
Rapid Generation of Murine Bispecific Antibodies Using FAST-IgTM for Preclinical Screening of HER2/CD3 T-Cell Engagers
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of FAST-Ig Antibodies
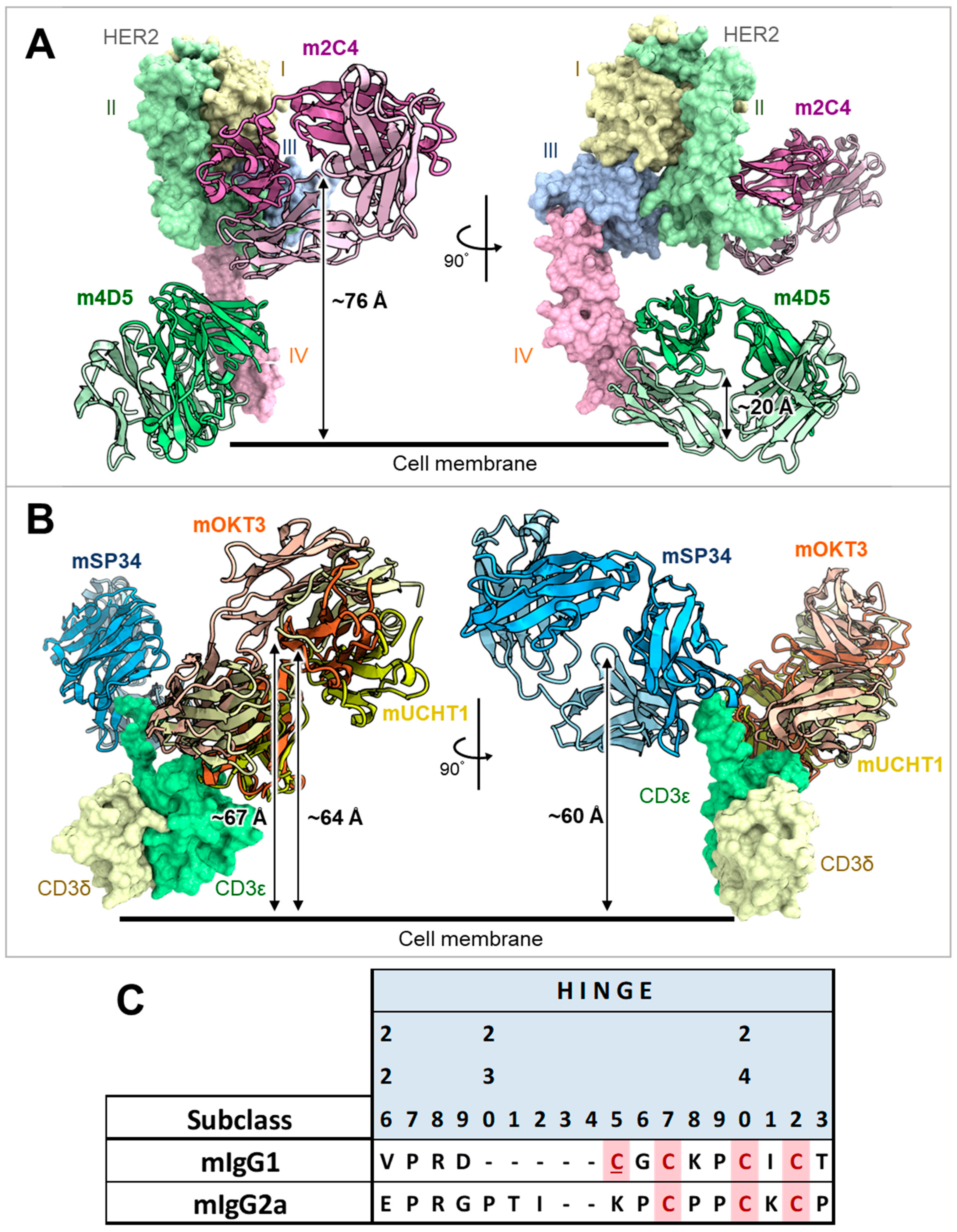
2.2. Antibody Homology Model Generation
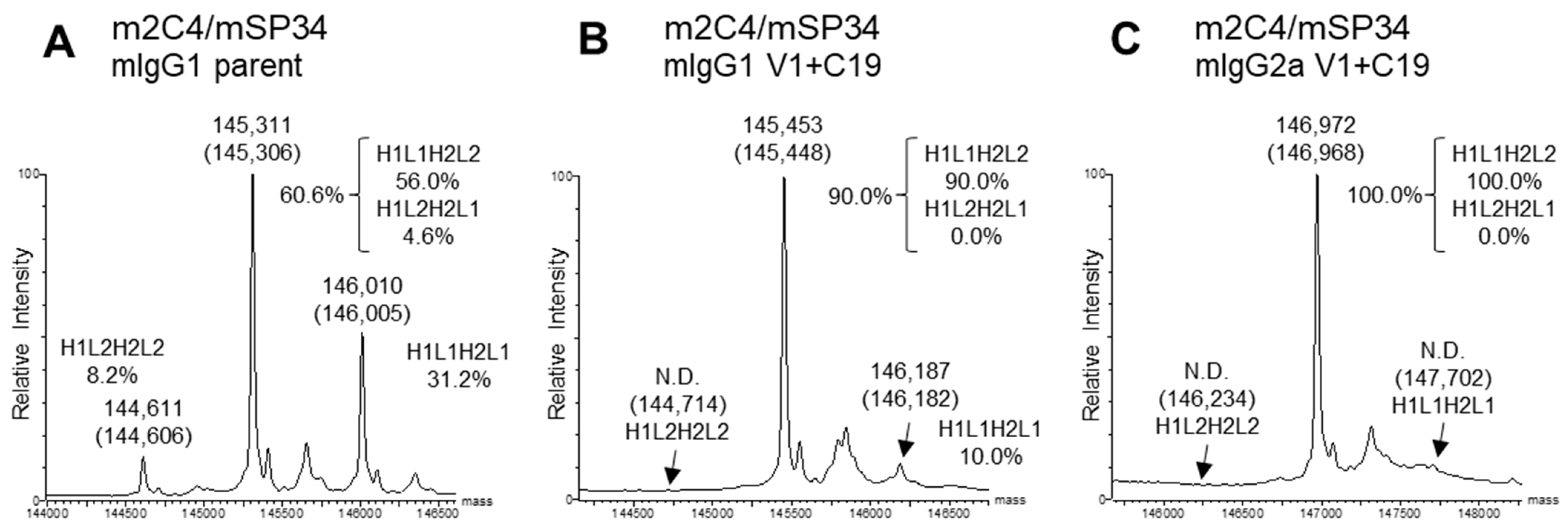
2.3. Estimation of Percentage of BsAb by Mass Spectrometry
2.4. Estimation of Percentage of BsAb by Cation Exchange Chromatography
2.5. Surface Plasmon Resonance (SPR)
2.6. Size Exclusion Chromatography
2.7. Thermal Shift Assay (TSA)
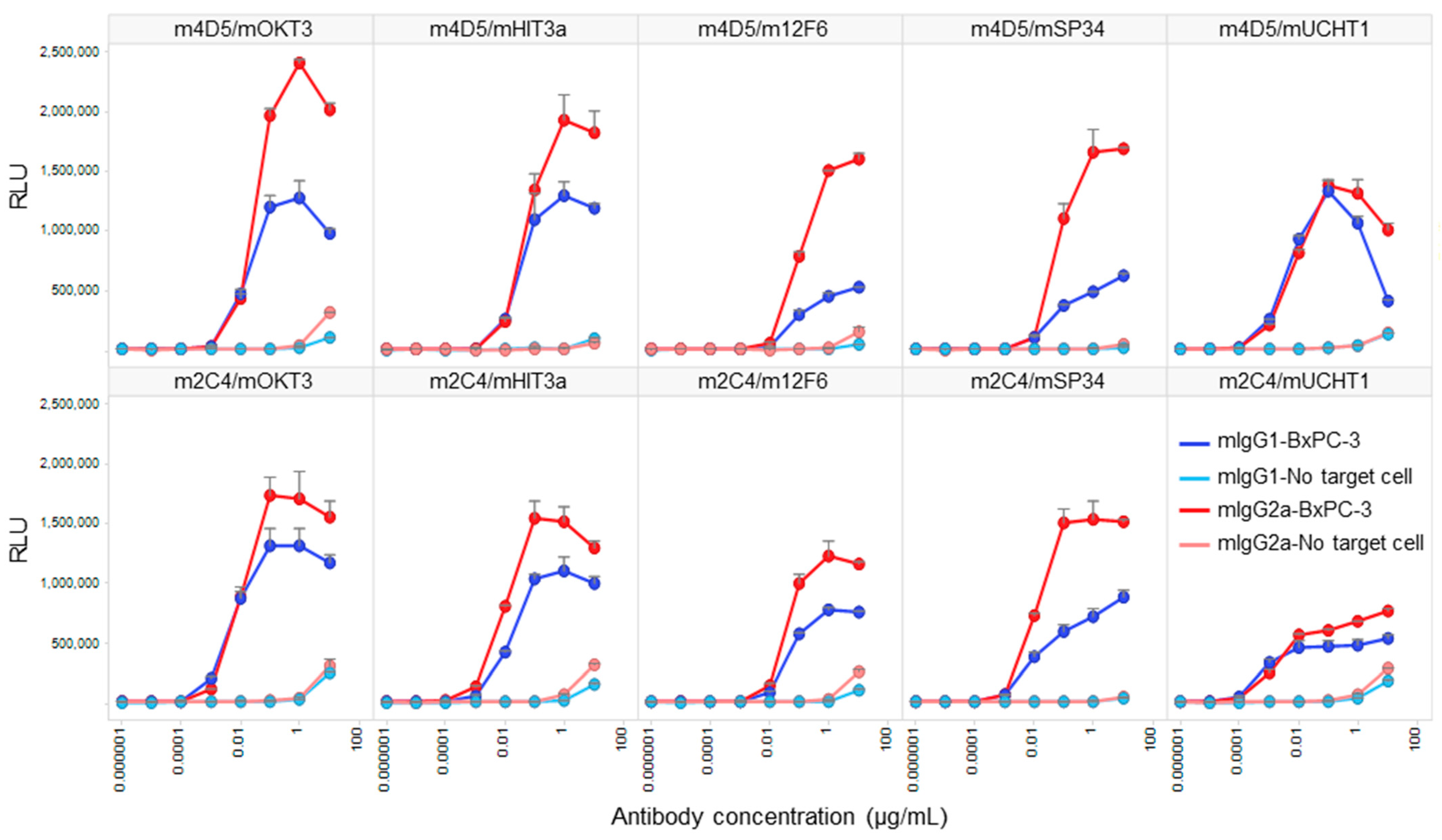
2.8. Jurkat NFAT-Luciferase Reporter Cell Assay
2.9. SDS-PAGE
3. Results
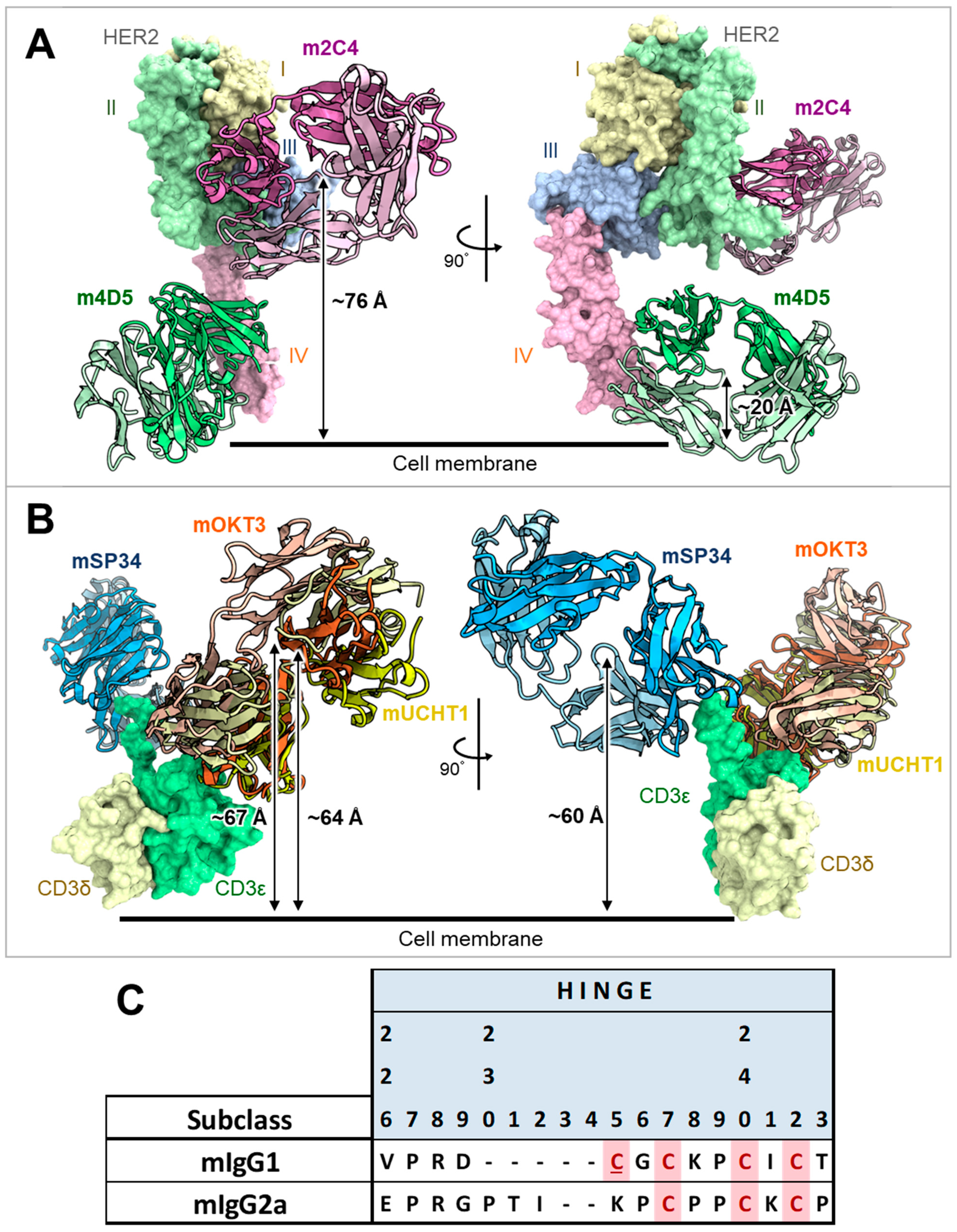
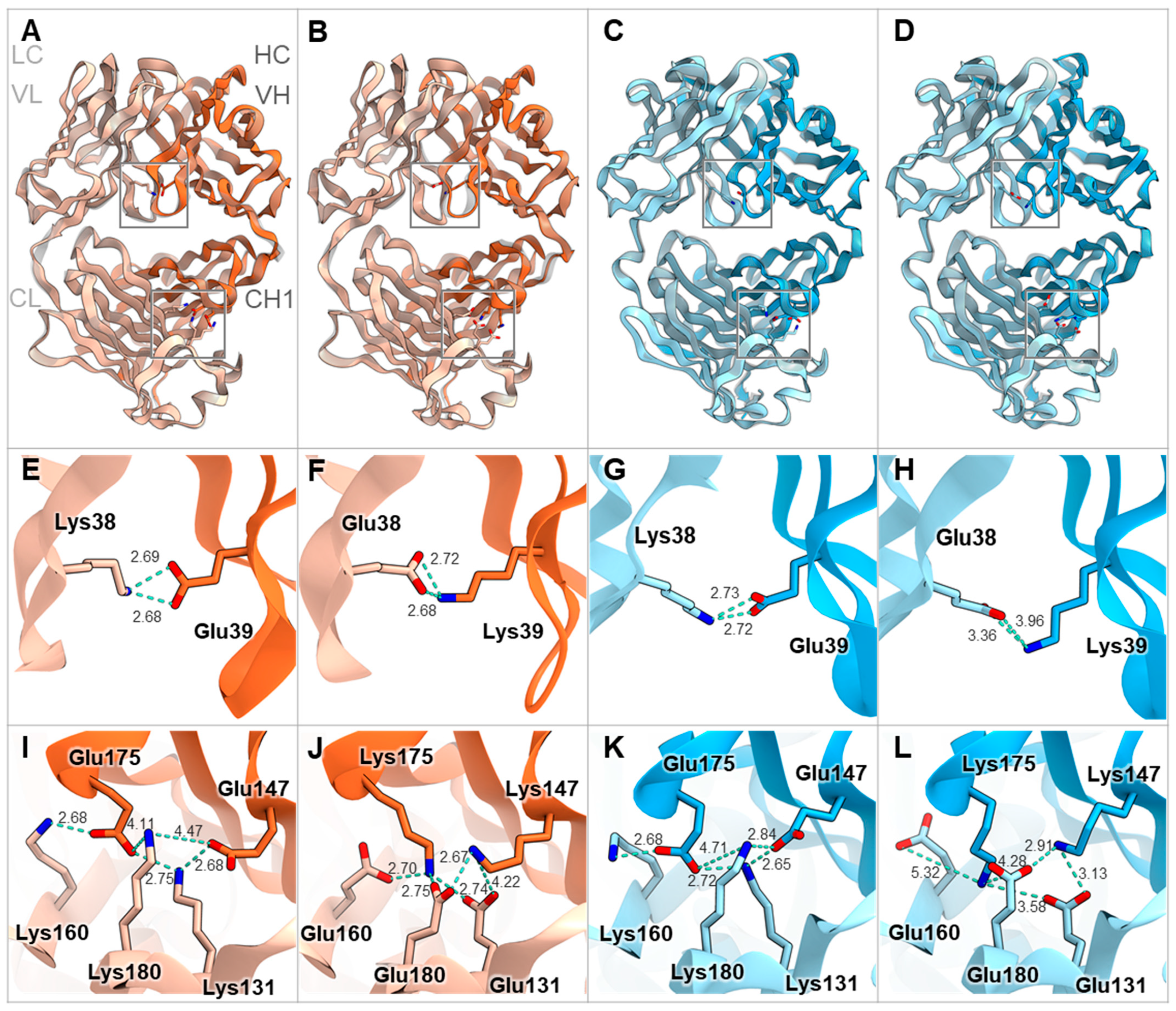
3.1. Comparison of Mouse and Human Antibody Amino Acid Sequences and Structures at the FAST-Ig Mutation Positions in the HC/LC Interface
3.2. Application of V1+C19 to Anti-HER2/CD3 Bispecific Antibodies
3.3. Retention of Parental Physicochemical Properties in Mouse BsAbs with V1+C19
3.4. Comparative Analysis of Target-Dependent T Cell Activation by HER2/CD3 BsAbs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, J.B.; Presta, L.G.; Carter, P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996, 9, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An efficient route to human bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Igawa, T.; Tsunoda, H. Methods for Producing Polypeptides by Regulating Polypeptide Association. Patent WO2006106905, 12 October 2006. [Google Scholar]
- Gunasekaran, K.; Pentony, M.; Shen, M.; Garrett, L.; Forte, C.; Woodward, A.; Ng, S.B.; Born, T.; Retter, M.; Manchulenko, K.; et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: Applications to bispecific molecules and monovalent IgG. J. Biol. Chem. 2010, 285, 19637–19646. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, W.; Regula, J.T.; Bähner, M.; Schanzer, J.; Croasdale, R.; Dürr, H.; Gassner, C.; Georges, G.; Kettenberger, H.; Imhof-Jung, S.; et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 11187–11192. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.M.; Wu, X.; Pustilnik, A.; Sereno, A.; Huang, F.; Rick, H.L.; Guntas, G.; Leaver-Fay, A.; Smith, E.M.; Ho, C.; et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat. Biotechnol. 2014, 32, 191–198. [Google Scholar] [CrossRef]
- Mazor, Y.; Oganesyan, V.; Yang, C.; Hansen, A.; Wang, J.; Liu, H.; Sachsenmeier, K.; Carlson, M.; Gadre, D.V.; Borrok, M.J.; et al. Improving target cell specificity using a novel monovalent bispecific IgG design. MAbs 2015, 7, 377–389. [Google Scholar] [CrossRef]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Bornstein, G.G.; Klakamp, S.L.; Andrews, L.; Boyle, W.J.; Tabrizi, M. Surrogate approaches in development of monoclonal antibodies. Drug Discov. Today 2009, 14, 1159–1165. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Meesters, J.I.; Bunce, M.; Armstrong, A.A.; Somani, S.; Nesspor, T.C.; Chiu, M.L.; Altintaş, I.; Verploegen, S.; Schuurman, J.; et al. Efficient Generation of Bispecific Murine Antibodies for Pre-Clinical Investigations in Syngeneic Rodent Models. Sci. Rep. 2017, 7, 2476. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Tsai, J.C.; Davis, J.H.; Chau, B.; Dong, J.; West, S.M.; Hogan, J.M.; Wheeler, M.L.; Bee, C.; Morishige, W.; et al. Design and characterization of mouse IgG1 and IgG2a bispecific antibodies for use in syngeneic models. MAbs 2020, 12, 1685350. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.; Kim, H.S.; Tong, R.K.; Bainbridge, T.W.; Vernes, J.M.; Zhang, Y.; Lin, Y.L.; Chung, S.; Dennis, M.S.; Zuchero, Y.J.; et al. Effector-attenuating Substitutions That Maintain Antibody Stability and Reduce Toxicity in Mice. J. Biol. Chem. 2017, 292, 3900–3908. [Google Scholar] [CrossRef] [PubMed]
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476–4481. [Google Scholar] [CrossRef] [PubMed]
- Dillon, M.; Yin, Y.; Zhou, J.; McCarty, L.; Ellerman, D.; Slaga, D.; Junttila, T.T.; Han, G.; Sandoval, W.; Ovacik, M.A.; et al. Efficient production of bispecific IgG of different isotypes and species of origin in single mammalian cells. MAbs 2017, 9, 213–230. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Yamano, T.; Betancur, J.; Nagatomo, S.; Ikeda, Y.; Yamaguchi, K.; Nabuchi, Y.; Sato, K.; Teranishi-Ikawa, Y.; Sato, M.; et al. Efficient production of bispecific antibody by FAST-Ig(TM) and its application to NXT007 for the treatment of hemophilia A. MAbs 2023, 15, 2222441. [Google Scholar] [CrossRef] [PubMed]
- Iwayanagi, Y.; Igawa, T.; Maeda, A.; Haraya, K.; Wada, N.A.; Shibahara, N.; Ohmine, K.; Nambu, T.; Nakamura, G.; Mimoto, F.; et al. Inhibitory FcγRIIb-Mediated Soluble Antigen Clearance from Plasma by a pH-Dependent Antigen-Binding Antibody and Its Enhancement by Fc Engineering. J. Immunol. 2015, 195, 3198–3205. [Google Scholar] [CrossRef]
- Ha, D.; Tanaka, A.; Kibayashi, T.; Tanemura, A.; Sugiyama, D.; Wing, J.B.; Lim, E.L.; Teng, K.W.W.; Adeegbe, D.; Newell, E.W.; et al. Differential control of human Treg and effector T cells in tumor immunity by Fc-engineered anti-CTLA-4 antibody. Proc. Natl. Acad. Sci. USA 2019, 116, 609–618. [Google Scholar] [CrossRef]
- Carter, P.; Presta, L.; Gorman, C.M.; Ridgway, J.B.; Henner, D.; Wong, W.L.; Rowland, A.M.; Kotts, C.; Carver, M.E.; Shepard, H.M. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. USA 1992, 89, 4285–4289. [Google Scholar] [CrossRef]
- Adams, C.W.; Allison, D.E.; Flagella, K.; Presta, L.; Clarke, J.; Dybdal, N.; McKeever, K.; Sliwkowski, M.X. Humanization of a recombinant monoclonal antibody to produce a therapeutic HER dimerization inhibitor, pertuzumab. Cancer Immunol. Immunother. 2006, 55, 717–727. [Google Scholar] [CrossRef]
- Kjer-Nielsen, L.; Dunstone, M.A.; Kostenko, L.; Ely, L.K.; Beddoe, T.; Mifsud, N.A.; Purcell, A.W.; Brooks, A.G.; McCluskey, J.; Rossjohn, J. Crystal structure of the human T cell receptor CD3 epsilon gamma heterodimer complexed to the therapeutic mAb OKT3. Proc. Natl. Acad. Sci. USA 2004, 101, 7675–7680. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhu, Z.; Xiong, D. CD3-Resisting Single Clone Antibody Heavy-Chain and Light-Chain Variable-Area Gene and Its Application. Patent CN1298020, 6 June 2001. [Google Scholar]
- Li, B.; Wang, H.; Dai, J.; Ji, J.; Qian, W.; Zhang, D.; Hou, S.; Guo, Y. Construction and characterization of a humanized anti-human CD3 monoclonal antibody 12F6 with effective immunoregulation functions. Immunology 2005, 116, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Ollier, R.; Hou, S.; Lissilaa, R.; Skegro, D.; Back, J. CD3/CD38 T Cell Retargeting Hetero-Dimeric Immunoglobulins and Methods of Their Production. Patent WO2016071355, 12 May 2016. [Google Scholar]
- Arnett, K.L.; Harrison, S.C.; Wiley, D.C. Crystal structure of a human CD3-epsilon/delta dimer in complex with a UCHT1 single-chain antibody fragment. Proc. Natl. Acad. Sci. USA 2004, 101, 16268–16273. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.K.; Labute, P. Assessment of fully automated antibody homology modeling protocols in molecular operating environment. Proteins 2014, 82, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Han, G.; Zhou, J.; Dillon, M.; McCarty, L.; Gavino, L.; Ellerman, D.; Spiess, C.; Sandoval, W.; Carter, P.J. Precise quantification of mixtures of bispecific IgG produced in single host cells by liquid chromatography-Orbitrap high-resolution mass spectrometry. MAbs 2016, 8, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Edelman, G.M.; Cunningham, B.A.; Gall, W.E.; Gottlieb, P.D.; Rutishauser, U.; Waxdal, M.J. The covalent structure of an entire gammaG immunoglobulin molecule. Proc. Natl. Acad. Sci. USA 1969, 63, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Kabat, E.W.P.; Gottesman, K.; Foeller, C. Sequences of Proteins of Immunological Interest; NIH: Bethesda, MD, USA, 1991.
- Junttila, T.T.; Li, J.; Johnston, J.; Hristopoulos, M.; Clark, R.; Ellerman, D.; Wang, B.E.; Li, Y.; Mathieu, M.; Li, G.; et al. Antitumor efficacy of a bispecific antibody that targets HER2 and activates T cells. Cancer Res. 2014, 74, 5561–5571. [Google Scholar] [CrossRef]
- Slaga, D.; Ellerman, D.; Lombana, T.N.; Vij, R.; Li, J.; Hristopoulos, M.; Clark, R.; Johnston, J.; Shelton, A.; Mai, E.; et al. Avidity-based binding to HER2 results in selective killing of HER2-overexpressing cells by anti-HER2/CD3. Sci. Transl. Med. 2018, 10, eaat5775. [Google Scholar] [CrossRef]
- Joshi, K.K.; Phung, W.; Han, G.; Yin, Y.; Kim, I.; Sandoval, W.; Carter, P.J. Elucidating heavy/light chain pairing preferences to facilitate the assembly of bispecific IgG in single cells. MAbs 2019, 11, 1254–1265. [Google Scholar] [CrossRef]
- Hao, Y.; Yu, X.; Bai, Y.; McBride, H.J.; Huang, X. Cryo-EM Structure of HER2-trastuzumab-pertuzumab complex. PLoS ONE 2019, 14, e0216095. [Google Scholar] [CrossRef]
- Fernandes, R.A.; Shore, D.A.; Vuong, M.T.; Yu, C.; Zhu, X.; Pereira-Lopes, S.; Brouwer, H.; Fennelly, J.A.; Jessup, C.M.; Evans, E.J.; et al. T cell receptors are structures capable of initiating signaling in the absence of large conformational rearrangements. J. Biol. Chem. 2012, 287, 13324–13335. [Google Scholar] [CrossRef] [PubMed]
- Zorn, J.A.; Wheeler, M.L.; Barnes, R.M.; Kaberna, J.; Morishige, W.; Harris, M.; Huang, R.Y.; Lohre, J.; Chang, Y.C.; Chau, B.; et al. Humanization of a strategic CD3 epitope enables evaluation of clinical T-cell engagers in a fully immunocompetent in vivo model. Sci. Rep. 2022, 12, 3530. [Google Scholar] [CrossRef]
- Liu, C.Y.; Ahonen, C.L.; Brown, M.E.; Zhou, L.; Welin, M.; Krauland, E.M.; Pejchal, R.; Widboom, P.F.; Battles, M.B. Structure-based engineering of a novel CD3ε-targeting antibody for reduced polyreactivity. MAbs 2023, 15, 2189974. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Stagg, N.J.; Johnston, J.; Harris, M.J.; Menzies, S.A.; DiCara, D.; Clark, V.; Hristopoulos, M.; Cook, R.; Slaga, D.; et al. Membrane-Proximal Epitope Facilitates Efficient T Cell Synapse Formation by Anti-FcRH5/CD3 and Is a Requirement for Myeloma Cell Killing. Cancer Cell 2017, 31, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yang, F.; Wang, C.; Narula, J.; Pascua, E.; Ni, I.; Ding, S.; Deng, X.; Chu, M.L.; Pham, A.; et al. One size does not fit all: Navigating the multi-dimensional space to optimize T-cell engaging protein therapeutics. MAbs 2021, 13, 1871171. [Google Scholar] [CrossRef] [PubMed]
- Kapelski, S.; Cleiren, E.; Attar, R.M.; Philippar, U.; Häsler, J.; Chiu, M.L. Influence of the bispecific antibody IgG subclass on T cell redirection. MAbs 2019, 11, 1012–1024. [Google Scholar] [CrossRef]
- Tashiro, D.; Suetaka, S.; Sato, N.; Ooka, K.; Kunihara, T.; Kudo, H.; Inatomi, J.; Hayashi, Y.; Arai, M. Intron-Encoded Domain of Herstatin, An Autoinhibitor of Human Epidermal Growth Factor Receptors, Is Intrinsically Disordered. Front. Mol. Biosci. 2022, 9, 862910. [Google Scholar] [CrossRef]
- Zuch de Zafra, C.L.; Fajardo, F.; Zhong, W.; Bernett, M.J.; Muchhal, U.S.; Moore, G.L.; Stevens, J.; Case, R.; Pearson, J.T.; Liu, S.; et al. Targeting Multiple Myeloma with AMG 424, a Novel Anti-CD38/CD3 Bispecific T-cell-recruiting Antibody Optimized for Cytotoxicity and Cytokine Release. Clin. Cancer Res. 2019, 25, 3921–3933. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Antigen | CL | VH | VL |
|---|---|---|---|---|
| m4D5 [20] | HER2 | kappa | QVQLQQSGPELVKPGASLKLSCTASGFNIKDTYIHWVKQRPEQGLEWIGRIYPTNGYTRYDPKFQDKATITADTSSNTAYLQVSRLTSEDTAVYYCSRWGGDGFYAMDYWGQGASVTVSS | DIVMTQSHKFMSTSVGDRVSITCKASQDVNTAVAWYQQKPGHSPKLLIYSASFRYTGVPDRFTGSRSGTDFTFTISSVQAEDLAVYYCQQHYTTPPTFGGGTKVEIK |
| m2C4 [21] | HER2 | kappa | EVQLQQSGPELVKPGTSVKISCKASGFTFTDYTMDWVKQSHGKSLEWIGDVNPNSGGSIYNQRFKGKASLTVDRSSRIVYMELRSLTFEDTAVYYCARNLGPSFYFDYWGQGTTLTVSS | DTVMTQSHKIMSTSVGDRVSITCKASQDVSIGVAWYQQRPGQSPKLLIYSASYRYTGVPDRFTGSGSGTDFTFTISSVQAEDLAVYYCQQYYIYPYTFGGGTKLEIK |
| mOKT3 [22] | CD3 | kappa | QVQLQQSGAELARPGASVKMSCKASGYTFTRYTMHWVKQRPGQGLEWIGYINPSRGYTNYNQKFKDKATLTTDKSSSTAYMQLSSLTSEDSAVYYCARYYDDHYCLDYWGQGTTLTVSS | QIVLTQSPAIMSASPGEKVTMTCSASSSVSYMNWYQQKSGTSPKRWIYDTSKLASGVPAHFRGSGSGTSYSLTISGMEAEDAATYYCQQWSSNPFTFGSGTKLEIN |
| mHIT3a [23] | CD3 | kappa | QVQLQESGAELARPGASVKMSCKASGYTFTRYTMHWVKQRPGQGLEWIGYINPSRGYTNYNQKFKDKATLTTDKSSSTAYMELTRLTSEDSAVYYCARYYDDHYCLDYWGQGTTVTVSS | DIELTQSPAIMSASPGEKVTMTCSASSSVSYMNWYQQKSGTSPKRWIYDTSKLASGVPARFSGSGSGTSYSLTISGMEAEDAATYYCQQWSSNPFTFGSGTKLELK |
| m12F6 [24] | CD3 | kappa | QVQLQQSGAELARPGASVKMSCKASGYTFTSYTMHWVKQRPGQGLEWIGYINPSSGYTKYNQKFKDKATLTADKSSSTAYMQLSSLTSEDSAVYYCARWQDYDVYFDYWGQGTTLTVSS | QIVLSQSPAILSASPGEKVTMTCRASSSVSYMHWYQQKPGSSPKPWIYATSNLASGVPARFSGSGSGTSYSLTISRVEAEDAATYYCQQWSSNPPTFGGGTKLETK |
| mSP34 [25] | CD3 | lambda | EVQLVESGGGLVQPKGSLKLSCAASGFTFNTYAMNWVRQAPGKGLEWVARIRSKYNNYATYYADSVKDRFTISRDDSQSILYLQMNNLKTEDTAMYYCVRHGNFGNSYVSWFAYWGQGTLVTVSA | QAVVTQESALTTSPGETVTLTCRSSTGAVTTSNYANWVQEKPDHLFTGLIGGTNKRAPGVPARFSGSLIGDKAALTITGAQTEDEAIYFCALWYSNLWVFGGGTKLTVL |
| mUCHT1 [26] | CD3 | kappa | EVQLQQSGPELVKPGASMKISCKASGYSFTGYTMNWVKQSHGKNLEWMGLINPYKGVSTYNQKFKDKATLTVDKSSSTAYMELLSLTSEDSAVYYCARSGYYGDSDWYFDVWGQGTTLTVFS | DIQMTQTTSSLSASLGDRVTISCRASQDIRNYLNWYQQKPDGTVKLLIYYTSRLHSGVPSKFSGSGSGTDYSLTISNLEQEDIATYFCQQGNTLPWTFAGGTKLEIK |
| Chain Name | V1+C19 Mutations [17] | |
|---|---|---|
| Anti-HER2 HC (H1) | VH | CH1 |
| Q39K | K147/Q175K | |
| Anti-HER2 LC (L1) | VL | CL |
| Q38E | S131E/Q160E/T180E | |
| Anti-CD3 HC (H2) | VH | CH1 |
| Q39E | K147E/Q175E | |
| Anti-CD3 LC (L2) | VL | CL |
| Q38K (E38K) | S131K/L160K/T180K (T131K/E160K/T180K) | |
| Subclass | FAST-Ig | BsAb Yield (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| m4D5/mOKT3 | m4D5/mHIT3a | m4D5/m12F6 | m4D5/mSP34 | m4D5/mUCHT1 | m2C4/mOKT3 | m2C4/mHIT3a | m2C4/m12F6 | m2C4/mSP34 | m2C4/mUCHT1 | ||
| mIgG1 | - | 31.5 | 96.2 | 72.9 | 15.1 | 58.7 * | 51.1 | 98.2 | 49.0 | 56.0 | 21.9 * |
| mIgG1 | V1+C19 | 100.0 | 94.5 | 100.0 | 100.0 | 100.0 * | 100.0 | 100.0 | 98.6 | 90.0 | 100.0 * |
| mIgG2a | V1+C19 | 100.0 | 100.0 | 100.0 | 100.0 | 100.0 * | 81.0 ** | 100.0 | 100.0 | 100.0 | 100.0 * |
| Subclass | FAST-Ig | KD (mol/L) | ||||||
|---|---|---|---|---|---|---|---|---|
| Anti-HER2 | Anti-CD3 | |||||||
| m4D5 | m2C4 | mOKT3 | mHIT3a | m12F6 | mSP34 | mUCHT1 | ||
| mIgG1 | - | 1.03 × 10−9 | 7.59 × 10−10 | 4.82 × 10−6 | 7.54 × 10−6 | 1.68 × 10−5 | 2.09 × 10−8 | 1.45 × 10−9 |
| mIgG1 | V1+C19 | 1.27 × 10−9 | 7.84 × 10−10 | 4.48 × 10−6 | 8.32 × 10−6 | 1.61 × 10−5 | 5.69 × 10−8 | 1.32 × 10−9 |
| mIgG2a | - | 1.14 × 10−9 | 1.08 × 10−9 | 6.58 × 10−6 | 1.04 × 10−5 | 1.67 × 10−5 | 1.60 × 10−8 | 1.02 × 10−9 |
| mIgG2a | V1+C19 | 9.60 × 10−10 | 1.36 × 10−9 | 7.68 × 10−6 | 1.04 × 10−5 | 1.64 × 10−5 | 2.57 × 10−8 | 1.32 × 10−9 |
| Subclass | FAST-Ig | Tm1 (°C) | ||||||
|---|---|---|---|---|---|---|---|---|
| Anti-HER2 | Anti-CD3 | |||||||
| m4D5 | m2C4 | mOKT3 | mHIT3a | m12F6 | mSP34 | mUCHT1 | ||
| mIgG1 | - | 72.6 | 75.6 | 75.6 | 73.2 | 76.2 | 67.2 | 75.0 |
| mIgG1 | V1+C19 | 69.0 | 74.4 | 75.0 | 72.6 | 76.2 | 67.8 | 74.4 |
| mIgG2a | - | 70.2 | 71.4 | 75.0 | 71.4 | 74.4 | 66.6 | 73.8 |
| mIgG2a | V1+C19 | 64.2 | 67.8 | 73.8 | 70.2 | 73.2 | 63.0 | 70.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koga, H.; Kuroi, H.; Hirano, R.; Hirayama, H.; Nabuchi, Y.; Kuramochi, T. Rapid Generation of Murine Bispecific Antibodies Using FAST-IgTM for Preclinical Screening of HER2/CD3 T-Cell Engagers. Antibodies 2024, 13, 3. https://doi.org/10.3390/antib13010003
Koga H, Kuroi H, Hirano R, Hirayama H, Nabuchi Y, Kuramochi T. Rapid Generation of Murine Bispecific Antibodies Using FAST-IgTM for Preclinical Screening of HER2/CD3 T-Cell Engagers. Antibodies. 2024; 13(1):3. https://doi.org/10.3390/antib13010003
Chicago/Turabian StyleKoga, Hikaru, Haruka Kuroi, Rena Hirano, Hiroyuki Hirayama, Yoshiaki Nabuchi, and Taichi Kuramochi. 2024. "Rapid Generation of Murine Bispecific Antibodies Using FAST-IgTM for Preclinical Screening of HER2/CD3 T-Cell Engagers" Antibodies 13, no. 1: 3. https://doi.org/10.3390/antib13010003