Enzyme-Based Labeling Strategies for Antibody–Drug Conjugates and Antibody Mimetics

Abstract
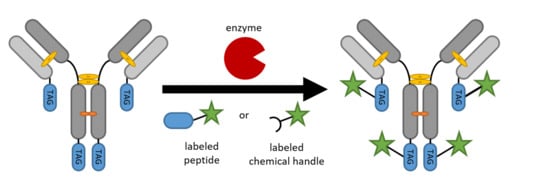
:
1. Introduction
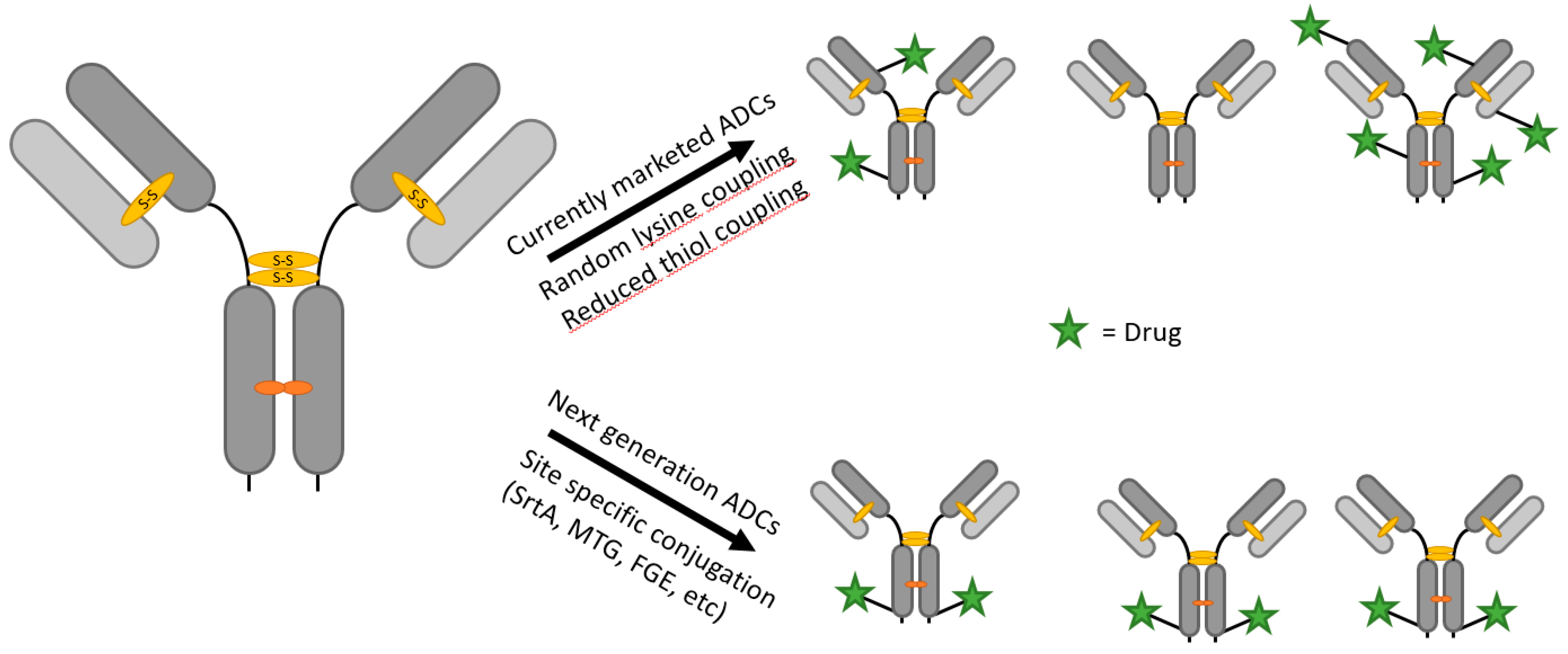
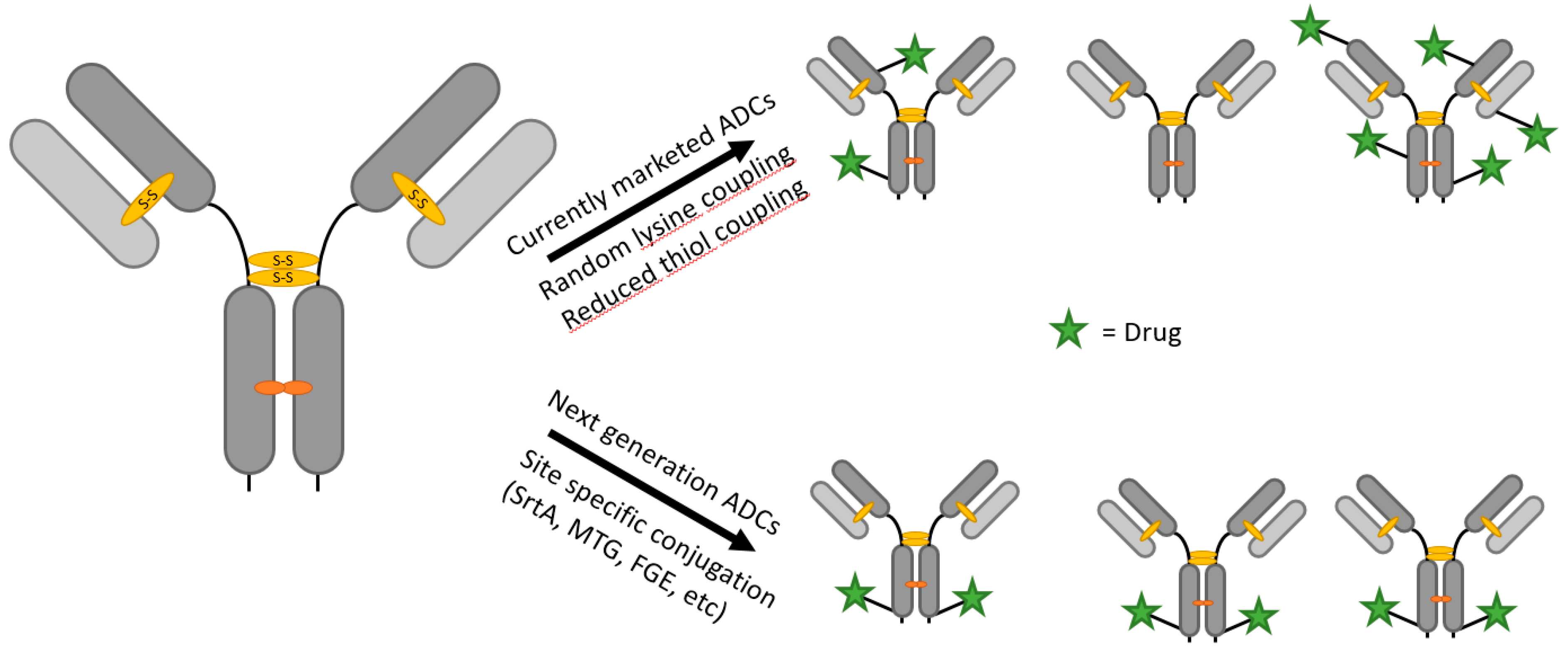
1.1. Antibody–Drug Conjugates
1.2. Antibody Fragments and Mimetics
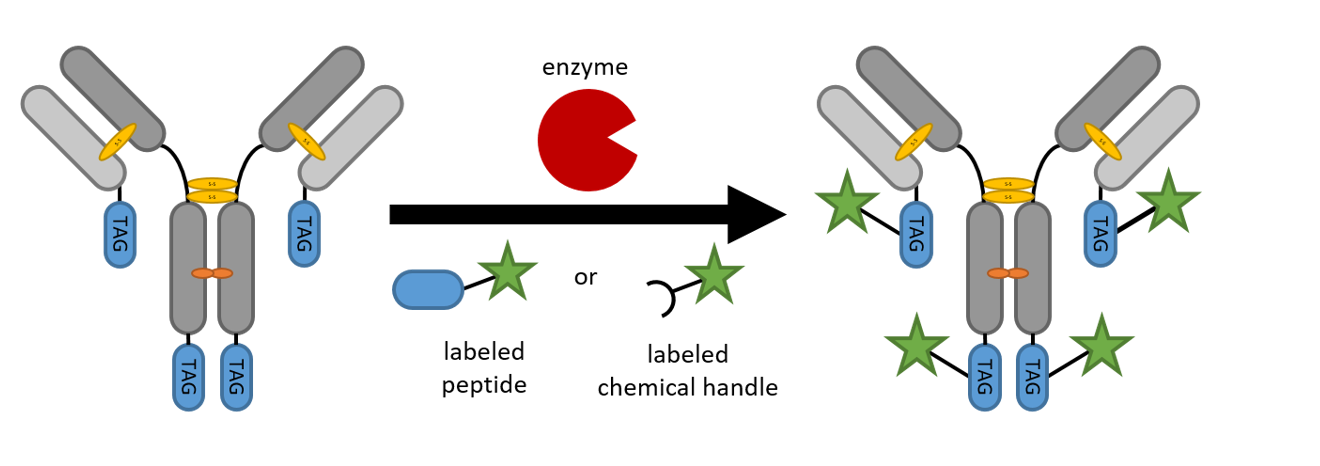
2. Site-Specific Protein Modification Strategies
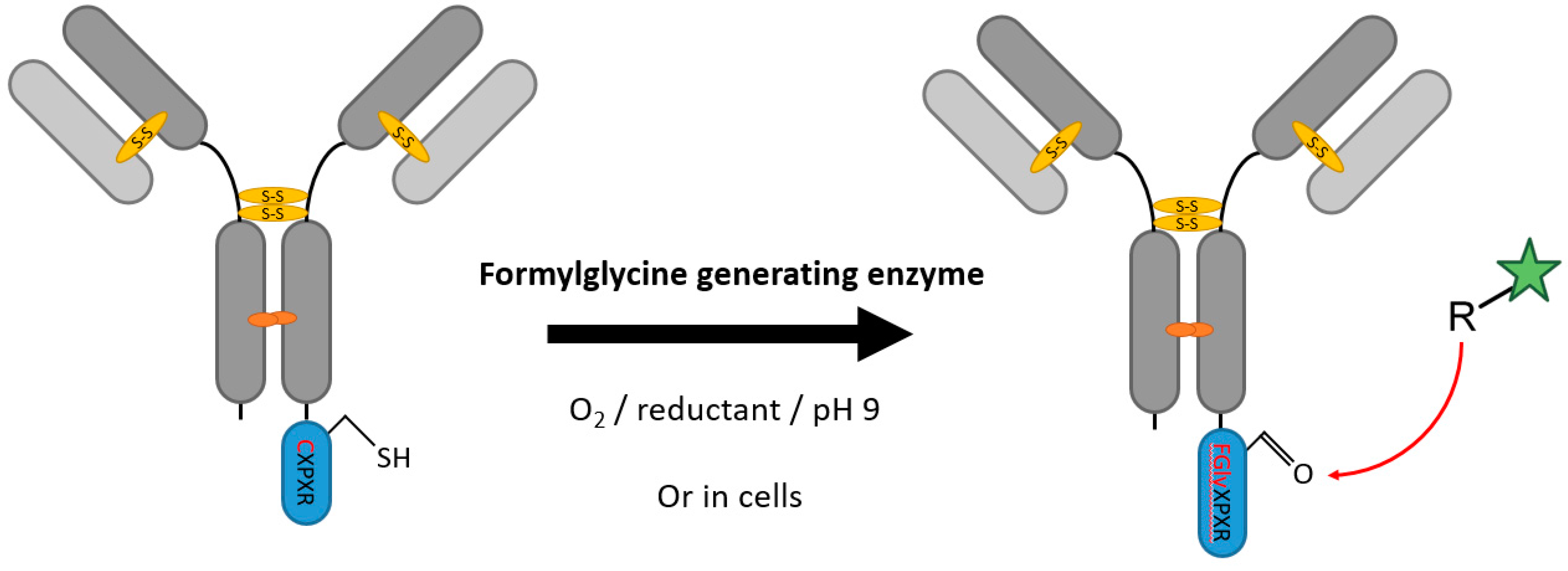
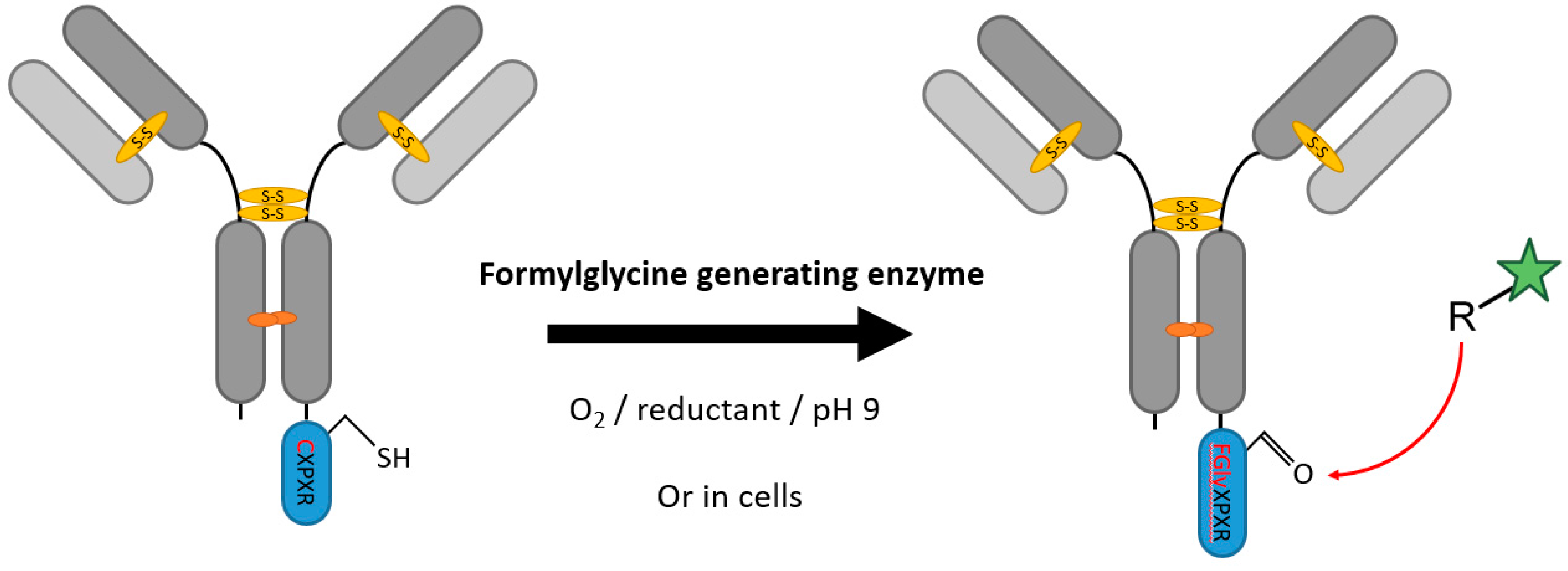
2.1. Formylglycine-Generating Enzymes
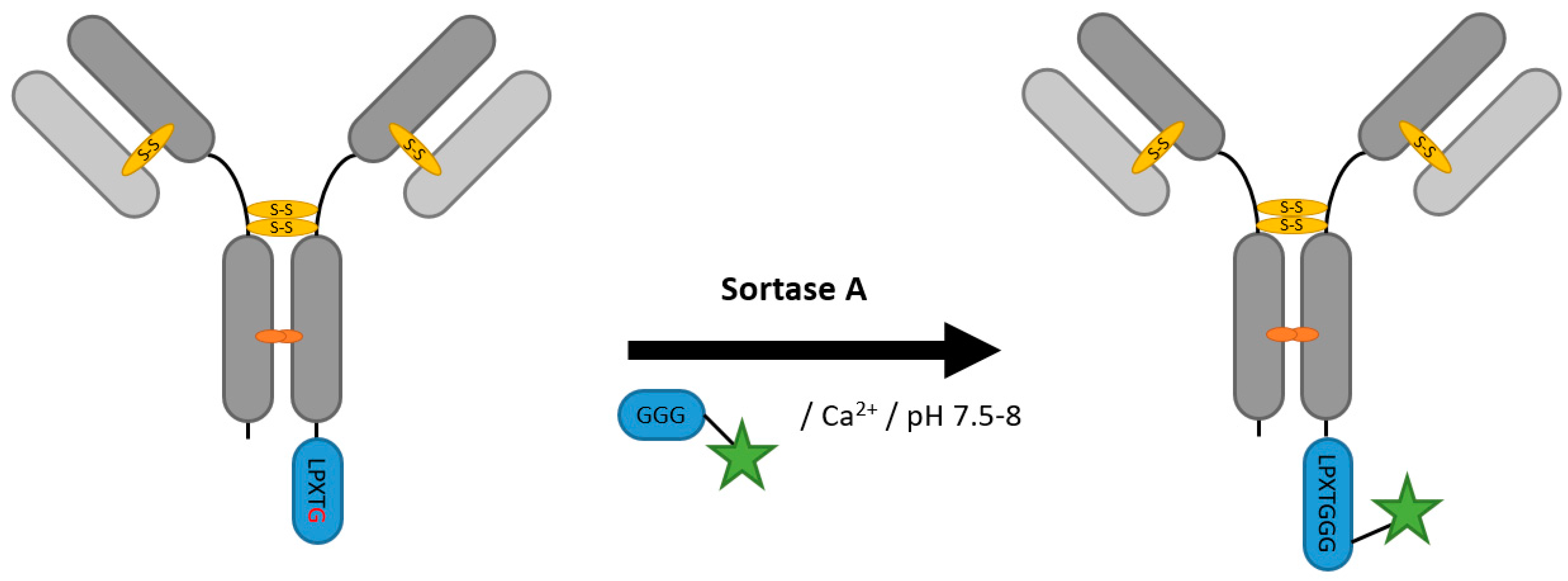
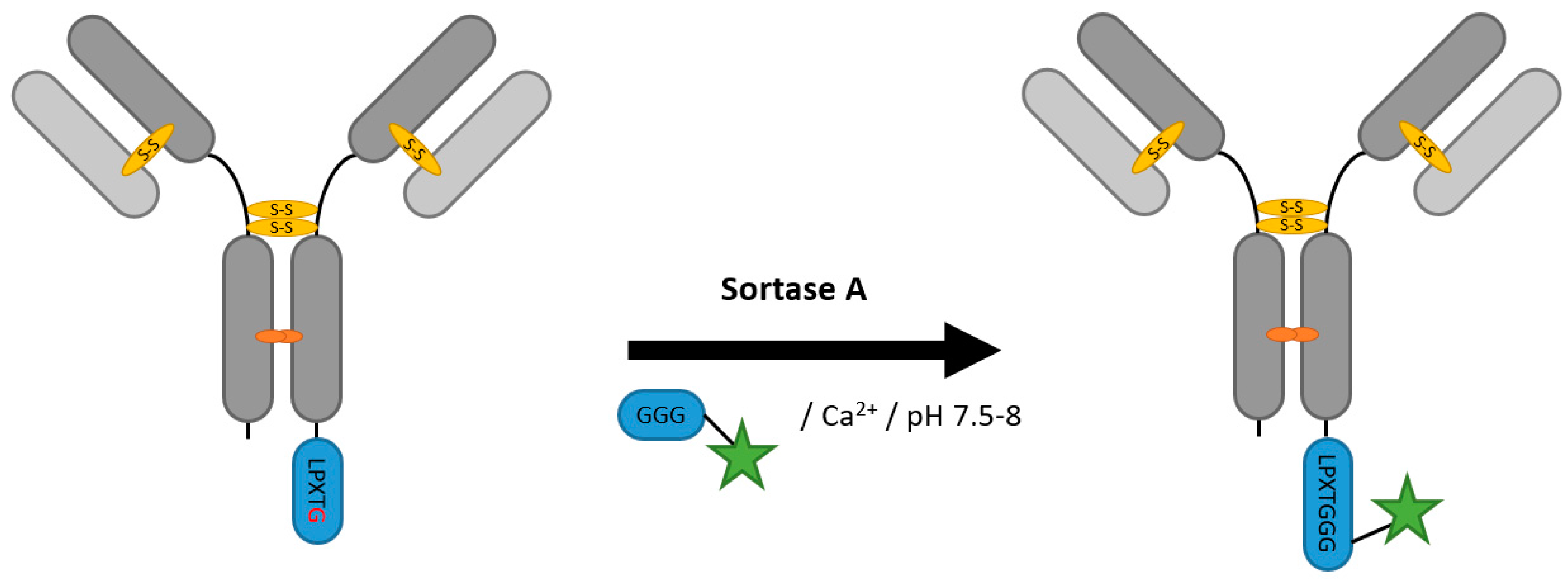
2.2. Sortases
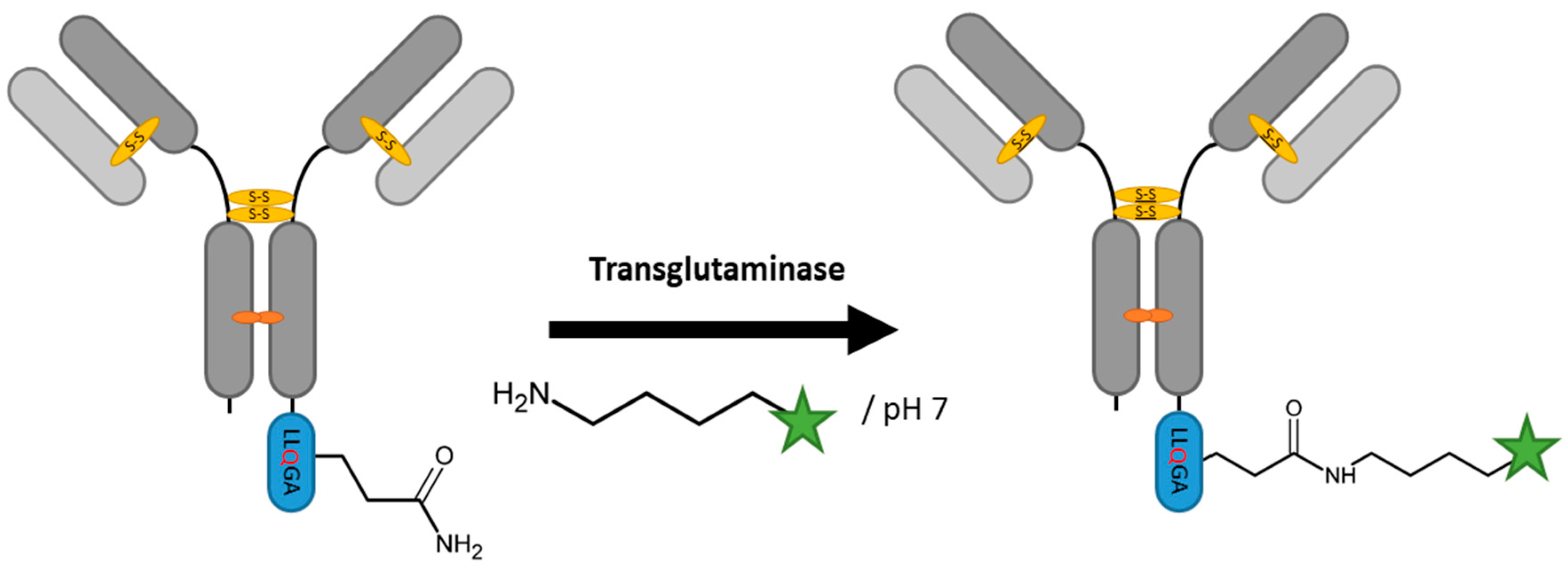
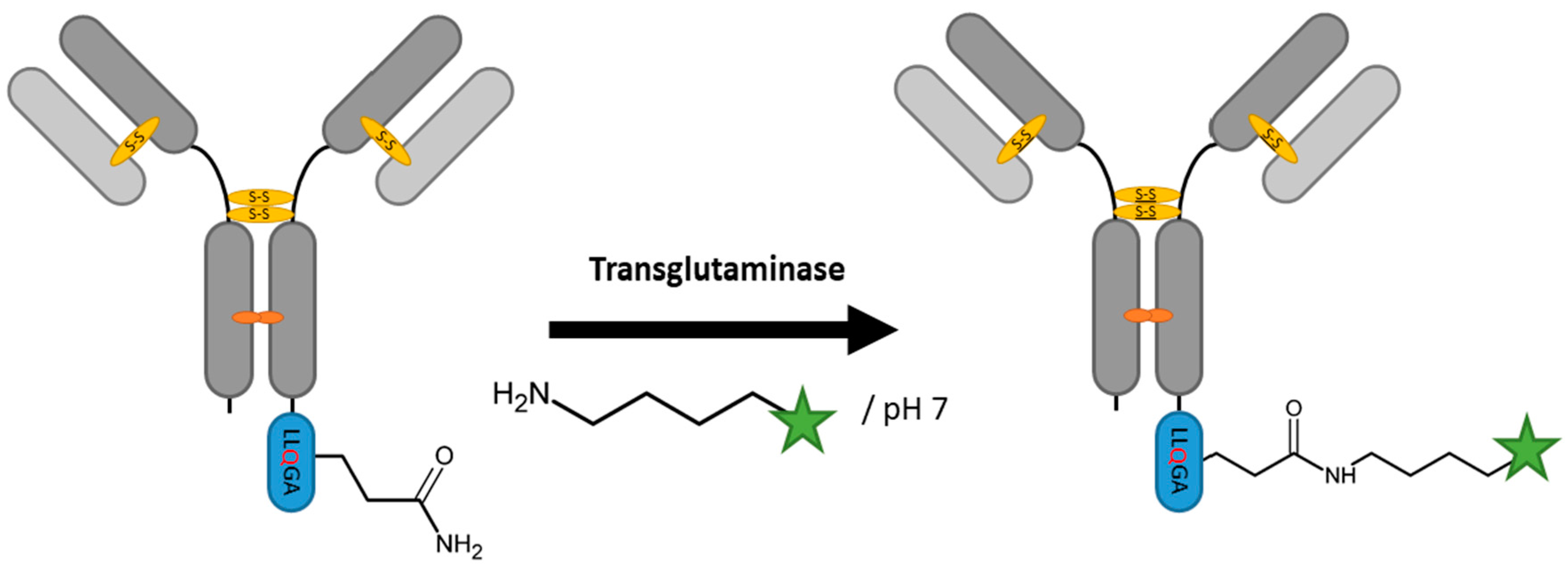
2.3. Transglutaminases
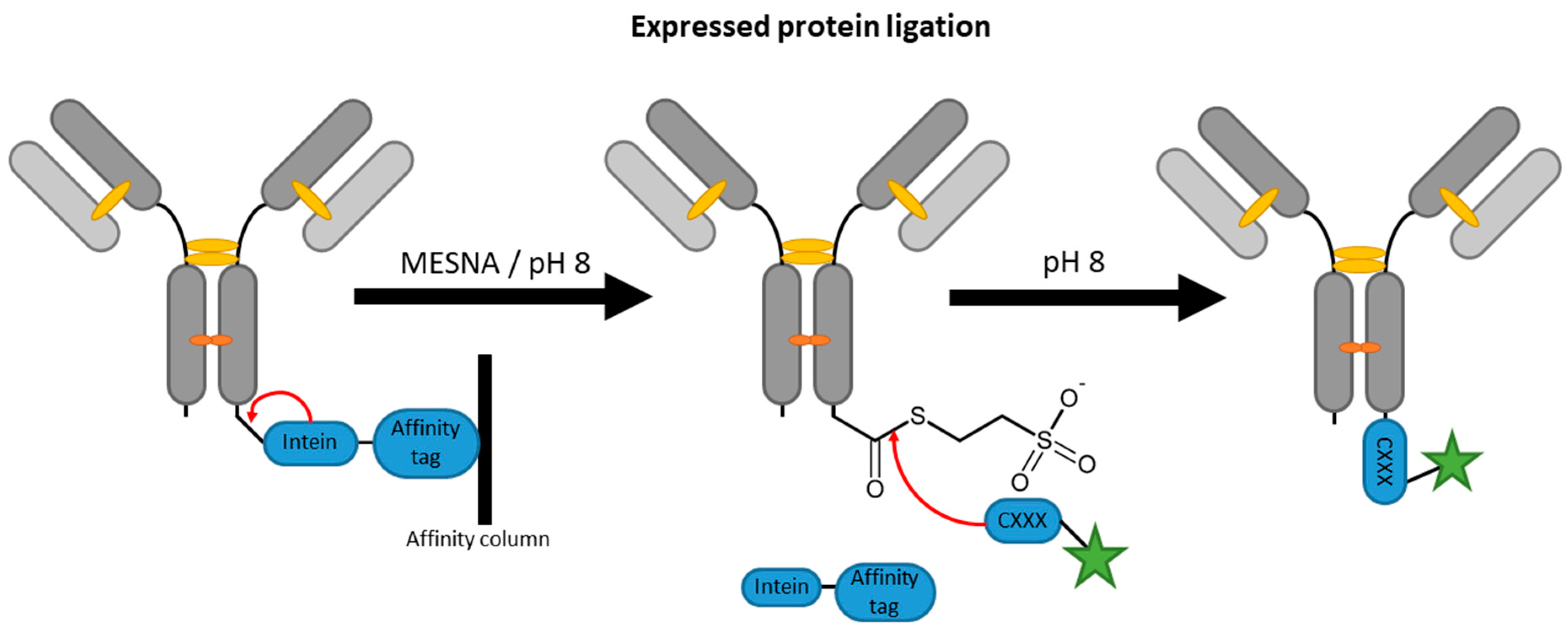
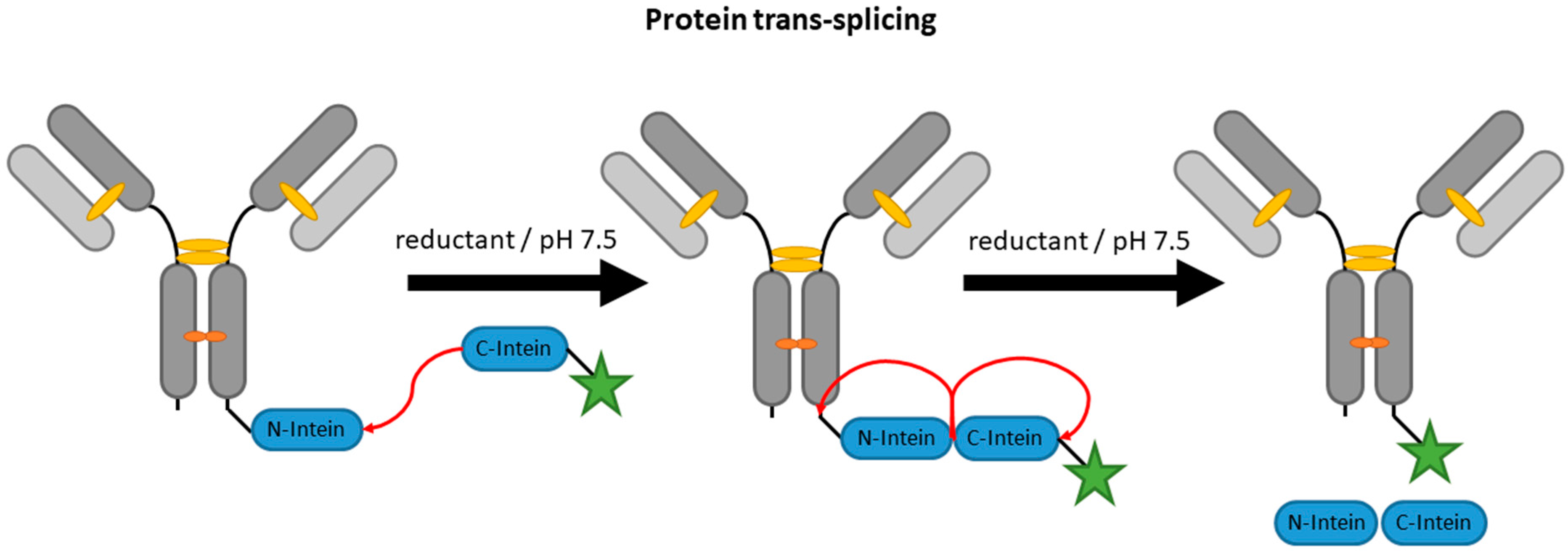
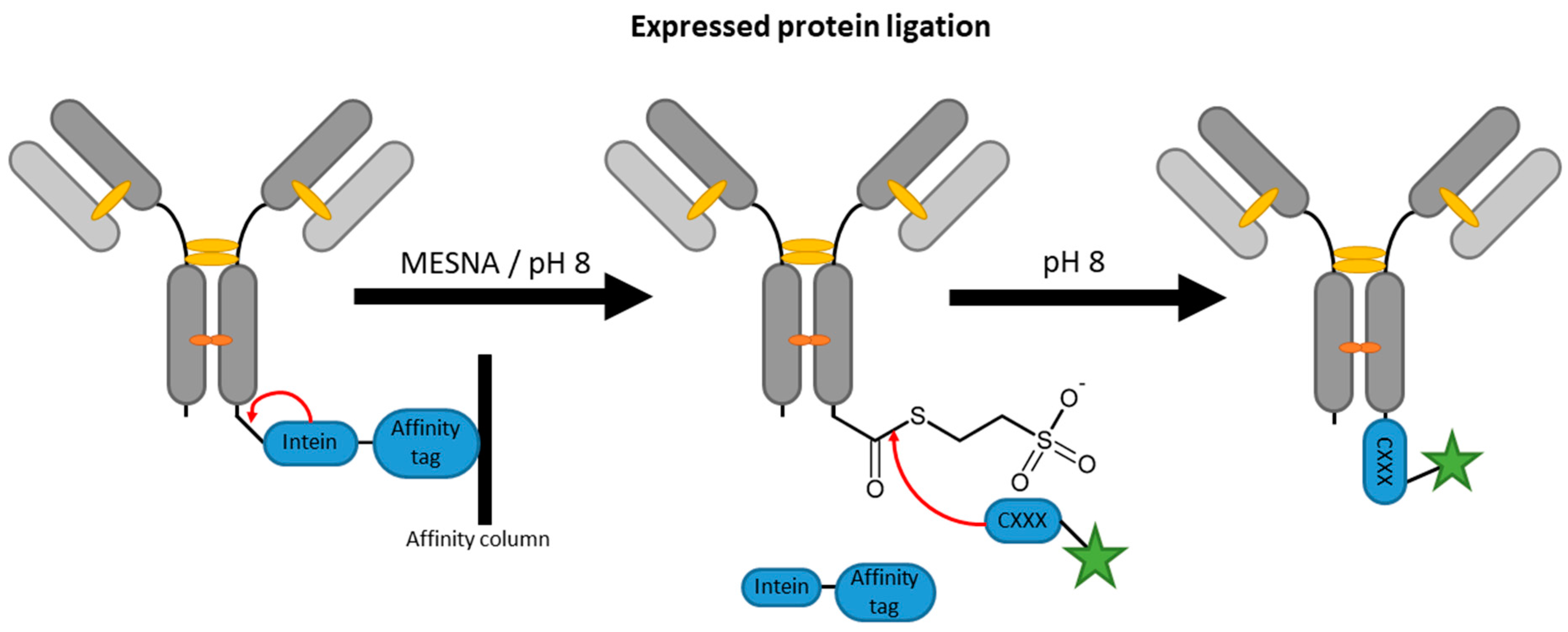
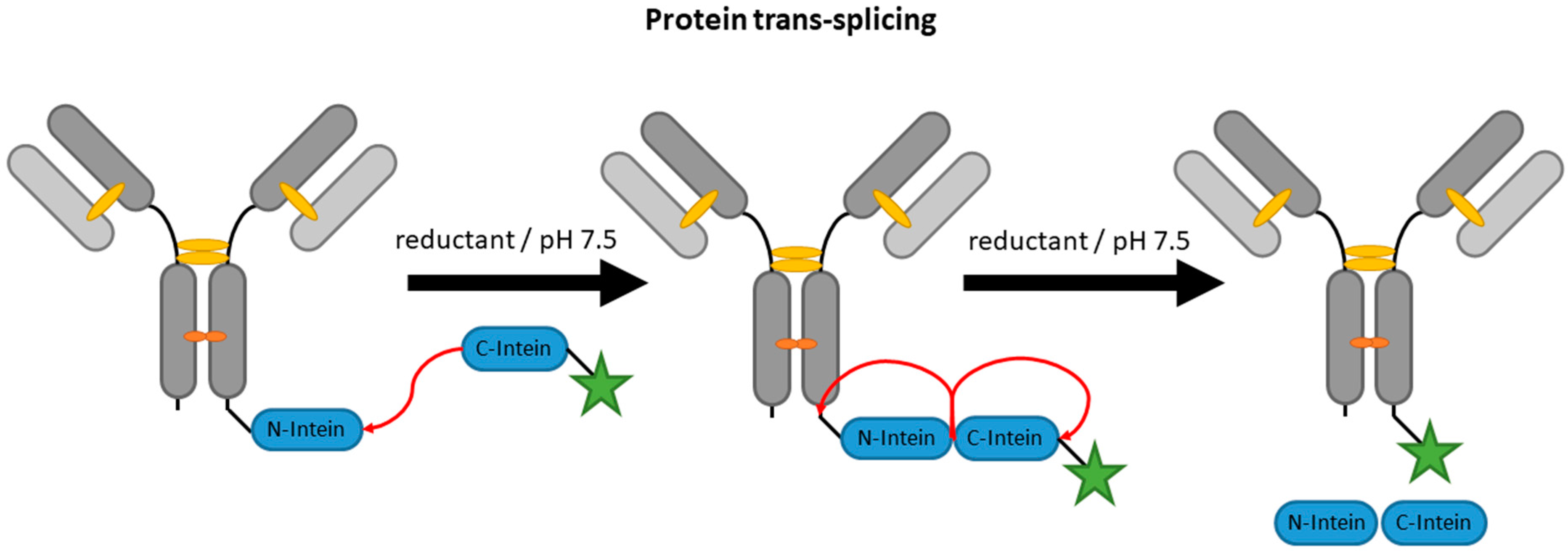
2.4. Inteins
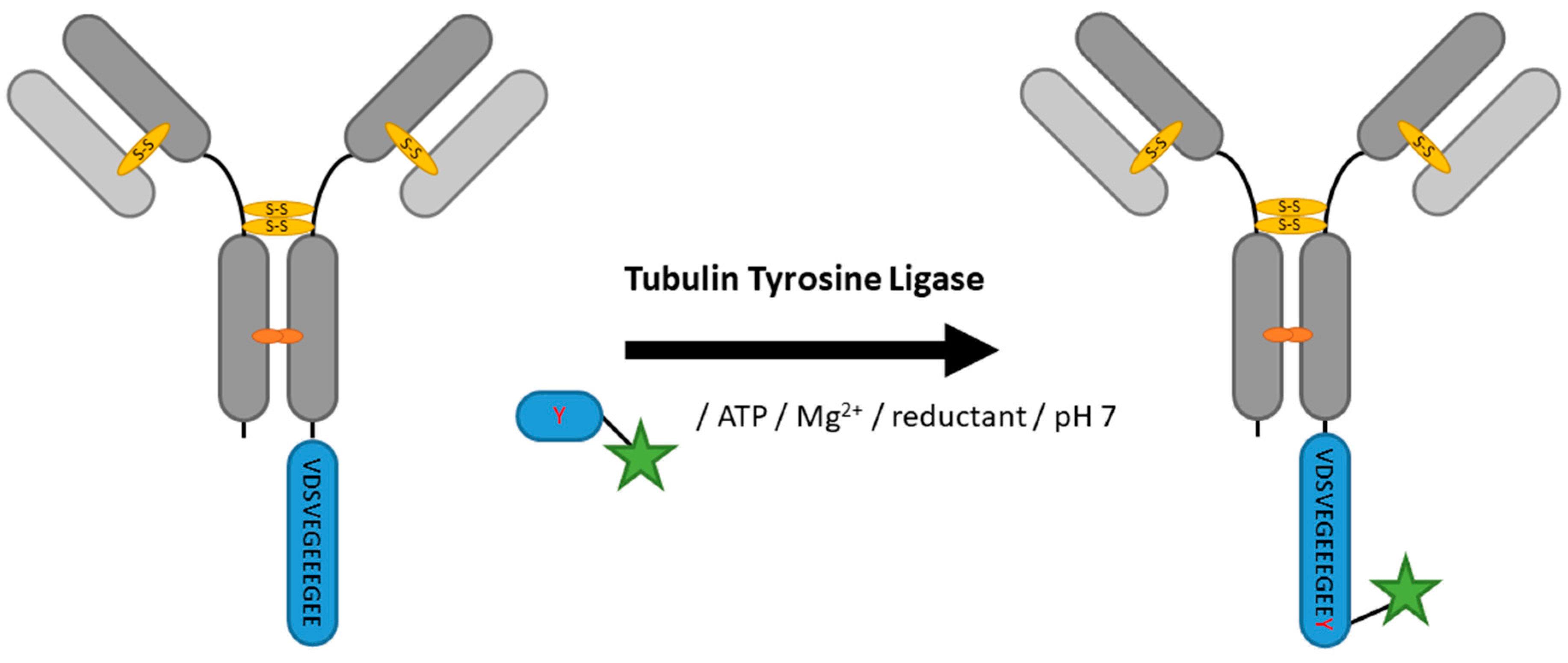
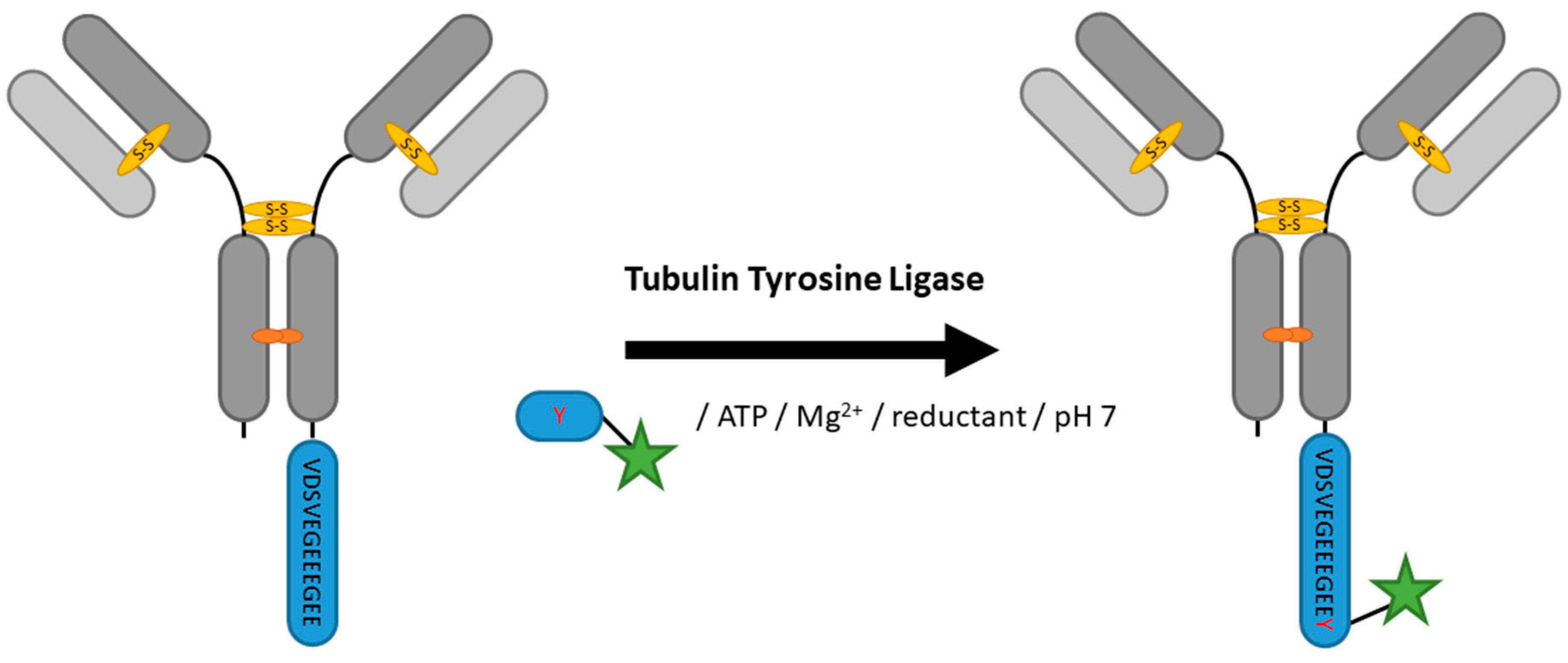
2.5. Tubulin Tyrosine Ligase
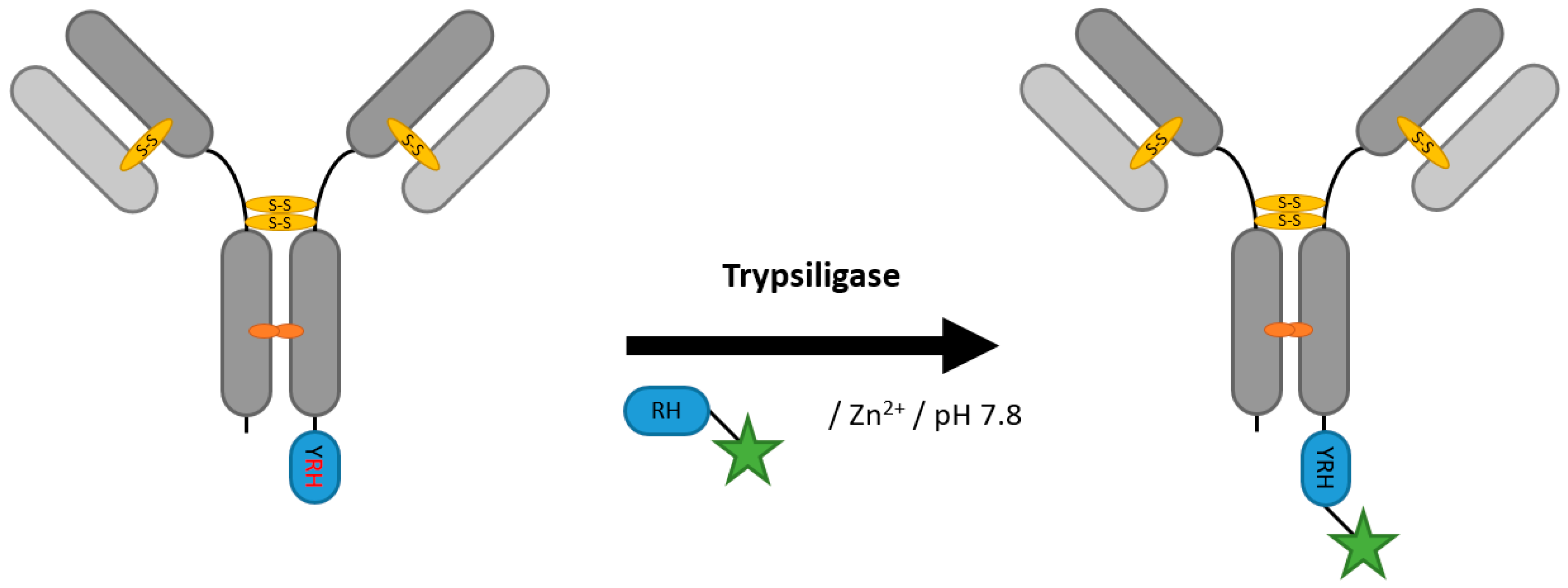
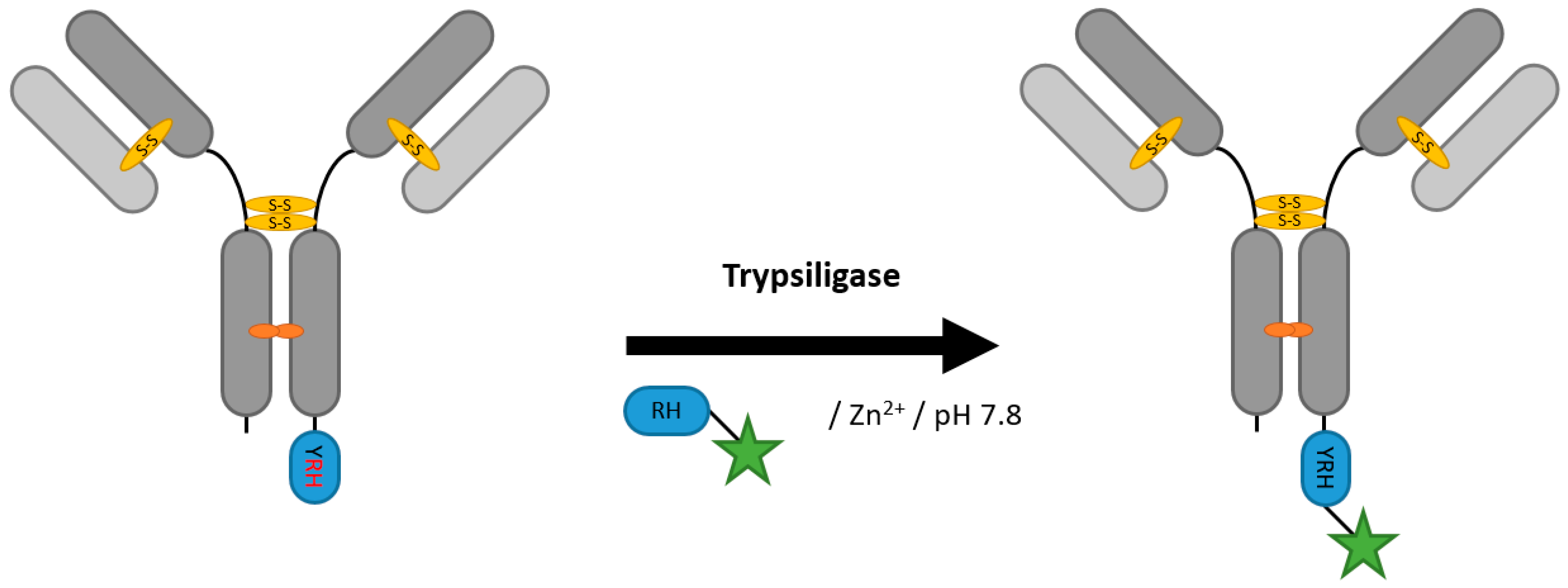
2.6. Proteases (Trypsiligase and Subtiligase)
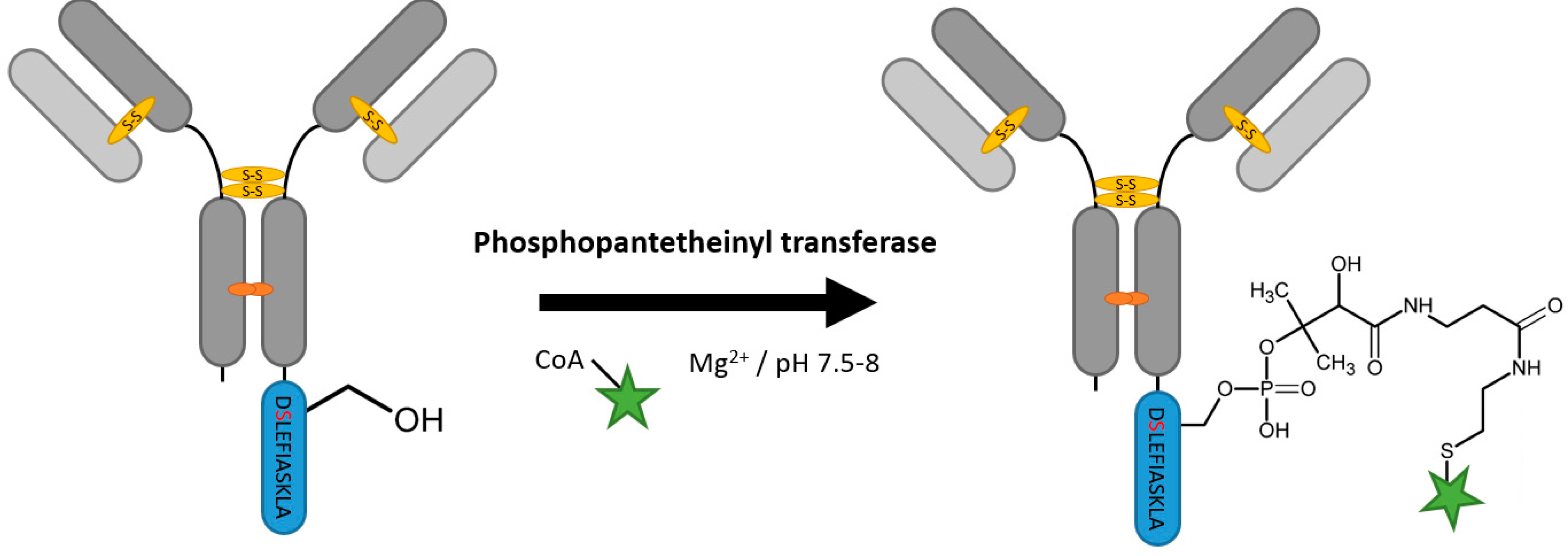
2.7. Phosphopantetheinyl Transferase
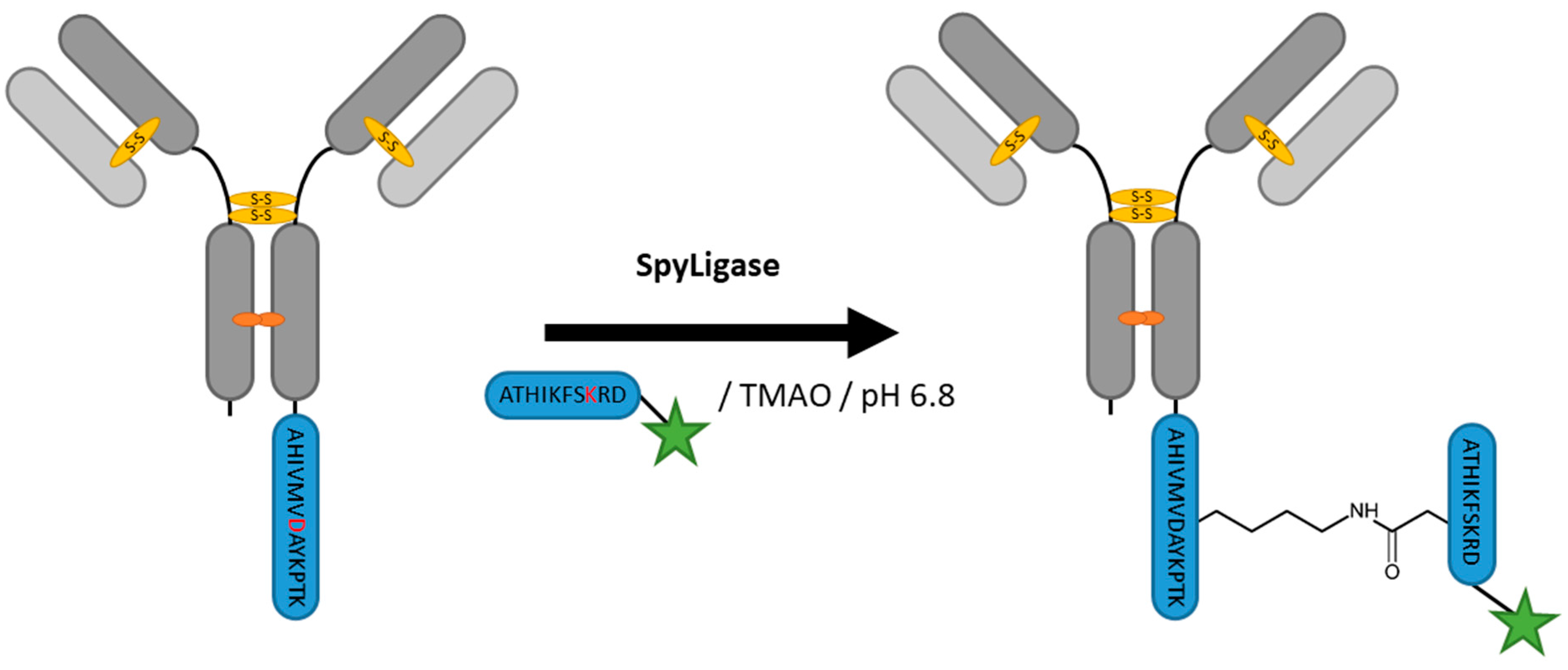
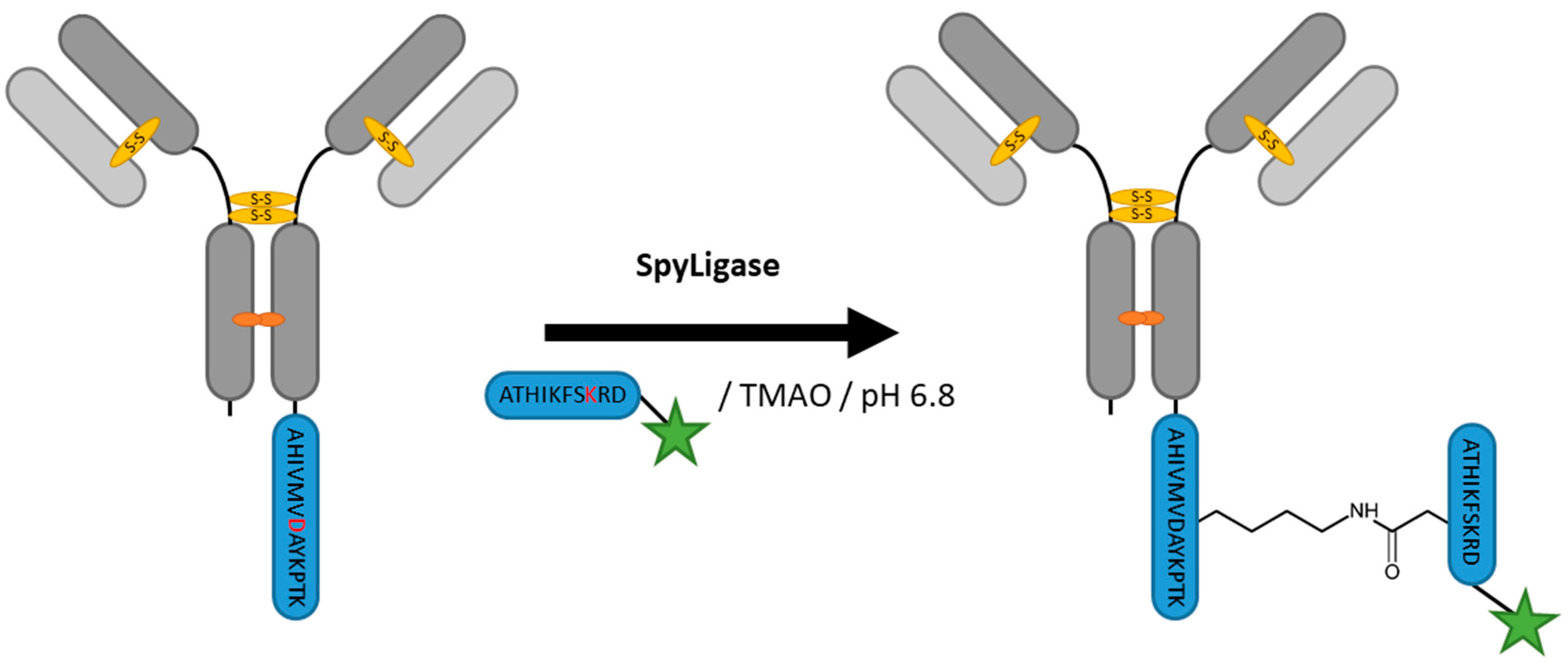
2.8. SpyLigase
2.9. Other Strategies
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. MAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Guided Missiles: Antibody–Drug Conjugates|BioPharma Dealmakers. Available online: https://biopharmadealmakers.nature.com/users/9880-biopharma-dealmakers/posts/19999-guided-missiles-antibody-drug-conjugates (accessed on 29 September 2017).
- Lambert, J.M.; Morris, C.Q. Antibody–Drug Conjugates (ADCs) for Personalized Treatment of Solid Tumors: A Review. Adv. Ther. 2014, 34, 1015–1035. [Google Scholar] [CrossRef] [PubMed]
- Junttila, T.T.; Li, G.; Parsons, K.; Phillips, G.L.; Sliwkowski, M.X. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res. Treat. 2011, 128, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Strop, P.; Liu, S.-H.; Dorywalska, M.; Delaria, K.; Dushin, R.G.; Tran, T.-T.; Ho, W.-H.; Farias, S.; Casas, M.G.; Abdiche, Y.; et al. Location Matters: Site of Conjugation Modulates Stability and Pharmacokinetics of Antibody Drug Conjugates. Chem. Biol. 2013, 20, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Sievers, E.L.; Senter, P.D. Antibody-Drug Conjugates in Cancer Therapy. Annu. Rev. Med. 2013, 64, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Reichert, J.M. Antibody-drug conjugates. MAbs 2014, 6, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Acchione, M.; Kwon, H.; Jochheim, C.M.; Atkins, W.M. Impact of linker and conjugation chemistry on antigen binding, Fc receptor binding and thermal stability of model antibody–drug conjugates. MAbs 2012, 4, 362–372. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, C.F.; Turcott, E.; Westendorf, L.; Webster, J.B.; Alley, S.C.; Kim, K.; Andreyka, J.; Stone, I.; Hamblett, K.J.; Francisco, J.A.; et al. Engineered antibody–drug conjugates with defined sites and stoichiometries of drug attachment. Protein Eng. Des. Sel. 2006, 19, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Flagella, K.M.; Graham, R.A.; Parsons, K.L.; Ha, E.; Raab, H.; Bhakta, S.; Nguyen, T.; Dugger, D.L.; Li, G.; et al. Engineered thio-trastuzumab-DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2-positive breast cancer. Clin. Cancer Res. 2010, 16, 4769–4778. [Google Scholar] [CrossRef] [PubMed]
- Pillow, T.H.; Tien, J.; Parsons-Reponte, K.L.; Bhakta, S.; Li, H.; Staben, L.R.; Li, G.; Chuh, J.; Fourie-O’donohue, A.; Darwish, M.; et al. Site-Specific Trastuzumab Maytansinoid Antibody−Drug Conjugates with Improved Therapeutic Activity through Linker and Antibody Engineering. J. Med. Chem. 2014, 57, 7890–7899. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Welborn, M.; Zhu, T.; Yang, N.J.; Santos, M.S.; Van Voorhis, T.; Pentelute, B.L. π-Clamp-mediated cysteine conjugation. Nat. Chem. 2016, 8, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Cortez-Retamozo, V.; Backmann, N.; Senter, P.D.; Wernery, U.; De Baetselier, P.; Muyldermans, S.; Revets, H. Efficient Cancer Therapy with a Nanobody-Based Conjugate. Cancer Res. 2004, 64, 2853–2857. [Google Scholar] [CrossRef] [PubMed]
- Vosjan, M.J.W.D.; Vercammen, J.; Kolkman, J.A.; Stigter-van Walsum, M.; Revets, H.; van Dongen, G.A.M.S. Nanobodies Targeting the Hepatocyte Growth Factor: Potential New Drugs for Molecular Cancer Therapy. Mol. Cancer Ther. 2012, 11, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Lyu, M.A.; Kurzrock, R.; Rosenblum, M.G. The immunocytokine scFv23/TNF targeting HER-2/neu induces synergistic cytotoxic effects with 5-fluorouracil in TNF-resistant pancreatic cancer cell lines. Biochem. Pharmacol. 2008, 75, 836–846. [Google Scholar] [CrossRef] [PubMed]
- Siegemund, M.; Seifert, O.; Zarani, M.; Džinić, T.; De Leo, V.; Göttsch, D.; Münkel, S.; Hutt, M.; Pfizenmaier, K.; Kontermann, R.E. An optimized antibody-single-chain TRAIL fusion protein for cancer therapy. MAbs 2016, 8, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.P.; Schier, R.; McCall, A.M.; Simmons, H.H.; Horak, E.M.; Alpaugh, R.K.; Marks, J.D.; Weiner, L.M. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001, 61, 4750–4755. [Google Scholar] [PubMed]
- Zahnd, C.; Kawe, M.; Stumpp, M.T.; de Pasquale, C.; Tamaskovic, R.; Nagy-Davidescu, G.; Dreier, B.; Schibli, R.; Binz, H.K.; Waibel, R.; et al. Efficient tumor targeting with high-affinity designed ankyrin repeat proteins: Effects of affinity and molecular size. Cancer Res. 2010, 70, 1595–1605. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-C.; Park, K.; Han, J.; Lee, J.; Kim, H.J.; Hong, S.; Heu, W.; Kim, Y.J.; Ha, J.-S.; Lee, S.-G.; et al. Design of a binding scaffold based on variable lymphocyte receptors of jawless vertebrates by module engineering. Proc. Natl. Acad. Sci. USA 2012, 109, 3299–3304. [Google Scholar] [CrossRef] [PubMed]
- Nord, K.; Gunneriusson, E.; Ringdahl, J.; Ståhl, S.; Uhlén, M.N.P. Binding proteins selected from combinatorial libraries of an alpha- helical bacterial receptor domain. Nat. Biotechnol. 1997, 15, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, K.; Covell, D.G.; Fletcher, J.E.; Weinstein, J.N. A modeling analysis of monoclonal antibody percolation through tumors: A binding-site barrier. J. Nucl. Med. 1990, 31, 1191–1198. [Google Scholar] [PubMed]
- Cortez-Retamozo, V.; Lauwereys, M.; Hassanzadeh Gh, G.; Gobert, M.; Conrath, K.; Muyldermans, S.; De Baetselier, P.; Revets, H. Efficient tumor targeting by single-domain antibody fragments of camels. Int. J. Cancer 2002, 98, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Baxter, L.T. Mechanisms of Heterogeneous Distribution of Monoclonal Antibodies and Other Macromolecules in Tumors: Significance of Elevated Interstitial Pressure. Cancer Res. 1988, 48, 7022–7032. [Google Scholar] [PubMed]
- Dierks, T.; Schmidt, B.; von Figura, K. Conversion of cysteine to formylglycine: A protein modification in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 1997, 94, 11963–11968. [Google Scholar] [CrossRef] [PubMed]
- Dierks, T.; Lecca, M.R.; Schmidt, B.; Von Figura, K. Conversion of cysteine to formylglycine in eukaryotic sulfatases occurs by a common mechanism in the endoplasmic reticulum. FEBS Lett. 1998, 423, 61–65. [Google Scholar] [CrossRef]
- Selmer, T.; Hallmann, A.; Schmidt, B.; Sumper, M.; Figura, K. The Evolutionary Conservation of a Novel Protein Modification, the Conversion of Cysteine to Serinesemialdehyde in Arylsulfatase from Volvox carteri. Eur. J. Biochem. 1996, 238, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Dierks, T.; Lecca, M.R.; Schlotterhose, P.; Schmidt, B.; von Figura, K. Sequence determinants directing conversion of cysteine to formylglycine in eukaryotic sulfatases. EMBO J. 1999, 18, 2084–2091. [Google Scholar] [CrossRef] [PubMed]
- Dierks, T.; Schmidt, B.; Borissenko, L.V.; Peng, J.; Preusser, A.; Mariappan, M.; von Figura, K. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell 2003, 113, 435–444. [Google Scholar] [CrossRef]
- Cosma, M.P.; Pepe, S.; Annunziata, I.; Newbold, R.F.; Grompe, M.; Parenti, G.; Ballabio, A. The Multiple Sulfatase Deficiency Gene Encodes an Essential and Limiting Factor for the Activity of Sulfatases the turnover and degradation of sulfated compounds, mostly complex molecules that are hydrolyzed in lyso- somes in concert with acidic glycosida. Cell 2003, 113, 445–456. [Google Scholar] [CrossRef]
- Preusser-Kunze, A.; Mariappan, M.; Schmidt, B.; Gande, S.L.; Mutenda, K.; Wenzel, D.; von Figura, K.; Dierks, T. Molecular characterization of the human Calpha-formylglycine-generating enzyme. J. Biol. Chem. 2005, 280, 14900–14910. [Google Scholar] [CrossRef] [PubMed]
- Berteau, O.; Guillot, A.; Benjdia, A.; Rabot, S. A new type of bacterial sulfatase reveals a novel maturation pathway in prokaryotes. J. Biol. Chem. 2006, 281, 22464–22470. [Google Scholar] [CrossRef] [PubMed]
- Rush, J.S.; Bertozzi, C.R. New aldehyde tag sequences identified by screening formylglycine generating enzymes in vitro and in vivo. J. Am. Chem. Soc. 2008, 130, 12240–12241. [Google Scholar] [CrossRef] [PubMed]
- Carrico, I.S.; Carlson, B.L.; Bertozzi, C.R. Introducing genetically encoded aldehydes into proteins. Nat. Chem. Biol. 2007, 3, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Shui, W.; Carlson, B.L.; Hu, N.; Rabuka, D.; Lee, J.; Bertozzi, C.R. Site-specific chemical modification of recombinant proteins produced in mammalian cells by using the genetically encoded aldehyde tag. Proc. Natl. Acad. Sci. USA 2009, 106, 3000–3005. [Google Scholar] [CrossRef] [PubMed]
- Rabuka, D.; Rush, J.S.; deHart, G.W.; Wu, P.; Bertozzi, C.R. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat. Protoc. 2012, 7, 1052–1067. [Google Scholar] [CrossRef] [PubMed]
- Drake, P.M.; Albers, A.E.; Baker, J.; Banas, S.; Barfield, R.M.; Bhat, A.S.; De Hart, G.W.; Garofalo, A.W.; Holder, P.; Jones, L.C.; et al. Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconjug. Chem. 2014, 25, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- York, D.; Baker, J.; Holder, P.G.; Jones, L.C.; Drake, P.M.; Barfield, R.M.; Bleck, G.T.; Rabuka, D. Generating aldehyde-tagged antibodies with high titers and high formylglycine yields by supplementing culture media with copper(II). BMC Biotechnol. 2016, 16, 23. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Alam, S.; Radhakrishnan, K.; Mariappan, M.; Rudolph, M.G.; May, C.; Dierks, T.; Von Figura, K.; Schmidt, B. Eukaryotic formylglycine-generating enzyme catalyses a monooxygenase type of reaction. FEBS J. 2015, 282, 3262–3274. [Google Scholar] [CrossRef] [PubMed]
- Roeser, D.; Preusser-Kunze, A.; Schmidt, B.; Gasow, K.; Wittmann, J.G.; Dierks, T.; von Figura, K.; Rudolph, M.G. A general binding mechanism for all human sulfatases by the formylglycine-generating enzyme. Proc. Natl. Acad. Sci. USA 2006, 103, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Knop, M.; Engi, P.; Lemnaru, R.; Seebeck, F.P. In Vitro Reconstitution of Formylglycine-Generating Enzymes Requires Copper(I). ChemBioChem 2015, 16, 2147–2150. [Google Scholar] [CrossRef] [PubMed]
- Holder, P.G.; Jones, L.C.; Drake, P.M.; Barfield, R.M.; Bañas, S.; de Hart, G.W.; Baker, J.; Rabuka, D. Reconstitution of Formylglycine-generating Enzyme with Copper(II) for Aldehyde Tag Conversion. J. Biol. Chem. 2015, 290, 15730–15745. [Google Scholar] [CrossRef] [PubMed]
- Knop, M.; Dang, T.Q.; Jeschke, G.; Seebeck, F.P. Copper is a Cofactor of the Formylglycine-Generating Enzyme. ChemBioChem 2017, 18, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.L.; Ballister, E.R.; Skordalakes, E.; King, D.S.; Breidenbach, M.A.; Gilmore, S.A.; Berger, J.M.; Bertozzi, C.R. Function and structure of a prokaryotic formylglycine-generating enzyme. J. Biol. Chem. 2008, 283, 20117–20125. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Liu, G.; Ton-That, H.; Schneewind, O. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science 1999, 285, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Ton-That, H.; Schneewind, O. Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus. Mol. Microbiol. 2001, 40, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Aulabaugh, A.; Ding, W.; Kapoor, B.; Alksne, L.; Tabei, K.; Ellestad, G. Kinetic mechanism of Staphylococcus aureus sortase SrtA. Biochemistry 2003, 42, 11307–11315. [Google Scholar] [CrossRef] [PubMed]
- Frankel, B.A.; Kruger, R.G.; Robinson, D.E.; Kelleher, N.L.; McCafferty, D.G. Staphylococcus aureus sortase transpeptidase SrtA: Insight into the kinetic mechanism and evidence for a reverse protonation catalytic mechanism. Biochemistry 2005, 44, 11188–11200. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Hart, S.A.; Schink, A.; Pollok, B.A. Sortase-Mediated Protein Ligation: A New Method for Protein Engineering. J. Am. Chem. Soc. 2004, 126, 2670–2671. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, R.; Subramanian, S.; Boder, E.T. Sortase a as a novel molecular “stapler” for sequence-specific protein conjugation. Bioconjug. Chem. 2007, 18, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Popp, M.W.; Antos, J.M.; Grotenbreg, G.M.; Spooner, E.; Ploegh, H.L. Sortagging: A versatile method for protein labeling. Nat. Chem. Biol. 2007, 3, 707–708. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Nagamune, T. Expansion of the sortase-mediated labeling method for site-specific N-terminal labeling of cell surface proteins on living cells. Chem. Commun. 2009, 7, 1022–1024. [Google Scholar] [CrossRef] [PubMed]
- Antos, J.M.; Chew, G.L.; Guimaraes, C.P.; Yoder, N.C.; Grotenbreg, G.M.; Popp, M.W.L.; Ploegh, H.L. Site-specific N- and C-terminal labeling of a single polypeptide using sortases of different specificity. J. Am. Chem. Soc. 2009, 131, 10800–10801. [Google Scholar] [CrossRef] [PubMed]
- Möhlmann, S.; Mahlert, C.; Greven, S.; Scholz, P.; Harrenga, A. In vitro Sortagging of an Antibody Fab Fragment: Overcoming Unproductive Reactions of Sortase with Water and Lysine Side Chains. ChemBioChem 2011, 12, 1774–1780. [Google Scholar] [CrossRef] [PubMed]
- Ta, D.T.; Redeker, E.S.; Guedens, W.; Adriaensens, P. Sortase A-mediated functionalization of nanobodies toward surface coupling. Biochem. Protein Sci. Vet. Immunol. Immunop. Biotechnol. Lett. 2004, 43, 1541–1551. [Google Scholar]
- Madej, M.P.; Coia, G.; Williams, C.C.; Caine, J.M.; Pearce, L.A.; Attwood, R.; Bartone, N.A.; Dolezal, O.; Nisbet, R.M.; Nuttall, S.D.; et al. Engineering of an anti-epidermal growth factor receptor antibody to single chain format and labeling by sortase A-mediated protein ligation. Biotechnol. Bioeng. 2012, 109, 1461–1470. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Theile, C.S.; Chen, G.-Y.; Bilate, A.M.; Duarte, J.N.; Avalos, A.M.; Fang, T.; Barberena, R.; Sato, S.; Ploegh, H.L. Fluorophore-Conjugated Holliday Junctions for Generating Super-Bright Antibodies and Antibody Fragments. Angew. Chem. Int. Ed. 2015, 54, 11706–11710. [Google Scholar] [CrossRef] [PubMed]
- Paterson, B.M.; Alt, K.; Jeffery, C.M.; Price, R.I.; Jagdale, S.; Rigby, S.; Williams, C.C.; Peter, K.; Hagemeyer, C.E.; Donnelly, P.S. Enzyme-mediated site-specific bioconjugation of metal complexes to proteins: Sortase-mediated coupling of copper-64 to a single-chain antibody. Angew. Chem. Int. Ed. 2014, 53, 6115–6119. [Google Scholar] [CrossRef] [PubMed]
- Rashidian, M.; Keliher, E.J.; Dougan, M.; Juras, P.K.; Cavallari, M.; Wojtkiewicz, G.R.; Jacobsen, J.T.; Edens, J.G.; Tas, J.M.J.; Victora, G.; et al. Use of 18F-2-fluorodeoxyglucose to label antibody fragments for immuno-positron emission tomography of pancreatic cancer. ACS Cent. Sci. 2015, 1, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Kornberger, P.; Skerra, A. Sortase-catalyzed in vitro functionalization of a HER2-specific recombinant Fab for tumor targeting of the plant cytotoxin gelonin. MAbs 2014, 6, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.; Duarte, J.N.; Ling, J.; Li, Z.; Guzman, J.S.; Ploegh, H.L. Structurally Defined αMHC-II Nanobody-Drug Conjugates: A Therapeutic and Imaging System for B-Cell Lymphoma. Angew. Chem. Int. Ed. 2016, 55, 2416–2420. [Google Scholar] [CrossRef] [PubMed]
- Massa, S.; Vikani, N.; Betti, C.; Ballet, S.; Vanderhaegen, S.; Steyaert, J.; Descamps, B.; Vanhove, C.; Bunschoten, A.; van Leeuwen, F.W.B.; et al. Sortase A-mediated site-specific labeling of camelid single-domain antibody-fragments: A versatile strategy for multiple molecular imaging modalities. Contrast Media Mol. Imaging 2016, 11, 328–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerli, R.R.; Hell, T.; Merkel, A.S.; Grawunder, U. Sortase Enzyme-Mediated Generation of Site-Specifically Conjugated Antibody Drug Conjugates with High In Vitro and In Vivo Potency. PLoS ONE 2015, 10, e0131177. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.; Kwakkenbos, M.J.; Claassen, Y.B.; Maijoor, K.; Böhne, M.; van der Sluijs, K.F.; Witte, M.D.; van Zoelen, D.J.; Cornelissen, L.A.; Beaumont, T.; et al. Bispecific antibody generated with sortase and click chemistry has broad antiinfluenza virus activity. Proc. Natl. Acad. Sci. USA 2014, 111, 16820–16825. [Google Scholar] [CrossRef] [PubMed]
- Piotukh, K.; Geltinger, B.; Heinrich, N.; Gerth, F.; Beyermann, M.; Freund, C.; Schwarzer, D. Directed evolution of sortase a mutants with altered substrate selectivity profiles. J. Am. Chem. Soc. 2011, 133, 17536–17539. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.J.; Webb, M.E.; Turnbull, W.B. Depsipeptide substrates for sortase-mediated N-terminal protein ligation. Nat. Protoc. 2014, 9, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.J.; Fascione, M.A.; Webb, M.E.; Turnbull, W.B. Efficient N-terminal labeling of proteins by use of sortase. Angew. Chem. Int. Ed. 2012, 51, 9377–9380. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, C.S.; Birckbichler, P.J.; Rice, R.H. Transglutaminases: Multifunctional cross-linking enzymes that stabilize tissues. FASEB J. 1991, 5, 3071–3077. [Google Scholar] [PubMed]
- Motoki, M.; Seguro, K. Transglutaminase and its use for food processing. Trends Food Sci. Technol. 1998, 9, 204–210. [Google Scholar] [CrossRef]
- Lorand, L.; Parameswaran, K.N.; Stenberg, P.; Tong, Y.S.; Velasco, P.T.; Jonsson, N.; Mikiver, L.; Moses, P. Specificity of Guinea Pig Liver Transglutaminase for Amine Substrates. Biochemistry 1979, 18, 1756–1765. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, T.; Umezawa, Y.; Nio, N.; Kubota, K. Comparison of Deamidation Activity of Transglutaminases. J. Food Sci. 2001, 66, 25–29. [Google Scholar] [CrossRef]
- Sugimura, Y.; Yokoyama, K.; Nio, N.; Maki, M.; Hitomi, K. Identification of preferred substrate sequences of microbial transglutaminase from Streptomyces mobaraensis using a phage-displayed peptide library. Arch. Biochem. Biophys. 2008, 477, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Ting, A.Y. Transglutaminase-catalyzed site-specific conjugation of small-molecule probes to proteins in vitro and on the surface of living cells. J. Am. Chem. Soc. 2006, 128, 4542–4543. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Hayashi, E.; Yamada, N.; Yatagai, M.; Takahara, Y. Further Studies on the Site-Specific Protein Modification by Microbial Transglutaminase. Bioconjug. Chem. 2001, 12, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Farias, S.E.; Strop, P.; Delaria, K.; Galindo Casas, M.; Dorywalska, M.; Shelton, D.L.; Pons, J.; Rajpal, A. Mass spectrometric characterization of transglutaminase based site-specific antibody–drug conjugates. Bioconjug. Chem. 2014, 25, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, V.; Schmelz, S.; Dickgiesser, S.; Beck, J.; Ebenig, A.; Fittler, H.; Frauendorf, H.; Piater, B.; Betz, U.A.K.; Avrutina, O.; et al. Locked by Design: A Conformationally Constrained Transglutaminase Tag Enables Efficient Site-Specific Conjugation. Angew. Chem. Int. Ed. 2015, 54, 13420–13424. [Google Scholar] [CrossRef] [PubMed]
- Josten, A.; Haalck, L.; Spener, F.; Meusel, M. Use of microbial transglutaminase for the enzymatic biotinylation of antibodies. J. Immunol. Methods 2000, 240, 47–54. [Google Scholar] [CrossRef]
- Dorywalska, M.; Strop, P.; Melton-Witt, J.A.; Hasa-Moreno, A.; Farias, S.E.; Galindo Casas, M.; Delaria, K.; Lui, V.; Poulsen, K.; Loo, C.; et al. Effect of attachment site on stability of cleavable antibody drug conjugates. Bioconjug. Chem. 2015, 26, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Lhospice, F.; Brégeon, D.; Belmant, C.; Dennler, P.; Chiotellis, A.; Fischer, E.; Gauthier, L.; Boëdec, A.; Rispaud, H.; Savard-Chambard, S.; et al. Site-Specific Conjugation of Monomethyl Auristatin E to Anti-CD30 Antibodies Improves Their Pharmacokinetics and Therapeutic Index in Rodent Models. Mol. Pharm. 2015, 12, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Puthenveetil, S.; Musto, S.; Loganzo, F.; Tumey, L.N.; O’Donnell, C.J.; Graziani, E. Development of Solid-Phase Site-Specific Conjugation and Its Application toward Generation of Dual Labeled Antibody and Fab Drug Conjugates. Bioconjug. Chem. 2016, 27, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Q.; Perler, F.B. The mechanism of protein splicing and its modulation by mutation. EMBO J. 1996, 15, 5146–5153. [Google Scholar] [PubMed]
- Muir, T.W.; Sondhi, D.; Cole, P.A. Expressed protein ligation: A general method for protein engineering. Proc. Natl. Acad. Sci. USA 1998, 95, 6705–6710. [Google Scholar] [CrossRef] [PubMed]
- Wood, R.J.; Pascoe, D.D.; Brown, Z.K.; Medlicott, E.M.; Kriek, M.; Neylon, C.; Roach, P.L. Optimized Conjugation of a Fluorescent Label to Proteins via Intein-Mediated Activation and Ligation. Bioconjug. Chem. 2004, 15, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Thom, J.; Anderson, D.; McGregor, J.; Cotton, G. Recombinant protein hydrazides: Application to site-specific protein PEGylation. Bioconjug. Chem. 2011, 22, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Möhlmann, S.; Bringmann, P.; Greven, S.; Harrenga, A. Site-specific modification of ED-B-targeting antibody using intein-fusion technology. BMC Biotechnol. 2011, 11, 76. [Google Scholar] [CrossRef] [PubMed]
- Vila-Perelló, M.; Liu, Z.; Shah, N.H.; Willis, J.A.; Idoyaga, J.; Muir, T.W. Streamlined expressed protein ligation using split inteins. J. Am. Chem. Soc. 2013, 135, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Hu, Z.; Liu, X.-Q. Protein trans-splicing by a split intein encoded in a split DnaE gene of Synechocystis sp. PCC6803. Biochemistry 1998, 95, 9226–9231. [Google Scholar] [CrossRef]
- Giriat, I.; Muir, T.W. Protein Semi-Synthesis in Living Cells. J. Am. Chem. Soc. 2003, 125, 7180–7181. [Google Scholar] [CrossRef] [PubMed]
- Appleby, J.H.; Zhou, K.; Volkmann, G.; Liu, X.-Q. Novel split intein for trans-splicing synthetic peptide onto C terminus of protein. J. Biol. Chem. 2009, 284, 6194–6199. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Li, M.; Song, H.; Xu, L.; Meng, Q.; Liu, X.Q. Protein Trans-Splicing of Multiple Atypical Split Inteins Engineered from Natural Inteins. PLoS ONE 2013, 8, e59516. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Vallejos, P.; Pallissé, R.; Mootz, H.D.; Schmidt, S.R. Unprecedented rates and efficiencies revealed for new natural split inteins from metagenomic sources. J. Biol. Chem. 2012, 287, 28686–28696. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.J.; Sekar, G.; Shah, N.H.; Mostafavi, A.Z.; Cowburn, D.; Muir, T.W. A promiscuous split intein with expanded protein engineering applications. Proc. Natl. Acad. Sci. USA 2017, 114, 8538–8543. [Google Scholar] [CrossRef] [PubMed]
- Kurpiers, T.; Mootz, H.D. Site-specific chemical modification of proteins with a prelabelled cysteine tag using the artificially split Mxe GyrA intein. ChemBioChem 2008, 9, 2317–2325. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, A.L.; Mootz, H.D. N-terminal chemical protein labeling using the naturally split GOS-TerL intein. J. Pept. Sci. 2017, 23, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Chen, J.; Ding, K.; Zong, H.; Xie, Y.; Jiang, H.; Zhang, B.; Lu, H.; Yin, W.; Gilly, J.; et al. Efficient generation of bispecific IgG antibodies by split intein mediated protein trans-splicing system. Sci. Rep. 2017, 7, 8360. [Google Scholar] [CrossRef] [PubMed]
- Ersfeld, K.; Web, J.; Plessmann, U.; Dodemont, H.; Gerke, V.; Weber, K. Characterization of the Tubulin-Tyrosine Ligase. Angew. Chem. Int. Ed. 1993, 120, 725–732. [Google Scholar] [CrossRef]
- Banerjee, A.; Panosian, T.D.; Mukherjee, K.; Ravindra, R.; Gal, S.; Sackett, D.L.; Bane, S. Site-specific orthogonal labeling of the carboxy terminus of α-tubulin. ACS Chem. Biol. 2010, 5, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Helma, J.; Mann, F.A.; Pichler, G.; Natale, F.; Krause, E.; Cardoso, M.C.; Hackenberger, C.P.R.; Leonhardt, H. Versatile and Efficient Site-Specific Protein Functionalization by Tubulin Tyrosine Ligase. Angew. Chem. Int. Ed. 2015, 54, 13787–13791. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Lemke, O.; Helma, J.; Gerszonowicz, L.; Waller, V.; Stoschek, T.; Durkin, P.M.; Budisa, N.; Leonhardt, H.; Keller, B.G.; et al. Broad substrate tolerance of tubulin tyrosine ligase enables one-step site-specific enzymatic protein labeling. Chem. Sci. 2017, 8, 3471–3478. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.Y.; Burnier, J.; Quan, C.; Stanley, M.; Tom, J.; Wells, J.A. A designed peptide ligase for total synthesis of ribonuclease A with unnatural catalytic residues. Science 1994, 266, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Atwell, S.; Wells, J.A. Selection for improved subtiligases by phage display. Proc. Natl. Acad. Sci. USA 1999, 96, 9497–9502. [Google Scholar] [CrossRef] [PubMed]
- Liebscher, S.; Schöpfel, M.; Aumüller, T.; Sharkhuukhen, A.; Pech, A.; Höss, E.; Parthier, C.; Jahreis, G.; Stubbs, M.T.; Bordusa, F. N-terminal protein modification by substrate-activated reverse proteolysis. Angew. Chem. Int. Ed. 2014, 53, 3024–3028. [Google Scholar] [CrossRef] [PubMed]
- Liebscher, S.; Kornberger, P.; Fink, G.; Trost-Gross, E.M.; Höss, E.; Skerra, A.; Bordusa, F. Derivatization of antibody fab fragments: A designer enzyme for native protein modification. ChemBioChem 2014, 15, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Liebscher, S.; Bordusa, F. Selective Coupling of Click Anchors to Proteins via Trypsiligase. Bioconjug. Chem. 2016, 27, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.K.; Zhang, L.; Rangan, V.S.; Smith, S. Cloning, expression, and characterization of a human 4′-phosphopantetheinyl transferase with broad substrate specificity. J. Biol. Chem. 2003, 278, 33142–33149. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Straight, P.D.; McLoughlin, S.M.; Zhou, Z.; Lin, A.J.; Golan, D.E.; Kelleher, N.L.; Kolter, R.; Walsh, C.T. Genetically encoded short peptide tag for versatile protein labeling by Sfp phosphopantetheinyl transferase. Proc. Natl. Acad. Sci. USA 2005, 102, 15815–15820. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Lin, A.J.; Golan, D.E.; Walsh, C.T. Site-specific protein labeling by Sfp phosphopantetheinyl transferase. Nat. Protoc. 2006, 1, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Cironi, P.; Lin, A.J.; Xu, Y.; Hrvatin, S.; Golan, D.E.; Silver, P.A.; Walsh, C.T.; Yin, J. Genetically encoded short peptide tags for orthogonal labelling by Sfp and AcpS phosphopantetheinyl transferases. ACS Chem. Biol. 2007, 2, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, J.; Klock, H.E.; Cellitti, S.E.; Bursulaya, B.; McMullan, D.; Jones, D.H.; Chiu, H.P.; Wang, X.; Patterson, P.; Zhou, H.; et al. Efficient Preparation of Site-Specific Antibody–Drug Conjugates Using Phosphopantetheinyl Transferases. Bioconjug. Chem. 2015, 26, 2554–2562. [Google Scholar] [CrossRef] [PubMed]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef] [PubMed]
- Fierer, J.O.; Veggiani, G.; Howarth, M. SpyLigase peptide-peptide ligation polymerizes affibodies to enhance magnetic cancer cell capture. Proc. Natl. Acad. Sci. USA 2014, 111, E1176–E1181. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, V.; Piater, B.; Zakeri, B.; Eichhorn, T.; Fischer, F.; Deutsch, C.; Becker, S.; Toleikis, L.; Hock, B.; Betz, U.A.K.; et al. Spontaneous Isopeptide Bond Formation as a Powerful Tool for Engineering Site-Specific Antibody–Drug Conjugates. Sci. Rep. 2016, 6, 39291. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Choi, H.J.; Yun, M.; Kang, Y.; Jung, J.E.; Ryu, Y.; Kim, T.Y.; Cha, Y.J.; Cho, H.S.; Min, J.J.; et al. Enzymatic Prenylation and Oxime Ligation for the Synthesis of Stable and Homogeneous Protein-Drug Conjugates for Targeted Therapy. Angew. Chem. Int. Ed. 2015, 54, 12020–12024. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Zou, P.; Ting, A.Y. Site-Specific Protein Modification Using Lipoic Acid Ligase and Bis-Aryl Hydrazone Formation. ChemBioChem 2012, 13, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.; Howarth, M.; Lin, W.; Ting, A.Y. Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat. Methods 2005, 2, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Howarth, M.; Takao, K.; Hayashi, Y.; Ting, A.Y. Targeting quantum dots to surface proteins in living cells with biotin ligase. Proc. Natl. Acad. Sci. USA 2005, 102, 7583–7588. [Google Scholar] [CrossRef] [PubMed]
- Heller, K.; Ochtrop, P.; Albers, M.F.; Zauner, F.B.; Itzen, A.; Hedberg, C. Covalent Protein Labeling by Enzymatic Phosphocholination. Angew. Chem. Int. Ed. 2015, 54, 10327–10330. [Google Scholar] [CrossRef] [PubMed]
- Van Geel, R.; Wijdeven, M.A.; Heesbeen, R.; Verkade, J.M.M.; Wasiel, A.A.; van Berkel, S.S.; van Delft, F.L. Chemoenzymatic Conjugation of Toxic Payloads to the Globally Conserved N-Glycan of Native mAbs Provides Homogeneous and Highly Efficacious Antibody–Drug Conjugates. Bioconjug. Chem. 2015, 26, 2233–2242. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strategy | Substrate | Reaction Buffer | Duration | Labeling Strategy | Locus | Yields | Applications | Drawbacks | Enzyme/Substrate (Molar) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Formylglycine-generating enzymes | CXPXR | 25 mM TEAM pH 9, 50 mM NaCl, 1 mM β-ME³ | 16 h at 18 °C | aldehyde coupling chemistry (HIPS-Ligation, trapped-Knoevenagel ligation etc.) | most surface accessible sites | 85–95% a [44] ~95% b [40] | whole antibodies [37,39,40,44] | -long downstream coupling reaction -in vitro conversion at alkaline pH and reductive environment | 0.1 | |
| Sortase A | LPXTG (Gn) | 50 mM Tris/HCl pH 7.9, 150 mM NaCl, 10 mM CaCl2 | 4 h at 42 °C | labeled peptide (Gn or LPXTG) | N- and C-terminal | ~80% [65] | whole antibody [65,66] Fab [62] | -side reactions with proteins or peptides with terminal glycines | ~1–3 | |
| Transglutaminase | (1) LLQGA or (2) GECTYFQAYGCTE | (1) 10 mM Phosphate buffer pH 7, 150 mM NaCl or (2) 100 mM HEPES pH 7 | (1) 16 h at 37 °C or (2) 3 h at 25 °C | labeled alkyl- or oligo-amine | most surface accessible sites | ~80–90% [78,79,81] | whole antibody [78,79,81] | -crosslinking via side chain lysine -deamidation of glutamine | (1) ~0.15–0.5 or (2) 1 | |
| Inteins | EPL | C-terminal intein (~100–150 aa) | 50 mM HEPES/NaOH pH 8, 500 mM NaCl, 50 mM MESNA | 22 h | labeled peptide with N-terminal cysteine | C-terminal | ~60% [87] | whole antibody [87,88], VHH [96] | -long fusion tags -premature extein cleavage during expression reduces yields | N/A c |
| PTS | terminal intein (~100–150 aa) | 50 mM HEPES/NaOH pH 7.5, 500 mM NaCl, 5 mM DTT | 24 h | labeled short complementary intein (6–12 aa) | N- and C-terminal | ~75% [87] | whole antibody [87,97] | -long fusion tags -hydrolysis or thiolysis during splicing yields side products | N/A c | |
| Tubulin Tyrosine Ligase | VDSVEGEEEGEE | 20 mM MES/K pH 7, 100 mM KCl, 10 mM MgCl2, 2.5 mM ATP, 5 mM DTT | 5 h at 37 °C | labeled tyrosine | C-terminal | 99% [100] | VHH [100] | -limited to C-terminus -long tag | 0.2 | |
| Trypsiligase | YRH | 100 mM HEPES/NaOH pH 7.8, 0.1 mM ZnCl2, 100 mM NaCl, 10 mM CaCl2 | 1 h at 20 °C | labeled RH-peptide | C-terminal | 70% [106] | Fab [105,106] | -remaining proteolytic activity generates side products | 0.1 | |
| Phosphopantetheinyl transferase | DSLEFIASKLA | 50 mM HEPES pH 7.5–8, 10 mM MgCl2 | 16 h at 20 °C | labeled Coenzyme A | N-, C-terminal and flexible loops | 95% [111] | whole antibody [111] | -large linker -rather hydrophobic tag | 0.4 | |
| SpyLigase | AHIVMVDAYKPTK | 40 mM Na2HPO4, 20 mM Citric acid pH 6.8, 1.5 M Trimethylamine N-oxide (TMAO) | 24 h at 4 °C | iso-peptide bond formation with labeled ATHIKFSKRD peptide | C- or N-terminal | ~80% [114] | whole antibody [114], affibody [113] | -enzyme excess needed | ~3 | |
| Farnesyltransferase | CVIM | 50 mM Tris/HCl pH 7.4, 5 mM MgCl2, 10 µM ZnCl2, 5 mM DTT | 12 h at 30 °C | attachment of aldehyde or keto functionalized prenyl pyrophosphate, subsequent oxime ligation | C-terminal | ~95% [115] | repebody [115] | -long downstream coupling reaction at low pH -enzyme excess needed | ~2 | |
| AnkX | TITSSYYR | 20 mM HEPES pH 7.5, 50 mM NaCl, 1 mM MgCl2, 1 mM DTE | 3 h at 25 °C | labeled cytidine diphosphate choline | N-, C-terminal and in internal loops | 70% d [119] | -lower yields -reductive environment | 0.02 | ||
| Biotin ligase | GLNDIFEAQKIEWHE | 50 mM Bicine pH 8.3, 5 mM Mg-acetate, 4 mM ATP | 3 h at 30 °C | ligation of Biotin to side-chain of lysine and subsequent labeling of ketone group | N-, C-terminal and internal loops | ~50% e [117] | -long Tag -lower yields | 0.065–0.13 | ||
| GlycoConnect | N-glycans | (1) 25 mM Tris pH 8, (2) 25 mM Tris/HCl pH 8, 10 mM MnCl2 | (1) 16 h at 37 °C (2) 16 h at 30 °C | (1) trimming of glycan with endoglycosidase and (2) attachment of a conjugable GalNAc derivative by glycosyltransferase | N-glycans | >95% [120] | whole antibody [120] | -long incubation at >30 °C -disruption of N-glycans | (1) ~0.02 (2) ~0.015 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falck, G.; Müller, K.M. Enzyme-Based Labeling Strategies for Antibody–Drug Conjugates and Antibody Mimetics. Antibodies 2018, 7, 4. https://doi.org/10.3390/antib7010004
Falck G, Müller KM. Enzyme-Based Labeling Strategies for Antibody–Drug Conjugates and Antibody Mimetics. Antibodies. 2018; 7(1):4. https://doi.org/10.3390/antib7010004
Chicago/Turabian StyleFalck, Georg, and Kristian M. Müller. 2018. "Enzyme-Based Labeling Strategies for Antibody–Drug Conjugates and Antibody Mimetics" Antibodies 7, no. 1: 4. https://doi.org/10.3390/antib7010004
APA StyleFalck, G., & Müller, K. M. (2018). Enzyme-Based Labeling Strategies for Antibody–Drug Conjugates and Antibody Mimetics. Antibodies, 7(1), 4. https://doi.org/10.3390/antib7010004