
Systemic and Mucosal Antibody Responses to Soluble and Nanoparticle-Conjugated Antigens Administered Intranasally

Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and Reagents
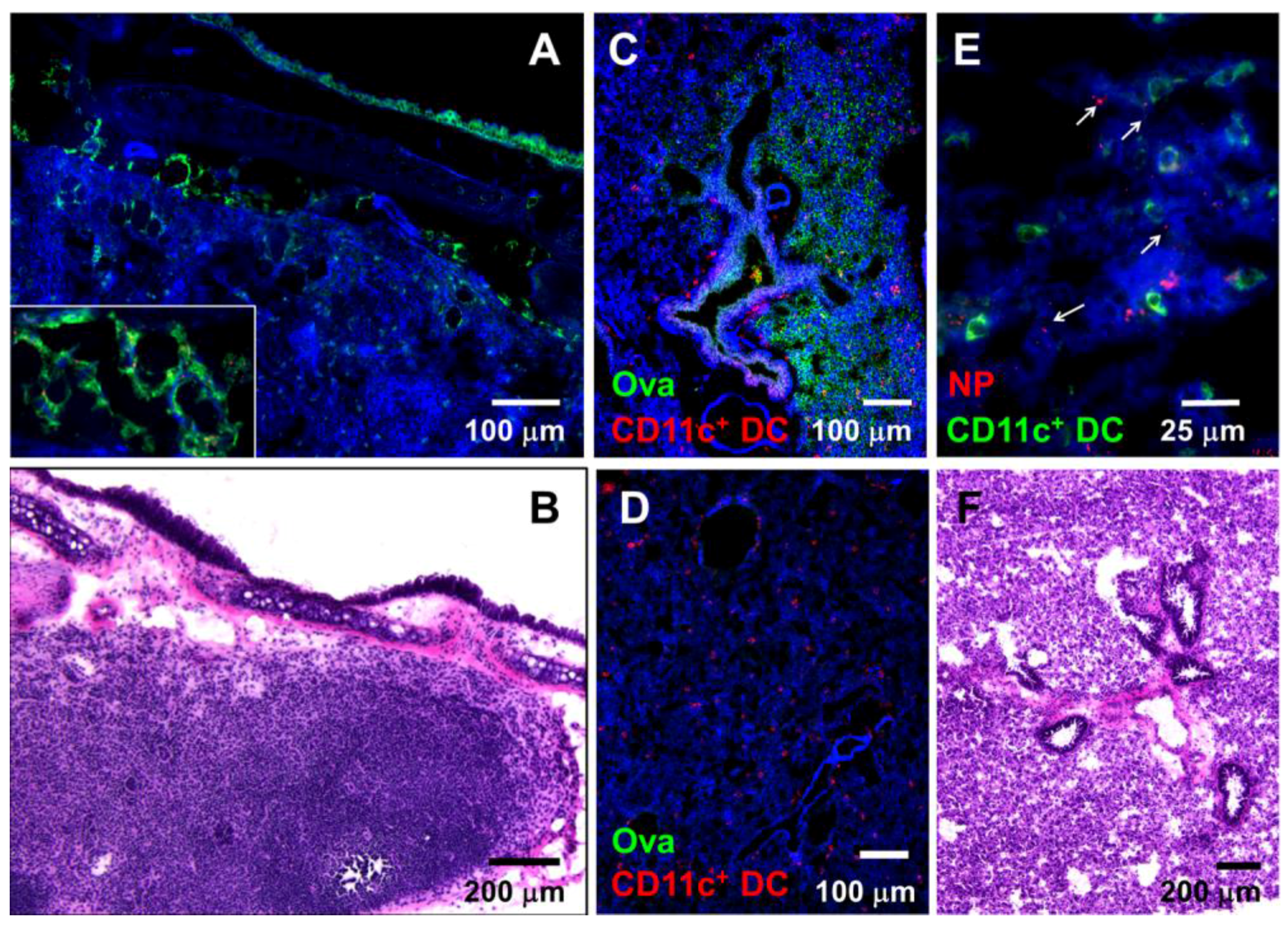
2.3. Analysis of Antigen Internalization by Immunofluorescence Microscopy (IFM)
2.4. I.n. Immunization with Soluble Ova and NP-Ova
2.5. Sample Collection and Determination of Ova-Specific Antibody Titers in Sera and Mucosal Secretions Using ELISA
2.6. Statistical Analysis
3. Results
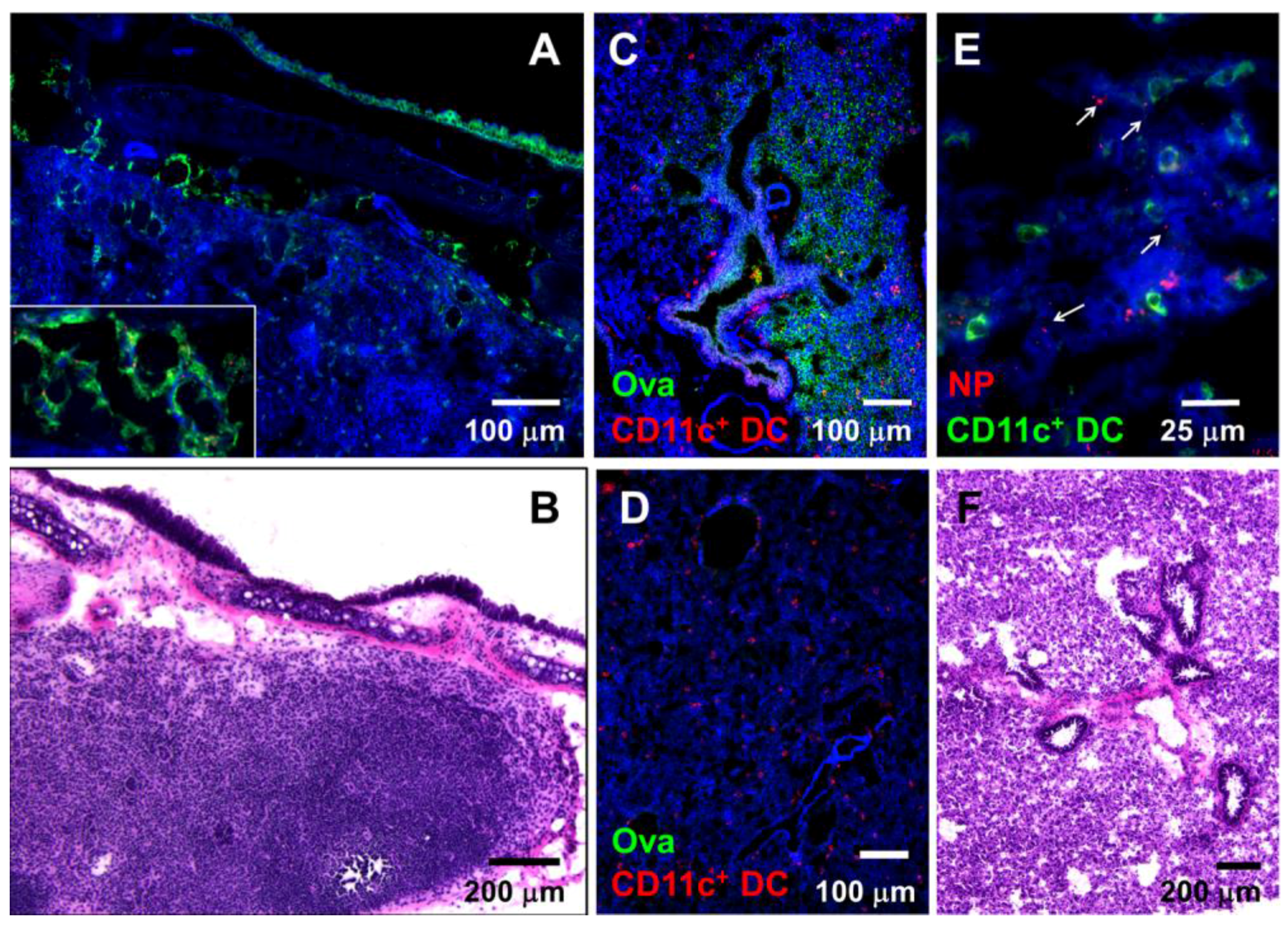
3.1. Nasally-Applied 20 nm NPs, Ova, and Dextran Reach the Respiratory Lymph Nodes (LNs) and Lung Tissue of Anesthetized Mice
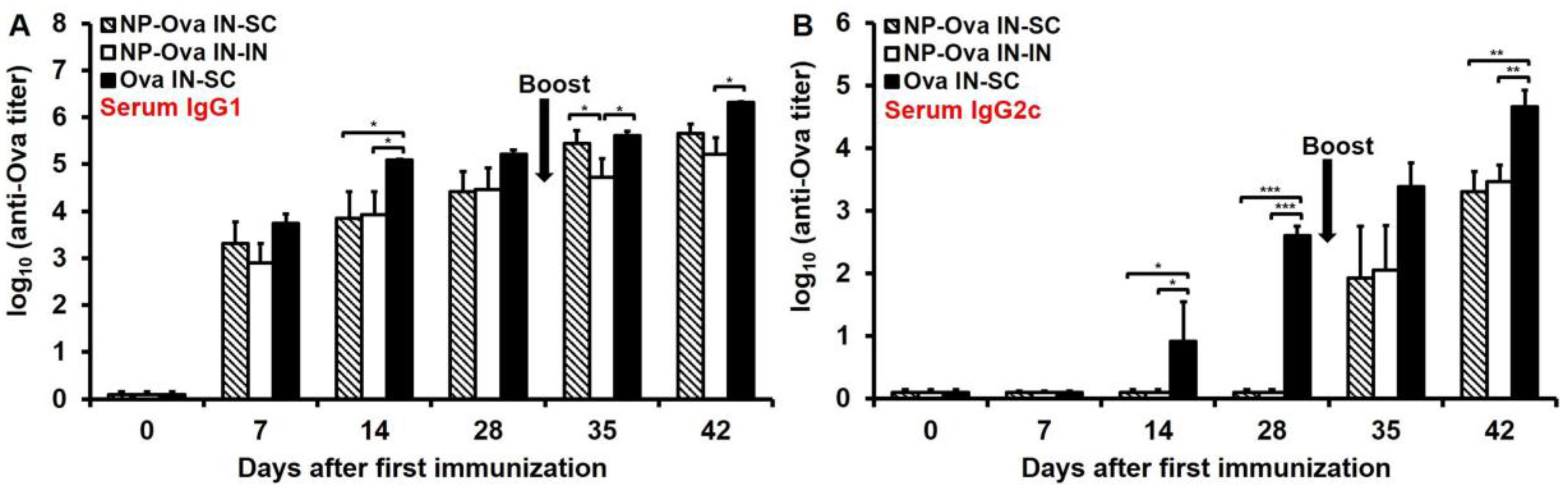
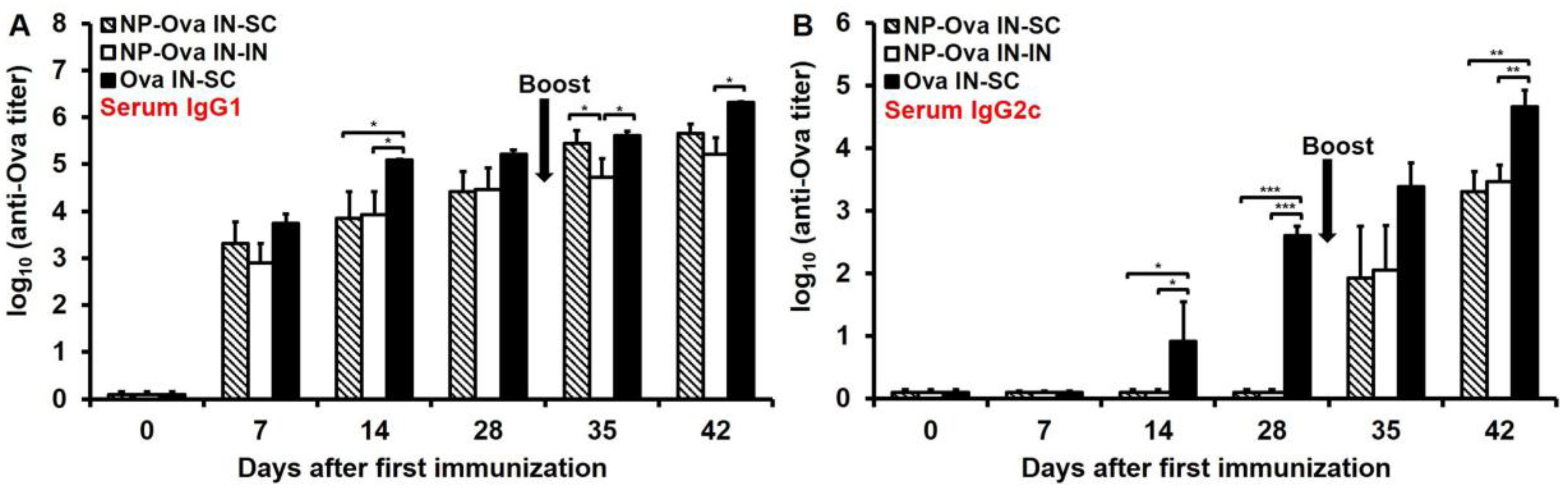
3.2. Nasal Administration of Soluble Ova Induces Serum IgG1/IgG2c Antibodies, While Administration of NP-Ova Prompts an IgG1-Dominated Response Prior to Boost
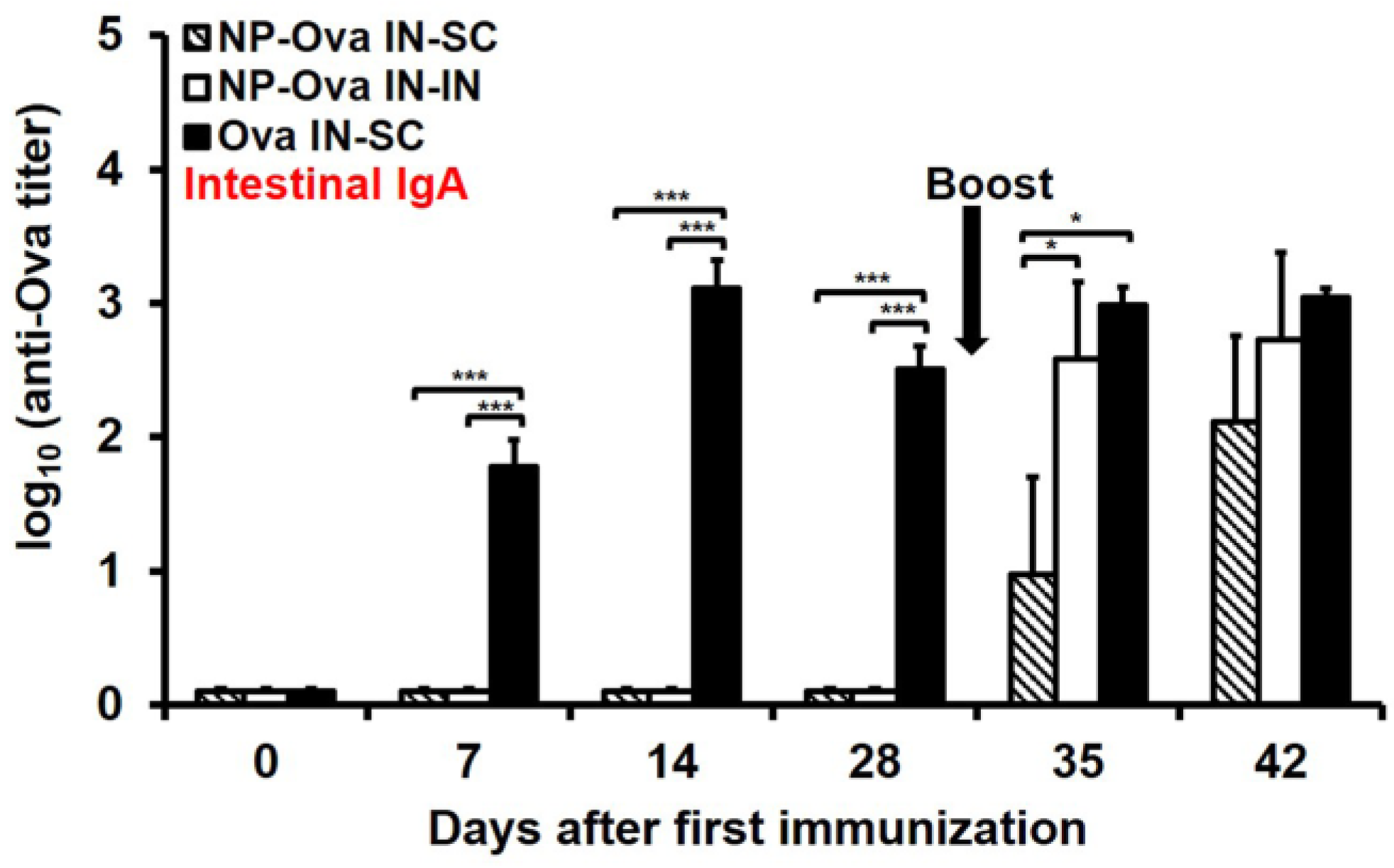
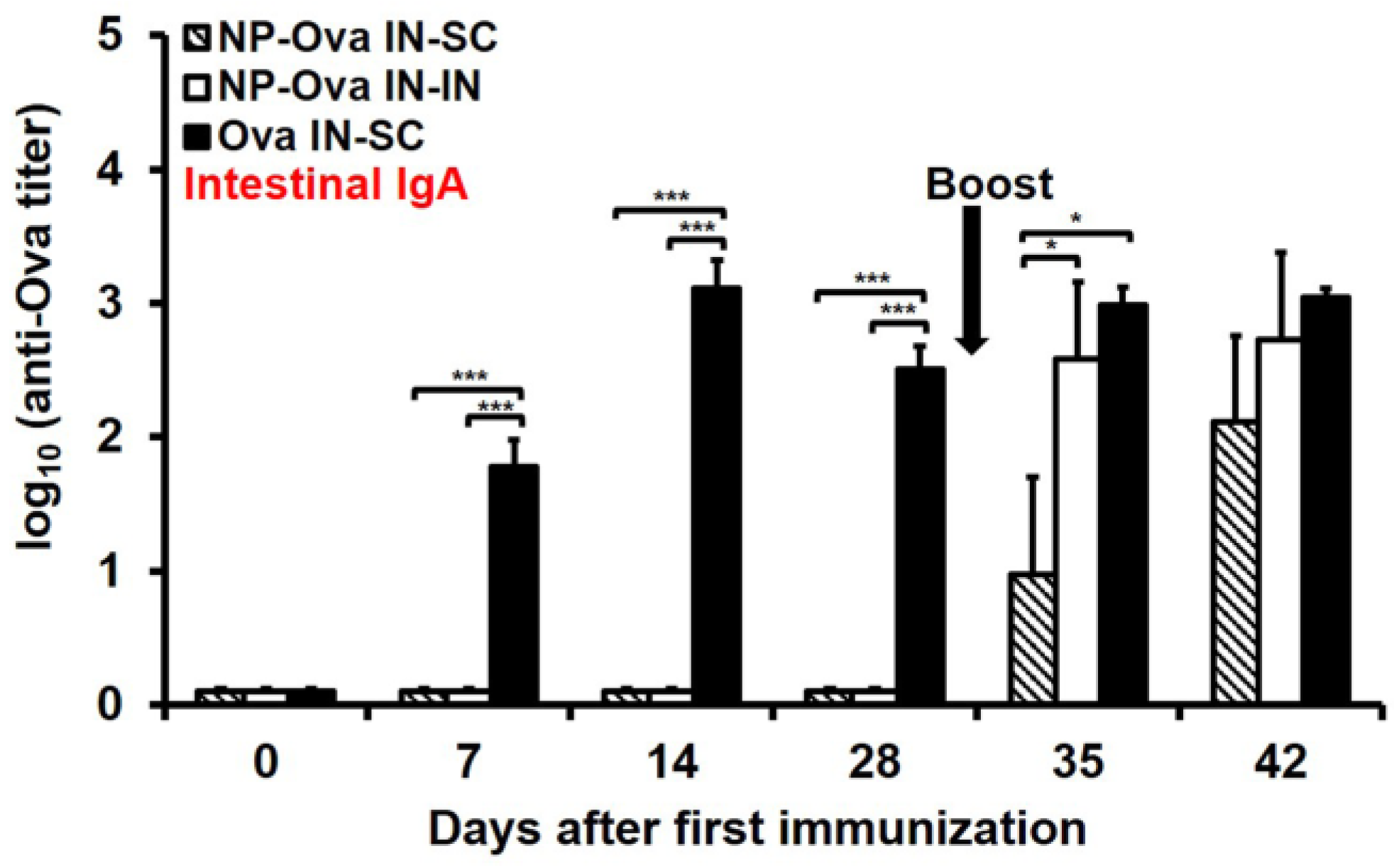
3.3. I.n. Priming with High Dose of Ova, Unlike NP-Ova, Induces Intestinal IgA Which Is Not Significantly Elevated Following s.c. Boosting with Ova+CFA
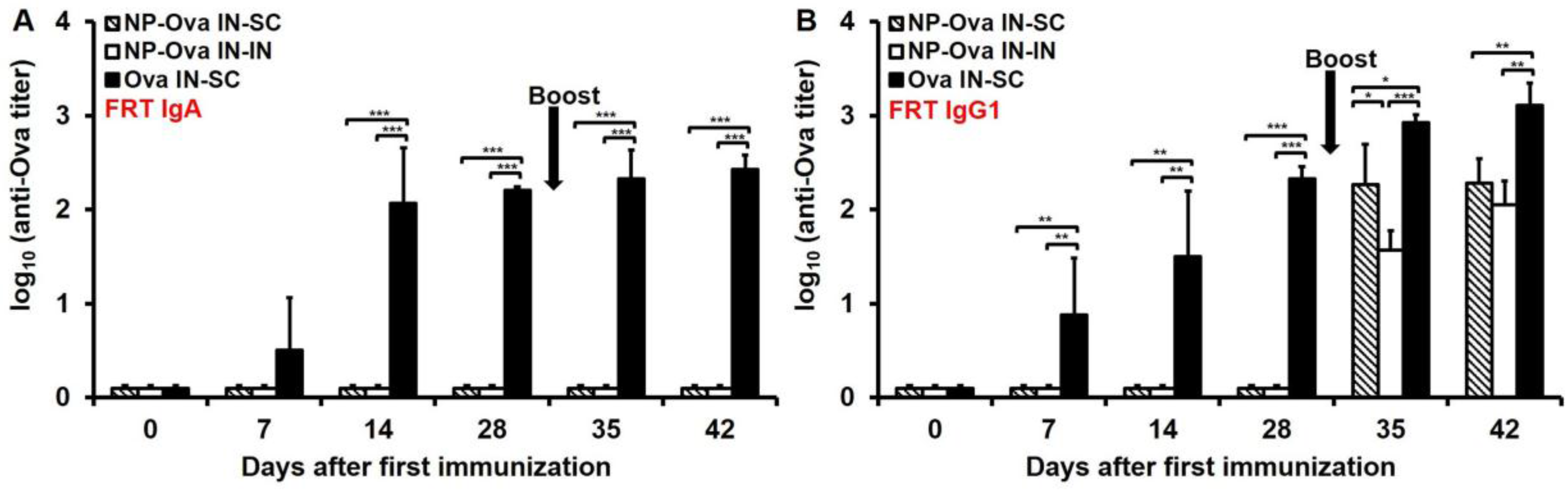
3.4. Nasal Priming with Ova Induces IgA and IgG1 in Vaginal Secretions, While i.n. Priming Followed by s.c. Boosting with NP-Ova Induces IgG1 and no IgA
3.5. Systemic (s.c.) Boosting Immunization Following i.n. Priming with NP-Ova Induces Superior Humoral Immune Memory Compared to Mucosal (i.n.) Boosting
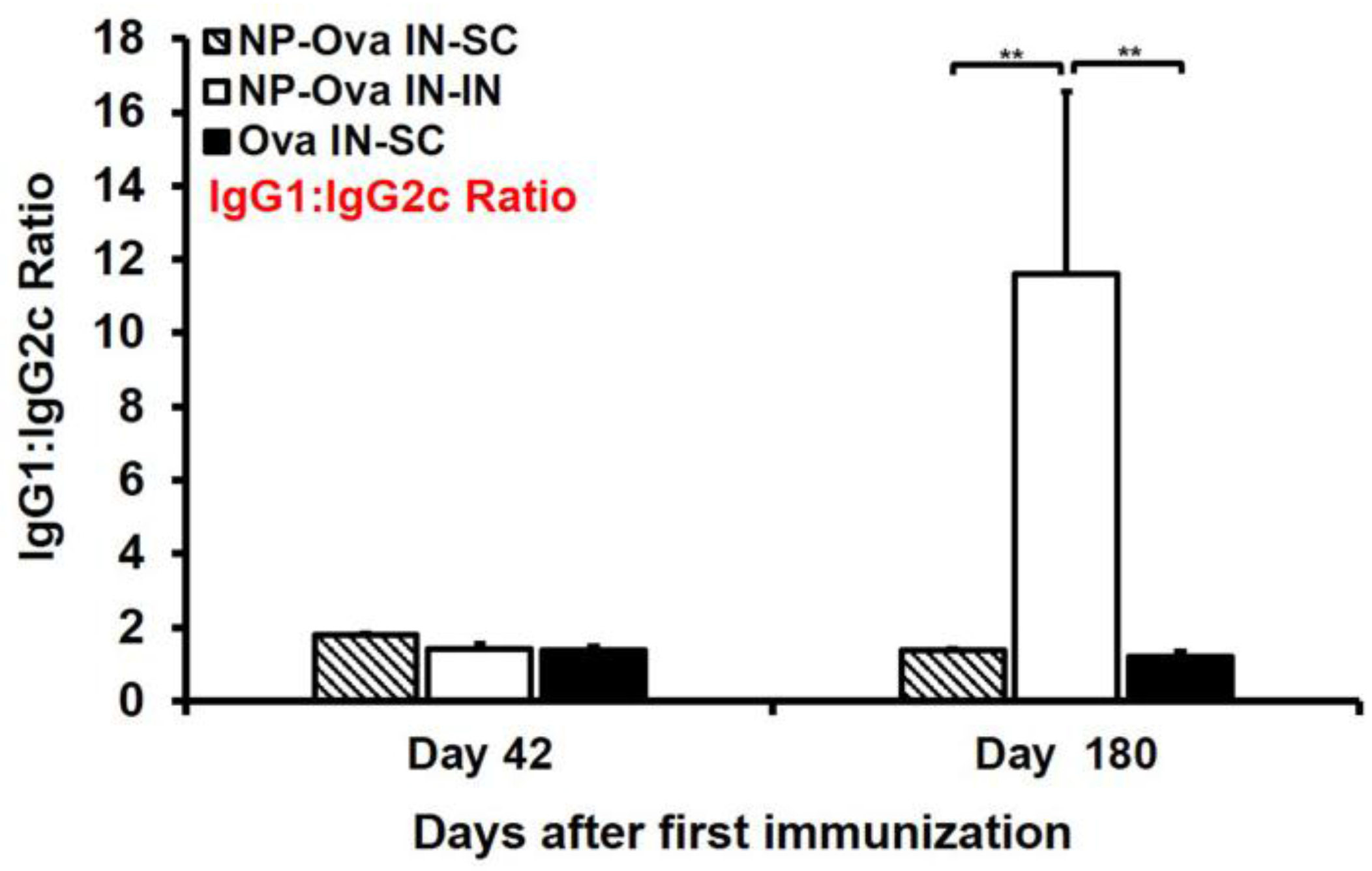
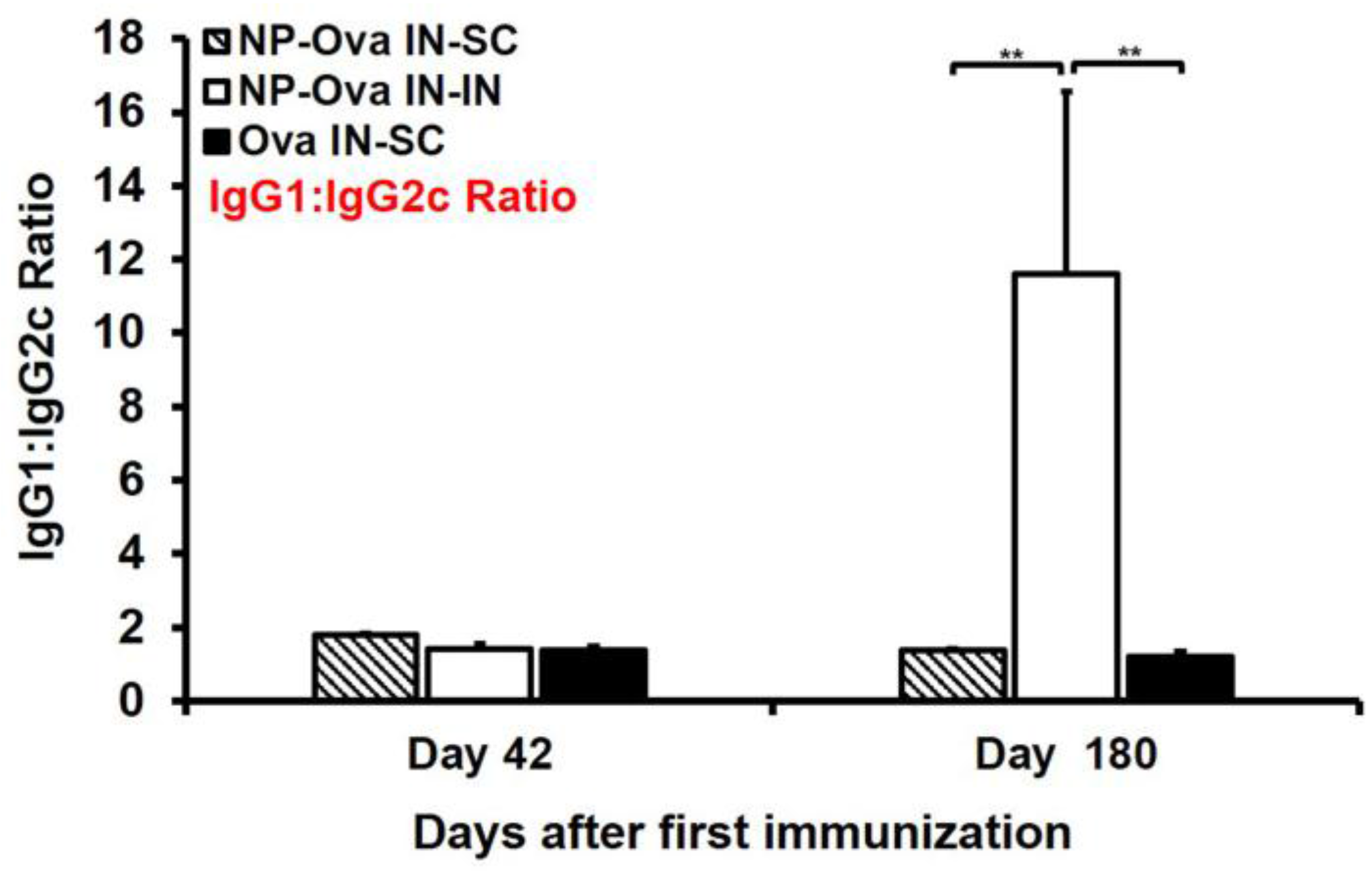
3.6. Mice Primed i.n. with Ova or NP-Ova Require a Secondary s.c. Boosting Immunization for Maintaining a Long-Lasting Mixed Th1/Th2 Immune Response
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| I.n. | intranasal |
| S.c. | subcutaneous |
| P.o. | per oral |
| NPs | nanoparticles |
| Ova | ovalbumin |
| NP-Ova | Ova conjugated 20 nm NPs |
| Ova+CFA | Ova with complete Freund’s adjuvant |
| NALT | nasal associated lymphoid tissue |
| DC | dendritic cells |
| LNs | mediastinal lymph nodes |
| FRT | female reproductive tract |
| CT | cholera toxin |
| OPV | Oral polio vaccine |
| IPV | Inactivated polio vaccine |
| DTH | Delayed Type Hypersensitivity |
References
- Ghendon, Y. The immune response of humans to live and inactivated influenza vaccines. Adv. Exp. Med. Biol. 1989, 257, 37–45. [Google Scholar] [PubMed]
- Barackman, J.D.; Ott, G.; O'Hagan, D.T. Intranasal immunization of mice with influenza vaccine in combination with the adjuvant lt-r72 induces potent mucosal and serum immunity which is stronger than that with traditional intramuscular immunization. Infect. Immun. 1999, 67, 4276–4279. [Google Scholar] [PubMed]
- Bender, B.S.; Johnson, M.P.; Small, P.A. Influenza in senescent mice: Impaired cytotoxic t-lymphocyte activity is correlated with prolonged infection. Immunology 1991, 72, 514–519. [Google Scholar] [PubMed]
- Renegar, K.B.; Small, P.A. Immunoglobulin a mediation of murine nasal anti-influenza virus immunity. J. Virol. 1991, 65, 2146–2148. [Google Scholar] [PubMed]
- Knuschke, T.; Sokolova, V.; Rotan, O.; Wadwa, M.; Tenbusch, M.; Hansen, W.; Staeheli, P.; Epple, M.; Buer, J.; Westendorf, A.M. Immunization with biodegradable nanoparticles efficiently induces cellular immunity and protects against influenza virus infection. J. Immunol. 2013, 190, 6221–6229. [Google Scholar] [CrossRef] [PubMed]
- Langermann, S.; Palaszynski, S.; Sadziene, A.; Stover, C.K.; Koenig, S. Systemic and mucosal immunity induced by bcg vector expressing outer-surface protein a of borrelia burgdorferi. Nature 1994, 372, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Y.; Nguyen, H.H.; Russell, M.W. Nasal lymphoid tissue (nalt) as a mucosal immune inductive site. Scand. J. Immunol. 1997, 46, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Zuercher, A.W. Upper respiratory tract immunity. Viral Immunol. 2003, 16, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Vadolas, J.; Davies, J.K.; Wright, P.J.; Strugnell, R.A. Intranasal immunization with liposomes induces strong mucosal immune responses in mice. Eur. J. Immunol. 1995, 25, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Johansson, E.L.; Wassen, L.; Holmgren, J.; Jertborn, M.; Rudin, A. Nasal and vaginal vaccinations have differential effects on antibody responses in vaginal and cervical secretions in humans. Infect. Immun. 2001, 69, 7481–7486. [Google Scholar] [CrossRef] [PubMed]
- Dahl, R.; Mygind, N. Anatomy, physiology and function of the nasal cavities in health and disease. Adv. Drug Deliv. Rev. 1998, 29, 3–12. [Google Scholar] [PubMed]
- Kang, M.L.; Cho, C.S.; Yoo, H.S. Application of chitosan microspheres for nasal delivery of vaccines. Biotechnol. Adv. 2009, 27, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Ugwoke, M.I.; Verbeke, N.; Kinget, R. The biopharmaceutical aspects of nasal mucoadhesive drug delivery. J. Pharm. Pharmacol. 2001, 53, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Ugwoke, M.I.; Agu, R.U.; Verbeke, N.; Kinget, R. Nasal mucoadhesive drug delivery: Background, applications, trends and future perspectives. Adv. Drug Deliv. Rev. 2005, 57, 1640–1665. [Google Scholar] [CrossRef] [PubMed]
- Dombu, C.; Carpentier, R.; Betbeder, D. Influence of surface charge and inner composition of nanoparticles on intracellular delivery of proteins in airway epithelial cells. Biomaterials 2012, 33, 9117–9126. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Atkinson, M.A.; Clare-Salzler, M.; Herschenfeld, A.; Forsthuber, T.; Lehmann, P.V.; Kaufman, D.L. Nasal administration of glutamate decarboxylase (GAD65) peptides induces Th2 responses and prevents murine insulin-dependent diabetes. J. Exp. Med. 1996, 183, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Samsom, J.N. Regulation of antigen-specific regulatory T-cell induction via nasal and oral mucosa. Crit. Rev. Immunol. 2004, 24, 157–177. [Google Scholar] [CrossRef] [PubMed]
- Czerkinsky, C.; Anjuere, F.; McGhee, J.R.; George-Chandy, A.; Holmgren, J.; Kieny, M.P.; Fujiyashi, K.; Mestecky, J.F.; Pierrefite-Carle, V.; Rask, C.; et al. Mucosal immunity and tolerance: Relevance to vaccine development. Immunol. Rev. 1999, 170, 197–222. [Google Scholar] [CrossRef] [PubMed]
- Redhead, K.; Sesardic, D.; Yost, S.E.; Attwell, A.M.; Watkins, J.; Hoy, C.S.; Plumb, J.E.; Corbel, M.J. Combination of dtp and haemophilus influenzae type b conjugate vaccines can affect laboratory evaluation of potency and immunogenicity. Biologicals 1994, 22, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Fujihashi, K.; Koga, T.; van Ginkel, F.W.; Hagiwara, Y.; McGhee, J.R. A dilemma for mucosal vaccination: Efficacy versus toxicity using enterotoxin-based adjuvants. Vaccine 2002, 20, 2431–2438. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kiyono, H.; Yamamoto, M.; Imaoka, K.; Fujihashi, K.; Van Ginkel, F.W.; Noda, M.; Takeda, Y.; McGhee, J.R. A nontoxic mutant of cholera toxin elicits Th2-type responses for enhanced mucosal immunity. Proc. Natl. Acad. Sci. USA 1997, 94, 5267–5272. [Google Scholar] [CrossRef] [PubMed]
- Samsom, J.N.; van Berkel, L.A.; van Helvoort, J.M.; Unger, W.W.; Jansen, W.; Thepen, T.; Mebius, R.E.; Verbeek, S.S.; Kraal, G. Fc gamma RIIB regulates nasal and oral tolerance: A role for dendritic cells. J. Immunol. 2005, 174, 5279–5287. [Google Scholar] [CrossRef] [PubMed]
- Slütter, B.; Jiskoot, W. Dual role of cpg as immune modulator and physical crosslinker in ovalbumin loaded n-trimethyl chitosan (TMC) nanoparticles for nasal vaccination. J. Control. Release 2010, 148, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.E.; Titball, R.; Williamson, D. Vaccine delivery using nanoparticles. Front. Cell. Infect. Microbiol. 2013, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Csaba, N.; Garcia-Fuentes, M.; Alonso, M.J. Nanoparticles for nasal vaccination. Adv. Drug Deliv. Rev. 2009, 61, 140–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zheng, X.; Zhang, C.; Shao, X.; Zhang, X.; Zhang, Q.; Jiang, X. Antigen-conjugated n-trimethylaminoethylmethacrylate chitosan nanoparticles induce strong immune responses after nasal administration. Pharm. Res. 2015, 32, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Gizurarson, S.; Tamura, S.; Kurata, T.; Hasiguchi, K.; Ogawa, H. The effect of cholera toxin and cholera toxin b subunit on the nasal mucosal membrane. Vaccine 1991, 9, 825–832. [Google Scholar] [CrossRef]
- Van Ginkel, F.W.; Jackson, R.J.; Yoshino, N.; Hagiwara, Y.; Metzger, D.J.; Connell, T.D.; Vu, H.L.; Martin, M.; Fujihashi, K.; McGhee, J.R. Enterotoxin-based mucosal adjuvants alter antigen trafficking and induce inflammatory responses in the nasal tract. Infect. Immun. 2005, 73, 6892–6902. [Google Scholar] [CrossRef] [PubMed]
- Stano, A.; Nembrini, C.; Swartz, M.A.; Hubbell, J.A.; Simeoni, E. Nanoparticle size influences the magnitude and quality of mucosal immune responses after intranasal immunization. Vaccine 2012, 30, 7541–7546. [Google Scholar] [CrossRef] [PubMed]
- Jani, P.; Halbert, G.W.; Langridge, J.; Florence, A.T. Nanoparticle uptake by the rat gastrointestinal mucosa: Quantitation and particle size dependency. J. Pharm. Pharmacol. 1990, 42, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.P.; Labhasetwar, V.; Amidon, G.L.; Levy, R.J. Gastrointestinal uptake of biodegradable microparticles: Effect of particle size. Pharm. Res. 1996, 13, 1838–1845. [Google Scholar] [CrossRef] [PubMed]
- Olmsted, S.S.; Padgett, J.L.; Yudin, A.I.; Whaley, K.J.; Moench, T.R.; Cone, R.A. Diffusion of macromolecules and virus-like particles in human cervical mucus. Biophys. J. 2001, 81, 1930–1937. [Google Scholar] [CrossRef]
- Chithrani, B.D.; Ghazani, A.A.; Chan, W.C. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006, 6, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.E.; Konjufca, V.H. Protein-coated nanoparticles are internalized by the epithelial cells of the female reproductive tract and induce systemic and mucosal immune responses. PLoS ONE 2014, 9, e114601. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.E.; Konjufca, V.H. Per-oral immunization with antigen-conjugated nanoparticles followed by sub-cutaneous boosting immunization induces long-lasting mucosal and systemic antibody responses in mice. PLoS ONE 2015, 10, e0118067. [Google Scholar] [CrossRef] [PubMed]
- Worbs, T.; Bode, U.; Yan, S.; Hoffmann, M.W.; Hintzen, G.; Bernhardt, G.; Förster, R.; Pabst, O. Oral tolerance originates in the intestinal immune system and relies on antigen carriage by dendritic cells. J. Exp. Med. 2006, 203, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Grassly, N.C.; Jafari, H.; Bahl, S.; Sethi, R.; Deshpande, J.M.; Wolff, C.; Sutter, R.W.; Aylward, R.B. Waning intestinal immunity after vaccination with oral poliovirus vaccines in India. J. Infect. Dis. 2012, 205, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.E.; Lickteig, D.J.; Plunkett, K.N.; Ryerse, J.S.; Konjufca, V. The uptake of soluble and particulate antigens by epithelial cells in the mouse small intestine. PLoS ONE 2014, 9, e86656. [Google Scholar] [CrossRef] [PubMed]
- Rosche, K.L.; Aljasham, A.T.; Kipfer, J.N.; Piatkowski, B.T.; Konjufca, V. Infection with salmonella enterica serovar typhimurium leads to increased proportions of F4/80+ Red pulp macrophages and decreased proportions of B and T lymphocytes in the spleen. PLoS ONE 2015, 10, e0130092. [Google Scholar] [CrossRef] [PubMed]
- Wanner, A.; Salathé, M.; O’Riordan, T.G. Mucociliary clearance in the airways. Am. J. Respir. Crit. Care Med. 1996, 154, 1868–1902. [Google Scholar] [CrossRef] [PubMed]
- Christopher, A.B.; Ochoa, S.; Krushansky, E.; Francis, R.; Tian, X.; Zahid, M.; Muñoz, R.; Lo, C.W. The effects of temperature and anesthetic agents on ciliary function in murine respiratory epithelia. Front. Pediatr. 2014, 2, 111. [Google Scholar] [CrossRef] [PubMed]
- Raphael, J.H.; Selwyn, D.A.; Mottram, S.D.; Langton, J.A.; O’Callaghan, C. Effects of 3 MAC of halothane, enflurane and isoflurane on cilia beat frequency of human nasal epithelium in vitro. Br. J. Anaesth. 1996, 76, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.; Stannard, W.; Passant, C.; O’Callaghan, C.; Banerjee, A. What effect does isoflurane have upon ciliary beat pattern: An in vivo study. Clin. Otolaryngol. Allied Sci. 2004, 29, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Slütter, B.; Bal, S.; Keijzer, C.; Mallants, R.; Hagenaars, N.; Que, I.; Kaijzel, E.; van Eden, W.; Augustijns, P.; Löwik, C.; et al. Nasal vaccination with n-trimethyl chitosan and plga based nanoparticles: Nanoparticle characteristics determine quality and strength of the antibody response in mice against the encapsulated antigen. Vaccine 2010, 28, 6282–6291. [Google Scholar] [CrossRef] [PubMed]
- Akagi, T.; Kawamura, M.; Ueno, M.; Hiraishi, K.; Adachi, M.; Serizawa, T.; Akashi, M.; Baba, M. Mucosal immunization with inactivated HIV-1-capturing nanospheres induces a significant HIV-1-specific vaginal antibody response in mice. J. Med. Virol. 2003, 69, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.M. Immunogenicity and efficacy of oral vaccines in developing countries: Lessons from a live cholera vaccine. BMC Biol. 2010, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- McCluskie, M.J.; Weeratna, R.D.; Payette, P.J.; Davis, H.L. Parenteral and mucosal prime-boost immunization strategies in mice with hepatitis B surface antigen and CpG DNA. FEMS Immunol. Med. Microbiol. 2002, 32, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Barr, T.A.; Brown, S.; Mastroeni, P.; Gray, D. B cell intrinsic MyD88 signals drive IFN-gamma production from T cells and control switching to IgG2c. J. Immunol. 2009, 183, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Ruane, D.; Chorny, A.; Lee, H.; Faith, J.; Pandey, G.; Shan, M.; Simchoni, N.; Rahman, A.; Garg, A.; Weinstein, E.G.; et al. Microbiota regulate the ability of lung dendritic cells to induce IgA class-switch recombination and generate protective gastrointestinal immune responses. J. Exp. Med. 2016, 213, 53–73. [Google Scholar] [CrossRef] [PubMed]
- Miyake, A.; Akagi, T.; Enose, Y.; Ueno, M.; Kawamura, M.; Horiuchi, R.; Hiraishi, K.; Adachi, M.; Serizawa, T.; Narayan, O.; et al. Induction of HIV-specific antibody response and protection against vaginal SHIV transmission by intranasal immunization with inactivated SHIV-capturing nanospheres in macaques. J. Med. Virol. 2004, 73, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Peterson, E.M.; de la Maza, L.M. Intranasal immunization induces long-term protection in mice against a chlamydia trachomatis genital challenge. Infect. Immun. 1996, 64, 5341–5348. [Google Scholar] [PubMed]
- Jafari, H.; Deshpande, J.M.; Sutter, R.W.; Bahl, S.; Verma, H.; Ahmad, M.; Kunwar, A.; Vishwakarma, R.; Agarwal, A.; Jain, S.; et al. Polio eradication. Efficacy of inactivated poliovirus vaccine in India. Science 2014, 345, 922–925. [Google Scholar] [CrossRef] [PubMed]
- Organization, W.H. Polio vaccines: WHO position paper. In The Weekly Epidemiological Record; WHO: Geneva, Switzerland, 2014; Volume 91, pp. 73–92. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immunization Strategy | Isotype | Log10 Ova-specific antibody titer by ELISA a | ||
|---|---|---|---|---|
| Day 42 | Day 180 | p | ||
| 1° NP-Ova I.N 2° Ova+CFA S.C. | Serum IgG1 | 5.66 ± 0.4 | 5.71 ± 0.5 | 0.8 |
| Serum IgG2c | 3.30 ± 0.7 | 4.30 ± 0.8 | 0.04 | |
| Intestinal IgA | 2.12 ± 1.3 | 2.15 ± 1.0 | 0.9 | |
| FRT IgA | 0.00 ± 0.0 | 0.00 ± 0.0 | 1.0 | |
| FRT IgG1 | 2.28 ± 0.5 | 3.41 ± 1.3 | 0.07 | |
| 1° NP-Ova I.N. 2° NP-Ova I.N. | Serum IgG1 | 5.21 ± 0.7 | 4.32 ± 0.9 | 0.08 |
| Serum IgG2c | 3.47 ± 0.5 | 1.00 ± 1.6 | 0.001 | |
| Intestinal IgA | 2.73 ± 1.3 | 2.87 ± 0.9 | 0.9 | |
| FRT IgA | 0.00 ± 0.0 | 0.00 ± 0.0 | 1.0 | |
| FRT IgG1 | 2.05 ± 0.5 | 1.90 ± 0.4 | 0.7 | |
| 1° Ova I.N. 2° Ova+CFA S.C | Serum IgG1 | 6.31 ± 0.0 | 5.73 ± 0.3 | 0.01 |
| Serum IgG2c | 4.67 ± 0.5 | 4.71 ± 0.5 | 0.9 | |
| Intestinal IgA | 3.04 ± 0.1 | 3.04 ± 0.4 | 1.0 | |
| FRT IgA | 2.43 ± 0.3 | 0.82 ± 1.2 | 0.03 | |
| FRT IgG1 | 3.10 ± 0.5 | 2.87 ± 1.2 | 0.7 | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Howe, S.E.; Sowa, G.; Konjufca, V. Systemic and Mucosal Antibody Responses to Soluble and Nanoparticle-Conjugated Antigens Administered Intranasally. Antibodies 2016, 5, 20. https://doi.org/10.3390/antib5040020
Howe SE, Sowa G, Konjufca V. Systemic and Mucosal Antibody Responses to Soluble and Nanoparticle-Conjugated Antigens Administered Intranasally. Antibodies. 2016; 5(4):20. https://doi.org/10.3390/antib5040020
Chicago/Turabian StyleHowe, Savannah E., Gavin Sowa, and Vjollca Konjufca. 2016. "Systemic and Mucosal Antibody Responses to Soluble and Nanoparticle-Conjugated Antigens Administered Intranasally" Antibodies 5, no. 4: 20. https://doi.org/10.3390/antib5040020