3. Discussion
Here we describe the discovery and development of a potent anti-P-cadherin/anti-CD3 bispecific DART protein, PF-06671008, capable of redirecting CD3-positive effector cells to kill P-cadherin expressing cells. We provide an assessment of its preclinical in vitro and in vivo pharmacologic properties, as well as its manufacturability characteristics. We have engineered an extended half-life bispecific molecule that demonstrates antibody-like stability and that can be manufactured under conventional bioprocessing techniques without many of the challenges normally associated with bispecific antibodies.
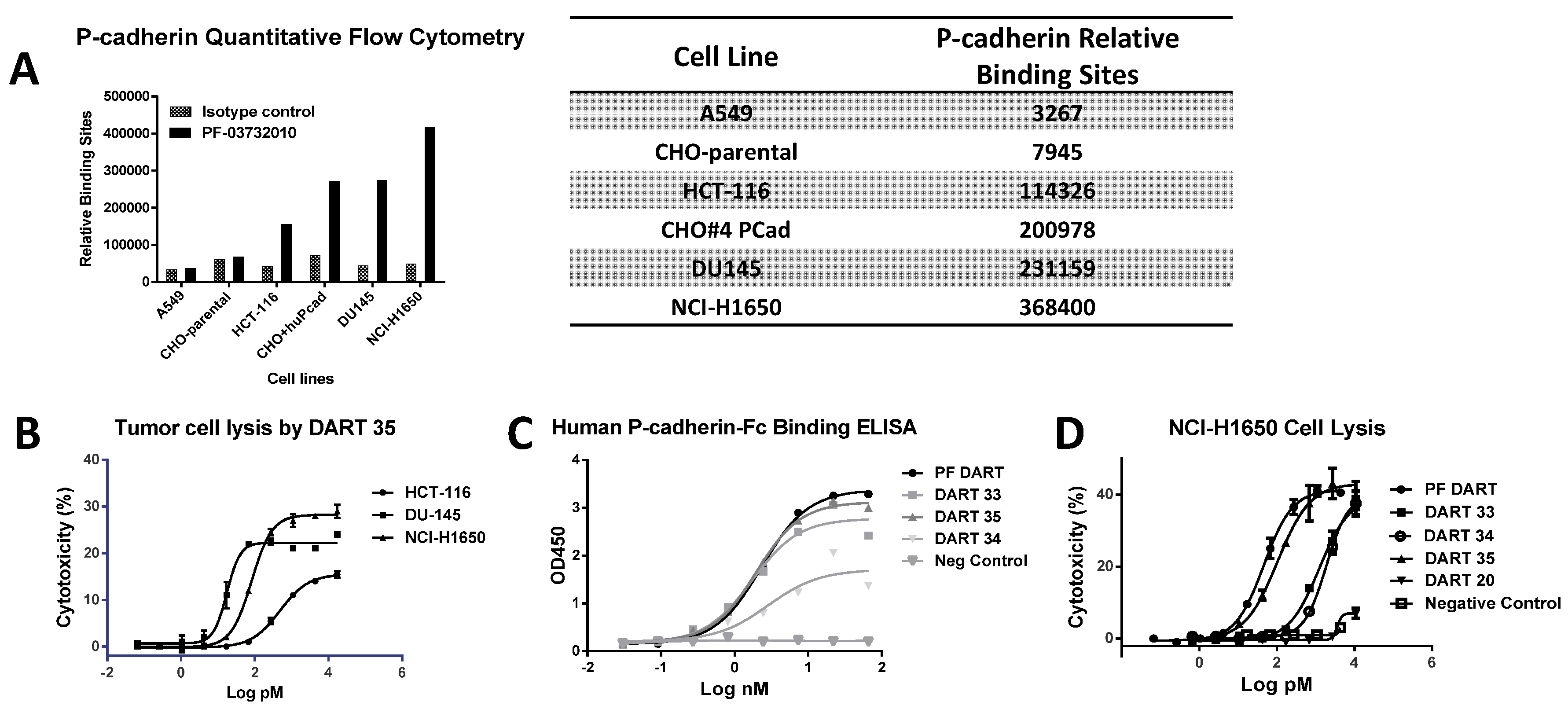
P-cadherin has been extensively studied as a target for cancer therapy, and while its role may vary across different cancer types, increased P-cadherin expression is well documented in multiple tumor types and often correlates with poor survival. We showed that a bispecific antibody (bsAb) composed of variable regions from a P-cadherin antibody and variable regions from an anti-CD3 antibody had potent T cell-dependent cytotoxic activity on P-cadherin-expressing tumor cells. Using quantitative flow cytometry, we determined the expression level of several tumor lines and demonstrated that the potency of P-cadherin-dependent cell lysis tracked with P-cadherin expression. We observed potent cytotoxicity even with cells expressing relatively low copy number suggesting T cell retargeting through bsAbs offers a potential therapeutic modality to target tumors with moderate to low target expression levels.
A panel of specific anti-P-cadherin scFv was discovered through phage display selections with a naïve human library targeting recombinant P-cadherin protein and cell lines engineered to overexpress P-cadherin. These small antibody fragments, already expressed and functional in the scFv format, were easily re-cloned into the diabody-based DART format. Cell and protein binding studies of this panel of anti-P-cadherin/anti-CD3 DART proteins demonstrated specific reactivity to human and cynomolgus monkey P-cadherin but in some cases not murine P-cadherin.
T cell-directed lysis of tumor cells expressing P-cadherin by these novel anti-P-cadherin/anti-CD3 DART molecules showed moderate cell killing activity (EC
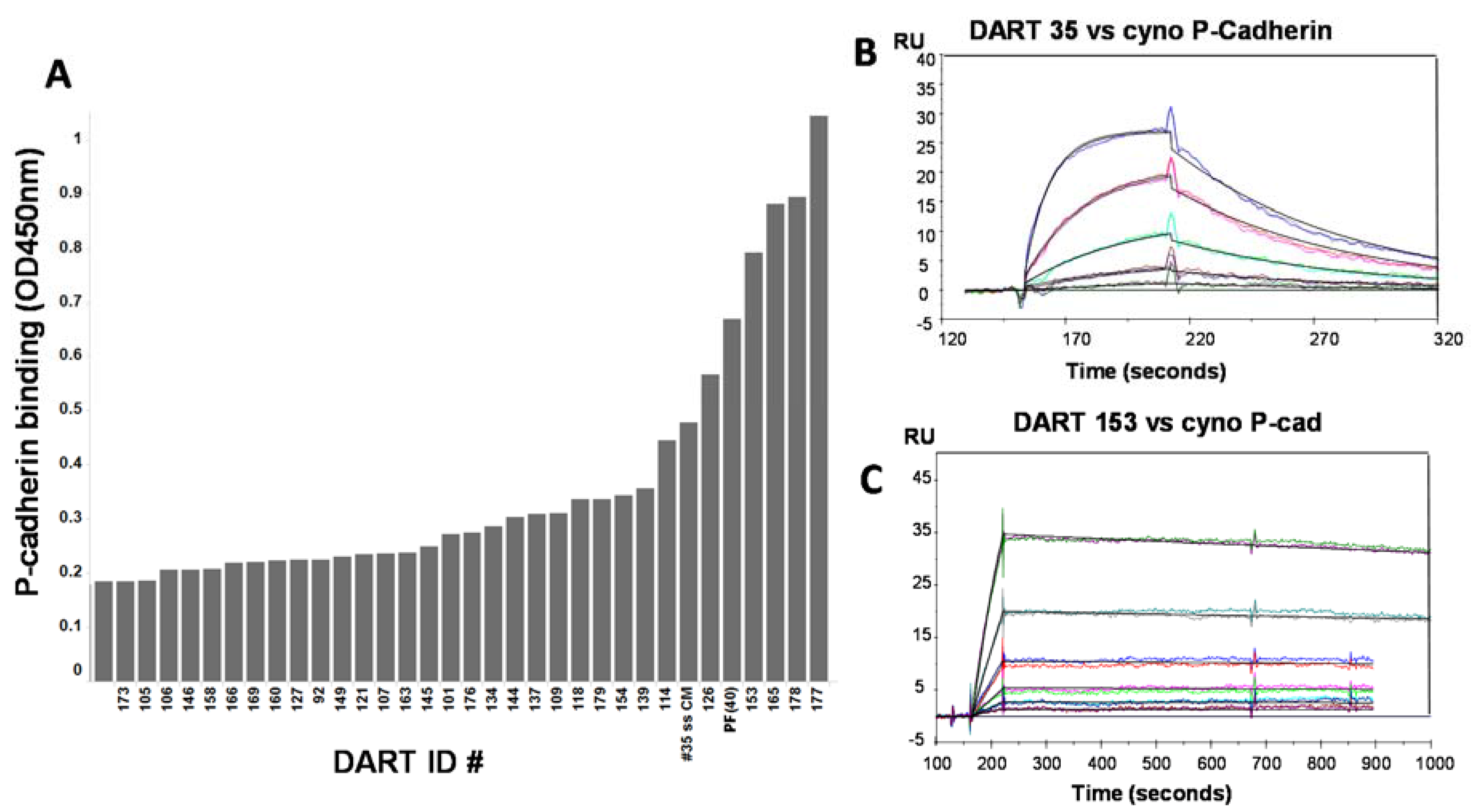
50 ~0.3–3.6 nM) for some but not all DARTs tested. These data supported further optimization to determine if improved affinity could increase cytotoxic activity. Mutagenesis techniques designed to introduce small numbers of amino acid changes to CDRs of either of the closely-related anti-P-cadherin clones 34 or 35 were combined with stringent phage selection methods to yield a panel of affinity optimized clones. Changes of only one or two amino acids in the CDR regions led to significant improvements in equilibrium dissociation constants above the 43.4 nM affinity of the starting clone, sometimes over 100-fold (
Table 3). For example, optimized DARTs 153 and 165 showed K
D values of 231 and 370 pM, respectively. For DART 153, a 187-fold improvement in binding affinity to P-cadherin translated to an increase of over 400-fold in CTL-directed potency in the HCT-116 colorectal tumor model, with an EC
50 value of 0.5 pM. Improvement in affinity generally correlated well with improvement in CTL activity. DARTs showing only a modest improvement in affinity such as 154 (11-fold) and 179 (10-fold) showed the smallest improvement in CTL activity (~12-fold and 5-fold respectively). Sequence analysis of the optimized clones identified a serine to asparagine change at position 99 of the CDR-H3 as a common mutation that emerged from the selections and was present in clones from both phage optimization strategies. In addition, the phage selections revealed a strong preference for the parental light chain sequence: from a library in which the mutagenized VH chain of clone 35 was paired with a collection of random naïve human VL chains from an unselected phage library, all hit molecules retained the parental VL framework (VL1b.366F5/DPL5), with only minor CDR sequence variations. Since the affinity optimization was conducted with P-cadherin binding domains in scFv format, in the absence of the anti-CD3 domain, it is likely that these observed improvements reflect increases in binding to P-cadherin and not alterations to the DART structure. This is further supported by the fact that binding to CD3 remained largely the same following optimization.
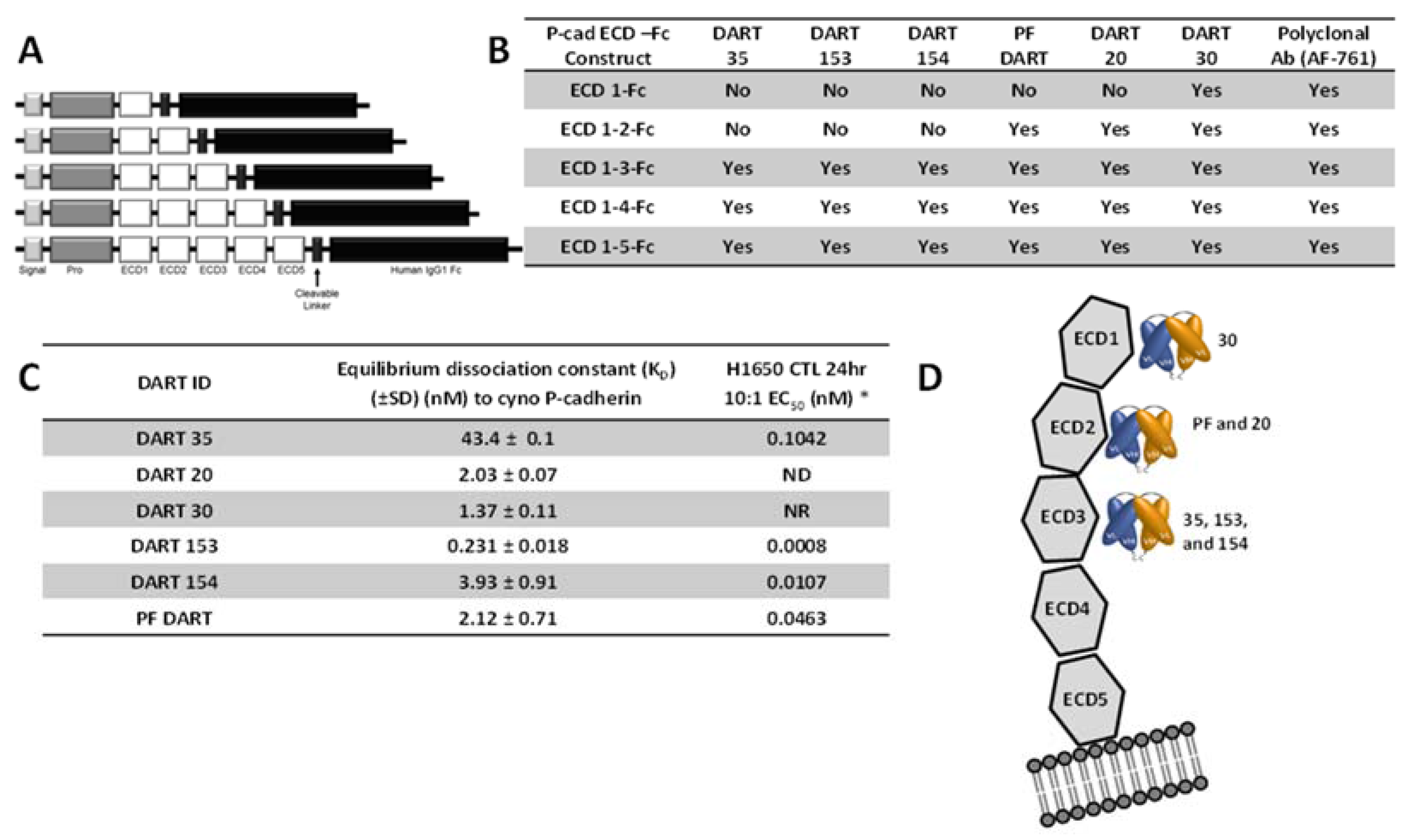
In addition to affinity, the epitope on the P-cadherin extracellular domain had a major influence on the potency of effector cell directed-tumor cell lysis. A common structural configuration shared across classical, type II and desmosomal cadherins is a series of five extracellular domains termed ECD1 (membrane distal) to ECD5 (membrane proximal), separated by calcium-binding domains (reviewed by Shapiro and Weis, 2009 [
29]). We used a set of truncated P-cadherin ECD constructs to show that DART 30 bound to ECD1, while DARTs 20 and PF bound to ECD2, and DART 35 and its optimized derivatives (153 and 154) bound more membrane-proximally, in ECD3. While DARTs 30, 20, PF, and 154 have similar binding affinity (1.4–3.9 nM), the cytotoxic potency is highest with the most membrane-proximal (DART 154, EC
50 0.01 nM), intermediate for the ECD 2-binding DARTs PF and 20 (0.4 and 34 nM, respectively), and absent for the most distal DART, 30. These results are consistent with those reported by Bluemel
et al., who showed that the distance of an epitope from the cell surface recognized by BiTE molecules to melanoma-associated chondroitin sulfate proteoglycan (MCSP), all with similar equilibrium dissociation constants, greatly influenced BiTE redirected lysis [
24]. They postulate that epitopes closer to the cell surface allow for more efficient formation of the cytolytic synapse and delivery of perforins and granzymes [
24].
Although the structure of P-cadherin is not known, the structure of C-cadherin has been solved to 3.1 Å [
30]. This structure shows an elongated, slightly curved ectodomain stretching away from the cell surface, with an approximate total length of 193 Å (19.3 nm; [
30], or ~3.9 nm for each domain. If P-cadherin has a similar structure, ECD3 would extend between approximately 11.6 and 15.4 nm from the cell surface, similar to the reported length of the TCR/peptide-MHC complex (~14 nm or 140 Å; [
24]). Binding to ECD3 via a compact DART protein to the membrane-proximal CD3 molecule may provide the optimal distance for formation of the T cell-target cell synapse, while epitopes in more distal domains would reduce the likelihood of synapse formation. While these observations are based on the crystal structure of a recombinant cadherin-Fc fusion protein,
in vitro CTL activity on tumor lines supports our hypothesis.
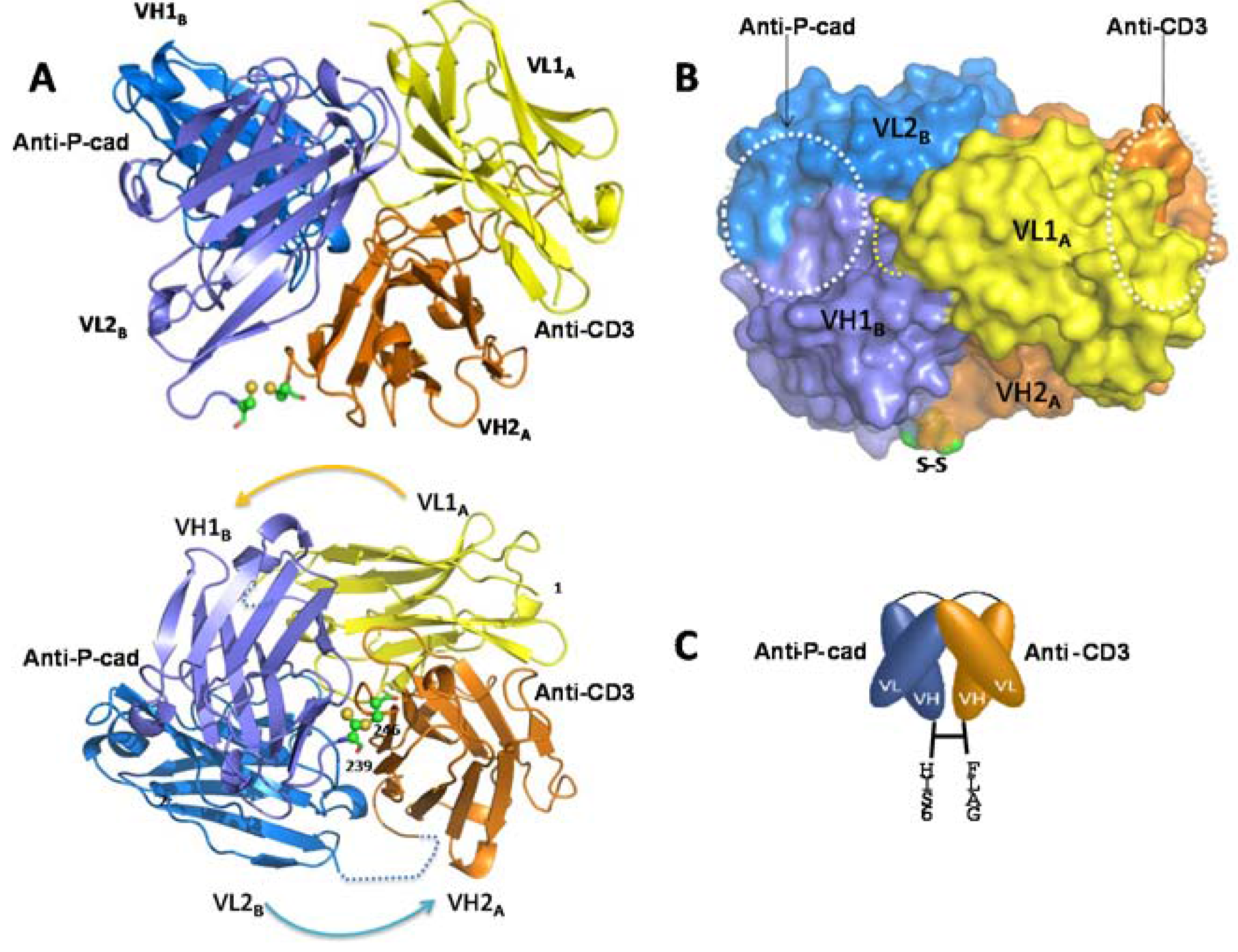
Here we report for the first time a crystal structure of a disulfide-constrained diabody. Diabodies can be engineered with V domains of different Fv regions mixed on opposite chains and can be separated by short linkers that prevent intra-chain VH-VL folding [
31]. The 2 Å resolution crystal structure of coil-less DART 35, arranged in the VL-VH format and separated by a 9-residue intra-chain linker on chain 1 (CD3VL-P-cadVH) and an 8-residue linker on chain 2 (PcadVL-CD3VH), shows that this DART construct assembles into a tightly packed spherical structure that differs considerably from the previously published diabody structures (
Figure 5A,B). The VH-VL diabody reported by Perisic
et al. [
25] (PDB accession No. 1LMK) consisted of short 5-residue intra-chain linkers and showed a symmetrical yet flexible structure. The anti-CEA diabody in VL-VH format, T84.66 (consisting of two identical scFv separated by longer, eight residue intra-chain linkers), takes on an asymmetrical shape as reported by Carmichael
et al. [
26] (PDB accession No. 1MOE).
The previously-published structures are elongated, with a wide 42 Å–60 Å gap separating the distal (non-CDR-containing) ends of the two Fv domains [
25,
26]. In contrast, the DART 35 structure reported here shows that the disulfide bond formed between the two chains keeps the diabody in a compact configuration and reduces the distance between the two domains to 5.3 Å. Addition of a
C-terminal Gly-Gly-Cys to the T84.66 diabody allowed the formation of a stable, covalent disulfide-linked diabody [
32,
33], and Olafsen
et al. proposed a structure in which the Fvs were rotated to bring the distal ends close enough for the disulfide bond formation, similar to the observed DART 35 structure. While the intra-chain linkers in the DART 35 are similar in size to those in T84.66, we did not observe these residues in our structure, likely due to disorder of the linkers.
The four subunits, VL1A, VH1B, VL2B, and VH2A shown in
Figure 5A all participate in the diabody interface, with the most contribution coming from the framework amino acid residues of both chains. The centers of the two paratopes are separated from each other by approximately 30 Å and are facing ~90° apart. The orientation and short distance between the antigen binding domains of the DART protein may allow for more efficient T cell-target cell synapse formation and contribute to the increase in potency of T cell directed lysis observed with DART molecules compared to other bispecific antibody formats [
17]. The anti-CD3 CDR region is positioned remotely from the subunit interface, whereas the anti-P-cadherin CDR region lies adjacent to the interface (
Figure 5B). This observation suggests that swapping the order of the binding domains or introducing a different binding domain to either P-cadherin or CD3 may impact orientation and therefore bispecific binding activity. Key mutations in the anti-P-cadherin CDR which show significant change in affinity are at Kabat position H99 in the CDR-H3, and to a lesser extent Kabat positions L96 and L97 in CDR-L3 (
Table 3). The H99 mutation to asparagine increases the affinity by between 10 and 187-fold depending on concomitant mutations and is predicted to either alter a potential surface interaction with P-cadherin or stabilize the loop conformation across the VH/VL interface of the P-cadherin-binding portion of the diabody. The L96 and L97 mutations which alter the affinity to a lesser extent are at the base of the CDR-L3 loop and likely support the loop conformation or stabilize the VH/VL interface. The significant effect of these mutations suggests that the paratope lies across both the VH and VL, encompassing at least CDR-H3 and CDR-L3.
While bispecific diabodies or bispecific single-chain Fvs are efficient at re-targeting cytotoxic T lymphocytes to tumor cells, they are cleared from circulation with a terminal half-life within a few hours due to their small size [
16,
34,
35]. This rapid clearance can impact therapeutic administration, requiring constant infusion in the case of blinatumomab [
35,
36]. To extend circulating half-life, we engineered an Fc fusion DART molecule by replacing the E and K coils with human IgG1 Fc domains. Incorporation of an Fc increases molecular weight and extends the serum half-life through interaction with the neonatal Fc receptor FcRn [
27]. In addition to half-life extension, Fc fusion frequently offers other benefits such as increasing recombinant protein expression, simplifying purification through Protein A affinity chromatography, and improving solubility and stability [
37]. We have observed these improvements in the fusion of DART 35 and its variants to Fc, discussed below.
To replace the heterodimerization function of the E and K coils, the knobs-into-holes design strategy was employed, introducing a set of Fc mutations that favor heterodimerization over the normal Fc homodimerization [
18,
19]. Additional modifications were made to the Fc to prevent ADCC through FcγR binding and inadvertent activation of CD3 in the absence of target, using mutations in the lower hinge region that have previously been demonstrated to abrogate ADCC activity [
28]. To identify the Fc fusion structure that supports optimal DART functional and biophysical properties, we generated eight Fc-DART variants of DART 35, with varying fusion linker lengths and hinge region lengths. While all LP DART proteins demonstrated an improvement in transient protein expression compared to the non LP-DART format (4–7-fold), an increase in HMMS was detected for all constructs following Protein A capture (33%–45%
vs. 7% for the non LP-DART format). This increase in HMMS is commonly seen with Fc-fusion proteins and can be easily removed with conventional chromatography. Binding activity for all LP-DART proteins to both P-cadherin and CD3 showed a modest decrease compared to the non-Fc DART format (1.5–3-fold), as did T cell-directed cytotoxicity on tumor cells (2.2–8.6-fold). The linker-hinge design that demonstrated the lowest impact on binding and CTL activity, LP-DART 5, contained a single glycine separating the
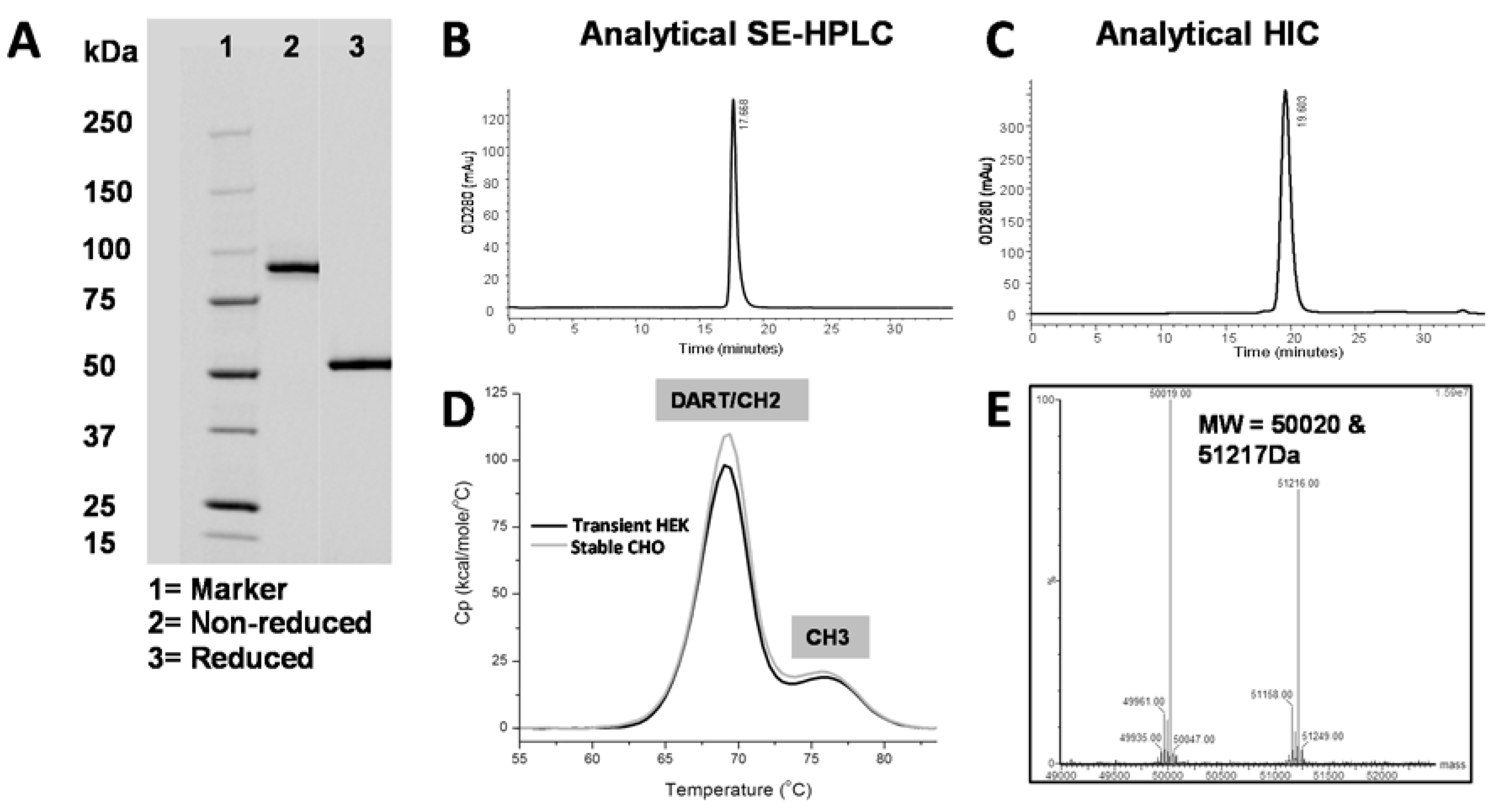
C-terminal serine from the VH domain and a truncated hinge region. We hypothesize that this short linker and truncated hinge minimize flexibility of the Fc domain and thereby reduce interference with antigen binding. When stably transfected into CHO cells, the affinity-optimized and Fc-engineered DART PF-06671008 (DART 153) was expressed at 1.3 grams/liter following Protein A purification. Using conventional bioprocessing chromatographic techniques, we were able to remove aggregates and inactive homodimer species, resulting in high purity (
Table 5,
Figure 6). The molecule exhibited antibody-like thermostability, with a T
m1 value ≥ 68°C.
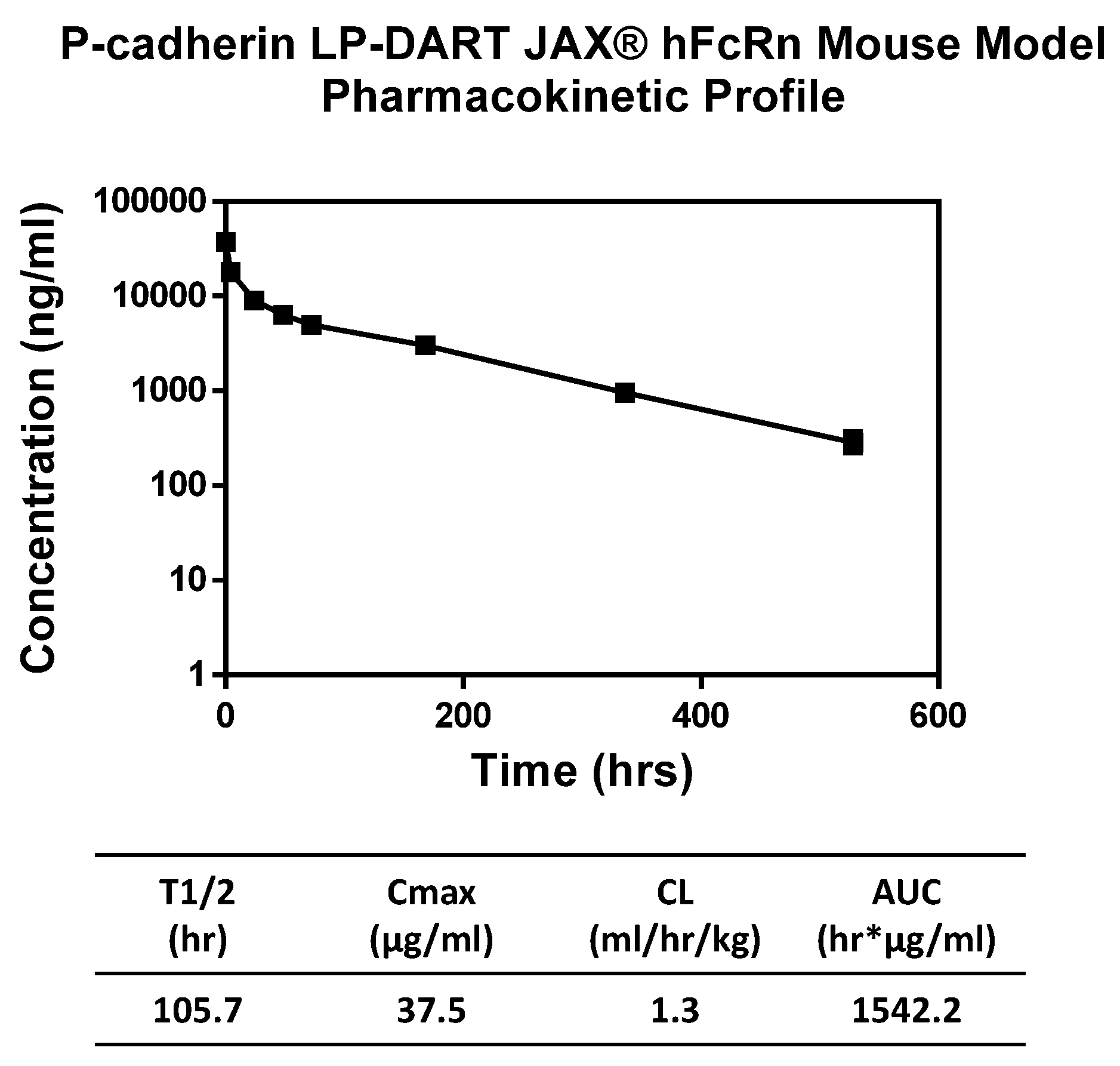
Pharmacokinetic properties of the anti-P-cadherin/anti-CD3 LP-DART protein were assessed in a human FcRn knock-in mouse model. Since the LP-DART molecule tested does not recognize mouse P-cadherin or mouse CD3, pharmacokinetics in this model were unaffected by target binding. The LP-DART protein exhibited a biphasic elimination curve with a clearance rate of 1.3 mL/h/kg and a terminal half-life of 105.7 h. Although not quite as long as some IgG, this increase to ~4.4 days from just a few hours for DART proteins without Fc [
16] represents a major improvement in serum half-life and will allow for less frequent therapeutic administration compared to standard DARTs and BiTEs.
ELISA, Biacore, and KinExa analyses demonstrated that appending an Fc domain maintained adequate antigen binding properties. The equilibrium dissociation constant of PF-06671008 against cynomolgus monkey P-cadherin determined by Biacore was 0.521 nM
versus 0.231 nM for the DART version of 153 (
Table 5). Kinetic binding analysis with CHO cells over-expressing human P-cadherin using KinExA demonstrated an equilibrium dissociation constant of 0.176 nM
versus 0.352 nM for cynomolgus monkey protein, indicating a similar level of binding between human and cynomolgus monkey P-cadherin.
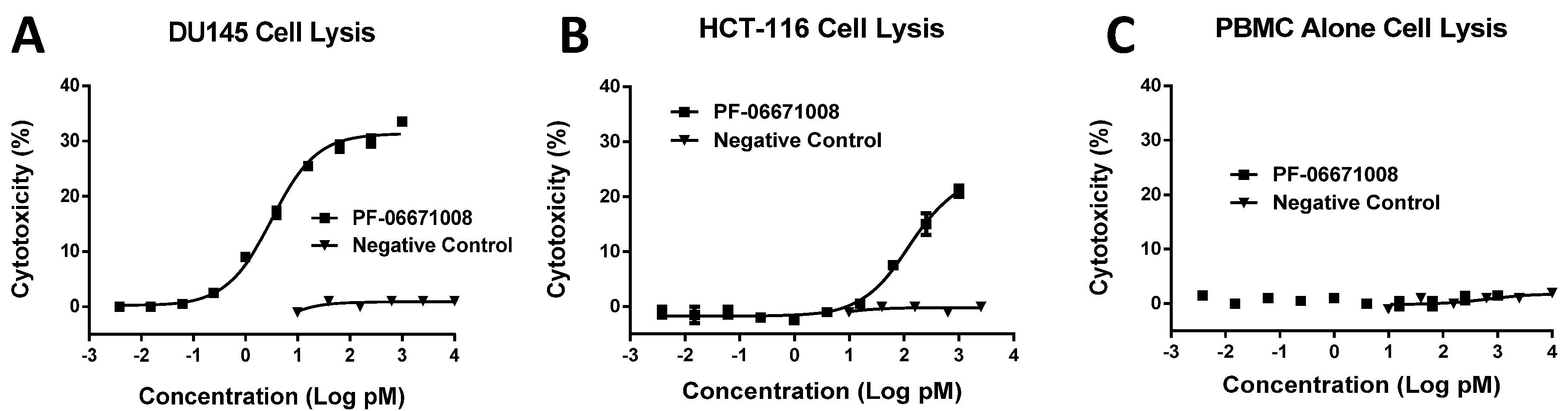
Consistent with its high-affinity binding, PF-06671008 exhibited potent redirected T cell-mediated killing of P-cadherin-positive cell lines with a range of P-cadherin expression levels. The EC50 values of 3.1 pM for DU145 cells and 122.6 pM for HCT-116 cells tracked with their P-cadherin expression levels. Human PBMCs alone, which do not express P-cadherin, were not killed in the presence of PF-06671008, and a control DART capable of engaging CD3 on T cells (4420-LP) did not mediate redirected killing of target cells, suggesting there is a strict requirement for P-cadherin expression for activity. PF-06671008-mediated redirected target cell killing was associated with a concomitant dose- and target-dependent induction of cytokine release by human T cells, as represented by increased levels of TNF-α, IFN-γ, IL-10, IL-6, IL-4, and IL-2 in supernatants. Similar to cytotoxicity, the sensitivity to PF-06671008-mediated cytokine release (as reflected by lower EC50 values) and maximal levels of cytokine release (Emax) induced by PF-06671008 showed lower EC50 and higher Emax values for cells with higher expression of P-cadherin.
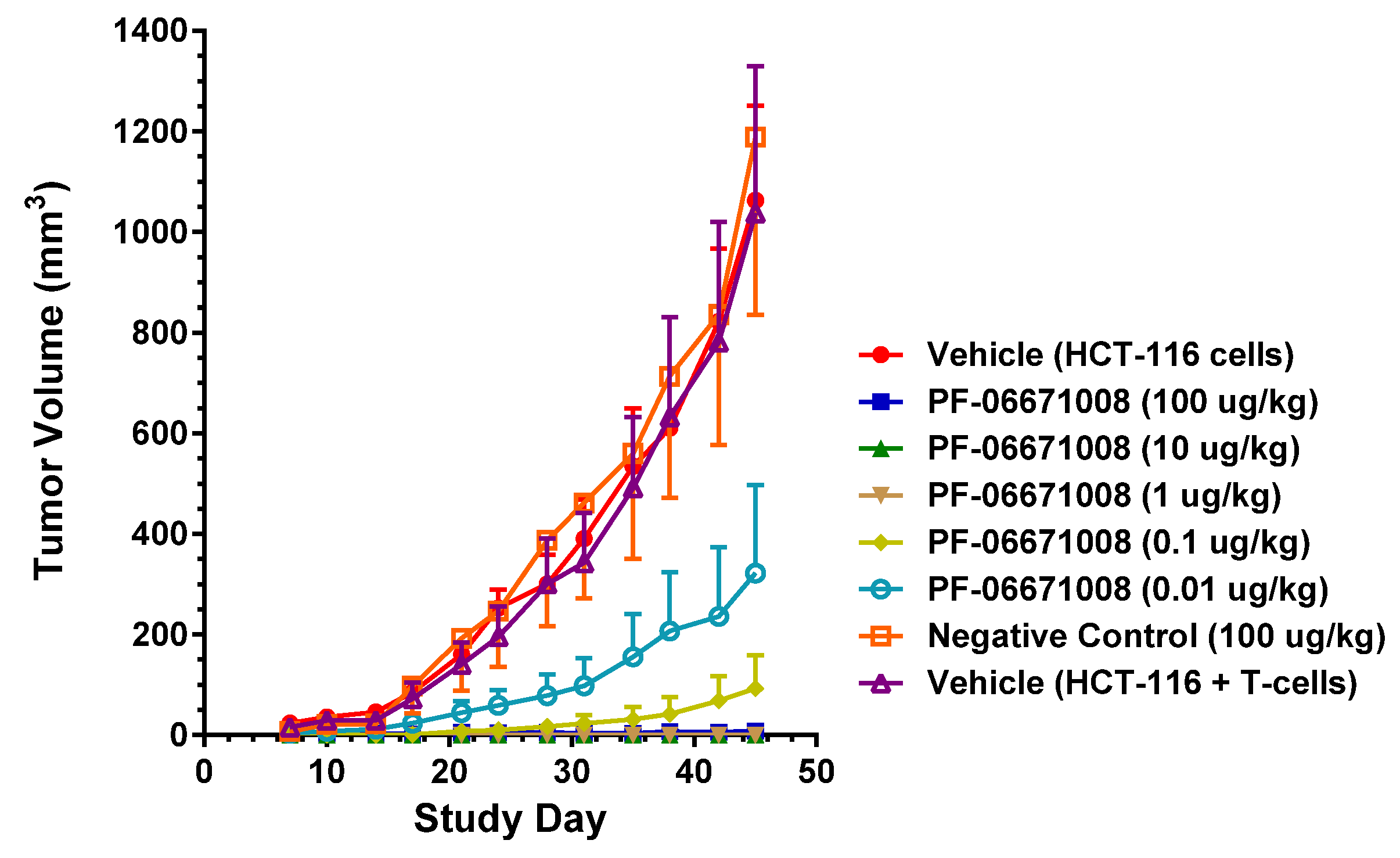
PF-06671008 demonstrated potent in vivo tumor growth inhibition in a colorectal carcinoma tumor model. Treatment with PF-06671008 administered as a single agent IV once daily for four doses resulted in a dose-dependent inhibition of tumor growth when HCT-116 adenocarcinoma tumor cells were implanted SC in the presence of activated human T cells. Inhibition of tumor growth was observed at PF-06671008 doses ≥ 0.01 µg/kg, while no tumor growth was observed in mice treated with doses ≥ 1 µg/kg.
Clinical dosing of this class of T-cell re-directing therapy has been extraordinarily low due to a combination of potency and adverse events associated with cytokine release syndrome (CRS) [
35], [
36]. P-cadherin expression is upregulated in multiple tumor types, whereas healthy tissues that do express endogenous P-cadherin have been documented to express much lower levels [
6]. Therefore targeting P-cadherin with this type of therapy may offer a reasonable therapeutic window at the expected low clinical dose range. Further investigation is underway to better understand the safety profile of PF-06671008.
4. Experimental Section
4.1. Enzyme-Linked Immunosorbent Assay (ELISA) to Measure Binding of scFv/DART Proteins to Recombinant Cadherin or CD3 Proteins
P-cadherin-Fc protein (R & D Systems, Minneapolis, MN) or P-cadherin His or negative control (E-cadherin-Fc and VE-cadherin-Fc; R & D Systems, Minneapolis MN) or CD3 epsilon-delta protein was coated overnight at 4 °C on 96-well Nunc Maxisorp plates (Thermo Fisher Scientific, Madison, CT, USA) at a concentration of 1 or 2 µg/mL in PBS. Plates were washed three times using PBS + Ca2+ and Mg2+ and blocked for 1 h at room temperature in 3% milk/PBS + Ca2+ and Mg2+. Samples prepared in block buffer were added to the blocked plates for 1 h at room temperature. Plates were washed three times with PBS + Ca2+ and Mg2+ prior to the addition of secondary antibody (either anti-human IgG-HRP 1:4000 (Southern Biotech, Birmingham, AL, USA) or anti His 1:2000 (Qiagen, Valencia, CA, USA). In some cases, prior to the addition of secondary antibody, plates were allowed to incubate in wash buffer overnight at 4 °C in an effort to detect improvements in off-rate. Plates were incubated for a further 1 h at room temperature and washed three times with PBS + Ca2+ and Mg2+. Signal was developed using TMB (SurModics, Eden Prairie, MN, USA), the reaction stopped with H2SO4, and the absorbance read at 450 nm.
4.2. Bi-Specific ELISA to P-Cadherin and CD3
Unpurified conditioned medium containing DART proteins as well as purified DART proteins were screened by ELISA for the ability to bind P-cadherin and CD3 simultaneously. Bispecific ELISA methods were previously described [
17] with the following modifications: CD3 epsilon/delta heterodimer was coated as described above, followed by incubation of test sample diluted in block buffer (3% milk/PBS) for 1 h at room temperature. Plates were then treated with sequential addition of 2 µg/mL P-cadherin-Fc-His (R & D Systems, Minneapolis, MN) for 1 h in 3% milk/1% bovine serum albumin (BSA)/PBS + Ca
2+ and Mg
2+ and anti His-HRP at 1:2000 (Qiagen, Valencia, CA, USA) with intervening plate washing. Bound complex was detected with TMB, the reaction stopped with H
2SO
4, and the absorbance read at 450 nm.
4.3. Quantitative Flow Cytometry to Measure P-Cadherin Expression
The anti-P-cadherin monoclonal antibody PF-06671003 [
20] was labeled with phycoerythrin with a fluorophore to protein ratio of 1:1 (eBioscience, San Diego, CA, USA) and used to measure P-cadherin expression by flow cytometry. Tumor cells (listed in
Figure 2A) were added to assay plates at 10
6 cells/mL and labeled with PF-06671008-PE in 100 μL/well of FACS buffer. The plates were incubated in the dark at 4 °C for 30 min and then 150 μL of FACS buffer was added to each well, followed by centrifugation at 311×
g (1200 rpm) for 5 min. The supernatant was removed and the cell pellets were washed once with 200 μL/well FACS buffer. After centrifugation, the cell pellets were resuspended in 100 μL/well FACS buffer for cell event collection by FACS Calibur flow cytometer equipped with acquisition software CellQuest Pro Version 5.2.1 (BD Biosciences, San Jose, CA, USA). Data analysis was performed using Flowjo v9.3.3 software (Treestar, Inc., Ashland, OR, USA).
4.4. ELISA to Measure Binding of DART Proteins to P-Cadherin-Expressing Cells
NCI-H1650 cells expressing endogenous P-cadherin or parental CHO-DUKX cells were seeded at 4 × 104 cells/well in a 96-well tissue culture plate on Day 1 and incubated at 37 °C/5% CO2 overnight until a confluent monolayer was observed. Cells were washed three times with PBS + Ca2+ and Mg2+and blocked for 1 h at room temperature with 3% milk/PBS + Ca2+ and Mg2+. Test samples diluted in block buffer were transferred to the plates and incubated for 1 h at room temperature. Plates were washed three times with PBS + Ca2+ and Mg2+ prior to the addition of secondary antibody (either anti-human IgG-HRP 1:4000 (Southern Biotech, Birmingham, AL, USA) or anti His 1:2000 (Qiagen, Valencia, CA, USA). Plates were incubated for a further 1 h at room temperature and washed four times with PBS + Ca2+ and Mg2+. Signal was developed using TMB, the reaction stopped with H2SO4, and the absorbance read at 450 nm.
4.5. Control Anti-P-Cadherin/Anti-CD3 DART Generation
Positive (PF-DART) and negative (4420-DART) control were generated from PF-03732010, a monoclonal anti-P-cadherin antibody [
20], and from an anti-fluorescein antibody, 4420 ([
17,
38], using methods previously described [
16,
17]. These DART constructs were engineered with the XR32 anti-CD3 epsilon antibody [
39,
40].
4.6. Selection of P-Cadherin-Specific scFv by Phage Display Using Naïve Library
ScFv that bind to the ECD of P-cadherin were identified following three rounds of selection using Pfizer’s in-house naïve phage display library. Selection strategies are outlined in
Figure S1. The library is composed of VH and VL domains derived from non-immunized human donors, randomly paired and cloned into a phagemid vector upstream of C terminal His
6 and c-Myc tags. Phage library rescue and phage selection were performed as previously described [
41].
4.7. Cloning, Sequencing and Reformatting to DARTs
ScFv DNA was sequenced on both strands (Genewiz, Cambridge, MA, USA or Wyzer, Cambridge, MA, USA) as either unpurified bacterial glycerol stock or as purified plasmid using conventional methods. ScFv fragments demonstrating strong binding to P-cadherin or cell surface (>3× background or negative cell binding) were selected for reformatting as DARTs. DART design and cloning methods were previously described [
16,
17,
40]. Methods for adapting novel P-cadherin scFv from Pfizer’s in-house naïve library and mutant libraries are as follows: Fragments were amplified by standard polymerase chain reaction (PCR) with primers annealing to human germline V and J sequences and incorporating restriction sites for DART cloning (BamHI/BspEI for VH and BssHII/BamHI for VL). Fragments were digested with corresponding restriction enzymes according to the manufacturer’s specifications (New England Biolabs, Ipswich, MA, USA). Anti-P-cadherin VH or VL fragments were gel-purified (Gel Purification Kit, Qiagen, Valencia, CA, USA) and ligated separately into Pfizer proprietary pre-digested mammalian expression vectors containing either the VL or VH, respectively, of anti-CD3 scFv as well as either of two alternative carboxy-terminal heterodimerization domains termed the E-coil and K-coil domains (
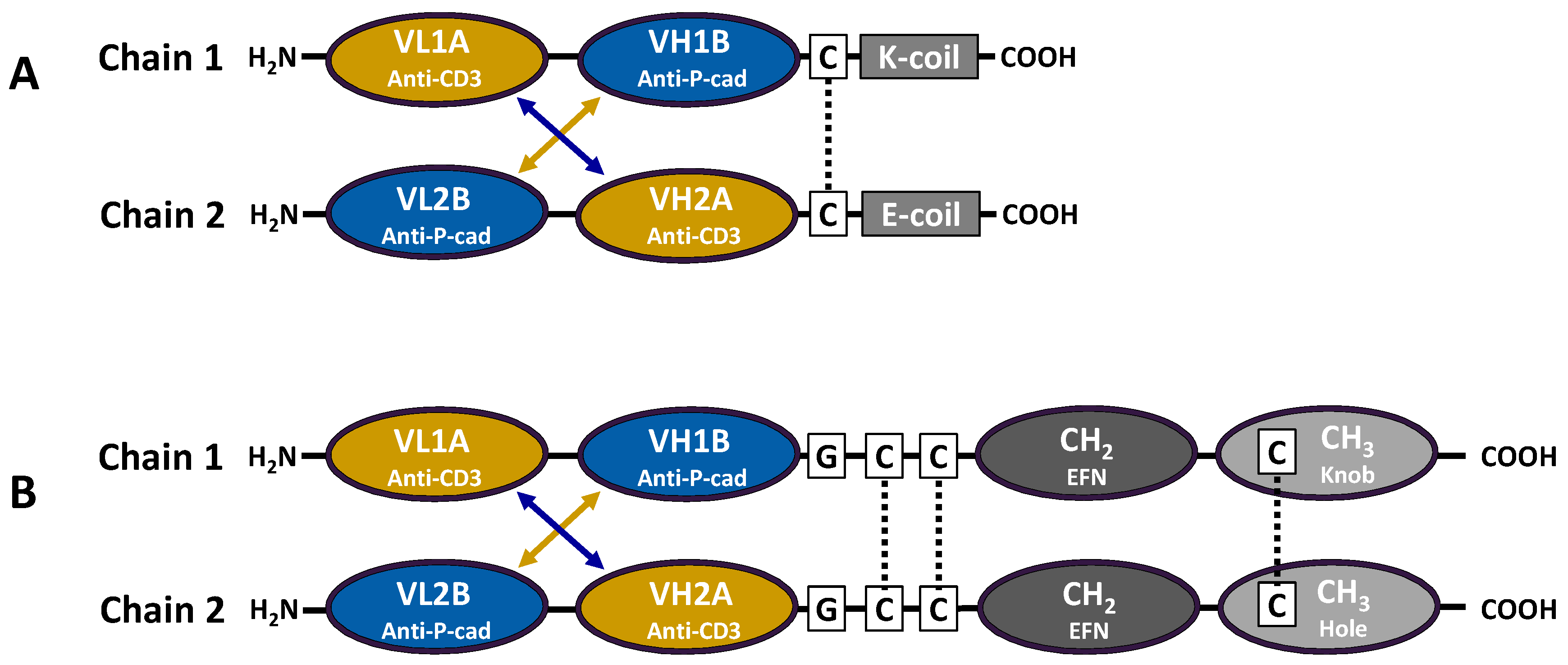
Figure 1A).
4.8. Affinity Maturation Library Construction and Identification of Optimized scFv
Variants of anti-P-cadherin scFvs with increased affinity were isolated from mutagenized libraries using two approaches. The first approach focused on parental clone 35 using random mutagenesis of the heavy chain coupled with light chain shuffling. Random mutations were introduced into the VH domain clone 35 by error-prone PCR using the GeneMorph II kit according to the manufacturer’s protocol (Agilent Technologies, Santa Clara, CA, USA). Mutagenized VH pools were digested with BssHII and XhoI and sub-cloned into a pool of phagemid vectors containing approximately 1 x1010 variable light (VL) genes from non-immunized human donors.
Mutant phage libraries were rescued as described previously [
41]. Two selection strategies were performed with these mutant libraries, including a two round solution-phase selection approach termed Hammer-hug [
23] and a cell-based selection approach described as follows. Approximately 4 × 10
7 cells not expressing P-cadherin (de-selection cells) were collected using cell dissociation buffer (PBS/5 mM EDTA) and washed twice with PBS. Cells were blocked with 3% milk/1% BSA/PBS for 1 h at 4 °C on a rotary mixer (20 rpm). Adherent cells expressing P-cadherin (capture cells) were plated 1 day prior at 1.2 × 10
6 cells/6-well plate, grown overnight at 37 °C, then washed with PBS + Ca
2+ and Mg
2+ and subsequently blocked with 3% milk/1% BSA/PBS for 1 h at room temperature. De-selection cells were collected by centrifugation, re-suspended in blocked phage and incubated at 4 °C as before. De-selection cells were pelleted and the phage supernatant was transferred to the blocked capture cells and allowed to incubate for 2 h at room temperature. The capture cells were washed five times with cold PBS + Ca
2+ and Mg
2+/0.1% Tween 20 and ten times with cold PBS + Ca
2+ and Mg
2+. Phage were eluted by incubating the cells in 100 mM triethylamine (TEA) solution for 10 min at room temperature, then harvesting all cell and media from each well. Eluted phage were harvested in the supernatant following centrifugation of cells. Phage were then recovered by infecting an ER2738
E. coli host and rescued as previously described [
41].
The second affinity optimization approach focused on parental clone 34, using a soft randomization approach targeting all CDRs except CDR-L2, as previously described [
23]. Phage selections were split across two branches and over three rounds of increasing stringency, with branch 1 starting at 50 nM and branch 2 starting at 10 nM. Each branch decreased in target concentration 10-fold over each successive round. Phage selections and screening were performed as previously described [
23,
41].
4.9. Binding Affinity Analysis by Surface Plasmon Resonance
The Biacore 3000 and T-200 instruments (GE Healthcare, Piscataway, NJ, USA) were used to determine affinity and kinetics of recombinant human CD3 epsilon-delta, human P-cadherin-Fc, and mouse and cynomolgus monkey P-cadherin ECD proteins binding to DART protein. P-cadherin or human CD3 proteins were immobilized on CM5 Biacore sensor chips using routine amine coupling. The immobilization buffer was 10 mM acetate buffer pH 4.5 with 2.0 mM CaCl2. Surface densities ranged from ~75–300 RU. A non-derivatized flow cell was used as a reference surface. Two-fold titration series of DART samples in running buffer were prepared at concentrations ranging from 100 to 6.25 nM, and running buffer alone was included as a zero reference. Samples were injected in duplicate for 60 s at a flow rate of 30 µL/min across the flow cells using TBS-P+ (10 mM Tris pH 7.4, 150 mM NaCl, 2 mM CaCl2, 0.1 mg/mL BSA 0.05% P20) as running buffer. Dissociation of the DART proteins was monitored for 180 s followed by regeneration with a 10 second injection of 10 mM glycine-HCl pH 1.5 at 30 µL/min. The data were fit globally to a 1:1 Langmuir binding model using BIAevaluation software (GE Healthcare, Piscataway, NJ, USA).
4.10. Binding Affinity Analysis by KinExA
A Kinetics Exclusion Assay (KinExA) instrument (model 3200, Sapidyne, Boise, ID, USA) was used to compare the DART protein binding affinity to recombinant cynomolgus monkey P-cadherin and human P-cadherin expressed on the surface of CHO cells. Binding experiments were performed in PBS buffer pH 7.4 containing MgCl2, CaCl2 and 1 mg/mL BSA using PMMA beads (Sapidyne, Boise, ID, USA, catalog number 440176) at 4 °C for 2 h. Receptor binding concentration was measured using the SACy5 fluor at 13 time points in 500 µL volume.
4.11. Fc-DART Protein Engineering
For Fc fusion linker and hinge assessment, fragments were prepared as before and ligated separately into pre-cut mammalian expression vectors containing either the VL or VH of anti CD3 scFv, either of two modified human IgG1 Fc domains, and varying linker/hinge sequences listed in
Table 4. These undigested expression vectors were synthesized at an external vendor and digested with appropriate restriction enzymes as described above prior to ligation. The “knob” Fc variant was constructed by replacement of a small amino acid with a larger one, e.g., T366W. The “hole” Fc variant was constructed by replacement of a large residues with a smaller ones e.g., T366S, L368A, and Y407V. Additionally, two cysteines (Y349C on the knob chain and S354C on the hole chain) were added for the introduction of a disulfide bond for added stability. All constructs were confirmed by DNA sequencing and transiently transfected into FreeStyle™ 293 HEK cells (Life Technologies, Grand Island, NY, USA) according to the manufacturer’s method and expressed over 5–7 days.
4.12. Stable CHO Cell Line Development and Expression
The two chains of PF-06671008 were cloned into a mammalian expression vector containing dual promoters and engineered for integration into a single recombination site engineered into the host CHO cell genome. The PF-06671008-Knob construct was cloned into the first cloning site, referred to as cassette 1. The PF-06671008-Hole construct was cloned into the second cloning site, cassette 2. The resulting plasmid was stably transfected into CHO host cells. A 10 L working volume Wavebag (GE Healthcare, Piscataway, NJ, USA) was seeded with 1.5 L cell culture in 8 L of Pfizer’s proprietary cell culture medium. The culture was grown at a temperature of 36.5 °C in a 5% CO2 environment. Cultures were fed with a proprietary feed at various time points. Conditioned medium was harvested on day 12 by filtration through a 20” 5 µm Pall Profile II filter (Port Washington, NY, USA) and a 10” 0.22 µm Pall Supor filter (Port Washington, NY, USA).
4.13. Purification of PF-06671008
Clarified conditioned medium was purified using conventional chromatographic techniques. The final pool was analyzed by OD280 (NanoDrop™, Thermo Fisher Scientific, Madison, CT), SDS-PAGE (BioRad Laboratories, Hercules, CA, Stain Free 4%–15%), analytical SEC (TSK G3000), and analytic HIC. CHO host-cell protein and Protein A leaching were quantified using ELISA kits following the manufacturer’s protocol (Cygnus Technologies, Southport, NC, USA). Analytical SEC was performed on an Agilent 1200 series HPLC (Agilent Technologies, Santa Clara, CA, USA) using either a Superdex200 10/30 column (GE Healthcare, Piscataway NJ, USA) or a TSKgel G3000SWxl column (Tosoh Bioscience, King of Prussia, PA, USA) according to the manufacturer’s protocol. An analytical HIC-HPLC assay was used to assess protein heterogeneity. Using an Agilent Infinity 1290 UHLPC (Agilent Technologies, Santa Clara, CA, USA), approximately 20 to 30 µg of protein was injected at a flow rate of 1 mL/min and protein was detected by absorption at 280 nm.
4.14. Mass Spectrometric Analysis
Purified LP-DART protein was analyzed by liquid chromatography/mass spectrometry (LC/MS) analysis on an Agilent 1100 capillary HPLC coupled with Water Xevo G2 Q-TOF mass spectrometer (Santa Clara, CA, USA). The analytes were loaded onto a ZORBAX Poroshell 300SB C8 column (Agilent Technologies, Santa Clara, CA, USA, 0.5 mm × 75 mm, maintained at 80 °C) with 0.1% formic acid, and eluted using a gradient of 20%–40% buffer B (80% acetonitrile, 18% 1-propanol, 2% water with 0.1% formic acid) at a flow rate of 20 µL/min over 5.5 min. Mass spectrometric detection was carried out in positive, sensitivity mode with capillary voltage set at 3.3 kV. Data analyses were performed with MaxEnt 1 function in MassLynx (Waters, Milford, MA, USA).
4.15. Differential Scanning Calorimetry
Purified DART samples were diluted in PBS to 1 mg/mL in a volume of 400 µL. PBS was used as a buffer blank in the reference cell. Samples were dispensed into the sample tray of a MicroCal VP-Capillary differential scanning calorimeter (DSC) with Autosampler (GE Healthcare, Piscataway, NJ, USA). Samples at either 0.3 mg/mL or 1 mg/mL were equilibrated for 5 min at 10 °C and then scanned up to 110 °C at a rate of either 60 °C/h or 100 °C/hr. A filtering period of 16 s was selected. Raw data were baseline-corrected, and the protein concentration was normalized. Origin Software 7.0 (OriginLab Corporation, Northampton, MA, USA) was used to fit the data to an MN2-State Model with an appropriate number of transitions.
4.16. Epitope Mapping of DART Proteins on the Human P-Cadherin Extracellular Domain
To identify the binding epitope of the anti-P-cadherin/anti-CD3 DART molecules, soluble P-cadherin ECD-Fc fusion protein constructs were generated. Each P-cadherin-Fc construct comprised a signal peptide, pro-peptide and a P-cadherin ECD subdomain region (either ECD1, ECD1–2, ECD1–3, ECD1–4, or ECD1–5) genetically fused to the hinge and CH2 and CH3 domains of human IgG1 via cleavable linker, as shown in
Figure 4. All constructs were confirmed by DNA sequencing and transiently transfected into FreeStyle™ 293 HEK cells (Life Technologies, Grand Island, NY, USA) according to the manufacturer’s method and expressed over 5–7 days. For enhanced processing of the pro-peptide, an expression vector containing the PACE cleavage enzyme was co-transfected along with the P-cadherin-containing vector. Purified protein was characterized for purity and activity by binding ELISA using commercially-available anti-human P-cadherin monoclonal and polyclonal antibodies.
4.17. Crystallography of Coil-Less DART 35
For crystallization trials, a modified version of DART 35 was generated with either a His
6 tag or a FLAG tag at the
C-terminus of each DART subunit chain to aid in purification (
Figure 5C). The constructs were transiently transfected into FreeStyle™ 293 HEK cells as described above. 10 mL anti-FLAG M2 resin (Sigma, St. Louis, MO, USA) was pre-equilibrated in TBS and allowed to batch-bind in 1.4 L conditioned medium for 2 h at 4 °C. The resin was then collected and packed into a column for anti-FLAG M2 chromatography, washed to baseline with TBS (20 CVs) and initially eluted with 0.1 M FLAG peptide buffer and finally eluted with 0.1 M glycine pH 3.0. The eluted protein was immediately neutralized with 10% 1.0 M Tris pH 8.0. Fractions with the highest purity were pooled, and the eluted protein was then further purified by Ni-NTA chromatography. Five mL of Ni-NTA resin pre-equilibrated in TBS was allowed to batch-bind to the protein for 1 h at 4 °C on an orbital mixer. The resin was then collected and placed in an Applied Biosystems column (Life Technologies, Grand Island, NY, USA). Resin was first washed to baseline with Buffer A (50 mM sodium phosphate, 300 mM sodium chloride pH 8.0), then with five CVs of Buffer A supplemented with 20 mM imidazole and finally eluted with Buffer A + 250 mM imidazole. Fractions with the highest purity were then pooled and further purified by size exclusion chromatography using a Superdex200 column (GE Healthcare, Piscataway, NJ, USA) and stored in TBS.
The purified HIS/FLAG-DART was concentrated to 9.6 mg/mL in a protein solution containing TBS. The crystals were obtained by hanging-drop vapor-diffusion method from a condition containing 15% PEG 8K and 0.5 M lithium sulfate. The hexagonal plate-like crystals had symmetry consistent with trigonal space group P321 with cell parameters a = b = 142.81 Å; c = 62.69 Å and with one coil-less DART molecule in the crystallographic asymmetric unit. The crystals were cryo-protected using a reservoir solution containing 25% ethylene glycol and were flash frozen in liquid nitrogen. A data set to a 2.0 Å resolution was collected from a single frozen crystal at IMCA beamline 17-ID at the Argonne National Laboratory (Lemont, IL, USA, APS). The data were processed and scaled using autoPROC (Global Phasing Ltd., Cambridge, UK) and SCALA (École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland). The final data set was 96.8% complete with average redundancy of 9.9 and with Rsym of 14.2%.
The structure was solved by molecular replacement with PHASER starting with the single chain Fv fragment models prepared from the Brookhaven PDB entry code, 1MOE. The solution was obtained by searching for each of the four subunits of the DART molecule separately. Several iterative rounds of manual adjustment and model rebuilding using COOT and crystallographic refinement using autoBUSTER yielded the final DART model with a crystallographic Rwork of 17.6% and Rfree of 20.5%, where Rwork= ||Fobs| − |Fcalc||/|Fobs| and Rfree is equivalent to Rwork, but calculated for a randomly chosen 5% of reflections omitted from the refinement process.
4.18. Isolation of PBMCs and T Cells from Human Whole Blood
Peripheral blood mononuclear cells (PBMCs) from healthy human donors were isolated from whole blood using Ficoll gradient centrifugation. Whole blood was diluted 1:1 with sterile Dulbecco’s phosphate buffered saline (DPBS). The diluted blood (35 mL) was layered onto 15 mL of Ficoll-Paque Plus in a 50 mL tube and the tubes were centrifuged at 400× g (1320 rpm) for 30 min with the brake off. The buffy coat layer between the two phases was collected into 50 mL tubes and centrifuged at 600× g (1620 rpm) for 5 min. The supernatant was discarded and the cell pellet was washed three times with DPBS by centrifuging the tubes at 600× g (1620 rpm) for 5 min. Viable cell count was determined using Trypan Blue dye to exclude non-viable cells. The PBMCs were resuspended in complete culture medium (RPMI 1640, 10% FBS, 1% pen/strep) and incubated at 37 °C with 5% CO2 overnight or were further processed to isolate human CD4+ or CD8+ T cells. T cells were isolated from PBMCs using the untouched human CD4+ T cell isolation kit or untouched human CD8+ T cell isolation kit (Life Technologies, Grand Island, NY, USA) according to the manufacturer’s instructions. After isolation, the T cells were resuspended in complete culture medium and incubated at 37 °C with 5% CO2 overnight.
4.19. CTL Cytotoxicity Assay (LDH Release Assay)
The CytoTox 96® Non-Radioactive Cytotoxicity Assay Kit (Promega, Madison, WI, USA) was used to measure cytotoxicity by quantitating the enzymatic activity of lactate dehydrogenase (LDH) released from lysed cells. The three target cancer cell lines were harvested by detaching cells with 0.25% Trypsin-EDTA solution and collected by centrifugation at 311× g (1200 rpm) for 5 min. The cells were washed once by DPBS and resuspended in assay medium (RPMI 1640 no phenol red, 10% FBS, 1% pen/strep). After counting and confirmation of viability (>85%), the cells were diluted in assay medium to a density of 4 × 105 cells/mL. 50 μL of the diluted cell suspension was added to a 96-well, U-bottom cell culture plate (BD Falcon). For each treatment, duplicate wells were included.
The effector cells (human PBMCs or purified human CD4+/CD8+ T cells) were washed once with assay media and resuspended in assay media at the appropriate cell density depending on the effector and target (E:T) cell ratio used in the assay. Effector cells (100 µL) were added to each well of the plate containing 50 µL target cancer cells. Test DART proteins were initially diluted to 4-times the highest concentration to be added to the assay plate and serially diluted 4-fold. 50 µL/well of the dilutions were added to the plate containing 100 μL effector cells/well and 50 μL target cells/well.
Three sets of controls to measure target cell spontaneous release (SR), antibody independent cellular cytotoxicity (AICC), and target cell maximal release (MR) were set up as follows: (1) SR: 50 µL target cells and 150 µL assay media without test molecules; (2) AICC: 50 µL target cells, 100 µL effector cells, and 50 μL assay media without test molecules; and (3) MR: 50 μL target cells and 120 μL assay media without test molecules to which lysis solution was added at the end of the experiment to determine maximal LDH release.
Plates were incubated at 37 °C with 5% CO2 for 24 to 48 h as indicated. Following incubation, 30 μL/well of 10× lysis solution was added to the maximum release wells, mixed by pipetting three times, and plates were incubated for 10 min to completely lyse the target cells. The plates were centrifuged at 311× g (1200 rpm) for 5 min and 40 µL/well of supernatant were transferred to a flat-bottom ELISA plate and 40 µL of LDH substrate solution was added to each well. Plates were incubated for 10–20 min at room temperature in the dark and then 40 µL of stop solution was added. The optical density (OD) was measured at 490 nm within 1 h on a Victor2 Multilabel plate reader. The percentage of cytotoxicity was calculated as described below and dose-response curves were generated using GraphPad Prism5 or 6 software by curve fitting the cytotoxicity values to the sigmoidal dose-response function.
Specific cell lysis was calculated from OD data using the following formula:
4.20. Determination of Cytokines in Supernatant
An ELISA was used to measure the levels of six cytokines in the supernatants (IFN-γ, TNF-α, IL-10, IL-6, IL-4, and IL-2). Cytokine detection kits were purchased from R & D Systems. The ELISA plate (Thermo Scientific, Madison, CT, USA) was coated with capture reagent from the kit diluted in 1× DPBS at the coating concentration suggested by the manufacturer and incubated at room temperature overnight. The next day the coated plate was washed with wash buffer containing 1× DPBS and 0.1% Tween20. The plate was then blocked with 200 µL/well of sample diluent containing 1× DPBS and 1% BSA for 1 h at room temperature and then washed. During the 1-h blocking step, test samples and standard samples (for generating standard curves) were prepared. After the blocking step, 100 µL/well of standard samples and test samples were added to the plate, followed by incubation for 2 h. The plate was incubated for an additional 2 h after 100 µL/well of diluted detecting reagent was added. Then the plate was washed again and 100 µL/well of streptavidin-horseradish peroxidase (SA-HRP) solution was added followed by incubation for 20 min. After washing, 100 µL/well of TMB One Component HRP Microwell Substrate was added followed by incubation for 15 to 20 min in the dark. The reaction was stopped by adding 1% sulfuric acid. The OD was measured at 450 nm within 15 min on a Victor 2 Multilabel plate reader. The data were exported as Microsoft Excel format for data analysis. The standard curves were generated using SoftMax Pro Version 5.4 (Molecular Devices) by fitting the data from each cytokine standard to a 4-parameter variable weight function with concentration of cytokine (x) as the independent variable and OD response (y) as the dependent variable:
In this expression, A refers to the left (lower) asymptote, D refers to the right (upper) asymptote, C refers to the cytokine concentration that produces 50% of the maximum response, and B is a scale parameter related to the shape and steepness of the curve. The curve fit is performed using the variable weight for each point option in SoftMax Pro with the formula 1/Y2.
The cytokine concentration was calculated from the 4-parameter curve using the following SoftMax command:
where X is the concentration of the analyte, Plot #N refers to the standard curve on Graph #N for Plate #N. Values are the mean OD value for the respective concentration of the cytokine.
4.21. Pharmacokinetic Property Assessment in a Human FcRn Knock-in Mouse Model
Female B6.Cg-Fcgrttm1Dcr Tg(CAG-FCGRT)276Dcr/DcrJ mice (Jackson Laboratories Inc., ID 004919, Bar Harbor, ME, USA) were dosed intravenously with anti-P-cadherin/anti-CD3 LP-DART at 2 mg/kg. Mice (n = 3/time point) were bled prior to dosing and at 5 min, 4 h, and on day 1, 2, 3, 7, 14 and 22. Serum was prepared and frozen at −80 °C until assayed.
Assay plates (MaxiSorp 96-well, Nunc, Rochester, NY, USA) were coated with a custom polyclonal goat anti-CD3 antibody overnight at 4 °C. Plates were washed three times using PBS + Ca2+ and Mg2+ and blocked for 1 h at room temperature in 0.5% BSA. Plates were then incubated with diluted test samples, a P-cadherin LP-DART calibration standard and appropriate controls. Plates were washed as before pre-, during and post sequential 1 h incubations with goat anti-human IgG Fc-biotin (Thermo Scientific, Madison, CT, USA) and streptavidin-horseradish peroxidase (SA-HRP; Thermo Scientific, Madison, CT, USA). Signal was developed using TMB (SurModics, Eden Prairie, MN, USA) and the reaction stopped with H2SO4. Plates were read at OD450 nm using a microplate reader (SpectraMax M2e, Molecular Device, Sunnyvale, CA, USA). The calibration standard curve was generated with the standard calibrators OD signals in the four-parameter logistic model using SoftMax Pro software (Version 5.4, Molecular Devices). Concentrations were determined from the interpolation of the samples’ OD signal data with the equation describing the standard curve. The lower limit of quantitation (LLOQ) for this assay was estimated to be 9.75 ng/mL. PK parameters were calculated using the WinNonlin non-compartment analysis (NCA) model.
4.22. In Vivo Tumor Growth Inhibition by PF-06671008
Human T cells were isolated from heparinized whole blood according to the manufacturer’s protocol provided in the RosetteSep T cell isolation kit. The purified T cells were subsequently activated by exposing the cells to anti-CD3 (OKT-3; 1 µg/mL) and anti-CD28 (66 µg/mL) antibodies for a period of 48 h. Following stimulation, the cells were grown in RPMI 1640 medium with 10% FBS and 1% penicillin/streptomycin in the presence of IL-2 (7.6 ng/mL) for up to 3 weeks. The HCT-116 cells were maintained in vitro in RPMI 1640 medium with 10% FBS and 1% penicllin/streptomycin. The human T cells and tumor cells were combined at a ratio of 1:5 (1 × 106 and 5 × 106 cells, respectively) and suspended in 200 µL of sterile Ham’s F12 and injected SC on Day 0.
Vehicle control (sterile saline containing 0.5% BSA), PF-06671008, or 4420-hXR32-LP control DART was administered IV via tail vein injections (100 µL) once daily for 4 days on Days 0, 1, 2, and 3 (see
Table 7). Individual animal weights were recorded twice weekly beginning at the time of tumor cell injection until study completion. Animals were observed twice weekly for general moribundity and daily for mortality. Animal deaths were to be assessed as drug-related or technical based on factors including gross observation and weight loss; animal deaths were to be recorded daily. Individual tumor dimensions (length × width) were measured using calipers and were recorded periodically throughout the study beginning on Day 7 and continuing through study completion. Tumor volume was estimated as follows:
Animals that died for any reason were to be censored from the data calculations at the time of death. Tumor growth inhibition (TGI) values were calculated for each group containing treated animals using the formula:
Individual mice lacking palpable tumors were classified as undergoing a complete response (CR). Statistical analyses were carried out between treated and control groups comparing tumor volumes. For these analyses, a two-way analyses of variance (ANOVA) followed by a Bonferroni post-test were employed. All analyses were performed using GraphPad Prism software (Version 6.03). Weight and tumor data from individual animals that died for any reason were to be censored from analysis (at the time of death). However, tumor data from animals reporting partial or complete responses were included in these calculations.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}