Diving through Membranes: Molecular Cunning to Enforce the Endosomal Escape of Antibody-Targeted Anti-Tumor Toxins


Abstract
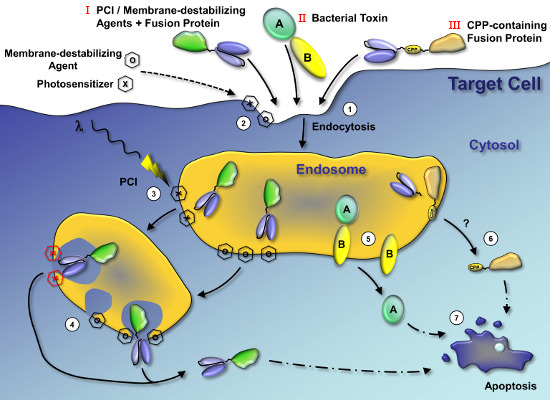
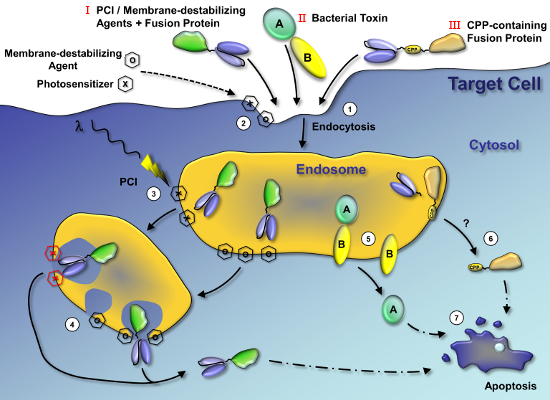
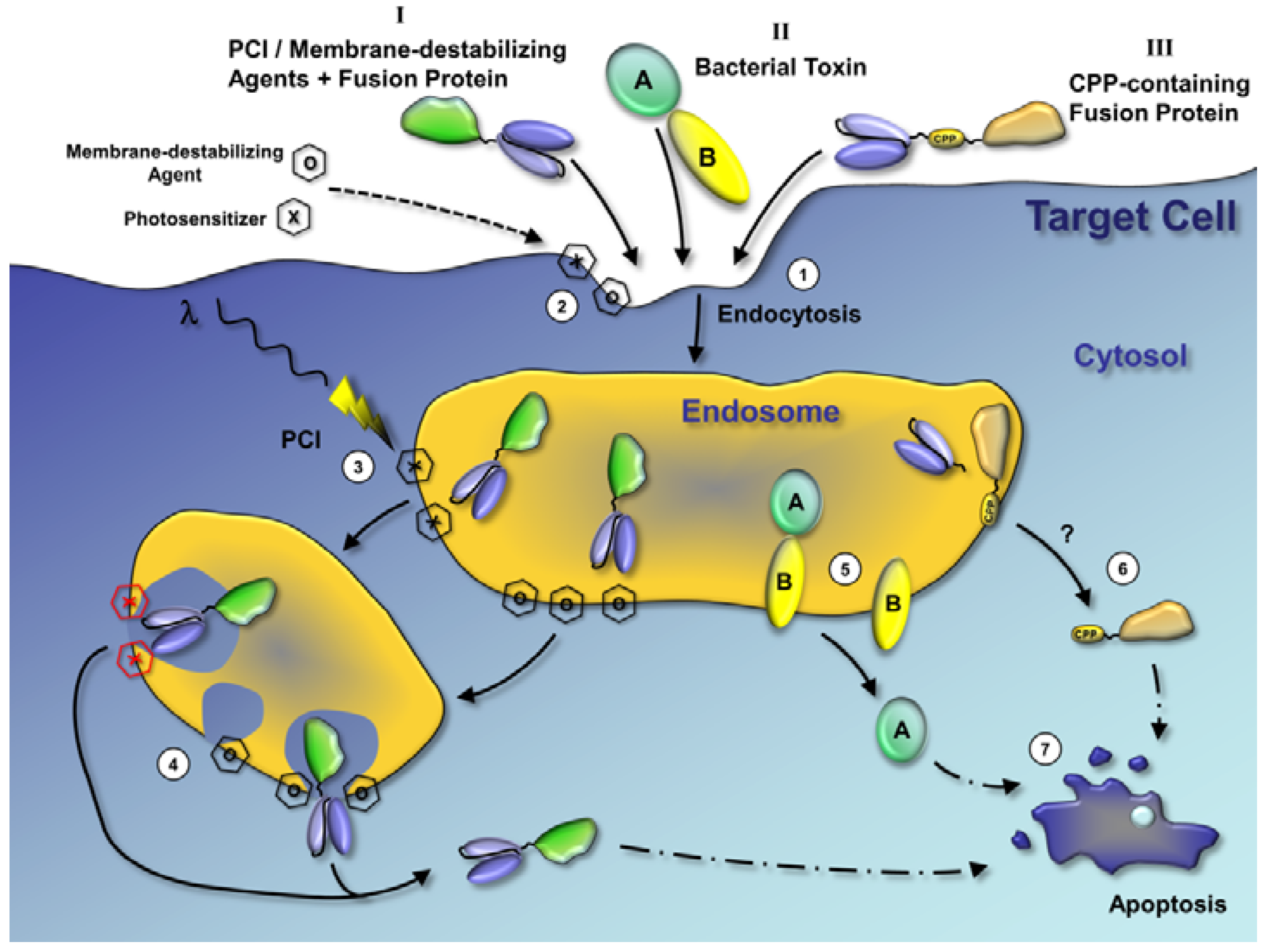
:
{kind=link}
{kind=link}
{kind=link}
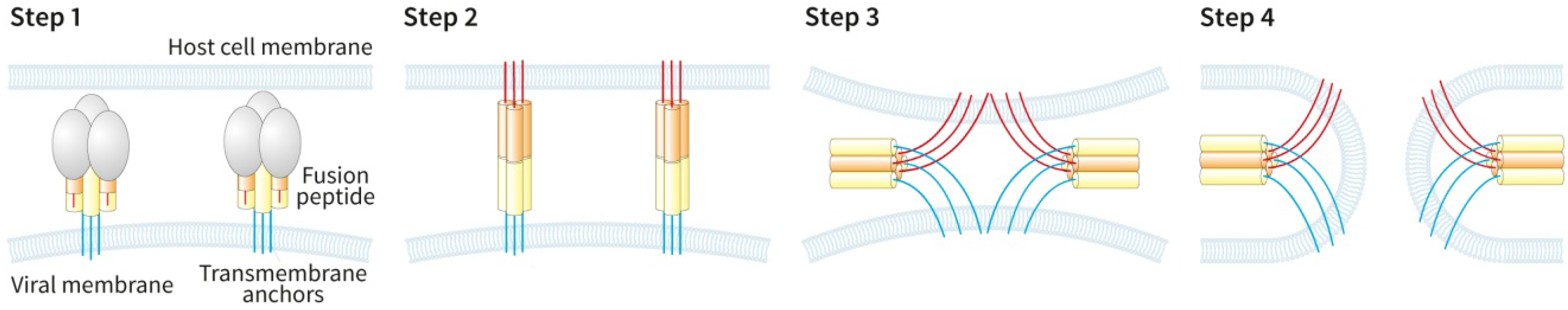{kind=link}
1. Introduction
2. Endosomal Escape
3. Molecular Ferries
3.1. Identifying Cell Penetrating Peptides from Various Organisms to Mediate Cytosolic Uptake
3.2. Exploiting Viruses to Augment Targeted Toxin Entry into Target Cells
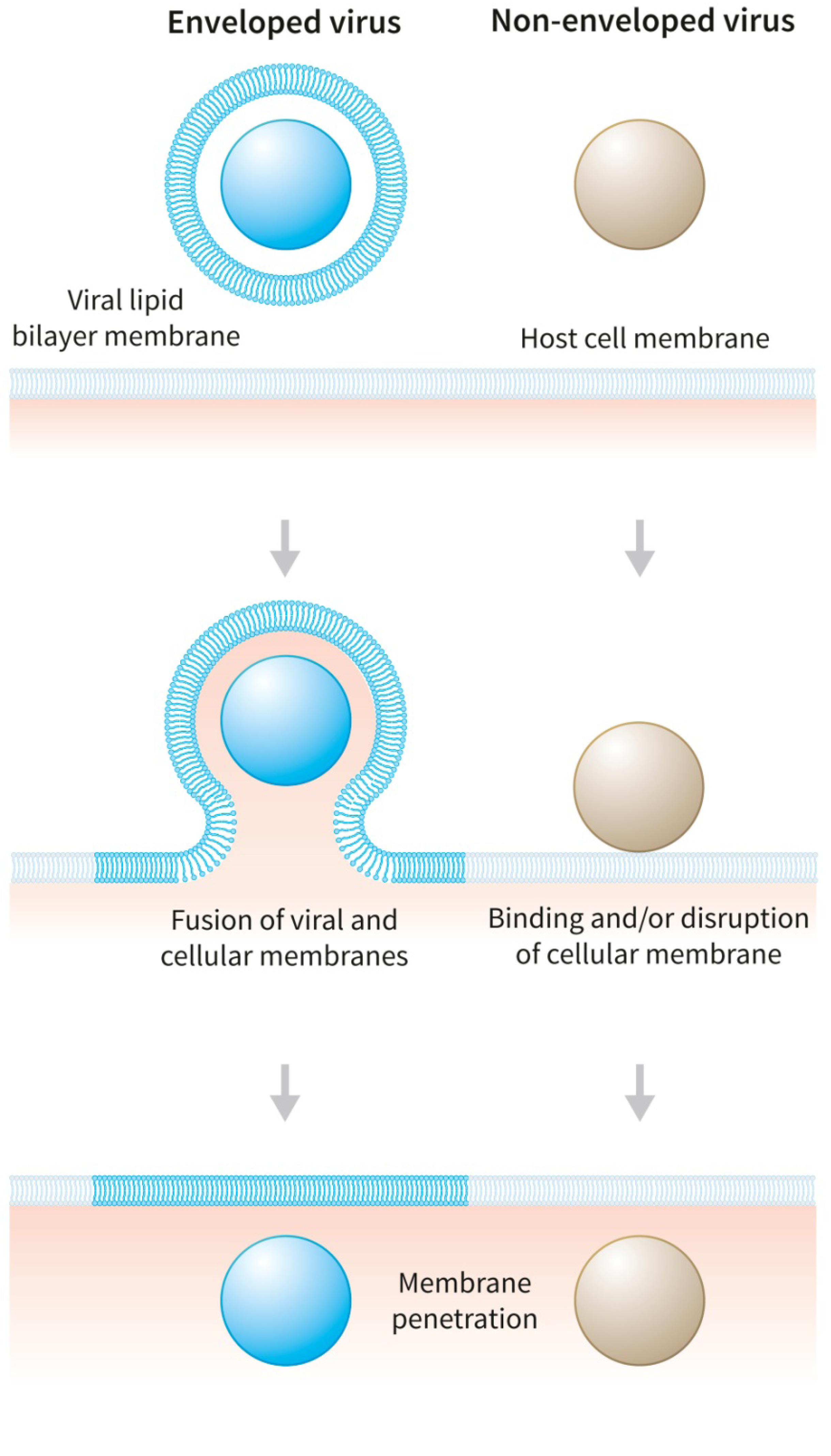

4. Leakage-Inducing Molecules
5. Physicochemical Techniques
6. Discussion
7. Conclusions
Acknowledgments
References
- Adair, J.R.; Howard, P.W.; Hartley, J.A.; Williams, D.G.; Chester, K.A. Antibody-drug conjugates—A perfect synergy. Expert Opin. Biol. Ther. 2012, 12, 1191–1206. [Google Scholar] [CrossRef]
- Govindan, S.V.; Goldenberg, D.M. Designing immunoconjugates for cancer therapy. Expert Opin. Biol. Ther. 2012, 12, 873–890. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Sharkey, R.M. Using antibodies to target cancer therapeutics. Expert Opin. Biol. Ther. 2012, 12, 1173–1190. [Google Scholar] [CrossRef]
- Chu, T.C.; Marks, J.W., 3rd; Lavery, L.A.; Faulkner, S.; Rosenblum, M.G.; Ellington, A.D.; Levy, M. Aptamer: Toxin conjugates that specifically target prostate tumor cells. Cancer Res. 2006, 66, 5989–5992. [Google Scholar] [CrossRef]
- Fuchs, H.; Bachran, C. Targeted tumor therapies at a glance. Curr. Drug Targets 2009, 10, 89–93. [Google Scholar] [CrossRef]
- Martin-Killias, P.; Stefan, N.; Rothschild, S.; Pluckthun, A.; Zangemeister-Wittke, U. A novel fusion toxin derived from an EpCAM-specific designed ankyrin repeat protein has potent antitumor activity. Clin. Cancer Res. 2011, 17, 100–110. [Google Scholar] [CrossRef]
- Vallera, D.A.; Chen, H.; Sicheneder, A.R.; Panoskaltsis-Mortari, A.; Taras, E.P. Genetic alteration of a bispecific ligand-directed toxin targeting human CD19 and CD22 receptors resulting in improved efficacy against systemic B cell malignancy. Leuk. Res. 2009, 33, 1233–1242. [Google Scholar] [CrossRef]
- Oh, S.; Stish, B.J.; Sachdev, D.; Chen, H.; Dudek, A.Z.; Vallera, D.A. A novel reduced immunogenicity bispecific targeted toxin simultaneously recognizing human epidermal growth factor and interleukin-4 receptors in a mouse model of metastatic breast carcinoma. Clin. Cancer Res. 2009, 15, 6137–6147. [Google Scholar] [CrossRef]
- Fuchs, H.; Bachran, C. Design of targeted protein toxins. In Drug Delivery in Oncology—From Basic Research to Cancer Therapy; Kratz, F., Senter, P., Steinhagen, H., Eds.; Wiley-VCH: Weinheim, Germany, 2011; Volume 3, pp. 1443–1487. [Google Scholar]
- Hetzel, C.; Bachran, C.; Tur, M.K.; Fuchs, H.; Stocker, M. Improved immunotoxins with novel functional elements. Curr. Pharm. Des. 2009, 15, 2700–2711. [Google Scholar] [CrossRef]
- Weldon, J.E.; Xiang, L.; Chertov, O.; Margulies, I.; Kreitman, R.J.; FitzGerald, D.J.; Pastan, I. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood 2009, 113, 3792–3800. [Google Scholar] [CrossRef]
- McGrath, M.S.; Rosenblum, M.G.; Philips, M.R.; Scheinberg, D.A. Immunotoxin resistance in multidrug resistant cells. Cancer Res. 2003, 63, 72–79. [Google Scholar]
- Moolten, F.L.; Cooperband, S.R. Selective destruction of target cells by diphtheria toxin conjugated to antibody directed against antigens on the cells. Science 1970, 169, 68–70. [Google Scholar]
- Liu, S.; Milne, G.T.; Kuremsky, J.G.; Fink, G.R.; Leppla, S.H. Identification of the proteins required for biosynthesis of diphthamide, the target of bacterial ADP-ribosylating toxins on translation elongation factor 2. Mol. Cell. Biol. 2004, 24, 9487–9497. [Google Scholar] [CrossRef]
- Kanellos, J.; MacKenzie, I.F.; Pietersz, G.A. In vivo studies of whole ricin monoclonal antibody immunoconjugates for the treatment of murine tumours. Immunol. Cell Biol. 1988, 66, 403–415. [Google Scholar] [CrossRef]
- Blythman, H.E.; Casellas, P.; Gros, O.; Gros, P.; Jansen, F.K.; Paolucci, F.; Pau, B.; Vidal, H. Immunotoxins: Hybrid molecules of monoclonal antibodies and a toxin subunit specifically kill tumour cells. Nature 1981, 290, 145–146. [Google Scholar]
- Weldon, J.E.; Pastan, I. A guide to taming a toxin—Recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011, 278, 4683–4700. [Google Scholar] [CrossRef]
- Polito, L.; Bortolotti, M.; Pedrazzi, M.; Bolognesi, A. Immunotoxins and other conjugates containing saporin-s6 for cancer therapy. Toxins (Basel) 2011, 3, 697–720. [Google Scholar] [CrossRef]
- Puri, M.; Kaur, I.; Perugini, M.A.; Gupta, R.C. Ribosome-inactivating proteins: Current status and biomedical applications. Drug Discov. Today 2012, 17, 774–783. [Google Scholar]
- Stirpe, F. Ribosome-inactivating proteins. Toxicon 2004, 44, 371–383. [Google Scholar] [CrossRef]
- Endo, Y.; Mitsui, K.; Motizuki, M.; Tsurugi, K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 S ribosomal RNA caused by the toxins. J. Biol. Chem. 1987, 262, 5908–5912. [Google Scholar]
- Rosenblum, M.G.; Barth, S. Development of novel, highly cytotoxic fusion constructs containing granzyme B: Unique mechanisms and functions. Curr. Pharm. Des. 2009, 15, 2676–2692. [Google Scholar] [CrossRef]
- Gao, X.; Xu, Z. Mechanisms of action of angiogenin. Acta Biochim. Biophys. Sin. (Shanghai) 2008, 40, 619–624. [Google Scholar] [CrossRef]
- Mahmud, H.; Dalken, B.; Wels, W.S. Induction of programmed cell death in ErbB2/HER2-expressing cancer cells by targeted delivery of apoptosis-inducing factor. Mol. Cancer Ther. 2009, 8, 1526–1535. [Google Scholar]
- Li, H.; Ray, G.; Yoo, B.H.; Erdogan, M.; Rosen, K.V. Down-regulation of death-associated protein kinase-2 is required for beta-catenin-induced anoikis resistance of malignant epithelial cells. J. Biol. Chem. 2009, 284, 2012–2022. [Google Scholar]
- Tur, M.K.; Neef, I.; Jost, E.; Galm, O.; Jager, G.; Stocker, M.; Ribbert, M.; Osieka, R.; Klinge, U.; Barth, S. Targeted restoration of down-regulated DAPK2 tumor suppressor activity induces apoptosis in Hodgkin lymphoma cells. J. Immunother. 2009, 32, 431–441. [Google Scholar] [CrossRef]
- Pennarun, B.; Meijer, A.; de Vries, E.G.; Kleibeuker, J.H.; Kruyt, F.; de Jong, S. Playing the DISC: turning on TRAIL death receptor-mediated apoptosis in cancer. Biochim. Biophys. Acta 2010, 1805, 123–140. [Google Scholar]
- Pirie, C.M.; Hackel, B.J.; Rosenblum, M.G.; Wittrup, K.D. Convergent potency of internalized gelonin immunotoxins across varied cell lines, antigens, and targeting moieties. J. Biol. Chem. 2011, 286, 4165–4172. [Google Scholar] [CrossRef]
- Bachran, C.; Durkop, H.; Sutherland, M.; Bachran, D.; Muller, C.; Weng, A.; Melzig, M.F.; Fuchs, H. Inhibition of tumor growth by targeted toxins in mice is dramatically improved by saponinum album in a synergistic way. J. Immunother. 2009, 32, 713–725. [Google Scholar] [CrossRef]
- Alfano, R.W.; Leppla, S.H.; Liu, S.; Bugge, T.H.; Ortiz, J.M.; Lairmore, T.C.; Duesbery, N.S.; Mitchell, I.C.; Nwariaku, F.; Frankel, A.E. Inhibition of tumor angiogenesis by the matrix metalloproteinase-activated anthrax lethal toxin in an orthotopic model of anaplastic thyroid carcinoma. Mol. Cancer Ther. 2010, 9, 190–201. [Google Scholar]
- Fujisawa, T.; Nakashima, H.; Nakajima, A.; Joshi, B.H.; Puri, R.K. Targeting IL-13Ralpha2 in human pancreatic ductal adenocarcinoma with combination therapy of IL-13-PE and gemcitabine. Int. J. Cancer 2010, 128, 1221–1231. [Google Scholar]
- Hassan, R.; Williams-Gould, J.; Steinberg, S.M.; Liewehr, D.J.; Yokokawa, J.; Tsang, K.Y.; Surawski, R.J.; Scott, T.; Camphausen, K. Tumor-directed radiation and the immunotoxin SS1P in the treatment of mesothelin-expressing tumor xenografts. Clin. Cancer Res. 2006, 12, 4983–4988. [Google Scholar]
- Gupta, M.; Tiwari, S.; Vyas, S. Structuring polymers for delivery of DNA-based therapeutics: Updated insights. Crit. Rev. Ther. Drug Carrier Syst. 2012, 29, 447–485. [Google Scholar] [CrossRef]
- Du, J.; Jin, J.; Yan, M.; Lu, Y. Synthetic nanocarriers for intracellular protein delivery. Curr. Drug Metab. 2012, 13, 82–92. [Google Scholar] [CrossRef]
- Pavlin, M.; Miklavcic, D. Theoretical and experimental analysis of conductivity, ion diffusion and molecular transport during cell electroporation—Relation between short-lived and long-lived pores. Bioelectrochemistry 2008, 74, 38–46. [Google Scholar] [CrossRef]
- Polson, A.G.; Calemine-Fenaux, J.; Chan, P.; Chang, W.; Christensen, E.; Clark, S.; de Sauvage, F.J.; Eaton, D.; Elkins, K.; Elliott, J.M.; et al. Antibody-drug conjugates for the treatment of non-Hodgkin's lymphoma: Target and linker-drug selection. Cancer Res. 2009, 69, 2358–2364. [Google Scholar] [CrossRef]
- Gan, Q.; Lu, X.; Dong, W.; Yuan, Y.; Qian, J.; Li, Y.; Shi, J.; Liu, C. Endosomal pH-activatable magnetic nanoparticle-capped mesoporous silica for intracellular controlled release. J. Materials Chem. 2012, 22, 15960–15968. [Google Scholar]
- Lee, E.S.; Gao, Z.; Bae, Y.H. Recent progress in tumor pH targeting nanotechnology. J. Control Release 2008, 132, 164–170. [Google Scholar] [CrossRef]
- Reyes, A.G.; Anne, J.; Mejia, A. Ribosome-inactivating proteins with an emphasis on bacterial RIPs and their potential medical applications. Future Microbiol. 2012, 7, 705–717. [Google Scholar] [CrossRef]
- Potala, S.; Sahoo, S.K.; Verma, R.S. Targeted therapy of cancer using diphtheria toxin-derived immunotoxins. Drug Discov. Today 2008, 13, 807–815. [Google Scholar] [CrossRef]
- Young, J.A.; Collier, R.J. Anthrax toxin: Receptor binding, internalization, pore formation, and translocation. Annu. Rev. Biochem. 2007, 76, 243–265. [Google Scholar] [CrossRef]
- Liu, S.; Redeye, V.; Kuremsky, J.G.; Kuhnen, M.; Molinolo, A.; Bugge, T.H.; Leppla, S.H. Intermolecular complementation achieves high-specificity tumor targeting by anthrax toxin. Nat. Biotechnol. 2005, 23, 725–730. [Google Scholar] [CrossRef]
- Schafer, J.M.; Peters, D.E.; Morley, T.; Liu, S.; Molinolo, A.A.; Leppla, S.H.; Bugge, T.H. Efficient targeting of head and neck squamous cell carcinoma by systemic administration of a dual uPA and MMP-activated engineered anthrax toxin. PLoS One 2011, 6, e20532. [Google Scholar]
- Hu, H.; Leppla, S.H. Anthrax toxin uptake by primary immune cells as determined with a lethal factor-beta-lactamase fusion protein. PLoS One 2009, 4, e7946. [Google Scholar] [CrossRef]
- Liu, X.H.; Collier, R.J.; Youle, R.J. Inhibition of axotomy-induced neuronal apoptosis by extracellular delivery of a Bcl-XL fusion protein. J. Biol. Chem. 2001, 276, 46326–46332. [Google Scholar] [CrossRef]
- Fuchs, H.; Bachran, C.; Li, T.; Heisler, I.; Durkop, H.; Sutherland, M. A cleavable molecular adapter reduces side effects and concomitantly enhances efficacy in tumor treatment by targeted toxins in mice. J. Control Release 2007, 117, 342–350. [Google Scholar] [CrossRef]
- Heisler, I.; Keller, J.; Tauber, R.; Sutherland, M.; Fuchs, H. A cleavable adapter to reduce nonspecific cytotoxicity of recombinant immunotoxins. Int. J. Cancer 2003, 103, 277–282. [Google Scholar] [CrossRef]
- Del Gaizo, V.; Payne, R.M. A novel TAT-mitochondrial signal sequence fusion protein is processed, stays in mitochondria, and crosses the placenta. Mol. Ther. 2003, 7, 720–730. [Google Scholar]
- Chen, X.; Bai, Y.; Zaro, J.L.; Shen, W.-C. Design of an in vivo cleavable disulfide linker in recombinant fusion proteins. BioTechniques 2010, 49, 513–518. [Google Scholar] [CrossRef]
- Sawant, R.; Torchilin, V. Intracellular transduction using cell-penetrating peptides. Mol. Biosyst. 2010, 6, 628–640. [Google Scholar] [CrossRef]
- Snyder, E.L.; Saenz, C.C.; Denicourt, C.; Meade, B.R.; Cui, X.S.; Kaplan, I.M.; Dowdy, S.F. Enhanced targeting and killing of tumor cells expressing the CXC chemokine receptor 4 by transducible anticancer peptides. Cancer Res. 2005, 65, 10646–10650. [Google Scholar]
- Lorenzetti, I.; Meneguzzi, A.; Fracasso, G.; Potrich, C.; Costantini, L.; Chiesa, E.; Legname, G.; Menestrina, G.; Tridente, G.; Colombatti, M. Genetic grafting of membrane-acting peptides to the cytotoxin dianthin augments its ability to de-stabilize lipid bilayers and enhances its cytotoxic potential as the component of transferrin-toxin conjugates. Int. J. Cancer 2000, 86, 582–589. [Google Scholar] [CrossRef]
- Hetzel, C.; Bachran, C.; Fischer, R.; Fuchs, H.; Barth, S.; Stocker, M. Small cleavable adapters enhance the specific cytotoxicity of a humanized immunotoxin directed against CD64-positive cells. J. Immunother. 2008, 31, 370–376. [Google Scholar] [CrossRef]
- Olson, E.S.; Aguilera, T.A.; Jiang, T.; Ellies, L.G.; Nguyen, Q.T.; Wong, E.H.; Gross, L.A.; Tsien, R.Y. In vivo characterization of activatable cell penetrating peptides for targeting protease activity in cancer. Integr. Biol. (Camb.) 2009, 1, 382–393. [Google Scholar] [CrossRef]
- Fuchs, H.; Bachran, C.; Heisler, I.; Sutherland, M. A closer look at protein transduction domains as a tool in drug delivery. Curr. Nanosci. 2005, 1, 117–124. [Google Scholar] [CrossRef]
- Cizeau, J.; Grenkow, D.M.; Brown, J.G.; Entwistle, J.; MacDonald, G.C. Engineering and biological characterization of VB6-845, an anti-EpCAM immunotoxin containing a T-cell epitope-depleted variant of the plant toxin bouganin. J. Immunother. 2009, 32, 574–584. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, J.; Wang, T.; Yu, C.J.; Jia, L.T.; Duan, Y.Y.; Yao, L.B.; Chen, S.Y.; Yang, A.G. HER2-targeting recombinant protein with truncated pseudomonas exotoxin A translocation domain efficiently kills breast cancer cells. Cancer Biol. Ther. 2008, 7, 1226–1231. [Google Scholar] [CrossRef]
- Cao, Y.; Marks, J.D.; Marks, J.W.; Cheung, L.H.; Kim, S.; Rosenblum, M.G. Construction and characterization of novel, recombinant immunotoxins targeting the Her2/neu oncogene product: In vitro and in vivo studies. Cancer Res. 2009, 69, 8987–8995. [Google Scholar] [CrossRef]
- Curiel, D.T.; Agarwal, S.; Wagner, E.; Cotten, M. Adenovirus enhancement of transferrin-polylysine-mediated gene delivery. Proc. Natl. Acad. Sci. USA 1991, 88, 8850–8854. [Google Scholar] [CrossRef]
- Jahn, R.; Scheller, R.H. SNAREs—Engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 2006, 7, 631–643. [Google Scholar] [CrossRef]
- Tsai, B. Penetration of nonenveloped viruses into the cytoplasm. Annu. Rev. Cell Dev. Biol. 2007, 23, 23–43. [Google Scholar] [CrossRef]
- Weissenhorn, W.; Hinz, A.; Gaudin, Y. Virus membrane fusion. FEBS Lett. 2007, 581, 2150–2155. [Google Scholar] [CrossRef]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef]
- Vazquez-Calvo, A.; Saiz, J.C.; McCullough, K.C.; Sobrino, F.; Martin-Acebes, M.A. Acid-dependent viral entry. Virus Res. 2012, 167, 125–137. [Google Scholar]
- Han, X.; Bushweller, J.H.; Cafiso, D.S.; Tamm, L.K. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat. Struct. Biol. 2001, 8, 715–720. [Google Scholar] [CrossRef]
- Wharton, S.A.; Martin, S.R.; Ruigrok, R.W.; Skehel, J.J.; Wiley, D.C. Membrane fusion by peptide analogues of influenza virus haemagglutinin. J. Gen. Virol. 1988, 69, 1847–1857. [Google Scholar] [CrossRef]
- Wagner, E.; Plank, C.; Zatloukal, K.; Cotten, M.; Birnstiel, M.L. Influenza virus hemagglutinin HA-2 N-terminal fusogenic peptides augment gene transfer by transferrin-polylysine-DNA complexes: Toward a synthetic virus-like gene-transfer vehicle. Proc. Natl. Acad. Sci. USA 1992, 89, 7934–7938. [Google Scholar] [CrossRef] [Green Version]
- Hughson, F.M. Enveloped viruses: A common mode of membrane fusion? Curr. Biol. 1997, 7, R565–R569. [Google Scholar] [CrossRef]
- Chignola, R.; Anselmi, C.; Dalla Serra, M.; Franceschi, A.; Fracasso, G.; Pasti, M.; Chiesa, E.; Lord, J.M.; Tridente, G.; Colombatti, M. Self-potentiation of ligand-toxin conjugates containing ricin A chain fused with viral structures. J. Biol. Chem. 1995, 270, 23345–23351. [Google Scholar] [CrossRef]
- Tolstikov, V.V.; Cole, R.; Fang, H.; Pincus, S.H. Influence of endosome-destabilizing peptides on efficacy of anti-HIV immunotoxins. Bioconjug. Chem. 1997, 8, 38–43. [Google Scholar] [CrossRef]
- Greber, U.F.; Webster, P.; Weber, J.; Helenius, A. The role of the adenovirus protease on virus entry into cells. EMBO J. 1996, 15, 1766–1777. [Google Scholar]
- Medina-Kauwe, L.K. Endocytosis of adenovirus and adenovirus capsid proteins. Adv. Drug Deliv. Rev. 2003, 55, 1485–1496. [Google Scholar] [CrossRef]
- Wiethoff, C.M.; Wodrich, H.; Gerace, L.; Nemerow, G.R. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 2005, 79, 1992–2000. [Google Scholar] [CrossRef]
- Stewart, P.L.; Fuller, S.D.; Burnett, R.M. Difference imaging of adenovirus: Bridging the resolution gap between X-ray crystallography and electron microscopy. EMBO J. 1993, 12, 2589–2599. [Google Scholar]
- FitzGerald, D.J.; Padmanabhan, R.; Pastan, I.; Willingham, M.C. Adenovirus-induced release of epidermal growth factor and pseudomonas toxin into the cytosol of KB cells during receptor-mediated endocytosis. Cell 1983, 32, 607–617. [Google Scholar] [CrossRef]
- Seth, P.; Fitzgerald, D.; Ginsberg, H.; Willingham, M.; Pastan, I. Evidence that the penton base of adenovirus is involved in potentiation of toxicity of Pseudomonas exotoxin conjugated to epidermal growth factor. Mol. Cell. Biol. 1984, 4, 1528–1533. [Google Scholar]
- FitzGerald, D.J.; Trowbridge, I.S.; Pastan, I.; Willingham, M.C. Enhancement of toxicity of antitransferrin receptor antibody-Pseudomonas exotoxin conjugates by adenovirus. Proc. Natl. Acad. Sci. USA 1983, 80, 4134–4138. [Google Scholar] [CrossRef]
- Griffin, T.W.; Childs, L.R.; FitzGerald, D.J.; Levin, L.V. Enhancement of the cytotoxic effect of anti-carcinoembryonic antigen immunotoxins by adenovirus and carboxylic ionophores. J. Natl. Cancer Inst. 1987, 79, 679–685. [Google Scholar]
- Satyamoorthy, K.; Soballe, P.W.; Soans, F.; Herlyn, M. Adenovirus infection enhances killing of melanoma cells by a mitotoxin. Cancer Res. 1997, 57, 1873–1876. [Google Scholar]
- Goldmacher, V.S.; Blattler, W.A.; Lambert, J.M.; McIntyre, G.; Stewart, J. Cytotoxicity of gelonin conjugated to targeting molecules: Effects of weak amines, monensin, adenovirus, and adenoviral capsid proteins penton, hexon, and fiber. Mol. Pharmacol. 1989, 36, 818–822. [Google Scholar]
- Matthews, D.A.; Russell, W.C. Adenovirus protein-protein interactions: Hexon and protein VI. J. Gen. Virol. 1994, 75, 3365–3374. [Google Scholar] [CrossRef]
- Matthews, D.A.; Russell, W.C. Adenovirus protein-protein interactions: Molecular parameters governing the binding of protein VI to hexon and the activation of the adenovirus 23K protease. J. Gen. Virol. 1995, 76, 1959–1969. [Google Scholar] [CrossRef]
- Lai, C.Y.; Wiethoff, C.M.; Kickhoefer, V.A.; Rome, L.H.; Nemerow, G.R. Vault nanoparticles containing an adenovirus-derived membrane lytic protein facilitate toxin and gene transfer. ACS Nano 2009, 3, 691–699. [Google Scholar] [CrossRef]
- Kickhoefer, V.A.; Garcia, Y.; Mikyas, Y.; Johansson, E.; Zhou, J.C.; Raval-Fernandes, S.; Minoofar, P.; Zink, J.I.; Dunn, B.; Stewart, P.L.; et al. Engineering of vault nanocapsules with enzymatic and fluorescent properties. Proc. Natl. Acad. Sci. USA 2005, 102, 4348–4352. [Google Scholar] [CrossRef]
- Liu, X.; Wu, J.; Zhang, S.; Li, C.; Huang, Q. Novel strategies to augment genetically delivered immunotoxin molecular therapy for cancer therapy. Cancer Gene Ther. 2009, 16, 861–872. [Google Scholar] [CrossRef]
- Huang, H.W.; Chen, F.Y.; Lee, M.T. Molecular mechanism of Peptide-induced pores in membranes. Phys. Rev. Lett. 2004, 92, 198304. [Google Scholar]
- Jenssen, H.; Hamill, P.; Hancock, R.E. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef]
- Li, W.; Nicol, F.; Szoka, F.C., Jr. GALA: A designed synthetic pH-responsive amphipathic peptide with applications in drug and gene delivery. Adv. Drug Deliv. Rev. 2004, 56, 967–985. [Google Scholar] [CrossRef]
- Liang, W.; Lam, J.K.W. Endosomal escape pathways for non-viral nucleic acid delivery systems. In Molecular Regulation of Endocytosis; Ceresa, B., Ed.; InTech: Rijeka, Croatia, 2012; pp. 429–456. [Google Scholar]
- Fuchs, H.; Bachran, D.; Panjideh, H.; Schellmann, N.; Weng, A.; Melzig, M.F.; Sutherland, M.; Bachran, C. Saponins as tool for improved targeted tumor therapies. Curr. Drug Targets 2009, 10, 140–151. [Google Scholar] [CrossRef]
- Wu, M. Enhancement of immunotoxin activity using chemical and biological reagents. Br. J. Cancer 1997, 75, 1347–1355. [Google Scholar] [CrossRef]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control Release 2011, 151, 220–228. [Google Scholar] [CrossRef]
- Chollet, P.; Favrot, M.C.; Hurbin, A.; Coll, J.L. Side-effects of a systemic injection of linear polyethylenimine-DNA complexes. J. Gene Med. 2002, 4, 84–91. [Google Scholar]
- Zenke, M.; Steinlein, P.; Wagner, E.; Cotten, M.; Beug, H.; Birnstiel, M.L. Receptor-mediated endocytosis of transferrin-polycation conjugates: An efficient way to introduce DNA into hematopoietic cells. Proc. Natl. Acad. Sci. USA 1990, 87, 3655–3659. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Sawyer, G.J.; Dong, X.; Qiu, Y.; Collins, L.; Fabre, J.W. The in vivo use of chloroquine to promote non-viral gene delivery to the liver via the portal vein and bile duct. J. Gene Med. 2003, 5, 209–218. [Google Scholar] [CrossRef]
- Vago, R.; Marsden, C.J.; Lord, J.M.; Ippoliti, R.; Flavell, D.J.; Flavell, S.U.; Ceriotti, A.; Fabbrini, M.S. Saporin and ricin A chain follow different intracellular routes to enter the cytosol of intoxicated cells. FEBS J. 2005, 272, 4983–4995. [Google Scholar] [CrossRef]
- Davol, P.A.; Bizuneh, A.; Frackelton, A.R., Jr. Wortmannin, a phosphoinositide 3-kinase inhibitor, selectively enhances cytotoxicity of receptor-directed-toxin chimeras in vitro and in vivo. Anticancer Res. 1999, 19, 1705–1713. [Google Scholar]
- Ippoliti, R.; Ginobbi, P.; Lendaro, E.; D'Agostino, I.; Ombres, D.; Benedetti, P.A.; Brunori, M.; Citro, G. The effect of monensin and chloroquine on the endocytosis and toxicity of chimeric toxins. Cell. Mol. Life Sci. 1998, 54, 866–875. [Google Scholar] [CrossRef]
- Shaik, M.S.; Ikediobi, O.; Turnage, V.D.; McSween, J.; Kanikkannan, N.; Singh, M. Long-circulating monensin nanoparticles for the potentiation of immunotoxin and anticancer drugs. J. Pharm. Pharmacol. 2001, 53, 617–627. [Google Scholar] [CrossRef]
- Bachran, C.; Weng, A.; Bachran, D.; Riese, S.B.; Schellmann, N.; Melzig, M.F.; Fuchs, H. The distribution of saponins in vivo affects their synergy with chimeric toxins against tumours expressing human epidermal growth factor receptors in mice. Br. J. Pharmacol. 2010, 159, 345–352. [Google Scholar] [CrossRef]
- Heisler, I.; Sutherland, M.; Bachran, C.; Hebestreit, P.; Schnitger, A.; Melzig, M.F.; Fuchs, H. Combined application of saponin and chimeric toxins drastically enhances the targeted cytotoxicity on tumor cells. J. Control Release 2005, 106, 123–137. [Google Scholar] [CrossRef]
- Weng, A.; Thakur, M.; Beceren-Braun, F.; Bachran, D.; Bachran, C.; Riese, S.B.; Jenett-Siems, K.; Gilabert-Oriol, R.; Melzig, M.F.; Fuchs, H. The toxin component of targeted anti-tumor toxins determines their efficacy increase by saponins. Mol. Oncol. 2012, 6, 323–332. [Google Scholar] [CrossRef]
- Weng, A.; Thakur, M.; von Mallinckrodt, B.; Beceren-Braun, F.; Gilabert-Oriol, R.; Wiesner, B.; Eichhorst, J.; Bottger, S.; Melzig, M.F.; Fuchs, H. Saponins modulate the intracellular trafficking of protein toxins. J. Control Release 2012, 164, 74–86. [Google Scholar] [CrossRef]
- Bachran, D.; Schneider, S.; Bachran, C.; Weng, A.; Melzig, M.F.; Fuchs, H. The endocytic uptake pathways of targeted toxins are influenced by synergistically acting Gypsophila saponins. Mol. Pharm. 2011, 8, 2262–2272. [Google Scholar] [CrossRef]
- Thakur, M.; Weng, A.; Bachran, D.; Riese, S.B.; Bottger, S.; Melzig, M.F.; Fuchs, H. Electrophoretic isolation of saponin fractions from Saponinum album and their evaluation in synergistically enhancing the receptor-specific cytotoxicity of targeted toxins. Electrophoresis 2011, 32, 3085–3089. [Google Scholar] [CrossRef]
- Thakur, M.; Mergel, K.; Weng, A.; von Mallinckrodt, B.; Gilabert-Oriol, R.; Durkop, H.; Melzig, M.F.; Fuchs, H. Targeted tumor therapy by epidermal growth factor appended toxin and purified saponin: An evaluation of toxicity and therapeutic potential in syngeneic tumor bearing mice. Mol. Oncol. 2012, in press. [Google Scholar]
- Master, A.; Livingston, M.; Sen Gupta, A. Photodynamic nanomedicine in the treatment of solid tumors: Perspectives and challenges. J. Control Release 2013, in press. [Google Scholar]
- Bossu, E.; A'Amar, O.; Parache, R.M.; Notter, D.; Labrude, P.; Vigneron, C.; Guillemin, F. Determination of the maximal tumor/normal skin ratio after HpD or m-THPC administration in hairless mouse (SKh-1) by fluorescence spectroscopy—A non-invasive method. Anticancer Drugs 1997, 8, 67–72. [Google Scholar] [CrossRef]
- Adigbli, D.K.; MacRobert, A.J. Photochemical internalisation: The journey from basic scientific concept to the threshold of clinical application. Curr. Opin. Pharmacol. 2012, 12, 434–438. [Google Scholar] [CrossRef]
- Weyergang, A.; Selbo, P.K.; Berstad, M.E.; Bostad, M.; Berg, K. Photochemical internalization of tumor-targeted protein toxins. Lasers Surg. Med. 2011, 43, 721–733. [Google Scholar]
- Selbo, P.K.; Rosenblum, M.G.; Cheung, L.H.; Zhang, W.; Berg, K. Multi-modality therapeutics with potent anti-tumor effects: Photochemical internalization enhances delivery of the fusion toxin scFvMEL/rGel. PLoS One 2009, 4, e6691. [Google Scholar]
- Yip, W.L.; Weyergang, A.; Berg, K.; Tonnesen, H.H.; Selbo, P.K. Targeted delivery and enhanced cytotoxicity of cetuximab-saporin by photochemical internalization in EGFR-positive cancer cells. Mol. Pharm. 2007, 4, 241–251. [Google Scholar]
- Berstad, M.B.; Weyergang, A.; Berg, K. Photochemical internalization (PCI) of HER2-targeted toxins: Synergy is dependent on the treatment sequence. Biochim. Biophys. Acta 2012, 1820, 1849–1858. [Google Scholar]
- Mathews, M.S.; Blickenstaff, J.W.; Shih, E.C.; Zamora, G.; Vo, V.; Sun, C.H.; Hirschberg, H.; Madsen, S.J. Photochemical internalization of bleomycin for glioma treatment. J. Biomed. Opt. 2012, 17, 058001. [Google Scholar] [CrossRef]
- Mathews, M.S.; Vo, V.; Shih, E.C.; Zamora, G.; Sun, C.H.; Madsen, S.J.; Hirschberg, H. Photochemical internalization-mediated delivery of chemotherapeutic agents in human breast tumor cell lines. J. Environ. Pathol. Toxicol. Oncol. 2012, 31, 49–59. [Google Scholar] [CrossRef]
- Fretz, M.M.; Hogset, A.; Koning, G.A.; Jiskoot, W.; Storm, G. Cytosolic delivery of liposomally targeted proteins induced by photochemical internalization. Pharm. Res. 2007, 24, 2040–2047. [Google Scholar]
- Mathews, M.S.; Shih, E.C.; Zamora, G.; Sun, C.H.; Cho, S.K.; Kwon, Y.J.; Hirschberg, H. Glioma cell growth inhibition following photochemical internalization enhanced non-viral PTEN gene transfection. Lasers Surg. Med. 2012, 44, 746–754. [Google Scholar] [CrossRef]
- Symens, N.; Mendez-Ardoy, A.; Diaz-Moscoso, A.; Sanchez-Fernandez, E.; Remaut, K.; Demeester, J.; Fernandez, J.M.; De Smedt, S.C.; Rejman, J. Efficient Transfection of Hepatocytes Mediated by mRNA Complexed to Galactosylated Cyclodextrins. Bioconjug. Chem. 2012, 23, 1276–1289. [Google Scholar] [CrossRef]
- Jin, H.; Lovell, J.F.; Chen, J.; Lin, Q.; Ding, L.; Ng, K.K.; Pandey, R.K.; Manoharan, M.; Zhang, Z.; Zheng, G. Mechanistic insights into LDL nanoparticle-mediated siRNA delivery. Bioconjug. Chem. 2012, 23, 33–41. [Google Scholar] [CrossRef]
- Park, S.J.; Na, K. The transfection efficiency of photosensitizer-induced gene delivery to human MSCs and internalization rates of EGFP and Runx2 genes. Biomaterials 2012, 33, 6485–6494. [Google Scholar] [CrossRef]
- Garaiova, Z.; Strand, S.P.; Reitan, N.K.; Lelu, S.; Storset, S.O.; Berg, K.; Malmo, J.; Folasire, O.; Bjorkoy, A.; Davies Cde, L. Cellular uptake of DNA-chitosan nanoparticles: The role of clathrin- and caveolae-mediated pathways. Int. J. Biol. Macromol. 2012, 51, 1043–1051. [Google Scholar] [CrossRef]
- Berg, K.; Nordstrand, S.; Selbo, P.K.; Tran, D.T.; Angell-Petersen, E.; Hogset, A. Disulfonated tetraphenyl chlorin (TPCS2a), a novel photosensitizer developed for clinical utilization of photochemical internalization. Photochem. Photobiol. Sci. 2011, 10, 1637–1651. [Google Scholar] [CrossRef]
- Selbo, P.K.; Weyergang, A.; Eng, M.S.; Bostad, M.; Maelandsmo, G.M.; Hogset, A.; Berg, K. Strongly amphiphilic photosensitizers are not substrates of the cancer stem cell marker ABCG2 and provides specific and efficient light-triggered drug delivery of an EGFR-targeted cytotoxic drug. J. Control Release 2012, 159, 197–203. [Google Scholar] [CrossRef]
- Tu, J.; Wang, T.; Shi, W.; Wu, G.; Tian, X.; Wang, Y.; Ge, D.; Ren, L. Multifunctional ZnPc-loaded mesoporous silica nanoparticles for enhancement of photodynamic therapy efficacy by endolysosomal escape. Biomaterials 2012, 33, 7903–7914. [Google Scholar] [CrossRef]
- Krpetic, Z.; Nativo, P.; See, V.; Prior, I.A.; Brust, M.; Volk, M. Inflicting controlled nonthermal damage to subcellular structures by laser-activated gold nanoparticles. Nano Lett. 2010, 10, 4549–4554. [Google Scholar] [CrossRef]
- Omata, D.; Negishi, Y.; Hagiwara, S.; Yamamura, S.; Endo-Takahashi, Y.; Suzuki, R.; Maruyama, K.; Nomizu, M.; Aramaki, Y. Bubble liposomes and ultrasound promoted endosomal escape of TAT-PEG liposomes as gene delivery carriers. Mol. Pharm. 2011, 8, 2416–2423. [Google Scholar] [CrossRef]
- Omata, D.; Negishi, Y.; Hagiwara, S.; Yamamura, S.; Endo-Takahashi, Y.; Suzuki, R.; Maruyama, K.; Aramaki, Y. Enhanced gene delivery using Bubble liposomes and ultrasound for folate-PEG liposomes. J. Drug Target. 2012, 20, 355–363. [Google Scholar] [CrossRef]
- Lukianova-Hleb, E.Y.; Belyanin, A.; Kashinath, S.; Wu, X.; Lapotko, D.O. Plasmonic nanobubble-enhanced endosomal escape processes for selective and guided intracellular delivery of chemotherapy to drug-resistant cancer cells. Biomaterials 2012, 33, 1821–1826. [Google Scholar] [CrossRef]
- Curcio, A.; Marotta, R.; Riedinger, A.; Palumberi, D.; Falqui, A.; Pellegrino, T. Magnetic pH-responsive nanogels as multifunctional delivery tools for small interfering RNA (siRNA) molecules and iron oxide nanoparticles (IONPs). Chem. Commun. (Camb.) 2012, 48, 2400–2402. [Google Scholar] [CrossRef]
- Luo, Z.; Cai, K.; Hu, Y.; Li, J.; Ding, X.; Zhang, B.; Xu, D.; Yang, W.; Liu, P. Redox-responsive Molecular nanoreservoirs for controlled intracellular anticancer drug delivery based on magnetic nanoparticles. Adv. Mater. 2011, 21, 431–435. [Google Scholar]
- De Groot, A.S.; Scott, D.W. Immunogenicity of protein therapeutics. Trends Immunol. 2007, 28, 482–490. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, Y.; Wang, H.; Wang, J.; Shin, M.C.; Byun, Y.; He, H.; Liang, Y.; Yang, V.C. Curb challenges of the "Trojan Horse" approach: Smart strategies in achieving effective yet safe cell-penetrating peptide-based drug delivery. Adv. Drug Deliv. Rev. 2013, in press. [Google Scholar]
- Xia, H.; Gu, G.; Hu, Q.; Liu, Z.; Jiang, M.; Kang, T.; Miao, D.; Song, Q.; Yao, L.; Tu, Y.; et al. Activatable cell penetrating peptide-conjugated nanoparticles with enhanced permeability for site-specific targeting delivery of anticancer drug. Bioconjug. Chem. 2013, 24, 419–430. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fuchs, H.; Bachran, C.; Flavell, D.J. Diving through Membranes: Molecular Cunning to Enforce the Endosomal Escape of Antibody-Targeted Anti-Tumor Toxins. Antibodies 2013, 2, 209-235. https://doi.org/10.3390/antib2020209
Fuchs H, Bachran C, Flavell DJ. Diving through Membranes: Molecular Cunning to Enforce the Endosomal Escape of Antibody-Targeted Anti-Tumor Toxins. Antibodies. 2013; 2(2):209-235. https://doi.org/10.3390/antib2020209
Chicago/Turabian StyleFuchs, Hendrik, Christopher Bachran, and David J. Flavell. 2013. "Diving through Membranes: Molecular Cunning to Enforce the Endosomal Escape of Antibody-Targeted Anti-Tumor Toxins" Antibodies 2, no. 2: 209-235. https://doi.org/10.3390/antib2020209
APA StyleFuchs, H., Bachran, C., & Flavell, D. J. (2013). Diving through Membranes: Molecular Cunning to Enforce the Endosomal Escape of Antibody-Targeted Anti-Tumor Toxins. Antibodies, 2(2), 209-235. https://doi.org/10.3390/antib2020209