
Episomal Vectors for Stable Production of Recombinant Proteins and Engineered Antibodies



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
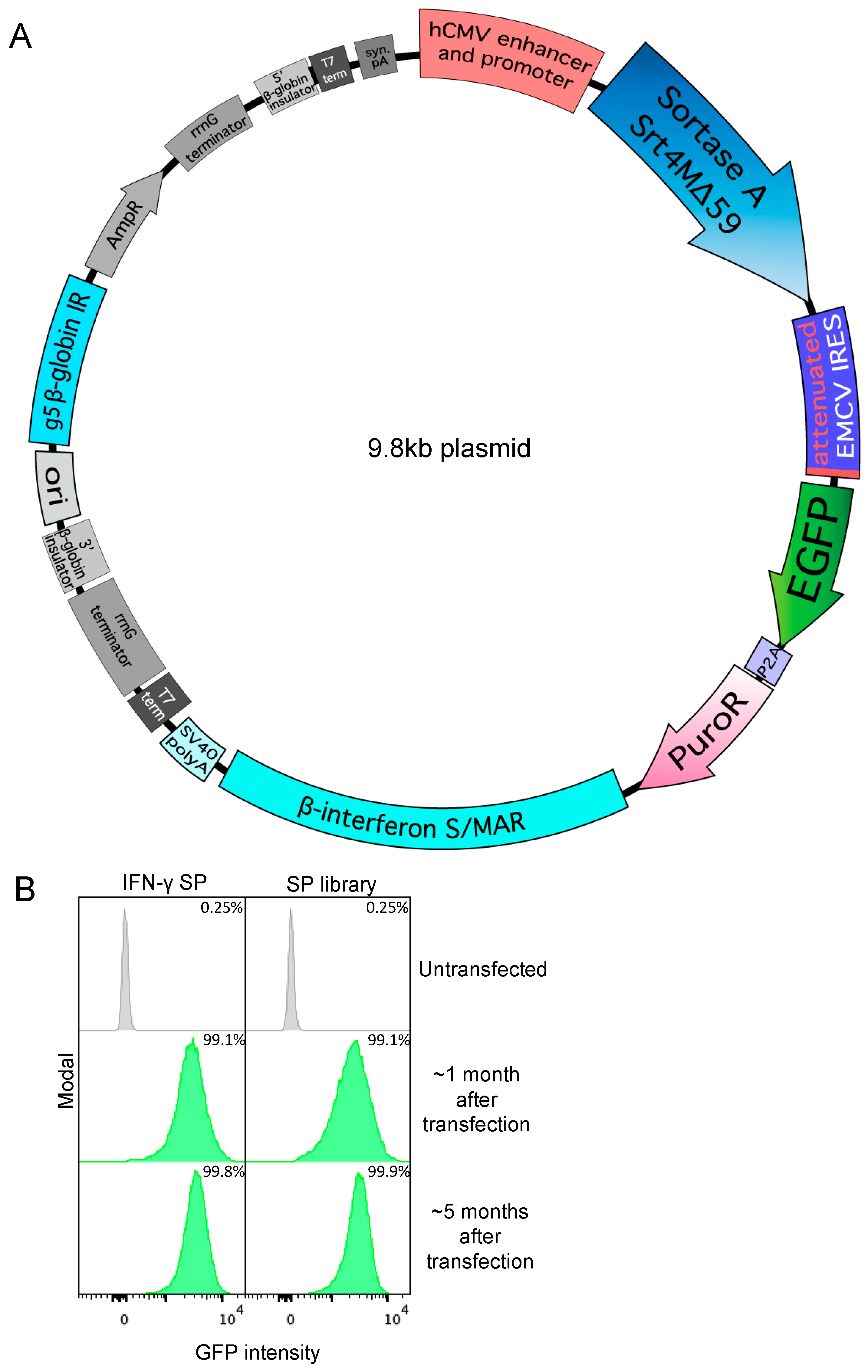
2.1. Vector Construction
2.2. Cloning of Single Proteins (e.g., Srt4M) Plasmids
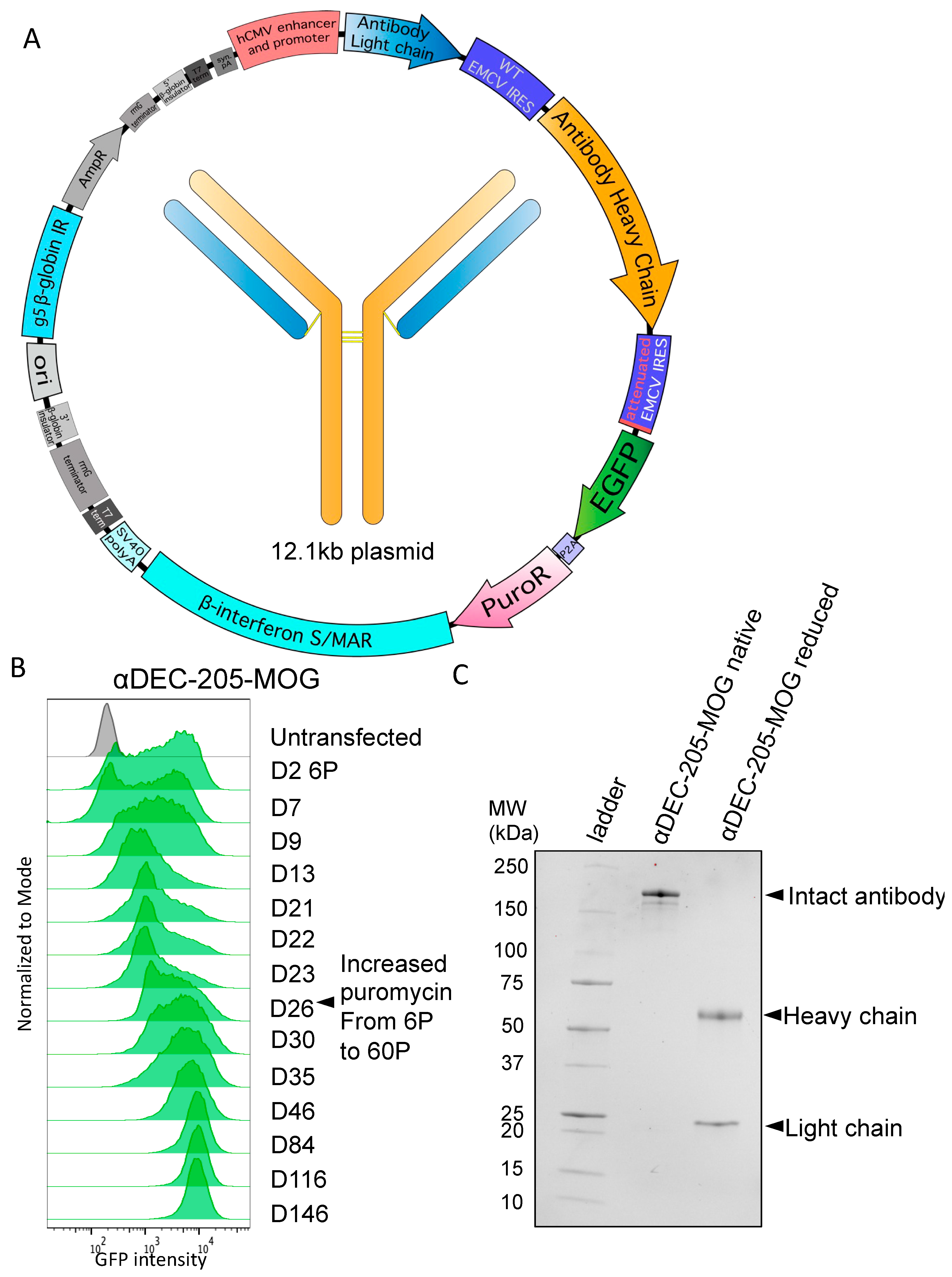
2.3. Cloning of Antibody Plasmids
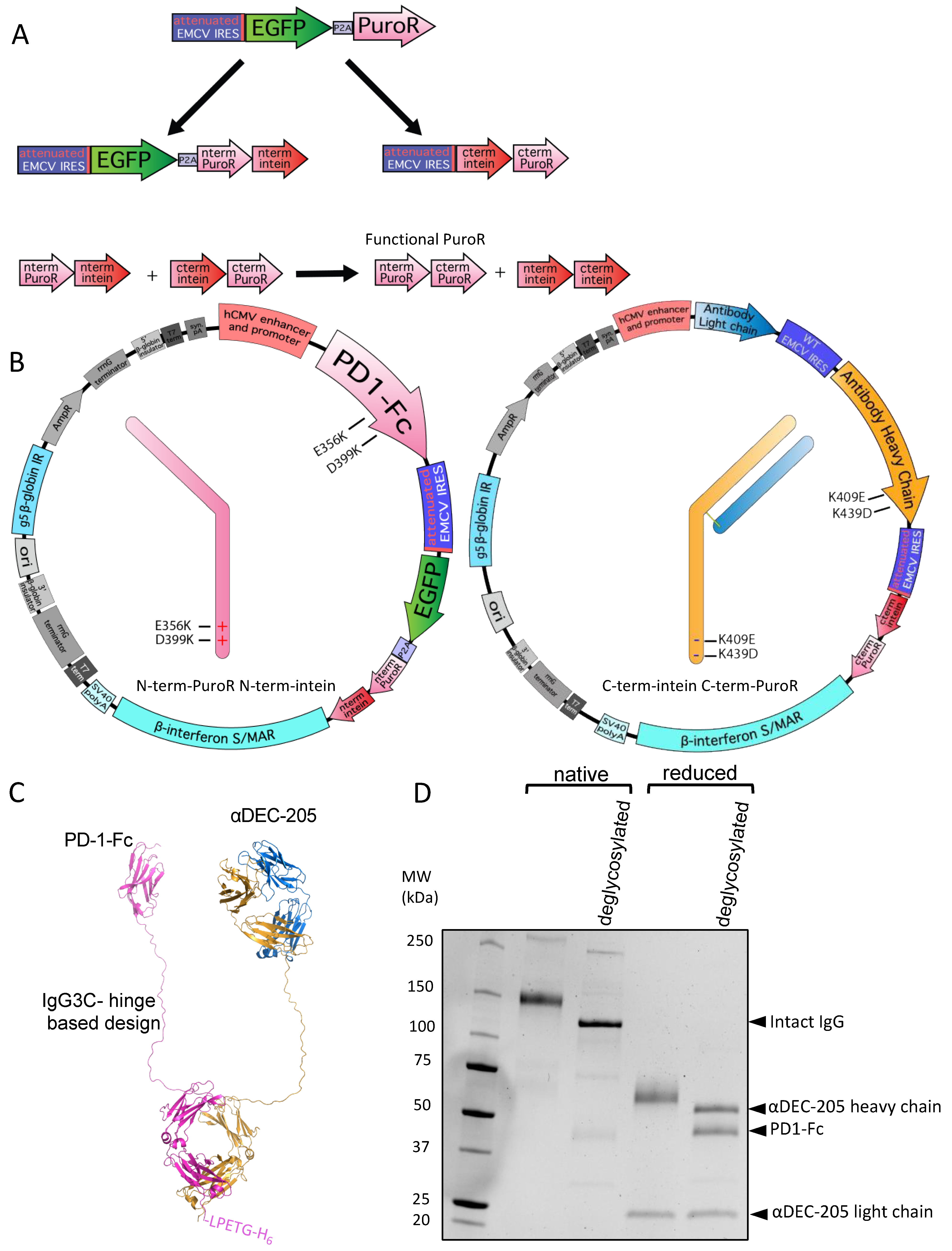
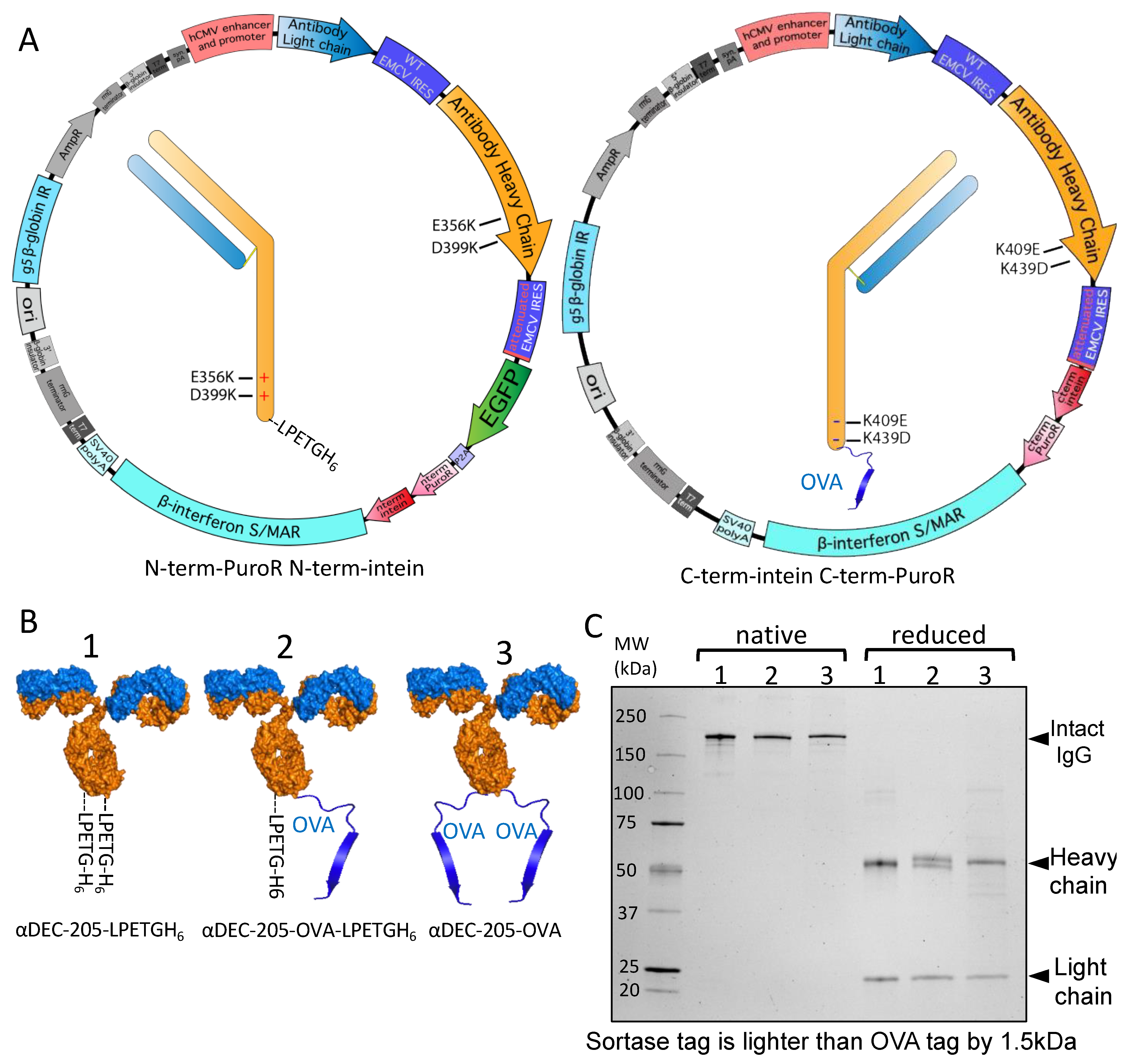
2.4. Single Plasmid for Bispecific Production
2.5. Cells, Transfection, and Selection
2.6. Antibody Expression and Protein Purification
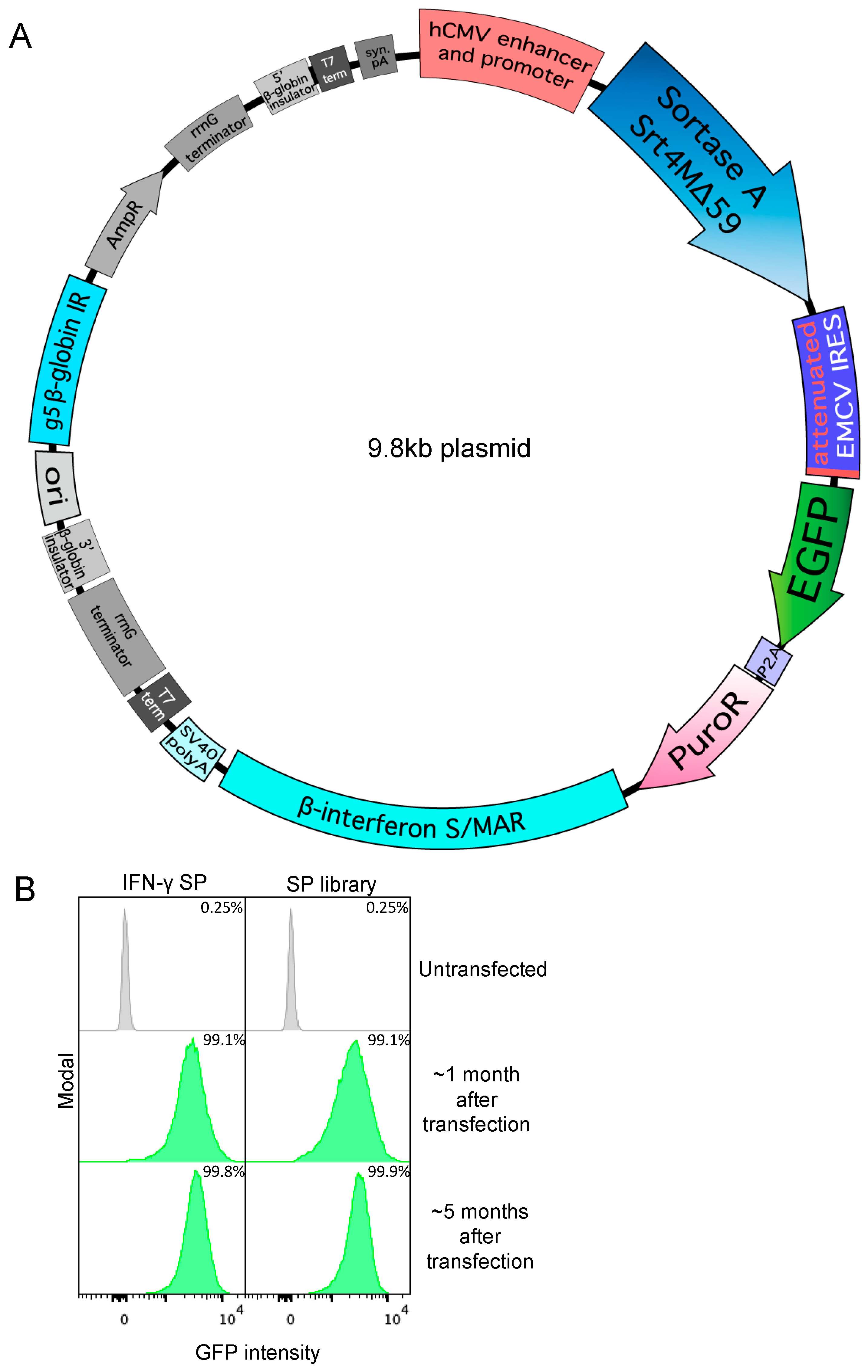
2.7. Sortase Signal Peptide Library Construction
2.8. Signal Peptide Library PCR Amplification and Sequencing
2.9. Antibody and Protein–Fc Constructs
2.10. siRNA
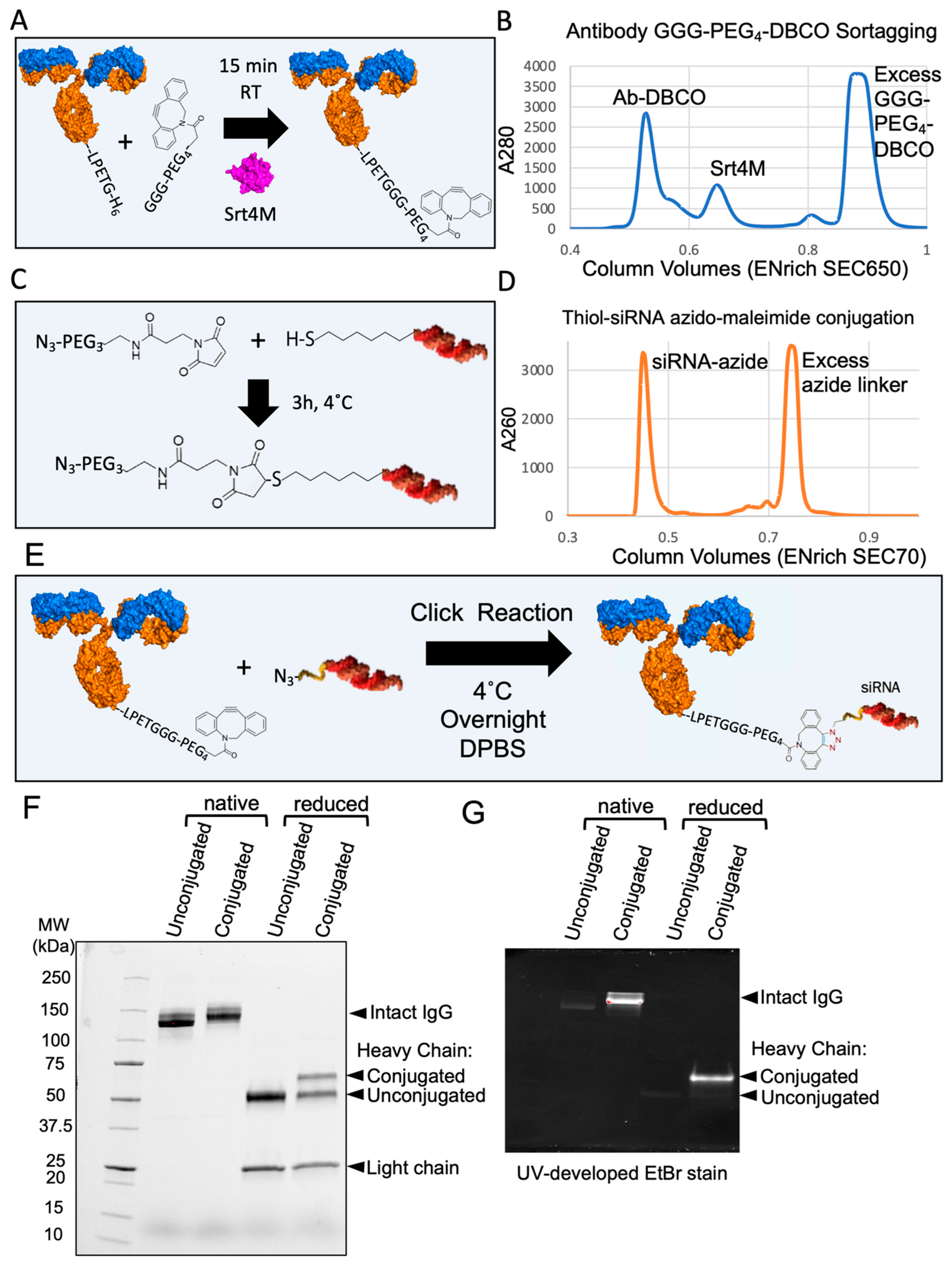
2.11. Antibody–siRNA Conjugation
3. Results
3.1. Secreted Expression of Sortase A Δ59 Variant Srt4M in Expi293
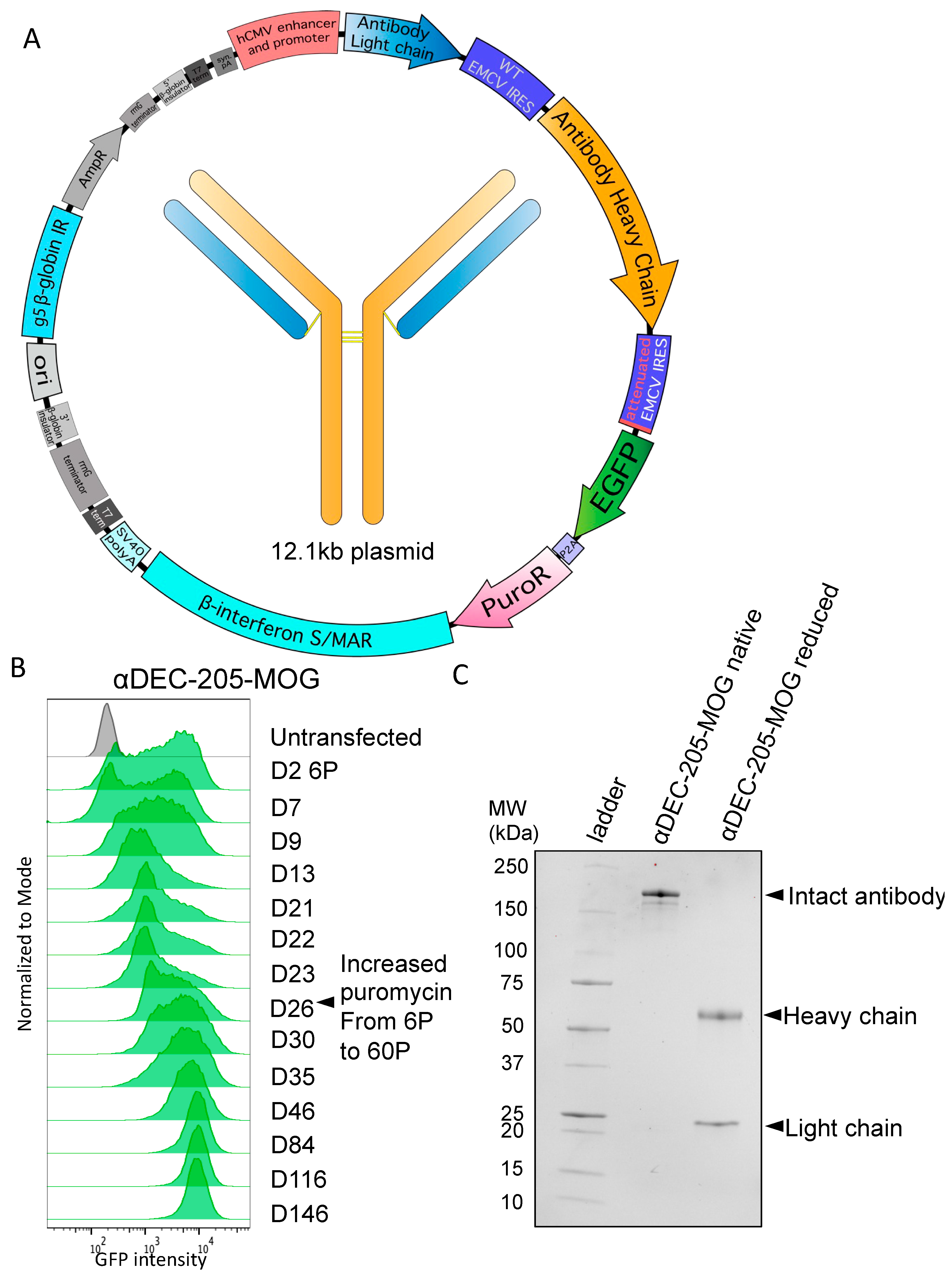
3.2. Stable Production of Antibodies with Heavy-Chain Tags
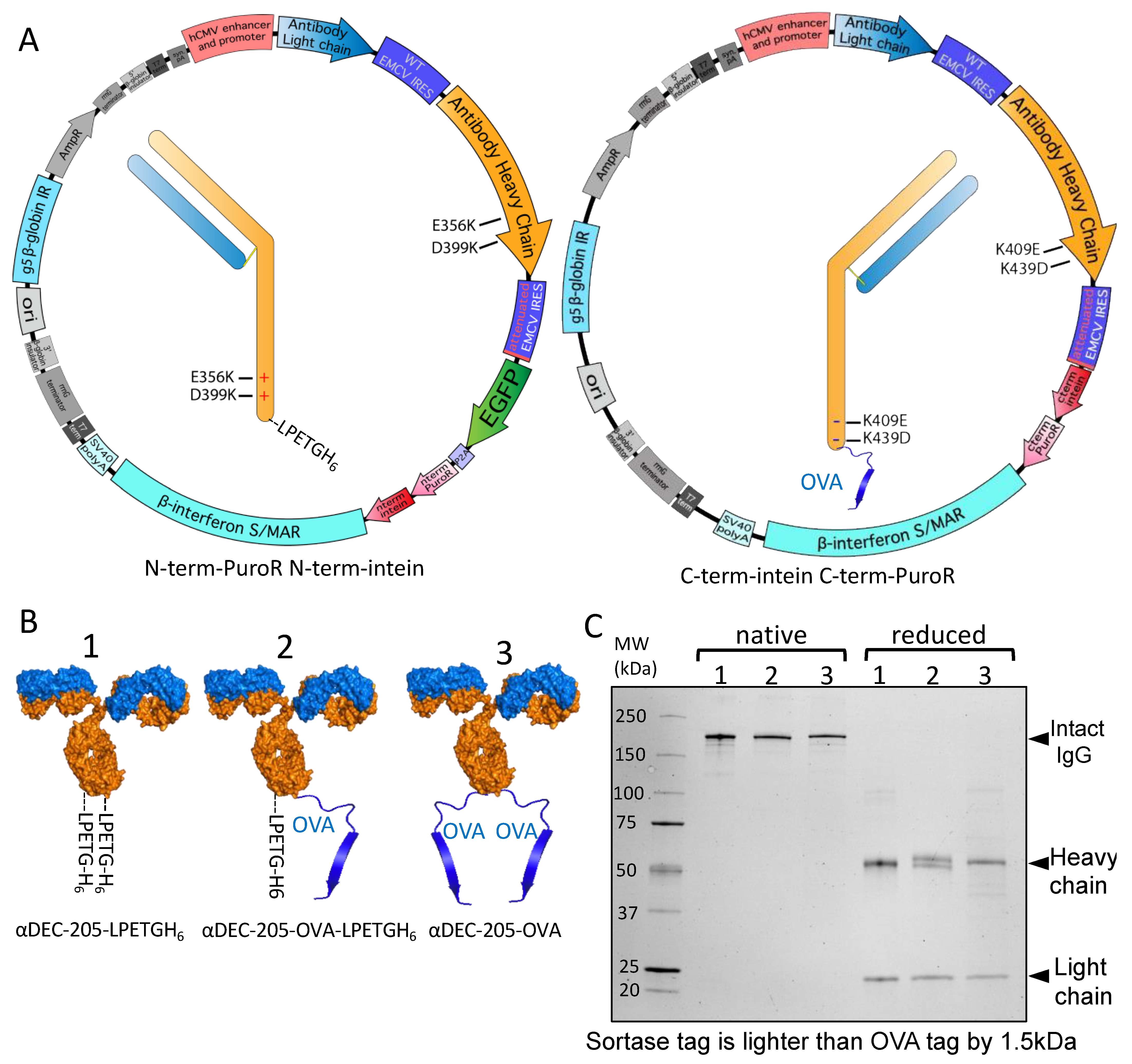
3.3. Utilizing Inteins for Production of a Bispecific Antibody-like Molecule
3.4. Split Inteins for Stable Production of Defined Multi-Tag Antibodies
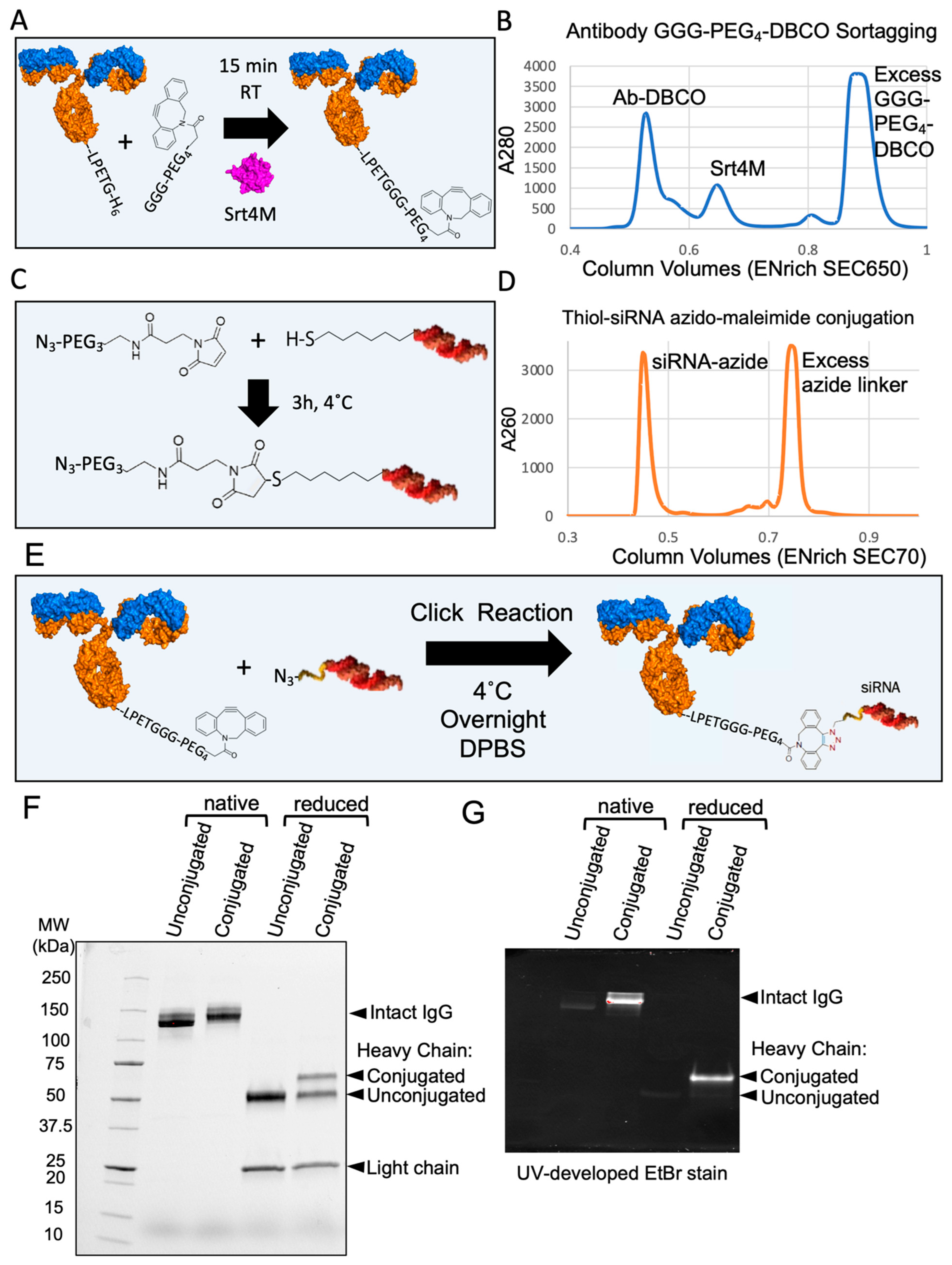
3.5. Site-Specific DAR1 siRNA–Antibody Conjugate
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, R.; Oldham, R.J.; Teal, E.; Beers, S.A.; Cragg, M.S. Fc-Engineering for Modulated Effector Functions-Improving Antibodies for Cancer Treatment. Antibodies 2020, 9, 64. [Google Scholar] [CrossRef]
- Abdeldaim, D.T.; Schindowski, K. Fc-Engineered Therapeutic Antibodies: Recent Advances and Future Directions. Pharmaceutics 2023, 15, 2402. [Google Scholar] [CrossRef]
- Tan, E.; Chin, C.S.H.; Lim, Z.F.S.; Ng, S.K. HEK293 Cell Line as a Platform to Produce Recombinant Proteins and Viral Vectors. Front. Bioeng. Biotechnol. 2021, 9, 796991. [Google Scholar] [CrossRef]
- Thompson, N.; Wakarchuk, W. O-glycosylation and its role in therapeutic proteins. Biosci. Rep. 2022, 42, 94. [Google Scholar] [CrossRef] [PubMed]
- Croset, A.; Delafosse, L.; Gaudry, J.P.; Arod, C.; Glez, L.; Losberger, C.; Begue, D.; Krstanovic, A.; Robert, F.; Vilbois, F.; et al. Differences in the glycosylation of recombinant proteins expressed in HEK and CHO cells. J. Biotechnol. 2012, 161, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Bosques, C.J.; Collins, B.E.; Meador, J.W., 3rd; Sarvaiya, H.; Murphy, J.L.; Dellorusso, G.; Bulik, D.A.; Hsu, I.H.; Washburn, N.; Sipsey, S.F.; et al. Chinese hamster ovary cells can produce galactose-α-1,3-galactose antigens on proteins. Nat. Biotechnol. 2010, 28, 1153–1156. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, A.; Mukuria, C.J.; Suzuki, E.; Naiki, M. Immunogenicity of N-glycolylneuraminic acid-containing carbohydrate chains of recombinant human erythropoietin expressed in Chinese hamster ovary cells. J. Biochem. 1995, 117, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.; Xie, F.; Beyl, R.N.; Chen, L.; Lewis, J.D.; Saag, K.G.; Curtis, J.R. Risk of Hypersensitivity to Biologic Agents Among Medicare Patients with Rheumatoid Arthritis. Arthritis Care Res. 2017, 69, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- Greven, J.A.; Brett, T.J. Production of Eukaryotic Glycoproteins for Structural and Functional Studies Using Expi293F Cells. Curr. Protoc. 2022, 2, e512. [Google Scholar] [CrossRef]
- 100044202. Gibco/ThermoFisher Scientific: Website. 2017. Available online: https://www.thermofisher.com/order/catalog/product/100044202 (accessed on 27 February 2024).
- Agrawal, V.; Slivac, I.; Perret, S.; Bisson, L.; St-Laurent, G.; Murad, Y.; Zhang, J.; Durocher, Y. Stable Expression of Chimeric Heavy Chain Antibodies in CHO Cells. In Single Domain Antibodies: Methods and Protocols; Saerens, D., Muyldermans, S., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 287–303. [Google Scholar] [CrossRef]
- Tandon, N.; Thakkar, K.N.; LaGory, E.L.; Liu, Y.; Giaccia, A.J. Generation of Stable Expression Mammalian Cell Lines Using Lentivirus. Bio Protoc. 2018, 8, e3073. [Google Scholar] [CrossRef] [PubMed]
- Kowarz, E.; Löscher, D.; Marschalek, R. Optimized Sleeping Beauty transposons rapidly generate stable transgenic cell lines. Biotechnol. J. 2015, 10, 647–653. [Google Scholar] [CrossRef]
- Napoleone, A.; Laurén, I.; Linkgreim, T.; Dahllund, L.; Persson, H.; Andersson, O.; Olsson, A.; Hultqvist, G.; Frank, P.; Hall, M.; et al. Fed-batch production assessment of a tetravalent bispecific antibody: A case study on piggyBac stably transfected HEK293 cells. New Biotechnol. 2021, 65, 9–19. [Google Scholar] [CrossRef]
- Li, Z.; Michael, I.P.; Zhou, D.; Nagy, A.; Rini, J.M. Simple piggyBac transposon-based mammalian cell expression system for inducible protein production. Proc. Natl. Acad. Sci. USA 2013, 110, 5004–5009. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, S.; Lu, M.; Tinberg, C.E.; Alba, B.M. Protein production from HEK293 cell line-derived stable pools with high protein quality and quantity to support discovery research. PLoS ONE 2023, 18, e0285971. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Hamafuji, T.; Noguchi, C.; Shimizu, N. Efficient recombinant production in mammalian cells using a novel IR/MAR gene amplification method. PLoS ONE 2012, 7, e41787. [Google Scholar] [CrossRef] [PubMed]
- Ohira, T.; Miyauchi, K.; Uno, N.; Shimizu, N.; Kazuki, Y.; Oshimura, M.; Kugoh, H. An efficient protein production system via gene amplification on a human artificial chromosome and the chromosome transfer to CHO cells. Sci. Rep. 2019, 9, 16954. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, C.; Araki, Y.; Miki, D.; Shimizu, N. Fusion of the Dhfr/Mtx and IR/MAR gene amplification methods produces a rapid and efficient method for stable recombinant protein production. PLoS ONE 2012, 7, e52990. [Google Scholar] [CrossRef]
- Stavrou, E.F.; Simantirakis, E.; Verras, M.; Barbas, C.; Vassilopoulos, G.; Peterson, K.R.; Athanassiadou, A. Episomal vectors based on S/MAR and the β-globin Replicator, encoding a synthetic transcriptional activator, mediate efficient γ-globin activation in haematopoietic cells. Sci. Rep. 2019, 9, 19765. [Google Scholar] [CrossRef]
- Jenke, B.H.; Fetzer, C.P.; Stehle, I.M.; Jönsson, F.; Fackelmayer, F.O.; Conradt, H.; Bode, J.; Lipps, H.J. An episomally replicating vector binds to the nuclear matrix protein SAF-A in vivo. EMBO Rep. 2002, 3, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lin, C.M.; Brooks, S.; Cimbora, D.; Groudine, M.; Aladjem, M.I. The human beta-globin replication initiation region consists of two modular independent replicators. Mol. Cell Biol. 2004, 24, 3373–3386. [Google Scholar] [CrossRef]
- Shimizu, N.; Hashizume, T.; Shingaki, K.; Kawamoto, J.K. Amplification of plasmids containing a mammalian replication initiation region is mediated by controllable conflict between replication and transcription. Cancer Res. 2003, 63, 5281–5290. [Google Scholar] [PubMed]
- Jenke, A.C.; Stehle, I.M.; Herrmann, F.; Eisenberger, T.; Baiker, A.; Bode, J.; Fackelmayer, F.O.; Lipps, H.J. Nuclear scaffold/matrix attached region modules linked to a transcription unit are sufficient for replication and maintenance of a mammalian episome. Proc. Natl. Acad. Sci. USA 2004, 101, 11322–11327. [Google Scholar] [CrossRef] [PubMed]
- Kingston, R.E.; Kaufman, R.J.; Bebbington, C.R.; Rolfe, M.R. Amplification using CHO cell expression vectors. Curr. Protoc. Mol. Biol. 2002, 16, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Stavrou, E.F.; Lazaris, V.M.; Giannakopoulos, A.; Papapetrou, E.; Spyridonidis, A.; Zoumbos, N.C.; Gkountis, A.; Athanassiadou, A. The β-globin Replicator greatly enhances the potential of S/MAR based episomal vectors for gene transfer into human haematopoietic progenitor cells. Sci. Rep. 2017, 7, 40673. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.C.; Bardor, M.; Feng, H.; Tong, Y.W.; Song, Z.; Yap, M.G.; Yang, Y. IRES-mediated Tricistronic vectors for enhancing generation of high monoclonal antibody expressing CHO cell lines. J. Biotechnol. 2012, 157, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Yeo, J.H.M.; Mariati; Yang, Y. An IRES-Mediated Tricistronic Vector for Efficient Generation of Stable, High-Level Monoclonal Antibody Producing CHO DG44 Cell Lines. Methods Mol. Biol. 2018, 1827, 335–349. [Google Scholar] [CrossRef]
- Rees, S.; Coote, J.; Stables, J.; Goodson, S.; Harris, S.; Lee, M.G. Bicistronic vector for the creation of stable mammalian cell lines that predisposes all antibiotic-resistant cells to express recombinant protein. Biotechniques 1996, 20, 102–104, 106, 108–110. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Soubias, O.; Khago, D.; Chao, F.A.; Li, Y.; Shaw, K.; Byrd, R.A. Optimization of sortase A ligation for flexible engineering of complex protein systems. J. Biol. Chem. 2020, 295, 2664–2675. [Google Scholar] [CrossRef]
- Chen, I.; Dorr, B.M.; Liu, D.R. A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. USA 2011, 108, 11399–11404. [Google Scholar] [CrossRef]
- Güler-Gane, G.; Kidd, S.; Sridharan, S.; Vaughan, T.J.; Wilkinson, T.C.; Tigue, N.J. Overcoming the Refractory Expression of Secreted Recombinant Proteins in Mammalian Cells through Modification of the Signal Peptide and Adjacent Amino Acids. PLoS ONE 2016, 11, e0155340. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, P.; Mistry, R.K.; Brown, A.J.; James, D.C. Protein-Specific Signal Peptides for Mammalian Vector Engineering. ACS Synth. Biol. 2023, 12, 2339–2352. [Google Scholar] [CrossRef] [PubMed]
- Barash, S.; Wang, W.; Shi, Y. Human secretory signal peptide description by hidden Markov model and generation of a strong artificial signal peptide for secreted protein expression. Biochem. Biophys. Res. Commun. 2002, 294, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Hawiger, D.; Inaba, K.; Dorsett, Y.; Guo, M.; Mahnke, K.; Rivera, M.; Ravetch, J.V.; Steinman, R.M.; Nussenzweig, M.C. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 2001, 194, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Bonifaz, L.C.; Bonnyay, D.P.; Charalambous, A.; Darguste, D.I.; Fujii, S.; Soares, H.; Brimnes, M.K.; Moltedo, B.; Moran, T.M.; Steinman, R.M. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med. 2004, 199, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Bourque, J.; Hawiger, D. Applications of Antibody-Based Antigen Delivery Targeted to Dendritic Cells In Vivo. Antibodies 2022, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Iberg, C.A.; Bourque, J.; Fallahee, I.; Son, S.; Hawiger, D. TNF-α sculpts a maturation process in vivo by pruning tolerogenic dendritic cells. Cell Rep. 2022, 39, 110657. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Sznol, M.; Zhao, B.; Wang, D.; Carvajal, R.D.; Keohan, M.L.; Chuang, E.; Sanborn, R.E.; Lutzky, J.; Powderly, J.; et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci. Transl. Med. 2014, 6, 232ra251. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.A.; Srivastava, P.; Matsuzaki, J.; Brumberger, Z.; Wang, E.S.; Kocent, J.; Miller, A.; Roloff, G.W.; Wong, H.Y.; Paluch, B.E.; et al. NY-ESO-1 Vaccination in Combination with Decitabine Induces Antigen-Specific T-lymphocyte Responses in Patients with Myelodysplastic Syndrome. Clin. Cancer Res. 2018, 24, 1019–1029. [Google Scholar] [CrossRef]
- Palanisamy, N.; Degen, A.; Morath, A.; Ballestin Ballestin, J.; Juraske, C.; Öztürk, M.A.; Sprenger, G.A.; Youn, J.-W.; Schamel, W.W.; Di Ventura, B. Split intein-mediated selection of cells containing two plasmids using a single antibiotic. Nat. Commun. 2019, 10, 4967. [Google Scholar] [CrossRef]
- Agard, N.J.; Prescher, J.A.; Bertozzi, C.R. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 2004, 126, 15046–15047. [Google Scholar] [CrossRef]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef]
- Jin, C.; Fotaki, G.; Ramachandran, M.; Nilsson, B.; Essand, M.; Yu, D. Safe engineering of CAR T cells for adoptive cell therapy of cancer using long-term episomal gene transfer. EMBO Mol. Med. 2016, 8, 702–711. [Google Scholar] [CrossRef]
- Fuertes Marraco, S.A.; Grosjean, F.; Duval, A.; Rosa, M.; Lavanchy, C.; Ashok, D.; Haller, S.; Otten, L.A.; Steiner, Q.G.; Descombes, P.; et al. Novel murine dendritic cell lines: A powerful auxiliary tool for dendritic cell research. Front. Immunol. 2012, 3, 331. [Google Scholar] [CrossRef]
- Fang, X.T.; Sehlin, D.; Lannfelt, L.; Syvänen, S.; Hultqvist, G. Efficient and inexpensive transient expression of multispecific multivalent antibodies in Expi293 cells. Biol. Proced. Online 2017, 19, 11. [Google Scholar] [CrossRef]
- Hawiger, D.; Wan, Y.Y.; Eynon, E.E.; Flavell, R.A. The transcription cofactor Hopx is required for regulatory T cell function in dendritic cell–mediated peripheral T cell unresponsiveness. Nat. Immunol. 2010, 11, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Hawiger, D.; Masilamani, R.F.; Bettelli, E.; Kuchroo, V.K.; Nussenzweig, M.C. Immunological unresponsiveness characterized by increased expression of CD5 on peripheral T cells induced by dendritic cells in vivo. Immunity 2004, 20, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Bournazos, S.; Gazumyan, A.; Seaman, M.S.; Nussenzweig, M.C.; Ravetch, J.V. Bispecific Anti-HIV-1 Antibodies with Enhanced Breadth and Potency. Cell 2016, 165, 1609–1620. [Google Scholar] [CrossRef]
- Katakowski, J.A.; Mukherjee, G.; Wilner, S.E.; Maier, K.E.; Harrison, M.T.; DiLorenzo, T.P.; Levy, M.; Palliser, D. Delivery of siRNAs to Dendritic Cells Using DEC205-Targeted Lipid Nanoparticles to Inhibit Immune Responses. Mol. Ther. 2016, 24, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, R.; Subramanian, S.; Boder, E.T. Sortase A as a Novel Molecular “Stapler” for Sequence-Specific Protein Conjugation. Bioconjugate Chem. 2007, 18, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.W.; Antos, J.M.; Grotenbreg, G.M.; Spooner, E.; Ploegh, H.L. Sortagging: A versatile method for protein labeling. Nat. Chem. Biol. 2007, 3, 707–708. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, H.; Schmittner, M.; Duschl, A.; Horejs-Hoeck, J. Residual endotoxin contaminations in recombinant proteins are sufficient to activate human CD1c+ dendritic cells. PLoS ONE 2014, 9, e113840. [Google Scholar] [CrossRef]
- Schneier, M.; Razdan, S.; Miller, A.M.; Briceno, M.E.; Barua, S. Current technologies to endotoxin detection and removal for biopharmaceutical purification. Biotechnol. Bioeng. 2020, 117, 2588–2609. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, O.; Wall, J.B.J.; Zheng, M.; Zhou, Y.; Wang, L.; Ruth Vaseghi, H.; Qian, L.; Liu, J. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 2017, 7, 2193. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, S.R.; Li, L.H.; Park, H.J.; Park, J.H.; Lee, K.Y.; Kim, M.K.; Shin, B.A.; Choi, S.Y. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS ONE 2011, 6, e18556. [Google Scholar] [CrossRef] [PubMed]
- Piechaczek, C.; Fetzer, C.; Baiker, A.; Bode, J.; Lipps, H.J. A vector based on the SV40 origin of replication and chromosomal S/MARs replicates episomally in CHO cells. Nucleic Acids Res. 1999, 27, 426–428. [Google Scholar] [CrossRef] [PubMed]
- Okada, N.; Shimizu, N. Dissection of the Beta-Globin Replication-Initiation Region Reveals Specific Requirements for Replicator Elements during Gene Amplification. PLoS ONE 2013, 8, e77350. [Google Scholar] [CrossRef] [PubMed]
- Dorr, B.M.; Ham, H.O.; An, C.; Chaikof, E.L.; Liu, D.R. Reprogramming the specificity of sortase enzymes. Proc. Natl. Acad. Sci. USA 2014, 111, 13343–13348. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Hong, H.; Cheng, X.; Liu, S.; Deng, T.; Guo, Z.; Wu, Z. One-step purification and immobilization of extracellularly expressed sortase A by magnetic particles to develop a robust and recyclable biocatalyst. Sci. Rep. 2017, 7, 6561. [Google Scholar] [CrossRef] [PubMed]
- Kock, K.; Ahlers, C.; Schmale, H. Structural organization of the genes for rat von Ebner’s gland proteins 1 and 2 reveals their close relationship to lipocalins. Eur. J. Biochem. 1994, 221, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Mathelin, C.; Tomasetto, C.; Rio, M.C. [Trefoil factor 1 (pS2/TFF1), a peptide with numerous functions]. Bull. Cancer 2005, 92, 773–781. [Google Scholar] [PubMed]
- Gupta, R.; Brunak, S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomput. 2002, 3, 10–322. [Google Scholar]
- Zong, Y.; Bice, T.W.; Ton-That, H.; Schneewind, O.; Narayana, S.V. Crystal structures of Staphylococcus aureus sortase A and its substrate complex. J. Biol. Chem. 2004, 279, 31383–31389. [Google Scholar] [CrossRef]
- Iberg, C.A.; Hawiger, D. Advancing immunomodulation by in vivo antigen delivery to DEC-205 and other cell surface molecules using recombinant chimeric antibodies. Int. Immunopharmacol. 2019, 73, 575–580. [Google Scholar] [CrossRef]
- Wang, F.; Tsai, J.C.; Davis, J.H.; Chau, B.; Dong, J.; West, S.M.; Hogan, J.M.; Wheeler, M.L.; Bee, C.; Morishige, W.; et al. Design and characterization of mouse IgG1 and IgG2a bispecific antibodies for use in syngeneic models. MAbs 2020, 12, 1685350. [Google Scholar] [CrossRef]
- Shah, N.H.; Muir, T.W. Inteins: Nature’s Gift to Protein Chemists. Chem. Sci. 2014, 5, 446–461. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Richard, E.; Michael, O.N.; Alexander, P.; Natasha, A.; Andrew, S.; Tim, G.; Augustin, Ž.; Russ, B.; Sam, B.; Jason, Y.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2022. [Google Scholar] [CrossRef]
- Regula, J.T.; Imhof-Jung, S.; Mølhøj, M.; Benz, J.; Ehler, A.; Bujotzek, A.; Schaefer, W.; Klein, C. Variable heavy-variable light domain and Fab-arm CrossMabs with charged residue exchanges to enforce correct light chain assembly. Protein Eng. Des. Sel. 2018, 31, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Schaefer, W.; Regula, J.T. The use of CrossMAb technology for the generation of bi- and multispecific antibodies. MAbs 2016, 8, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, W.; Regula, J.T.; Bähner, M.; Schanzer, J.; Croasdale, R.; Dürr, H.; Gassner, C.; Georges, G.; Kettenberger, H.; Imhof-Jung, S.; et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 11187–11192. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.C.L.; Koh, E.Y.C.; van Beers, M.; Mueller, M.; Wan, C.; Teo, G.; Song, Z.; Tong, Y.W.; Bardor, M.; Yang, Y. Control of IgG LC:HC ratio in stably transfected CHO cells and study of the impact on expression, aggregation, glycosylation and conformational stability. J. Biotechnol. 2013, 165, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Pasqual, G.; Angelini, A.; Victora, G.D. Triggering positive selection of germinal center B cells by antigen targeting to DEC-205. Methods Mol. Biol. 2015, 1291, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.F. Endosomal escape of RNA therapeutics: How do we solve this rate-limiting problem? Rna 2023, 29, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Hemu, X.; Zhang, X.; Chang, H.Y.; Poh, J.E.; Tam, J.P. Consensus design and engineering of an efficient and high-yield peptide asparaginyl ligase for protein cyclization and ligation. J. Biol. Chem. 2023, 299, 102997. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Wong, Y.H.; Nguyen, G.K.T.; Tam, J.P.; Lescar, J.; Wu, B. Engineering a Catalytically Efficient Recombinant Protein Ligase. J. Am. Chem. Soc. 2017, 139, 5351–5358. [Google Scholar] [CrossRef]
- Le Gall, C.M.; van der Schoot, J.M.S.; Ramos-Tomillero, I.; Khalily, M.P.; van Dalen, F.J.; Wijfjes, Z.; Smeding, L.; van Dalen, D.; Cammarata, A.; Bonger, K.M.; et al. Dual Site-Specific Chemoenzymatic Antibody Fragment Conjugation Using CRISPR-Based Hybridoma Engineering. Bioconjug. Chem. 2021, 32, 301–310. [Google Scholar] [CrossRef]
- Anami, Y.; Tsuchikama, K. Transglutaminase-Mediated Conjugations. Methods Mol. Biol. 2020, 2078, 71–82. [Google Scholar] [CrossRef]
- Swee, L.K.; Guimaraes, C.P.; Sehrawat, S.; Spooner, E.; Barrasa, M.I.; Ploegh, H.L. Sortase-mediated modification of αDEC205 affords optimization of antigen presentation and immunization against a set of viral epitopes. Proc. Natl. Acad. Sci. USA 2013, 110, 1428–1433. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fallahee, I.; Hawiger, D. Episomal Vectors for Stable Production of Recombinant Proteins and Engineered Antibodies. Antibodies 2024, 13, 18. https://doi.org/10.3390/antib13010018
Fallahee I, Hawiger D. Episomal Vectors for Stable Production of Recombinant Proteins and Engineered Antibodies. Antibodies. 2024; 13(1):18. https://doi.org/10.3390/antib13010018
Chicago/Turabian StyleFallahee, Ian, and Daniel Hawiger. 2024. "Episomal Vectors for Stable Production of Recombinant Proteins and Engineered Antibodies" Antibodies 13, no. 1: 18. https://doi.org/10.3390/antib13010018
APA StyleFallahee, I., & Hawiger, D. (2024). Episomal Vectors for Stable Production of Recombinant Proteins and Engineered Antibodies. Antibodies, 13(1), 18. https://doi.org/10.3390/antib13010018