romagné, F.; Vivier, E.; Ugolini, S. Genetic and antibody-mediated reprogramming of natural killer cell missing-self recognition in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 12879–12884. [Google Scholar]
- Sola, C.; André, P.; Lemmers, C.; Fuseri, N.; Bonnafous, C.; Bléry, M.; Wagtmann, N.R.; Romagné, F.; Vivier, E.; Ugolini, S. Genetic and antibody-mediated reprogramming of natural killer cell missing-self recognition in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 12879–12884. [Google Scholar]
- Pende, D.; Marcenaro, S.; Falco, M.; Martini, S.; Bernardo, M.E.; Montagna, D.; Romeo, E.; Cognet, C.; Martinetti, M.; Maccario, R.; et al. Anti-leukemia activity of alloreactive NK cells in KIR ligand-mismatched haploidentical HSCT for pediatric patients: Evaluation of the functional role of activating KIR and redefinition of inhibitory KIR specificity. Blood 2009, 113, 3119–3129. [Google Scholar]
- Moesta, A.K.; Norman, P.J.; Yawata, M.; Yawata, N.; Gleimer, M.; Parham, P. Synergistic Polymorphism at Two Positions Distal to the Ligand-Binding Site Makes KIR2DL2 a Stronger Receptor for HLA-C than KIR2DL3. J. Immol. 2008, 180, 3969–3979. [Google Scholar]
- Stern, M.; Ruggeri, L.; Capanni, M.; Mancusi, A.; Velardi, A. Human leukocyte antigens A23, A24, and A32 but not A25 are ligands for KIR3DL1. Blood 2008, 112, 708–710. [Google Scholar]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar]
- Ruggeri, L.; Mancusi, A.; Burchielli, E.; Capanni, M.; Carotti, A.; Aloisi, T.; Aversa, F.; Martelli, M.F.; Velardi, A. NK cell alloreactivity and allogeneic hematopoietic stem cell transplantation. Blood Cells Mol. Dis. 2008, 40, 84–90. [Google Scholar] [CrossRef]
- Leung, W.; Iyengar, R.; Turner, V.; Lang, P.; Bader, P.; Conn, P.; Niethammer, D.; Handgretinger, R. Determinants of antileukemia effects of allogeneic NK cells. J. Immunol. 2004, 172, 644–650. [Google Scholar]
- Hsu, K.C.; Keever-Taylor, C.A.; Wilton, A.; Pinto, C.; Heller, G.; Arkun, K.; O'Reilly, R.J.; Horowitz, M.M.; Dupont, B. Improved outcome in HLA-identical sibling hematopoietic stem-cell transplantation for acute myelogenous leukemia predicted by KIR and HLA genotypes. Blood 2005, 105, 4878–4884. [Google Scholar]
- Leung, W.; Handgretinger, R.; Iyengar, R.; Turner, V.; Holladay, M.S.; Hale, G.A. Inhibitory KIR-HLA receptor-ligand mismatch in autologous haematopoietic stem cell transplantation for solid tumour and lymphoma. Br. J.Cancer 2007, 97, 539–542. [Google Scholar] [CrossRef]
- Venstrom, J.M.; Zheng, J.; Noor, N.; Danis, K.E.; Yeh, A.W.; Cheung, I.Y.; Dupont, B.; O'Reilly, R.J.; Cheung, N.K.; Hsu, K.C. KIR and HLA genotypes are associated with disease progression and survival following autologous hematopoietic stem cell transplantation for high-risk neuroblastoma. Clin. Cancer Res. 2009, 15, 7330–7334. [Google Scholar]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar]
- Bachanova, V.; Burns, L.J.; McKenna, D.H.; Curtsinger, J.; Panoskaltsis-Mortari, A.; Lindgren, B.R.; Cooley, S.; Weisdorf, D.; Miller, J.S. Allogeneic natural killer cells for refractory lymphoma. Cancer Immunol. Immun. 2010, 59, 1739–1744. [Google Scholar]
- Geller, M.A.; Cooley, S.; Judson, P.L.; Ghebre, R.; Carson, L.F.; Argenta, P.A.; Jonson, A.L.; Panoskaltsis-Mortari, A.; Curtsinger, J.; McKenna, D.; et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011, 13, 98–107. [Google Scholar]
- Olson, J.A.; Leveson-Gower, D.B.; Gill, S.; Baker, J.; Beilhack, A.; Negrin, R.S. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood 2010, 115, 4293–4301. [Google Scholar]
- Delgado, D.C.; Hank, J.A.; Kolesar, J.; Lorentzen, D.; Gan, J.; Seo, S.; Kim, K.M.; Shusterman, S.; Gillies, S.D.; Reisfeld, R.A.; et al. Genotypes of NK cell KIR receptors, their ligands, and Fcγ receptors in the response of neuroblastoma patients to Hu14.18-IL2 immunotherapy. Cancer Res. 2010, 70, 9554–9661. [Google Scholar]
- Gillies, S.D.; Lan, Y.; Williams, S.; Carr, F.; Forman, S.; Raubitschek, A.; Lo, K.M. An anti-CD20-IL-2 immunocytokine is highly efficacious in a SCID mouse model of established human B lymphoma. Blood 2005, 105, 3972–3978. [Google Scholar] [CrossRef]
- Gubbels, J.A.; Gadbaw, B.; Buhtoiarov, I.N.; Horibata, S.; Kapur, A.K.; Patel, D.; Hank, J.A.; Gillies, S.D.; Sondel, P.M.; Patankar, M.S.; et al. Ab-IL2 fusion proteins mediate NK cell immune synapse formation by polarizing CD25 to the target cell-effector cell interface. Cancer Immunol. Immun. 2011, 60, 1789–1800. [Google Scholar]
- Buhtoiarov, I.N.; Neal, Z.C.; Gan, J.; Buhtoiarova, T.N.; Patankar, M.S.; Gubbels, J.A.A.; Hank, J.A.; Yamane, B.; Rakhmilevich, A.L.; Reisfeld, R.A.; et al. Differential internalization of hu14.18-IL2 immunocytokine by NK and tumor cells: Impact on conjugation, cytotoxicity and targeting. J. Leuk. Biol. 2011, 89, 625–638. [Google Scholar]
- Albertini, M.R.; Hank, J.A.; Gadbaw, B.; Kostlevy, J.; Haldeman, J.; Schalch, H.; Kim, K.M.; Eickhoff, J.; Gillies, S.D.; Sondel, P.M. Phase II Trial of Hu14.18-IL2 for Patients with Metastatic Melanoma. Cancer Immunol. Immun. 2012, in press.. [Google Scholar]
- Gillies, S.D.; Young, D.; Lo, K.-M.; Roberts, S. Biological activity and in vivo clearance of antitumor antibody/cytokine fusion proteins. Bioconjug. Chem. 1993, 4, 230–235. [Google Scholar] [CrossRef]
- Griffon-Etienne, G.; Boucher, Y.; Brekken, C.; Suit, H.D.; Jain, R.K. Taxane-induced apoptosis decompresses blood vessels and lowers interstitial fluid pressure in solid tumors: Clinical implications. CancerRes. 1999, 59, 3776–3782. [Google Scholar]
- Johnson, E.E.; Lum, H.D.; Rakhmilevich, A.L.; Schmidt, B.E.; Furlong, M.; Buhtoiarov, I.N.; Hank, J.A.; Raubitschek, A.; Colcher, D.; Reisfeld, R.A.; et al. Intratumoral Immunocytokine Treatment Results in Enhanced Antitumor Activity. Cancer Immunol. Immun. 2008, 57, 1891–1902. [Google Scholar]
- Weide, B.; Derhovanessian, E.; Pflugfelder, A.; Eigentler, T.K.; Radny, P.; Zelba, H.; Pföhler, C.; Pawelec, G.; Garbe, C. High response rate after intratumoral treatment with interleukin-2: Results from a phase 2 study in 51 patients with metastasized melanoma. Cancer 2010, 116, 4139–4146. [Google Scholar]
- Weide, B.; Eigentler, T.K.; Pflugfelder, A.; Leiter, U.; Meier, F.; Bauer, J.; Schmidt, D.; Radny, P.; Pföhler, C.; Garbe, C. Survival after intratumoral interleukin-2 treatment of 72 melanoma patients and response upon the first chemotherapy during follow-up. Cancer Immunol. Immun. 2011, 60, 487–493. [Google Scholar]
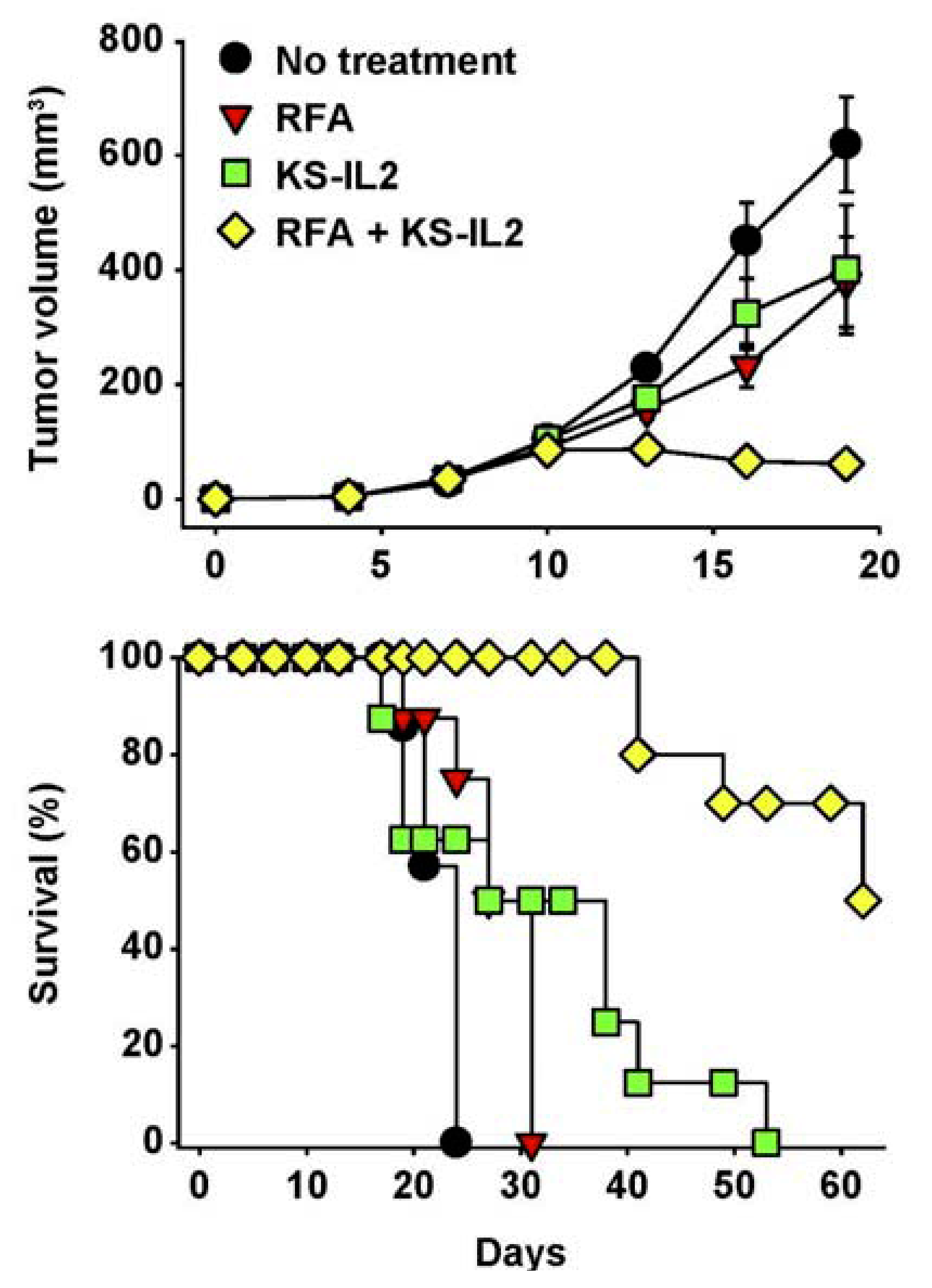
- Johnson, E.E.; Yamane, B.H.; Lum, H.D.; Buhtoiarov, I.N.; Rakhmilevich, A.L.; Mahvi, D.M.; Gillies, S.D.; Sondel, P.M. Radiofrequency Ablation Combined with KS-IL2 Immunocytokine (EMD 273066) Results in an Enhanced Anti-tumor Effect Against Murine Colon Adenocarcinoma. Clin. Cancer Res. 2009, 15, 4875–4884. [Google Scholar]
- Gillies, S.D. unpublished work, 2012; Provenance Biopharmaceuticals Corp.: Burlington, MA, USA.
- Gillies, S.D.; Lan, Y.; Lo, K.M.; Super, M.; Wesolowski, J. Improving the efficacy of antibody-interleukin 2 fusion proteins by reducing their interaction with Fc receptors. Cancer Res. 1999, 59, 2159–2166. [Google Scholar]
- Gillies, S.D.; Lo, K.M.; Burger, C.; Lan, Y.; Dahl, T.; Wong, W.K. Improved circulating half-life and efficacy of an antibody-interleukin 2 immunocytokine based on reduced intracellular proteolysis. Clin. Cancer Res. 2002, 8, 210–216. [Google Scholar]
- Wahlin, B.E.; Aggarwal, M.; Montes-Moreno, S.; Gonzalez, L.F.; Roncador, G.; Sanchez-Verde, L.; Christensson, B.; Sander, B.; Kimby, E. A unifying microenvironment model in follicular lymphoma: outcome is predicted by programmed death-1—Positive, regulatory, cytotoxic, and helper T cells and macrophages. Clin. Cancer Res. 2010, 16, 637–650. [Google Scholar]
- Shanafelt, A.B.; Lin, Y.; Shanafelt, M.C.; Forte, C.P.; Dubois-Stringfellow, N.; Carter, C.; Gibbons, J.A.; Cheng, S.L.; Delaria, K.A.; Fleischer, R.A. T-cell-selective interleukin 2 mutein exhibits potent antitumor activity and is well tolerated in vivo. Nat. Biotechnol. 2000, 18, 1197–1202. [Google Scholar]
- Gillies, S.D.; Lan, Y.; Hettmann, T.; Brunkhorst, B.; Sun, Y.; Mueller, S.O.; Lo, K.M. A low-toxicity IL-2-based immunocytokine retains antitumor activity despite its high degree of IL-2 receptor selectivity. Clin. Cancer Res. 2011, 17, 3673–3685. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sondel, P.M.; Gillies, S.D. Current and Potential Uses of Immunocytokines as Cancer Immunotherapy. Antibodies 2012, 1, 149-171. https://doi.org/10.3390/antib1020149
Sondel PM, Gillies SD. Current and Potential Uses of Immunocytokines as Cancer Immunotherapy. Antibodies. 2012; 1(2):149-171. https://doi.org/10.3390/antib1020149
Chicago/Turabian StyleSondel, Paul M., and Stephen D. Gillies. 2012. "Current and Potential Uses of Immunocytokines as Cancer Immunotherapy" Antibodies 1, no. 2: 149-171. https://doi.org/10.3390/antib1020149