
Facial Asymmetry: A Narrative Review of the Most Common Neurological Causes


Abstract
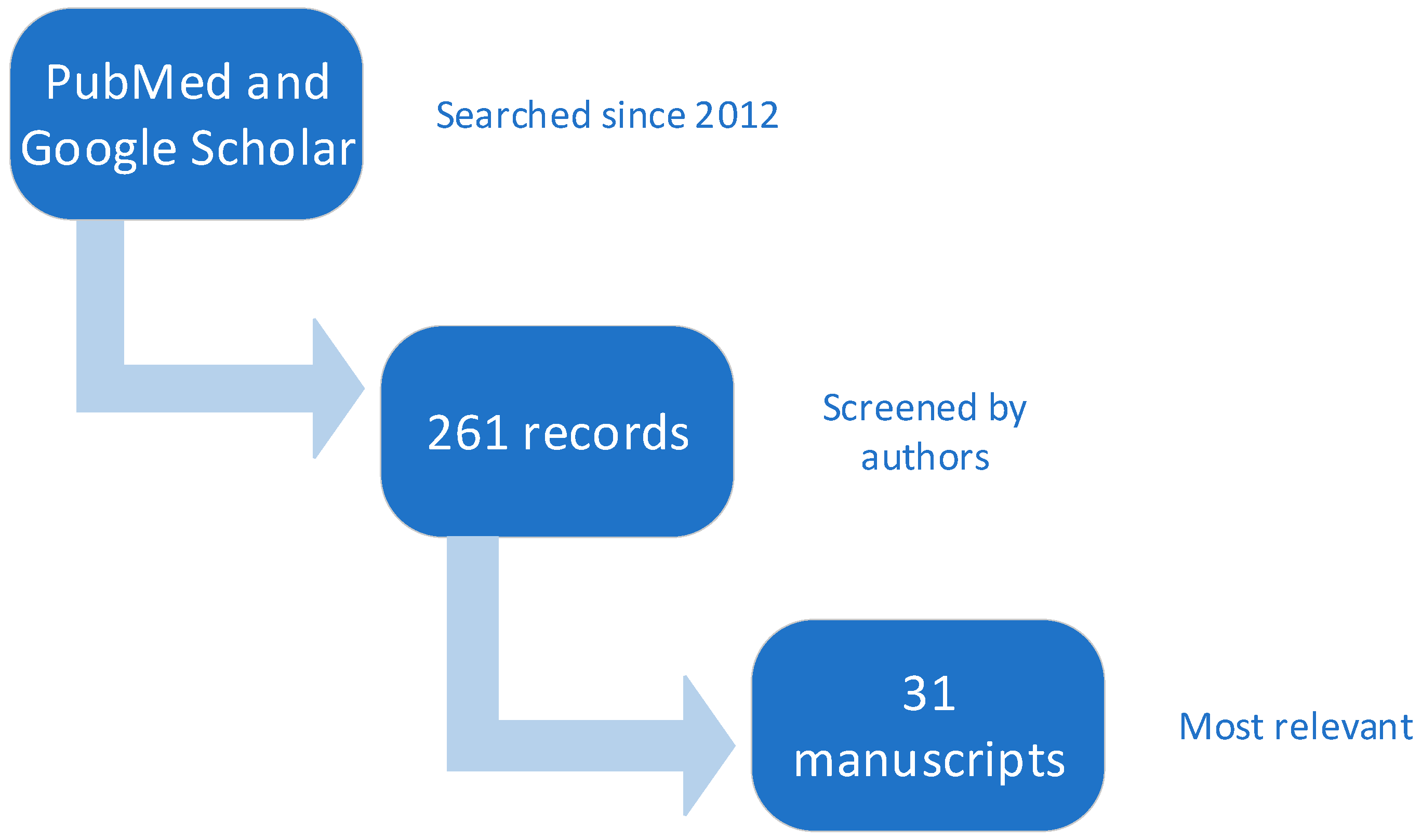
:1. Introduction
2. Methods
3. Results
3.1. Cranial Nerves
3.1.1. Nerve III (Oculomotor Nerve)
3.1.2. Nerve IV (Trochlear Nerve)
3.1.3. Nerve V (Trigeminal Nerve)
3.1.4. Nerve VII (Facial Nerve)
Bell’s Palsy
Central Palsy
Moebius Syndrome
Ramsay Hunt Syndrome
Melkersson–Rosenthal Syndrome
Hemifacial Spasm
Miller Fisher Syndrome
3.2. Developmental Disorders
3.2.1. Parry–Romberg Syndrome (PRS)
3.2.2. Asymmetric Crying Facies (ACF)
3.2.3. Dyke–Davidoff–Masson Syndrome
3.2.4. Harlequin Syndrome
3.2.5. Klippel–Feil Syndrome
3.2.6. CHARGE Syndrome
3.2.7. HOXA1 Syndromes: Athabascan Brain Stem Dysgenesis Syndrome (ABDS) and Bosley–Salih–Alorainy Syndrome (BSAS)
3.2.8. Isolated Hereditary Congenital Facial Paresis (HCFP)
3.3. Myopathies
3.3.1. Facioscapulohumeral Muscular Dystrophy
3.3.2. Titinopathy
3.3.3. Myotonic Dystrophy (MD) Type 1
3.3.4. Carey–Fineman–Ziter Syndrome (CFZS)
3.3.5. Nemaline Myopathy
3.4. Headache
3.4.1. Cluster Headache
3.4.2. Masticatory Muscles Headache
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Stedman, J.K. Stedman’s Medical Dictionary, 28th ed.; Lippincott Williams & Wilkins (LWW), 2006; Available online: http://www.stedmans.com/ (accessed on 28 November 2021).
- Choi, K.Y. Analysis of Facial Asymmetry. Arch. Craniofacial Surg. 2015, 16, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ortensi, L.; Vitali, T.; Bonfiglioli, R.; Grande, F. New Tricks in the Preparation Design for Prosthetic Ceramic Laminate Veeners. Prosthesis 2019, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- Mertens, I.; Siegmund, H.; Grüsser, O.-J. Gaze motor asymmetries in the perception of faces during a memory task. Neuropsychology 1993, 31, 989–998. [Google Scholar] [CrossRef]
- Komori, M.; Kawamura, S.; Ishihara, S. Averageness or symmetry: Which is more important for facial attractiveness? Acta Psychol. 2009, 131, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Berlin, N.F.; Berssenbrügge, P.; Runte, C.; Wermker, K.; Jung, S.; Kleinheinz, J.; Dirksen, D. Quantification of facial asymmetry by 2D analysis—A comparison of recent approaches. J. Cranio-Maxillofac. Surg. 2014, 42, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Ptosis: Causes, Presentation, and Management. Aesthetic Plast. Surg. 2003, 27, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Bacharach, J.; Lee, W.W.; Harrison, A.R.; Freddo, T.F. A review of acquired blepharoptosis: Prevalence, diagnosis, and current treatment options. Eye 2021, 35, 2468–2481. [Google Scholar] [CrossRef]
- Kim, T.; Nam, K.; Kwon, B.S. Isolated Oculomotor Nerve Palsy in Mild Traumatic Brain Injury. Am. J. Phys. Med. Rehab. 2020, 99, 430–435. [Google Scholar] [CrossRef]
- Kim, K.; Noh, S.R.; Kang, M.S.; Jin, K.H. Clinical Course and Prognostic Factors of Acquired Third, Fourth, and Sixth Cranial Nerve Palsy in Korean Patients. Korean J. Ophthalmol. 2018, 32, 221. [Google Scholar] [CrossRef] [PubMed]
- Kung, N.; Van Stavern, G. Isolated Ocular Motor Nerve Palsies. Semin. Neurol. 2015, 35, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Laine, F.J. Cranial Nerves III, IV, and VI. Top. Magn. Reson Imaging 1996, 8, 111. [Google Scholar] [CrossRef]
- Morillon, P.; Bremner, F. Trochlear nerve palsy. Br. J. Hosp. Med. 2017, 78, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.R.; Nejad, M.K.; Askarizadeh, F.; Pour, F.F.; Pazooki, M.R.; Moeinitabar, M.R. Facial asymmetry in ocular torticollis. J. Curr. Ophthalmol. 2015, 27, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.J.; Memmen, J.E.; Katz, N.N.K.; Parks, M.M. Familial Congenital Superior Oblique Palsy. Ophthalmology 1986, 93, 88–90. [Google Scholar] [CrossRef]
- Tollefson, M.M.; Mohney, B.G.; Diehl, N.N.; Burke, J.P. Incidence and Types of Childhood Hypertropia: A Population-Based Study. Ophthalmology 2006, 113, 1142–1145. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-K.; Lee, E.-H.; Hwang, M. Pure trigeminal motor neuropathy: A case report. Arch. Phys. Med. Rehab. 2000, 81, 995–998. [Google Scholar] [CrossRef]
- Chia, L.-G. Pure trigeminal motor neuropathy. BMJ 1988, 296, 609–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.; Hodgson, E.; Felstead, A. Focal atrophy of the masticatory muscles caused by pure trigeminal motor neuropathy: Case report. Br. J. Oral Maxillofac. Surg. 2016, 54, e13–e14. [Google Scholar] [CrossRef]
- Braun, J.S.; Hahn, K.; Bauknecht, H.-C.; Schielke, E. Progressive Facial Asymmetry due to Trigeminal Motor Neuropathy. Eur. Neurol. 2006, 55, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Andonopoulos, A.P.; Lagos, G.; Drosos, A.A.; Moutsopoulos, H.M. The Spectrum of Neurological Involvement in Sjögren’s Syndrome. Rheumatology 1990, 29, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Kämppi, A.; Kämppi, L.; Kemppainen, P.; Kanerva, M.; Toppila, J.; Auranen, M. Focal atrophy of the unilateral masticatory muscles caused by pure trigeminal motor neuropathy: Case report. Clin. Case Rep. 2018, 6, 939–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takezawa, K.; Townsend, G.; Ghabriel, M. The facial nerve: Anatomy and associated disorders for oral health professionals. Odontology 2018, 106, 103–116. [Google Scholar] [CrossRef]
- Williams, O.; Ulane, C. Facial Nerve (Cranial Nerve VII). In Encyclopedia of the Neurological Sciences; Elsevier: Amsterdam, The Netherlands, 2014; pp. 263–268. Available online: https://linkinghub.elsevier.com/retrieve/pii/B9780123851574006576 (accessed on 28 November 2021).
- George, E.; Richie, M.B.; Glastonbury, C.M. Facial Nerve Palsy: Clinical Practice and Cognitive Errors. Am. J. Med. 2020, 133, 1039–1044. [Google Scholar] [CrossRef]
- Myers, E.N.; De Diego, J.I.; Prim, M.P.; Madero, R.; Gavil$Aan, J. Seasonal Patterns of Idiopathic Facial Paralysis: A 16-Year Study. Otolaryngol. Neck Surg. 1999, 120, 269–271. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, L.; Luo, T.; Wu, F.; Zhao, B.; Li, X. The etiology of Bell’s palsy: A review. J. Neurol. 2020, 267, 1896–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahdab, R.; Saade, H.; Kikano, R.; Ferzli, J.; Tarcha, W.; Riachi, N. Pure ipsilateral central facial palsy and contralateral hemiparesis secondary to ventro-medial medullary stroke. J. Neurol. Sci. 2013, 332, 154–155. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, M.; Ono, T.; Lam, O.L.T.; Müller, F. Oro-facial impairment in stroke patients. J. Oral Rehabil. 2017, 44, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Sands, K.A.; Shahripour, R.B.; Kumar, G.; Barlinn, K.; Lyerly, M.J.; Haršány, M.; Cure, J.; Yakov, Y.L.; Alexandrov, A.W.; Alexandrov, A.V. Acute Isolated Central Facial Palsy as Manifestation of Middle Cerebral Artery Ischemia: Isolated Central Facial Palsy with MCA Ischemia. J. Neuroimaging 2016, 26, 499–502. [Google Scholar] [CrossRef]
- Wolf, M.E.; Rausch, H.-W.; Eisele, P.; Habich, S.; Platten, M.; Alonso, A. Acute Corticonuclear Tract Ischemic Stroke with Isolated Central Facial Palsy. J. Stroke Cerebrovasc. Dis. 2019, 28, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Picciolini, O.; Porro, M.; Cattaneo, E.; Castelletti, S.; Masera, G.; Mosca, F.; Bedeschi, M.F. Moebius syndrome: Clinical features, diagnosis, management and early intervention. Ital. J. Pediatr. 2016, 42, 56. [Google Scholar] [CrossRef] [Green Version]
- Rucker, J.C.; Webb, B.D.; Frempong, T.; Gaspar, H.; Naidich, T.P.; Jabs, E.W. Characterization of ocular motor deficits in congenital facial weakness: Moebius and related syndromes. Brain 2014, 137, 1068–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, A.; Madhavi, M.R.; Nagasudha, M.; Bhavi, S. A rare case of Moebius sequence. Indian J. Ophthalmol. 2012, 60, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.H.; Jamal, S.; Rashid, M.A.; Javaid, U.; Butt, N.H. Moebius Syndrome with Hypoglossal Palsy, Syndactyly, Brachydactyly, and Anisometropic Amblyopia. Cureus 2018, 10, e2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domeshek, L.F.; Zuker, R.M.; Borschel, G.H. Management of Bilateral Facial Palsy. Otolaryngol. Clin. N. Am. 2018, 51, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Ananthapadmanabhan, S.; Soodin, D.; Sritharan, N.; Sivapathasingam, V. Ramsay Hunt syndrome with multiple cranial neuropathy: A literature review. Eur. Arch. Oto-Rhino-Laryngol. 2021, 1–6. Available online: https://link.springer.com/10.1007/s00405-021-07136-2 (accessed on 1 November 2021). [CrossRef]
- Jeon, Y.; Lee, H. Ramsay Hunt syndrome. J. Dent. Anesth. Pain Med. 2018, 18, 333. [Google Scholar] [CrossRef]
- Sweeney, C.J. Nosological Entities?: Ramsay Hunt syndrome. J. Neurol. Neurosurg. Psychiatry 2001, 71, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Crouch, A.E.; Andaloro, C. Ramsay Hunt Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: http://www.ncbi.nlm.nih.gov/books/NBK557409/ (accessed on 22 November 2021).
- Ostwal, S.; Salins, N.; Deodhar, J.; Muckaden, M. Management of ramsay hunt syndrome in an acute palliative care setting. Indian J. Palliat. Care 2015, 21, 79–81. [Google Scholar] [CrossRef] [PubMed]
- Casper, J.; Mohammad-Khani, S.; Schmidt, J.J.; Kielstein, J.T.; Lenarz, T.; Haller, H.; Wagner, A.D. Melkersson–Rosenthal syndrome in the context of sarcoidosis: A case report. J. Med. Case Rep. 2021, 15, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jamil, R.T.; Agrawal, M.; Gharbi, A.; Sonthalia, S. Cheilitis Granulomatosa. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: http://www.ncbi.nlm.nih.gov/books/NBK470396/ (accessed on 22 November 2021).
- Scully, C.; Langdon, J.; Evans, J. Marathon of eponyms: 13 Melkersson-Rosenthal syndrome: Marathon of eponyms. Oral Dis. 2010, 16, 707–708. [Google Scholar] [CrossRef] [PubMed]
- Ziem, P.E.; Pfrommer, C.; Goerdt, S.; Orfanos, C.E.; Blume-Peytavi, U. Melkersson-Rosenthal syndrome in childhood: A challenge in differential diagnosis and treatment: Melkersson-Rosenthal Syndrome in Childhood. Br. J. Dermatol. 2000, 143, 860–863. [Google Scholar] [CrossRef]
- Vaughan, C.L.; Goetz, C.G. Hemifacial Spasm. In Encyclopedia of the Neurological Sciences; Elsevier: Amsterdam, The Netherlands, 2014; p. 545. Available online: https://linkinghub.elsevier.com/retrieve/pii/B9780123851574000221 (accessed on 1 November 2021).
- Chopade, T.R.; Bollu, P.C. Hemifacial Spasm. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: http://www.ncbi.nlm.nih.gov/books/NBK526108/ (accessed on 22 November 2021).
- Lefaucheur, J.-P.; Ben Daamer, N.; Sangla, S.; Le Guerinel, C. Diagnosis of primary hemifacial spasm. Neurochirurgie 2018, 64, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Hermier, M. Imaging of hemifacial spasm. Neurochirurgie 2018, 64, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Rocha Cabrero, F.; Morrison, E.H. Miller Fisher Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: http://www.ncbi.nlm.nih.gov/books/NBK507717/ (accessed on 23 November 2021).
- Berlit, P.; Rakicky, J. The Miller Fisher syndrome. Review of the literature. J. Clin. Neuroophthalmol. 1992, 12, 57–63. [Google Scholar]
- Al Othman, B.; Raabe, J.; Kini, A.; Lee, A.G. Update: The Miller Fisher variants of Guillain–Barré syndrome. Curr. Opin. Ophthalmol. 2019, 30, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.-Y.; Yuki, N.; Shahrizaila, N. Delayed facial palsy in Miller Fisher syndrome. J. Neurol. Sci. 2015, 358, 409–412. [Google Scholar] [CrossRef]
- Willison, H.J.; Veitch, J.; Paterson, G.; Kennedy, P.G. Miller Fisher syndrome is associated with serum antibodies to GQ1b ganglioside. J. Neurol. Neurosurg. Psychiatry 1993, 56, 204–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.A.; Kumar, R.A.; Shantha, G.P.S.; Aloogopinathan, G. Progressive hemi facial atrophy—Parry Romberg syndrome pre-senting as severe facial pain in a young man: A case report. Cases J. 2009, 2, 6776. [Google Scholar] [CrossRef] [Green Version]
- Deshingkar, S.A.; Barpande, S.R.; Bhavthankar, J.D.; Humbe, J.G. Progressive hemifacial atrophy (Parry-Romberg Syndrome). Contemp. Clin. Dent. 2012, 3, 78–81. [Google Scholar] [CrossRef]
- Stone, J. Parry–Romberg syndrome: A global survey of 205 patients using the Internet. Neurology 2003, 61, 674–676. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.-J.; Liu, W.; Yang, B.; Shi, L.; Yin, L.; Zhang, Z.-Y. Parry–Romberg syndrome with rare maxillofacial deformities: A report on two cases. J. Cranio-Maxillofac. Surg. 2014, 42, 780–783. [Google Scholar] [CrossRef]
- Renault, F. Facial electromyography in newborn and young infants with congenital facial weakness. Dev. Med. Child. Neurol. 2007, 43, 421–427. [Google Scholar] [CrossRef]
- Dubnov-Raz, G.; Merlob, P.; Geva-Dayan, K.; Blumenthal, D.; Finkelstein, Y. Increased rate of major birth malformations in infants with neonatal “asymmetric crying face”: A hospital-based cohort study. Am. J. Med Genet. Part A 2007, 143, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Abdul Rashid, A.M.; Noh, M.S.F. Dyke-Davidoff-Masson syndrome: A case report. BMC Neurol. 2018, 18, 76. [Google Scholar] [CrossRef]
- Diestro, J.D.B.; Dorotan, M.K.C.; Camacho, A.C.; Perez-Gosiengfiao, K.T.; Cabral-Lim, L.I. Clinical spectrum of Dyke-Davidoff-Masson syndrome in the adult: An atypical presentation and review of literature. BMJ Case Rep. 2018, 2018, bcr-2018-224170. [Google Scholar] [CrossRef] [PubMed]
- Gökçe, E.; Beyhan, M.; Sade, R. Radiological imaging findings of Dyke–Davidoff–Masson syndrome. Acta Neurol. Belg. 2017, 5, 469–893. [Google Scholar] [CrossRef] [PubMed]
- Atalar, M.H.; Icagasioglu, D.; Tas, F. Cerebral hemiatrophy (Dyke?Davidoff?Masson syndrome) in childhood: Clinicoradiological analysis of 19 cases. Pediatr. Int. 2007, 49, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jiang, W.; Yan, W.; Tian, J.; Xu, J.; Li, Y.; Zhao, Y.; Dai, Y.; Cheng, G.; Hou, G. Clinical characteristics and neuroimaging findings of seven patients with Dyke Davidoff Masson syndrome. BMC Neurol. 2021, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bremner, F.; Smith, S. Pupillographic Findings in 39 Consecutive Cases of Harlequin Syndrome. J. Neuro-Ophthalmol. 2008, 28, 171–177. [Google Scholar] [CrossRef]
- Willaert, W.I.M.; Scheltinga, M.R.M.; Steenhuisen, S.F.; Hiel, J.A.P. Harlequin syndrome: Two new cases and a management proposal. Acta Neurol. Belg. 2009, 109, 214–220. [Google Scholar] [PubMed]
- Elboukhari, K.; Baybay, H.; Elloudi, S.; Douhi, Z.; Mernissi, F.Z. Idiopathic harlequin syndrome: A case report and literature review. Pan Afr. Med. J. 2019, 33, 141. [Google Scholar] [CrossRef] [PubMed]
- Frikha, R. Klippel-Feil syndrome: A review of the literature. Clin. Dysmorphol. 2020, 29, 35–37. [Google Scholar] [CrossRef] [PubMed]
- Menger, R.P.; Rayi, A.; Notarianni, C. Klippel Feil Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: http://www.ncbi.nlm.nih.gov/books/NBK493157/ (accessed on 22 November 2021).
- Goto, M.; Nishimura, G.; Nagai, T.; Yamazawa, K.; Ogata, T. Familial Klippel–Feil anomaly and t(5;8)(q35.1;p21.1) translocation. Am. J. Med. Genet. Part A 2006, 140, 1013–1015. [Google Scholar] [CrossRef] [PubMed]
- Tassabehji, M.; Fang, Z.M.; Hilton, E.N.; McGaughran, J.; Zhao, Z.; de Bock, C.E.; Howard, E.; Malass, M.; Donnai, D.; Diwan, A.; et al. Mutations in GDF6 are associated with vertebral segmentation defects in Klippel-Feil syndrome. Hum. Mutat. 2008, 29, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- McGaughran, J.M.; Oates, A.; Donnai, D.; Read, A.P.; Tassabehji, M. Mutations in PAX1 may be associated with Klippel–Feil syndrome. Eur. J. Hum. Genet. 2003, 11, 468–474. [Google Scholar] [CrossRef] [Green Version]
- Karaca, E.; Yuregir, O.O.; Bozdogan, S.T.; Aslan, H.; Pehlivan, D.; Jhangiani, S.N.; Akdemir, Z.C.; Gambin, T.; Bayram, Y.; Atik, M.M.; et al. Rare variants in the notch signaling pathway describe a novel type of autosomal recessive Klippel-Feil syndrome. Am. J. Med Genet. Part A 2015, 167, 2795–2799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovankovičová, A.; Jakubíková, J.; Ďurovčíková, D. A case of Klippel–Feil syndrome with congenital enlarged Eustachian tube. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Litrenta, J.; Bi, A.S.; Dryer, J.W. Klippel-Feil Syndrome: Pathogenesis, Diagnosis, and Management. J. Am. Acad. Orthop. Surg. 2021, 29, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Lagravère, M.O.; Barriga, M.I.; Valdizán, C.; Saldarriaga, A.; Pardo, J.F.; Flores, M. The Klippel-Feil syndrome: A case report. J. Can. Dent. Assoc. 2004, 70, 685–688. [Google Scholar]
- Hsu, P.; Ma, A.; Wilson, M.; Williams, G.; Curotta, J.; Munns, C.F.; Mehr, S. CHARGE syndrome: A review of CHARGE syndrome. J. Paediatr. Child. Health 2014, 50, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Pauli, S.; Bajpai, R.; Borchers, A. CHARGEd with neural crest defects. Am. J. Med. Genet. Part C Semin. Med Genet. 2017, 175, 478–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usman, N.; Sur, M. CHARGE Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: http://www.ncbi.nlm.nih.gov/books/NBK559199/ (accessed on 22 November 2021).
- van Ravenswaaij-Arts, C.; Martin, D.M. New insights and advances in CHARGE syndrome: Diagnosis, etiologies, treatments, and research discoveries. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 397–406. [Google Scholar] [CrossRef] [PubMed]
- de Geus, C.M.; Free, R.H.; Verbist, B.M.; Sival, D.A.; Blake, K.D.; Meiners, L.C.; van Ravenswaaij-Arts, C.M.A. Guidelines in CHARGE syndrome and the missing link: Cranial imaging. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 450–464. [Google Scholar] [CrossRef] [Green Version]
- Zentner, G.E.; Layman, W.S.; Martin, D.M.; Scacheri, P.C. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am. J. Med. Genet. A 2010, 152, 674–686. [Google Scholar] [CrossRef] [Green Version]
- Blake, K.D.; Hartshorne, T.S.; Lawand, C.; Dailor, A.N.; Thelin, J.W. Cranial nerve manifestations in CHARGE syndrome. Am. J. Med Genet. Part A 2008, 146, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Chetty, M.; Roberts, T.S.; Elmubarak, M.; Bezuidenhout, H.; Smit, L.; Urban, M. CHARGE syndrome: Genetic aspects and dental challenges, a review and case presentation. Head Face Med. 2020, 16, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Bedeschi, M.F.; Crippa, B.L.; Colombo, L.; Buscemi, M.; Rossi, C.; Villa, R.; Gangi, S.; Picciolini, O.; Cinnante, C.; Fergnani, V.G.C.; et al. A case series of CHARGE syndrome: Identification of key features for a neonatal diagnosis. Ital. J. Pediatr. 2020, 46, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Holve, S.; Friedman, B.; Hoyme, H.; Tarby, T.J.; Johnstone, S.J.; Erickson, R.P.; Clericuzio, C.L.; Cunniff, C. Athabascan brainstem dysgenesis syndrome. Am. J. Med. Genet. 2003, 120, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Rankin, J.K.; Andrews, C.; Chan, W.-M.; Engle, E.C. HOXA1 mutations are not a common cause of Möbius syndrome. J. Am. Assoc. Pediatr. Ophthalmol. Strabismus 2010, 14, 78–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alrashdi, I.S.; Rich, P.; Patton, M.A. A family with hereditary congenital facial paresis and a brief review of the literature. Clin. Dysmorphol. 2010, 19, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Gutowski, N.J.; Chilton, J.K. The congenital cranial dysinnervation disorders. Arch. Dis. Child. 2015, 100, 678–681. [Google Scholar] [CrossRef]
- Vogel, M.; Velleuer, E.; Schmidt-Jiménez, L.F.; Mayatepek, E.; Borkhardt, A.; Alawi, M.; Kutsche, K.; Kortüm, F. HomozygousHOXB1loss-of-function mutation in a large family with hereditary congenital facial paresis. Am. J. Med. Genet. Part A 2016, 170, 1813–1819. [Google Scholar] [CrossRef] [PubMed]
- Terzis, J.K.; Anesti, K. Developmental facial paralysis: A review. J. Plast. Reconstr. Aesthetic Surg. 2011, 64, 1318–1333. [Google Scholar] [CrossRef]
- Webb, B.D.; Shaaban, S.; Gaspar, H.; Cunha, L.F.; Schubert, C.R.; Hao, K.; Robson, C.D.; Chan, W.-M.; Andrews, C.; MacKinnon, S.; et al. HOXB1 Founder Mutation in Humans Recapitulates the Phenotype of Hoxb1−/− Mice. Am. J. Hum. Genet. 2012, 91, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacconi, S.; Salviati, L.; Desnuelle, C. Facioscapulohumeral muscular dystrophy. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2015, 1852, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Fecek, C.; Emmady, P.D. Facioscapulohumeral Muscular Dystrophy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: http://www.ncbi.nlm.nih.gov/books/NBK559028/ (accessed on 23 November 2021).
- Lemmers, R.J.L.F.; O’Shea, S.; Padberg, G.W.; Lunt, P.W.; van der Maarel, S.M. Best practice guidelines on genetic diagnostics of Faci-oscapulohumeral muscular dystrophy: Workshop 9th June 2010, LUMC, Leiden, The Netherlands. Neuromuscul. Disord. 2012, 22, 463–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mul, K.; Lassche, S.; Voermans, N.C.; Padberg, G.W.; Horlings, C.G.; van Engelen, B.G. What’s in a name? The clinical features of facioscapulohumeral muscular dystrophy. Pract. Neurol. 2016, 16, 201–207. [Google Scholar] [CrossRef]
- Tawil, R. Facioscapulohumeral Muscular Dystrophy. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 541–548. Available online: https://linkinghub.elsevier.com/retrieve/pii/B9780444640765000351 (accessed on 1 November 2021).
- Loonen, T.G.J.; Horlings, C.G.C.; Vincenten, S.C.C.; Beurskens, C.H.G.; Knuijt, S.; Padberg, G.W.A.M.; Statland, J.M.; Voermans, N.C.; Maal, T.J.J.; van Engelen, B.G.M.; et al. Characterizing the face in facioscapulohumeral muscular dystrophy. J. Neurol. 2020, 268, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Savarese, M.; Maggi, L.; Vihola, A.; Jonson, P.H.; Tasca, G.; Ruggiero, L.; Bello, L.; Magri, F.; Giugliano, T.; Torella, A.; et al. Interpreting Genetic Variants in Titin in Patients with Muscle Disorders. JAMA Neurol. 2018, 75, 557–565. [Google Scholar] [CrossRef] [Green Version]
- Tskhovrebova, L.; Trinick, J. Roles of Titin in the Structure and Elasticity of the Sarcomere. J. Biomed. Biotechnol. 2010, 2010, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrin, A.; Metay, C.; Villanova, M.; Carlier, R.; Pegoraro, E.; Morales, R.J.; Stojkovic, T.; Richard, I.; Richard, P.; Romero, N.B.; et al. A new congenital multicore titinopathy associated with fast myosin heavy chain deficiency. Ann. Clin. Transl. Neurol. 2020, 7, 846–854. [Google Scholar] [CrossRef]
- Sasaki, R.; Ohta, Y.; Tadokoro, K.; Matsumoto, N.; Nomura, E.; Omote, Y.; Takemoto, M.; Hishikawa, N.; Yamashita, T.; Kumutpongpanich, T.; et al. TTN missense variants in two siblings with asymmetric facial and limb weakness. J. Neurol. Sci. 2020, 415, 116885. [Google Scholar] [CrossRef]
- Oates, E.C.; Jones, K.J.; Donkervoort, S.; Charlton, A.; Brammah, S.; Smith, J.E., 3rd; Ware, J.S.; Yau, K.S.; Swanson, L.C.; Whiffin, N.; et al. Congenital Titinopathy: Comprehensive characterization and pathogenic insights. Ann. Neurol. 2018, 83, 1105–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vydra, D.G.; Rayi, A. Myotonic Dystrophy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: http://www.ncbi.nlm.nih.gov/books/NBK557446/ (accessed on 23 November 2021).
- Thornton, C.A. Myotonic Dystrophy. Neurol. Clin. 2014, 32, 705–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moxley, R.T.; Ciafaloni, E.; Guntrum, D. Myotonic Dystrophy. In Neuromuscular Disorders of Infancy, Childhood, and Adolescence; Elsevier: Amsterdam, The Netherlands, 2015; pp. 697–718. Available online: https://linkinghub.elsevier.com/retrieve/pii/B9780124170445000378 (accessed on 1 November 2021).
- Cacucci, L.; Ricci, B.; Moretti, M.; Gasparini, G.; Pelo, S.; Grippaudo, C. Surgical Orthodontic Treatment of a Patient Affected by Type 1 Myotonic Dystrophy (Steinert Syndrome). Case Rep. Dent. 2017, 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Camacho, A.; Martínez, B.; Alvarez, S.; Gil-Fournier, B.; Ramiro, S.; Hernández-Laín, A.; Noemía, N.; Rogelio, S. Carey-Fineman-Ziter Syndrome: A MYMK-Related Myopathy Mimicking Brainstem Dysgenesis. J. Neuromuscul. Dis. 2020, 7, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Moebius Syndrome Research Consortium; Di Gioia, S.A.; Connors, S.; Matsunami, N.; Cannavino, J.; Rose, M.F.; Gilette, N.M.; Artoni, P.; de Macena Sobreira, N.L.; Chan, W.-M.; et al. A defect in myoblast fusion underlies Carey-Fineman-Ziter syndrome. Nat. Commun. 2017, 8, 16077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanchi, P.P.D.; Edmonds, L.K.; Koh, T.H.H.G. Carey-Fineman-Ziter syndrome: A spectrum of presentations. Case Rep. Périnat. Med. 2012, 1, 99–102. [Google Scholar] [CrossRef]
- Sewry, C.A.; Laitila, J.M.; Wallgren-Pettersson, C. Nemaline myopathies: A current view. J. Muscle Res. Cell Motil. 2019, 40, 111–126. [Google Scholar] [CrossRef] [Green Version]
- Dubowitz, V.; Sewry, C.A.; Oldfors, A. Muscle Biopsy E-Book: A Practical Approach; Elsevier Health Sciences: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Amburgey, K.; McNamara, N.; Bennett, L.R.; McCormick, M.E.; Acsadi, G.; Dowling, J.J. Prevalence of congenital myopathies in a representative pediatric united states population. Ann. Neurol. 2011, 70, 662–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehtokari, V.-L.; Gardberg, M.; Pelin, K.; Wallgren-Pettersson, C. Clinically variable nemaline myopathy in a three-generation family caused by mutation of the skeletal muscle alpha-actin gene. Neuromuscul. Disord. 2018, 28, 323–326. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.; Schnell, C.; Strickland, C.D.; Shield, L.K.; Morgan, G.; Iannaccone, S.T.; Laing, N.G.; Begges, A.H.; North, K.N. Nemaline myopathy: A clinical study of 143 cases. Ann. Neurol. 2001, 50, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Bahra, A.; May, A.; Goadsby, P.J. Cluster headache: A prospective clinical study with diagnostic implications. Neurology 2002, 58, 354–361. [Google Scholar] [CrossRef] [PubMed]
- May, A. Diagnosis and Clinical Features of Trigemino-Autonomic Headaches. Headache J. Head Face Pain 2013, 53, 1470–1478. [Google Scholar] [CrossRef] [PubMed]
- Baseer, M.; Alqhtani, N.; Alshammery, D.; AlOtaibi, N.; AlZamil, F.; Allaboon, A.; AlTuwaijri, D. Correlations between mandibular asymmetries and temporomandibular disorders: A systematic review. J. Int. Soc. Prev. Community Dent. 2021, 11, 481–489. [Google Scholar] [CrossRef]
- Murphy, M.K.; MacBarb, R.F.; Wong, M.E.; Athanasiou, K.A. Temporomandibular Disorders: A Review of Etiology, Clinical Management, and Tissue Engineering Strategies. Int. J. Oral Maxillofac. Implant. 2013, 28, e393–e414. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Leung, Y. Temporomandibular Disorders: Current Concepts and Controversies in Diagnosis and Management. Diagn. 2021, 11, 459. [Google Scholar] [CrossRef] [PubMed]
- Toh, A.Q.J.; Chan, J.L.H.; Leung, Y.Y. Mandibular asymmetry as a possible etiopathologic factor in temporomandibular disorder: A prospective cohort of 134 patients. Clin. Oral Investig. 2021, 25, 4445–4450. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Okamoto, K.; Maruyama, T. Facial asymmetry in patients with cervicobrachial pain and headache. J. Oral Rehabil. 2001, 28, 1009–1014. [Google Scholar] [CrossRef]
- Ahn, S.-J.; Baek, S.-H.; Kim, T.-W.; Nahm, D.-S. Discrimination of internal derangement of temporomandibular joint by lateral cephalometric analysis. Am. J. Orthod. Dentofac. Orthop. 2006, 130, 331–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trpkova, B.; Major, P.; Nebbe, B.; Prasad, N. Craniofacial asymmetry and temporomandibular joint internal derangement in female adolescents: A posteroanterior cephalometric study. Angle Orthod. 2000, 70, 81–88. [Google Scholar] [CrossRef]
- Chan, B.H.; Leung, Y. SPECT bone scintigraphy for the assessment of condylar growth activity in mandibular asymmetry: Is it accurate? Int. J. Oral Maxillofac. Surg. 2018, 47, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Chew, M.T. Spectrum and management of dentofacial deformities in a multiethnic Asian population. Angle Orthod. 2006, 76, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Chew, M.T. Soft and hard tissue changes after bimaxillary surgery in Chinese Class III patients. Angle Orthod. 2005, 75, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.M. Hemispheric and facial asymmetry: Gender differences. Laterality Asymmetries Body Brain Cogn. 2000, 5, 251–258. [Google Scholar] [CrossRef]
- Ferrario, V.F.; Sforza, C.; Miani, A.; Tartaglia, G. Craniofacial morphometry by photographic evaluations. Am. J. Orthod. Dentofac. Orthop. 1993, 103, 327–337. [Google Scholar] [CrossRef]
- Haraguchi, S.; Iguchi, Y.; Takada, K. Asymmetry of the Face in Orthodontic Patients. Angle Orthod. 2008, 78, 421–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ercan, I.; Ozdemir, S.T.; Etoz, A.; Sigirli, D.; Tubbs, R.S.; Loukas, M.; Guney, I. Facial asymmetry in young healthy subjects evaluated by statistical shape analysis. J. Anat. 2008, 213, 663–669. [Google Scholar] [CrossRef]
- Shaner, D.J.; Peterson, A.E.; Beattie, O.B.; Bamforth, J.S. Assessment of soft tissue facial asymmetry in medically normal and syndrome-affected individuals by analysis of landmarks and measurements. Am. J. Med. Genet. 2000, 93, 143–154. [Google Scholar] [CrossRef]
- Lee, M.-S.; Chung, D.H.; Lee, J.-W.; Cha, K.-S. Assessing soft-tissue characteristics of facial asymmetry with photographs. Am. J. Orthod. Dentofac. Orthop. 2010, 138, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Thiesen, G.; Gribel, B.F.; Freitas, M.P. Facial asymmetry: A current review. Dent. Press J. Orthod. 2015, 20, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Bailey, L.J.; Haltiwanger, L.H.; Blakey, G.H.; Proffit, W.R. Who seeks surgical-orthodontic treatment: A current review. Int. J. Adult Orthod. Orthognath. Surg. 2001, 16, 280–292. [Google Scholar]
- Sándor, G.K.; McGuire, T.P.; Ylikontiola, L.P.; Serlo, W.S.; Pirttiniemi, P.M. Management of Facial Asymmetry. Oral Maxillofac. Surg. Clin. N. Am. 2007, 19, 395–422. [Google Scholar] [CrossRef]
- Iyer, J.; Hariharan, A.; Cao, U.M.N.; Tran, S.D. Acquired Facial, Maxillofacial, and Oral Asymmetries—A Review Highlighting Diagnosis and Management. Symmetry 2021, 13, 1661. [Google Scholar] [CrossRef]
- Cheong, Y.-W.; Lo, L.-J. Facial asymmetry: Etiology, evaluation, and management. Chang. Gung. Med. J. 2011, 34, 341–351. [Google Scholar]
- Urban, S.D.; Waite, P.D. Management of Facial Asymmetry. Am. J. Cosmet. Surg. 2005, 22, 249–259. [Google Scholar] [CrossRef]
- Bishara, S.E.; Burkey, P.S.; Kharouf, J.G. Dental and facial asymmetries: A review. Angle Orthod. 1994, 64, 89–98. [Google Scholar] [CrossRef]
- Reis, V.A.; Zaidel, D.W. Functional asymmetry in the human face: Perception of health in the left and right sides of the face. Laterality Asymmetries Brain Behav. Cogn. 2001, 6, 225–231. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| cranial nerves | oculomotor nerve trochlear nerve trigeminal nerve facial nerve |
| developmental disorders | Parry–Romberg syndrome (PRS) Asymmetric crying facies (ACF) Dyke–Davidoff–Masson syndrome Harlequin syndrome Klippel–Feil syndrome CHARGE syndromeHOXA1 syndromes Isolated Hereditary Congenital Facial Paresis (HCFP) |
| myopathies | Facioscapulohumeral muscular dystrophy Titinopathy Myotonic dystrophy (MD) type 1 Nemaline myopathy Carey–Fineman–Ziter syndrome (CFZS) |
| headache | cluster headache masticatory muscles headache |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chojdak-Łukasiewicz, J.; Paradowski, B. Facial Asymmetry: A Narrative Review of the Most Common Neurological Causes. Symmetry 2022, 14, 737. https://doi.org/10.3390/sym14040737
Chojdak-Łukasiewicz J, Paradowski B. Facial Asymmetry: A Narrative Review of the Most Common Neurological Causes. Symmetry. 2022; 14(4):737. https://doi.org/10.3390/sym14040737
Chicago/Turabian StyleChojdak-Łukasiewicz, Justyna, and Bogusław Paradowski. 2022. "Facial Asymmetry: A Narrative Review of the Most Common Neurological Causes" Symmetry 14, no. 4: 737. https://doi.org/10.3390/sym14040737