1. Introduction
TATA-box binding protein-associated factor 1 (TAF1) is the largest subunit of the transcription factor IID complex (TFIID), which is composed of TBP and 13 different TAFs [
1,
2]. Binding of TFIID to the core promoter elements is required for assembly of a functional transcription initiation complex. TFIID also serves as a co-activator by directly transmitting signals from sequence-specific activators to other components of the basal transcription machinery [
3,
4,
5]. Moreover, TAFs function to directly activate selected genes in vivo [
6,
7,
8]. TAF1 appears to function as a major scaffold by which TBP and other TAFs interact in the assembly of TFIID, and TAF1 also plays a critical role in the regulation of cell growth [
6]. TAF1 possesses intrinsic protein kinase activity [
9], histone acetyltransferase activity [
10] and ubiquitin-activating and conjugating activity [
11]. Previous work has demonstrated that the kinase activity of TAF1 is important for progression through the G1 phase [
12].
We previously found that the
TAF1 gene is the causative gene of X-linked recessive dystonia-parkinsonism (XDP/DYT3; MIM314250), which is characterized by severe neurodegeneration in the striatum [
13]. The novel, neuron-specific isoform of the
TAF1 gene (
N-TAF1) had the complete translation frame of the major form of
TAF1 with an insertion of two amino acid residues, alanine and lysine, in the carboxyl terminal kinase domain. The expression of
N-TAF1 is localized in the brain and is significantly decreased in the brain of patients with XDP/DYT3, suggesting that it may have an essential role in neuronal survival in the striatum. Although we identified the mouse homologue of
N-TAF1 (
mN-Taf1) and demonstrated the expression pattern of N-Taf1 mRNA in mouse embryo head and brain [
14], little is known about distribution of N-TAF1 protein. We report here the characterization of a monoclonal antibody, 3A-11F, which specifically recognizes N-TAF1.
2. Results and Discussion
Of three hybridomas that were successfully isolated, one positive clone (3A-11F) which reacted strongly to N-TAF1 peptide, but not to TAF1 peptide, was selected (data not shown).
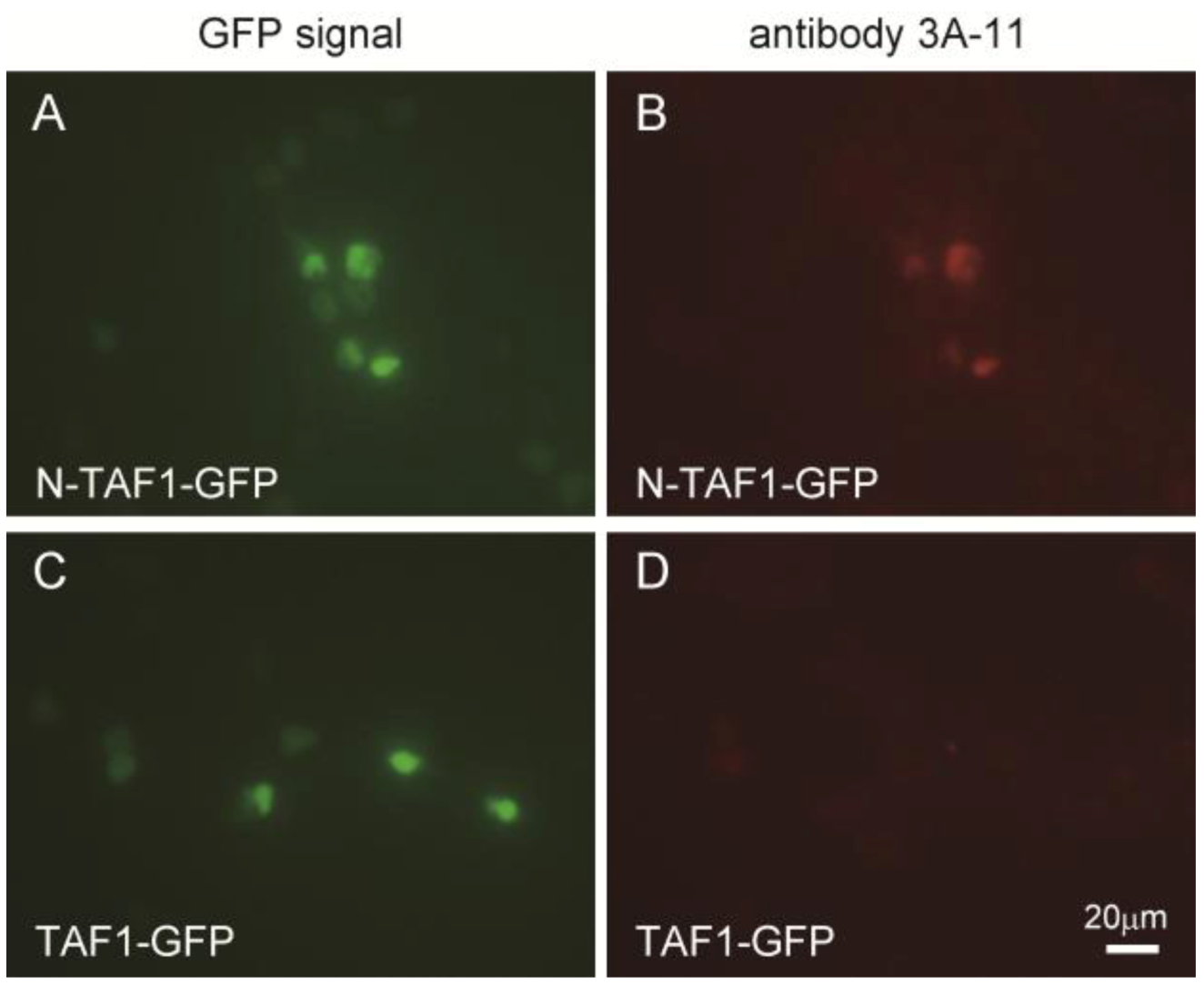
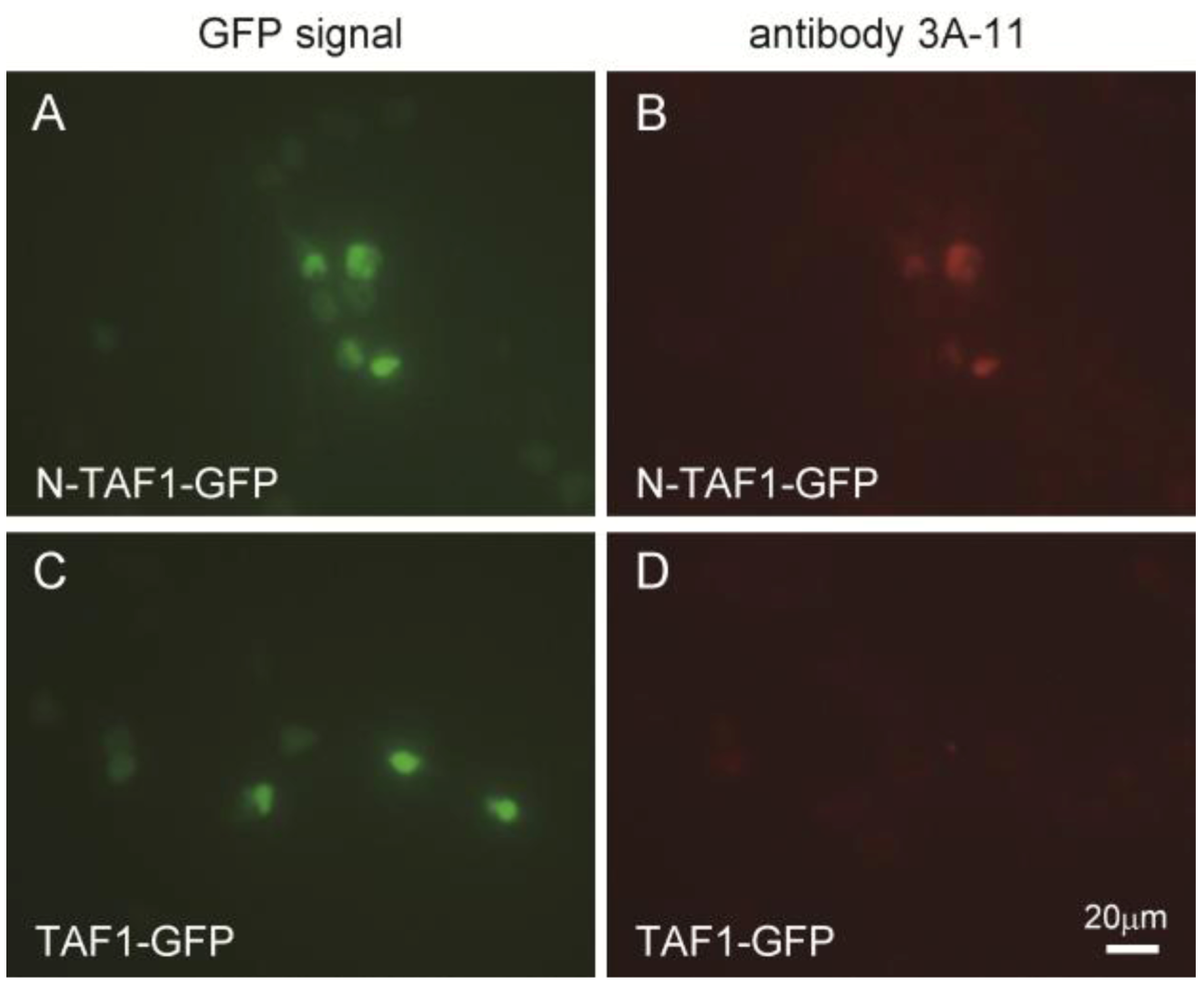
Figure 1 shows the immunocytochemistry results of HEK293 cells expressing GFP-tagged N-TAF1 or GFP-tagged TAF1 probed with 3A-11F. GFP-tagged N-TAF1 (
A) and TAF1 (
C) proteins were observed in green color. Red colored signals from immunostaining with 3A-11F and Alexa 555-conjugated anti-mouse IgG were observed in HEK293 cells expressing mN-TAF1-GFP (
B) but were not seen in cells expressing mTAF1-GFP (
D). The 3A-11F monoclonal antibody clearly stained N-TAF protein tagged with GFP, but did not stain TAF-1 protein tagged with GFP.
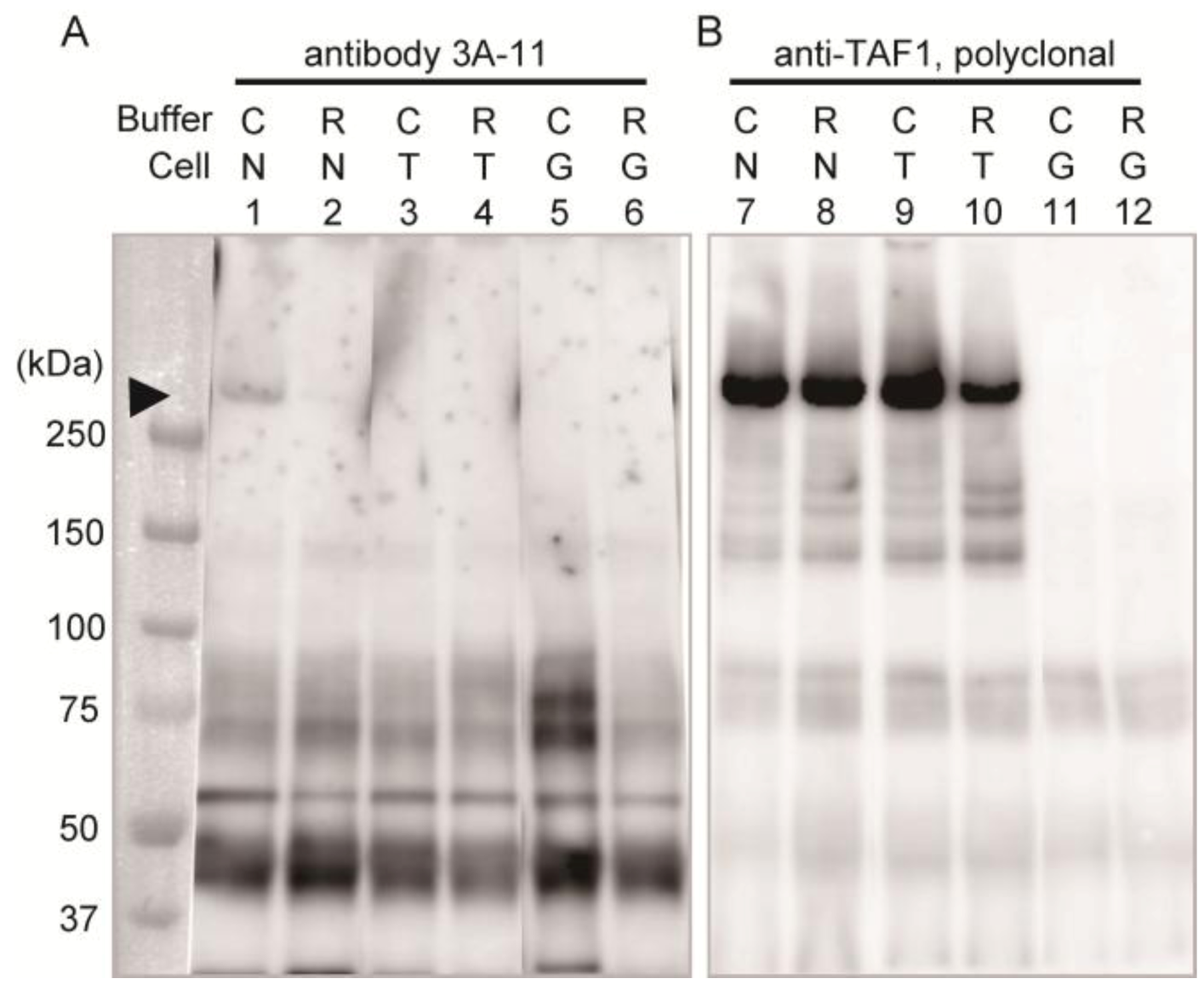
Immunoprecipitation assay with homogenates of these cells was performed to confirm the specificity of 3A-11F.
Figure 2 shows the results of the immunoprecipitation assay using 3A-11F (lanes 1–6 in
Figure 2) and the polyclonal antibody to TAF1 (lanes 7–12 in
Figure 2). The detection antibody was a GFP specific antibody and non 3A-11F or TAF1, which were used for the enrichment of the antigens by immunoprecipitation. A weak band of about 250 kDa (equivalent to molecular weight of N-Taf1) was detected in the 3A-11F immunoprecipitated sample of cells expressing GFP-tagged N-TAF1 solubilized with Co-IP buffer (lane 1,
Figure 2) but not with RIPA buffer (lane 2,
Figure 2). Co-IP buffer makes more gentle condition than RIPA buffer and other typical homogenization buffers. Therefore, 3A-11 appeared to have the conformation-sensitive character. On the other hand, the cells expressing TAF1 or GFP (lanes 3–6,
Figure 2) did not show this 250 kDa band. Additional bands are visible, one between 37 and 50 kDa, one shortly above 50 kDa, and two at about 75 kDa. These bands may be non-specific caused by long exposure time. In the TAF1 antibody immunoprecipitated samples, a strong band signal of about 250 kDa was detected from cells expressing N-TAF1 (lanes 7 and 8,
Figure 2) and TAF-1 (lanes 9 and 10,
Figure 2). These results suggest that the transgene expression levels in the transfected cells are enough and 3A-11 captured N-TAF1 protein specifically, less efficient it is.
Figure 1.
Immunocytochemistry of cultured Human embryonic kidney cells (HEK293) that express mouse neuron-specific TATA-binding protein-associated factor 1 (mN-TAF1) tagged with green fluorescent protein (GFP) (A, B) or mTAF1-GFP (C, D), using the monoclonal antibody, 3A-11F. Scale bar = 20 µm.
Figure 1.
Immunocytochemistry of cultured Human embryonic kidney cells (HEK293) that express mouse neuron-specific TATA-binding protein-associated factor 1 (mN-TAF1) tagged with green fluorescent protein (GFP) (A, B) or mTAF1-GFP (C, D), using the monoclonal antibody, 3A-11F. Scale bar = 20 µm.
Figure 2.
Immunoprecipitation using neuron-specific TATA-binding protein-associated factor 1 (N-TAF1) monoclonal antibody, 3A-11F (A), and TAF1 polyclonal antibody (B). HEK293 cells were transiently transfected with green fluorescent protein (GFP)-tagged mouse N-TAF1 (N), mouse TAF1-GFP (T) and GFP only (G) as indicated. The detection antibody was a GFP specific antibody. C indicates that cells were solubilized in Co-IP buffer and R indicates that cells were solubilized in RIPA buffer. Exposure time is seven (A) or one (B) minutes.
Figure 2.
Immunoprecipitation using neuron-specific TATA-binding protein-associated factor 1 (N-TAF1) monoclonal antibody, 3A-11F (A), and TAF1 polyclonal antibody (B). HEK293 cells were transiently transfected with green fluorescent protein (GFP)-tagged mouse N-TAF1 (N), mouse TAF1-GFP (T) and GFP only (G) as indicated. The detection antibody was a GFP specific antibody. C indicates that cells were solubilized in Co-IP buffer and R indicates that cells were solubilized in RIPA buffer. Exposure time is seven (A) or one (B) minutes.
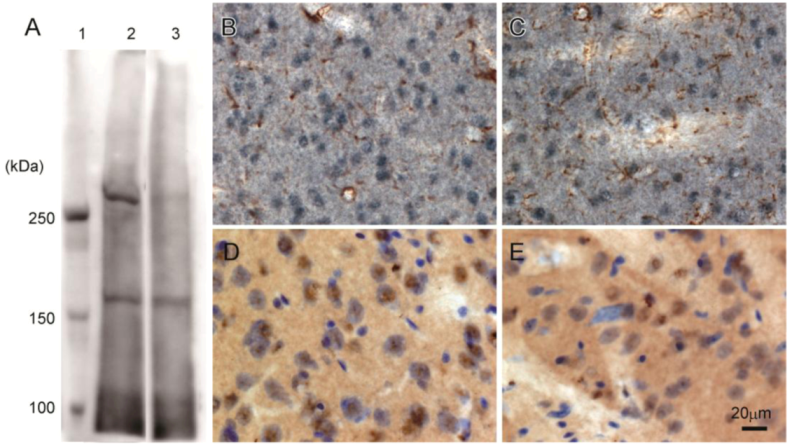
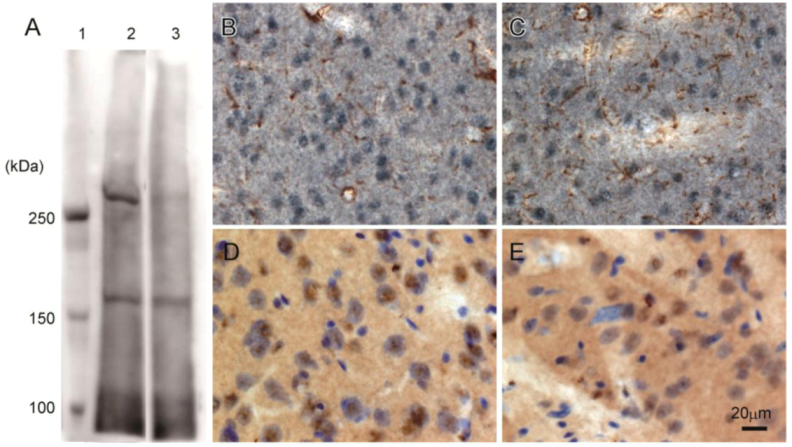
Western blot analysis of rat brain confirmed that 3A-11F stained a protein of about 250 kDa, corresponding to the size of N-TAF1 (
Figure 3A). The 250 kDa signal is visible with soluble fraction (lane 2,
Figure 3A), but not with membrane fraction (lane 3,
Figure 3A). Immunohistochemistry showed that 3A-11F stained nuclei in cells (
Figure 3B–E). Double-staining for 3A-11F and GFAP demonstrated that 3A-11F-positive cells were a separate population from GFAP-positive astrocytes (
Figure 3B,C). Double-staining for 3A-11F and Nissl stain showed that 3A-11F-positive nuclei were localized to neurons (
Figure 3C,D). These results support previous findings that TAF1 is nuclear DNA-binding protein.
Figure 3.
Western blot analysis (A) and immunohistochemistry (B–E) using rat brain probed with monoclonal antibody, 3A-11F. A: Fifty µg of rat brain soluble fraction (lane 2) or membrane fraction (lane 3) as well as a protein marker (lane 1) were electrophoresed on a 4% polyacrylamide gel. B and C: Double-immunostaining for N-TAF1 (purple) and glial fibrillary acidic protein (GFAP) (brown). D and E: Double-staining for N-TAF1 (brown) and Nissl stain (purple). Scale bar = 20 µm.
Figure 3.
Western blot analysis (A) and immunohistochemistry (B–E) using rat brain probed with monoclonal antibody, 3A-11F. A: Fifty µg of rat brain soluble fraction (lane 2) or membrane fraction (lane 3) as well as a protein marker (lane 1) were electrophoresed on a 4% polyacrylamide gel. B and C: Double-immunostaining for N-TAF1 (purple) and glial fibrillary acidic protein (GFAP) (brown). D and E: Double-staining for N-TAF1 (brown) and Nissl stain (purple). Scale bar = 20 µm.
We have reported the immunohistochemical staining of N-TAF1 in the rat striatum using 3F-11 [
15]. This study confirms the data and further demonstrates immunocytochemistry of cultured cells and immunoprecitation assay using 3A-11F, as compared with polyclonal antibody to TAF1.
Despite a difference of only two amino acids between N-TAF1 and TAF-1, we have isolated and characterized a monoclonal antibody that specifically binds only N-TAF1 on both immunocytochemistry and immunoprecipitation assays. 3A-11F specifically stained the nuclear region of neurons in the brain, consistent with our previous study demonstrating that
N-TAF1 mRNA was expressed in neuroblastoma cells but not in glioblastoma cells [
13]. XDP/DYT3 is the first reported example of a non-polyQ disease that supports the transcriptional dysregulation hypothesis in dystonia [
16,
17]. The role of the
N-TAF1 in non-dividing neuronal cells is still unclear. But the trace of its molecular behavior is sure to provide a way to investigate possible mechanisms of neuronal loss and the development of dystonic movements. Recently, we found the differences in the expression pattern between
mTaf1 and
mN-Taf1 [
14]. These findings suggest that
N-TAF1 may have an important role, such as protectoral function in differentiated neurons, rather than in cell division and proliferation during neurogenesis and further support an important role for
N-TAF1 in the regulation of many neuron-specific genes. Thus, TAF1 has been considered to have universal expression profile and functions in various tissues, but the presence of tissue-specific isoforms of TAF1 will help us unravel how the basic transcription factor affect the limited region, such as a specific site of the nervous system, and how they can be responsible for the disease. Our newly developed N-TAF1 monoclonal antibody, 3A-11F, promises to be a useful tool that will contribute to a better understanding of neuron-specific gene expression in future studies. Those results may lead to new perspectives not only to elucidate the importance of gene transcription in neuronal cells, but to study gene transcriptional regulation itself in eukaryotic cells.
3. Experimental Section
3.1. Synthetic Peptide Design and Generation of Monoclonal Antibodies
Since N-TAF1 consists of the common form of TAF1 plus the insertion of two amino acids, A and K, the peptide sequence peculiar to N-TAF1 is the region extending over the two amino acids. A 15-mer synthetic peptide (NH2–TPGPYTPQAKPPDLY–COOH) was designed based on the mouse N-Taf1 (mN-Taf1) sequence published in the DNA Databank of Japan (DDBJ; accession No. AB299229.1). This sequence is identical to that of rat and human N-TAF1 and was predicted to be highly soluble and thus likely to be accessible with an antibody. The peptide was synthesized by TAKARA BIO INC. (Otsu, Japan) and was chemically linked to the carrier protein keyhole limpet hemocyanin (KLH). KLH-conjugated N-TAF1 peptide was injected into two C57BL6 mice. Lymph nodes were then taken from the mice and processed for B cell hybridoma fusions. Hybridoma cloning was performed using ELISA methods to detect positive signals for N-TAF1 peptide but not for TAF1 peptide. Three clones were obtained and from them we selected one hybridoma, 3A-11, that displayed the highest titer for N-TAF1. The hybridoma cells were inoculated into the abdominal lumen of Scid mice and ascitic fluid was obtained. The monoclonal antibody was purified by ammonium sulfate precipitation and affinity chromatography using a protein A column. The purified antibody was stored in 10 mM phosphate buffered saline (PBS), pH 7.4, containing 0.1% sodium azide.
3.2. Plasmids Construction, Immunocytochemistry and Immunoprecipitation
To construct GFP-fused mN-Taf1 and mTaf1, we subcloned PCR fragments into the Bgl II and BamH1 site of the pEGFP-N1 using an In-Fusion Advantage PCR Cloning Kit (TAKARA BIO). In-Fusion cloning sites were introduced into the mN-TAF1 or mTAF1 cDNA [
16] by PCR using the forward primer (5′-TACCGGACTCAGATCCACCATGGGCCCGGGGTG-3′) and reverse primer (5′-GGCGACCGGTGGATCTTCATCAGAGTCCAAATCAGTGTCTCCG-3′). We transfected mN-TAF1-GFP or mTAF1-GFP into Human embryonic kidney cells (HEK293) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. To confirm the expression of mN-TAF1 and mTAF1 in HEK293 cells, immunocytochemistry was performed using different antibodies. After rinsing twice with cold PBS, the cells were fixed for 30 minutes with 4% paraformaldehyde in 100 mM phosphate buffer at 4 °C. The fixed cells were rinsed gently three times with 100 mM PBS containing 0.3% Triton X-100 (PBST). They were incubated overnight in PBST at 4 °C with mN-TAF1 monoclonal antibody (diluted to 1 μg/mL) or TAF1 goat polyclonal antibody (1 μg/mL; sc-17134, Santa Cruz Biotechnology Inc., Santa Cruz, CA). After three 10-minute washes with PBST, the cells were then incubated for 2 hours with a combination of secondary antibodies (Invitrogen), consisting of Alexa Fluor 555 donkey anti-mouse IgG (H+L) and anti-goat IgG (H+L) (diluted 1:500) at room temperature. Cells were rinsed three times with PBST (10 minutes each rinse), and then observed under a Biozero Microscope (Keyence Corporation, Osaka, Japan) with the appropriate filter sets.
For the immunoprecipitation, HEK293 cells were transiently transfected with mN-TAF1-GFP and mTAF1-GFP. Cells were solubilized by sonic treatment in 10 volumes of RIPA buffer (20 mM HEPES buffer pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% NP-40, 1% Sodium Deoxycholate, 0.1% SDS and Complete Mini Protease Inhibitor Cocktail) or Co-IP buffer (20 mM HEPES buffer pH 7.4, 120 mM NaCl, 2 mM EDTA, 1% Triton X-100, 10% Glycerol and Complete Mini Protease Inhibitor Cocktail). Lysates were immunoprecipitated by anti-N-TAF1 antibody or anti-TAF1 antibody using the Dynabeads Protein G Immunoprecipitation Kit (Invitrogen). Eluate was resolved in a 5–20% gradient SDS-polyacrylamide gel. Electrophoresed fractions were transferred to a PVDF membrane, followed by blocking for 1 hour with 5% skim milk dissolved in 25 mM TBS buffer containing 0.1% Tween 20. The membrane was probed overnight at 4 °C by anti-GFP rabbit polyclonal antibody (diluted 1:2,000; Medical & Biological Laboratories Co., Ltd., Nagoya, Japan) and then incubated for 30 minutes at room temperature with a peroxidase-labeled anti-mouse IgG (diluted 1:20,000; Jackson ImmunoResearch Laboratories, Inc.). West Pico Substrate was used as described above to detect chemiluminescent signals on the membranes.
3.3. Animals, Western Blotting and Immunohistochemistry
Three adult male Wistar rats, weighing 270–390 g were used in this study, which was performed in accordance with the PHS Policy on Humane Care and Use of Laboratory Animals, the NIH Guide for the Care and Use of Laboratory Animals, and the Animal Welfare Act (7 U.S.C. et seq.). The Institutional Animal Care and Use Committee of the Shiga University of Medical Science approved all experimental protocols using animals.
One male Wistar rat (CLEA Japan, Tokyo, Japan) weighing 390 g was used for Western blotting. Under deep anesthesia with pentobarbital (80 mg/kg), the rat was killed by decapitation. The striatum was dissected out and homogenized in 5 volumes of ice-cold 50 mM Tris-HCl (pH 7.4) containing 0.5% Triton X-100 and Complete Mini protease inhibitor mixture tablets (one tablet/10 mL; Roche Diagnostics, Mannheim, Germany). The homogenate was separated into soluble and membrane fraction by centrifugation at 12,000 g for 20 minutes at 4 °C. Fifty μg of proteins was electrophoresed on a 4% SDS-polyacrylamide gel. Fractions were then transferred onto polyvinylidene fluoride (PVDF) membranes (Immobilon-P, Millipore, Bedford, MA, USA) which were soaked for 1 hour with 5% skim milk dissolved in 10 mM phosphate buffer containing 0.3% Triton X-100 (TBST). The membrane was incubated overnight at 4 °C with mN-TAF1 monoclonal antibody (diluted 1:1,000) and for 1 hour with a peroxidase-labeled anti-mouse IgG (diluted 1:20,000; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). After washing with TBST, the membranes were incubated for 5 minutes with SuperSignal West Pico Substrate (Thermo Fisher Scientific Inc., Rockford, IL, USA) to observe chemiluminescent signals. Detection was performed with a LAS-4000 image reader (FUJIFILM, Tokyo, Japan).
For the immunohistochemistry, three male Wistar rats weighing 270–560 g were used. Under pentobarbital anesthesia (80 mg/kg), the animals were perfusion-fixed prior to processing for immunohistochemistry as described previously [
18]. Cryostat-cut 20 mm-thick sections of the brain were incubated overnight in a free-floating state with N-TAF1 antibody (diluted 1 μg/mL). The sections were incubated at room temperature for 1 hour with biotinylated anti-mouse IgG (diluted 1:1,000; Vector Laboratories, Burlingame, CA, USA), and then incubated for 1 hour with avidin-biotinylated peroxidase complex (diluted 1:4,000; Vector Laboratories), also at room temperature. PBST was used to dilute the antibodies and wash the sections between each step. A purple color was developed with 0.02% 3,3′-diaminobenzidine and 0.3% nickel ammonium sulfate in 50 mM Tris–HCl buffer (pH 7.6). After treatment for 30 minutes at room temperature with 0.5% hydrogen peroxide in PBST to eliminate the residual horseradish peroxidase activity, the sections underwent secondary immunostaining with rabbit anti-glial fibrillary acidic protein (GFAP) antibody (diluted 1:1,000; DAKO, Carpinteria, CA, USA). Some sections were used for double-staining for N-TAF1 (brown) and Nissl stain (purple). After the primary N-TAF1 immunoreaction, sections were soaked in the second antibody for 1 hour with Histofine Simple Stain MAX-PO (M) (diluted 1:10, NICHIREI BIOSCIENCES INC., Tokyo, Japan). Color was developed by reacting the sections for 20 minutes with 50 mM Tris-HCl buffer (pH 7.6) containing 0.02% 3,3′-diaminobenzidine and 0.005% hydrogen peroxide. After mounting on silane-coated glass slides, sections were stained with cresyl violet.



{kind=link}
{kind=link}
{kind=link}