

Catalytic Asymmetric Synthesis of C-Chiral Phosphonates

V.P. Kukhar’ Institute of Bioorganic Chemistry and Petrochemistry, NAS of Ukraine, Murmanska St. 1, 02094 Kyiv, Ukraine
*
Authors to whom correspondence should be addressed.
Symmetry 2022, 14(9), 1758; https://doi.org/10.3390/sym14091758
Submission received: 2 July 2022
/
Revised: 2 August 2022
/
Accepted: 10 August 2022
/
Published: 23 August 2022
(This article belongs to the Special Issue Asymmetric Centers in Optically Active Molecules Containing a Heteroatom)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The current review is devoted to the achievements in the development of methods for the catalytic asymmetric synthesis of phosphonates containing a chiral center in the side chain. C-chiral phosphonates are widely represented among natural compounds with various biological activities as insecticides, herbicides, antibiotics, and bioregulators. Synthetic representatives of this class have found practical application as biologically active compounds. The review summarizes methods of asymmetric metal complex catalysis and organocatalysis as applied to such reactions as phospha-aldol reaction, two-component and three-component phospha-Mannich reaction, phospha-Michael reaction, as well as hydrogenation of unsaturated phosphonates and phosphine oxides, ketophosphonates, and iminophosphonates. Methods for the asymmetric hydride reduction of C=X phosphonates (X=O, S, NR) are also discussed in detail. The review presents updated literature reports, as well as original research by the author.
1. Introduction
C-chiral phosphonates containing asymmetric centers in the side chain are the most numerous and interesting type of organophosphorus compounds in terms of biological activity [1,2]. Various representatives of C-chiral phosphonates have been found among natural compounds. In addition, a great number of optically active C-chiral functionalized phosphonates have been synthesized that are of great biological importance [3,4]. For example, various C-P-phosphonates, amino- and hydroxyphosphonates, hydroxy-2-aminoethylphosphonic acid HO-AEP, dihydroxyphosphonic acid FR-33289, 1,5-dihydroxy-2-oxopyrrolidiphosphonic acid SF-2312, phosphonotrixine (PTX), etc. Many of these compounds have attracted attention for their interesting pharmacological properties as antibacterial, antiviral, anticancer drugs, antibiotics, enzyme inhibitors, amino acid mimetics, pesticides [5,6]. The physiological activity of phosphonates containing a hydrolytically stable P–C bond was attributed, on the one hand, to their structural similarity to biologically important phosphates, in which the P–O bond is replaced by a P–C bond, and on the other hand, phosphonic acids are structural analogs of carboxylic acids. Replacing the carboxyl group in “normal” hydroxycarboxylic acids with a phosphonate group causes them to exhibit an inhibitory effect on enzymes or receptors to which natural amino or hydroxycarboxylic acids normally bind. Acting as antagonists of these acids, they inhibit enzymes involved in the metabolism of carboxylic acids and thus affect physiological processes.
The biological activity of phosphonic acids depends on the absolute configuration of the stereogenic α–carbon atom. For example, of the four diastereomers of the antibiotic alaphospholin, the diastereomer having the (S,R) configuration exhibits the highest activity against pathogenic microorganisms. Three other stereoisomers are significantly inferior to it in activity (see [5,6] and references cited therein). Derivatives of phosphinic acid, β–hydroxyphosphonates, α–hydroxy–β–aminophosphonates, polyhydroxyphosphonates, and difluoromethylene-phosphonates show high biological activity as enzyme inhibitors. Some hydroxyphosphonates with high antitumor activity are used to treat cancer. Bisphosphonates are attracting considerable interest as the most effective drugs in the treatment of osteoporosis. Recently, bis-phosphonates containing chiral centers in the side chain have been obtained. These compounds have shown interesting biological activity [7,8,9,10,11,12,13,14,15].
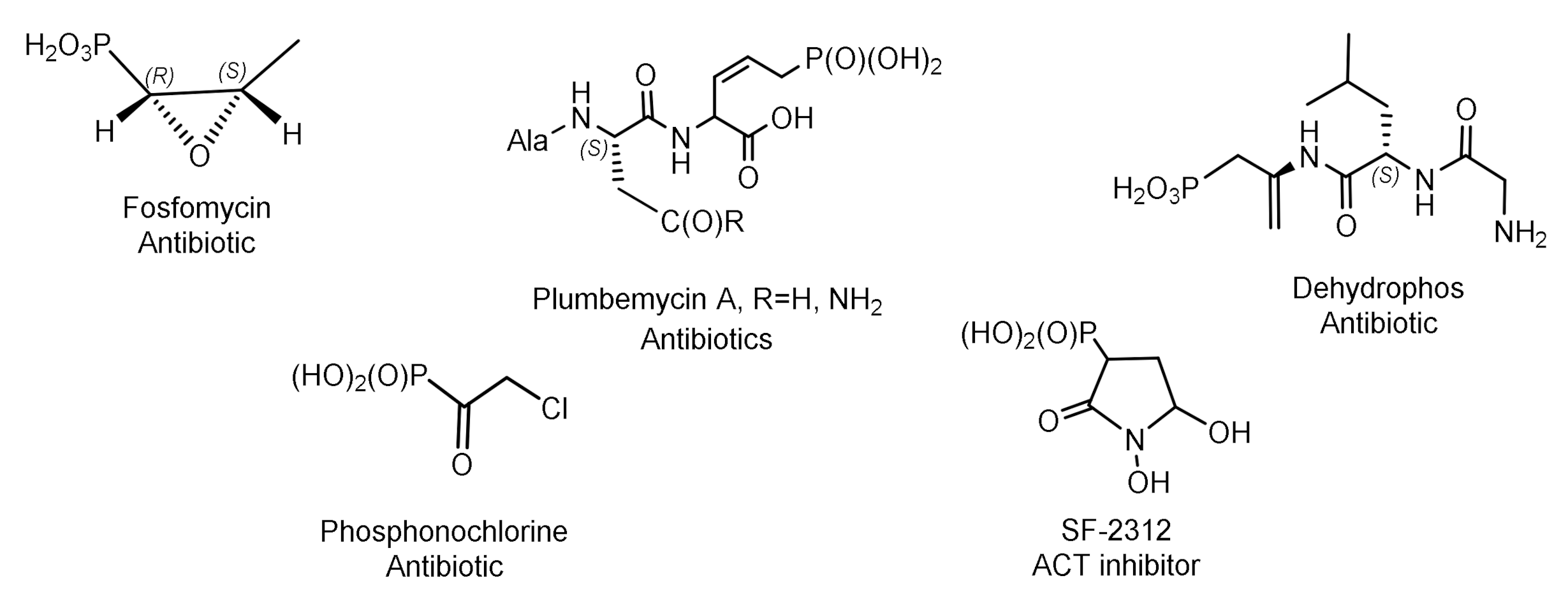
Some of natural phosphonates have found commercial applications in agriculture and medicine. For example, aminophosphonic acids and phosphonopeptides exhibit antibacterial, antitumor, antiviral, and antifungal activity [7]. Their representatives are herbicides and plant growth regulators. Various natural phosphonates have been found and isolated from bacteria, while the antibiotic phosphonochlorine has been found in soil-isolated fungal strains such as Fusarium avenaceum, Fusarium oxysporum, Fusarium tricinctum, and Talaromyces. flavus [8]. The phosphonate antibiotic SF-2312 was found in the actinomycete Micromonospora. SF-2312 is active under anaerobic conditions and is a potent low nanomolar enolase inhibitor [9]. It is also moderately active against some Gram-negative bacteria, and its synergistic effect with glucose-6-phosphate has been observed against Staphylococcus aureus and Escherichia coli (Figure 1).
Fosfomycin is used to treat urinary tract diseases and prostatitis; it is effective as an antibiotic against gram-positive and gram-negative MDR and XDR bacteria [10]. Fosmidomycin and FR-900098 have been proposed as drugs against Plasmodium, the causative agent of malaria [11]. Phosphinothricin was first synthesized as a racemate, which is used as the active ingredient in several commercial herbicides (Figure 2).
A number of ciliathrin derivatives are known that play an important role in the life of various organisms and exhibit interesting biological properties. Phosphonopyruvate is a key substrate in the synthesis of many naturally occurring phosphonates: ciliathrin, phosphonoalanine, (R)-2-amino-1-hydroxyethylphosphonic acid, phosphonoacetaldehyde, phosphonomethylmalic acid, and 2-keto-4-hydroxy-5-phosphonopentanoic acid [13,14]. Cyanophos, which has pesticidal properties, was found in Streptomyces regensis (Figure 3) [15].
Of great interest in recent applications is new technologies and approaches for the use of chiral biologically active compounds, such as, for example, a chiral switch, which significantly expands the field of chiral drugs and opens up new opportunities for their patenting. Of great interest are the available methods for the synthesis of chiral compounds of high enantiomeric purity. Particular attention is paid to catalytic methods for the synthesis of chiral phosphonates using metal complex catalysis and organocatalysis. In this review, we analyze progress in this area over the past 5–7 years.
2. Methods for the Synthesis of Chiral Phosphonates
Recent years have seen a steady growth in the use of chiral organophosphorus catalysts in asymmetric synthesis. To obtain enantiomerically pure C-chiral phosphonates, various methods can be used, including asymmetric metal complex catalysis, stereo-differentiation reactions, enzymatic kinetic resolution, chromatographic separation. Asymmetric catalysis using transition metal complexes containing chiral ligands PAMP, DIPAMP, DIOP, CHIRAPHOS, and others are widely used in the asymmetric synthesis of C-chiral phosphonates. [16,17,18,19,20,21]. Over the last few years, great success has been achieved in the asymmetric catalytic methods for the preparation of C-chiral phosphonates. The most important method for the synthesis of this type of organophosphorus is the phosphonylation of compounds containing a C=X group, i.e., carbonyl compounds, imines, and compounds with an activated C=C bond. There are several known methods of asymmetric phosphonylation of C=X compounds with dialkyl phosphites: phosphoaldol addition, phospha-Mannich reaction, phospha-Michael reaction, reaction of phosphonate carbanions with aldehydes, ketones, imines. Phospha-aldol reaction (the Abramov reaction) usually leads to the formation of hydroxyphosphonates, while the phospha-Mannich reaction is one of the most convenient methods for the synthesis of chiral α–aminoalkylphosphonates (Figure 4).
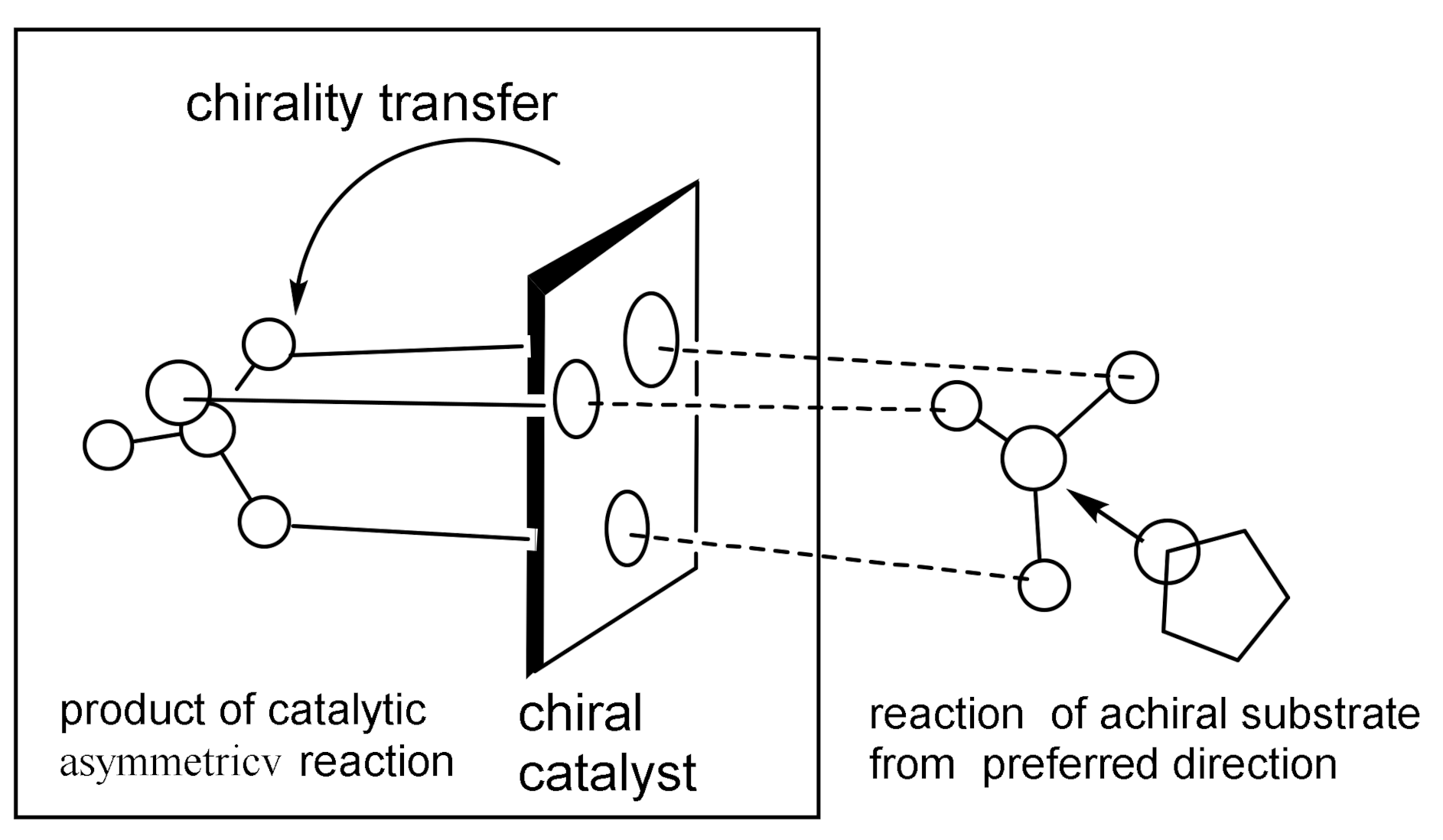
As shown in Figure 5, the coordination of a source of chirality (a chiral phosphine ligand) with a chiral catalyst creates an asymmetric center from which chirality is transferred to the reacting substrate, resulting in the formation of an optically active reaction product [18,19,20,21]. Therefore, the use of catalytic methods in the asymmetric synthesis of organic compounds is highly economical, since the use of a relatively small amount of a chiral catalyst makes it possible to obtain a largely superior amount of the reaction product.
2.1. Addition of Phosphorus Nucleophiles to C=X Compounds
The reactions of unsaturated C=X compounds with phosphorus nucleophiles, which donate their pair of electrons to form a new bond, are the most important in organophosphorus chemistry. Through these reactions, the largest number of organophosphorus compounds were synthesized, many of which have gained great practical importance in the chemistry of phosphorus. The most important place is occupied by the reactions of addition to C=X compounds: phospha-aldol reaction, phospha-Mannich reaction, phospha-Michael reaction.
The phospha-aldol reaction is a special case of the aldol reaction. The most common case of the P-aldol reaction is the addition of dialkyl phosphites to aldehydes or ketones catalyzed by Brønsted bases or Lewis acids (Figure 6) [22].
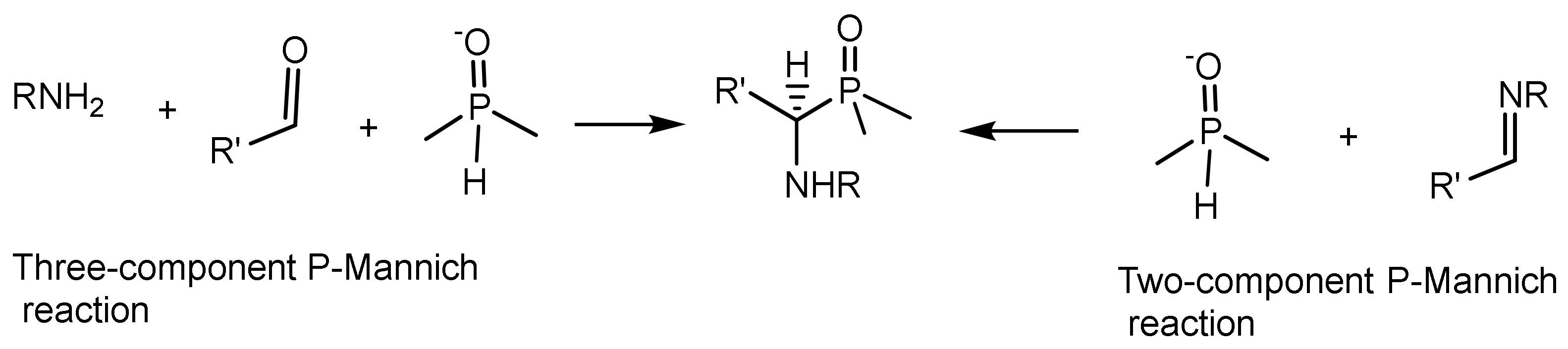
Phospha–Mannich reaction (or Kabachnik–Fields reaction). By analogy with the classical Mannich reaction, in this case, the reaction proceeds in the ternary system aldehyde, amine, and dialkyl phosphite, which is considered as an analog of the carbonyl component. Upon mixing reagents, primary amines or anilines often easily react with aldehydes to form aldimines, and the three-component phospha-Mannich reaction turns into a two-component one. Therefore, the addition reaction of dialkyl phosphites to aldimines can be considered as a phospha-Mannich reaction [22]. There are two variants of the P-Mannich reaction, which are (a) the two-component and (b) the three-component versions (Figure 7). Several variants of the phospha-Mannich reaction are possible: (a) the reaction of phosphites with imines; (b) reaction of phosphites with aldehydes and amines; (c) addition of secondary phosphines to imines in the presence of proton donor reagents or Lewis acids; and (d) adding secondary silylphosphines to imines.
The phospha–Michael reaction (or phospha–Michael addition) is a general addition reaction of phosphorus nucleophiles to an acceptor-substituted alkene or alkyne. A special case of the phospha-Michael reaction is the Pudovik reaction. which consists in the addition of H–phosphite to an alkene activated by an electronegative group under the catalytic action of bases. The phospha-Michael reaction is an important method for the synthesis of compounds with a P–C bond. There are several versions of such reactions, namely, asymmetric phosphonylation reactions proceeding with the transfer of chirality from a chiral reagent to an achiral substrate. Phosphonylation reactions proceed under the stereochemical control of a chiral substituent in the substrate or reagent and under the conditions of asymmetric metal complex catalysis or organocatalysis.
In this review, we consider the addition reactions of phosphorus nucleophiles to compounds containing an activated C=C bond, proceeding under the control of chiral catalysts or chiral transition metal complexes. The addition of nucleophilic phosphorus reagents to unsaturated substrates is catalyzed by both chiral bases and acids (Figure 8) [23].
2.1.1. Phospha-Aldol Reaction
The phospha-aldol reaction is a method of hydroxyphosphonylation of carbonyl compounds with trivalent phosphorus acid esters, usually in the presence of catalysts, which can be either Brønsted bases or Lewis acids. In the case of chiral catalysts, an asymmetric variant of the reaction is possible [7,22]. Metal complex catalysis, organocatalysis, and biocatalysis are used, leading to the formation of functionalized molecules with high enantiomeric purity and, therefore, having great potential in synthetic chemistry [24,25,26,27].
Several types of asymmetric catalysts have been proposed for the stereochemical control of the phospha-aldol reaction. For example, Shibazaki reported that heterobimetallic complexes containing chiral bis-dinaphthol ligands efficiently catalyze asymmetric hydrophosphorylation. The bimetallic binoyl complexes LLB and ALB catalyze the addition of dimethyl phosphite to aromatic aldehydes, giving α–hydroxyphosphonates whose optical purity reached 90% ee. The reaction proceeds through the formation of an intermediate complex LLB with phosphite and aldehyde, in which the nucleophilic attack of the phosphite on the aldehyde occurs. The first metal-complex enantioselective hydrophosphonylation of aldehydes was described by Shibazaki and coworkers using heterobimetallic complexes [AlLi(binaphthoxide)2] [27,28,29] (Figure 9). Shibuya used a Sharpless catalyst generated from diisopropyl L-tartrate and Ti(OPr-i)4 to initiate the P-aldol reaction. The reaction of para-substituted benzaldehyde with diethyl phosphite in the presence of 20 mol% catalyst in ether solution gave enantiomerically enriched (R)-hydroxyphosphonate with a yield of 75% and 53% ee. At the same time, LLB catalysis under the same conditions gave hydroxyphosphonates in 69–98% yield and 20–82% ee (Figure 9).
Nakajima used the atropisomeric ligand BINAPO as a phospha-alldol reaction catalyst for the enantioselective phosphonylation of aldehydes. The effectiveness of chiral BINAPO as a catalyst for enantioselective addition has been studied in detail [30,31,32]. Chiral phosphine oxides (Lewis bases) catalyze the enantioselective phosphonylation of aldehydes with trialkyl phosphites in combination with silicon tetrachloride, resulting in optically active hydroxyphosphonates (Figure 10). The reaction conditions were optimized using benzaldehyde as a model substrate in the presence of various trialkyl phosphites in dichloromethane at −78 °C. Among several Lewis bases tested, the atropisomeric diphosphine dioxide (S)-BINAPO, used in combination with diisopropylethylamine as a base and additive, was found to provide the best catalytic activity, yielding the desired α–hydroxyphosphonates in 91% yields and up to 52% enantioselectivity. The addition of SiCl4 to the reaction mixture is a key parameter to ensure high stereoselectivity of the reaction. This study also shows that higher yields are obtained with trialkyl phosphites, which carry smaller alkyl groups, while triphenyl phosphite does not react at all (Figure 10) [30].
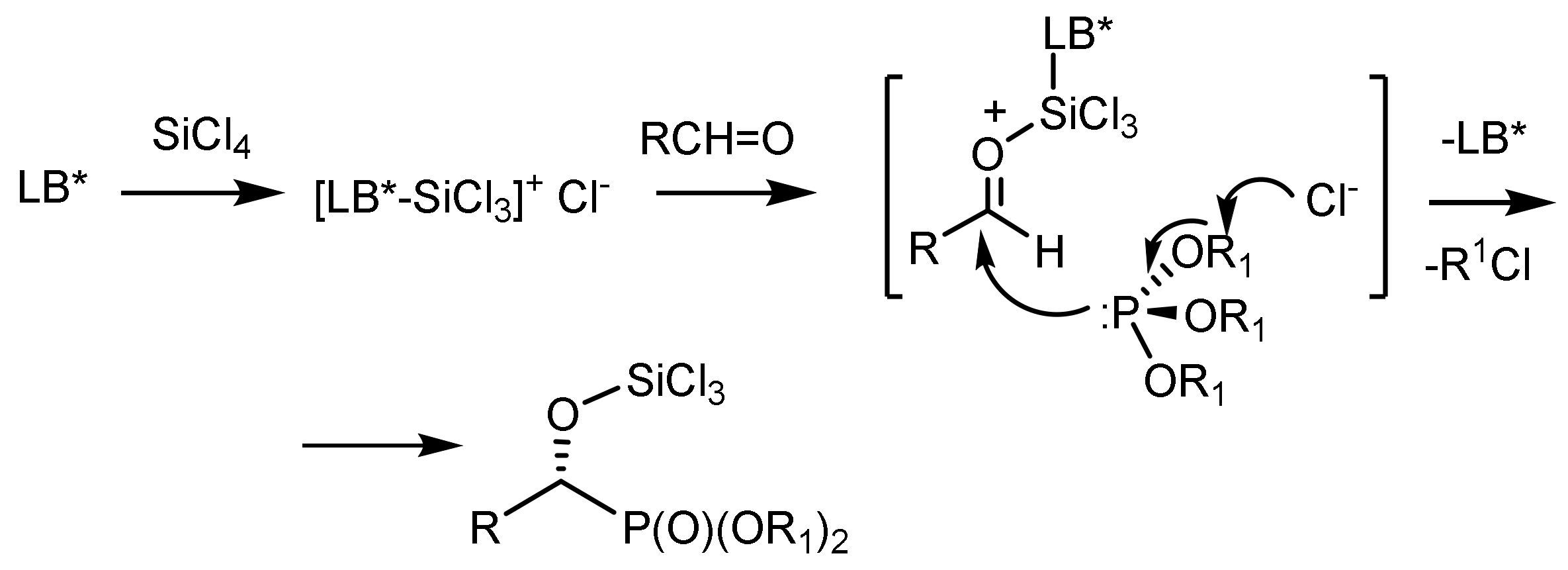
The reaction mechanism, confirmed by 31P{1H} NMR analysis, involves the coordination of a chiral phosphine oxide ligand (LB*) with silicon tetrachloride, which leads to the formation of the cationic complex [(LB*)/SiCl3]+Cl−. Subsequently, later reactive compounds can effectively activate prochiral aldehydes and therefore promote an enantioselective nucleophilic attack of the trialkyl phosphite to form a chiral trichlorosilylated α–hydroxyphosphonate with simultaneous release of the corresponding alkyl chloride via a nucleophilic substitution reaction to form a P=O group (Figure 11) [33,34,35].
Laborde, Sevrain et al. [33,34,35] demonstrated that 1,10–dibenzyl-bis-(triazolyl)diphenylphosphine is effective for SiCl4-mediated enantioselective hydrophosphonylation of aldehydes with trialkyl phosphites. After optimizing the reaction conditions, the bulkier diisopropyl phosphite proved to be a better phosphonylating agent than diethyl phosphite, yielding the corresponding enantioenriched α-hydroxyphosphonates in excellent yields and moderate-to-good selectivity.
The bifunctional chiral complex Al(III)-BINOL (or Ti(OPr-i)4-BINOL) containing two chiral centers exhibited very good enantioselectivity in phospha-aldol reaction (Figure 12) [36]. A number of aromatic, heteroaromatic, α,β-unsaturated, and aliphatic aldehydes were hydrophosphonylated using this catalyst to form α-hydroxyphosphonates in yields up to 99% and enantioselectivity up to 87% ee [36]. The effectiveness of this catalyst is probably explained by coordination of asymmetric inductions of the chiral Lewis base (cinchona alkaloid) with the chiral Lewis acid (the BINOL-Ti complex) (Figure 13). In this case, the asymmetric inductions act in one direction, which contributes to the achievement of high catalytic efficiency and enantioselectivity (Figure 14) [7,36].
Benjamin List et al. [37] used a chiral disulfonimide catalyst to carry out the catalytic enantioselective Abramov reaction. Several functionalized α-hydroxyphosphonates have been synthesized in good yields with excellent enantiomer ratios up to >99 ee (Figure 15).
Chiral phosphine oxides (Lewis bases) paired with silicon tetrachloride catalyze the enantioselective phosphonylation of aldehydes with trialkyl phosphites, which leads to optically active α–hydroxyphosphonates with moderate enantioselectivities. 31P NMR analysis of benzaldehyde phosphonylation with triethyl phosphite confirms the proposed reaction mechanism [38,39,40].
The strategy of synergistic activation by two or more reaction sites is a convenient approach to increase the stereoselectivity of asymmetric catalysis. Double stereoselectivity was achieved in the case of the reaction of chiral di(1R,2S,5R)-mentylphosphite with chiral 2,3-D-isopropylidene-(R)–glyceraldehyde. The reaction of glyceraldehyde with dimentyl phosphite in the presence of a chiral catalyst (R)-ALB proceeds under the stereochemical control of three chiral inductors and is the most stereoselective, which indicates the additivity of the stereoselectivities of chiral reagents under the conditions of a stereodifferentiating reaction. (Figure 16) [32].
Optically active aluminum-Salalen complexes are effective catalysts for the hydrophosphonylation of aldehydes, giving the corresponding α–hydroxyphosphonates with good enantioselectivity. The high enantioselectivity of the Salalen complex is attributed to its unique structure, which has a distorted triangular bipyramidal configuration, which allows the Salalen ligand to adopt a cisoid structure in which the chiral amino group is close to the aluminum atom [38,39,40]. The mechanism of the hydrophosphonylation reaction of dimethyl phosphite with benzaldehyde catalyzed by the Al(Salalen) complex was studied by DFT (discrete fourier transform) and ONIOM methods. The authors [40] came to a conclusion that the stereochemistry of the reaction catalyzed (own N–layered integrated molecular orbital and molecular mechanics) methods by the chiral Al(Salalen) complex is controlled by the steric repulsion between the ortho t-Bu groups of the ligand and dimethylphosphite, as well as the coordination mode of dimethylphosphite to the catalyst. The calculations reproduce the predominant (S)-product with high ee, confirming the experimental observations. The high enantioselectivity of the complex is explained by its unique structure, which has deformed trigonal-bipyramidal configuration. Such configuration allows the Salalen ligand to occupy a cisoid position in which the chiral amino group is located close to aluminum atom [39,40,41,42,43,44].
In another case, a chiral, trigonal-pyramidal aluminum(Salalen) complex was studied, in which the asymmetric nitrogen atom is bonded to a metal ion and the N-methyl group is cis to the chlorine ligand controlling the asymmetric reaction. The complex effectively catalyzes the enantioselective hydrophosphonylation of various aldehydes with dimethyl phosphite [42,43]. Electron-donor groups in the para-position to the OH group, as well as an increase in the volume of substituents R’ in the ortho position of the aromatic substituent of the Salalen complex (t-Bu, Ad, Et2MeC), increased the enantioselectivity of the reaction. Calculations confirmed the predominant formation of the (S) product formed during the reaction (Figure 17).
A new strategy for sequential asymmetric C-H oxidation/phosphonylation of primary alcohols has been developed [44]. This methodology involves a combination of oxidation with air oxygen catalyzed by Cu(I)/TEMPO/NMI (N-methylimidazole) with chiral enantioselective addition catalyzed by Al(Salalen). Various primary alcohols, including benzyl alcohols, allyl alcohols, propargyl alcohols, alkyl alcohols, and other alcohols containing oxygen-, sulfur-, and nitrogen-containing heterocycles, were readily converted to chiral α-hydroxyphosphonates in good yields and high enantioselectivity. Using this methodology, various biologically active compounds have been synthesized, including a CD45 inhibitor and plant growth regulators (Figure 18).
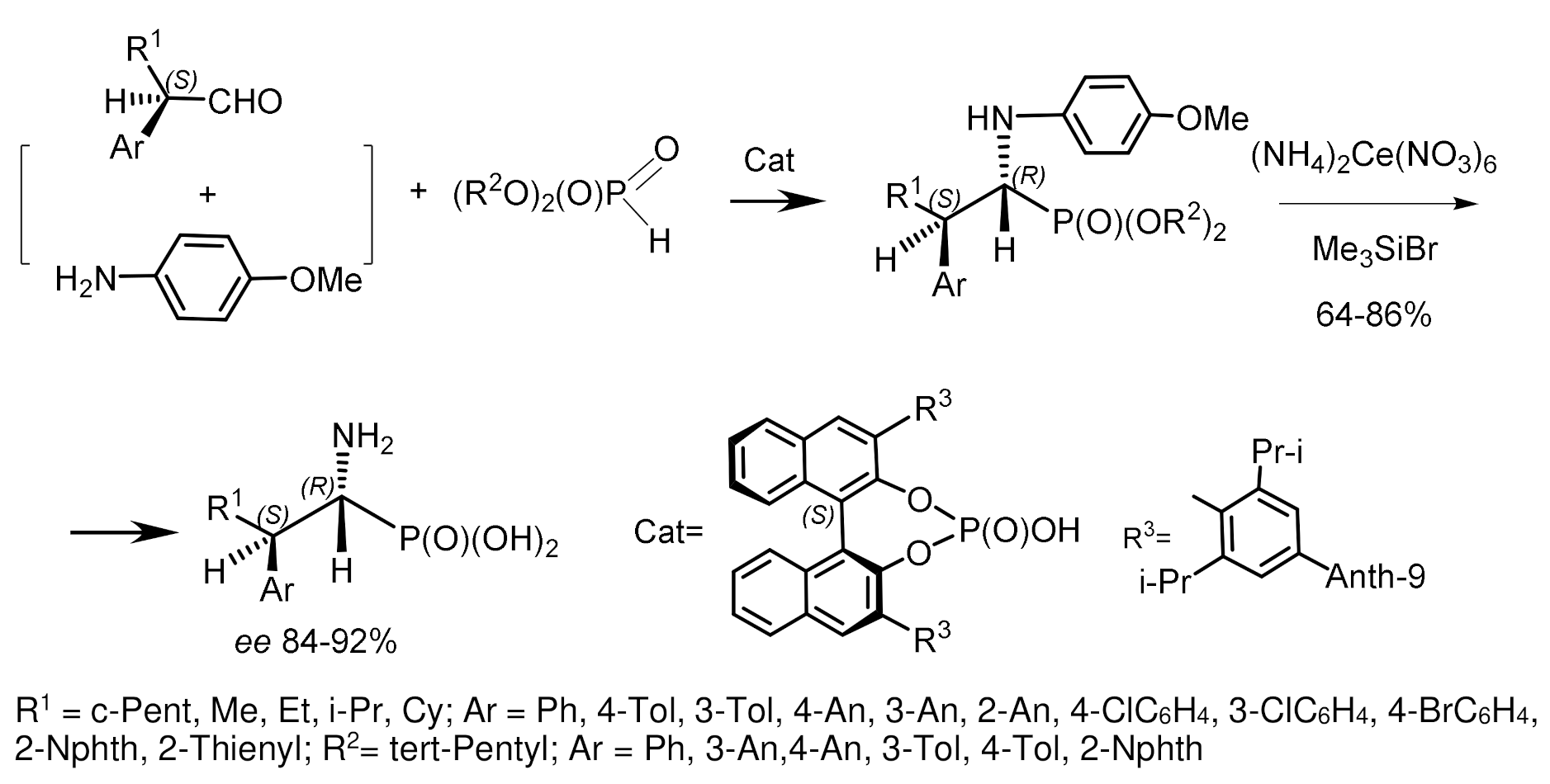
Chiral catalysts for the asymmetric hydrophosphonylation of aldehydes have been proposed, which are Al(III) complexes with tridentate Schiff bases. For example, the tridentate Schiff base obtained from L-Valinol and 3,5-di-tert-butylsalicylicaldehyde reacted in situ with Et2AlCl to form complexes that catalyzed the reaction of dialkyl phosphites with various alkyl and arylaldehydes and trifluoromethyl ketones [45,46,47,48,49,50] with high enantioselectivity; the reaction with acetophenone proceeded with low enantioselectivity, but with high yields. The counterions of Schiff’s base/Al(III) complexes had a significant effect on the enantioselectivity of the reaction. Yamamoto et al. synthesized α–hydroxy- and α–aminophosphonates in high yields and high enantioselectivity using bis(8-quinolinato)(TBOx) aluminum complex (0.5–1 mol%) [47]. Under optimized reaction conditions, it was found that electron-enriched aldehydes, compared with electron-deficient aromatic aldehydes, are more reactive and selective. Aliphatic aldehydes also reacted with satisfactory selectivity. All reactions proceeded very fast, with high yields and enantioselectivities. Reducing the catalyst loading to 0.5 mol% had no effect on enantioselectivity and yields. The best results were obtained with phosphites containing trifluoroethyl groups (Figure 19) [48].
Triiminophosphorane, generated in situ from chiral P-spiro-tetraaminophosphonium salt and potassium tert-butoxide, proved to be a very efficient catalyst for the phosphaaldol reaction. Iminophosphoranes with electron-donating substituents in the aromatic ring were catalytically active even at a loading of catalyst up to 1 mol% and a decrease in the temperature of the reaction mixture to −98 °C (Figure 20). The investigation of reaction mechanism showed that the catalyst molecule simultaneously interacts through hydrogen bonds with the nucleophile and electrophile, transferring a proton to the aldehyde along with the formation of a carbon–carbon bond. The most favorable transition structure results from the minimum energies of geometric distortion [49].
The reaction allows phosphonilation of aliphatic, heteroaromatic, and aromatic aldehydes with high yields of corresponding α–hydroxyphosphonates and optical purity reaching 99% ee. The authors suggested that the reaction proceeds through the formation of a highly active salt of dimethyl phosphite with a chiral tetraaminophosphonium cation, which is responsible for the addition stereochemistry.
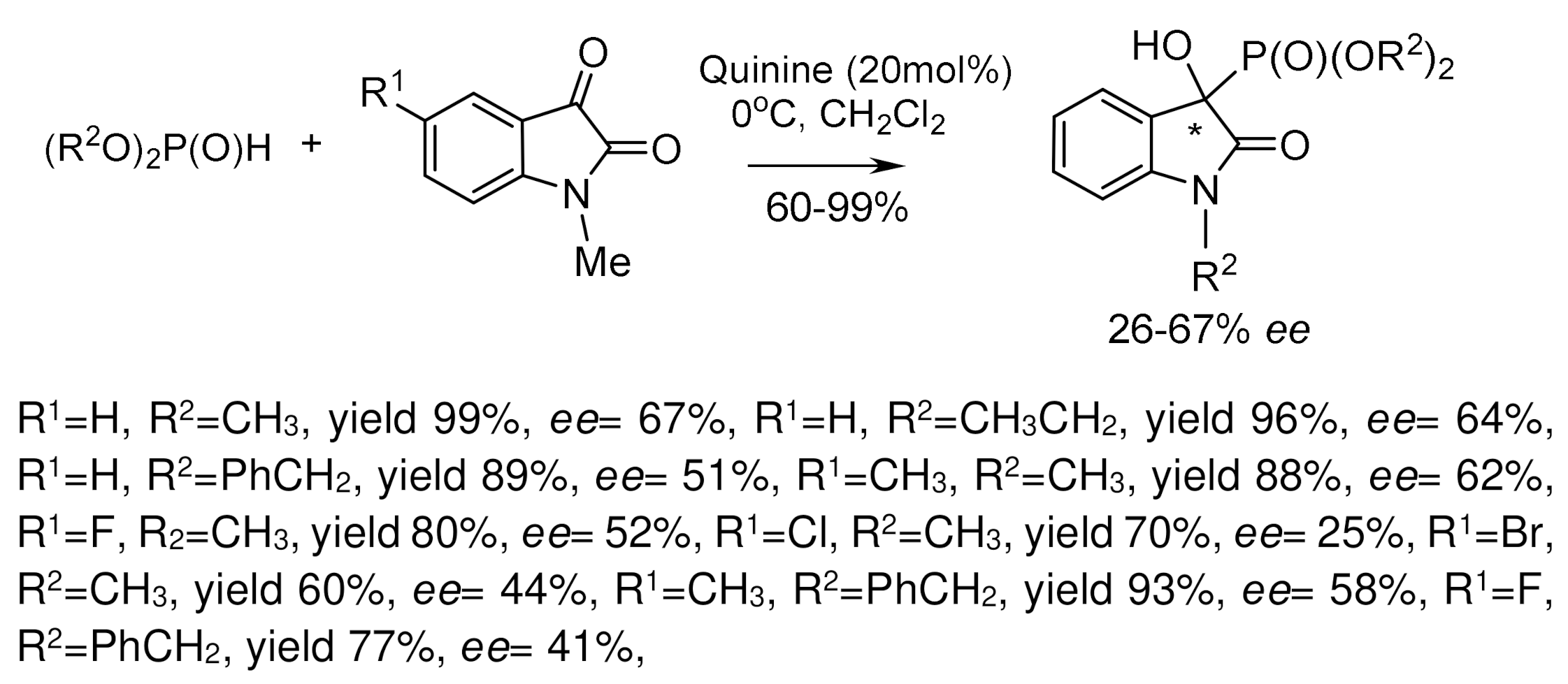
Along with metal-complex catalysts for the asymmetric phospha-aldol reaction, organo-catalysts such as quinine and other alkaloids were also used [50]. For example, one of the recently published articles shows the possibility of using quinine as a catalyst for the addition of H-phosphonates to activated ketones with satisfactory enantioselectivity (67% ee and high yields). Phospha-aldol reaction of diphenylphosphite to N-alkylated isatins catalyzed by quinine and quinidine proceeded successfully [39]. For example, crystallization of scalemic dimethylhydroxy-(2-nitrophenyl)methylphosphonate from diethyl ether gave an optically pure crystalline product having the (S)-absolute configuration, while the (R)-product remained in solution (Figure 21) [51].
Good results in the hydrophosphonylation of aldehydes were achieved, using a bifunctional catalyst that simultaneously activated the aldehyde (through interaction with the H-compound) and the H-phosphonate (through the polarization of the P-H bond), previously proved to be an effective catalyst for this reaction (Figure 22) [52].
Zhao et al. [53] reported an enantioselective cross-aldol reaction between enolizable aldehydes and α-ketophosphonates. This reaction is a convenient enantioselective method for the synthesis of tertiary β–formyl–α–hydroxyphosphonates. The present reaction is a convenient enantioselective method for the synthesis of tertiary β–formyl–α–hydroxyphosphonates. The use of quinine as an organocatalyst made it possible to achieve excellent enantioselectivity of the cross-aldol reaction. Some β–formyl–α–hydroxyphosphonates showed biologically active properties and inhibited the growth of malignant tumor cells. As a result of selection, 9-amino-9-deoxy-epiquinine in the presence of 4-methoxybenzoic acid (MBA) was chosen as the most effective catalyst. The reaction was carried out in toluene at 0 °C. It was found that the size of the ester alkyl group in the phosphonate does not affect the yields and enantioselectivity of the reaction [53]. The products were obtained in good yields and enantioselectivities using cinchonidine as a bifunctional catalyst and pyruvic nitrile as a source of the cyano group (Figure 23) [53,54].
Kong et al. [55] studied the enantioselective synthesis of tertiary α-hydroxyphosphonates. Reactions between acylphosphonates and trimethylsilyl cyanide were carried out in the presence of a bifunctional catalyst, a quinine derivative with thiourea, containing two chiral groups. The resulting Me3Si ester of cyanophosphonate was converted to cyanohydrinphosphonate by acid hydrolysis. The best results were obtained in the presence of p-nitrophenol. Modifications of the R1 group in the acylphosphonate, from aromatic radicals with electron-withdrawing groups to donor groups, had little effect on the yields and enantioselectivity. Interestingly, the enantioselectivity decreased with increasing volume of the R2 groups. In addition, the reaction between diethylcinamoylphosphonate and Me3SiCN has also been studied. The reaction product (R1 = PhCH=CH-) was obtained in good yield and high enantioselectivity. The asymmetric synthesis of the phosphonate was controlled by the thio group of the catalyst due to the formation of an H–bond. Also, p-nitrophenol generated HCN from Me3SiCN, followed by cyano-anion attack on the carbonyl group of activated α-ketophosphonate (Figure 24).
The reaction between aromatic ketoesters and dimethyl phosphite catalyzed by thiourethane derivatives of cinchona alkaloids resulted in the formation of hydroxyphosphonates in good yields and enantioselectivities. The best results were obtained with cinchonidine thiourethane organocatalysts [56,57]. For example, enantioselective phospha-Mannich reaction of diphenylphosphite with N-Boc-ketimines derived from N-substituted isatins, catalyzed by the bifunctional quinine derivative which is a Brønsted base (Figure 25). Under optimal reaction conditions, chiral addition products were obtained in high yields and enantioselectivity reaching 98% ee. Among the studied N–substituted imines, the reaction of N–benzyl-substituted ketimine with secondary phosphites led to the formation of 3-hydroxy-2-oxindolyl-3-phosphonate with high enantioselectivity and in 95% yield
The enantioselective synthesis of α-hydroxyphosphinates using the proline-catalyzed phosphaaldol reaction of L-α-acylphosphinates with acetone was described by Yuan [58]. The conversion of α-hydroxyphosphinates to α-hydroxy-H-phosphinates was achieved by treatment with trifluoroacetic acid (TFA). In addition, the redox reaction of both products, with the formation of phosphonates, occurred in the presence of trimethylchlorosilane and alcohol (Figure 26).
2.1.2. Phospha-Mannich Reaction
The phospha-Mannich reaction is a convenient method for the preparation of α-aminophosphonates, which can be readily converted to α-aminophosphonic acids via removal of alkoxy groups by hydrolysis or hydrogenolysis [59,60,61,62,63]. The phospha-Mannich reaction, involving the condensation of primary or secondary amines, oxo compounds (aldehydes and ketones), and >P(O)H compounds, especially dialkyl phosphites, is a convenient method for the synthesis of α-aminophosphonates, which are of great importance due to their biological activity [64]. Typically, a three-component Mannich reaction can proceed through the formation of an imine or α-hydroxyphosphonate intermediate. Monitoring of several Kabachnik–Fields reactions in situ using Fourier transform infrared spectroscopy (FTIR) showed the participation of an imine intermediate, which was confirmed by theoretical calculations [64,65].
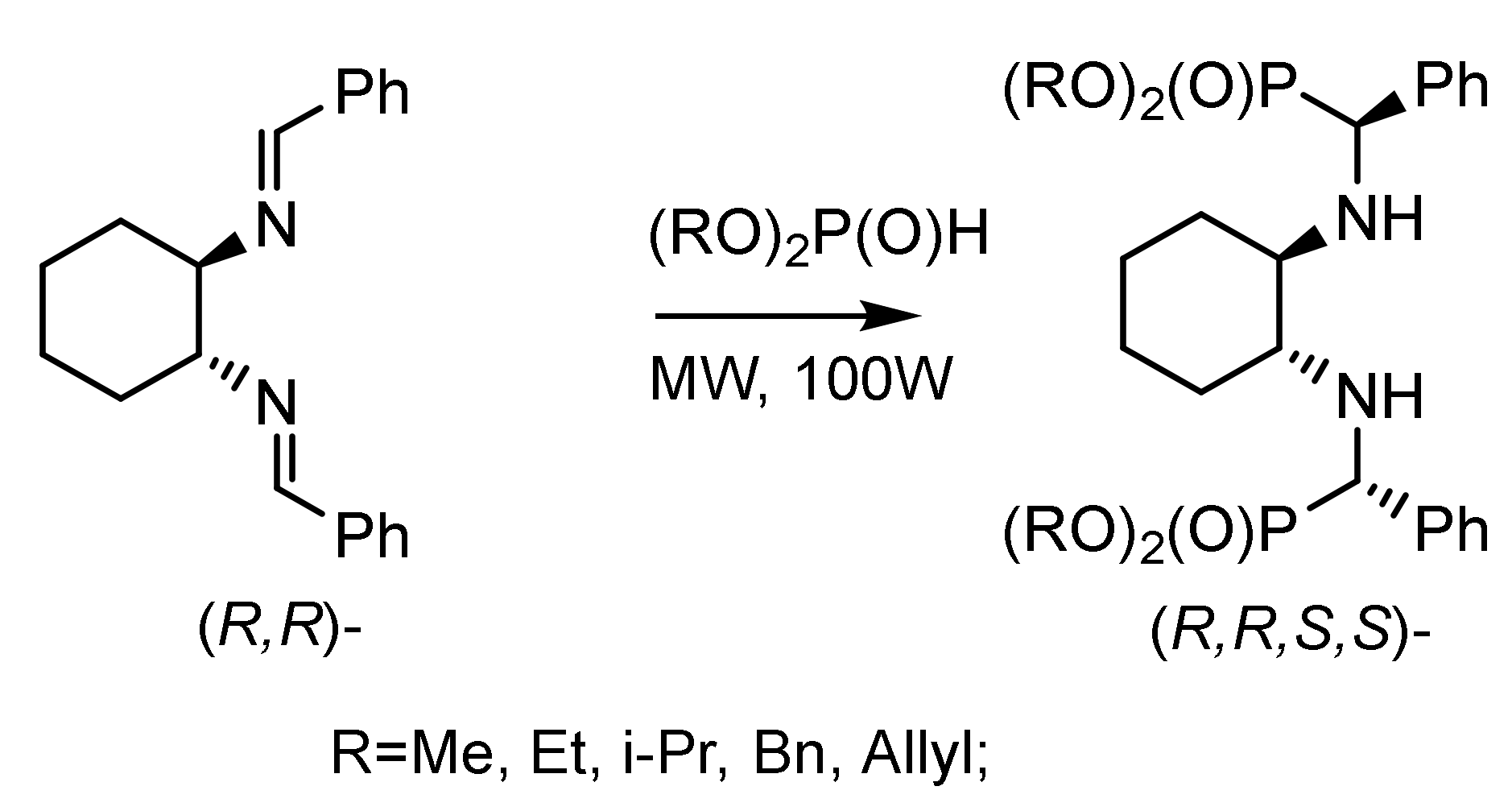
For example, nucleophilic addition of diethylphosphite to (S)-aldimine obtained from (9S)–amino-deoxyquinine and 4-chlorobenzaldehyde in toluene at 90 °C gave (S,S)-α-aminophosphonate in 60% yield and high diastereoselectivity. Salen-like compounds such as bis-aminophosphone systems containing a (R,R)-1,2-diaminocyclohexyl moiety have been synthesized by adding dialkyl phosphites to the azomethine bond of N,N’–dibenzylidene-1,2-diaminocyclohexane (Figure 27).
The synthesis of enantioenriched α-aminophosphonates using asymmetric catalytic hydrophosphonylation and asymmetric P-Mannich reaction has attracted great interest [59]. For catalytic asymmetric aminophosphonylation, chiral metal complexes, LLB, BINOL, etc. have been successfully used.
Fehn et al. reported that the N,N′-dioxide-Sc(III) complex initiates a three-component phospha-Mannich reaction, giving the corresponding α–aminophosphonates in good yields and high enantioselectivities (up to 87% ee) (Figure 28) [66].
Chiral N,N’-dioxide-Ytterbium(III)–complexes promoted the asymmetric addition of diethyl phosphite to aldehydes, giving the corresponding products with good yields and enantioselectivities [66]. The use of bicyclic iminium salts to prepare asymmetric cyclic α-aminophosphonates has been reported (Figure 29). Chiral cyclic imines are synthesized from diamine and ketoesters, and their subsequent treatment in toluene with dialkyl phosphites leads to tetrasubstituted α-aminophosphonates in high yields and diastereoselectivity [67]. However, if imines were activated with bromotrimethylsilane, they react with an iminium ion reactive towards tris(trimethylsilyl)phephite, after which α-aminophosphonic acid derivatives can be obtained in high yields (70–99%) and a diastereoselectivity of 70 to 98% de (Figure 29).
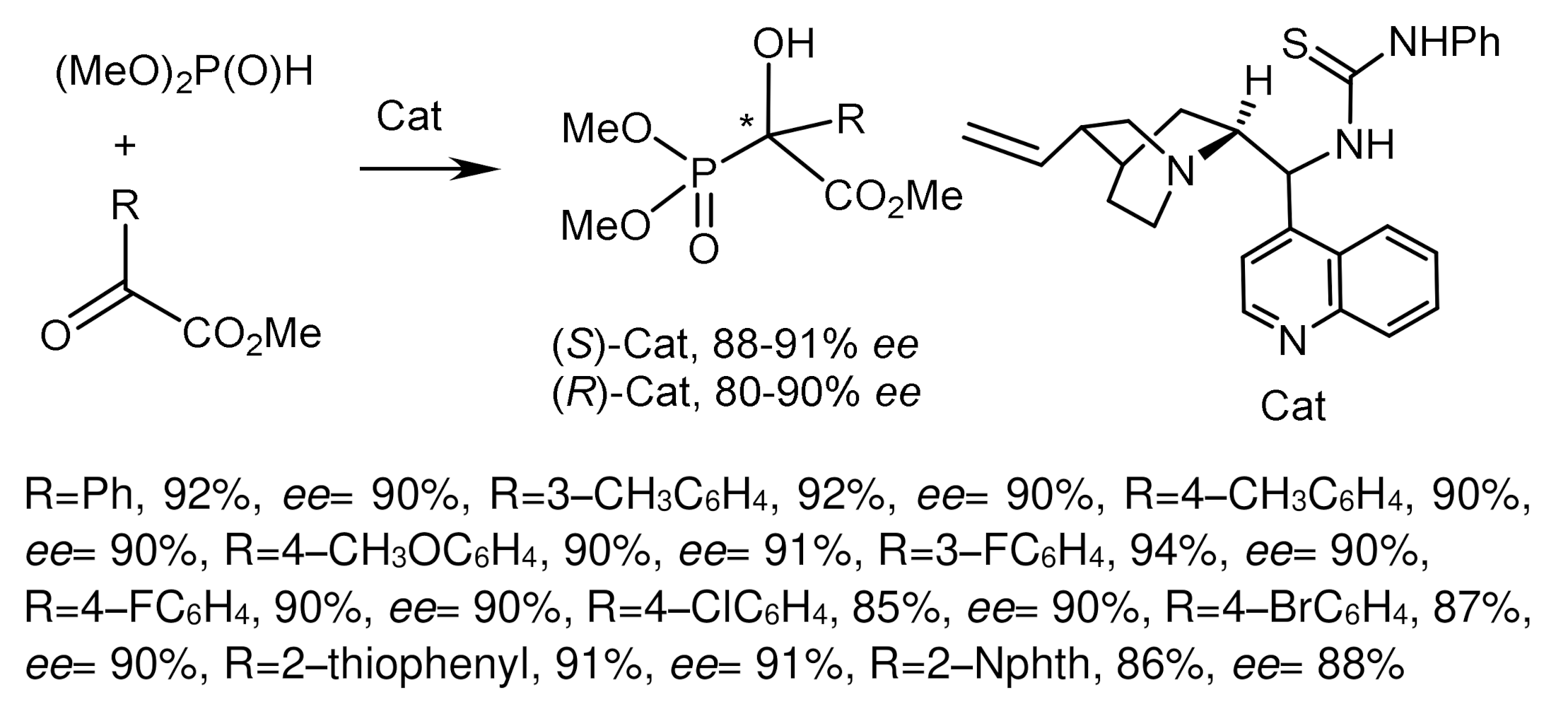
An interesting example of an asymmetric phospha–Mannich reaction was described by B. List et al. [68]. The reaction of a three-component mixture consisting of aldehyde, p-anisidine, and di-3-pentylphosphite, catalyzed by a chiral atropisomeric acid, a p-anthracenyl-substituted analog of TRIP, resulted in the formation of aminophosphonates in high yield and good diastereoselectivity and enantioselectivity. The Kabachnik–Fields reaction proceeded under conditions of dynamic kinetic resolution and catalysis by chiral phosphoric acid. It has been found that the volume of alkyl substituents of the aldehyde affects the stereochemical outcome of the reaction. Aldehydes containing branched alkyl substituents (isopropyl, cyclopentyl, cyclohexyl) reacted with high stereoselectivity. In contrast, aldehydes containing R=methyl or ethyl react with low stereoselectivity (Figure 30).
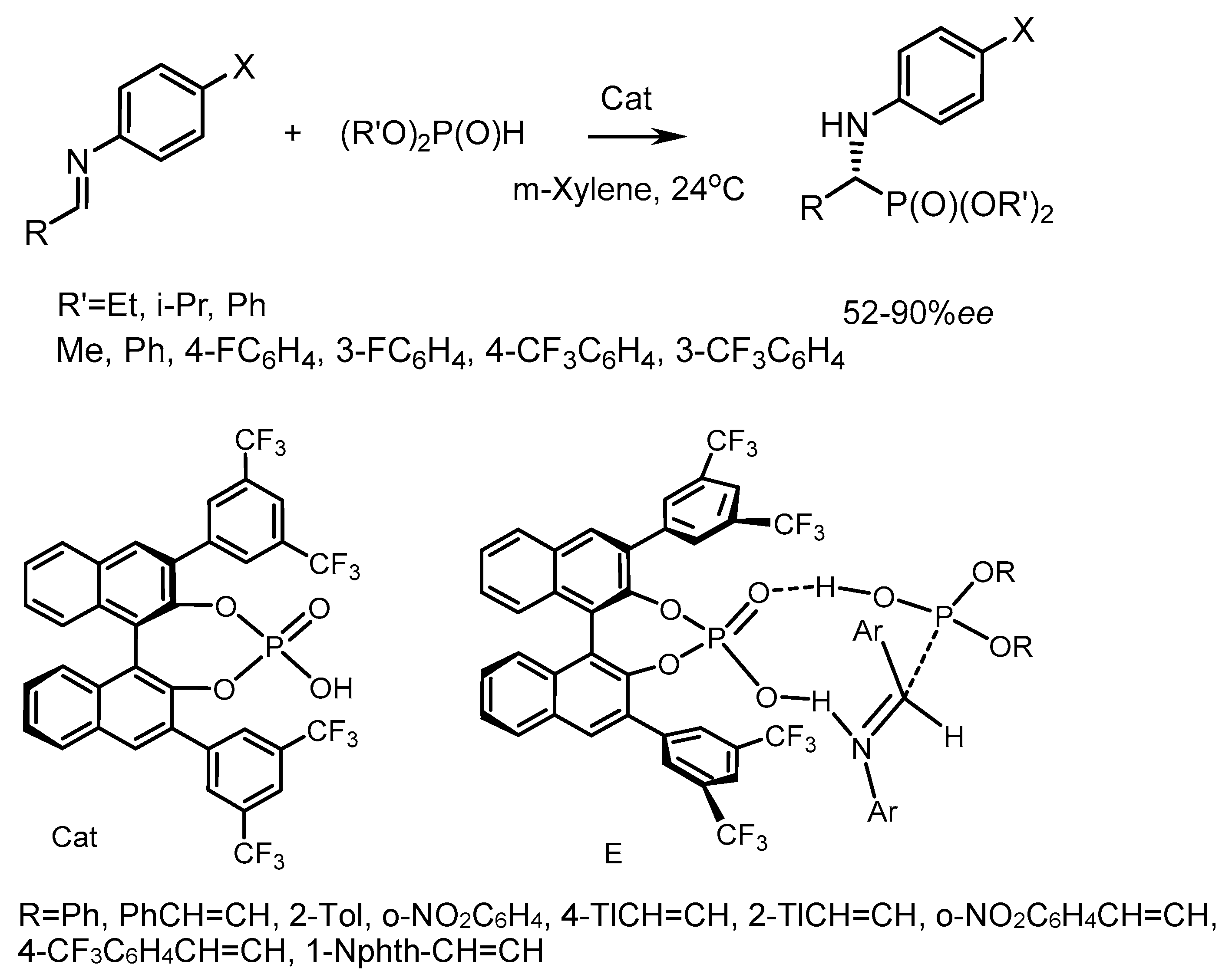
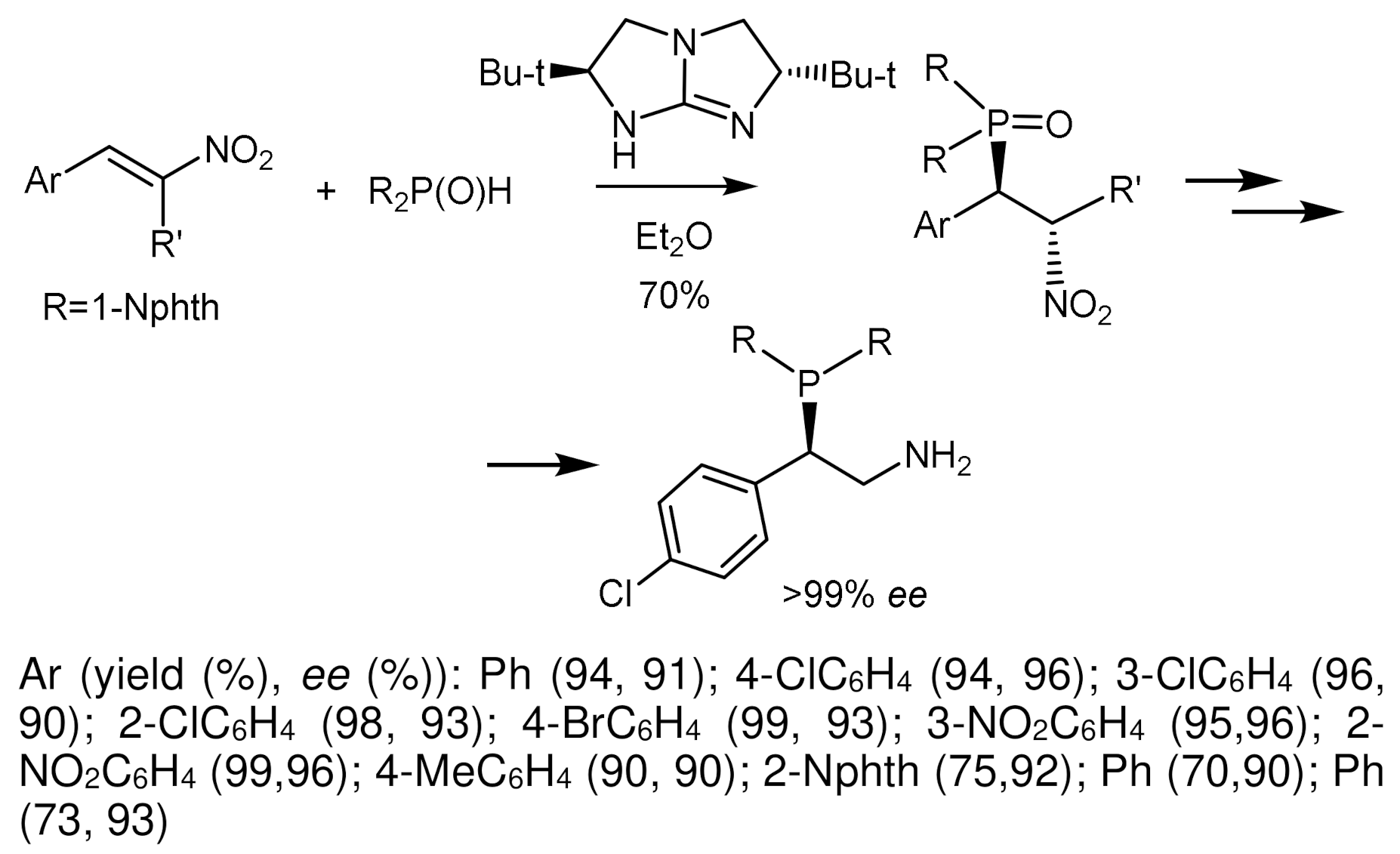
Akiyama et al. [69] studied the reaction of imines with dialkyl phosphites catalyzed by Brønsted’s chiral acid, 3,3′-bis(3,5-ditrifluoromethylphenyl)-1,1′-8-binaphthyl-2,2′-diyl. The reaction led to the formation of α–aminophosphonates with moderate yields and good enantioselectivities reaching 90% ee [70]. To explain the stereoselectivity of the reaction, the authors proposed a nine-membered transition state E (Figure 31). Probably, the catalyst, like Brønsted’s acid, activates the imine, and phosphoryl oxygen, like Brønsted’s base, forms a hydrogen bond with the OH group P(III) of the tautomeric form of phosphite, which provides a facial attack on the imine and promotes the growth of enantioselectivity due to the effect of convergence of reaction centers. The structure of the substrate is an important factor influencing the result of the reaction. The reactions of diisopropyl phosphite with aldimines, which are derivatives of cinnamaldehyde containing electron-withdrawing groups in the ortho-position of the aromatic ring (CF3, NO2, Cl), proceeded with the highest enantioselectivities. The asymmetric reaction of achiral imines with dialkyl phosphites in the presence of 10 mol% chiral Brønsted acid led to the formation of α-aminophosphonates in moderate yields (30–65%) and with good enantiomeric excesses (up to 90.6% ee) (Figure 31) [70].
Cramer et al. described an interesting example of asymmetric addition of phosphorus nucleophiles to imines with formation of tetrasubstituted α–aminophosphonates [70]. The reaction of imidoyl chloride in the presence of palladium catalyst (Cat) and CsOAc first led to the formation of chiral imine in good yield and with high enantiomeric excess (90%, 97% ee). Then, the hydrophosphonylation of chiral imine with diethylphosphite in the presence of boron trifluoride resulted in the α-aminophosphonate as a single diastereoisomer in good yield (Figure 32).
The one-pot Kabachnik–Fields reaction catalyzed by the Salalen complex, described by Katsuki [71], proceeded with satisfactory enantioselectivity. Optically active aluminum(Salalen) complexes are effective catalysts for Mannich hydrophosphonylation of aldehydes and aldimines, giving the corresponding α–hydroxy- and α–aminophosphonates with high enantioselectivity. The reaction proceeded with high enantioselectivity with aldimines derived from aliphatic or aromatic aldehydes. The efficient catalysis of the complex is explained by its unique structure: it has a distorted trigonal bipyramid configuration, which allows the Salalen ligand to adopt a cisoid structure in which the amine chiral group is located near the metal center. Particularly high enantioselectivities have been obtained with aldimines containing R=4-methoxy-3-methylphenyl at the nitrogen. The reaction of dimethylphosphite with alkynyl or alkenylaldimines derived from phenylpropargylaldehyde and diphenylmethylamine proceeded with high enantioselectivity. However, the reactions of aldimines derived from cinnamaldehyde and diphenylmethylamine or 4-methoxy-3-methylphenylaniline proceeded with low enantioselectivity (Figure 33 and Figure 34).
The aluminum catalyst (Cat) showed high enantioselectivity towards aldimines (Figure 35). Hydrophosphonylation of a number of aldimines substituted with various substituents and heteroatoms with this catalyst proceeded with high enantioselectivity. The enantiomeric excesses and yields of the reaction of aldimines with phosphites catalyzed by a sterically hindered 8-quinoline-bis (TBOx) aluminum complex depends on the volume of substituent at the nitrogen atom (Figure 35).
Petersen et al. [72] investigated the effect of various chiral bases on the P-Mannich reaction of N-Boc-protected arylimines with diethyl phosphite. They revealed the key role that the free hydroxyl group plays in the cinchona catalysts (Figure 36).
The first example of organocatalyzed enantioselective addition of phosphite to ketimines derived from isatins was described by Reddy et al. Enantioselective addition of diphenyl phosphite to isatin-ketimines has been achieved using a bifunctional squaramide catalyst [56,73]. The method works efficiently with ketimines to produce the corresponding 3–amino–2–oxoindolin-3-yl-phosphonates in very good yields and with enantioselectivity up to 98% ee [74]. Under optimal reaction conditions, chiral addition products were obtained in high yields and enantios1electivity reaching 98% ee. Among the studied N-substituted imines, the reaction of N–benzyl-substituted ketimine with secondary phosphites led to the formation of 3–hydroxy-2-oxindolyl-3-phosphonate with the highest enantioselectivity and in 95% yield (Figure 37).
The reaction of imines obtained from aromatic and aliphatic aldehydes with dialkylphosphites catalyzed by chiral thioureas resulted in the formation of aminophosphonates in high yields and very good enantioselectivities. The hydrophosphonylation products were then converted to α–aminophosphonic acids with the high optical purity achieved in the organocatalytic phospha-Mannich reaction retained (Figure 38) [75].
The enantioselective aldol reaction of acetylphosphonates with activated carbonyl compounds was studied with organocatalysts that are derivatives of quinine. The aldol formed at the previous stage was converted by methanolysis or aminolysis without isolation from the reaction mixture into the corresponding esters or amides [76]. Thiourethane quinines have also been used to initiate the enantioselective organocatalytic phospha–Mannich reaction with cyclic β–unsaturated ketones and diarylphosphine oxides. Optically active products having a stereocenter on the quaternary chiral carbon were obtained in high yields and enantioselectivities reaching 98% ee.
Tan et al. reported the phospha–Mannich reaction of -phosphinates with N-tosylimines to synthesize α-aminophosphine oxides and phosphinates. Guanidinium salts showed high catalytic activity in the P–C bond formation reaction. The reaction was carried out with a triple excess of H-phosphinates, and K2CO3 was added to increase the stereoselectivity and reaction rate. Aminophosphinates containing a stereogenic center at the phosphorus atom were formed as mixtures of diastereoisomers with a predominance of the syn-isomer (Figure 39) [77].
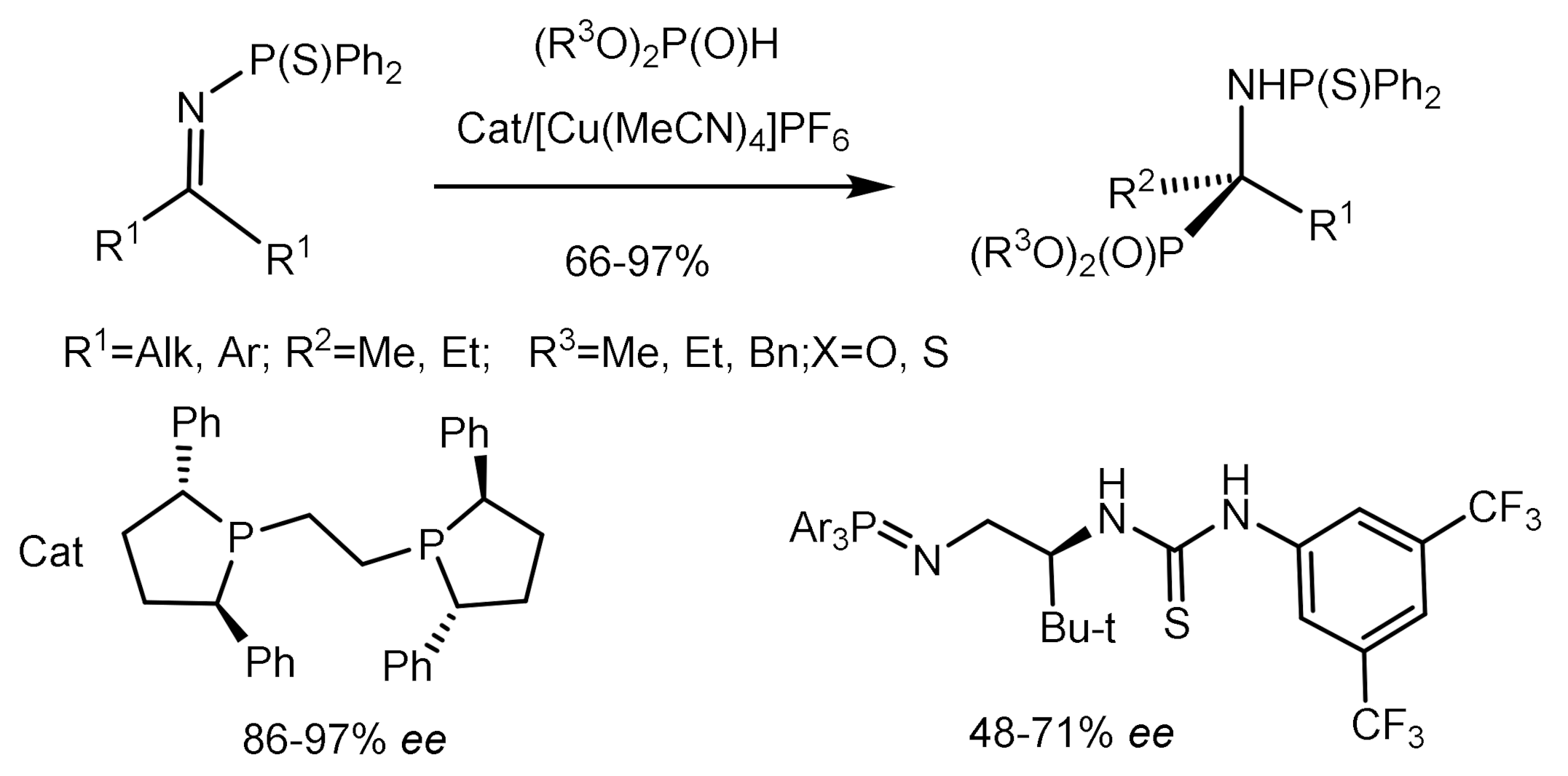
Shibasaki described the hydrophosphorylation reaction of N-thiophosphine imines with phosphites using copper complexes with chiral bis-phosphines as catalysts. As a result, tetrasubstituted α–aminophosphonates with high enantioselectivity (86–97% ee) were obtained [78]. The reaction gave good yields and enantioselectivity of aminophosphonates even with a small amount of catalyst (0.5–2%), which, moreover, could be reused (Figure 40).
Bhusare et al. [79] developed a one-pot method for the synthesis of optically active α-aminophosphonates in three-component reactions between aromatic aldehydes, aniline, and triethyl phosphite, initiated by an organo-catalyst. As a result of process optimization and selection of organo-catalysts, it was found that 1-acetyl-N-tosylpyrrolidine-2-carboxamide is the best catalyst for this reaction, providing the corresponding α-aminophosphonates in high yields (71–90%) and excellent enantioselectivity (73–92% ee). Various aromatic aldehydes and amines were reacted with triethyl phosphite in the presence of a catalyst in glacial acetic acid and ethanol. In all cases, α-aminophosphonates were obtained in good yields and enantioselectivities. (Figure 41).
Organo-catalysts based on chiral atropisomeric phosphoric acids initiate the asymmetric Kabachnik–Fields reaction [68,79,80]. Bulky alkyl substituents have been found to increase the stereoselectivity of the reaction. The reaction proceeded with high stereoselectivity in the case of aldehydes containing branched alkyl substituents (isopropyl, cyclopentyl, and cyclohexyl). In contrast, aldehydes containing substituents R = methyl or ethyl reacted with low stereoselectivity (Figure 42).
2.1.3. Phospha–Mukayama Reaction
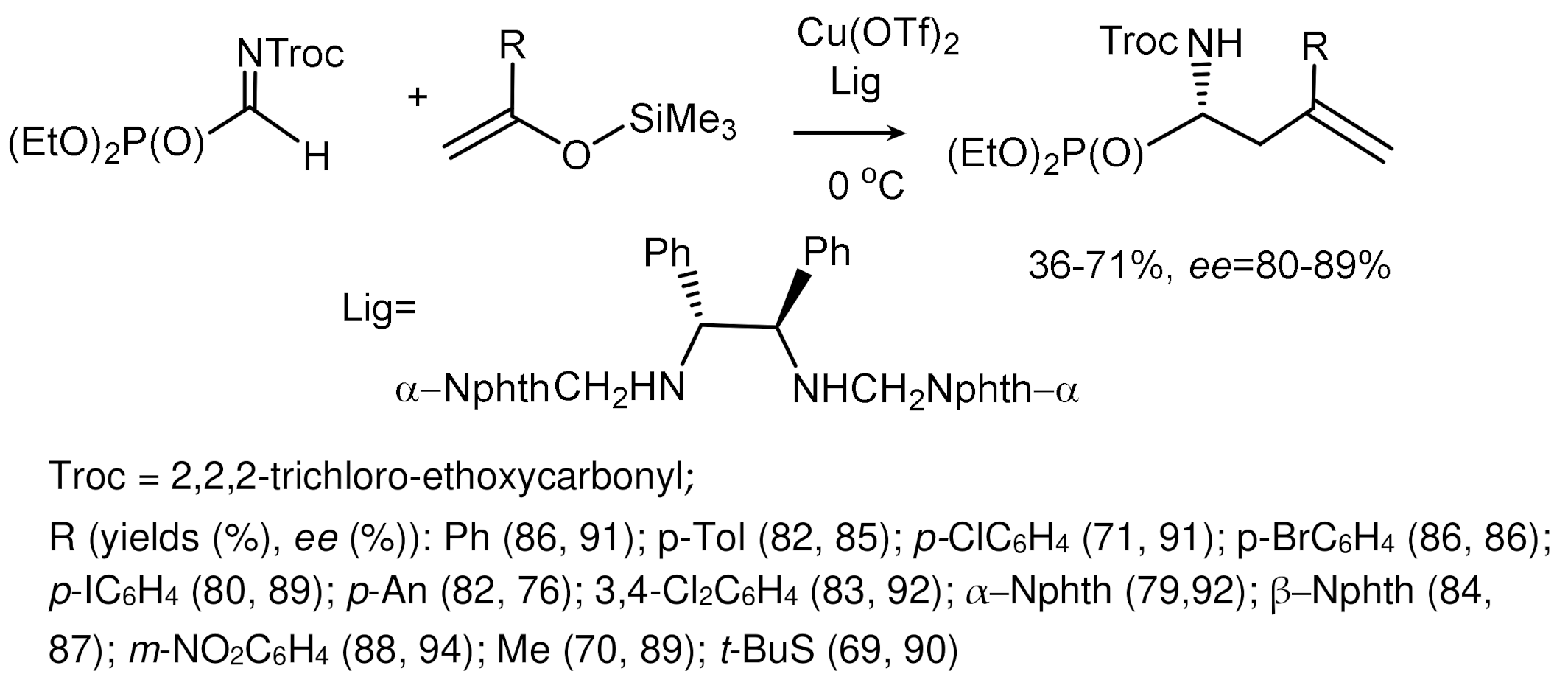
The reaction of silyl enolates with aldehydes catalyzed by titanium tetrachloride was discovered by Mukayama in 1973 and named after him [81]. This reaction is a convenient method for the synthesis of hydroxyphosphonates, in connection with which several papers have been published on the use of the Mukayama reaction in the asymmetric synthesis of hydroxyphosphonates. For example, TADDOL catalyzes the enantioselective Mukayama reaction of acylphosphonates with silylenolates to form hydroxyphosphonates with one quaternary carbon atom, forming with high diastereo- and enantioselectivity [82] (Figure 43). An enantioselective reaction of silyl enolates with N-acyl-α–iminophosphonates catalyzed by chiral copper complexes, resulting in chiral α–aminophosphonates, has also been described. Complexes of Cu(OTf)2 and diamine were effective catalysts for this reaction, providing the formation of aminophosphonates in high yields and enantioselectivities [83]. The enantioselective reaction of N-acyl-α–iminophosphonates silylenolates leads to the formation of chiral α–aminophosphonates (Figure 44).
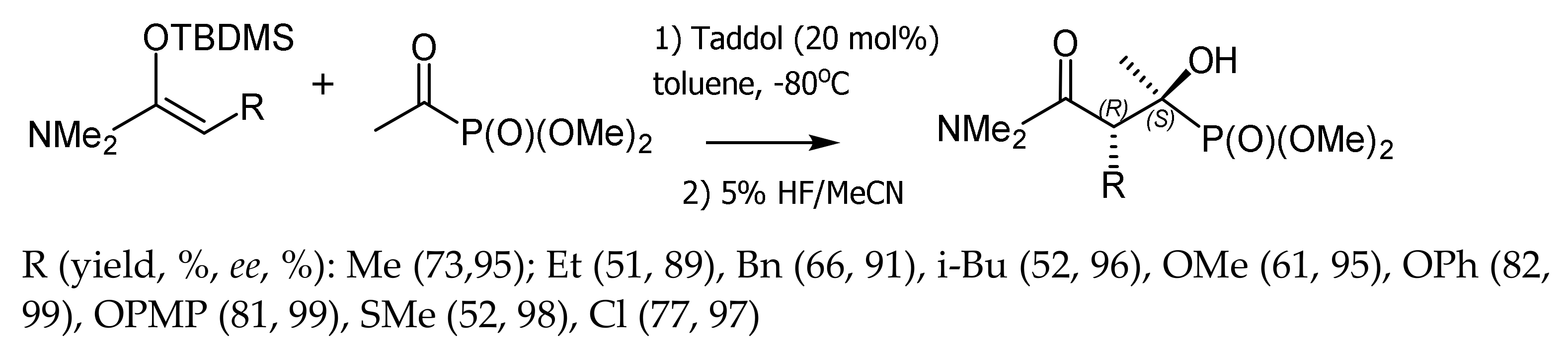
Rawal et al. [82] reported a highly stereoselective Mukayama reaction of diethyl α–ketophosphonate with N-acetal-O-ketenes catalyzed by commercially available TADDOL. The stereoselectivity of the reaction increased with decreasing temperature. The tert-butyldimethylsilyl (TBDMS) group could be easily removed from the final product by treatment with 5% HF in CH3CN. All tertiary alcohols with tertiary and quaternary stereogenic centers were obtained in good yields and with high enantio- and diastereoselectivities. This method gives access to optically active α,β–dihydroxyphosphonates (Figure 45).
2.1.4. Phospha-Michael Reaction
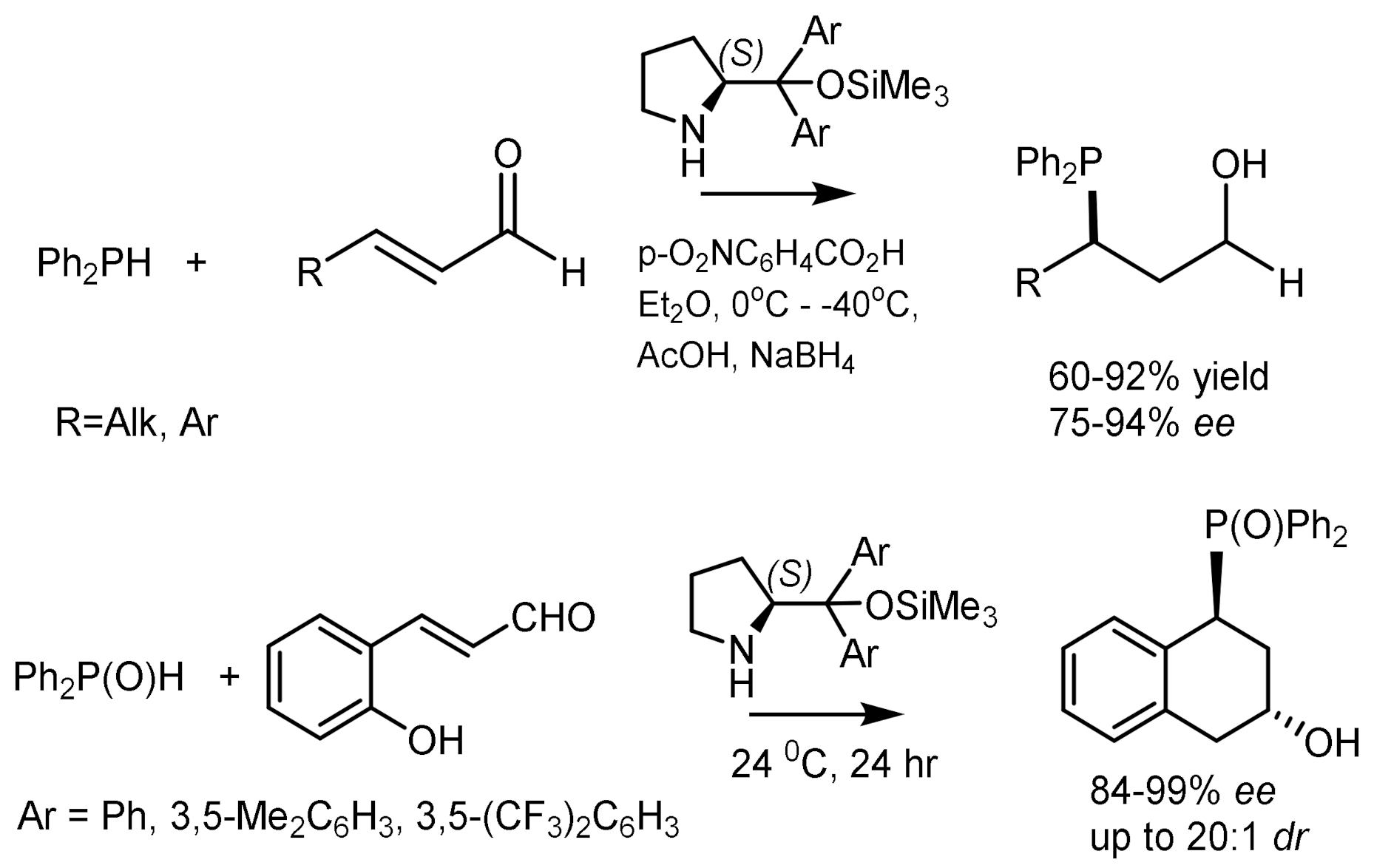
The asymmetric Michael addition of phosphorus nucleophiles (R2PO− or R2P− groups) to activated alkenes proceeds stereoselectively in the presence of chiral catalysts [84,85,86,87,88]. Trimethylsilyl ethers of diarylprolinol proved to be particularly effective catalysts, by means of which highly enantioselective hydrophosphination of α, β-unsaturated aldehydes with diphenylphosphine was carried out. As a result, the organocatalytic asymmetric β–functionalization of aldehydes with a P-centered nucleophile was developed, which is a simple method for obtaining highly enantioenriched tertiary phosphines (Figure 46) [84,85,86,87,88,89,90]. Asymmetric cascade cyclization reaction of o-hydroxycinnamic aldehydes and diphenylphosphine oxide catalyzed by the silyl ether of L-diarylprolinol was followed by cyclization with formation of highly enantioenriched 4-diphenylphosphinylchroman-2-ols with 84–99% ee. This catalytic method was successfully extended to the enantioselective β-phosphonylation of α,β-unsaturated aldehydes with trivalent trialkyl phosphites [91]. However, the asymmetric phospha-Michael addition of α,β-unsaturated aldehydes to diphenylphosphine proceeded with moderate enantioselectivity [92], although diarylphosphine oxides and diphenoxyphosphine successfully phosphonylated analogues of nitroalkenes in the presence of chiral guanidines [93,94], quinine [95], squaramids [96], etc. [97] (Figure 46).
Activated vinyl phosphonates containing an electron-withdrawing group have attracted great interest in asymmetric organo-catalysis [98,99,100,101,102,103,104,105,106]. For example, the reaction of α,β-unsaturated ketones and aldehydes with diarylphosphine oxides catalyzed by diarylprolinol, resulted in optically active products in high yields and with enantioselectivity up to 86–99% ee (Figure 47) [92].
The asymmetric Michael reaction of vinyl bis-phosphonates with enol compounds catalyzed by the pyrrolidine organocatalyst proceeded with satisfactory enantioselectivity (Figure 48). The reaction was completed in 12 h at room temperature in the presence of 20 mol% catalyst in CHCl3 with formation of Michael adducts in 80% yield and with enantioselectivities reaching 90% ee. Lowering temperature led to decrease of enantioselectivity from 90% to 80% ee [105,106]. Barros and Phillips [107] synthesized chiral keto-bis-phosphonates by Michael addition of cyclic ketones to vinyl bis-phosphonate catalyzed by 0.1 mol equiv. (S)–(+)-1-(2-pyrrolidinyl)-pyrrolidine and benzoic acid as a cocatalyst. All reactions proceeded with the formation of geminal bis-phosphonates in yields up to 86%, dr 1:99 and high enantioselectivities. While cyclohexanone and its derivatives gave monoalkyl products regioselectively, the reaction with cyclopentanone resulted in the formation of 2,5-dialkylated products with 99% ee (Figure 49).
The reaction mechanism based on stereochemical studies of the reaction was proposed. The mechanism postulated the Michael acceptor attack from the Si-face of E-enamine. The selectivity of the organocatalytic the asymmetric conjugate addition was explained by the authors as an acyclic synclinal transition state based on the Seebach model, in which there are electrostatic interactions between the enamine nitrogen and the phosphonate fragment. The bulky aryl and silyl groups favor the selective formation of E-enamine and the selective shielding of its Re-face (Figure 50) [105].
Enantioselective addition of cyclic 1,3-dicarbonyl compounds to ethyl (E)-but-2-enoylphosphonate proceeds easily in the presence of cinchonine thiourethanes (Figure 51) [101,102]. Chiral quinine squaramides were the most efficient catalysts for these reactions. The initially formed α-oxophosphonates were converted to the corresponding esters or benzylamides in basic media. The addition of 2-hydroxy-1,4-naphthoquinones to β,γ–unsaturated α-ketophosphonates was performed by cooling to –25 °C in a chloroform-toluene solution (Figure 52). Thiourethanes have shown themselves to be effective organocatalysts that provide high enantioselectivity. The resulting adducts were converted to β-(3-hydroxy-1,4-dioxo-1,4-dihydronaphthalen-2-yl) by treating the reaction mixtures with methanol in the presence of DBU (Figure 53) [96].
The enantioselective phospha–Michael reaction of diphenylphosphonate with nitroolefins was catalyzed by a guanidinium/bis-thiocarbamide organocatalyst. Nitroolefins containing various aromatic and aliphatic substituents reacted with H-phosphites to form addition products with 90–98% ee. Among a number of solvents studied, toluene provided the highest enantioselectivity (89% ee). The enantioselectivity of the reaction increased to 95% ee with the addition of water [102,103]. It was found that the reaction enantioselectively led to the formation of products having the (R)-absolute configuration (Figure 54).
Asymmetric addition of phosphonates to trans-crotonophenone and chalcone derivatives using diaminomethylenemalononitrile as an organo-catalyst led to the formation of the corresponding chiral γ–ketophosphonates in high yields and excellent enantioselectivity (up to 95% ee). The reaction resulted in the corresponding β-substituted carboxylates in high yields and with high enantioselectivity (94–99% ee) with subsequent neutralization of the resulting products by the action of DBU and MeOH as the second nucleophile (Figure 55) [104].
High stereoselectivity was achieved in the case of using iminium complexes, with the trans-configuration, creating the most effective shielding of the Re-stereoface side (R=Ar) of the iminium complex with the large chiral group of the protected diarylprolinol. Substituted enals containing aryl, heteroaryl, alkyl, and alkenyl groups could be phosphorylated by this method, yielding products with 75–94% ee and good yields. The highest enantioselectivities were obtained with diphenylphosphine and diarylprolinol containing 3,5-bis(trifluoro)phenyl groups in toluene or chloroform with 2-fluorobenzoic acid additives. In this case, depending on the substituent R’, yields of the products were 80–85%, and the enantioselectivity was 76–99% ee. Cordova et al. [90] proposed a mechanism for a chemo- and enantioselective organocatalytic hydrophosphination of α,β-unsaturated aldehydes based on theoretical calculations using the Gaussian-93 program. They came to the conclusion that the bulky substituent (R=Ar) shields the Re-stereoface side of the iminium ion, which favors the Si-stereoface attack (Figure 56) [105,106].
Terada et al. [94] synthesized β–nitrophosphonates using axially chiral guanidine derivatives as catalysts for the Michael addition of diphenyl phosphite to nitroalkenes. The enantioselectivity of the reaction increased with increasing substituent size. The reaction was carried out in the presence of 1 mol% catalyst, and low catalyst loading did not affect the enantioselectivity of addition. It was noted that the addition of α–nitrophosphonates to α,β–unsaturated ketones, catalyzed by quinine–thiourethane, proceeded with moderate enantioselectivity depending on the structure of the ketone. In the presence of electron-donating aromatic substituents, the enantioselectivity was at the level of 70% ee. On the contrary, the introduction of electronegative groups into the aromatic nucleus led to a decrease in enantioselectivity to 35–44% ee (Figure 57).
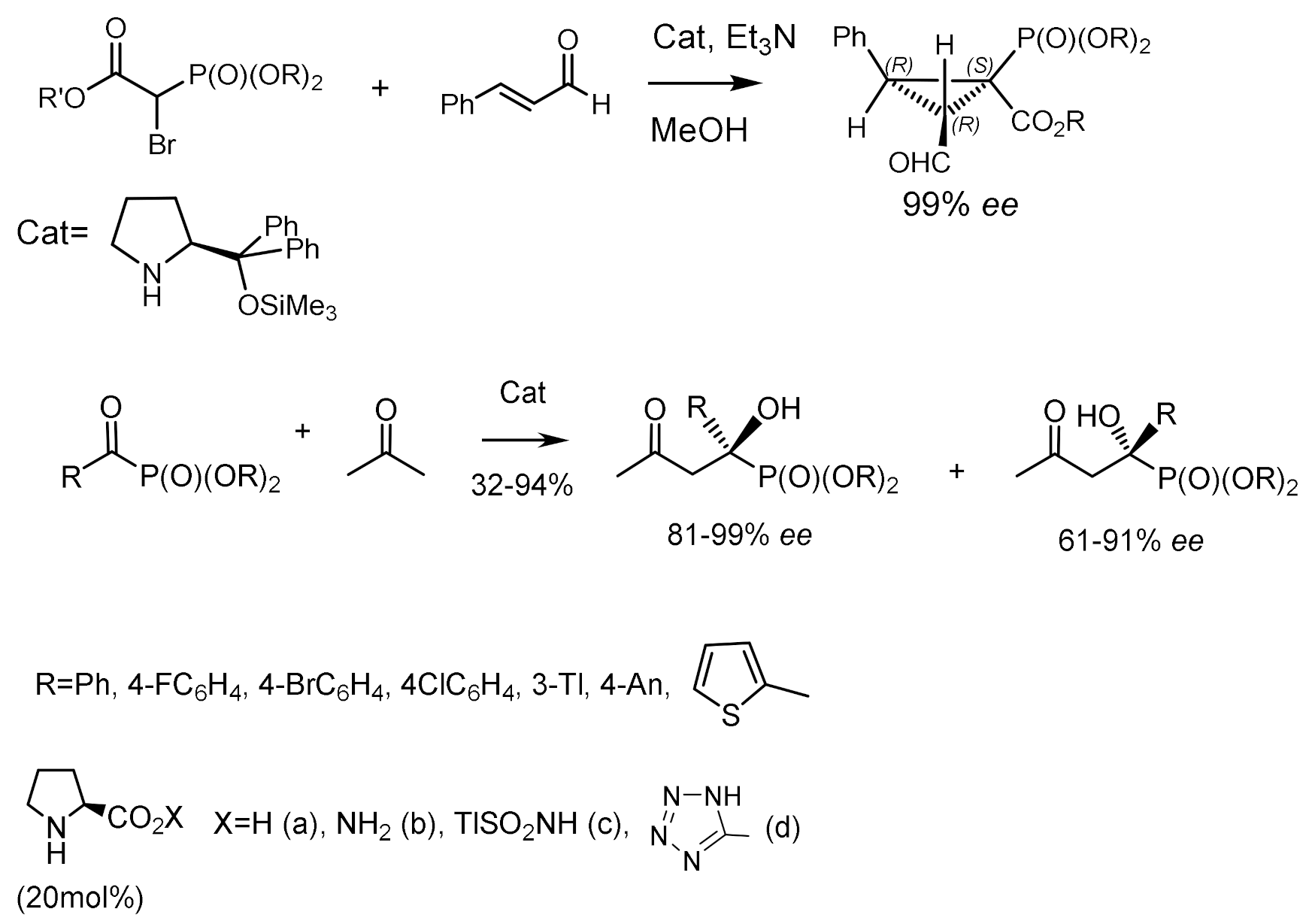
An organocatalytic domino reaction consisting of Michael addition/intramolecular alkylation between α,β-unsaturated aldehydes and bromophosphonoacetates was developed. The reaction consists of Michael addition and intramolecular alkylation of α,β-unsaturated aldehydes with bromophosphonoacetates. Highly functionalized cyclopropylphosphonates containing three chiral centers, one of them quaternary, were obtained with good diastereoselectivities of up to 83:17 and very high enantioselectivities of up to 99% [107]. Aromatic, heteroaromatic, and aliphatic nitroalkenes were phosphonylated using this reaction. The corresponding adducts were obtained with high yields and enantioselectivities. The authors used Michael adducts in the synthesis of biologically important β–aminophosphonates. Noteworthy are the works devoted to the study of the reaction of isatin-based ketimines with α-diazomethylphosphonates catalyzed by chiral binaphthanol-derived silver phosphate [108] and click reactions for protein functionalization [109]. Proline and its derivatives catalyze the reaction of ketophosphonates with enolizable ketones, which leads to the formation of chiral ketophosphonates [110]. The highest yields and enantioselectivities were obtained in the reaction of ketophosphonates with acetone and proline. It was found that methyl and isopropyl ethers of ketophosphonates in reaction with acetone give hydroxyphosphonates with the highest enantiomeric excess (ee). At the same time, the reaction of butanone and methoxyacetone with ketophosphonates, while yielding optically active phosphonates, presented relatively low enantioselectivity. The organocatalytic reaction of ketones with α–ketophosphonates having an asymmetrically substituted phosphorus atom was also studied. However, due to the stereogenic center on phosphorus, a mixture of two diastereomers of α–hydroxyphosphinates was obtained in a ~1:1 ratio, some of which were separated by crystallization. The optical purity of these diastereomers was very good (from 91% to 99% ee), and their absolute configurations were determined by X-ray analysis (Figure 58) [111,112].
L-Prolinamide catalyzes the reaction of diethyl formylphosphonates with ketones to form secondary hydroxyphosphonates in high yields (Figure 59). The authors suggested that the addition proceeds through the transition states, which have a nine-membered chair conformation, in which the phosphonate occupies a pseudoequatorial position. As a result, enamine undergoes addition from the Si-stereoface side of diethyl formylphosphonate, giving the observed product configuration (Figure 60) [112].
Jorgensen et al. [91] reported enantioselective phosphonylation of α,β–unsaturated aldehydes with trialkyl phosphites, in combination with a Brønsted acid and a nucleophile. Optically active β-aldehyde phosphonates formed as a result of β–phosphonylation of β-unsaturated aldehydes are universal chiral synthons for many syntheses. The organocatalytic enantioselective reaction of β-phosphonylation of aliphatic α,β-unsaturated aldehydes proceeded with good yields and enantioselectivities (84–85% ee). The use of chiral catalyst in this reaction allows for yields of enantiomeric product in 55% yield and with 82% ee. The same results were obtained for functionalized enals; however, yields were slightly lower. The reaction proceeded readily also with aromatic α, β-unsaturated aldehydes such as cinnamic aldehyde and its para-substituted derivatives. Using aromatic α, β-unsaturated aldehydes, the corresponding phosphonates were obtained in satisfactory yields and with enantioselectivities 74–88% ee (Figure 61).
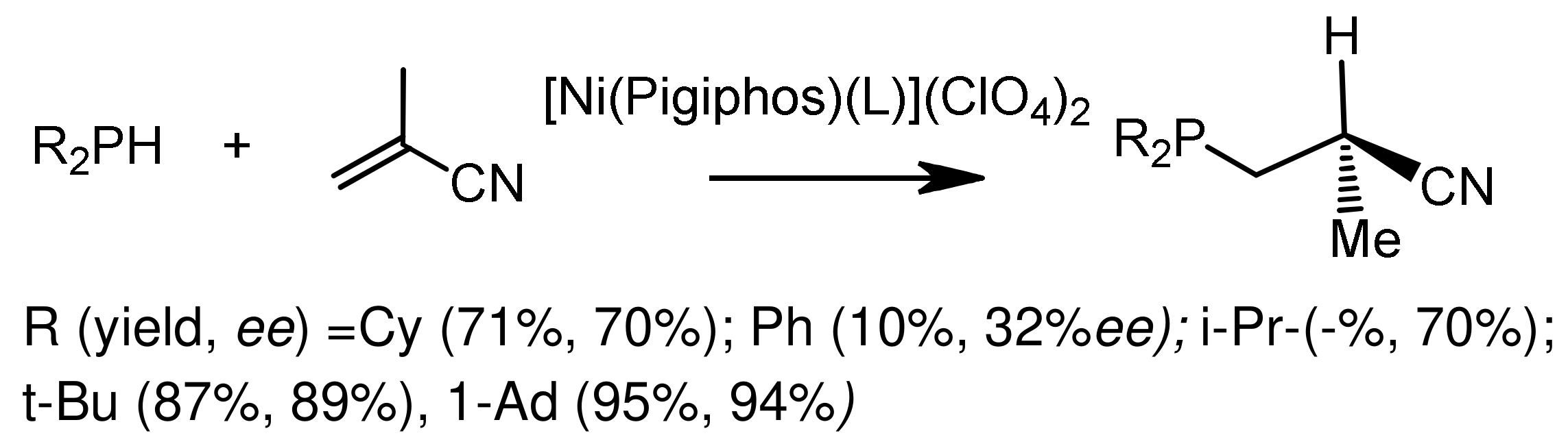
The asymmetric addition of dialkylphosphites or dialkylphosphine oxides to electron-deficient alkenes by the Michael reaction is successfully realized under the conditions of metal-complex catalysis [113,114,115,116,117]. For example, Gluck reported asymmetric hydrophosphination of activated olefins catalyzed by a platinum complex containing the (R,R)-Me-Duphos)(trans-stilbene) ligand, resulting in chiral phosphines in good yields and with moderate enantiselectivities [114] (Figure 62). Togni et al. [115,116] proposed a method for the asymmetric hydrophosphination of methacrylonitrile with secondary phosphines, catalyzed by a nickel complex containing the chiral Pigiphos ligand (Figure 63). The authors suggested that methacrylonitrile coordinates with the nickel atom through the nitrile nitrogen and increases the electrophilicity of the activated double bond. Increasing the volume of the substituents resulted in an increase in stereoselectivity (95% yield and 94% ee), although the reaction time was increased in some cases. The enantioselectivity of the reaction reached 89–94% ee. The addition of secondary phosphines proceeded more easily, since the methacrylonitrile ligand is in the chiral environment of bulky substituents that initiate chirality. The absolute configuration of the obtained enantiomers of tertiary phosphines was (S). The Pigiphos/nickel(II) complex catalyzed the reaction of secondary phosphines with methacrylonitrile to form chiral 2-cyanopropylphosphines in good yields and with high enantiomeric purity (up to 94% ee). A reaction mechanism was proposed that takes into account the coordination of methacrylonitrile with a dicationic nickel catalyst, accompanied by 1,4-addition of phosphines and proton transfer in the subsequent stage, which determines the reaction rate. Kinetic data, including the isotope effect, and DFT calculations supported the proposed mechanism. Features of this mechanism suggest stereospecific proton transfer, reversible format ion of the P-C bond, as well as the formation of an unusual Ni intermediate ketenimine [116] (Figure 64). Fehn et al. showed that claw-like (S,S)-complexes of palladium catalyze the asymmetric addition of diarylphosphines to various enones with formation of chiral phosphines in good yields and with high enantioselectivities (yields 63–93%, 90–99% ee) [117] (Figure 65).
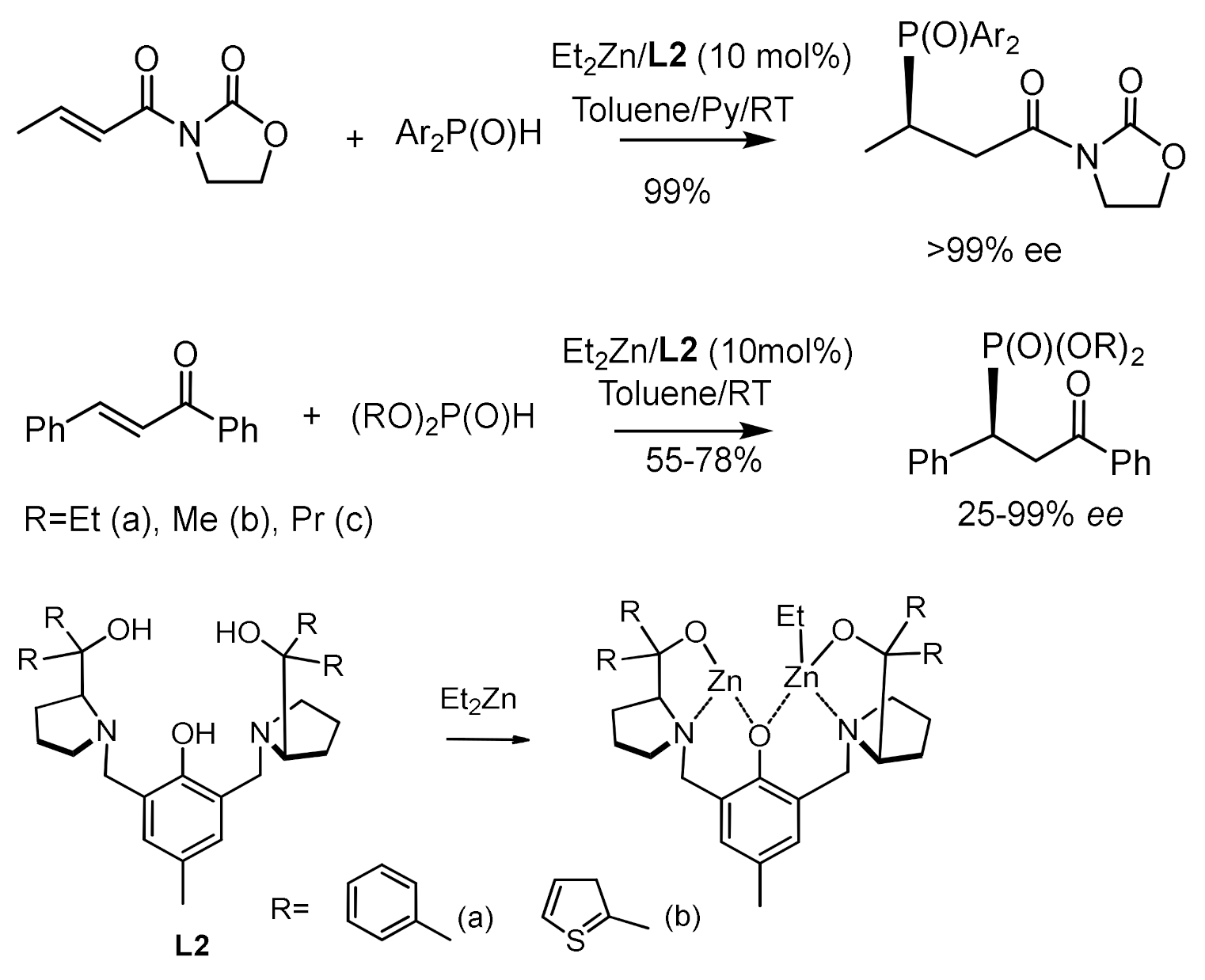
Wang et al. reported the asymmetric 1,4-addition reaction of diethyl phosphite with simple enones catalyzed by a binuclear zinc complex [118,119] with formation of ketophosphonates in high yields and very good enantioselectivity (up to 99% ee). This catalytic phospha-Michael reaction was tested on a range of enones containing aryl and alkyl substituents in the β–position which reacted with good enantioselectivities (Figure 66).
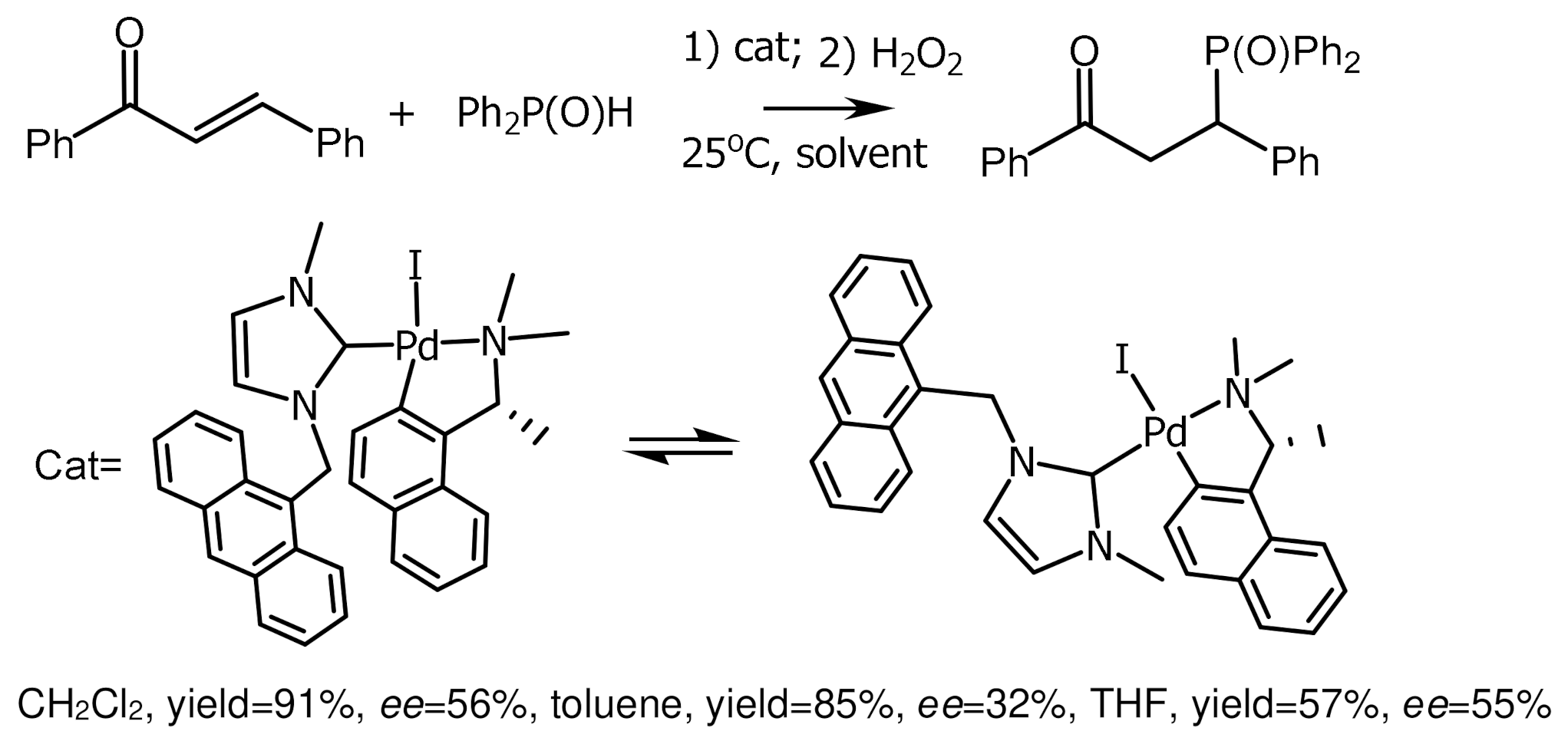
Chiral cyclic complexes of palladium with N-heterocyclic carbene ligands were synthesized starting from enantiomerically pure benzylamines [120]. These complexes exist as two atropisomers which have been isolated by column chromatography. The complexes were used as catalysts in 1,4-additions of diarylphosphines to α,β-unsaturated ketones. As a result, tertiary phosphine oxides were obtained in good yields but moderate enantioselectivities (Figure 66) [121]. Cyclic palladium complexes catalyzed the diastereo- and enantioselective addition of phenylphosphine to bis(enones). This reaction allows the intermolecular construction of chiral tertiary phosphorus heterocycles by the one-pot method in high yields [122,123,124,125]. The highly active, chemo- and enantioselective addition of diphenylphosphine to α,β-unsaturated imines, catalyzed by the cyclic (S)-palladium complex, was used to synthesize a number of chiral tertiary enaminophosphites in excellent yields (Figure 67) [120].
The cyclic palladium complex catalyzed the diastereo- and enantioselective addition of primary and secondary phosphines to enones and enamines. The reaction of bis-enones with phenylphosphine made it possible to obtain chiral phosphorus heterocycles in a one-pot synthesis with high yields and high enantioselectivities as shown in Figure 68. This chemo- and enantioselective reaction was also used to synthesize a number of chiral tertiary enamidophosphites (Figure 68) [122,123].
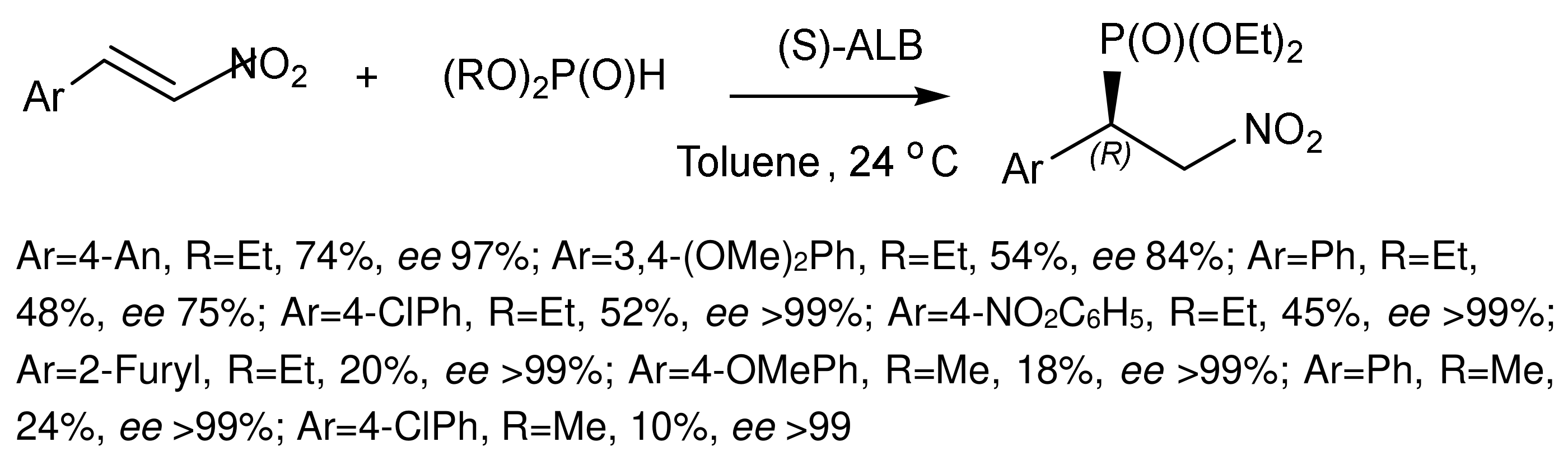
Michael addition of dialkyl phosphites to nitroalkenes in the presence of (S)-(−)- aluminum lithium bis(binaphthoxide) complex ALB gave β–nitrophosphonates, which are precursors of beta-aminophosphonic acid with good enantioselectivities. The dinaphthoxide complex (S)-ALB catalyzed the reaction giving addition products in good yields and with good enantioselectivities [126,127] (Figure 69).
Tessema et al. [97] used a salt of 4-N,N-dimethylaminopyridinium saccharinate (DMAP Hsac) and long-chain pyridine fluoride (4–Rf-CH2OCH2-Py, where Rf = C11F23) to catalyze the phospha–Michael reaction. The addition of diisopropyl-, dimethyl-, or triethylphosphites to β-unsaturated malonates in the presence of these catalysts proceeded easily at 80–100 °C in high yield and the possibility of catalyst reuse up to eight cycles. Addition products containing phosphorus and benzimidazole units were in most cases formed in high yields and with excellent enantioselectivity. Reduction of the phosphine oxide unit in the addition product gives the corresponding chiral phosphine, which is a potential chiral P,N-ligand based on benzimidazole, without loss of enantiomeric excess (Figure 70) [128].
Phospha-Michael addition in recent years has spread to new and unusual structures [128]. For example, Palacios [129,130,131,132] and Onys’ko [133] reported an asymmetric aza-Henry reaction with phosphonoketimines catalyzed by bifunctional thiourethane alkaloids. The reaction easy proceeds with compounds containing both electron-donating and electron-withdrawing substituents in the aromatic nucleus. As a result of the reaction, α–amino-p-nitrophosphonates were obtained with good yields and enantioselectivities (yields 82–87% and 80–84% ee). The same authors described an enantioselective reaction of aza-reformed non-cyclic ketimines using dialkylzinc reagents and a chiral ligand derived from BINOL. The reaction products were converted to aryl- and heteroarylketimines and alkyliodoacetates with high yields and enantioselectivities (76–92%, 93–99% ee) (Figure 71).
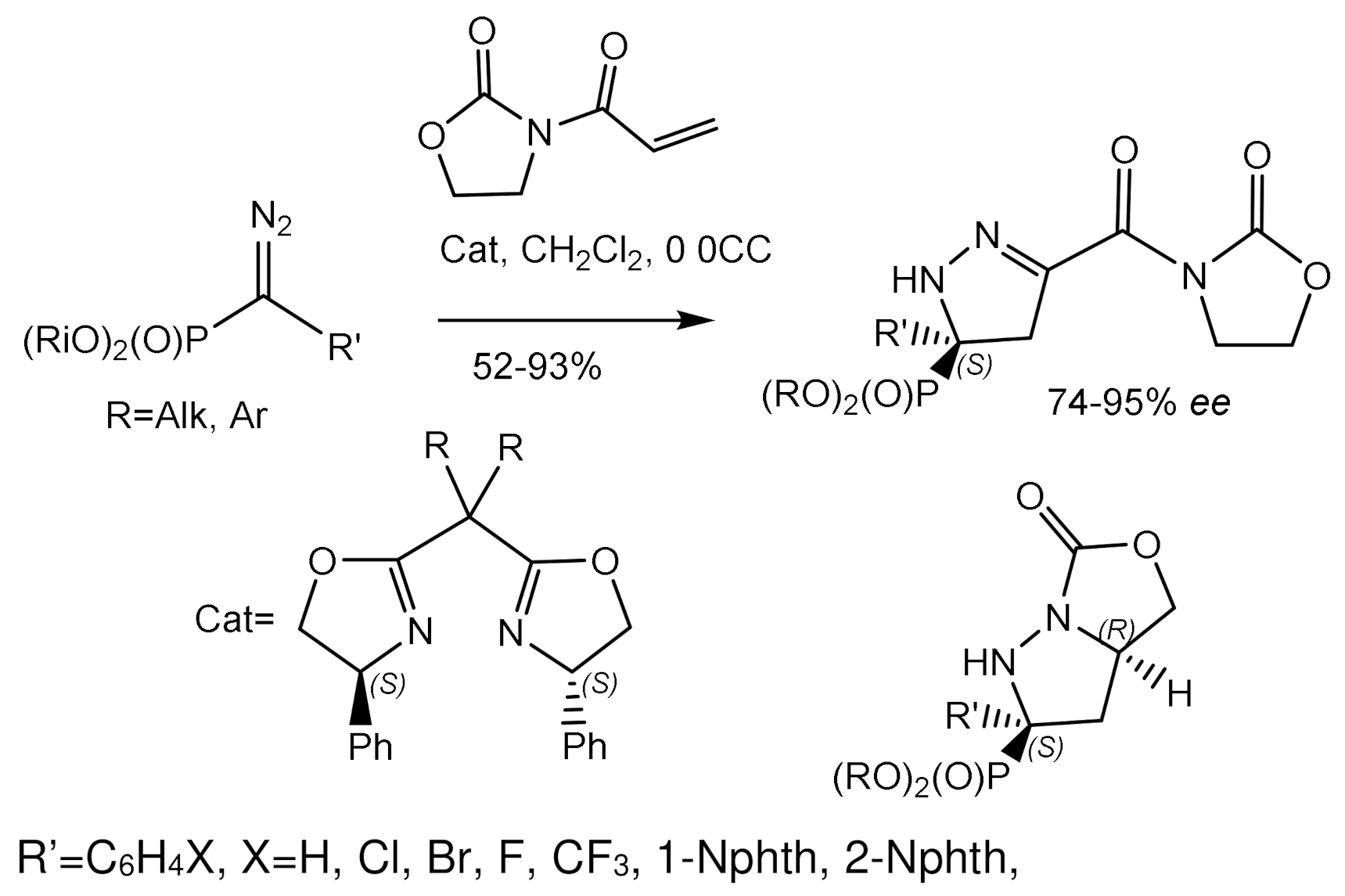
More recently, an example of a dipolar cycloaddition has been reported by Peng et al. [134]. The interaction of diazophosphonates with acryloyloxazolidones in the presence of the Mg- complex led to the formation of enantio-enriched pyrazolines in high yields and with very good enantiomeric excesses (Figure 72).
3. Asymmetric Hydrogenation of Unsaturated Phosphonates
The reactions of catalytic hydrogenation and reduction of unsaturated compounds are undoubtedly the most important methods of modern organic synthesis. An example of a significant industrial scale of enantioselective catalytic processes (more than 10,000 tons annually) is the production of (S)-Metolachlor, an herbicide widely used in agriculture on crops of cereals and soybeans [135]. Metolachlor is an atropisomeric compound, forming four stereoisomers, of which only two (S)-diastereomers are biologically active. The steric effects of 2,6-disubstituted aniline make it difficult to rotate around the Ar-C to N bond. Therefore, the (R)- and (S)-enantiomers exist as atropisomers. A key step in the production of metolachlor is the asymmetric hydrogenation of an intermediate imine using an iridium complex with the JOSIPHOS ferrocenyldiphosphine ligand [136]. Various examples of industrial use of catalytic asymmetric hydrogenation to obtain enantiomerically pure organic substances, including organophosphorus compounds, are presented in the literature [7,17].
3.1. Asymmetric Hydrogenation of Alkenephosphonates
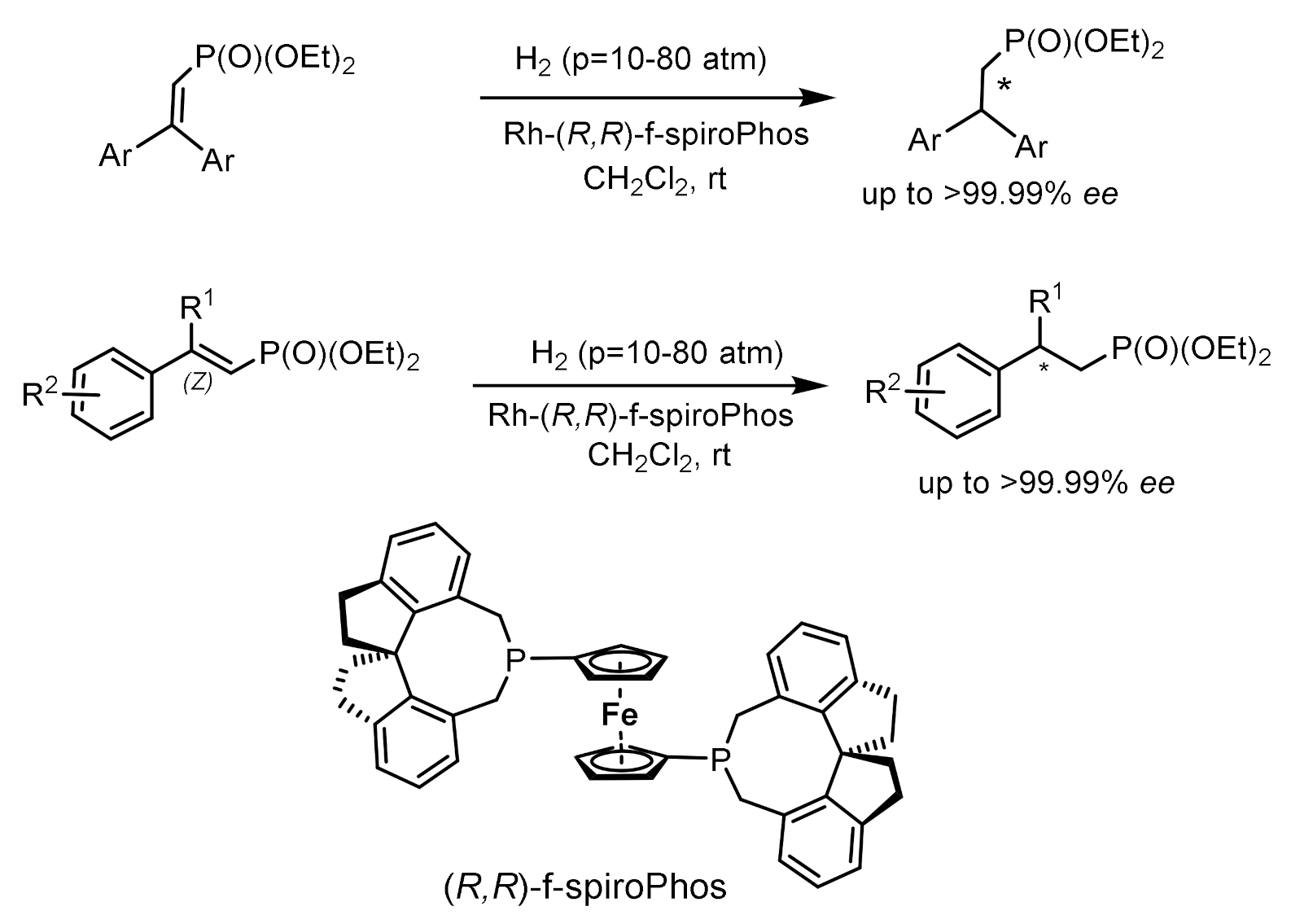
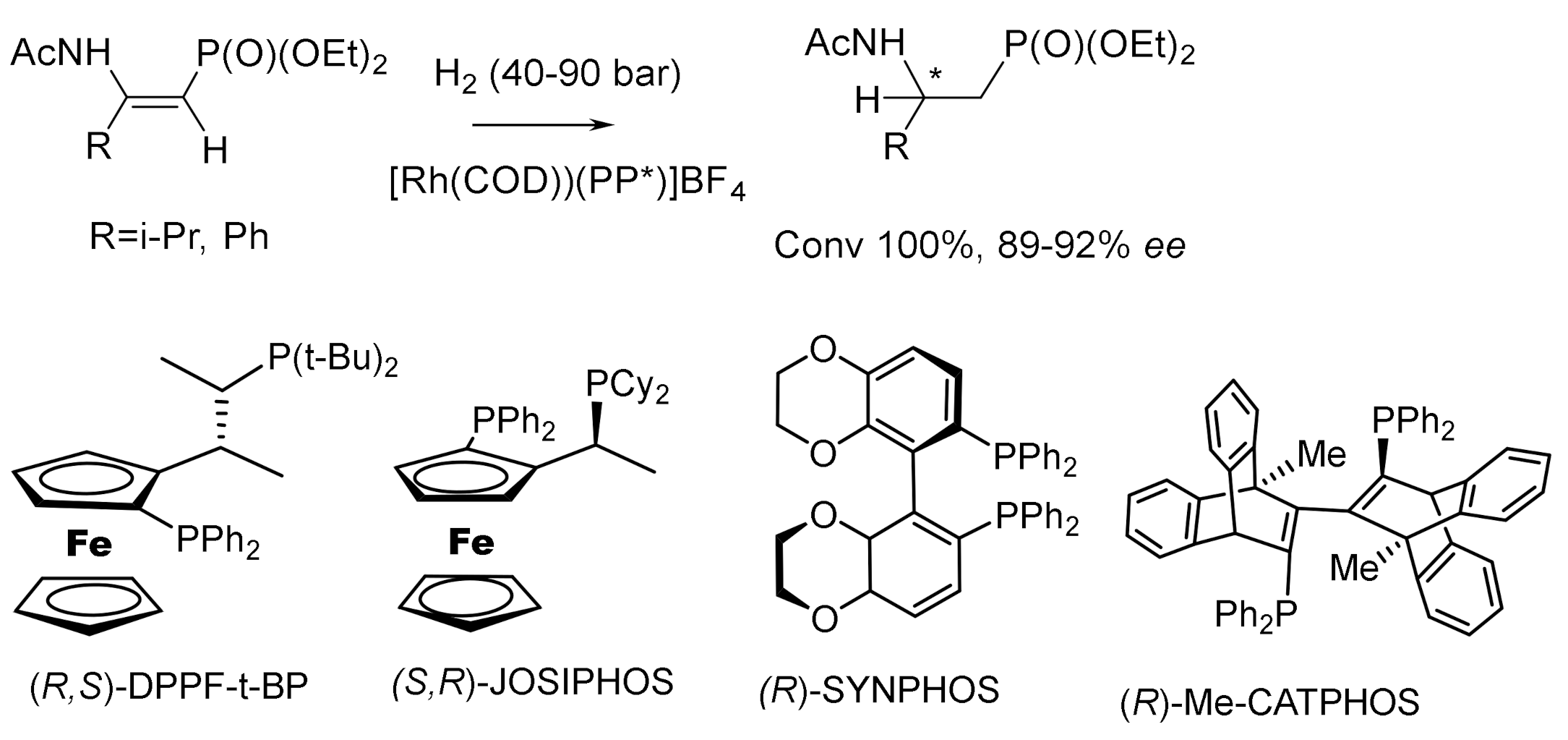
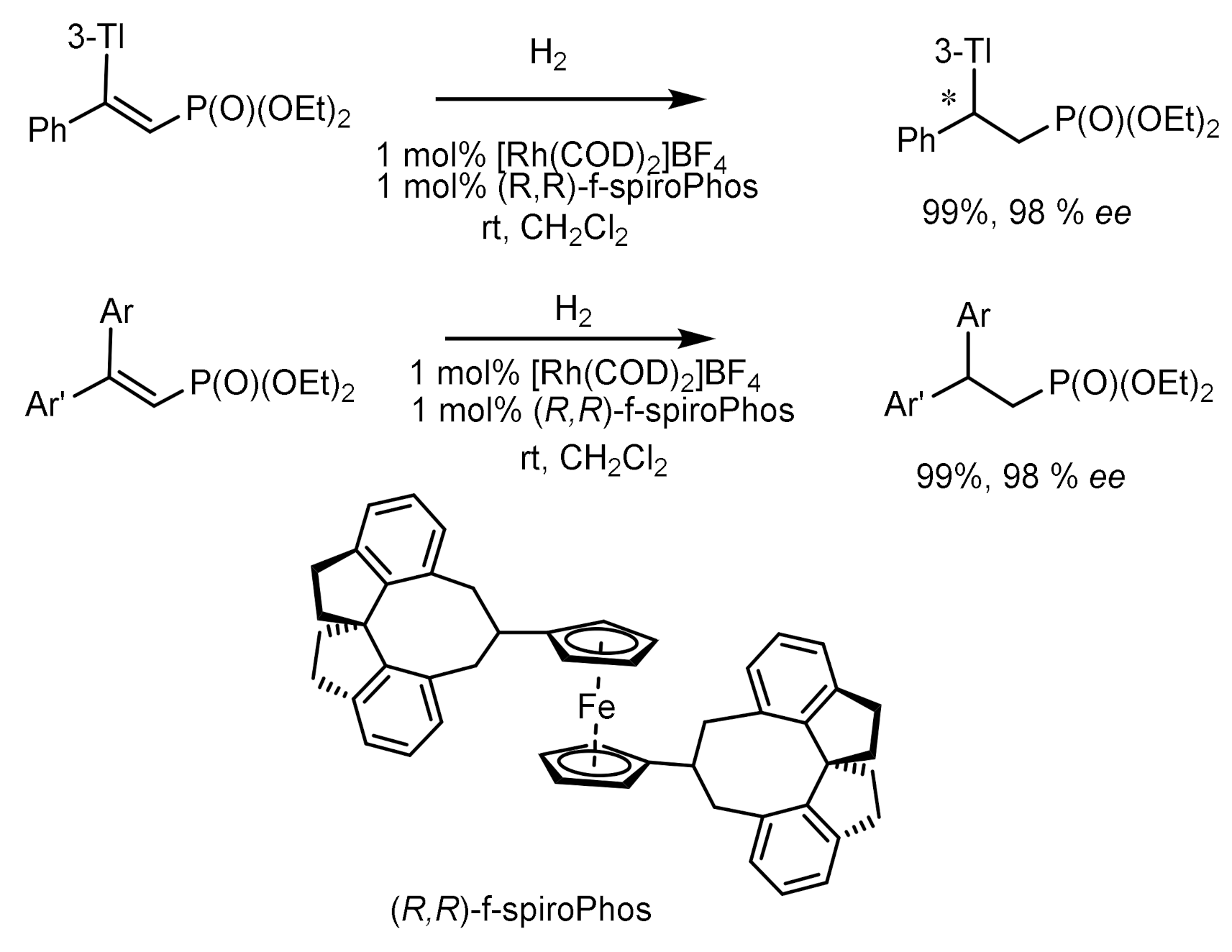
Asymmetric hydrogenation of unsaturated phosphonates in the presence of chiral transition metal complexes is an important method for the synthesis of functionalized phosphonates. For example, the hydrogenation of β,β-diarylvinylphosphonates catalyzed by the Rh-(R,R)-f-spiroPhos complex [127] made it possible to obtain chiral β,β-diarylethylphosphonates with 99.9% ee. In addition, this catalyst also demonstrates relatively excellent performance for β-aryl -β-alkyl unsaturated phosphonates, providing the corresponding chiral phosphonates with ee values up to 99.9%. This methodology provides direct access to the asymmetric synthesis of chiral phosphonates. Hydrogenation with the PROPRAPHOS ligand in both absolute configurations led to the formation of (R)- and (S)-enantiomers of α–aminophosphinates with enantioselectivities of 88–92% ee. Et-DuPhos-Rh(I) chiral complexes providing enantioselectivity reaching 94–94% ee are effective catalysts for the asymmetric hydrogenation of N-aryl and N-benzyloxycarbonyl-enamidophosphonates (Figure 73).
Unsaturated phosphonates have been implemented for the synthesis of β,β-diarylchiral phosphonates with excellent enantioselectivity (up to 99.9% ee) catalyzed by the Rh-(R,R)-f-spiroPhos complex. In addition, this catalyst also exhibits comparatively excellent performance for β-aryl-β-alkyl-unsaturated phosphonates, providing the corresponding chiral phosphonates with ee up to 99.9%. This methodology provides direct access to the asymmetric synthesis of chiral phosphonates (Figure 74).
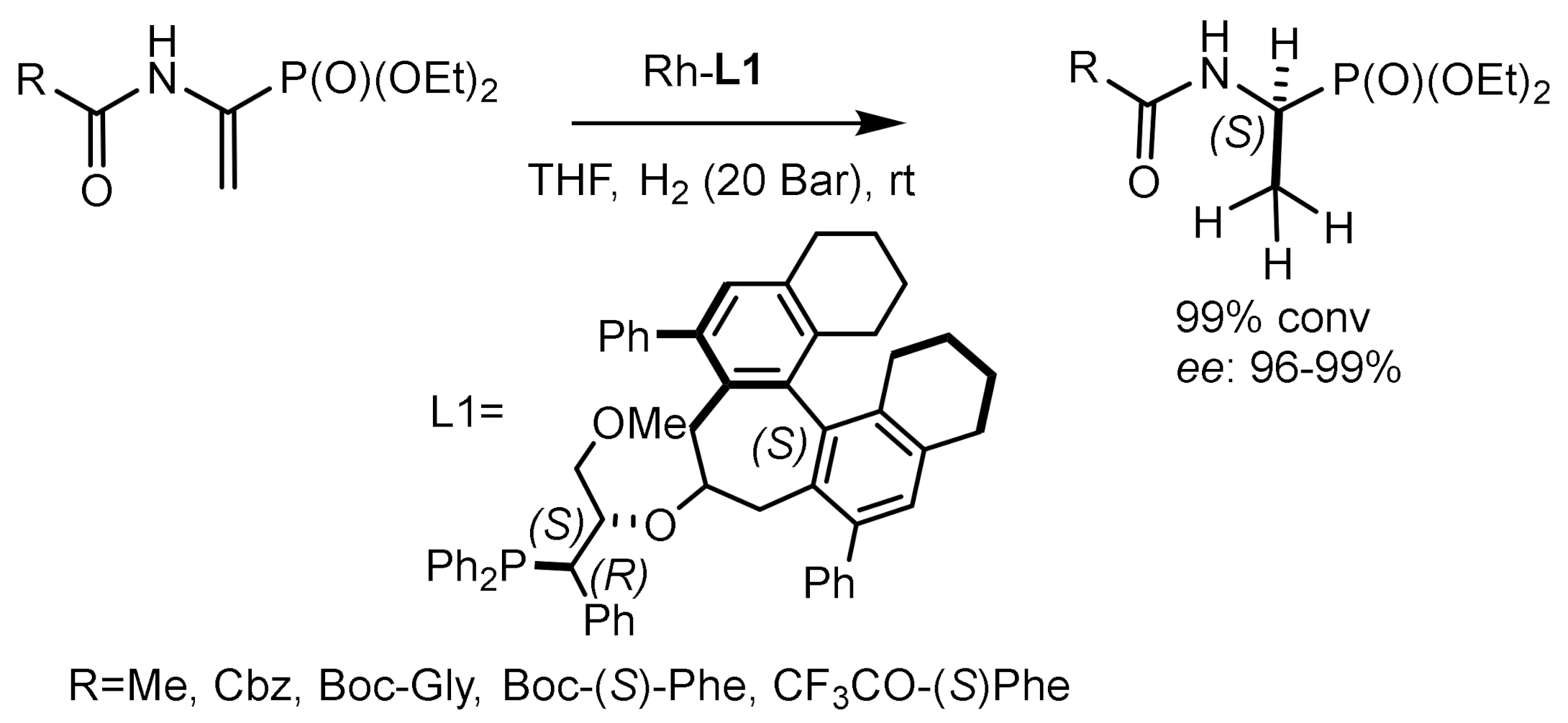
Wang et al. [137,138] used rhodium complexes containing chiral phosphine-aminophosphine ligands for the enantioselective catalytic hydrogenation of α–enol phosphonates and α–enamidophosphonates. Phosphine-aminophosphine ligands showed higher enantioselectivity compared to BoPhoz analogs. Very good enantioselectivity (93–97% ee) was achieved in the hydrogenation of various substrates catalyzed by the (S)-(L)/Rh(COD)]BF4 complex, indicating a high potential of phosphine-aminophosphine ligands in the production of optically active hydroxyphosphonates and α–aminophosphonates (Figure 75).
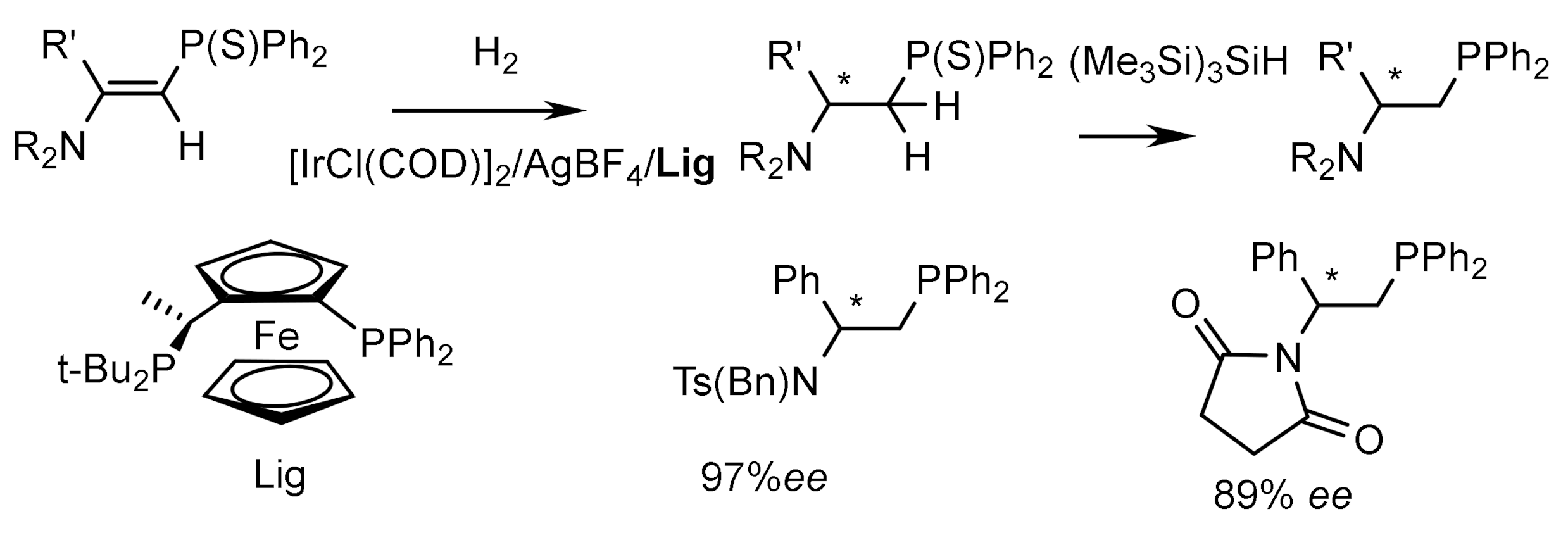
Iridium complexes containing chiral phosphine ligands are highly efficient catalysts for asymmetric hydrogenation. For example, the catalytic hydrogenation of vinylphosphonates using a rhodium complex containing a ferrocene ligand (Lig) proceeded with the formation of optically active (E)-2-amino-1-thiophosphinylalkanes in high yields and with high ee [139]. Subsequent desulfurization of the hydrogenation products led to the formation of 2-amino-1-phosphinoalkanes, which are chiral N,P ligands (Figure 76).
Hydrogenation of prochiral β–N-acetylamino-vinylphosphonates catalyzed by chiral rhodium complexes containing chiral ferrocene ligands led to the formation of chiral β–N-acetylaminophosphonates with almost 100% yields and high enantioselectivities (up to 92% ee). The highest enantioselectivities were achieved when the reaction was carried out in dichloromethane or THF. In some cases, during the hydrogenation of (Z)-alkenephosphonates, an inversion of the generated chirality was observed. The catalysts were prepared in situ by mixing [Rh(COD)2]BF4 with an equimolecular amount of a bidentate phosphorus ligand (Figure 77) [135].
Asymmetric hydrogenation of β,β-diarylunsaturated phosphonates catalyzed by the Rh-(R,R)-f-spiroPhos complex has been used to synthesize β,β-diarylchiral phosphonates with high enantioselectivity (up to 99.9% ee). In addition, this catalyst also exhibits excellent performance for β-aryl-β-alkyl unsaturated phosphonates, giving the corresponding chiral phosphonates with optical purity up to 99.9% ee [127] (Figure 78).
Doherty et al. [140] found that rhodium complexes containing the ligands (R,S)-JOSIPHOS and (R)-Me-CATPHOS are efficient enantioselective catalysts for the asymmetric hydrogenation of (E)- and (Z)-β–aryl–β–(enamido) phosphonates. Rhodium complexes with the ligands (R)-Me-CATPHOS and (R,S)-JOSIPHOS in most cases provided asymmetric hydrogenation of both (E)- and (Z)-β–aryl–β–enamidophosphonates in yields of 72–97% and with 99% ee and up [141]. Enantioselective hydrogenation of vinylphosphonates (70–94% ee) was achieved using iridium complexes with phosphinoxazoline ligands. For example, optically active 1-arylethylphosphinates, which are phosphorus analogues of naproxen, have been synthesized with 92–95% ee by hydrogenation of vinylphosphonates in methylene chloride, at +20 °C or low heat and a pressure of 5–60 bar (Figure 79) [142]. Chiral phosphine ligands, 1,2-bis(alkylmethylphosphino)ethanes (BisP) and bis(alkylmethyl) phosphinomethanes) (MiniPHOS) exhibited high enantioselectivity in the rhodium-catalyzed hydrogenation of vinylphosphonates (Figure 80) [143].
Chiral rhodium complexes containing P–OR ligands have proven to be effective catalysts for the enantioselective hydrogenation of enolphosphonates. The highest enantioselectivity (98% ee) was achieved with substrates containing an alkyl substituent in the β–position, while the hydrogenation of olefins containing an aromatic substituent proceeded with enantioselectivities not exceeding 92% ee [141,142,143,144] (Figure 81).
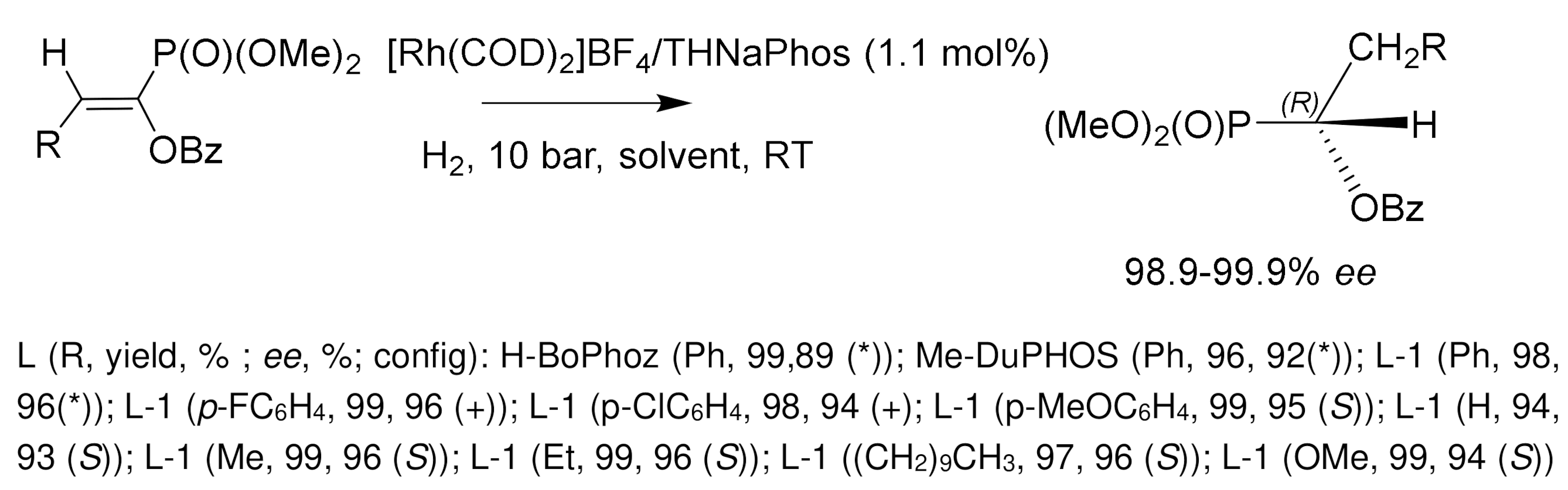
Much attention was paid to the development of an enantioselective method for the synthesis of α-hydroxyphosphonates by catalytic hydrogenation of β-aryl-, β-alkoxy-, and β-alkyl-substituted enolphosphonates. Wang [138] used for this purpose rhodium complexes containing unsymmetrical phosphine–amide–phosphite THNAPhos ligands. As a result of asymmetric hydrogenation, phosphonates with an optical purity of 99.9% ee were obtained. In another study [145], the hydrogenation of enolphosphonates in the presence of cationic rhodium catalysts L/Rh(COD)OTf containing C2-symmetric ligands L=DuPHOS and BPE also proceeded with high stereoselectivity 85–99.9% ee at room temperature and low hydrogen pressure (10 bar), in methylene chloride or isopropanol solution. Good results were also obtained with rhodium complexes containing phosphine-aminophosphine ligands (S)-HY–Phos (yields 95–99%, 93–96% ee), as well as with chiral (S)-HY–Phos ligand (98–99% ee) [142,143,145] (Figure 82).
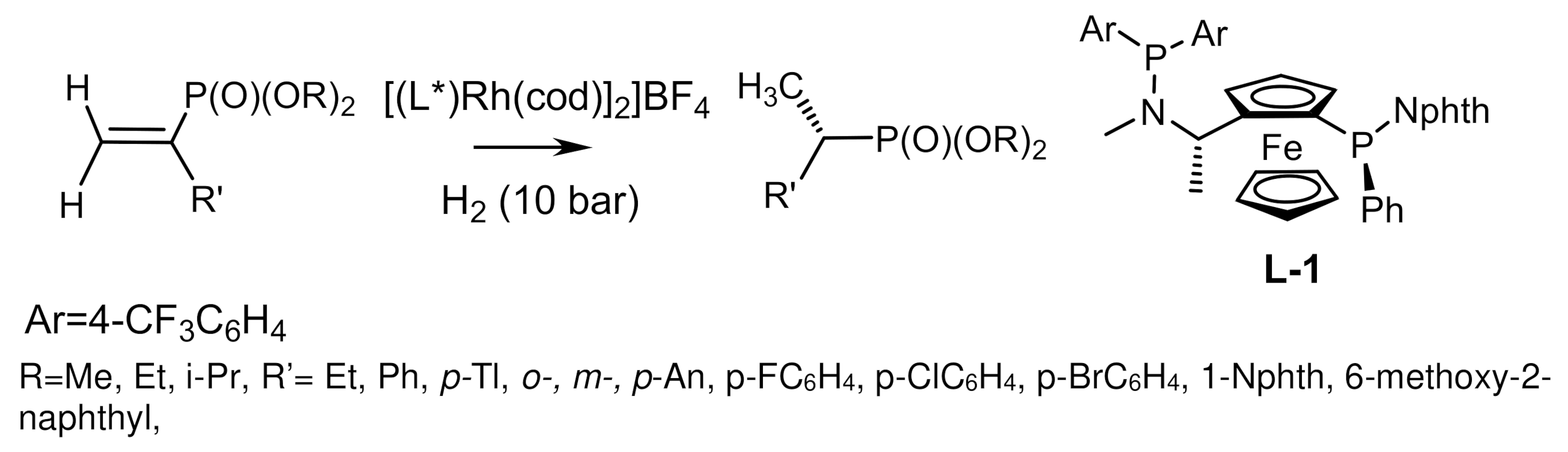
The Rh/ClickFerrophos complexes also proved to be efficient catalysts in the hydrogenation of β-dialkylphosphonates, (Z)-β-enolvinylphosphonates, and α-phenylethylenephosphonates, which were converted to the corresponding chiral phosphonates in good yields and enantioselectivities reaching 96% ee [141,144,146,147] (Figure 83).
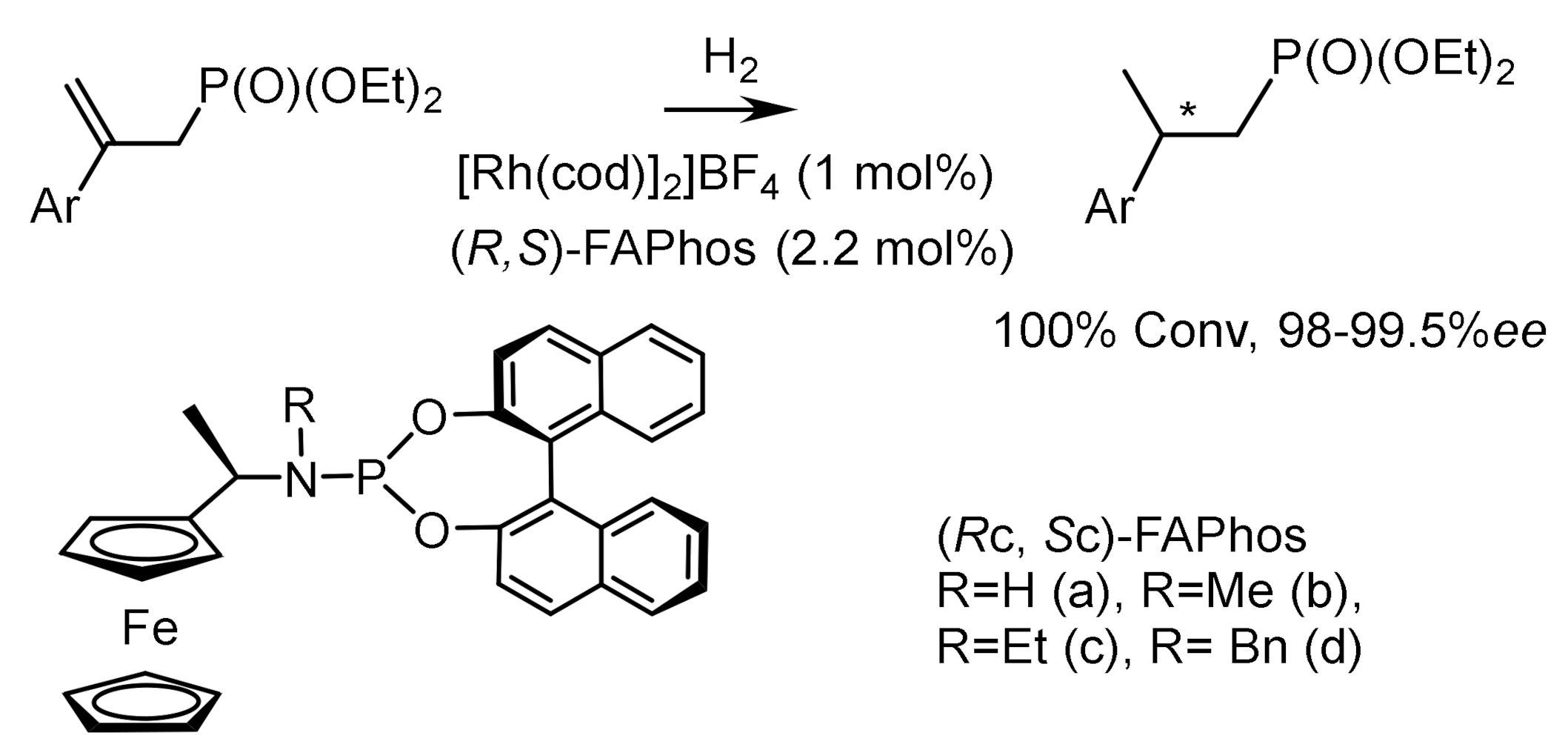
Hydrogenation of β–substituted α,β–unsaturated phosphonates catalyzed by rhodium complexes containing ferrocene ligands led to the formation of chiral β–substituted alkanephosphonates with high ee. Moreover, hydrogenation of (E)-substrates occurred with 100% conversions, enantioselectivities reaching 99.5% ee, and hydrogenation of (Z)-substrates with 98.0% ee. The absolute configurations of most compounds were not determined, although it was noted that hydrogenation catalyzed by a rhodium complex containing the (RC,SC)–FAPhos ligand proceeded to form compounds having the (R)-configuration. The chiral phosphine-amidophosphite ligand (S)–HY–Phos has been used for Rh-catalyzed asymmetric hydrogenation of α–acetamido)-cinnamates, enamides, and enolphosphonates with 98–99% ee [143,144,145] (Figure 84).
Asymmetric hydrogenation of 1-aryl- and 1-alkylethenylphosphonates in the presence of rhodium complexes containing P-chiral aminophosphine-phosphine ligands of the BoPhoz type led to the formation of chiral 1-aryl and 1-alkyl-substituted ethylphosphonates with 92–98% ee and yields up to 90–95% (Figure 85). Hydrogenation was carried out at the hydrogen pressure of 10 bar, room temperature, 1.1 mol% of catalyst and 0.25 mmol of substrate (Figure 86) [146].
Hydrogenation of electron-deficient carboxyethylvinylphosphonates also occurred with high enantioselectivity reaching 96% ee [144,147]. Diphenylvinylphosphine oxide and di- and trisubstituted vinylphosphonates were used as substrates in asymmetric hydrogenation catalyzed by an iridium complex. The highest enantioselectivity (up to 99% ee) was observed for substrates with aromatic and aliphatic groups at the prochiral carbon (Figure 87).
Kafarsky et al. performed the hydrogenation of substituted vinylphosphonates using rhodium complexes containing P–OP L1, ent–L1, or (R,R)-Me–DuPHOS ligands as catalysts. As a result, α-aminophosphonic acids of high enantiomeric purity (up to 99% ee and de) were obtained. Due to the high quality of the obtained amino acids, it was possible to use in the synthesis of phosphonopeptides, which are valuable building blocks for obtaining biologically relevant molecules [148] (Figure 88).
3.2. Asymmetric Hydrogenation and Reduction of Ketophosphonates
Chiral transition metal complexes catalyze the hydrogenation and hydrosilylation of prochiral ketones. From a practical point of view, the asymmetric hydrogenation of ketophosphonates is one of the convenient methods for the synthesis of chiral hydroxyphosphonates [21,149]. The asymmetric catalytic synthesis of α–amino and α–hydroxyphosphonates is of great interest due to the pharmaceutical activity of these compounds. Asymmetric hydrogenation uses various catalysts, in particular Ru(II)–BINAP complexes. Early works in this area include studies by Noyori et al. [149] who showed that Ru(II)–BINAP complexes (1 mol% RuCI2(R)-BINAP](dmf)n) successfully catalyze the hydrogenation of 1-(formamido) alkenylphosphonates in methanol at low pressure H2 and 30 °C to form α–formamidophosphonates with high yields and 94–98% ee. Phosphoalanine, phosphoethylglycine, and phosphophenylalanine were obtained by this method in high enantiomeric purity. It was found that the use of the (S)-BINAP–Ru(II) catalyst gave predominantly (R)-products, while the (R)-BINAP complexes resulted in the formation of products having the (S)-configuration. Genet reported the asymmetric hydrogenation of ketophosphonates catalyzed by chiral (R)-MeO–BIPHEP complexes and obtained hydroxyphosphonates with enantioselectivities up to 99% ee (Figure 89) (for detailed information see [7]. (Figure 89).
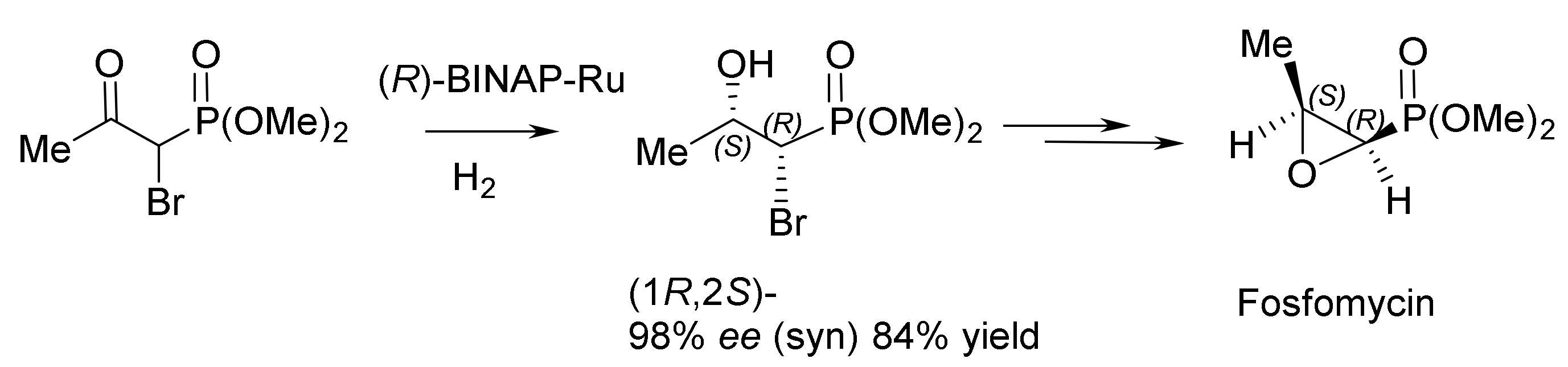
Noyori et al. developed a catalytic method for the synthesis of fosfomycin, an antibiotic used in clinics to treat patients. Starting from racemic (rac)-β-keto–α–bromophosphonate, fosfomycin was prepared in yield of 84%, with 98% ee and a syn:anti ratio of 90:10 (Figure 90 and Figure 91) [149].
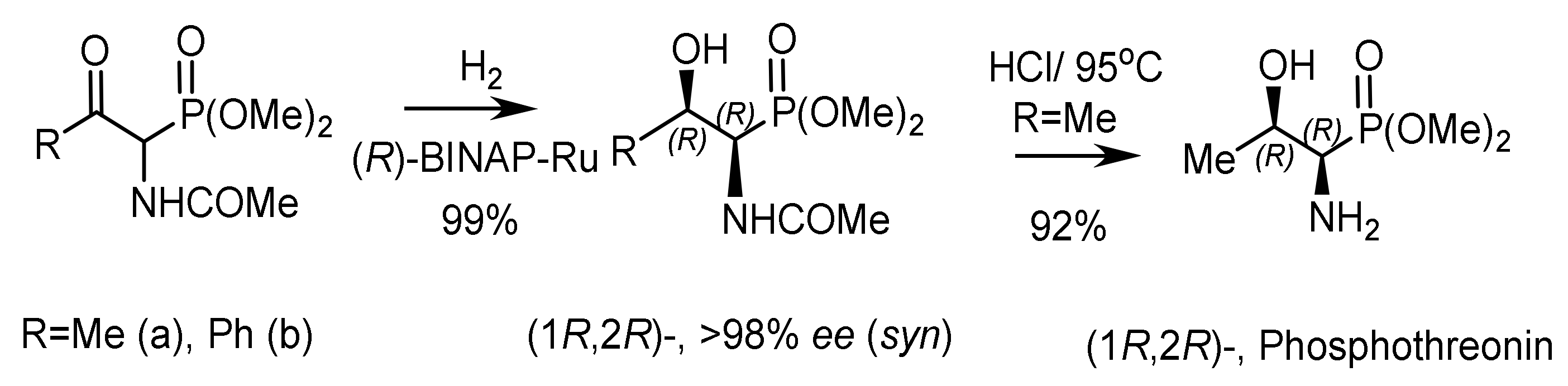
By this method, racemic α–acetamido-β–ketophosphonate was converted to (1R,2R)-hydroxyphosphonate with high diastereoselectivity (syn:anti = 97:3) and an enantioselectivity of 98% ee (Figure 92). Next, (1R,2R)-hydroxyphosphonate was converted to enantiomerically pure (1R,2R)-phosphothreonine in 92% yield. BINAP–ruthenium catalytic hydrogenation of labile ketophosphonates led to the formation of (R,R)- or (S,S)-α–amido-hydroxyphosphonates with high enantio- and diastereoselectivity (95% ee, syn:anti 98:2) (Figure 93).
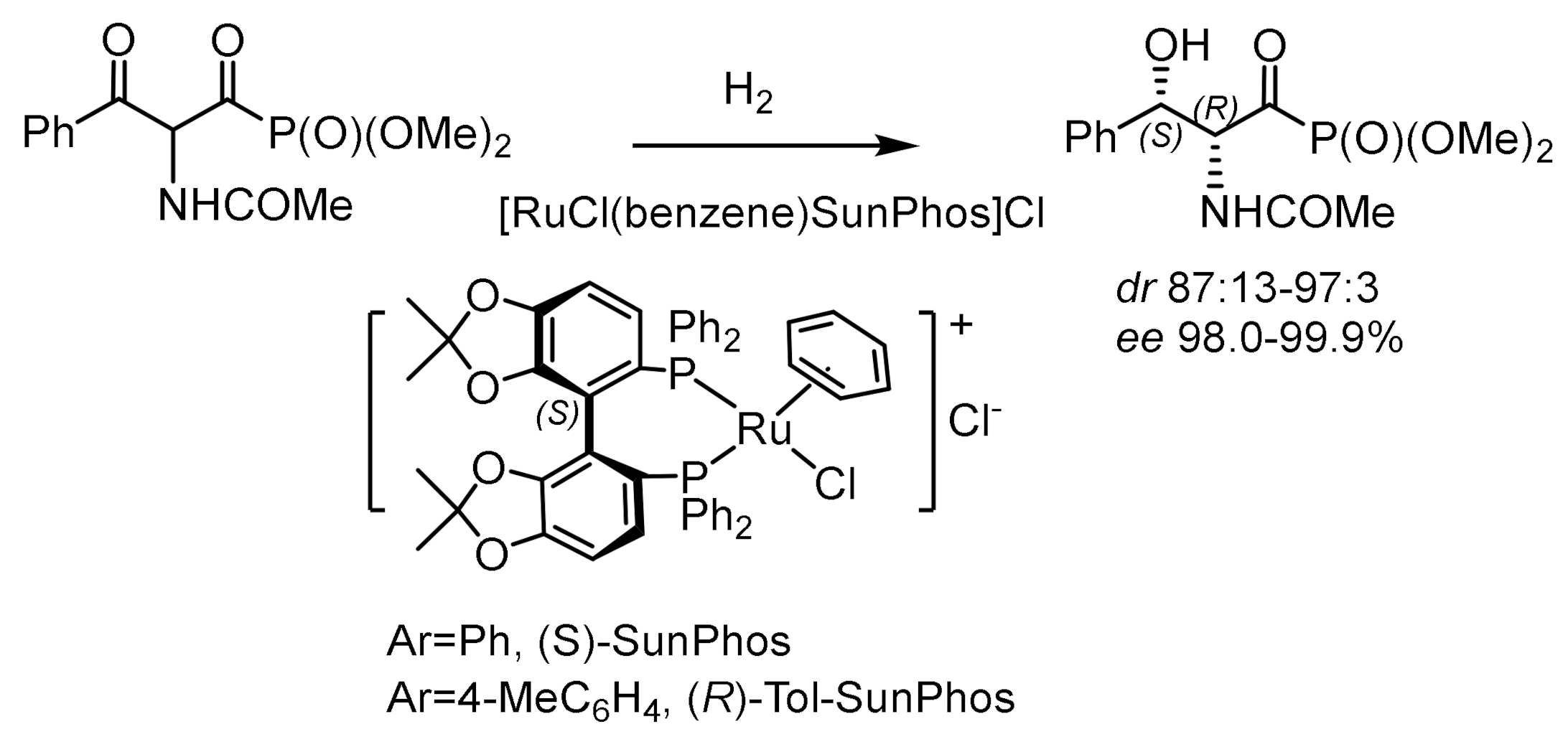
Dynamic kinetic resolution of α-amido-β-ketophosphonates by asymmetric hydrogenation in the presence of atropoisomeric ruthenium catalysts led to the formation of the corresponding β–hydroxy-α-amidophosphonates with high diastereo- and enantioselectivities (syn/anti = 99:1 and 99.8% ee). The catalysts were synthesized from atropisomeric (S)-SunPhos ligands and [RuCl2(benzene)]2. Hydrogenation was carried out under a hydrogen pressure at 10 bar and a temperature of 50 °C in methanol. As a result, chiral phosphonates were obtained with 98.0% ee and a M isomer ratio of 97:3, with corresponding β-hydroxy-(R)-amido phosphonates with high enantioselectivity [150,151]. As a result of hydrogenation of a series of methyl, ethyl, and isopropyl (2-oxo-2-phenylethyl)phosphonates, the corresponding alcohols were obtained with 99.7, 95.5, and 90.0% ee, respectively. Additives have been found to play an important role in increasing the diastereo- and enantioselectivity of the reaction. For example, the addition of CeCl3-7H2O increased the stereoselectivity of the reaction to syn/anti = 99:1 and 99.8% ee [152]. The electron-donating groups in the para-position of the phenyl group of ketophosphonates increased the stereoselectivity, while the electron-withdrawing groups decreased the chemical yields of the hydrogenation products. Ma et al. has recently reported the hydrogenation of β-ketophosphonates with chiral Ir/P,N,N-ligands (L) catalyst. A number of β-ketophosphonates were hydrogenated, and the corresponding β-hydroxyphosphonates were obtained in high yields with very good enantioselectivities under mild condition (Figure 94) [151].
Zhow et al. developed the asymmetric hydrogenation of a range of linear and cyclic α-iminophosphonates with a palladium catalyst, providing efficient access to optically active α-aminophosphonates with ee up to 99% (Figure 95) [153].
Enantioselective reduction of prochiral ketophosphonates is one of the important methods for obtaining enantio-enriched hydroxyphosphonates. The reduction with borohydrides in the presence of chiral catalysts (oxazaborolidines, alcaloids, chiral amines) is the most effective catalytic system. A number of biologically active compounds have been synthesized by oxazaborolidine-catalyzed reduction of ketones with boron hydrides at a key stage. The chiral-modified borohydrides fixed on a polymer support were reusable. Asymmetric CBS (Cory, Bakshi, Shibata) catalytic reduction is an efficient method for preparing various chiral alcohols [154,155]. This method has been applied for the synthesis of enantiomerically enriched hydroxyphosphonates via enantioselective catalytic reduction of α-ketophosphonates with catecholborane and oxazoborolidine catalyst. Meier et al., using this methodology, obtained hydroxyphosphonates with enantioselectivities up to >99% ee (Figure 96) [154]. The borohydride, coordinated to the nitrogen atom of oxazaborolidine, increases the acidity of the intracyclic boron atom, facilitating the coordination of the ketone that is being reduced. The mechanism of catalysis was investigated using ab initio MO calculations. An interesting example of asymmetric reduction catalysts developed on the basis of the oxazaborolidine structure is the oxazophospholidine borohydride complex compound, which reduced aromatic and aliphatic ketophosphonates when heated in toluene, giving hydroxyphosphonates with moderate enantioselectivity (Figure 96). Barco et al. have described a diastereoselective synthesis of β-amino-α-hydroxyphosphonates using borohydride reduction of β-phthalimido-α-ketophosphonates catalyzed by chiral oxazaborolidines. While reduction with borohydride-dimethyl sulfide in THF gave a mixture of (S,S)- and (S,R)-diastereomers (dr = 8:1–10:1), the reaction of ketophosphonates with catecholborane in the presence of a catalytic amount (12 mol.%) oxazaborolidine in toluene at −60 °C led to the formation of a single (S,S)-diastereomer in good yield [156].
The enantioselective reduction of NH-imines catalyzed by oxazaborolidines is a convenient method for the preparation of aminophosphonates. Reduction with catecholborane catalyzed by methyloxazaborolidine makes it possible to obtain aminophosphonates in high yields and enantioselectivity (Figure 97) [157,158]. Palacios et al. [159] described the asymmetric reduction of 2H-azirine-2-phosphonates. The key step in this reaction is the Neber reaction of p-toluenesulfonyl oximes derived from phosphonates. Various alkaloids such as sparteine (SP), quinidine (QN), hydroquinidine (HQ), or quinine (Q) were used for cyclization. Reduction of 2H-azirines with sodium borohydride in ethanol gave cis-aziridine phosphonates with moderate ee (Figure 98).
Enantiomerically pure carboxylic acids of natural origin have been used for the enantioselective reduction with borohydrides of ketophosphonates into hydroxyphosphonates. In particular, the reduction of ketophosphonates with NaBH4 catalyzed with L-proline afforded hydroxyphosphonates with 50–70% ee. This reducing system was applied for the preparation of a number of chiral hydroxyphosphonates (Figure 99) [160].
An interesting method for the enantioselective reduction of ketophosphonates was developed based on the reduction with sodium borohydride in the presence of (S,S)- or (R,R)-tartaric acid [161,162,163]. The reduction of α–ketophosphonates with this reagent was carried out by cooling to −30 °C in THF. The stereochemistry of reduction of α- and β-ketophosphonates with sodium borohydride/tartaric acid depended on the absolute configuration of tartaric acid. Reduction of α-ketophosphonates with a reagent derived from sodium borohydride and (R,R)-tartaric acid led to the formation of (S)-α-hydroxyphosphonates, and reduction with sodium borohydride/(S,S)-tartaric acid gave (R)-α -hydroxyphosphonates (Figure 100).
The reduction stereoselectivity of ketophosphonates containing chiral mentyl groups at the phosphorus atom with NaBH4-(R,R)-TA was higher than the stereoselectivity of ketophosphonates containing achiral methyl or ethyl groups at phosphorus. The higher stereoselectivity of reduction in the case of dimenthyl aryl ketophosphonates is explained by the effect of double asymmetric induction. The higher stereoselectivity of the reduction of dimentyl aryl ketophosphonates can be explained by the effect of double asymmetric induction, since in this case the asymmetric inductions of the menthyl groups and tartaric acid were summed up. At the same time, the reduction of dimentylketophosphonates with sodium borohydride/(S,S)-tartaric acid proceeded with lower stereoselectivity. For example, reduction of dimentylbenzoylphosphonate gave the corresponding hydroxyphosphonate with only 45% de. Obviously, the asymmetric inductions of (1R,2S,5R)-mentyl groups and (R,R)-tartaric acid act in concert in the same direction and therefore add up, increasing the resulting stereoselectivity. At the same time, the asymmetric inductions of (1R,2S,5R)-mentyl groups and (S,S)-tartaric acid act inconsistently in opposite directions and are therefore subtracted, reducing the resulting stereoselectivity.
The stereoisomers of dimenthyl (S)- and (R)-2-hydroxy-3-chloropropylphosphonate were obtained with enantioselectivity of 96% ee [161]. These compounds are useful chiral synthons (chirons) for the synthesis of enantiomerically pure β-hydroxyphosphonates. Through these chirons, various biologically important chiral β-hydroxyphosphonic acids, such as phosphono-carnitine and γ–amino-β–hydroxypropylphosphonic acid (phosphono-GABOB), have been obtained. The treatment with potash of (S)-2-hydroxy-3-chloropropylphosphonate in DMF in the presence of potassium iodide led to the formation of (R)-epoxide with 99% de. Reaction of epoxide with sodium azide in the methanol containing additive of ammonium chloride gave (R)-2-hydroxy-3-azidopropylphosphonate, which was converted into aziridine in high yield (Figure 101) [162,163].
The reduction of acylphosphonates with formic acid and triethylamine, catalyzed by RuCl [(S,S)-TsDPEN] (p-cymene) complex containing a chiral aminosulfonamide ligand, led to the formation of (R)-hydroxyphosphonate with high diastereo- and enantioselectivity up to 99% ee. Highly selective catalytic hydrogenation of acylphosphonates has been used for dynamically kinetically resolution of α-aryl-acylphosphonates with formation of β-stereogeneous α-hydroxyphosphonic acid derivatives. The absolute configurations of the products were established by X-ray diffraction analysis, showing anti-orientation of the groups OH и Ar (Figure 102) [164].
Silane in the presence of (S)-SegPhos/Cu(OAc) 2H2O (1–5 mol %) PMHS and t-BuOH complex reduced 3-aryl-4-phosphonobutenoates with enantioselectivities up to 94% ee. The enantioselectivity of the reduction was influenced by the spatial and electronic effects of the substrates [165]. Substrates having an electron-withdrawing group in the para-position of the phenyl ring were reduced with higher ee values than substrates with electron-donating groups (Figure 103). Mhamdi aand Touil reported a green and universal method for the preparation of γ-hydroxyphosphonates and phosphine oxides by means of reduction of γ-phosphonyl ketones with sodium borohydride deposited on alumina at room temperature in the absence of a solvent [166].
4. Conclusions
Metal-complex catalysis and organo-catalysis are important methods for the synthesis of C-chiral phosphonates, which are the most numerous and interesting type of organophosphorus compounds in terms of biological activity. In this review, we have considered several variants of asymmetric catalysis, which make it possible to obtain C-chiral phosphonates with a high enantiomeric excess (ee). It should be noted that, despite intensive studies of catalytic methods for the synthesis of C-chiral phosphonates, not all problems in this area have been solved. The prospects for the chemical modification of C-chiral phosphonates with the introduction of new, more complex groups into the side chain, including those with a specific configuration, are far from being exhausted. The exact absolute configuration can only be set successfully in a limited number of cases. In addition, only in a limited number of cases do the wide range of substrates and the cost of the catalyst meet the requirements necessary for industrial use. Therefore, the search for new, more efficient catalytic systems is an urgent task in this field of chemistry. In this regard, the development of highly efficient methods for the asymmetric organo-catalytic and metal-complex catalysis is an urgent task. It is not difficult to predict that the main efforts in studying the chemistry of chiral analogues of natural compounds will be concentrated in this direction, and new enantioselective reactions are promising for solving these problems.
Obviously, asymmetric catalysis by means of chiral bifunctional organocatalysts can be a very useful tool in the design of chiral phosphorus-containing compounds with a complex structure [167]. Organo-catalysis, which has shown its promise in many syntheses of C-chiral phosphonates, is necessary for the search of new catalysts and new reactions. It can be expected that catalytic methods for the synthesis of C-chiral phosphonates will remain the focus of intensive research, especially in the field of biologically active compounds. In the future, the development of various methods for applying catalysts, their modification, and the creation of new preparative forms of organo- and biocatalysts will serve as the basis for the creation of new asymmetric catalysts suitable for industrial application.
Author Contributions
O.I.K. and A.O.K. wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are openly available.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
Ala—Alanine; ALB—[AlLi(binaphthoxide)2]; An—Anisyl; Arg—Argenine; BINAPO—Boc—tert-Butoxycarbonyl; CHIRAPHOS—(2S,3S)-(–)-Bis(diphenylphosphino)butane; de—Diastereomeric excess; DIBAL-H—Diisobutylaluminum hydride; DIOP—O-Isopropylidene-2,3-dihydroxy-1,4-bis(diphenylphosphino)butane; DIPAMP—(R,R)-(Ethane-1,2-diyl)bis[(2-methoxyphenyl)(phenyl)phosphane]; DMSO—Dimethylsulfoxide; DiPT—Diisopropyl tartrate; DiPE—Diisopropyl ether; ee—Enantiomeric excess; Gly—Glycine; GTP—Guanosine-5′-triphosphate; i-Pr—iso-Propyl; Leu—Leucine; Mes—Mesyl; Mnt—(1R,2S,5R)-Menthyl; MTBE—Methyl tert-butyl ether; Nphth—Naphthyl; PAMP—phenylanisylmethylphosphine PEP—Phosphoenolpyruvate; rac—Racemate; TA—Tartaric acid; TBDMS—tert-Butyldimethylsilyl; TBDMS—Tert-butyl-dimethylsilan; Tl-Toluyl; TMS—Trimethylsilyl; THF—Tetrahydrofurane; TFA—Trifluoroacetic acid; TMS—Trimethylsilyl; Tol—Tolyl; Ts—toluenesulfonyl (tosyl); Val—Valine.
References
- Berlicki, L.; Kafarski, P. Computer-Aided Analysis and Design of Phosphonic and Phosphinic Enzyme Inhibitors as Potential Drugs and Agrochemicals. Curr. Org. Chem. 2005, 9, 1829–1850. [Google Scholar] [CrossRef]
- Gbubele, J.D.; Olszewski, T.K. Asymmetric synthesis of organophosphorus compounds using H–P reagents derived from chiral alcohols. Org. Biomol. Chem. 2021, 19, 2823–2846. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, M.; Kandatsu, M. Isolation of 2-Aminoethane. Phosphonic Acid from Rumen Protozoa. Nature 1959, 184, 901–902. [Google Scholar] [CrossRef] [PubMed]
- Hilderbrand, R.L. The Role of Phosphonates in Living Systems, 1st ed.; Taylor and Francis Group: Abingdon, UK; CRC Press: Boca Raton, FL, USA, 2017; ISBN1 10:1315897377. ISBN2 13:978-1315897370. [Google Scholar]
- Kafarski, P. Phosphonates: Their Natural Occurrence and Physiological Role. Contemporary Topics about Phosphorus in Biology and Materials; Lee, W., Abdillah, A., Palma, J., Churchill, D.G., Eds.; IntechOpen: London, UK, 2020; pp. 1–19. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Phosphorus compounds of natural origin: Prebiotic, Stereochemistry, Application. Symmetry 2021, 13, 889. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Asymmetric Synthesis in Organophosphorus Chemistry: Synthetic Methods, Catalysis and Application; Wiley-VCH: Weinheim, Germany, 2016; ISBN 978-3-527-34150-4. [Google Scholar]
- Takeuchi, M.; Nakajima, M.; Ogita, T.; Inukai, M.; Kodama, K.; Furuya, K.; Nagaki, H.; Haneishi, T. Fosfonochlorin, a new antibiotic with spheroplast forming activity. J. Antibiot. 1989, 42, 198–205. [Google Scholar] [CrossRef]
- Leonard, P.G.; Satani, N.; Maxwell, D.; Lin, Y.-H.; Hammoudi, N.; Peng, Z.F. SF2312 is a natural phosphonate inhibitor of enolase. Nat. Chem. Biol. 2016, 12, 1053–1058. [Google Scholar] [CrossRef]
- Falagas, M.E.; Vouloumanou, K.; Samonis, G.; Vardakas, K.Z. Fosfomycin. Clin. Microbiol. Rev. 2016, 29, 321–347. [Google Scholar] [CrossRef]
- Chofor, R.; Sooriyaarachchi, S.; Risseeuw, M.D.P.; Bergfors, T.; Pouyez, J.; Johny, C.; Haymon, A.; Everaert, A.; Dowd, C.S.; Maes, L.; et al. Synthesis and Bioactivity of β-Substituted Fosmidomycin Analogues Targeting 1-Deoxy-D-xylulose-5-phosphate Reductoisomerase. J. Med. Chem. 2015, 58, 2988–3001. [Google Scholar] [CrossRef]
- Kafarski, P. Phosphonopeptides containing free phosphonic groups: Recent advances. RSC Adv. 2020, 10, 25898–25910. [Google Scholar] [CrossRef]
- Hidaka, T.; Seto, H.; Imai, S. Biosynthesis mechanism of carbon-phosphorus bond formation. Isolation of carboxyphosphonoenolpyruvate and its conversion to phosphinopyruvate. J. Am. Chem. Soc. 1989, 111, 8012–8013. [Google Scholar] [CrossRef]
- Peck, S.C.; van der Donk, W. Phosphonate biosynthesis and catabolism: A treasure trove for unusual enzymology. Curr. Opin. Chem. Biol. 2013, 17, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T. The Chemical Biology of Phosphorus; RSC: Cambridge, UK, 2020; 546p. [Google Scholar] [CrossRef]
- Mikołajczyk, M. Phosphonate reagents and building blocks in the synthesis of bioactive compounds, natural products and medicines. Pure Appl. Chem. 2019, 91, 811–838. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I.; Kukhar, V.P.; Kolodiazhna, A.O. Asymmetric catalysis as a method for the synthesis of chiral organophosphorus compounds. Tetrahedron Asymmetry 2014, 25, 865–922. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I.; Kolodiazhna, A.O. Multiple stereoselectivity in organophosphorus chemistry. Phosph. Sulf. Silicon Relat. Elem. 2016, 191, 444–458. [Google Scholar] [CrossRef]
- Feringa, B.L. Chiral phosphorus compounds by catalytic asymmetric C–P coupling. Nat. Catal. 2022, 5, 8–9. [Google Scholar] [CrossRef]
- Numan, A.; Brichacek, M. Asymmetric Synthesis of Stereogenic Phosphorus P(V) Centers Using Chiral Nucleophilic Catalysis. Molecules 2021, 26, 3661. [Google Scholar] [CrossRef]
- Kolodyazhna, A.O.; Kolodyazhnyi, O.I. Stereoselective Synthesis of Organophosphorus Compounds; Naukova Dumka Pbl: Kyiv, Ukraine, 2017; 342p, ISBN 978-966-00-1599-9. [Google Scholar]
- Rádai, Z.; Keglevich, G. Synthesis and Reactions of Hydroxyphosphonates. Molecules 2018, 23, 1493. [Google Scholar] [CrossRef]
- Enders, D.; Saint-Dizier, A.; Lannou, M.-I.; Lenzen, A.; Lenzen, A. The Phospha-Michael Addition in Organic Synthesis. Eur. J. Org. Chem. 2006, 1, 29–49. [Google Scholar] [CrossRef]
- Guo, H.; Chiao, Y.; Zhanhu, F.; Yang, S.; Kwon, W.O. Phosphine Organocatalysis. Chem. Rev. 2018, 118, 10049–10293. [Google Scholar] [CrossRef]
- Phillips, A.M.F. Organocatalytic Asymmetric Synthesis of Chiral Phosphonates. Mini-Rev. Org. Chem. 2014, 11, 164–185. [Google Scholar] [CrossRef]
- Ordóñez, M.; Viveros-Ceballos, J.L.; Romero-Estudillo, I. Stereoselective Synthesis of α-Aminophosphonic Acids through Pudovik and Kabachnik-Fields Reaction; IntechOpen: Zagreb, Croatia, 2017. [Google Scholar]
- Shibazaki, M.; Sasai, H. Advances in Asymmetric Synthesis Direct Catalytic Asymmetric Three Component Kabachnik-Fields Reaction; Hassner, A., Ed.; Stamford; JAi Press: London, UK, 1998; Volume 3, pp. 191–233. [Google Scholar]
- Yamagishi, T.; Yokomatsu, T.; Suemune, K.; Shibuya, S. Enantioselective synthesis of a-hydroxyphosphinic acid derivatives through hydrophosphinylation of aldehydes catalyzed by Al-Li-BINOL complex. Tetrahedron 1999, 55, 12125–12136. [Google Scholar] [CrossRef]
- Nakajima, M.; Kotani, S.; Ishizuka, T.; Hashimoto, S. Chiral phosphine oxide BINAPO as a catalyst for enantioselective allylation of aldehydes with allyltrichlorosilanes. Tetrahedron Lett. 2005, 46, 157–159. [Google Scholar] [CrossRef]
- Nakanishi, K.; Kotani, S.; Sugiura, M.; Nakajima, M. First asymmetric Abramov-type phosphonylation of aldehydes with trialkyl phosphites catalyzed by chiral Lewis bases. Tetrahedron 2008, 64, 6415–6419. [Google Scholar] [CrossRef]
- Ayad, T.; Gernet, A.; Pirat, J.-L.; Virieux, D. Enantioselective reactions catalyzed by phosphine oxides. Tetrahedron 2019, 75, 4385–4418. [Google Scholar] [CrossRef]
- Kolodiazhna, A.O.; Kukhar, V.P.; Chernega, A.N.; Kolodiazhnyi, O.I. Double and triple asymmetric induction in phosphaaldol reactions. Tetrahedron Asymmetry 2004, 15, 1961–1963. [Google Scholar] [CrossRef]
- Laborde, C.; Wei, M.-M.; Van der Lee, A.; Deydier, E.; Daran, J.-C.; Volle, J.-N.; Poli, R.; Pirat, J.-L.; Manoury, E.; Virieux, D. Double [3 + 2]-dimerisation cascade synthesis of bis(triazolyl)bisphosphanes, a new scaffold for bidentate bisphosphanes. J. Chem. Soc. Dalton Trans. 2015, 44, 12539–12545. [Google Scholar] [CrossRef]
- Sevrain, N.; Volle, J.-N.; Pirat, J.-L.; Ayad, T.; Virieux, D. Chiral Bisdiphenylphosphine Dioxides Bearing a Bis(triazolyl) Backbone as Promising Lewis Bases for Asymmetric Organocatalysis. Eur. J. Org. Chem. 2018, 2018, 2267–2272. [Google Scholar] [CrossRef]
- Sevrain, N.; Volle, J.-N.; Pirat, J.-L.; Ayad, T.; Virieux, D. 1,1′-Dibenzyl-bis-(triazolyl)diphenylphosphine dioxide: A new efficient organocatalyst for silicon tetrachloride-mediated enantioselective Abramov-type phosphonylation of aldehydes with trialkyl phosphites. RSC Adv. 2017, 7, 52101–52104. [Google Scholar] [CrossRef]
- Gou, S.; Zhou, X.; Wang, J.; Liu, X.; Feng, X. Asymmetric hydrophosphonylation of aldehydes catalyzed by bifunctional chiral Al(III) complexes. Tetrahedron 2008, 64, 2864–2870. [Google Scholar] [CrossRef]
- Guin, J.; Wang, Q.; van Gemmeren, M.; List, B. The Catalytic Asymmetric Abramov Reaction. Angew. Chem. Int. Ed. 2015, 54, 355–358. [Google Scholar] [CrossRef]
- Yang, F.; Zhao, D.; Lan, J.; Xi, P.; Yang, L.; Xiang, S.; You, J. Self-Assembled Bifunctional Catalysis Induced by Metal Coordination Interactions: An Exceptionally Efficient Approach to Enantioselective Hydrophosphonylation. Angew. Chem. Int. Ed. 2008, 47, 5646–5649. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Wang, L.-L.; Baia, J.-F.; Ji, L.-N.; Yang, Q.-C.; Huang, Q.-C. Highly effective and nantioselective Phospho-Aldol reaction of diphenyl phosphite with N-alkylated isatins catalyzed by quinine. Tetrahedron Lett. 2011, 52, 1157–1160. [Google Scholar] [CrossRef]
- Suyama, K.; Sakai, Y.; Matsumoto, K.; Saito, B.; Katsuki, T. Highly Enantioselective Hydrophosphonylation of Aldehydes: Base-Enhanced Aluminum-salalen Catalysis. Angew. Chem. Int. Ed. 2010, 49, 797–799. [Google Scholar] [CrossRef] [PubMed]
- Guin, J.; Wang, Q.; Gemmeren, M.V.; List, B. Die katalytische asymmetrische Abramov-Reaktion. Angew. Chem. 2015, 127, 362–365. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, X.; Yang, X. Highly Enantioselective Hydrophosphonylation of Aldehydes Catalyzed by Tridentate Schiff Base Aluminum(III) Complexes. Angew. Chem. Int. Ed. 2008, 47, 392–394. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, Y.; Chang, L. Highly Efficient Synthesis of Quaternary a-Hydroxy Phosphonates via Lewis Acid-Catalyzed Hydrophosphonylation of Ketones. Adv. Synth. Catal. 2009, 351, 2567–2572. [Google Scholar] [CrossRef]
- Ying, S.; Huang, X.; Guo, X.; Yang, S.; Zhao, J.; Shang, D.; Liu, X.; Lin, L.; Feng, X. The sequential C–H oxidation/asymmetric phosphonylation of primary alcohols to synthesize α-hydroxyphosphonates. Green Synth. Catal. 2021, 2, 315–319. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, Q.; Hui, Y.; Chen, W.; Jiang, J.; Lin, L.; Liu, X.; Feng, X. Catalytic Asymmetric Synthesis of Quaternary a-Hydroxy Trifluoromethyl Phosphonate via Chiral Aluminum(III) Catalyzed Hydrophosphonylation of Trifluoromethyl Ketones. Org. Lett. 2010, 12, 4296–4299. [Google Scholar] [CrossRef]
- Wang, F.; Liu, X.; Cui, X.; Xiong, Y.; Zhou, X.; Feng, X. Asymmetric Hydrophosphonylation of α–Ketoesters Catalyzed by CinchonaDerived Thiourea Organocatalysts. Chem. Eur. J. 2009, 15, 589–592. [Google Scholar] [CrossRef]
- Abell, J.P.; Yamamoto, H. Catalytic Enantioselective Pudovik Reaction of Aldehydes and Aldimines with Tethered Bis(8-quinolinato) (TBOx) Aluminum Complex. J. Am. Chem. Soc. 2008, 130, 10521–10523. [Google Scholar] [CrossRef]
- Uraguchi, D.; Ito, T.; Ooi, T. Generation of Chiral Phosphonium Dialkyl Phosphite as a Highly Reactive P-Nucleophile: Application to Asymmetric Hydrophosphonylation of Aldehydes. J. Am. Chem. Soc. 2009, 131, 3836–3837. [Google Scholar] [CrossRef] [PubMed]
- Simón, L.; Paton, R.S. Origins of Asymmetric Phosphazene Organocatalysis: Computations Reveal a Common Mechanism for Nitro- and Phospho-Aldol Additions. J. Org. Chem. 2015, 80, 2756–2766. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Shi, M. Applications of Chiral Phosphine-Based Organocatalysts in Catalytic Asymmetric Reactions. Chem. Asian J. 2014, 9, 2720–2734. [Google Scholar] [CrossRef] [PubMed]
- Kolodyazhnaya, A.O.; Kukhar, V.P.; Kolodyazhnyi, O.I. Organic Catalysis of Phospha-Aldol Condensation. Russ. J. Gen. Chem. 2008, 78, 2043–2051. [Google Scholar] [CrossRef]
- Alegre-Requena, J.V.; Marques-Lopez, E.; Sanz Miguel, P.J.; Herrera, R.P. Organocatalytic enantioselective hydrophosphonylation of aldehydes. Org. Biomol. Chem. 2014, 12, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Perera, S.; Naganaboina, V.K.; Wang, L.; Zhang, B.; Guo, Q.; Rout, L.; Zhao, C.-G. Organocatalytic Highly Enantioselective Synthesis of β-Formyl-ahydroxyphosphonates. Adv. Synth. Catal. 2011, 353, 1729–1734. [Google Scholar] [CrossRef]
- Vicario, J.; Ezpeleta, J.M.; Palaciosa, F. Asymmetric Cyanation of a-Ketiminophosphonates Catalyzed by Cinchona Alkaloids: Enantioselective Synthesis of Tetrasubstituted a-Aminophosphonic Acid Derivatives from Trisubstituted α-Aminophosphonates. Adv. Synth. Catal. 2012, 354, 2641–2647. [Google Scholar] [CrossRef]
- Kong, S.; Fan, W.; Wu, G.; Miao, Z. Enantioselective Synthesis of Tertiary a-Hydroxy Phos. phonates Catalyzed by Carbohydrate I Cinchona Alkaloid Thiourea Organocatalysts. Angew. Chem. Int. Ed. 2012, 51, 8864–8867. [Google Scholar] [CrossRef]
- George, J.; Sridhar, B.; Reddy, B.V.S. First example of quinine-squaramide catalyzed enantioselective addition of diphenyl phosphite to ketimines derived from isatins. Org. Biomol. Chem. 2014, 12, 1595–1602. [Google Scholar] [CrossRef]
- Kumar, A.; Sharma, V.; Kaur, J.; Kumar, V.; Mahaja, S.; Mahajan, S. Cinchona-derived Thiourea Catalyzed Hydrophosphonylation of Ketirnines—An Enantioselective Synthesis of a-Amino Phosphonates. Tetrahedron Lett. 2014, 70, 7044–7049. [Google Scholar] [CrossRef]
- Yao, Q.; Yuan, C.A. A Synthetic Study of Chiral α–Hydroxy-H-Phosphinates Based on Proline Catalysis. Chem. Eur. J. 2013, 19, 6080–6088. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, I.; Mohammadi, L.; Heravi, M.M. Organocatalyzed Asymmetric Mannich Reaction: An Update. Chem. Sel. 2021, 6, 1008–1066. [Google Scholar] [CrossRef]
- Albrecht, Q.; Albrecht, A.; Krawczyk, H.; Jorgensen, K.A. Organocatalytic Asymmetric Synthesis of Organophosphorus Compounds. Chem. Eur. J. 2010, 16, 28–48. [Google Scholar] [CrossRef] [PubMed]
- Saranya, N.; Ann Harry, K.; Krishnan, K.; Anilkumar, G. Recent Developments and Perspectives in the Asymmetric Mannich Reaction. Asian J. Org. Chem. 2018, 7, 613–633. [Google Scholar] [CrossRef]
- Yin, Z.; Guo, J.; Zhang, R.; Hu, X.; Borovkov, V. Direct Asymmetric Three-Component Mannich Reaction Catalyzed by Chiral Counteranion-Assisted Silver. J. Org. Chem. 2020, 85, 10369–10377. [Google Scholar] [CrossRef] [PubMed]
- Kafarski, P.; Gorny vel Gorniak, M.; Andrasiak, I. Kabachnik-Fields Reaction Under Green Conditions—A Critical Overview. Curr. Green Chem. 2015, 2, 218–222. [Google Scholar] [CrossRef]
- Keglevich, G.; Bálint, E. The Kabachnik–Fields Reaction: Mechanism and Synthetic Use. Molecules 2012, 17, 12821. [Google Scholar] [CrossRef]
- Boratynski, P.J.; Skarzewski, J.; Sidorowicz, L. Stereochemistry of hydrophosphonylation of 9-aminoquinine Schiff bases. Arkivoc 2012, 4, 204–215. [Google Scholar] [CrossRef]
- Chen, W.; Hui, Y.; Zhou, X.; Jiang, J.; Cai, Y.; Liu, X.; Lin, L.; Feng, X. Chiral N,N′-dioxide-Yb(III) complexes catalyzed enantioselective hydrophosphonylation of aldehydes. Tetrahedron Lett. 2010, 51, 4175–4178. [Google Scholar] [CrossRef]
- Iwanejko, J.; Brol, A.; Szyja, B.M.; Daszkiewicz, M.; Wojaczynska, E.; Olszewski, T.K. Aminophosphonates and aminophosphonic acids with tetrasubstituted stereogenic center: Diastereoselective synthesis from cyclic ketimines. Org. Biomol. Chem. 2019, 17, 7352–7359. [Google Scholar] [CrossRef]
- Cheng, X.; Goddard, R.; Buth, G.; List, B. Direct Catalytic Asymmetric Three-Component Kabachnik-Fields Reaction. Angew. Chem. Int. Ed. 2008, 47, 5079–5081. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Morita, H.; Itoh, J.; Fuchibem, K. Chiral Brnnsted Acid Catalyzed Enantioselective Hydrophosphonylation of Imines: Asymmetric Synthesis of a-Amino Phosphonates. Org. Lett. 2005, 7, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Pedroni, J.; Cramer, N. Enantioselective C–H Functionalization—Addition Sequence Delivers Densely Substituted 3-Azabicyclo [3.1.0]hexanes. J. Am. Chem. Soc. 2017, 139, 12398–12401. [Google Scholar] [CrossRef] [PubMed]
- Saito, B.; Egami, H.; Katsuki, T. Synthesis of an Optically Active Al(salalen) Complex and Its Application to Catalytic Hydrophosphonylation of Aldehydes and Aldimines. J. Am. Chem. Soc. 2007, 129, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, D.; Marcolini, M.; Bernardi, L.; Fini, F.; Herrera, R.P.; Sgarzani, V.; Ricci, A. Direct Access to Enantiomerically Enriched α-Amino Phosphonic Acid Derivatives by Organocatalytic Asymmetric Hydrophosphonylation of Imines. J. Org. Chem. 2006, 71, 6269–6272. [Google Scholar] [CrossRef]
- Kadota, T.; Sawa, M.; Kondo, Y.; Morimoto, H.; Ohshima, T. Catalytic Enantioselective Strecker Reaction of Isatin-Derived N-Unsubstituted Ketimines. Org. Lett. 2021, 23, 4553–4558. [Google Scholar] [CrossRef]
- Cao, W.L.; Liu, X.L.; Feng, X. Chiral organobases: Properties and applications in asymmetric catalysis. Chin. Chem. Lett. 2018, 29, 1201–1208. [Google Scholar] [CrossRef]
- Joly, G.D.; Jacobsen, E.N. Thiourea-Catalyzed Enantioselective Hydrophosphonylation of Imines: Practical Access to Enantiomerically Enriched α-Amino Phosphonic Acids. J. Am. Chem. Soc. 2004, 126, 4102–4103. [Google Scholar] [CrossRef]
- Guang, J.; Guo, Q.; Zhao, J.C.-G. Acetylphosphonate as a Surrogate of Acetate or Acetamide in Organocatalyzed Enantioselective Aldol Reactions. Org. Lett. 2012, 14, 3174–3177. [Google Scholar] [CrossRef]
- Fu, X.; Loh, W.-T.; Zhang, Y.; Chen, T.; Ma, T.; Liu, H.; Wang, J.; Tan, C.-H. Chiral Guanidinium Salt Catalyzed Enantioselective Phospha-Mannich Reactions. Angew. Chem. Int. Ed. 2009, 48, 7387–7390. [Google Scholar] [CrossRef]
- Lin, S.; Otsuka, Y.; Yin, L.; Kumagai, N.; Shibasaki, M. Catalytic Enantioselective Addition of Diethyl Phosphite to N-Thiophosphinoyl Ketimines: Preparation of (R)-Diethyl (1-Amino-1-phenylethyl)phosphonate. Org. Synth. 2017, 94, 313–331. [Google Scholar] [CrossRef]
- Thorat, P.B.; Goswami, S.V.; Magar, R.L.; Patil, B.R.; Bhusare, S.R. An efficient organocatalysis: A one-pot highly enantioselective synthesis of α-aminophosphonates. Eur. J. Org. Chem. 2013, 24, 5509–5516. [Google Scholar] [CrossRef]
- Vellalath, S.; Čorić, I.; List, B. N-Phosphinyl Phosphoramide—A Chiral Brønsted Acid Motif for the Direct Asymmetric N,O-Acetalization of Aldehydes. Angew. Chem. Int. Ed. 2010, 49, 9749–9752. [Google Scholar] [CrossRef] [PubMed]
- Mukaiyama, T.; Narasaka, K.; Banno, K. New Aldol type reaction. Chem. Lett. 1973, 2, 1011–1014. [Google Scholar] [CrossRef]
- Gondi, V.B.; Hagihara, K.; Rawal, V.H. Diastereoselective and Enantioselective Synthesis of Tertiary α–Hydroxy Phosphonates through Hydrogen-Bond Catalysis. Angew. Chem. Int. Ed. 2009, 48, 776–779. [Google Scholar] [CrossRef]
- Kobayashi, S.; Kiyohara, H.; Nakamura, Y.; Matsubara, R. Catalytic Asymmetric Synthesis of α-Amino Phosphonates Using Enantioselective Carbon-Carbon Bond-Forming Reactions. J. Am. Chem. Soc. 2004, 126, 6558–6559. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Liu, W.; Zheng, Y.; He, Z. Organocatalytic asymmetric cascade cyclization reaction of o-hydroxy cinnamaldehydes with diphenylphosphine oxide. Chin. Chem. Lett. 2018, 29, 1625–1628. [Google Scholar] [CrossRef]
- Heravi, M.M.; Hajiabbasi, P. Recent advances in C-heteroatom bond forming by asymmetric Michael addition. Mol. Divers. 2014, 18, 411–439. [Google Scholar] [CrossRef]
- Zhao, D.; Mao, L.; Yang, D.; Wang, R. Zinc-mediated asymmetric additions of dialkylphosphine oxides to α,β-unsaturated ketones and N-sulfinylimines. J. Org. Chem. 2010, 75, 6756–6763. [Google Scholar] [CrossRef]
- Zhao, D.; Wang, Y.; Mao, L.; Wang, R. Highly enantioselective conjugate additions of phosphites to α,β-unsaturated N-acylpyrroles and imines: A practical approach to enantiomerically enriched amino phosphonates. Chem. Eur. J. 2009, 15, 10983–10987. [Google Scholar] [CrossRef]
- Bartoli, G.; Bosco, M.; Carlone, A.; Locatelli, M.; Mazzanti, A.; Sambri, L.; Melchiorre, P. Organocatalytic asymmetric hydrophosphination of nitroalkenes. J. Chem. Soc. Chem. Commun. 2007, 722–724. [Google Scholar] [CrossRef] [PubMed]
- Carlone, A.; Bartoli, G.; Bosco, M.; Sambri, L.; Melchiorre, P. Organocatalytic Asymmetric Hydrophosphination of α,β-unsaturated Aldehydes. Angew. Chem. Int. Ed. 2007, 46, 4504–4506. [Google Scholar] [CrossRef] [PubMed]
- Ibrahem, I.; Rios, R.; Vesely, J.; Hammar, P.; Eriksson, L.; Himo, F.; Cordova, A. Enantioselective organocatalytic hydrophosphination of α,β-unsaturated aldehydes. Angew. Chem. Int. Ed. 2007, 46, 4507–4510. [Google Scholar] [CrossRef] [PubMed]
- Maerten, E.; Cabrera, S.; Kjrsgaard, A.; Jorgensen, K.A. Organocatalytic asymmetric direct phosphonylation of alpha, beta-unsaturated aldehydes: Mechanism, scope, and application in synthesis. J. Org. Chem. 2007, 72, 8893–8903. [Google Scholar] [CrossRef] [PubMed]
- Ibrahem, I.; Hammar, P.; Vesely, J.; Rios, R.; Eriksson, L.; Córdov, A. Organocatalytic Asymmetric Hydrophosphination of α,β-Unsaturated Aldehydes: Development, Mechanism and DFT Calculations. Adv. Synth. Catal. 2008, 350, 1875–1884. [Google Scholar] [CrossRef]
- Fu, X.; Jiang, Z.; Tan, C.H. Bicyclic guanidine-catalyzed enantioselective phospha-Michael reaction: Synthesis of chiral β-aminophosphine oxides and β-aminophosphines. J. Chem. Soc. Chem. Commun. 2007, 2007, 5058–5060. [Google Scholar] [CrossRef]
- Terada, M.; Ikehara, T.; Ube, H. Enantioselective 1,4-addition reactions of diphenyl phosphite to nitroalkenes catalyzed by an axially chiral guanidine. J. Am. Chem. Soc. 2007, 129, 14112–14113. [Google Scholar] [CrossRef]
- Wang, J.; Heikkinen, L.D.; Li, H.; Zu, L.; Jiang, W.; Xie, H.; Wang, W. Quinine-catalyzed enantioselective Michael addition of diphenyl phosphite to nitroolefins: Synthesis of chiral precursors of α−substituted β-aminophosphonates. Adv. Synth. Catal. 2007, 349, 1052–1056. [Google Scholar] [CrossRef]
- Zhu, Y.; Malerich, J.P.; Rawal, V.H. Squaramide-catalyzed enantioselective Michael addition of diphenyl phosphite to nitroalkenes. Angew. Chem. 2010, 122, 157–160. [Google Scholar] [CrossRef]
- Tessema, E.; Elakkat, V.; Chiu, C.-F.; Zheng, J.-H.; Chan, K.L.; Shen, C.-R.; Zhang, P.; Lu, N. Recoverable Phospha-Michael Additions Catalyzed by a 4-N,N-Dimethylaminopyridinium Saccharinate Salt or a Fluorous Long-Chained Pyridine: Two Types of Reusable Base Catalysts. Molecules 2021, 26, 1159. [Google Scholar] [CrossRef]
- Sufcer-Mosse, S.; Tissot, M.; Alexakis, A. First Enantioselective Organocatalytic Conjugate Addition of Aldehydes to Vinyl Phosphonates. Org. Lett. 2007, 9, 3749–3752. [Google Scholar] [CrossRef] [PubMed]
- Sulzer-Moss, S.; Alexakis, A.; Mareda, J. Enantioselective Organocatalytic Conjugate Addition of Aldehydes to Vinyl Sulfones and Vinyl Phosphonates as Challenging Michael Acceptors. Chem. Eur. J. 2009, 15, 3204–3220. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.-W.; Liu, Y.-Y.; Ding, W.; Li, T.-R.; Shi, D.Q.; Chen, J.-R.; Xiao, W.-J. Chiral Squaramide Catalyzed Asymmetric Conjugate Additions of 3-Substituted Oxindoles to Vinylphosphonates. Synthesis 2013, 45, 1647–1653. [Google Scholar] [CrossRef]
- Wen, S.; Li, P.; Wu, H.; Yu, F.; Liang, X.; Ye, J. Enantioselective organocatalytic phospha-Michael reaction of α,β-unsaturated ketones. Chem. Commun. 2010, 46, 4806–4808. [Google Scholar] [CrossRef]
- Liu, T.; Wang, Y.; Wu, G. Organocatalyzed Enantioselective Michael Addition of 2-Hydroxy-1,4-naphtho-quinone to p,y-Unsaturated Ketophosphonates. J. Org. Chem. 2011, 76, 4119–4124. [Google Scholar] [CrossRef]
- Sohtome, Y.; Horitsugi, N.; Takagi, R.; Nagasawa, K. Enantioselective Phospha-Michael Reaction of Diphenyl Phosphonate with Nitroolefins Utilizing Conformationally Flexible Guanidinium/Bisthiourea Organocatalyst: Assembly-State Tunability in Asymmetric ganocatalysis. Adv. Synth. Cat. 2011, 353, 2631–2636. [Google Scholar] [CrossRef]
- Arai, R.; Hirashima, S.-I.; Nakano, T.; Kawada, M.; Akutsu, H.; Nakashima, K.; Miura, T. Asymmetric Conjugate Addition of Phosphonates to Enones Using Cinchona–Diaminomethylenemalononitrile Organocatalysts. J. Org. Chem. 2020, 85, 3872–3878. [Google Scholar] [CrossRef]
- Luo, X.; Zhou, Z.; Li, X.; Liang, X.; Ye, J. Enantioselective organocatalytic phospha-Michael reaction of α,β-unsaturated aldehydes. RSC Adv. 2011, 1, 698–705. [Google Scholar] [CrossRef]
- Albrecht, L.; Richter, B.; Vila, C.; Krawczyk, H.; Jørgensen, A.K. Organocatalytic Domino Michael-Knoevenagel Condensation Reaction for the Synthesis of Optically Active 3-Diethoxyphosphoryl-2-oxocyclohex-3-enecarboxylates. Chem. Eur. J. 2009, 15, 3093–3102. [Google Scholar] [CrossRef]
- Phillips, A.M.F.; Barros, M.T. Enantioselective organocatalytic synthesis of α-cyclopropylphosphonates through a domino Michael addition/intramolecular alkylation reaction. Eur. J. Org. Chem. 2014, 2014, 152–163. [Google Scholar] [CrossRef]
- Chen, J.; Wen, X.; Wang, Y.; Du, F.; Cai, L.; Peng, Y. Asymmetric Mannich Reaction of Isatin-Based Ketimines with α-Diazomethylphosphonates Catalyzed by Chiral Silver Phosphate. Org. Lett. 2016, 18, 4336–4339. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Kurra, Y.; Liu, W.R. Phospha-Michael Addition as a New Click Reaction for Protein Functionalization. Chembiochem 2016, 17, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Bera, K.; Namboothiri, I.N.N. Quinine-derived thiourea and squaramide catalyzed conjugate addition of nitrophosphonates to enones: Asymmetric synthesis of quaternary aminophosphonates. J. Org. Chem. 2015, 80, 1402–1413. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Perera, S.; Zhao, C.-G. Organocatalytic Enantioselective Synthesis of Both Diastereomers of Hydroxyphosphinates. J. Org. Chem. 2010, 75, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Dodda, R.; Zhao, C.G. Organocatalytic Highly Enantioselective Synthesis of Secondary R-Hydroxyphosphonates. Org. Lett. 2006, 8, 4911–4914. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Guo, L.; Xu, L.; Loh, T.-P. Asymmetric P−C Bond Formation: Diastereoselective Synthesis of Adjacent P,C-Stereogenic Allylic Phosphorus Compounds. Chem. Asian J. 2016, 9, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Scriban, C.; Glueck, D.S.; Zakharov, L.N. P-C and C-C Bond Formation by Michael Addition in Platinum-Catalyzed Hydrophosphination and in the Stoichiometric Reactions of Platinum Phosphido Complexes with Activated Alkenes. Organometallics 2006, 25, 5757–5767. [Google Scholar] [CrossRef]
- Sadow, A.D.; Haller, I.; Fadini, L.; Togni, A. Nickel(Il)-Catalyzed Highly Enantioselective Hydrophosphination of Methacrylonitrile. J. Am. Chem. Soc. 2004, 126, 14704–14705. [Google Scholar] [CrossRef]
- Sadow, A.D.; Togni, A. Enantioselective Addition of Secondary Phosphines to Methacrylonitrile: Catalysis and Mechanism. J. Am. Chem. Soc. 2005, 127, 17012–17024. [Google Scholar] [CrossRef]
- Feng, J.J.; Chen, X.F.; Shi, M.; Duan, W.L. Palladium-Catalyzed Asymmetric Addition of Diarylphosphines to Enones toward the Synthesis of Chiral Phosphines. J. Am. Chem. Soc. 2010, 132, 5562–5563. [Google Scholar] [CrossRef]
- Zhao, D.; Mao, L.; Wang, L. Catalytic asymmetric construction of tetrasubstituted carbon stereocenters by conjugate addition of dialkyl phosphine oxides to β,β–disubstituted a,b-unsaturated carbonyl compounds. J. Chem. Soc. Chem. Comm. 2012, 48, 889–891. [Google Scholar] [CrossRef] [PubMed]
- Shao, N.; Luo, Y.Y.; Lu, H.-J.; Hua, Y.-Z.; Wang, M.-C. Enantioselective phospha-Michael reaction of diethyl phosphonate with exocyclic α,β-unsaturated benzocyclic ketones catalyzed by a dinuclear zinc−AzePhenol catalyst. Tetrahedron 2018, 74, 2130–2142. [Google Scholar] [CrossRef]
- Sabater, S.; Mata, J.A.; Peris, E. Chiral Palladacycles with N-Heterocyclic Carbene Ligands as Catalysts for Asymmetric Hydrophosphination Chiral Palladacycles with N-Heterocyclic Carbene Ligands as Catalysts for Asymmetric Hydrophosphination. Organometallics 2013, 32, 1112–1120. [Google Scholar] [CrossRef]
- Russo, A.; Lattanzi, A. Asymmetric Organocatalytic Conjugate Addition of Diarylphosphane Oxides to Chalcones. Eur. J. Org. Chem. 2010, 2010, 6736–6739. [Google Scholar] [CrossRef]
- Huang, Y.R.; Chew, J.; Pullarkat, S.A. Asymmetric Synthesis of Enaminophosphines via Palladacycle-Catalyzed Addition of Ph2PH to α, β-Unsaturated Imines. J. Org. Chem 2012, 77, 6849–6854. [Google Scholar] [CrossRef]
- Nakamura, S.; Hayashi, M.; Hiramatsu, Y.; Shibata, N.; Funahashi, Y.; Toru, T. Catalytic Enantioselective Hydrophosphonylation of Ketimines Using Cinchona Alkaloids. J. Am. Chem. Soc. 2009, 131, 18240–18241. [Google Scholar] [CrossRef]
- Yadavalli, K.P.; Cummines, J.E.; Carlislea, C.J.; Lepore, S.D. Diastereoselective additions of H-phosphinates to alkenyl ketones under phase-transfer conditions. Chem. Commun. 2022, 58, 6441–6444. [Google Scholar] [CrossRef]
- Huang, Y.; Pullarkat, S.A.; Teong, S.; Chew, R.J.; Li, Y.; Leung, P.-H. Palladacycle-Catalyzed Asymmetric Intermolecular Construction of Chiral Tertiary P- Y. Heterocycles by Stepwise Addition of H-P-H Bonds to Bis(enones). Organometallics 2012, 31, 4871–4875. [Google Scholar] [CrossRef]
- Rai, V.; Namboothiri, I.N.N. Enantioselective conjugate addition of dialkyl phosphites to nitroalkenes. Tetrahedron Asymm. 2008, 19, 2335–2338. [Google Scholar] [CrossRef]
- Wang, C.; Xie, F.; Guo, Q.; Xie, C.; Zi, G.; Ye, W.; Zhou, Z.; Hou, G.; Zhang, Z. Enantioselective Synthesis of Chiral Phosphonates via Rh/f-spiroPhos Catalyzed Asymmetric Hydrogenation of β,β-Disubstituted Unsaturated Phosphonates. J. Org. Chem. 2021, 86, 12034–12045. [Google Scholar] [CrossRef]
- Hou, L.; Kikuchi, J.; Ye, H.; Bao, M.; Terada, M. Chiral Phosphoric Acid-Catalyzed Enantioselective Phospha-Michael-Type Addition Reaction of Diarylphosphine Oxides with Alkenyl Benzimidazoles. J. Org. Chem. 2020, 85, 14802–14809. [Google Scholar] [CrossRef] [PubMed]
- Maestro, A.; del Corte, X.; López-Francés, A.; de Marigorta, E.M.; Palacios, F.; Vicario, J. Asymmetric Synthesis of Tetrasubstituted α-Aminophosphonic Acid Derivatives. Molecules 2021, 26, 3202. [Google Scholar] [CrossRef] [PubMed]
- Vicario, J.; Ortiz, P.; Ezpeleta, J.M.; Palacios, F. Asymmetric synthesis of functionalized tetrasubstituted aminophosphonates through enantioselective aza-Henry reaction of phosphorylated ketimines. J. Org. Chem. 2015, 80, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Maestro, A.; del Corte, X.; Martinez de Marigorta, E.; Palacios, F.; Vicario, J. Enantioselective synthesis of functionalized -aminophosphonic acid derivatives. Phosph. Sulf. Silicon Relat. Elem. 2019, 194, 287–291. [Google Scholar] [CrossRef]
- Maestro, A.; Martinez de Marigorta, E.; Palacios, F.; Vicario, J. Enantioselective Aza-Reformatsky Reaction with Ketimines. Org. Lett. 2019, 21, 9473–9477. [Google Scholar] [CrossRef]
- Rassukana, Y.V.; Yelenich, I.P.; Vlasenko, Y.G.; Onys’ko, P.P. Asymmetric synthesis of phosphonotrifluoroalanine derivatives via proline-catalyzed direct enantioselective CC bond formation reactions of NH trifluoroacetimidoyl phosphonate. Tetrahedron Asymmetry 2014, 25, 1234–1238. [Google Scholar] [CrossRef]
- Huang, N.; Tong, X.; Zhou, S.; Guo, Q.; Peng, Y. Asymmetric [3 + 2] Cycloaddition Reactions of α-Substituted Diazophosphonates with 3-Acryloyl-2-oxazolidinone to Access Chiral Pyrazoline Derivatives with Phosphonyl at a Tetrasubstituted Stereogenic Center. Adv. Synth. Catal. 2019, 361, 4805–4810. [Google Scholar] [CrossRef]
- Kadyrov, R.; Holz, J.; Schaffner, B.; Zayas, O.; Almena, J.; Boerner, A. Synthesis of chiral β-aminophosphonates via Rh-catalyzed asymmetric hydrogenation of β-amido-vinylphosphonates. Tetrahedron Asymmetry 2008, 19, 1189–1192. [Google Scholar] [CrossRef]
- Blaser, H.U. The Chiral Switch of Metolachlor. In Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions; Blaser, H.-U., Schmidt, E., Eds.; Wiley: Basel, Switzerland, 2004; pp. 55–70. [Google Scholar] [CrossRef]
- Zhang, J.; Dong, K.; Wang, Z.; Ding, K. Asymmetric hydrogenation of α- or β-acyloxy α,β-unsaturated phosphonates catalyzed by a Rh(i) complex of monodentate phosphoramidite. Org. Biomol. Chem. 2012, 10, 1598–1601. [Google Scholar] [CrossRef]
- Wang, D.Y.; Hu, X.P.; Huang, J.D.; Deng, J.; Yu, S.B.; Duan, Z.C.; Xu, X.-F.; Zheng, Z. Readily available chiral phosphine−aminophosphine ligands for highly efficient Rh-catalyzed asymmetric hydrogenation of α-enol ester phosphonates and α-enamido phosphonates Z. J. Org. Chem. 2008, 73, 2011–2014. [Google Scholar] [CrossRef]
- Kondoh, A.; Yorimitsu, H.; Oshima, K. Regio- and Stereoselective Hydroamidation of 1-Alkynylphosphine Sulfides Catalyzed by Cesium Base. Org. Lett. 2008, 10, 3093–3095. [Google Scholar] [CrossRef] [PubMed]
- Doherty, S.; Knight, J.G.; Bell, A.L. Rhodium complexes of (R)-Me-CATPHOS and (R)-(S)-JOSIPHOS: Highlyenantioselective catalysts for the asymmetric hydrogenation of (E)- and (Z)- J3-aryl-J3-(enamido) phosphonates. Tetrahedron Asymmetry 2009, 20, 1437–1444. [Google Scholar] [CrossRef]
- Duan, Z.-C.; Hu, X.-P.; Wang, D.-Y.; Huang, J.-D.; Yu, S.-B.; Deng, J.; Zheng, Z. Enantioselective Synthesis of Optically Active Alkanephosphonates via Rhodium-Catalyzed Asymmetric Hydrogenation of β-Substituted α,β-Unsaturated Phosphonates with Ferrocene-Based Monophosphoramidite Ligands. Adv. Synth. Cat. 2008, 350, 1979–1983. [Google Scholar] [CrossRef]
- Goulioukina, N.S.; Dolgina, T.M.; Bondarenko, G.N.; Beletskaya, I.P.; MIlyin, M.; Davankov, V.A.; Pfaltz, A. Highly enantioselective hydrogenation of α,β-unsaturated phosphonates with iridium–phosphinooxazoline complex: Synthesis of a phosphorus analogue of naproxen. Tetrahedron Asymmetry 2003, 14, 1397–1401. [Google Scholar] [CrossRef]
- Duan, Z.C.; Hu, X.P.; Zhang, C.; Wang, D.Y.; Yu, S.B.; Zheng, Z. Highly enantioselective Rh-catalyzed hydrogenation of β,γ-unsaturated phosphonates with chiral ferrocene-based monophosphoramidite ligands. J. Org. Chem. 2009, 74, 9191–9194. [Google Scholar] [CrossRef]
- Oki, H.; Oura, I.; Nakamura, T.; Ogata, K.; Fukuzawa, S.I. Modular synthesis of the ClickFerrophos ligand family and their use in Rhodium- and ruthenium-catalyzed asymmetric hydrogenation. Tetrahedron Asymmetry 2009, 20, 2185–2191. [Google Scholar] [CrossRef]
- Rubio, M.A.; Suárez, A.; Alvarez, E.; Pizzano, A. Highly enantioselective hydrogenation of enol ester phosphonates catalyzed by rhodium phosphine-phosphite complexes. Chem. Commun. 2005, 2005, 628–630. [Google Scholar] [CrossRef]
- Duan, Z.C.; Hu, X.P.; Zhang, C.; Zheng, Z. Enantioselective Rh-Catalyzed Hydrogenation of 3-Aryl-4-phosphonobutenoates with a P-Stereogenic BoPhoz-Type Ligand. J. Org. Chem. 2010, 75, 8319–8321. [Google Scholar] [CrossRef]
- Cheruku, P.; Paptchikhine, A.; Church, T.L.; Andersson, P.G. Iridium-N,P-Ligand-Catalyzed Enantioselective Hydrogenation of Diphenylvinylphosphine Oxides and Vinylphosphonates. J. Am. Chem. Soc. 2009, 131, 8285–8289. [Google Scholar] [CrossRef]
- Fernández-Pérez, H.; Lenartowicz, P.; Carreras, L.; Grabulosa, A.; Kafarski, P.; Vidal-Ferran, A. Access to α-Aminophosphonic Acid Derivatives and Phosphonopeptides by [Rh(P–OP)]-Catalyzed Stereoselective Hydrogenation. J. Org. Chem. 2020, 85, 14779–14784. [Google Scholar] [CrossRef]
- Noyori, R. Asymmetric Catalysis: Science and Opportunities (Nobel Lecture). Angew. Chem. Int. Ed. 2002, 41, 2008–2022. [Google Scholar] [CrossRef]
- Tao, X.; Li, W.; Li, X.; Xie, X.; Zhang, Z. Diastereo- and Enantioselective Asymmetric Hydrogenation of α-Amido-β-keto Phosphonates via Dynamic Kinetic Resolution. Org. Lett. 2013, 15, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-F.; Hou, C.-J.; Wei, D.-Q.; He, X.; Chu, T.-T.; Chen, X.-sh.; Hu, X.-P. Iridium-atalyzed asymmetric hydrogenation of β-ketophosphonates with chiral ferrocenyl P,N,N-ligands. Appl. Organomet. Chem. 2021, 35, e6283. [Google Scholar] [CrossRef]
- Tao, X.; Li, W.; Ma, X.; Li, X.; Fan, W.; Zhu, L.; Xie, X.; Zhang, Z. Enantioselective Hydrogenation of β-Ketophosphonates with Chiral Ru(II) Catalysts. J. Org. Chem. 2012, 77, 8401–8409. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Wu, B.; Gao, X.; Chen, M.-W.; Zhou, Y.-G. Enantioselective Synthesis of α-Amino Phosphonates via Pd-Catalyzed Asymmetric Hydrogenation. Org. Lett. 2016, 18, 692–695. [Google Scholar] [CrossRef] [PubMed]
- Meier, C.; Laux, W.H.G.; Bats, J.W. Asymmetric synthesis of chiral, Nonra-cemic dialkyl-α-hydroxyalkylphosphonates via a (−)-Chlorodiisopino-campheylborane (Ipc2B-Cl) reduction. Tetrahedron Asymmetry 1996, 7, 89–94. [Google Scholar] [CrossRef]
- Heravi, M.M.; Asadi, S.; Nazari, N.; Lashkariani, B.M. Application of Corey–Bakshi–Shibata, Corey–Kim, Corey–Seebach, Corey–Winter, Corey–Link, and Corey–Ganem–Gilman in organic and total synthesis. Monatsh. Chem. Chemi. Month. 2016, 147, 961–987. [Google Scholar] [CrossRef]
- Barco, A.; Benetti, S.; Bergamini, P.; de Risi, C.; Marchetti, P.; Pollini, G.P.; Zanirato, V. Diastereoselective synthesis of β-amino-α-hydroxy phosphonates via oxazaborolidine catalyzed reduction of β-phthalimido-α-ketophosphonates. Tetrahedron Lett. 1999, 40, 7705–7708. [Google Scholar] [CrossRef]
- Rassukana, Y.V.; Onys’ko, P.P.; Kolotylo, M.V.; Sinitsa, A.D.; Lyzwa, P.; Mikoiajczyk, M. A new strategy for asymmetric synthesis of aminophosphonic acid derivatives: The first enantioselective catalytic reduction of C-phosphorylated imines. Tetrahedron Lett. 2009, 50, 288–290. [Google Scholar] [CrossRef]
- Onysko, P.P.; Chudakova, T.I.; Pirozhenko, V.V.; Rozhenko, A.B. α-Ketophosphonates in the synthesis of α-iminophosphonates. Curr. Green Chem. 2020, 7, 226–238. [Google Scholar] [CrossRef]
- Palacios, F.; Aparicio, D.; Ochoa de Retana, A.M.; de los Santos, J.M.; Gil, J.I.; Lopez de Munain, R. Asymmetric synthesis of 2H-aziridine phosphonates, and—Or -aminophosphonates from enantiomerically enriched 2H-azirines. Tetrahedron Asymmetry 2003, 14, 689–700. [Google Scholar] [CrossRef]
- Guliaiko, I.; Nesterov, V.; Sheiko, S.; Kolodiazhnyi, O.I.; Freytag, M.; Jones, P.G.; Schmutzler, R. Synthesis of Optically Active Hydroxyphosphonates. Heteroat. Chem. 2008, 19, 133–139. [Google Scholar] [CrossRef]
- Gryshkun, E.V.; Nesterov, V.; Kolodyazhnyi, O.I. Enantioselective reduction of ketophosphonates using adducts of chiral natural acids with sodium bo-rohydride. Arkivoc 2012, 2012, 100–117. [Google Scholar] [CrossRef]
- Nesterov, V.V.; Kolodiazhnyi, O.I. Efficient method for the asymmetric reduction of α– and β–ketophosphonates. Tetrahedron 2007, 63, 6720–6731. [Google Scholar] [CrossRef]
- Nesterov, V.V.; Kolodiazhnyi, O.I. Di(1R,2S,5R)-menthyl 2-hydroxy-3-chloropropyl-phosphonate as useful chiron for the synthesis of α– and β–hydroxyphosphonates. Synlett 2007, 15, 2400–2404. [Google Scholar] [CrossRef]
- Corbett, M.T.; Johnson, J.S. Diametric Stereocontrol in Dynamic Catalytic Reduction of Racemic Acyl Phosphonates: Divergence from α-Keto Ester Congeners. J. Am. Chem. Soc. 2013, 135, 594–597. [Google Scholar] [CrossRef]
- Guo, W.L.; Hou, C.J.; Duan, Z.C.; Hu, X.P. An enantioselective synthesis of 3-aryl-4-phosphonobutyric acid esters via Cu-catalyzed asymmetric conjugate reduction. Tetrahedron Asymmetry 2011, 22, 2161–2164. [Google Scholar] [CrossRef]
- Mhamdi, A.; Touil, S. An operationally simple, fast and green synthesis of novel γ-hydroxyphosphonate and phosphine oxide derivatives. Green Chem. Lett. Rev. 2018, 11, 12–17. [Google Scholar] [CrossRef]
- Chiminazzo, A.; Sperni, L.; Scarso, A.; Strukul, G. Organocatalytic Enantioselective Epoxidation of Some Aryl-Substituted Vinylidenebisphosphonate Esters: On the Way to Chiral Anti-Osteoporosis Drugs. Catalysts 2017, 7, 90. [Google Scholar] [CrossRef]
Figure 1.
Antibiotics of natural origin.
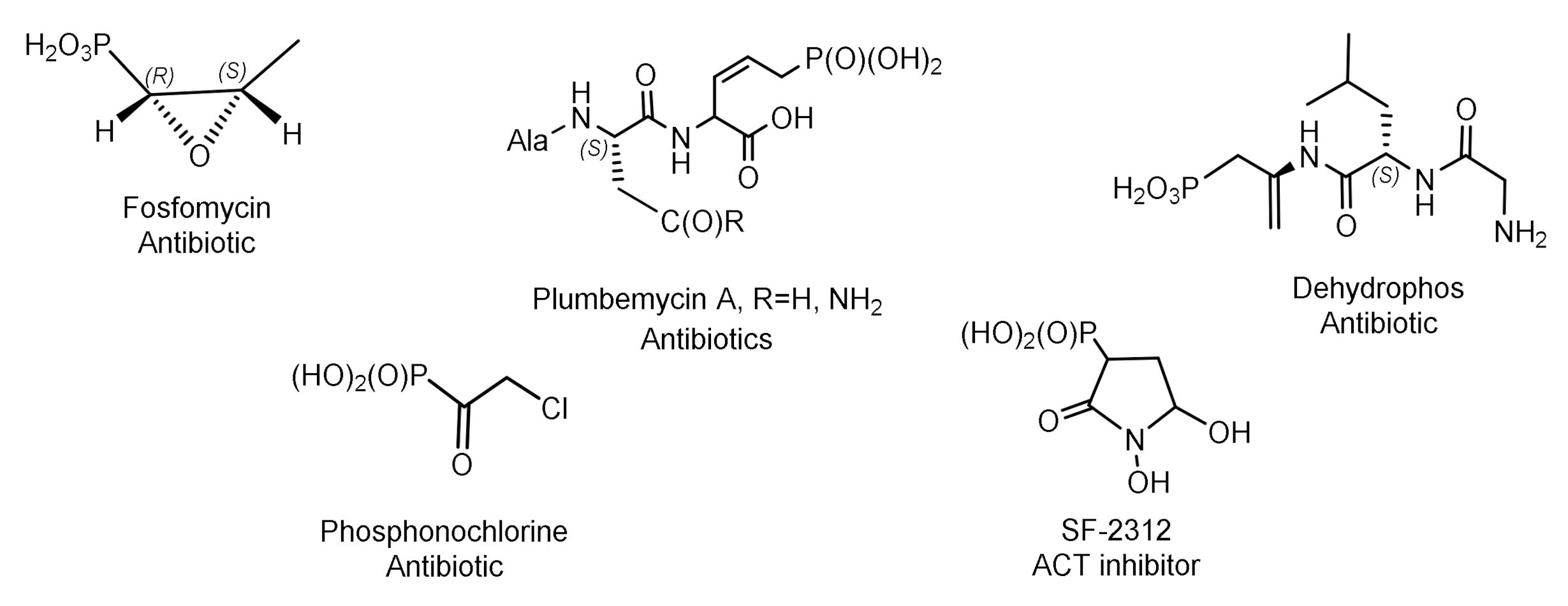
Figure 2.
Examples of natural herbicides.
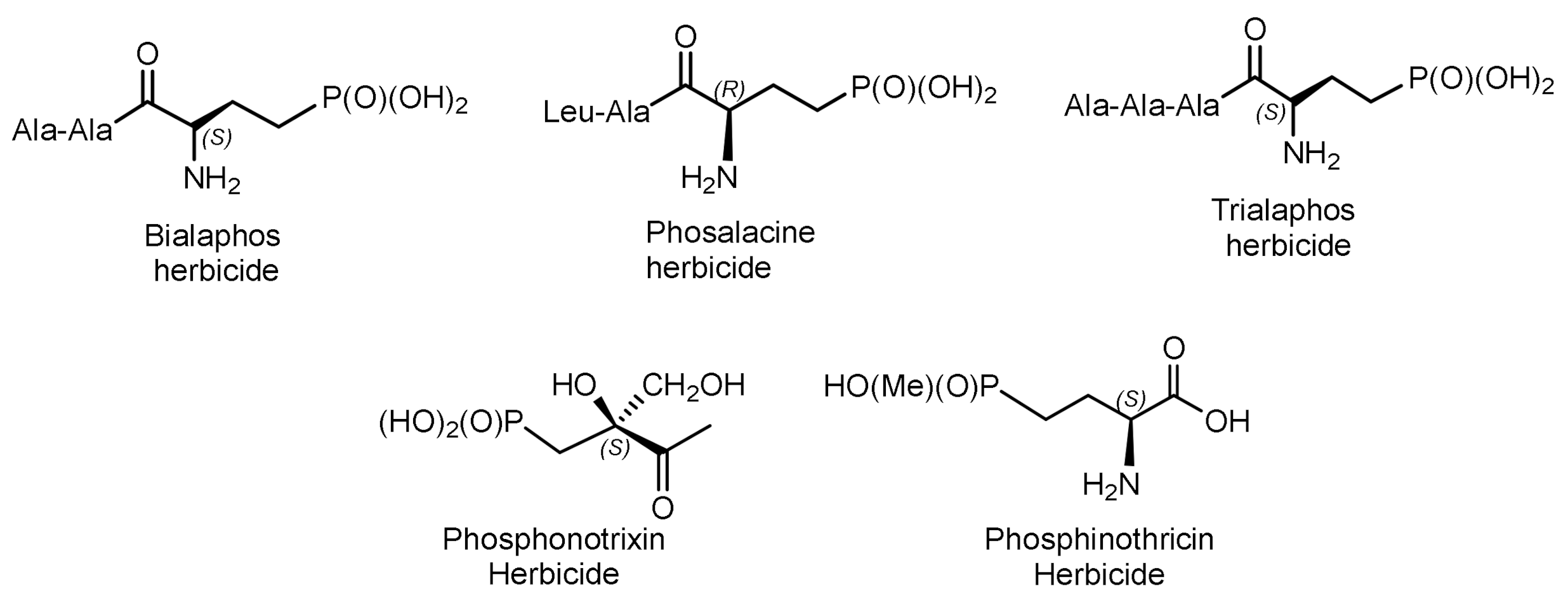
Figure 3.
Natural phosphonates of agrochemical interest.

Figure 4.
Various routes to achieve chiral functionalized phosphonates.
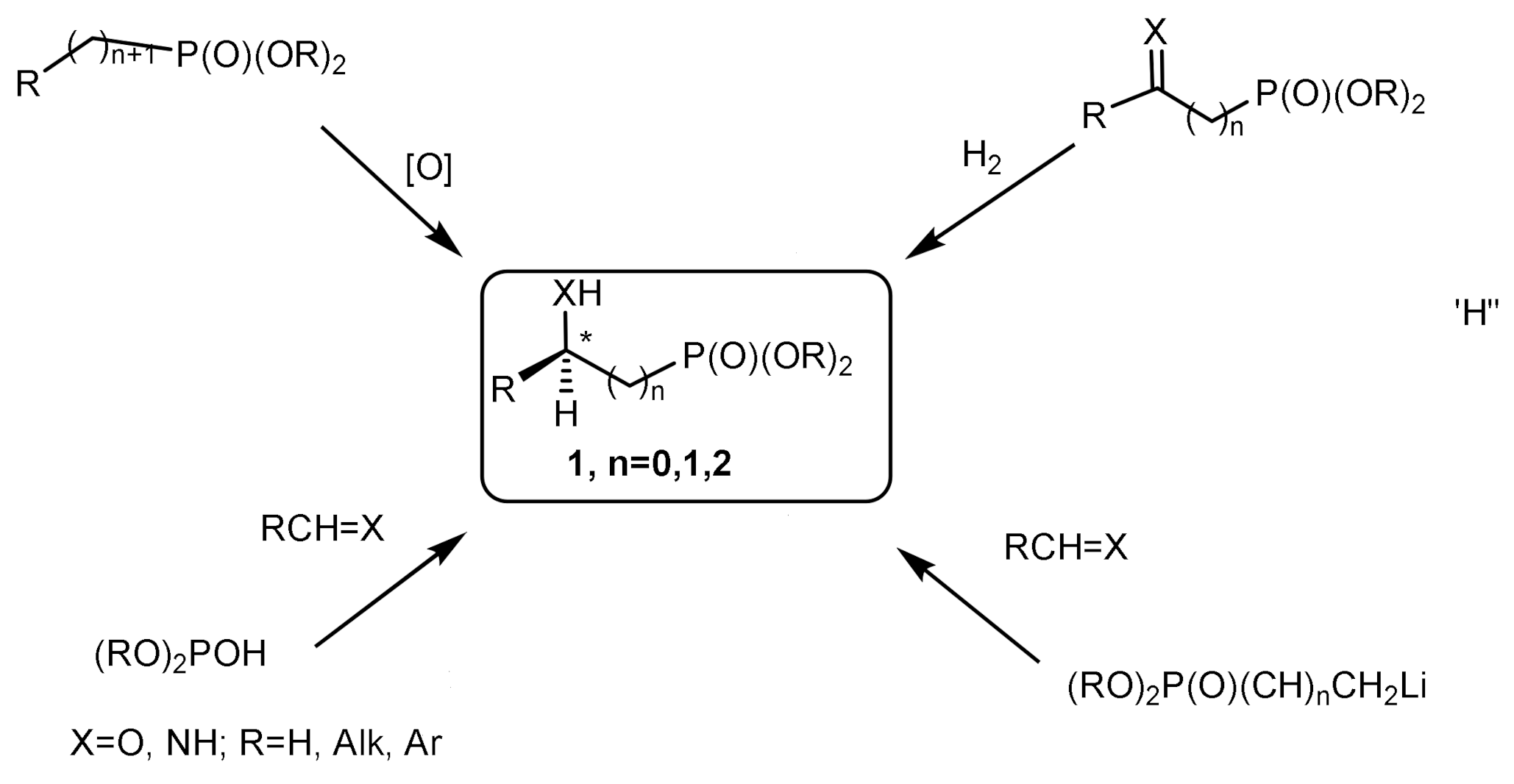
Figure 5.
Transfer of chirality to substrate from chiral catalyst.
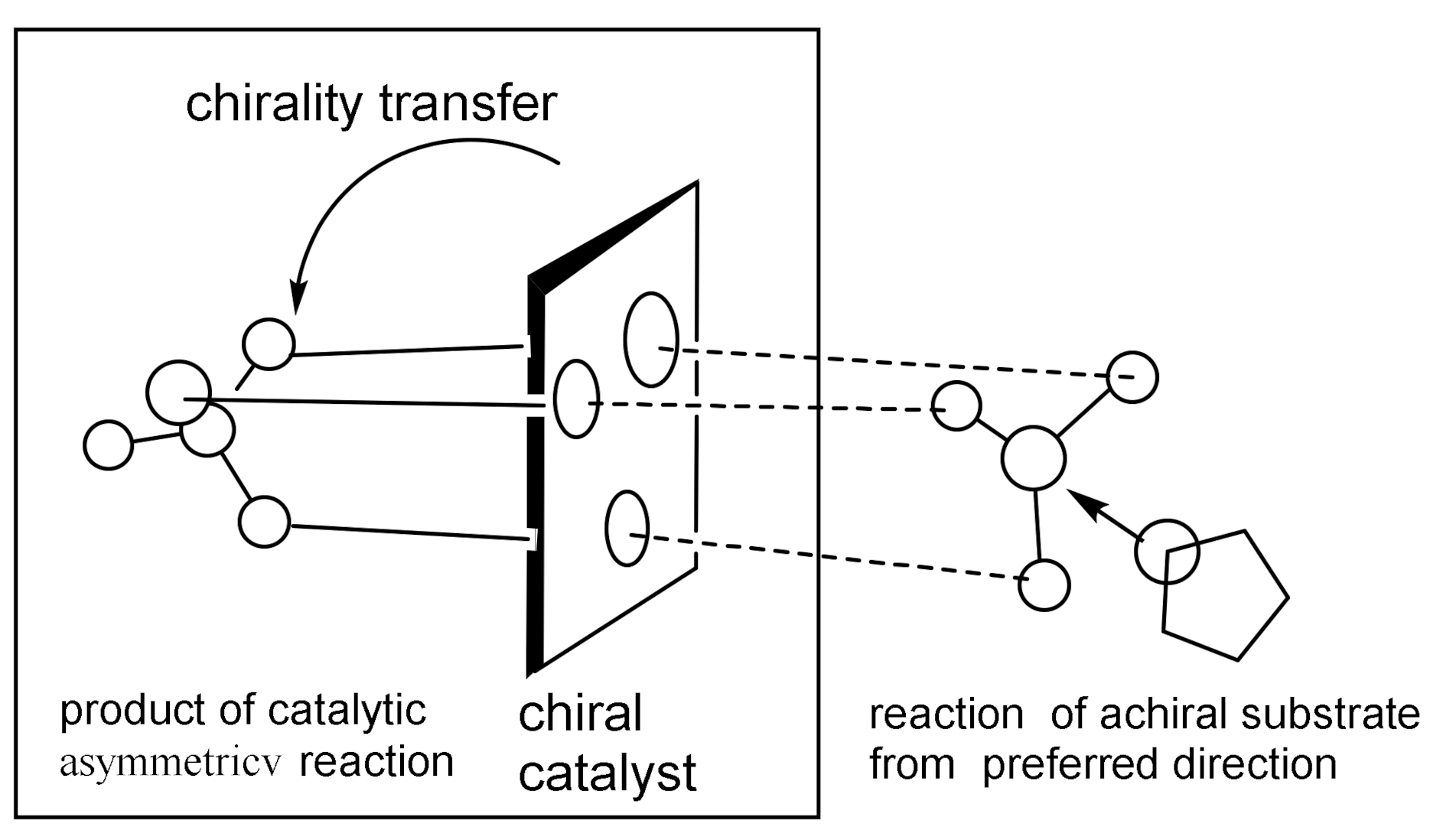
Figure 6.
Aldol and Phosphaaldol Reactions.

Figure 7.
Two- and three-component phospha–Mannich reaction.
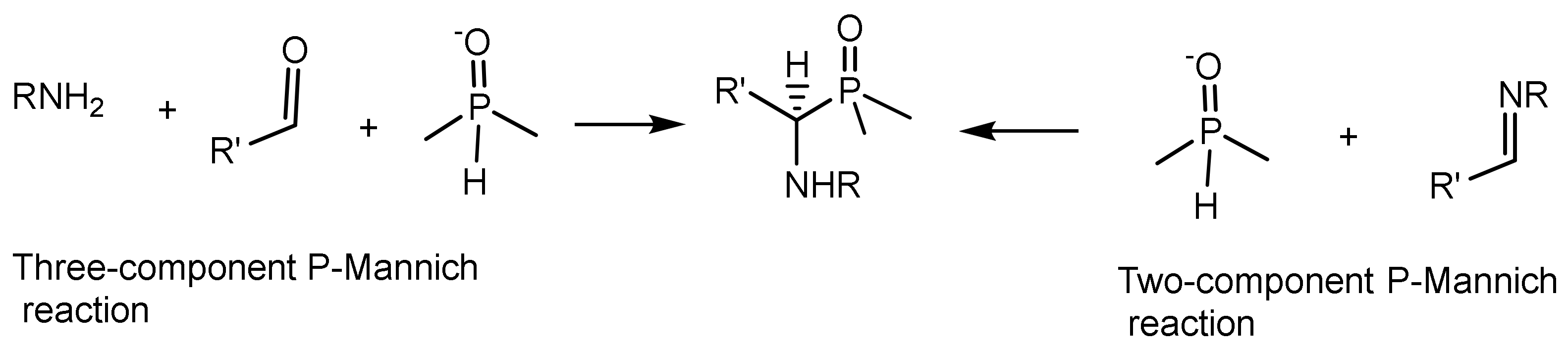
Figure 8.
Phospha–Michael reaction.
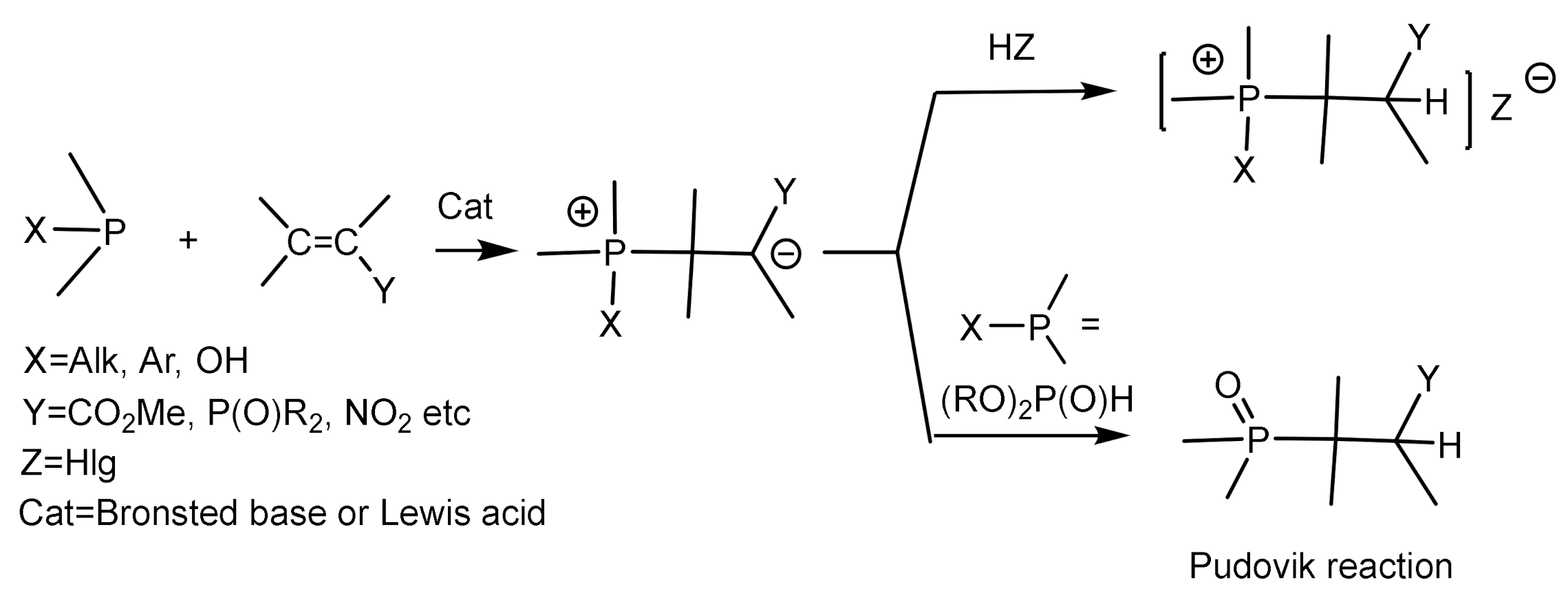
Figure 9.
Phospha–aldol reaction catalyzed by metallic complexes.

Figure 10.
Phospha–aldol reaction catalyzed by BINAPO/SiCl4.

Figure 11.
Mechanism of the reaction catalyzed by tetrachlorosilane.
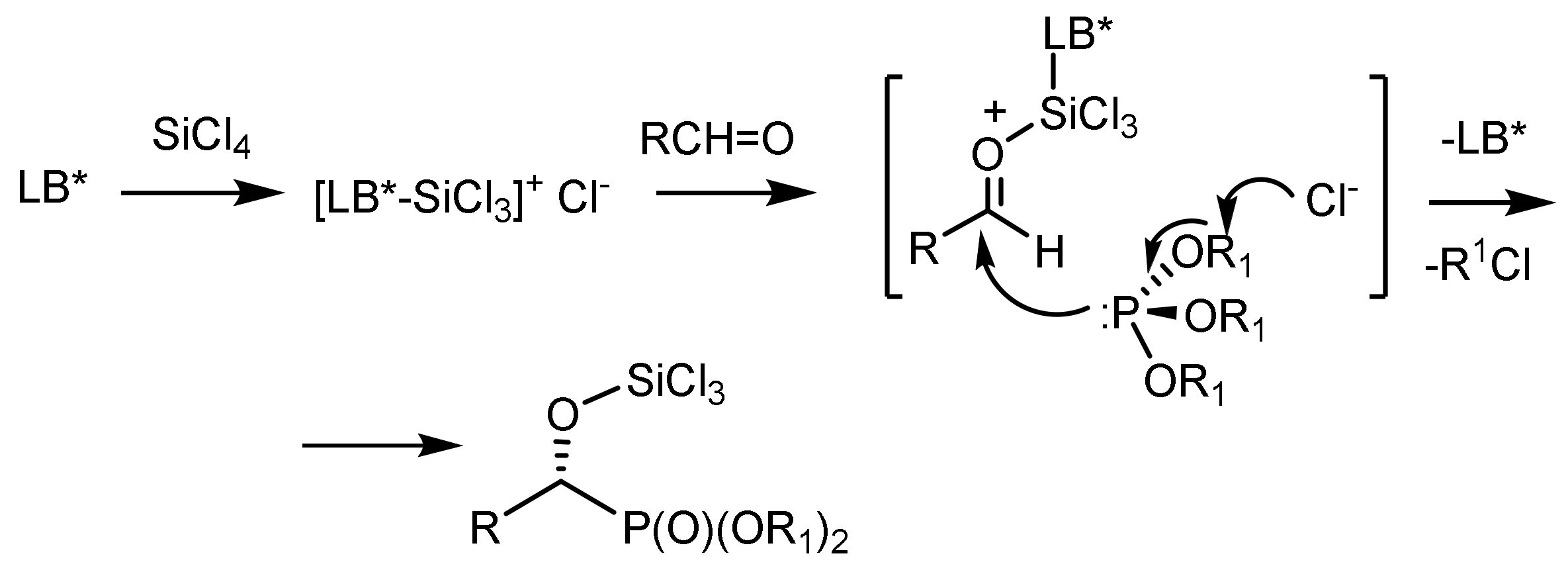
Figure 12.
Enantioselective hydrophosphonylation of aldehydes catalyzed Al(III)-BINOL.

Figure 13.
Structures of ligands used in the formation of bifunctional catalysts.

Figure 14.
The phospha–aldol reaction catalyzed by bifunctional catalysts and the proposed transition-state model.
Figure 14.
The phospha–aldol reaction catalyzed by bifunctional catalysts and the proposed transition-state model.
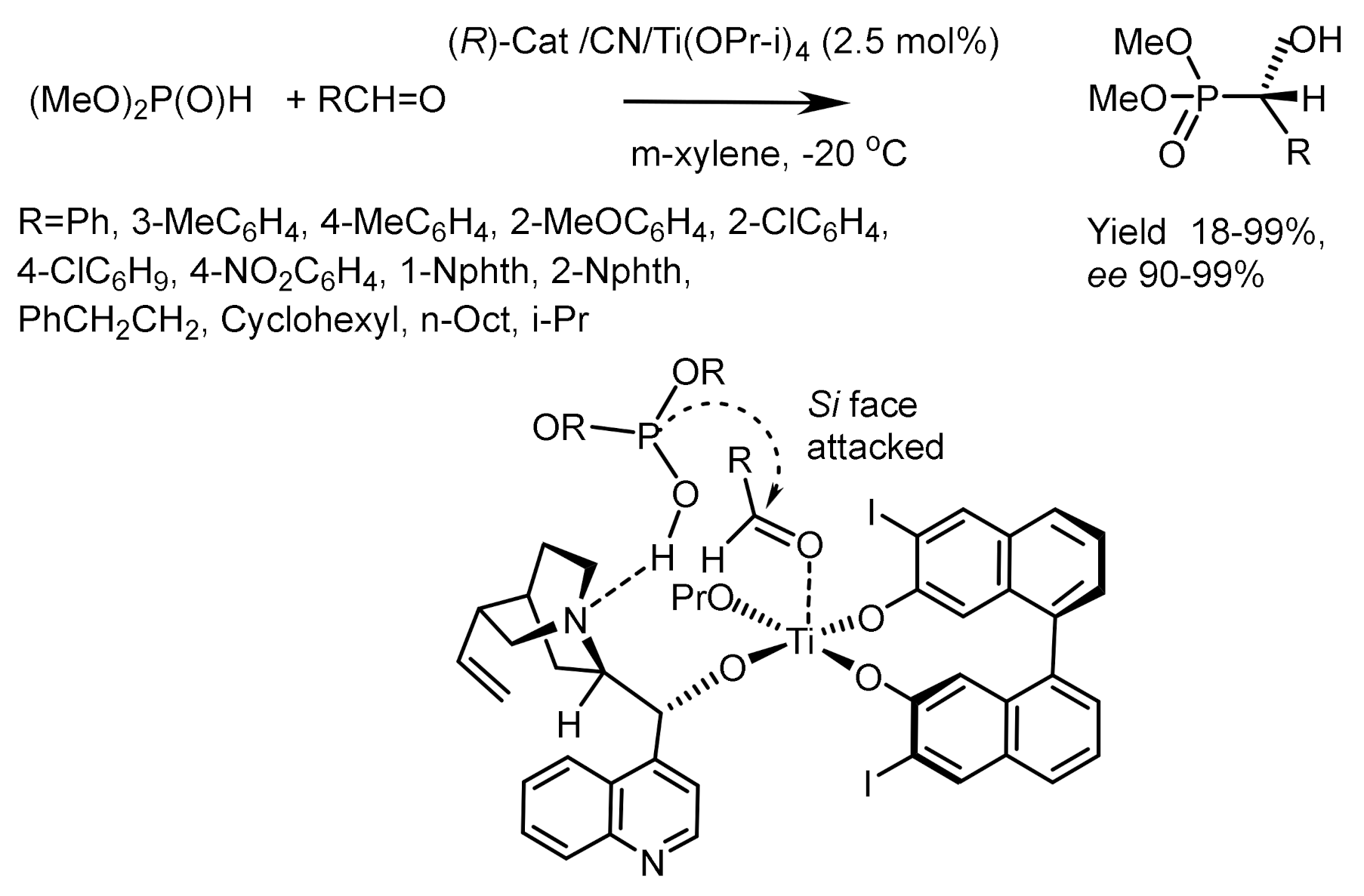
Figure 15.
The enantioselective P-aldol reaction initiated by chiral disulfonimide catalyst.
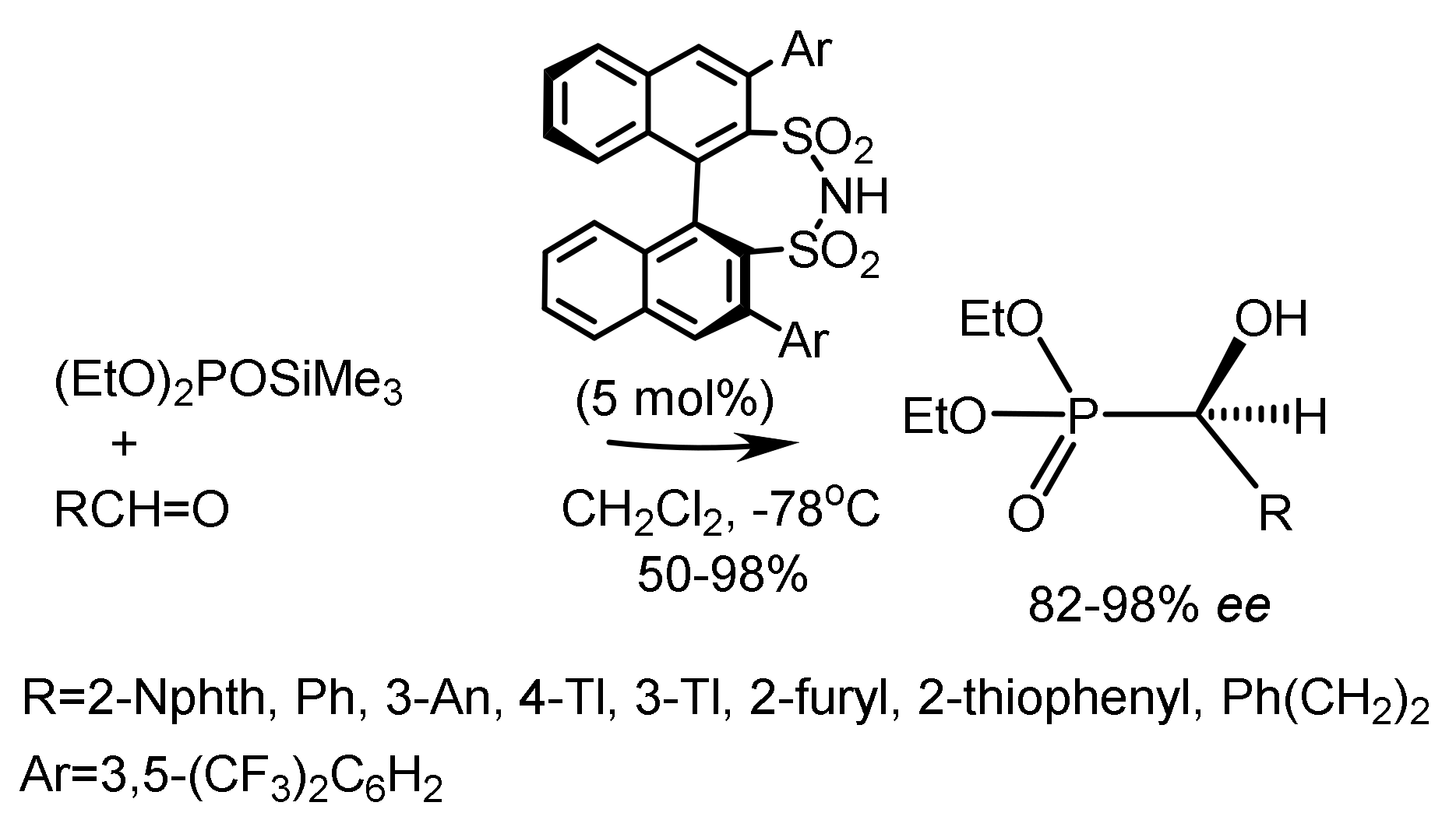
Figure 16.
The reaction of chiral di(1R,2S,5R)-mentylphosphite with chiral 2,3-D-isopropylidene-(R)-glyceraldehyde.
Figure 16.
The reaction of chiral di(1R,2S,5R)-mentylphosphite with chiral 2,3-D-isopropylidene-(R)-glyceraldehyde.
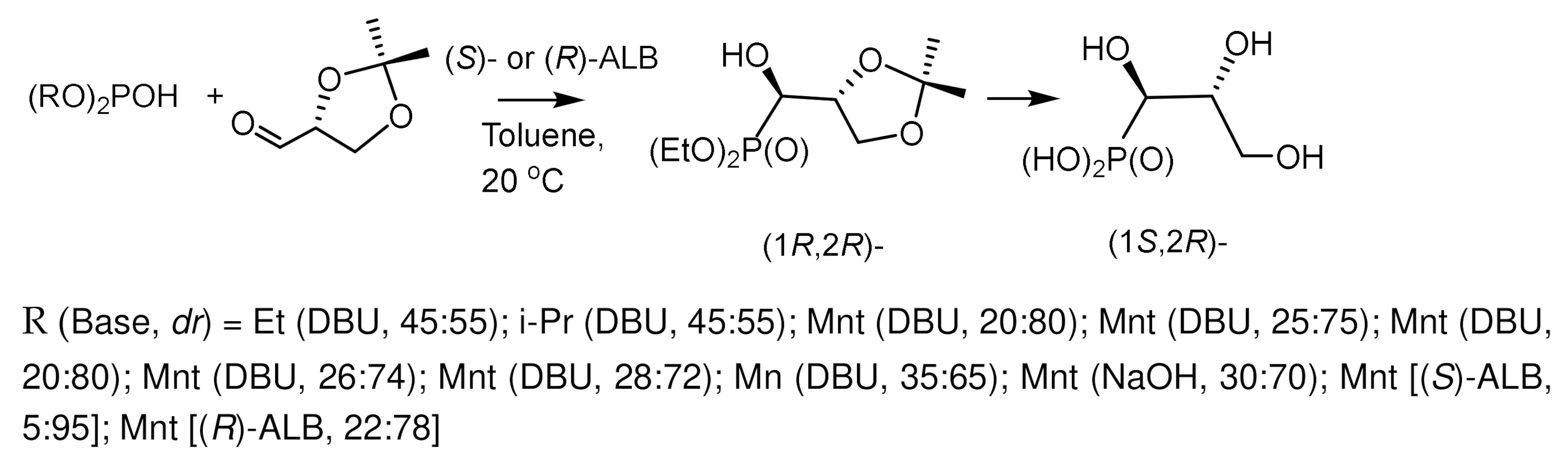
Figure 17.
Hydrophosphonylation of benzaldehyde with dimethyl phosphite catalyzed by Al(Salalen) complexes.
Figure 17.
Hydrophosphonylation of benzaldehyde with dimethyl phosphite catalyzed by Al(Salalen) complexes.
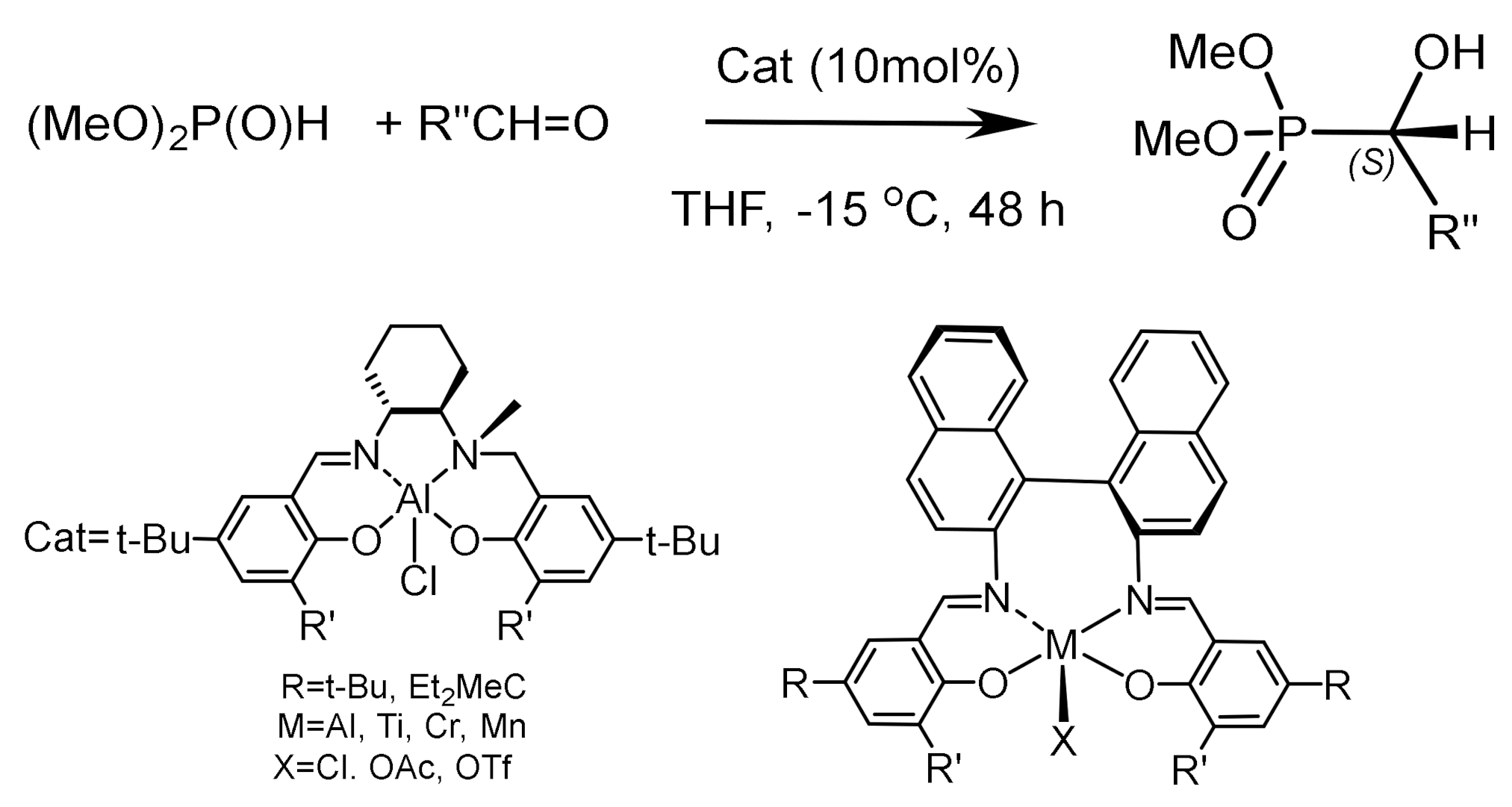
Figure 18.
Sequential C-H oxidation/asymmetric phosphonylation reaction of primary alcohols.

Figure 19.
Phospha–aldol reaction catalyzed by bis(8–quinolinato)(TBOx)aluminum complex.
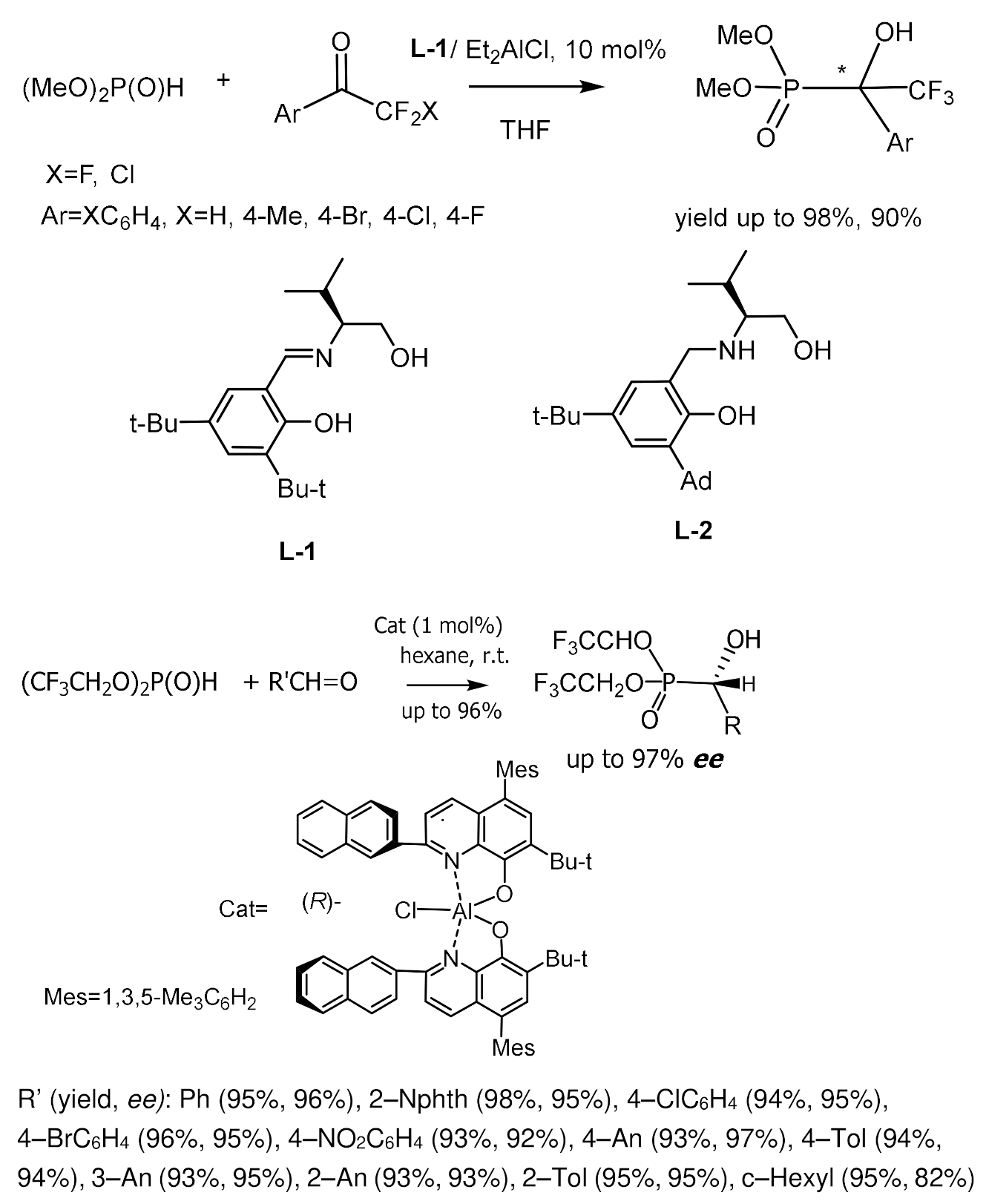
Figure 20.
Phospha–aldol reaction catalyzed by triiminophosphorane.
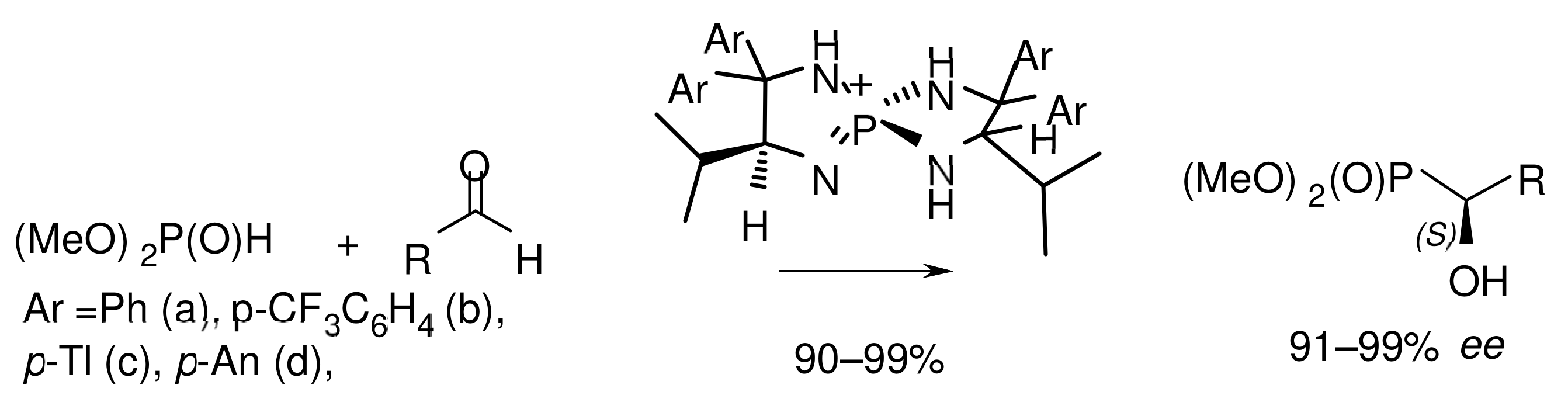
Figure 21.
Phospha-aldol reaction of diphenyl phosphite with N-alkylated isatins.

Figure 22.
Catalytic phospha-aldol reaction.
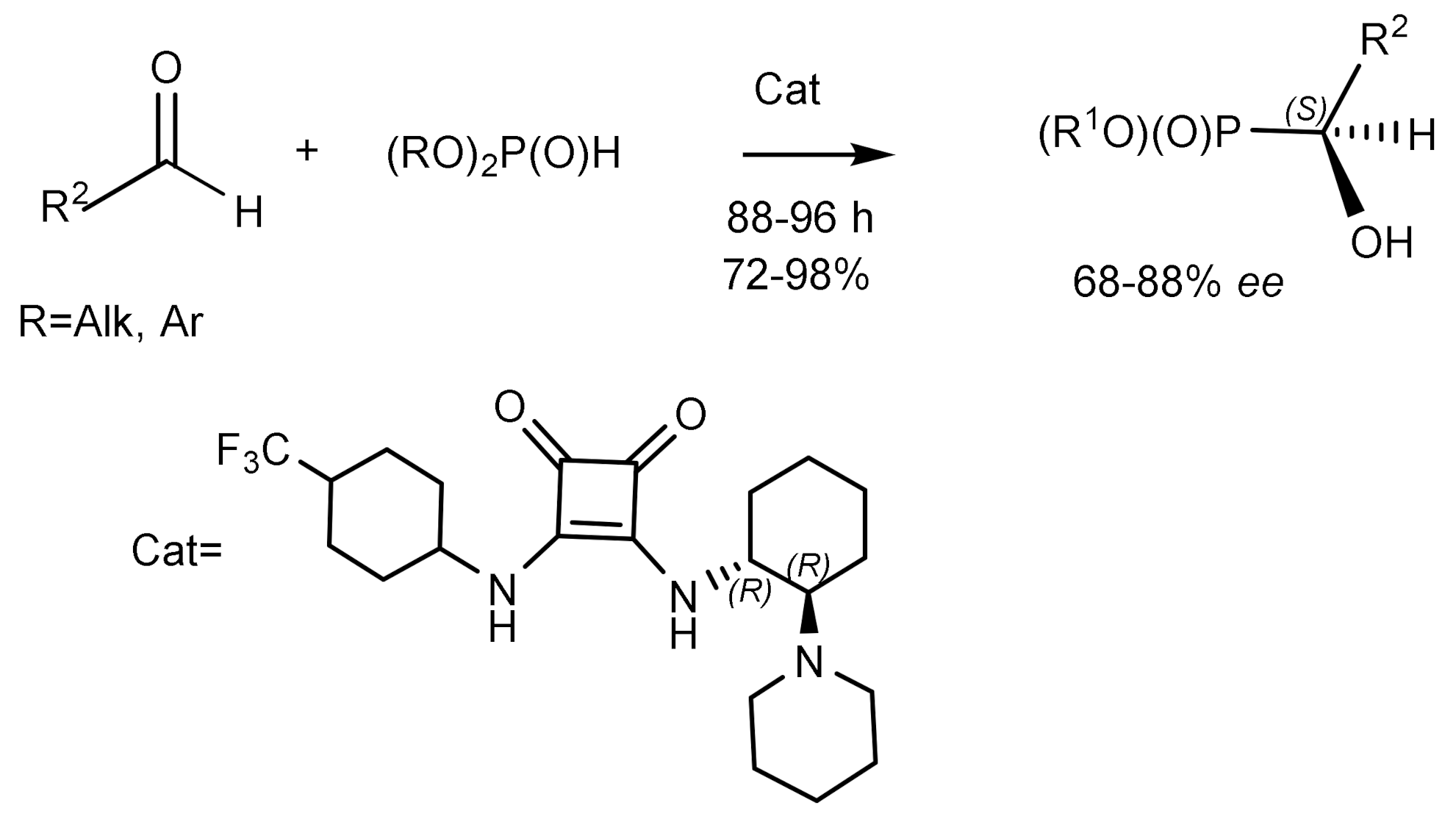
Figure 23.
Cross–aldol reaction of enolizable aldehydes with ketophosphonates.
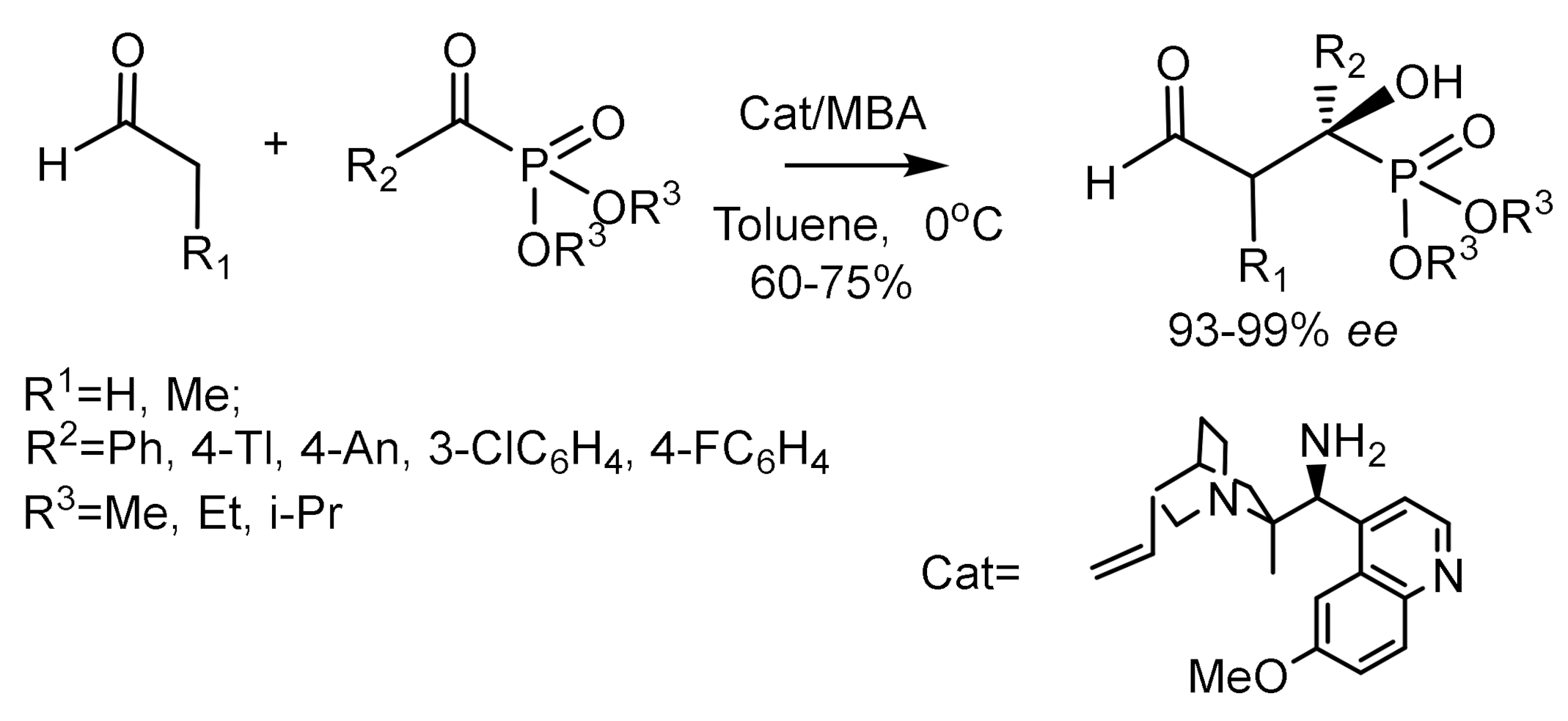
Figure 24.
Enantioselective synthesis of tertiary α–hydroxycyanophosphonates.
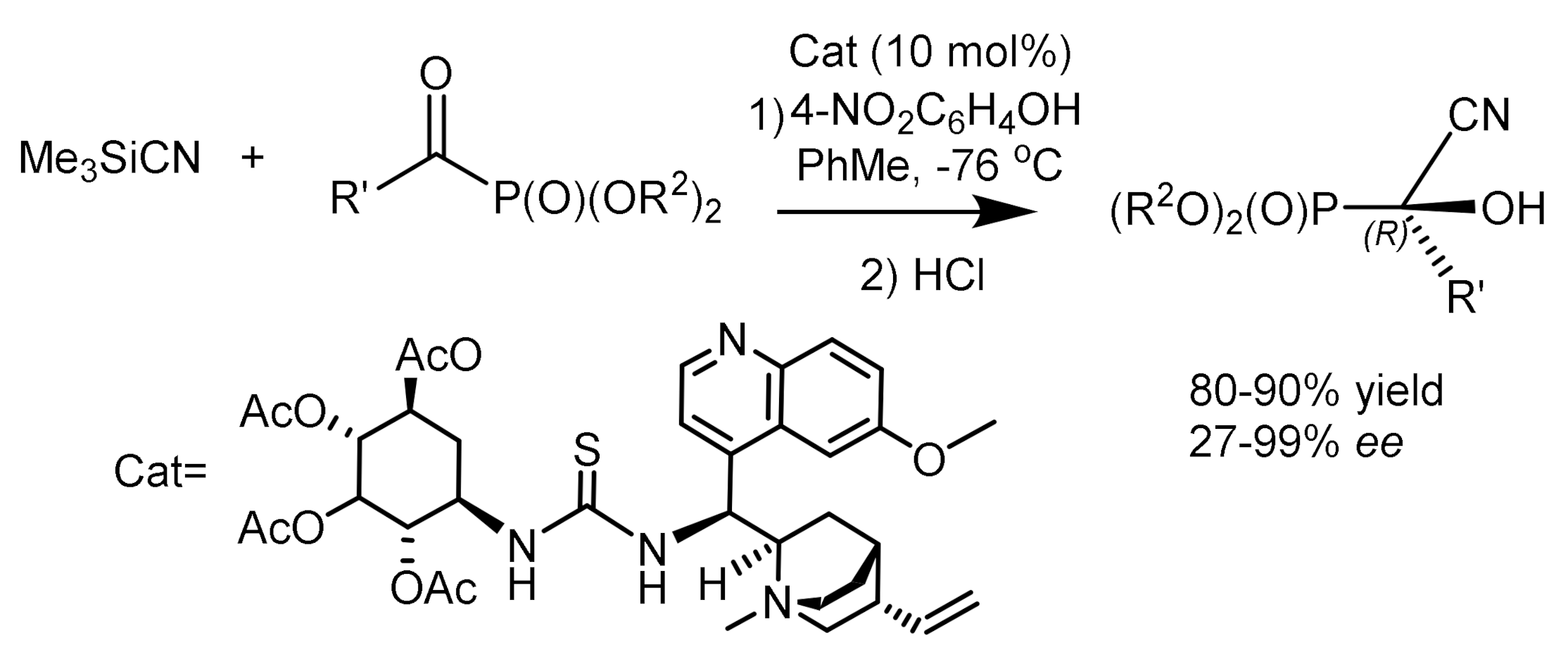
Figure 25.
Reaction of ketoesters and dimethyl phosphite catalyzed by thiourethane derivatives of cinchon alkaloids.
Figure 25.
Reaction of ketoesters and dimethyl phosphite catalyzed by thiourethane derivatives of cinchon alkaloids.

Figure 26.
Catalytic phospha-aldol reaction of L-α-acylphosphinates with acetone.

Figure 27.
Synthesis of bis-aminophosphonates containing a (R,R)-1,2-diaminocyclohexyl moiety.

Figure 28.
An example of a three-component Kabachnik–Fields reaction.
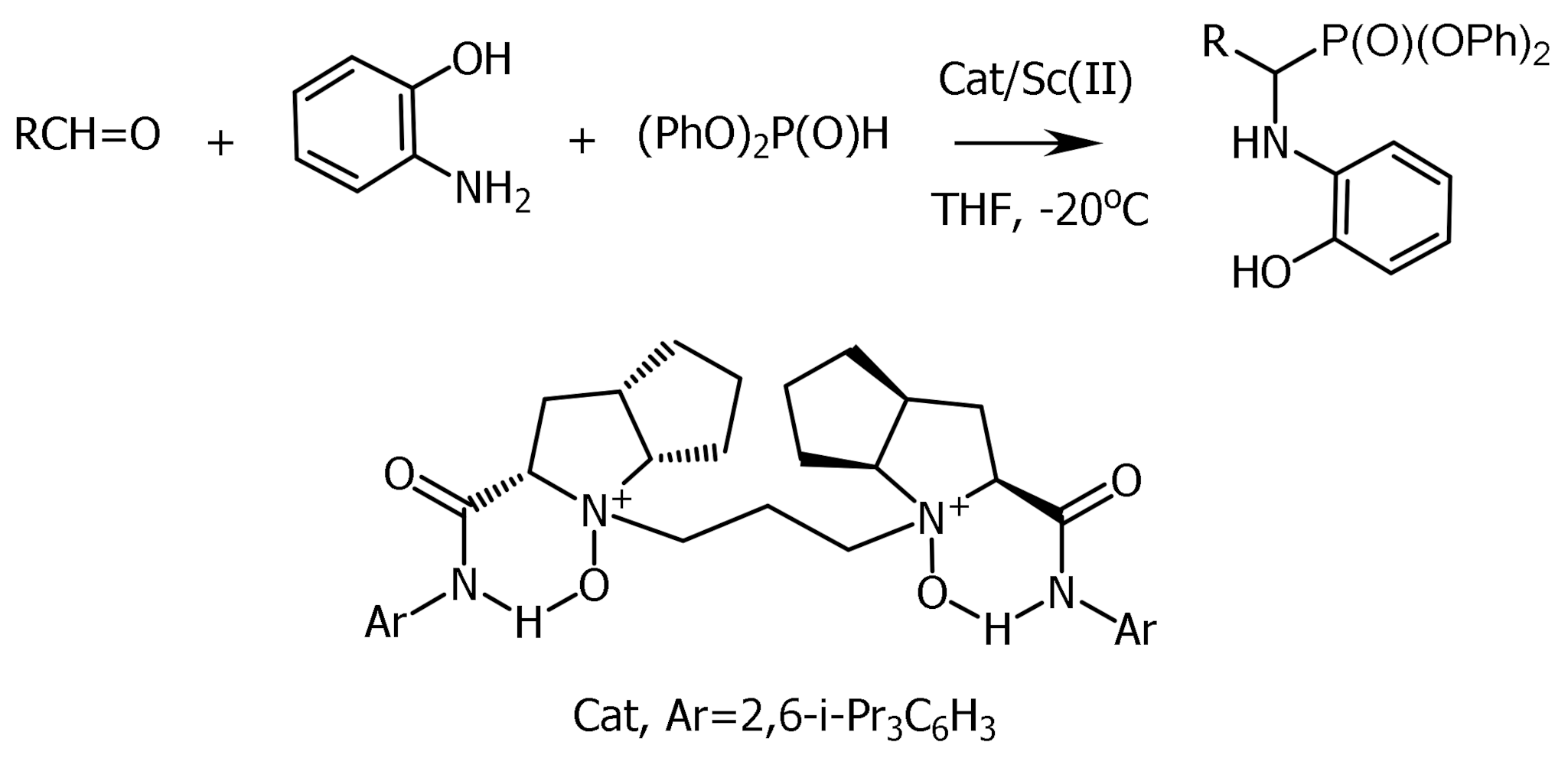
Figure 29.
Synthesis of bicyclic α–aminophosphonates and α–aminophosphonic acid derivatives.
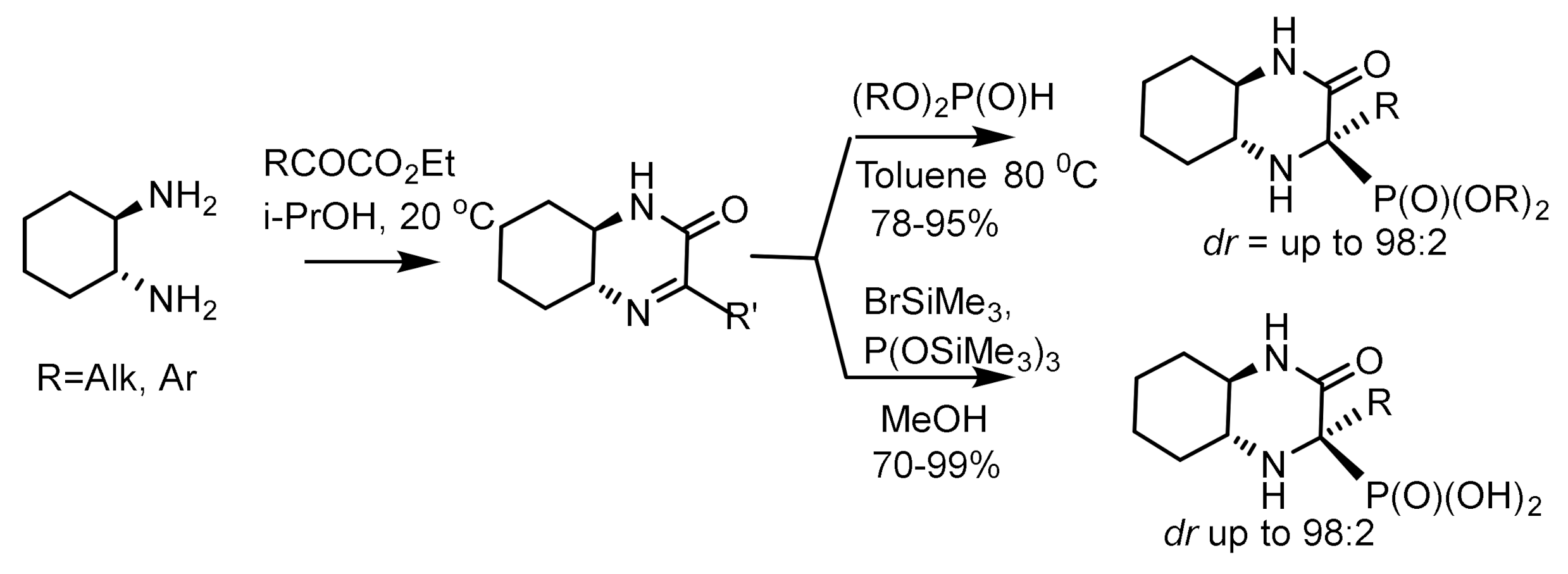
Figure 30.
The Kabachnik–Fields reaction proceeding under conditions of dynamic kinetic resolution.
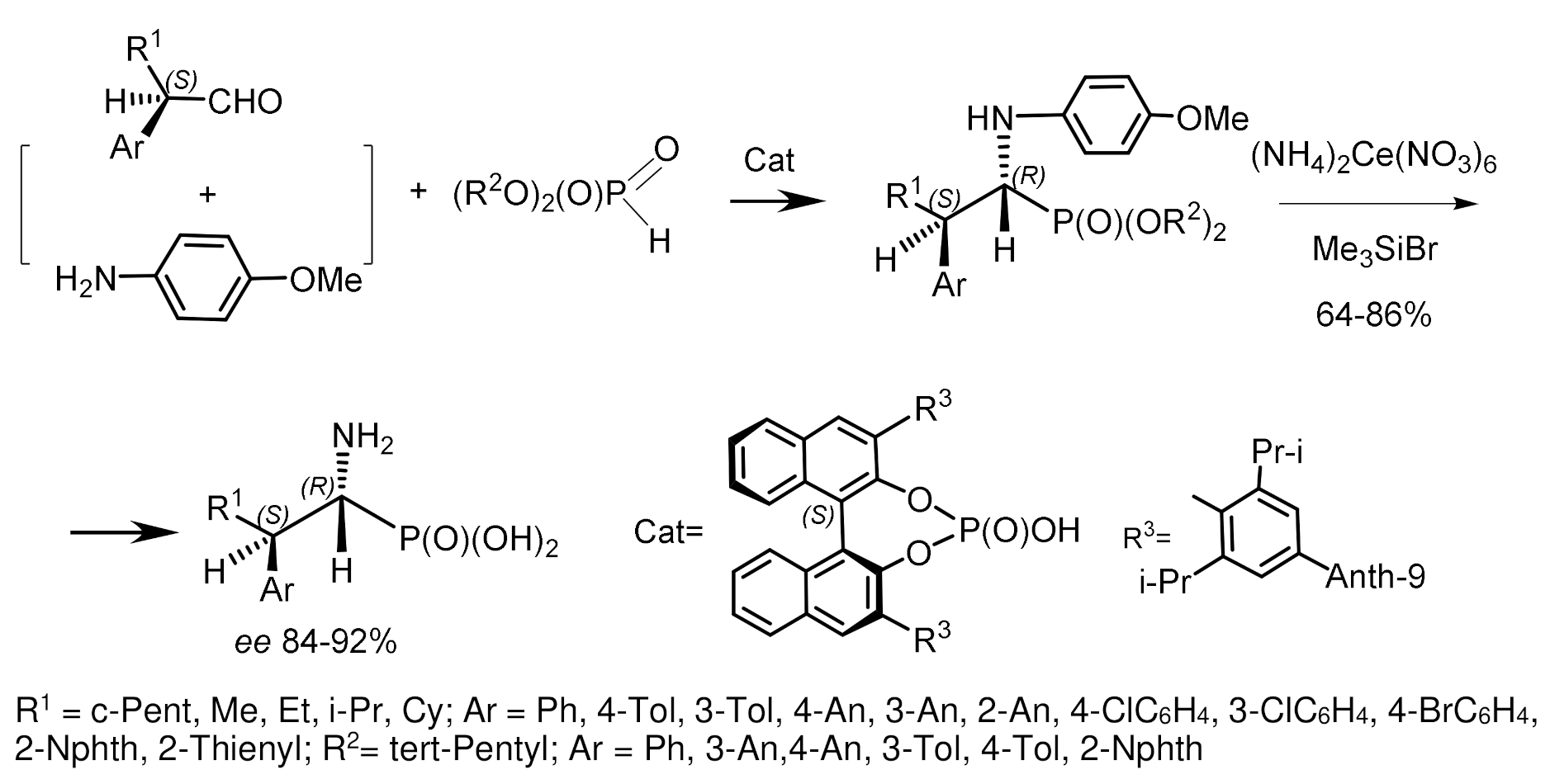
Figure 31.
Asymmetric reaction of achiral imines with dialkyl phosphites.

Figure 32.
Synthesis of α–aminophosphonates.
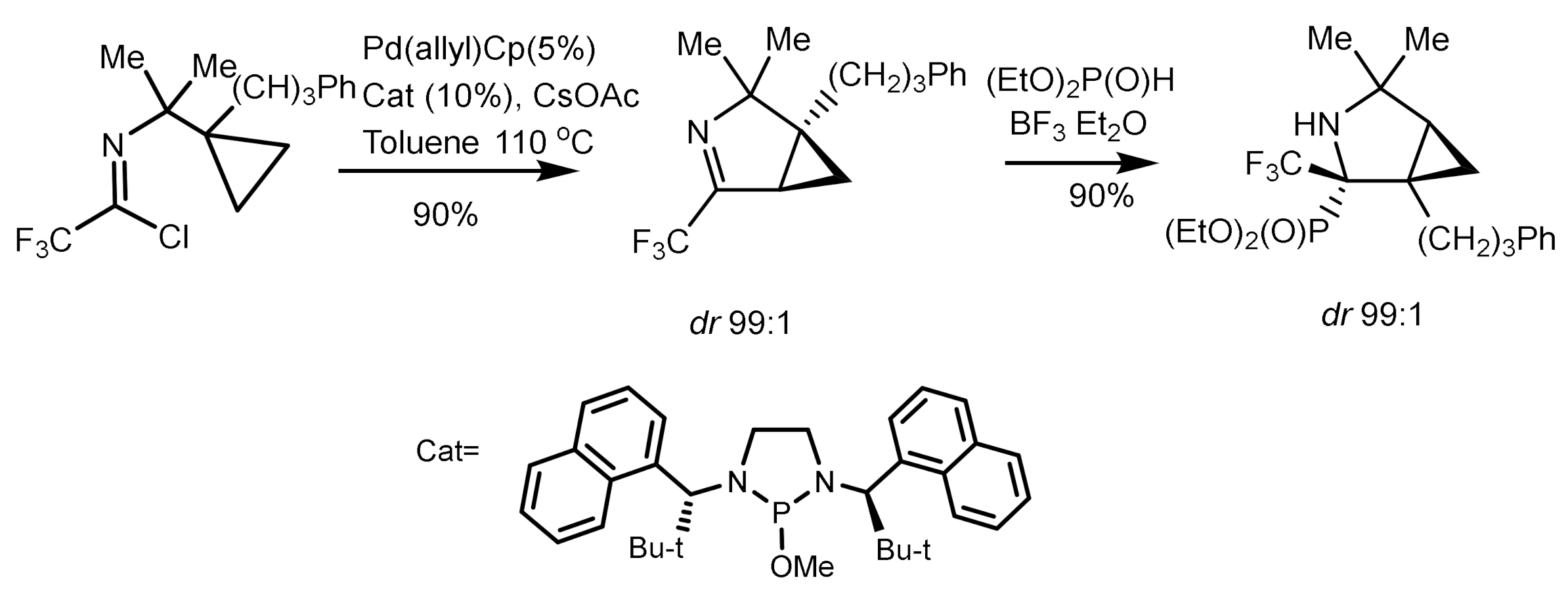
Figure 33.
Asymmetric hydrophosphonylation of aldimines.

Figure 34.
One-pot hydrophosphonylation of aldimines catalyzed by aluminum-Salalen complex (Cat).

Figure 35.
Reaction of aldimines with phosphites catalyzed by 8-quinoline-bis(TBOx) aluminum complex.
Figure 35.
Reaction of aldimines with phosphites catalyzed by 8-quinoline-bis(TBOx) aluminum complex.
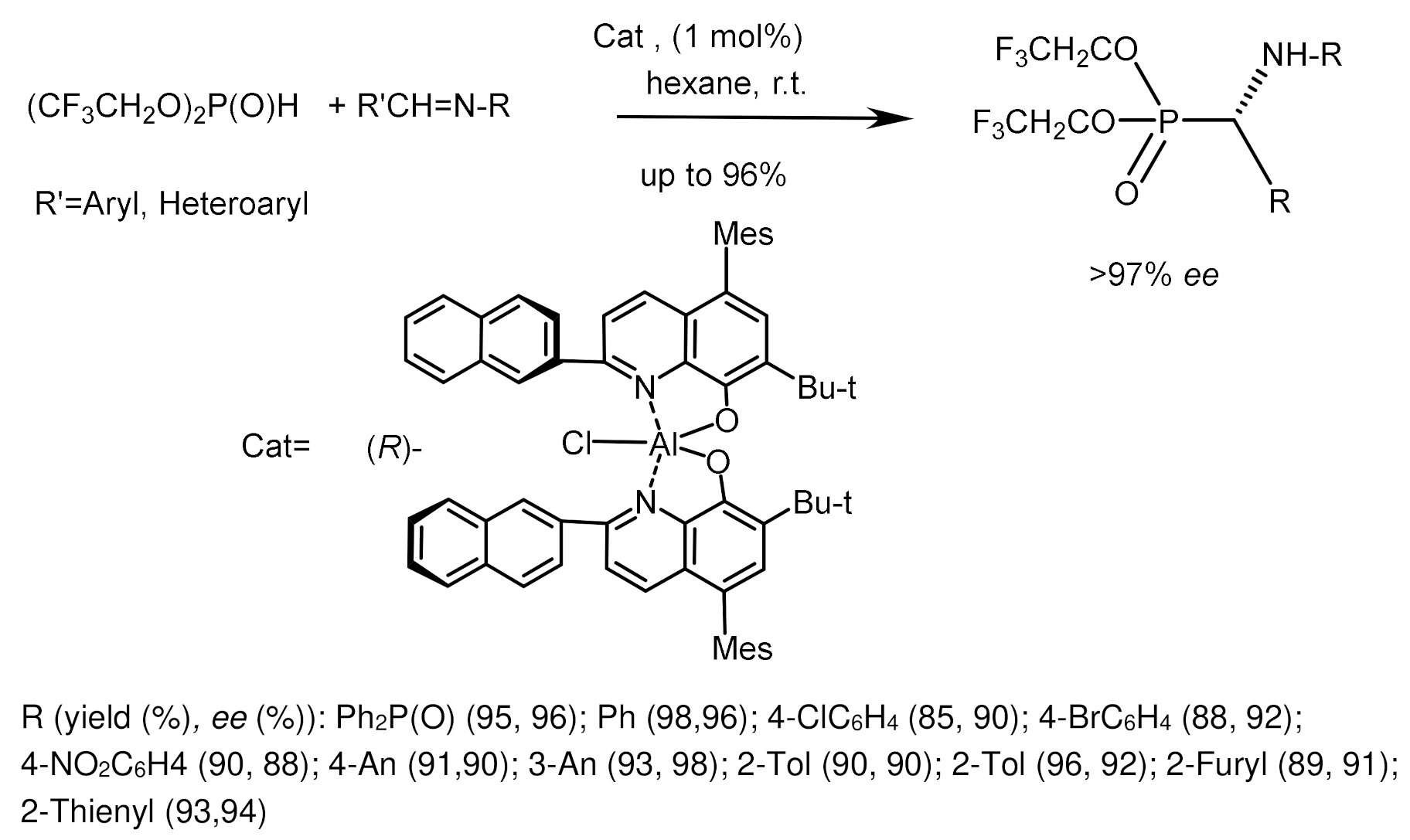
Figure 36.
Phospha–Mannich reaction catalyzed by Quinine.
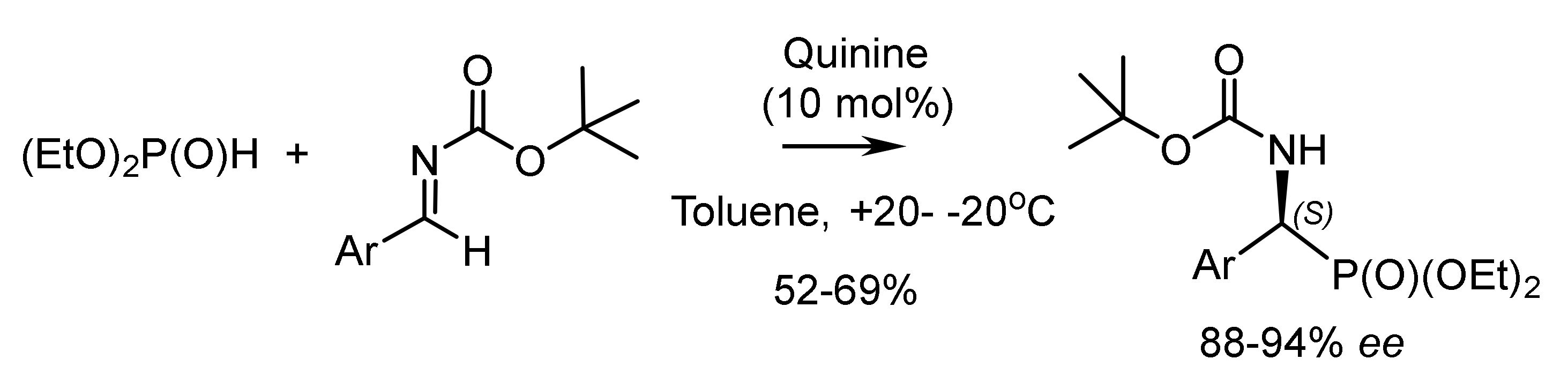

Figure 37.
Enantioselective addition of diphenyl phosphite to ketimines derived from isatins.

Figure 38.
Thiourea–catalyzed enantioselective hydrophosphonylation of imines.
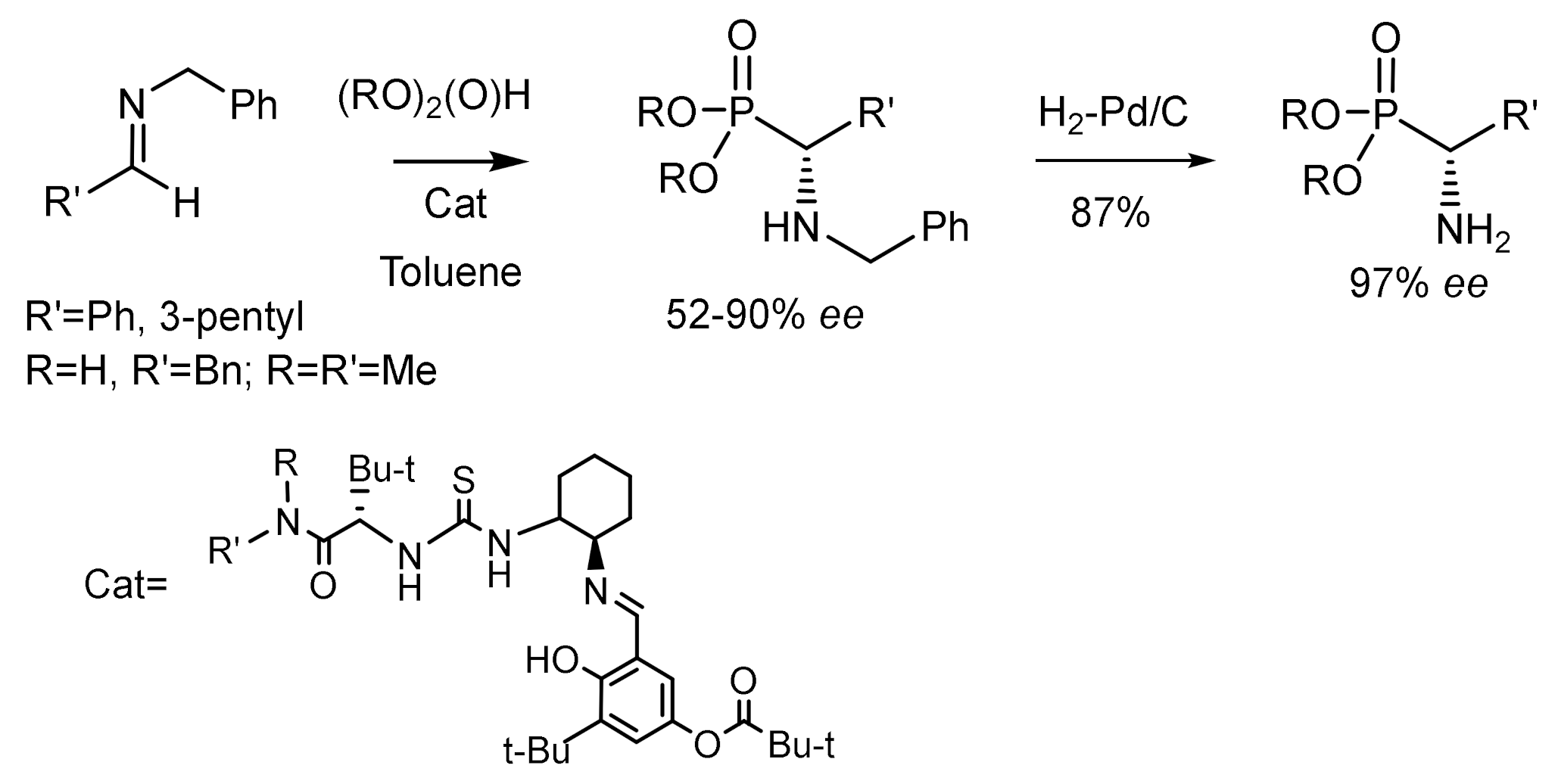
Figure 39.
Phospha–Mannich reaction of H-phosphinates with N–tosylimine.

Figure 40.
Catalytic asymmetric hydrophosphorylation of N–thiophosphinyl ketimines.
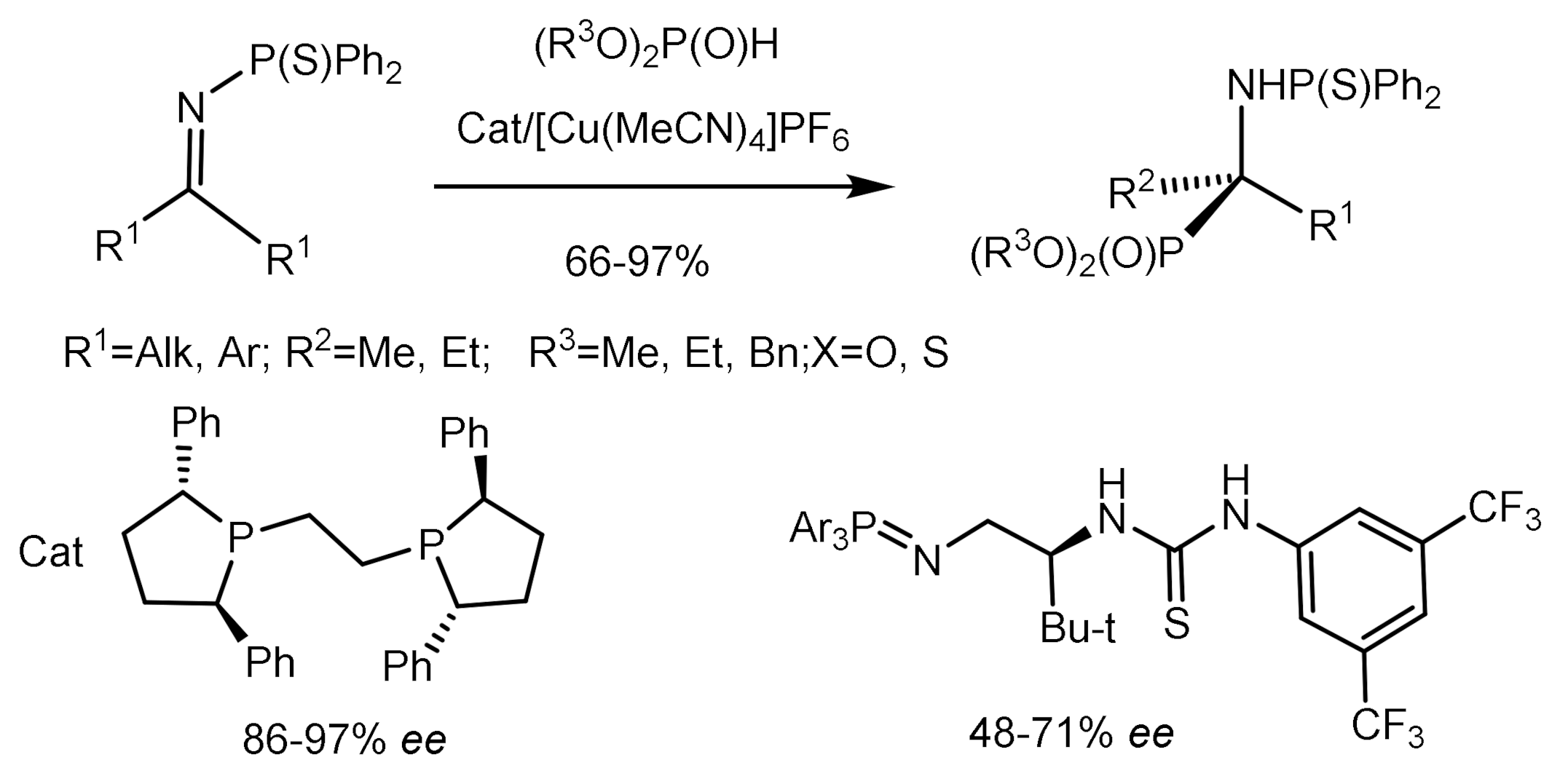
Figure 41.
Three-component reaction: aldehyde–aniline–triethylphosphite.
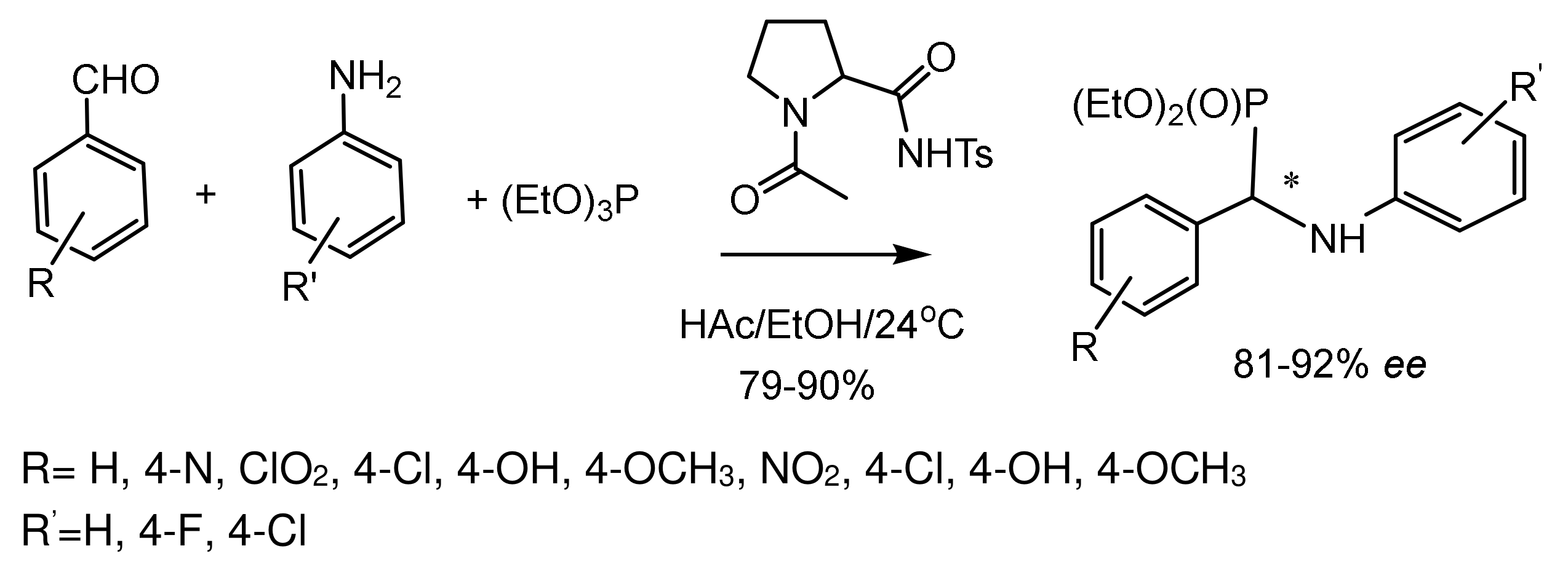
Figure 42.
Catalysis of the Kabachnik–Fields reaction with atropoisomeric phosphoric acid.
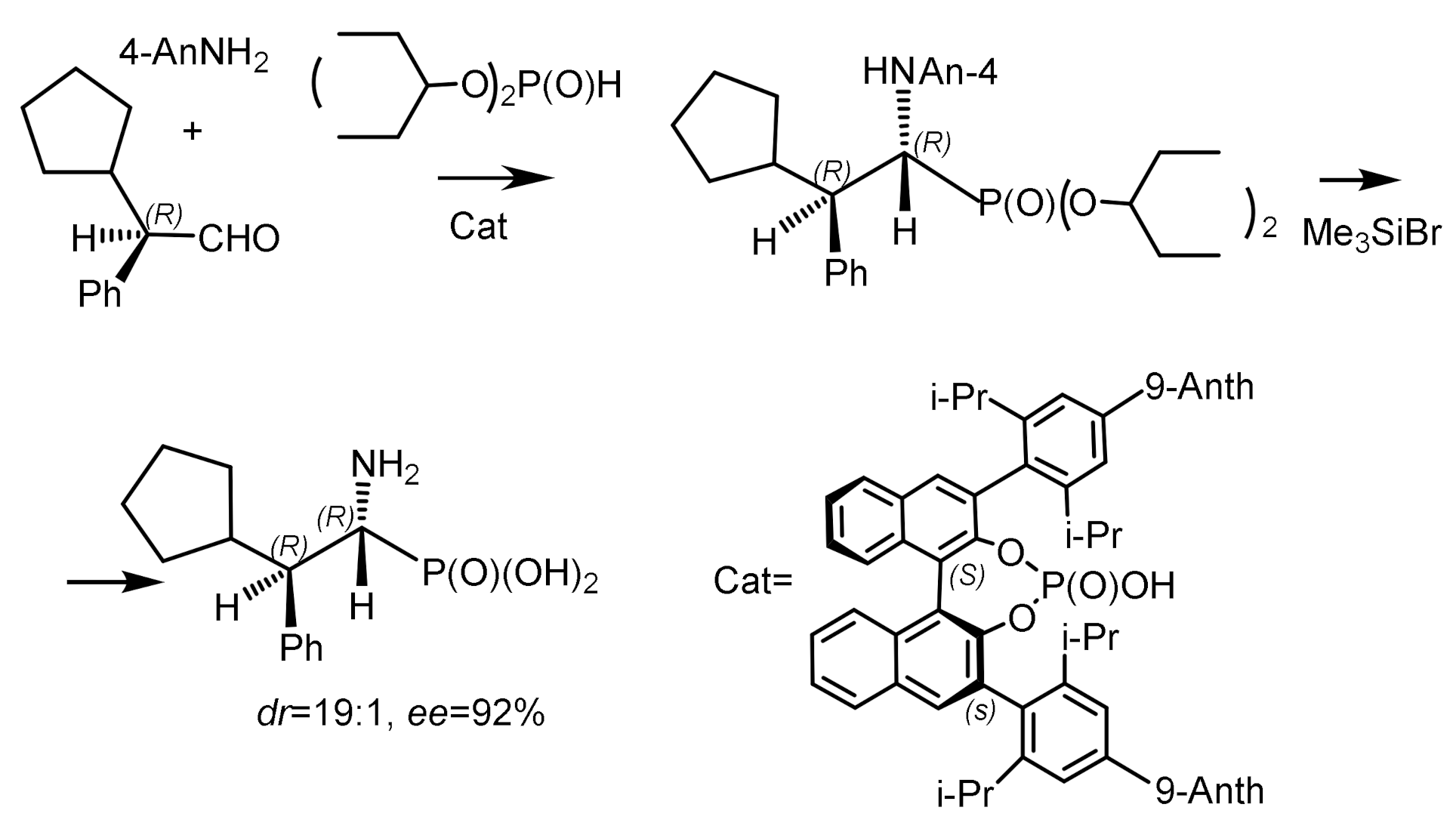
Figure 43.
Mukayama reaction of acylphosphonates with silylenolates.
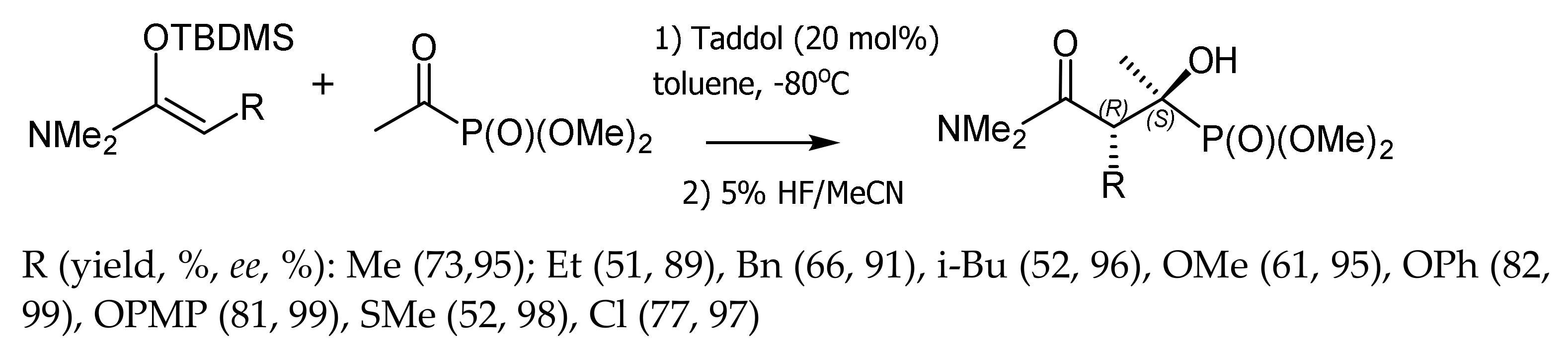
Figure 44.
Enantioselective reaction of silyl enolates with N-acyl–α–ιminophosphonates.
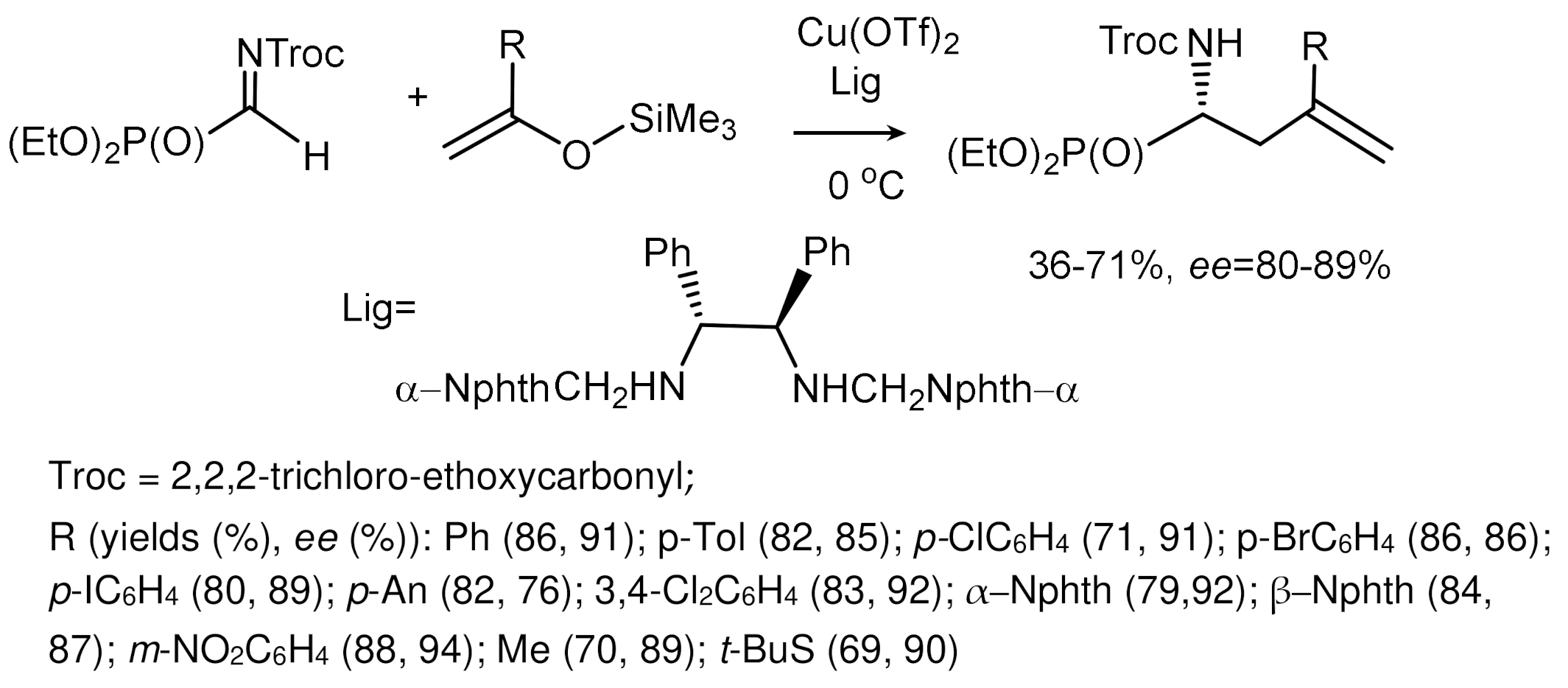
Figure 45.
The Mukayama reaction of diethyl α–ketophosphonate with N-acetal–O–ketenes.

Figure 46.
Phospha–Michael reaction catalyzed by a chiral diarylprolinol catalyst.
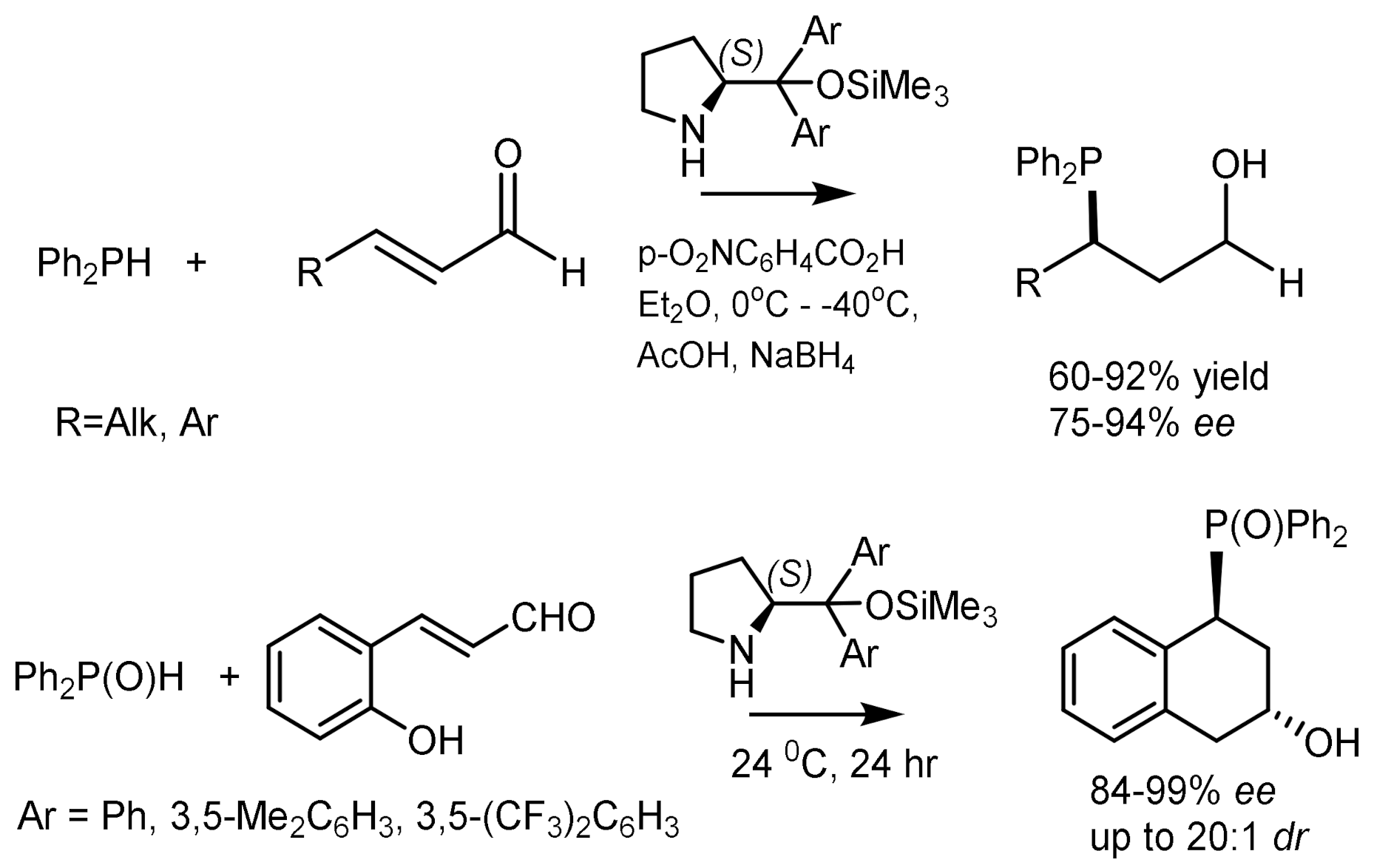
Figure 47.
Organocatalytic phospha-Michael reaction of α,β-unsaturated ketones with diarylphosphine oxides.
Figure 47.
Organocatalytic phospha-Michael reaction of α,β-unsaturated ketones with diarylphosphine oxides.
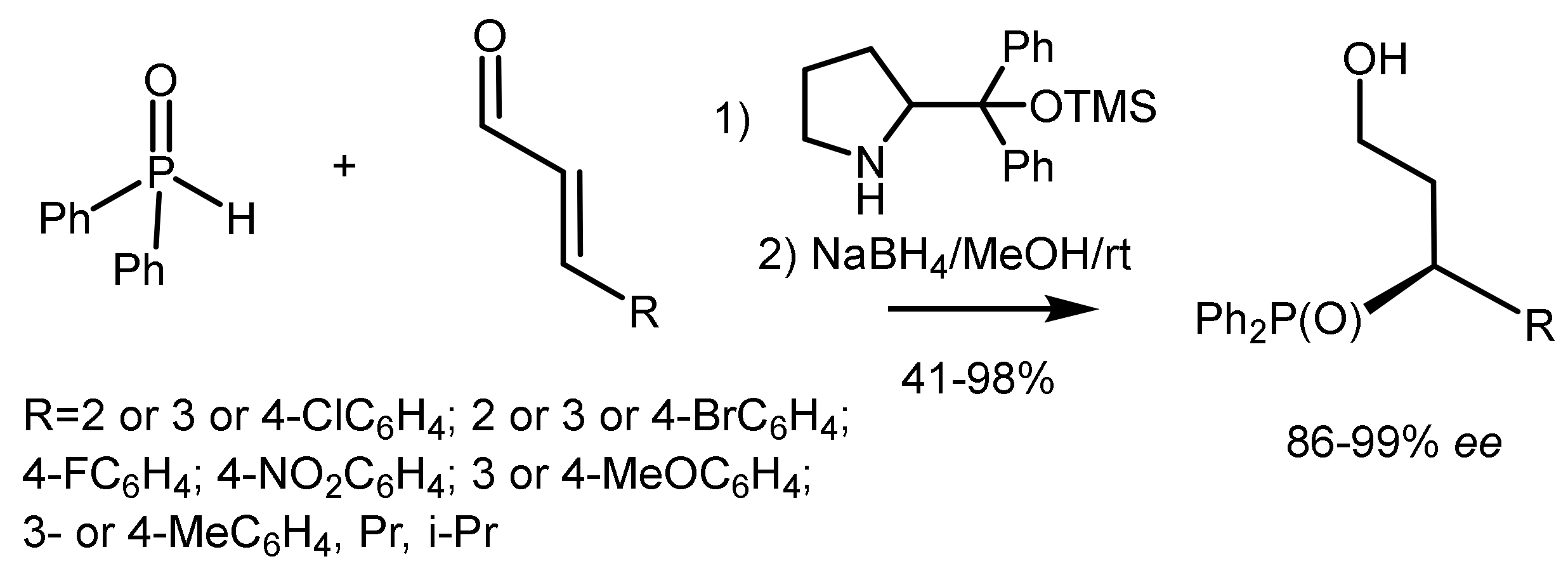
Figure 48.
Synthesis of chiral γ–keto-bis-phosphonates.
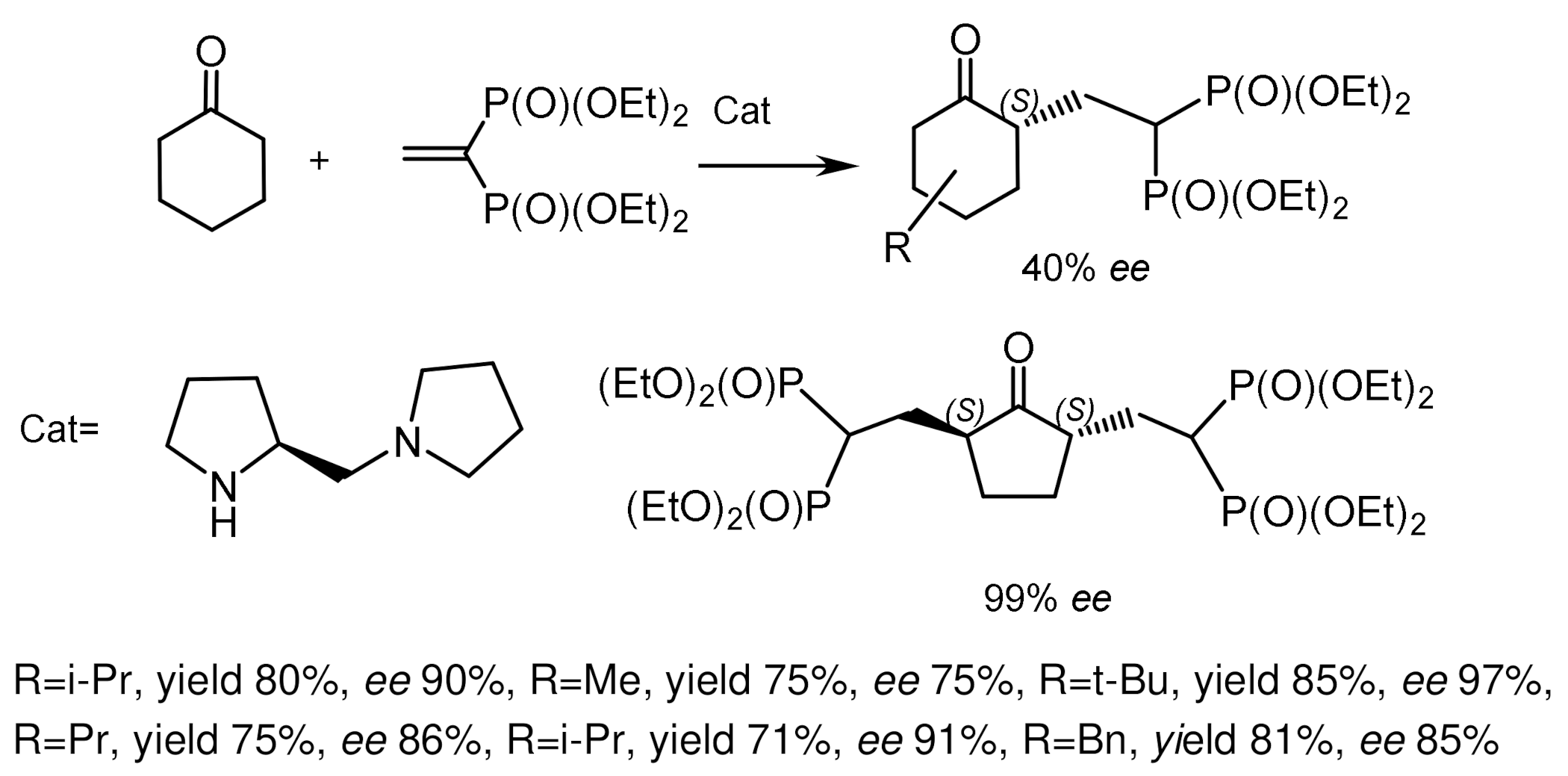
Figure 49.
Phospha-Michael reaction of aldehydes with vinyl bis-phosphonate.
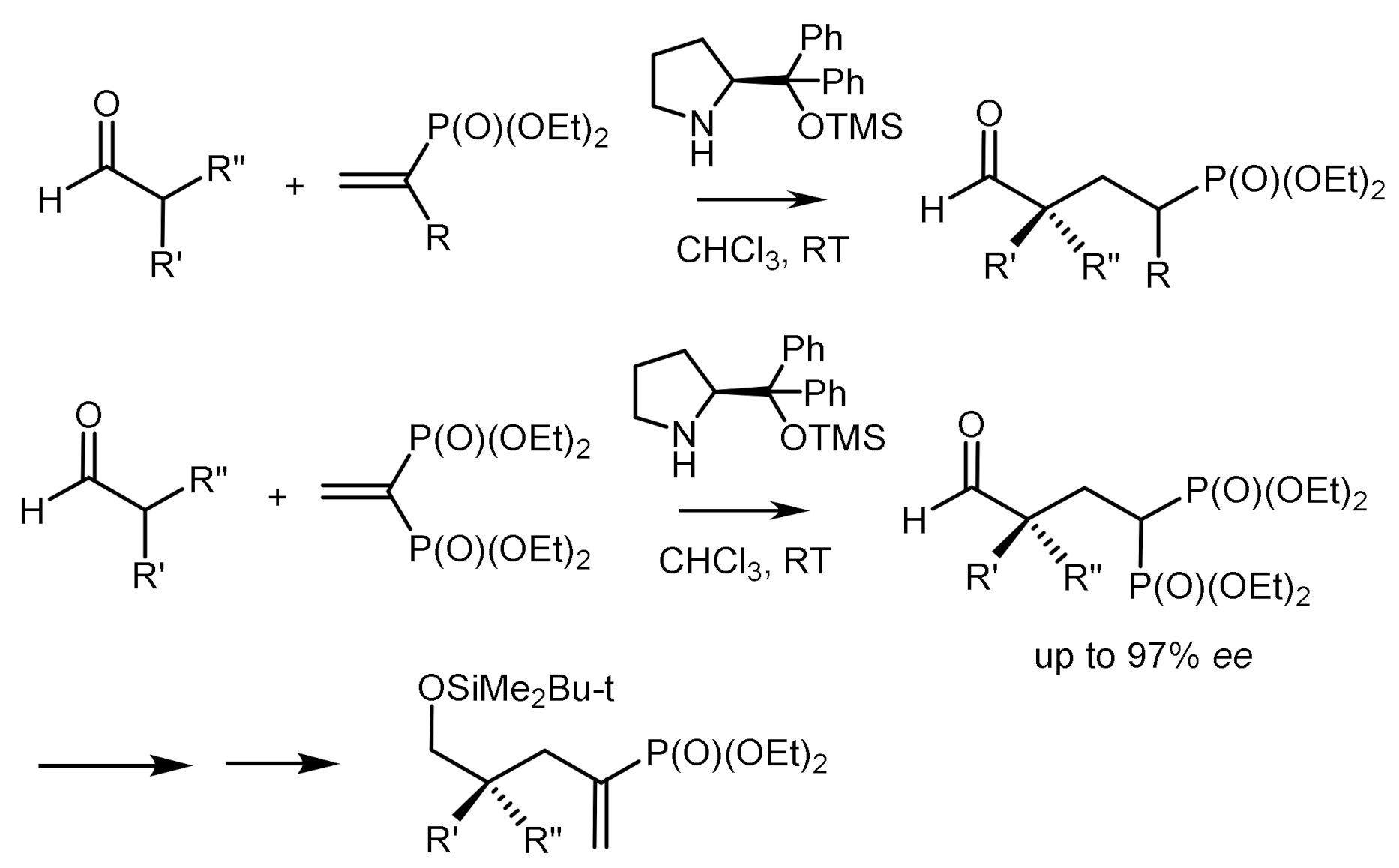
Figure 50.
The proposed transition state of Michael reaction catalyzed by the pyrrolidine organocatalyst.
Figure 50.
The proposed transition state of Michael reaction catalyzed by the pyrrolidine organocatalyst.
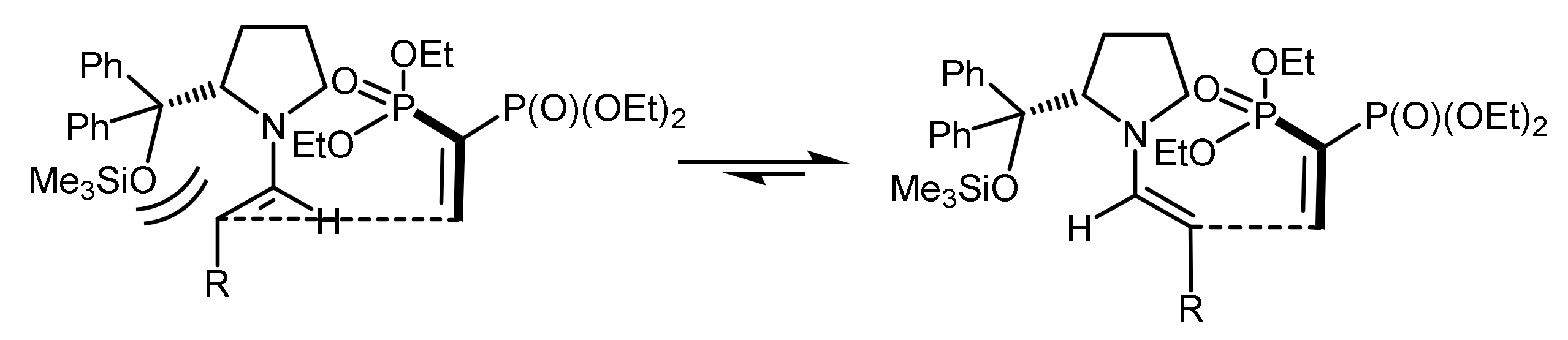
Figure 51.
The phospha-Michael reaction catalyzed by a chiral quinine thiourethane.
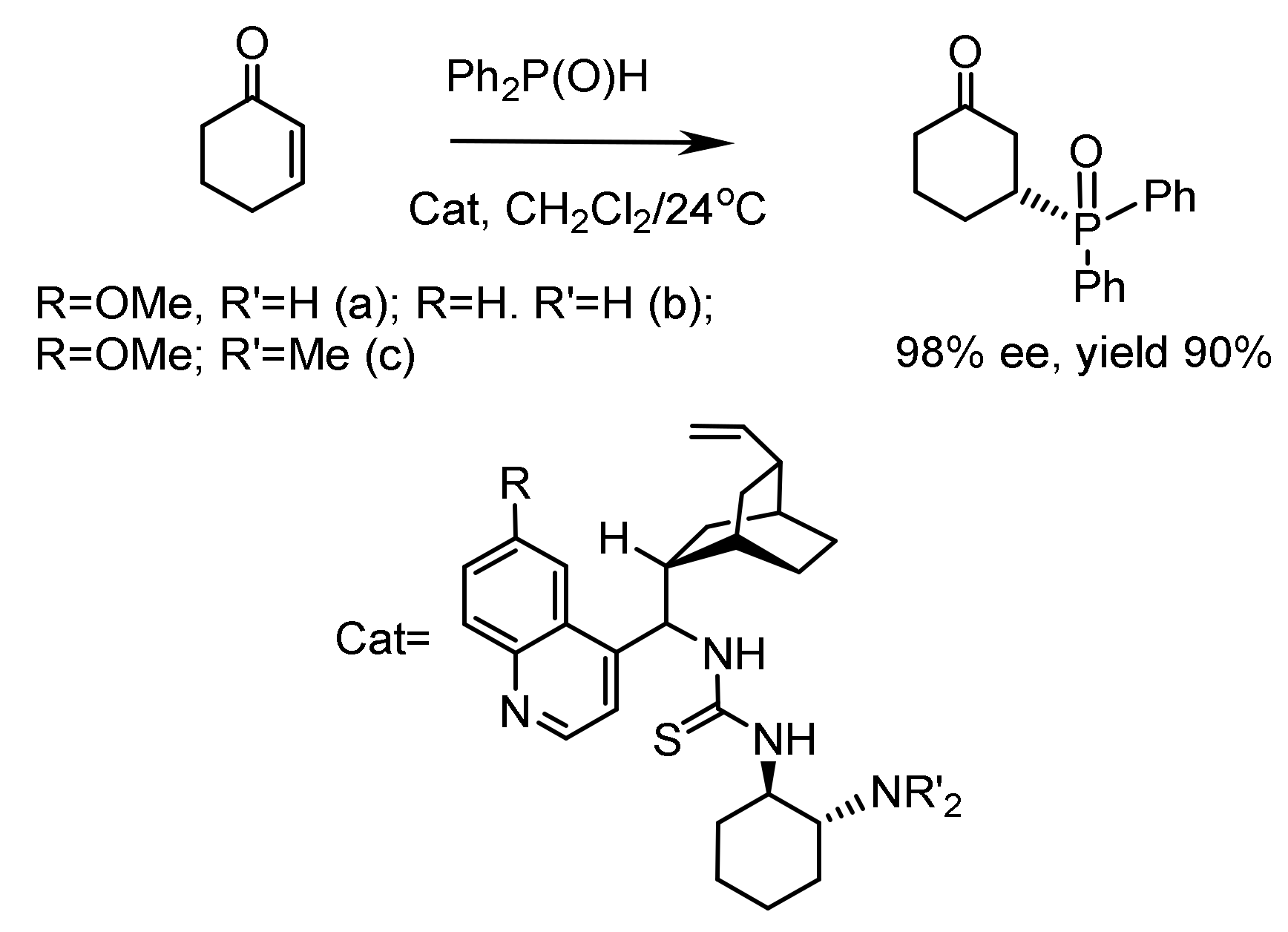
Figure 52.
Enantioselective addition of cyclic 1,3-dicarbonyl compounds to ethyl (E)-but-2-enoylphosphonate.
Figure 52.
Enantioselective addition of cyclic 1,3-dicarbonyl compounds to ethyl (E)-but-2-enoylphosphonate.
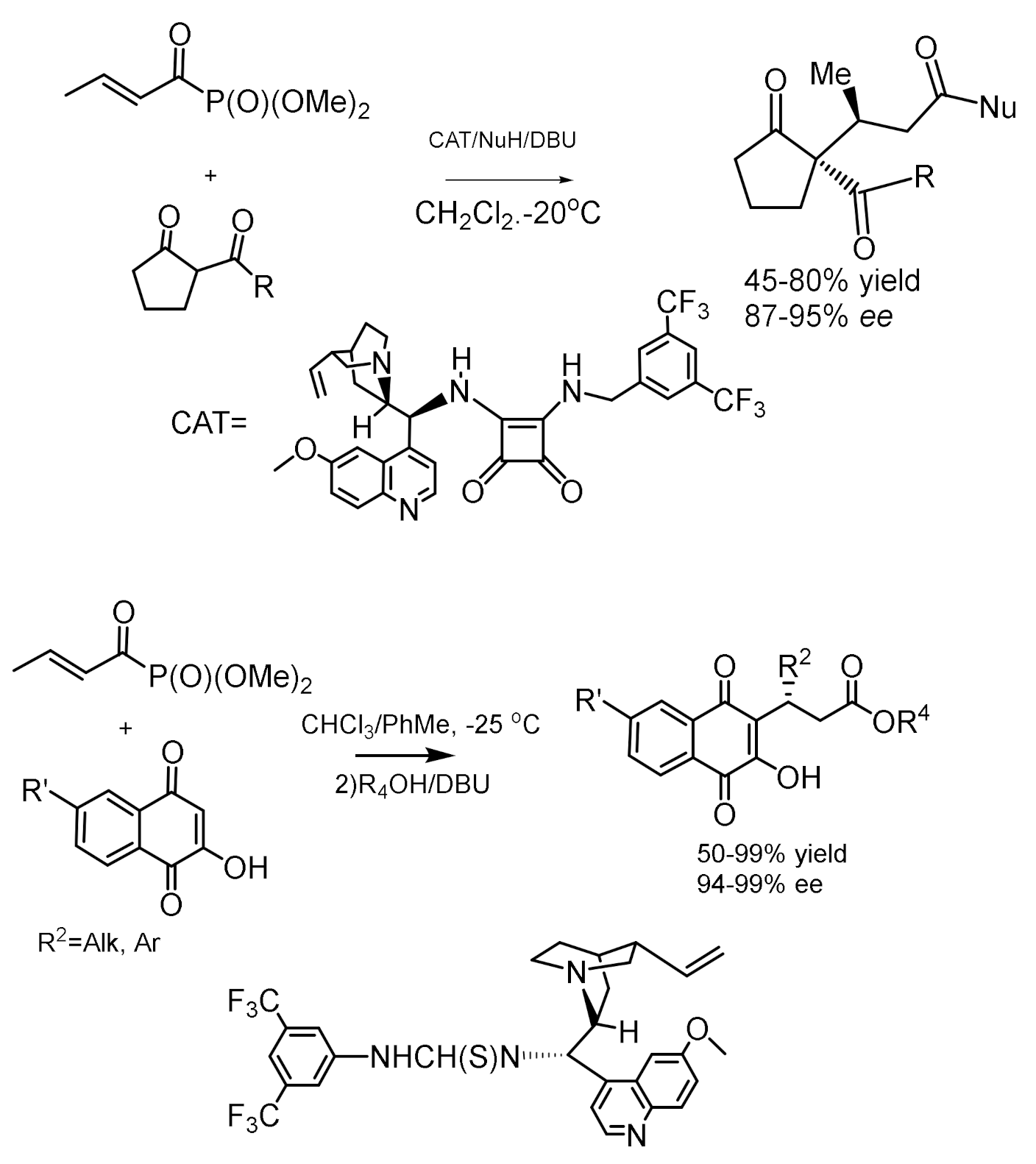
Figure 53.
The phospha–Michael reaction catalyzed by a squaramide catalyst.
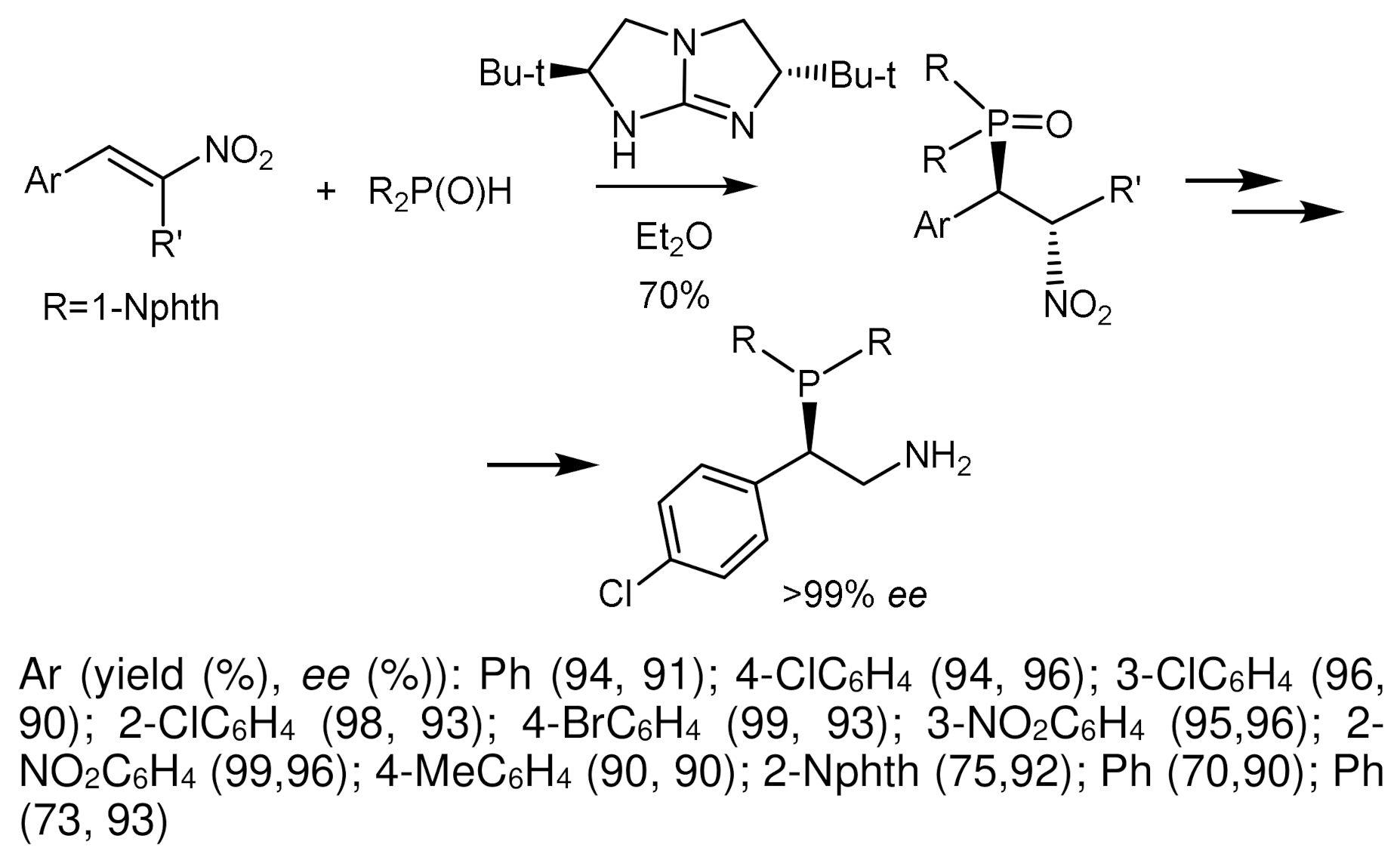
Figure 54.
The phospha-Michael reaction catalyzed by guanidinium/bis-thiocarbamide.

Figure 55.
Asymmetric additions of phosphonates to trans-crotonophenone derivatives.

Figure 56.
The reaction of enals with diphenylphosphine.

Figure 57.
Addition of diphenyl phosphite to nitroalkenes catalyzed by guanidine derivatives.

Figure 58.
Reaction of ketophosphonates with ketones catalyzed by L-proline derivatives.
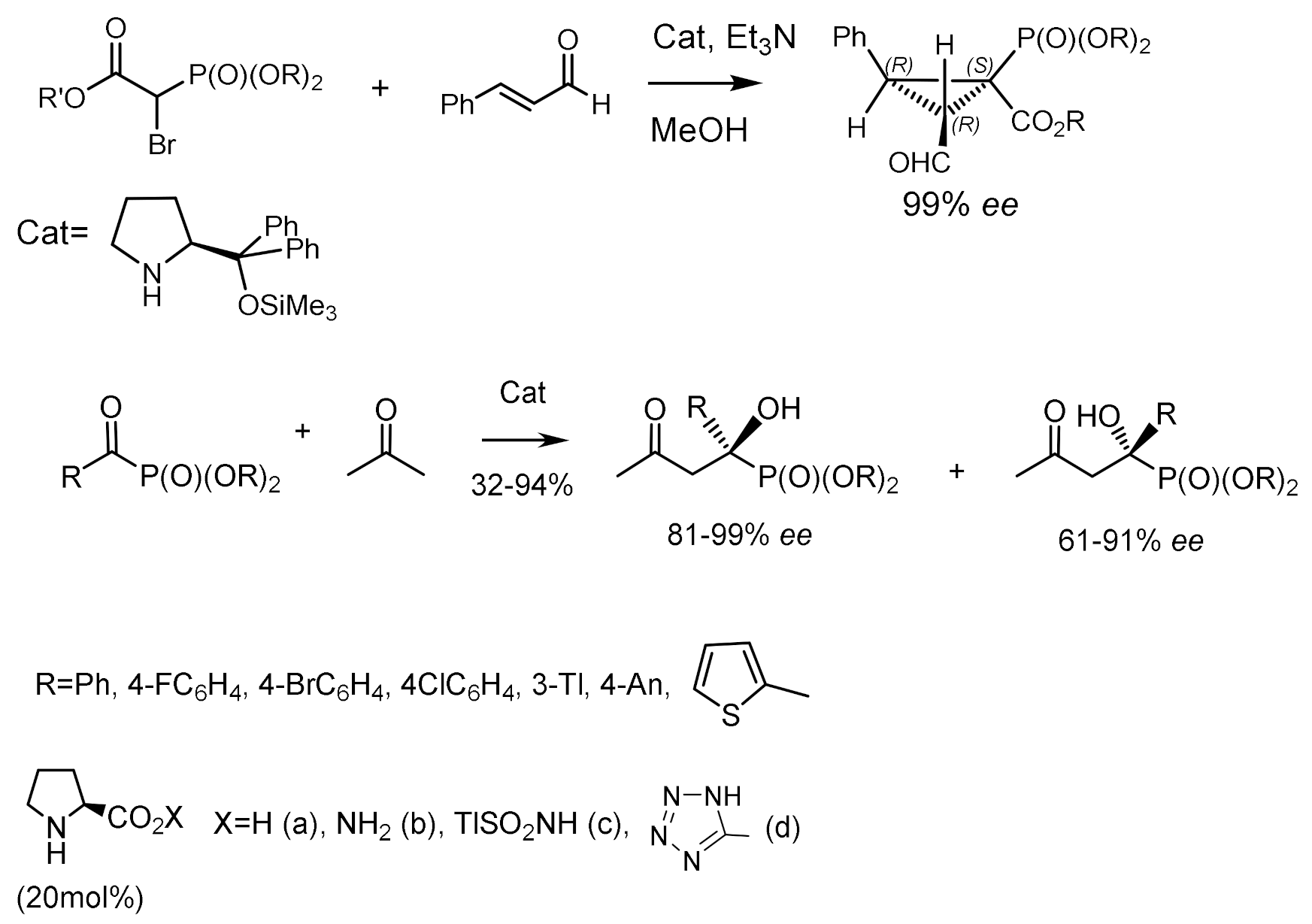
Figure 59.
Reaction of diethyl formylphosphonates with ketones catalyzed by L-prolinamide.
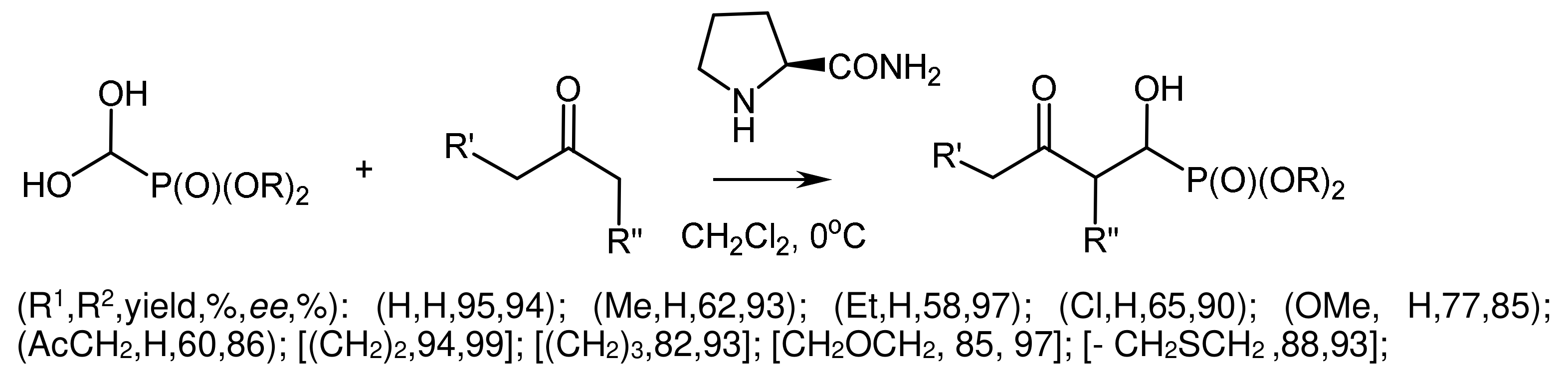
Figure 60.
The proposed by authors [75] transition state structures of the diethylformylphosphonate reaction with ketones.
Figure 60.
The proposed by authors [75] transition state structures of the diethylformylphosphonate reaction with ketones.
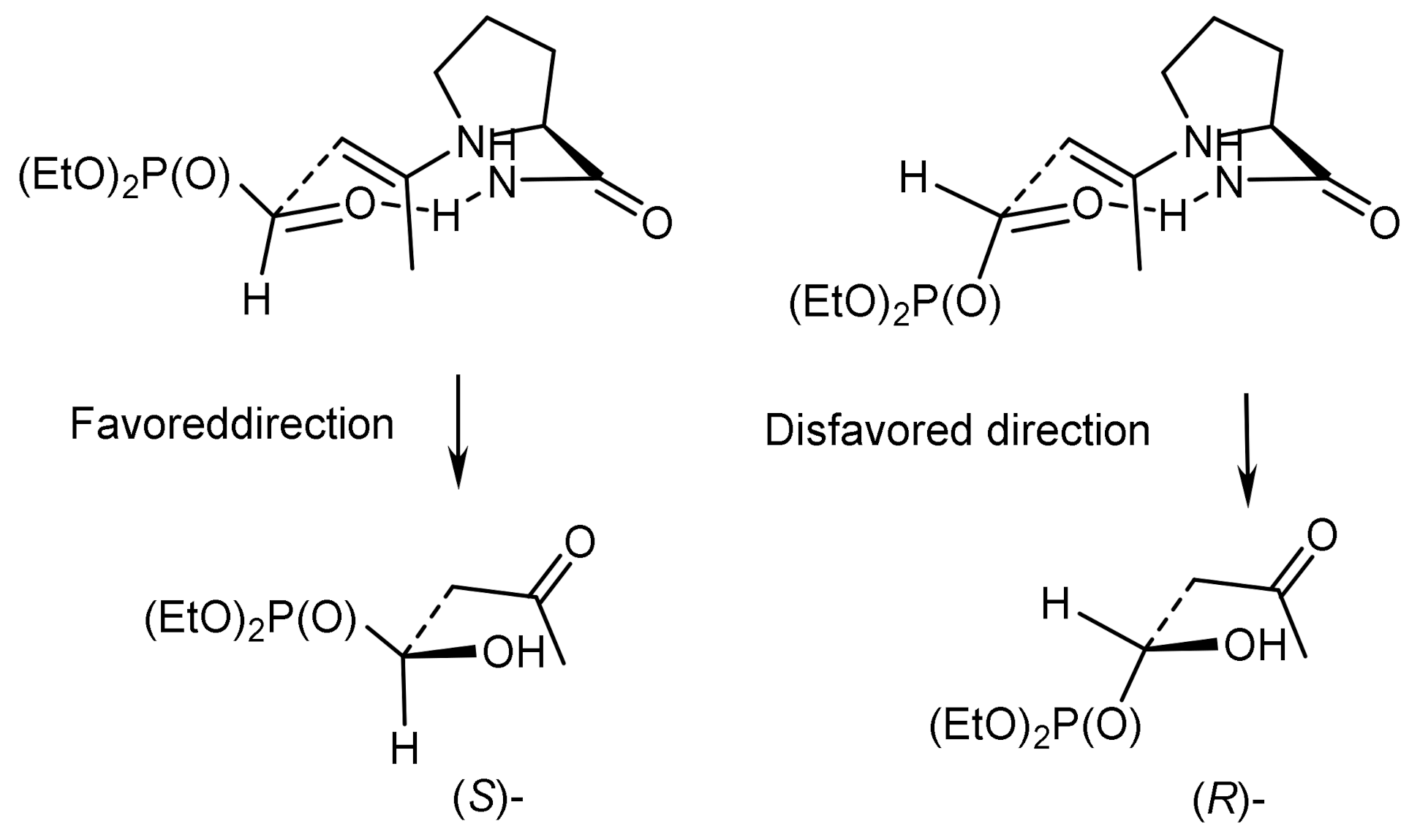
Figure 61.
Enantioselective phosphonylation of α,β–unsaturated aldehydes.
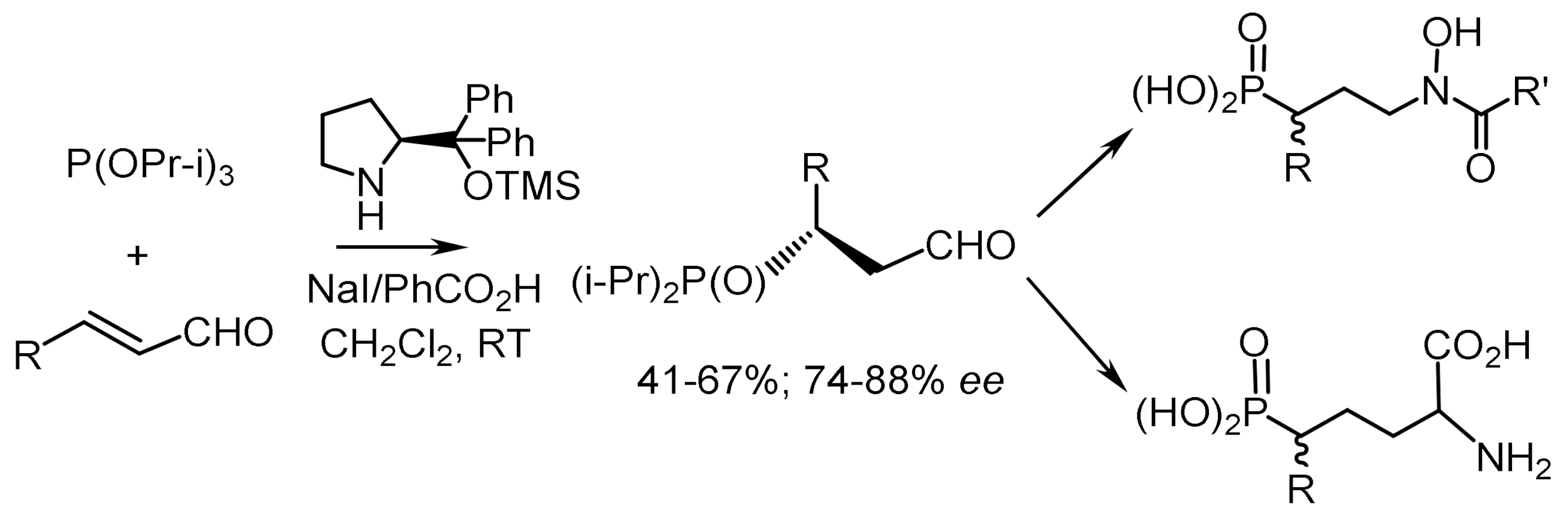
Figure 62.
Asymmetric hydrophosphination of activated olefins catalyzed by a platinum complex.

Figure 63.
Enantioselective phospha-Michael reaction of secondary phosphines catalyzed by the complex Pigiphos–Ni(II).
Figure 63.
Enantioselective phospha-Michael reaction of secondary phosphines catalyzed by the complex Pigiphos–Ni(II).
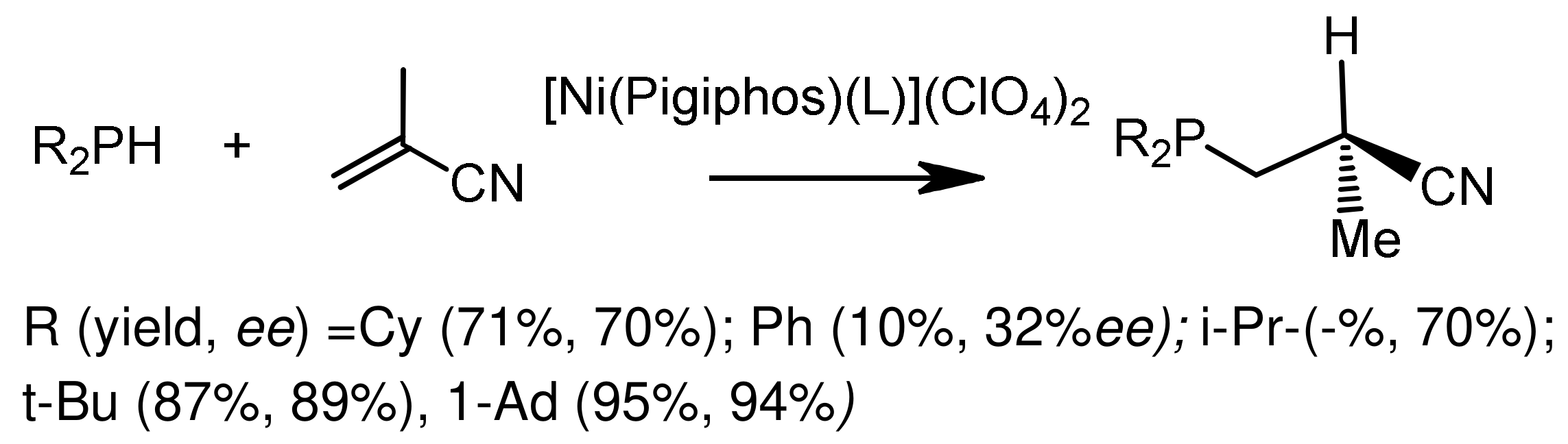
Figure 64.
Enantioselective addition of secondary phosphines catalyzed by organic nickel complex (A) Pigiphos–Ni(II) complex, (B) catalytic cycle of hydrophosphonylation of methacrylonitrile by the complex [Ni(3-Pigiphos) (NCMeCCH2)].
Figure 64.
Enantioselective addition of secondary phosphines catalyzed by organic nickel complex (A) Pigiphos–Ni(II) complex, (B) catalytic cycle of hydrophosphonylation of methacrylonitrile by the complex [Ni(3-Pigiphos) (NCMeCCH2)].
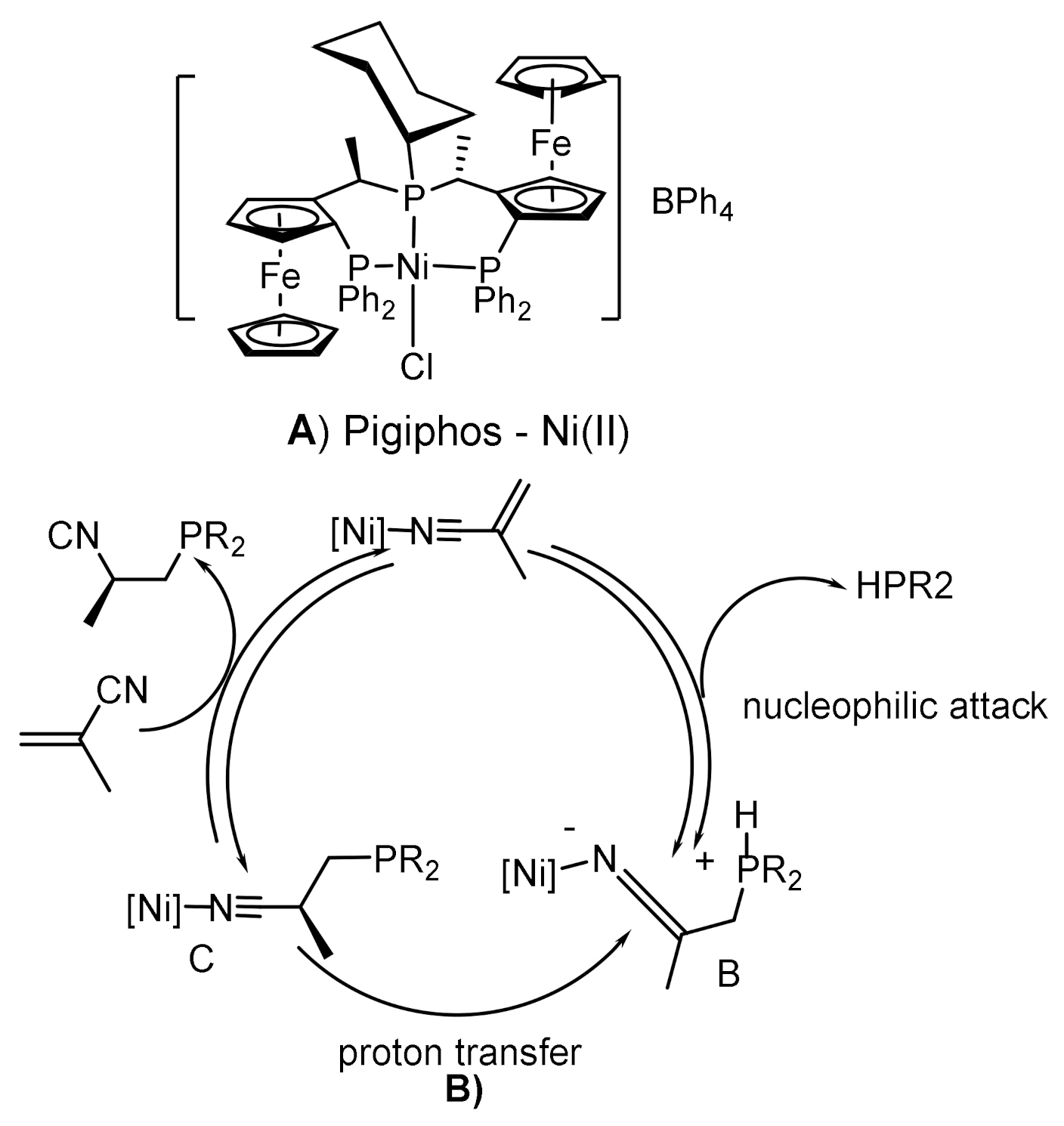
Figure 65.
Asymmetric addition of diarylphosphines to enones.
Figure 66.
Reaction of enones with P(O)H compounds.
Figure 67.
P-Michael addition catalyzed by a chiral palladium complex.
Figure 68.
Asymmetric synthesis of enaminophosphines via palladacycle-catalyzed addition of Ph2PH to α,β–unsaturated imines.
Figure 68.
Asymmetric synthesis of enaminophosphines via palladacycle-catalyzed addition of Ph2PH to α,β–unsaturated imines.
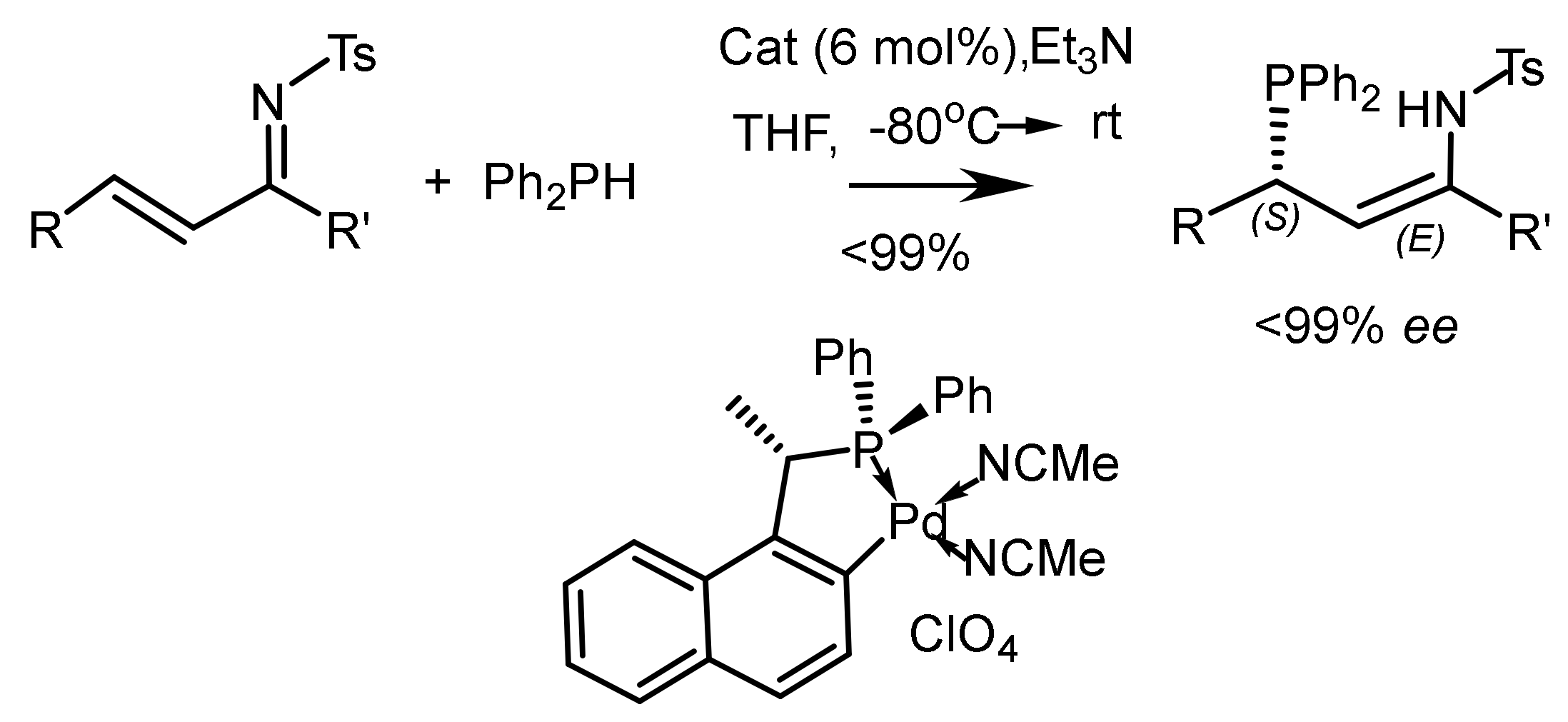
Figure 69.
The Michael addition of dialkyl phosphites to nitroalkenes.

Figure 70.
Preparation of potential chiral P,N-benzimidazole ligand using the Michael addition.

Figure 71.
Asymmetric aza-Henry reaction with phosphonoketimines catalyzed by bifunctional thiourethane alkaloids.
Figure 71.
Asymmetric aza-Henry reaction with phosphonoketimines catalyzed by bifunctional thiourethane alkaloids.

Figure 72.
Asymmetric [3 + 2] cycloadditions of α-diazophosphonates.

Figure 73.
Hydrogenation of β,β-diarylvinylphosphonates catalyzed by the Rh-(R,R)-f-spiroPhos complex.
Figure 73.
Hydrogenation of β,β-diarylvinylphosphonates catalyzed by the Rh-(R,R)-f-spiroPhos complex.
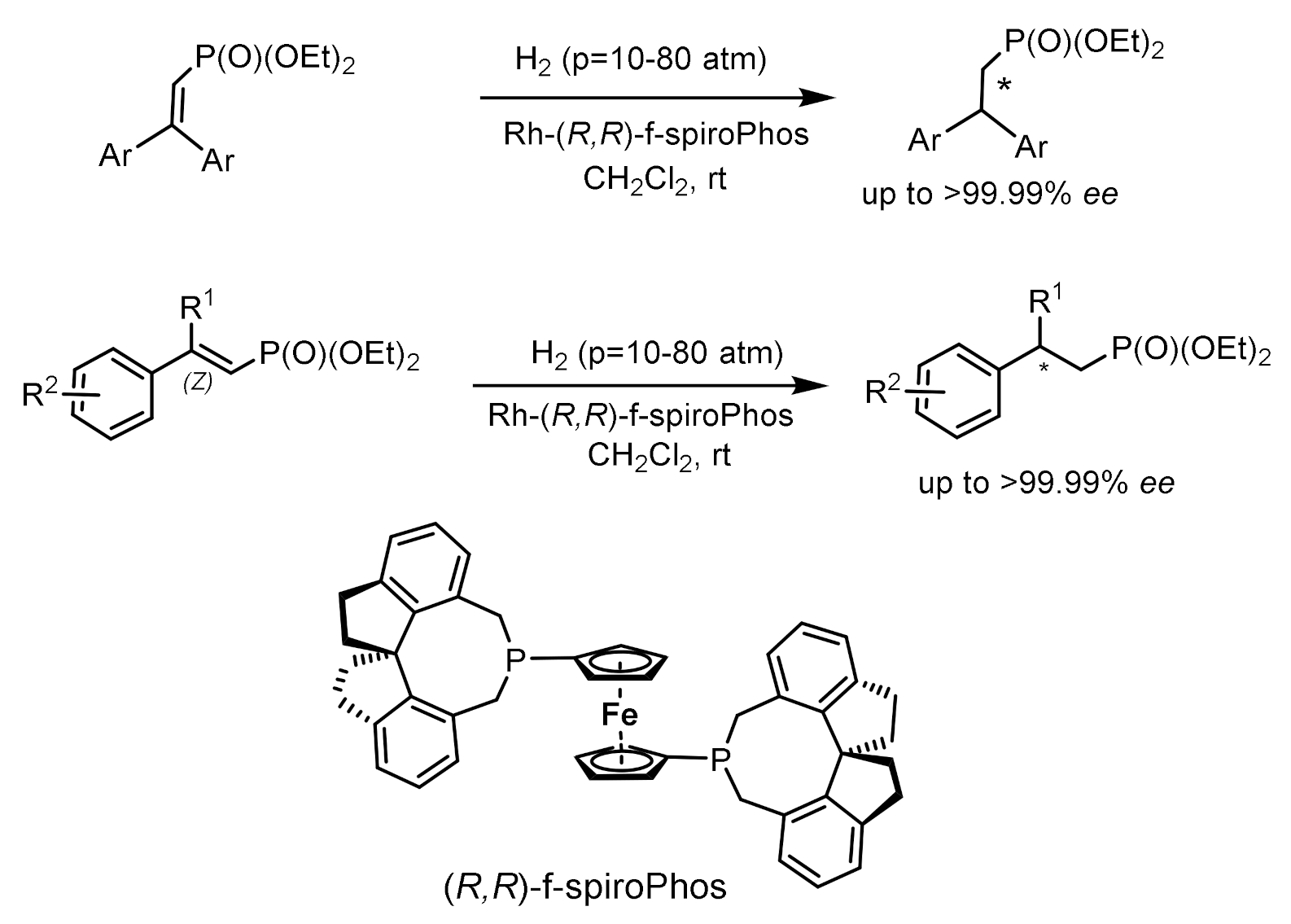
Figure 74.
Catalytic hydrogenation of N-[1-(dimethoxyphosphoryl)vinyl]formamide.
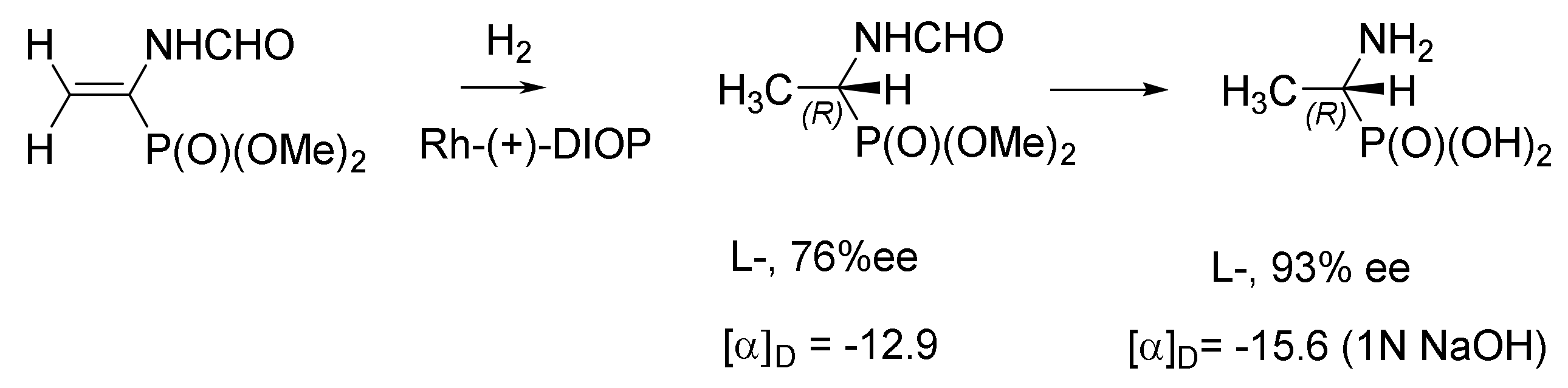
Figure 75.
Hydrogenation of α–enamidophosphonates catalyzed by the Rh/(S)-L1 complex.
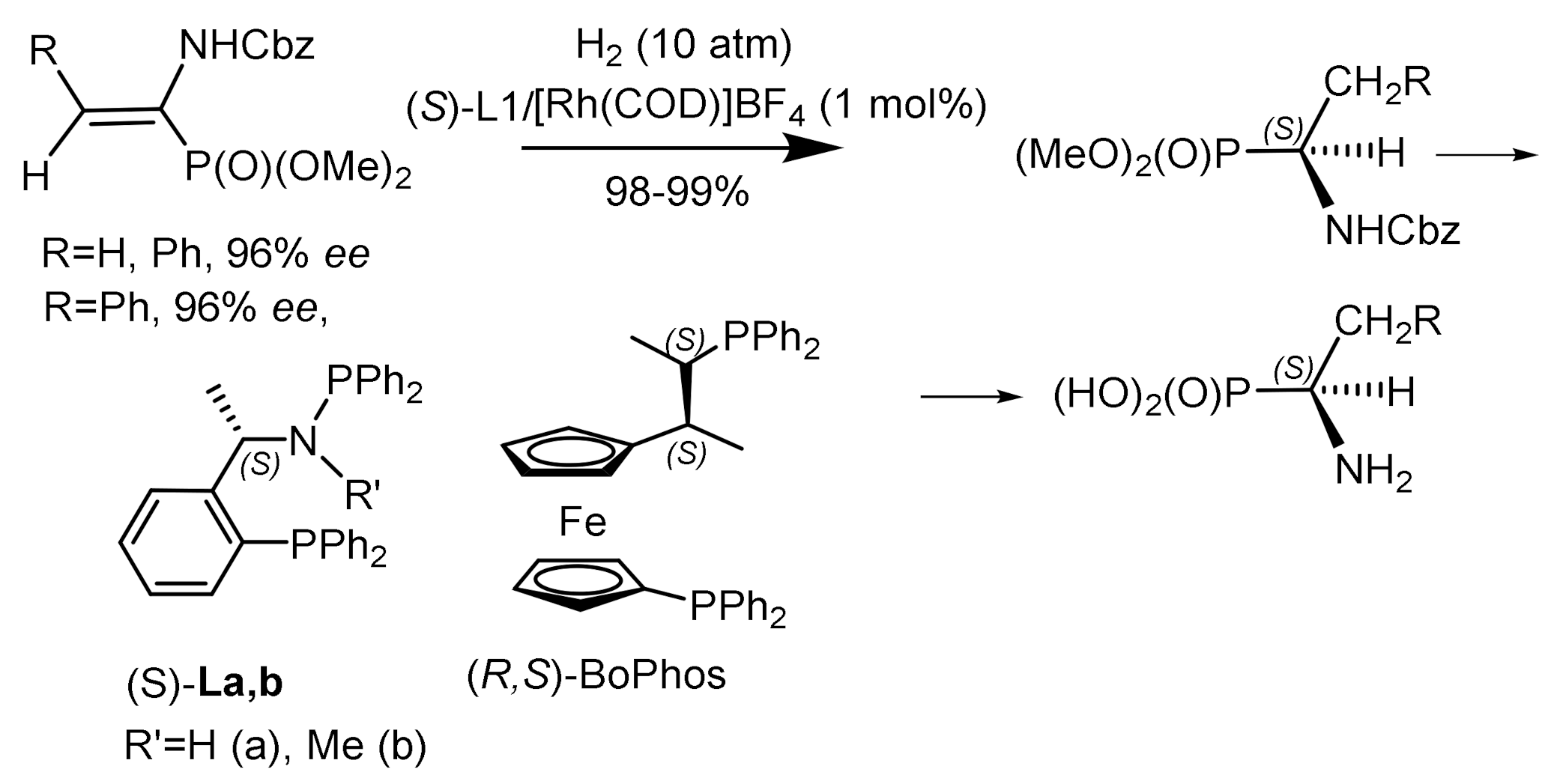
Figure 76.
Catalytic hydrogenation of vinylphosphonates using an iridium complex.
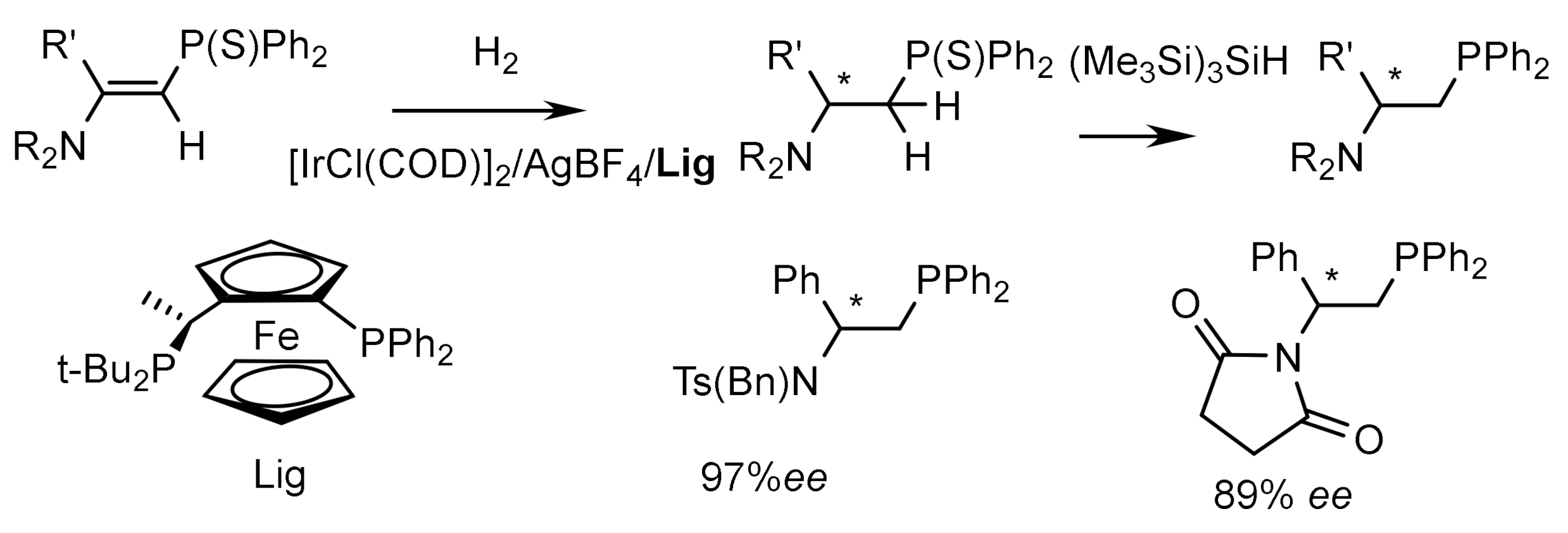
Figure 77.
Hydrogenation of prochiral β–N-acetylamino-vinylphosphonates catalyzed by chiral rhodium complexes.
Figure 77.
Hydrogenation of prochiral β–N-acetylamino-vinylphosphonates catalyzed by chiral rhodium complexes.

Figure 78.
Rh-catalyzed asymmetric hydrogenation of α,β–unsaturated phosphonates.
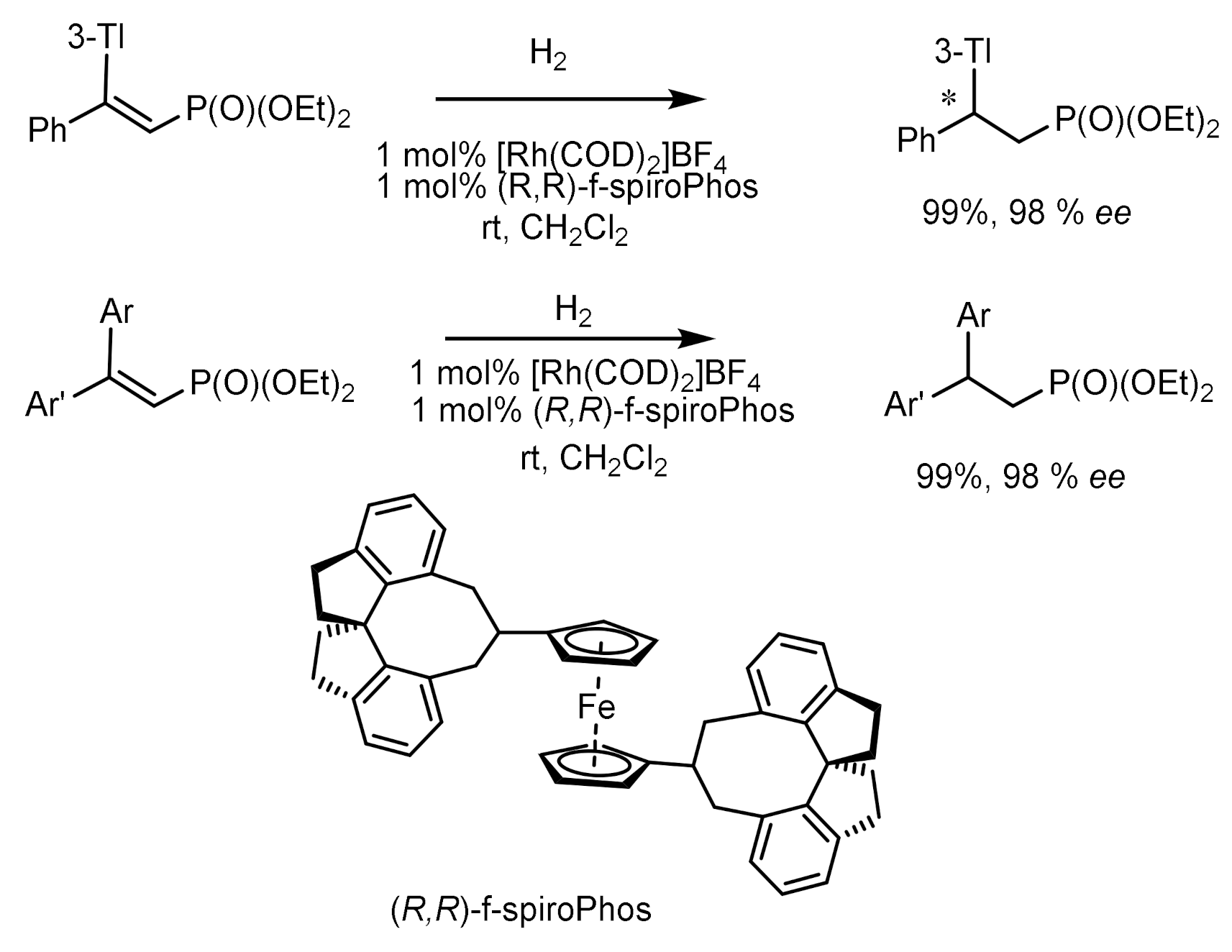
Figure 79.
Asymmetric hydrogenation of (E)- and (Z)–β–aryl-β–(enamido) phosphonates with chiral Iridium complex.
Figure 79.
Asymmetric hydrogenation of (E)- and (Z)–β–aryl-β–(enamido) phosphonates with chiral Iridium complex.

Figure 80.
Hydrogenation of β–arylalkenesphosphonates catalyzed by rhodium complexes.
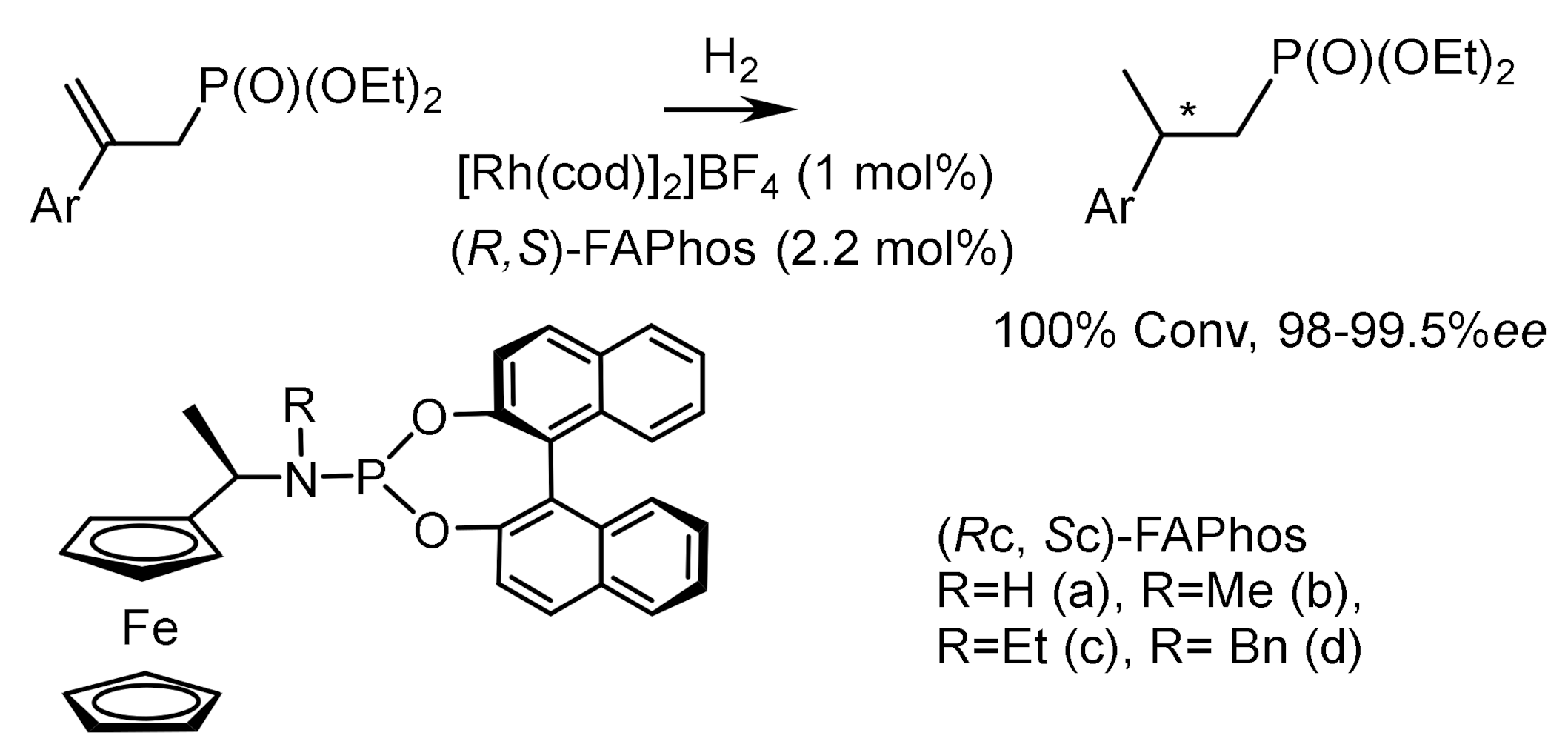
Figure 81.
Enantioselective hydrogenation of enolphosphonates catalyzed by chiral rhodium complexes containing P–OR ligands.
Figure 81.
Enantioselective hydrogenation of enolphosphonates catalyzed by chiral rhodium complexes containing P–OR ligands.

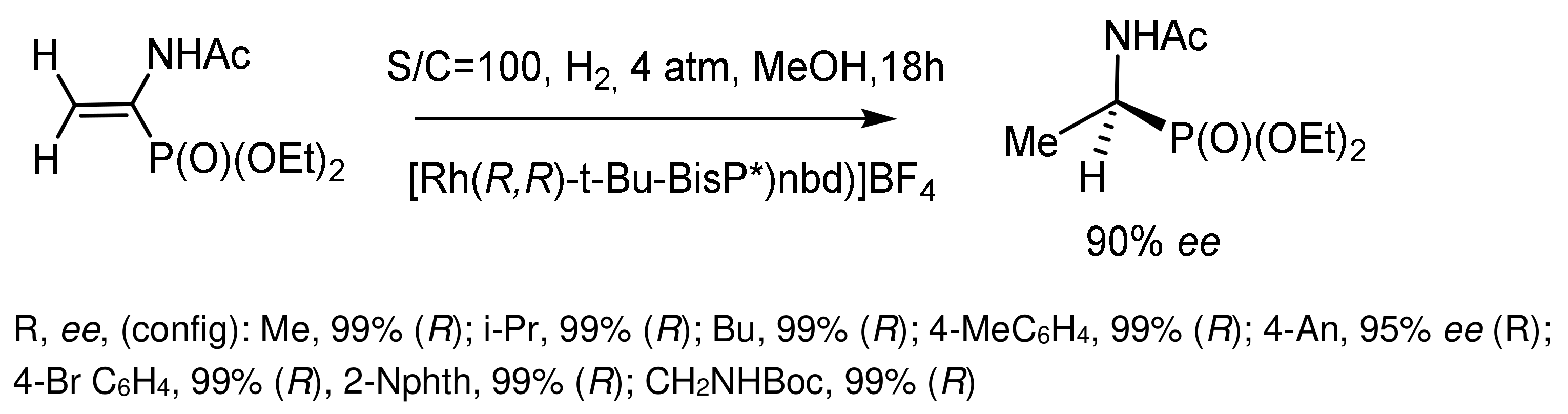
Figure 82.
Asymmetric hydrogenation of enolphosphonates.

Figure 83.
Catalytic hydrogenation of unsaturated phosphonates.

Figure 84.
Hydrogenation of β–substituted α,β–unsaturated phosphonates catalyzed by rhodium complexes containing ferrocene ligands.
Figure 84.
Hydrogenation of β–substituted α,β–unsaturated phosphonates catalyzed by rhodium complexes containing ferrocene ligands.
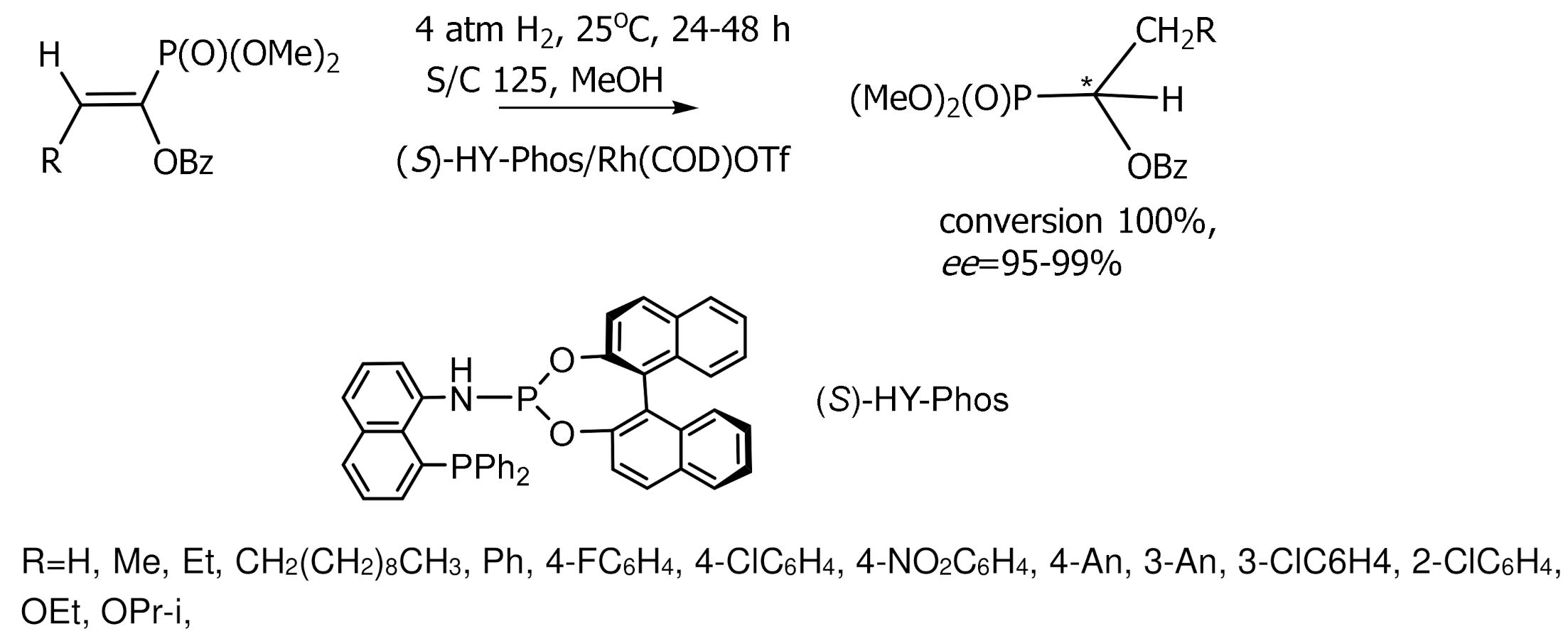
Figure 85.
Hydrogenation of vinyl phosphonates with Rh/(SC, RFc, RP) catalyst.
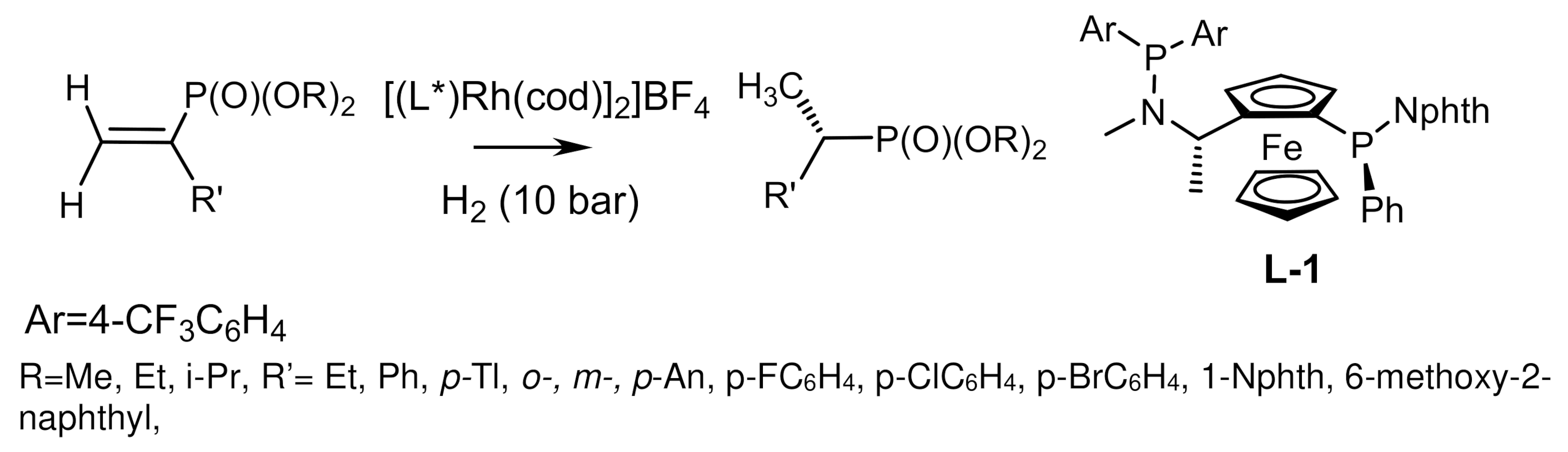
Figure 86.
Rh-catalyzed asymmetric hydrogenation of α–enolphosphonate.
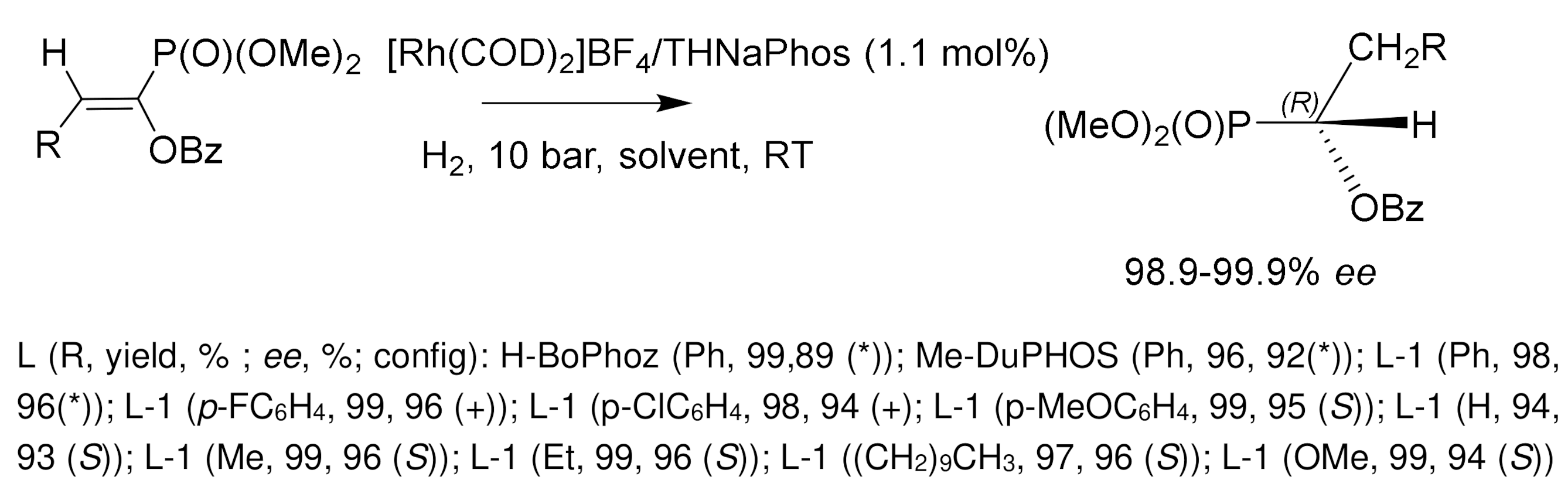
Figure 87.
Asymmetric hydrogenation of carboxyethylvinylphosphonates.

Figure 88.
Asymmetric hydrogenation of N-substituted vinylphosphonates.
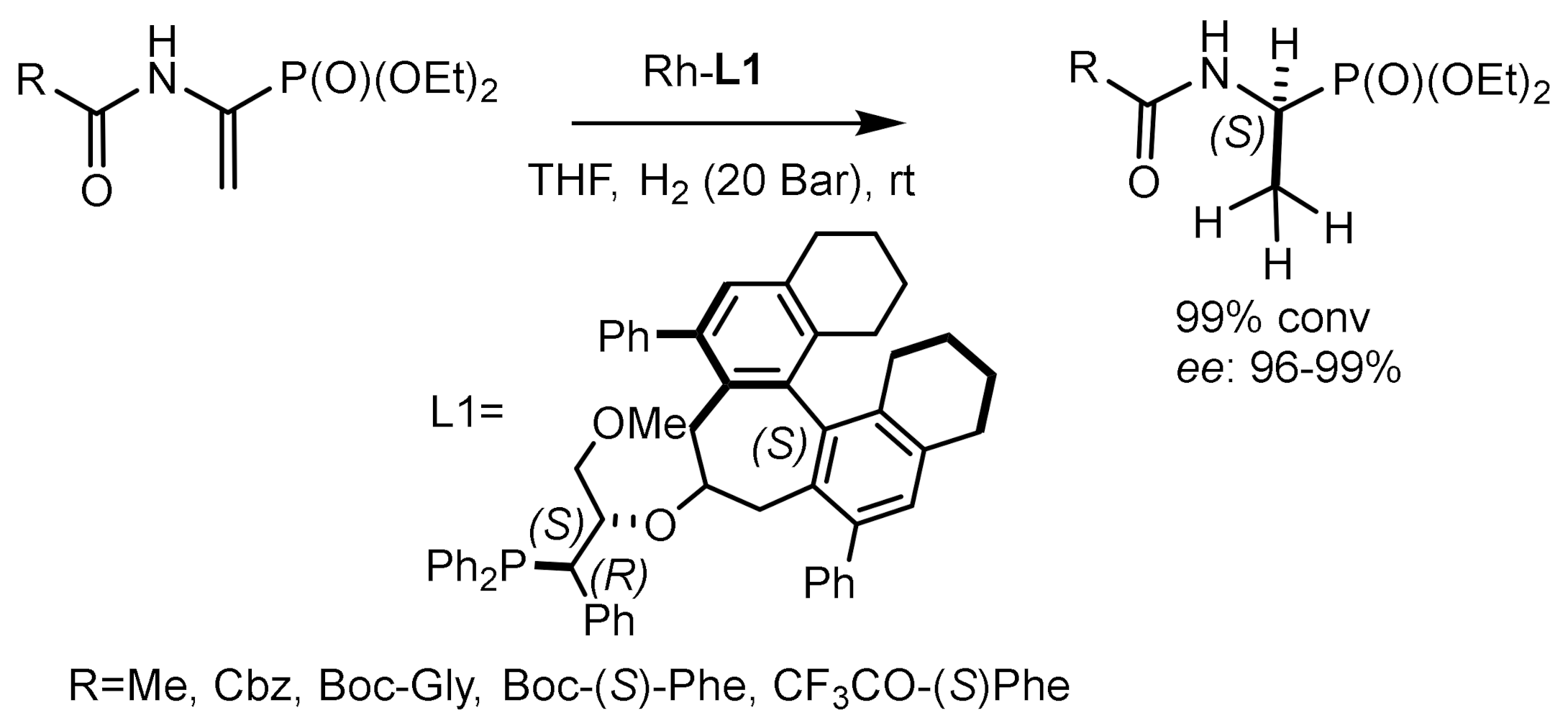
Figure 89.
BINAP- and BIPHEP-catalyzed ruthenium(II) asymmetric hydrogenation of β-ketophosphonates.
Figure 89.
BINAP- and BIPHEP-catalyzed ruthenium(II) asymmetric hydrogenation of β-ketophosphonates.
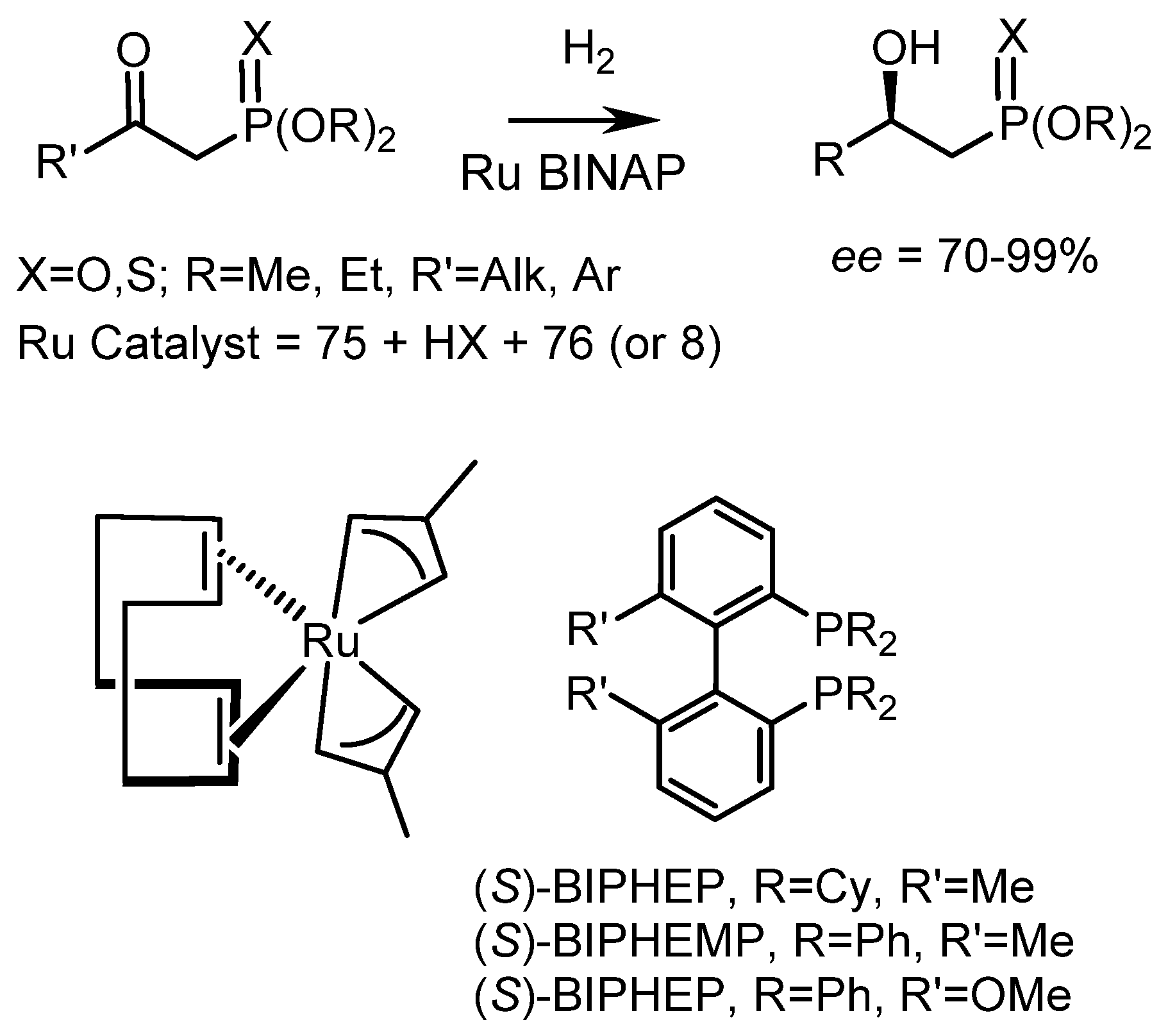
Figure 90.
Enantioselective synthesis of Fosfomycin.
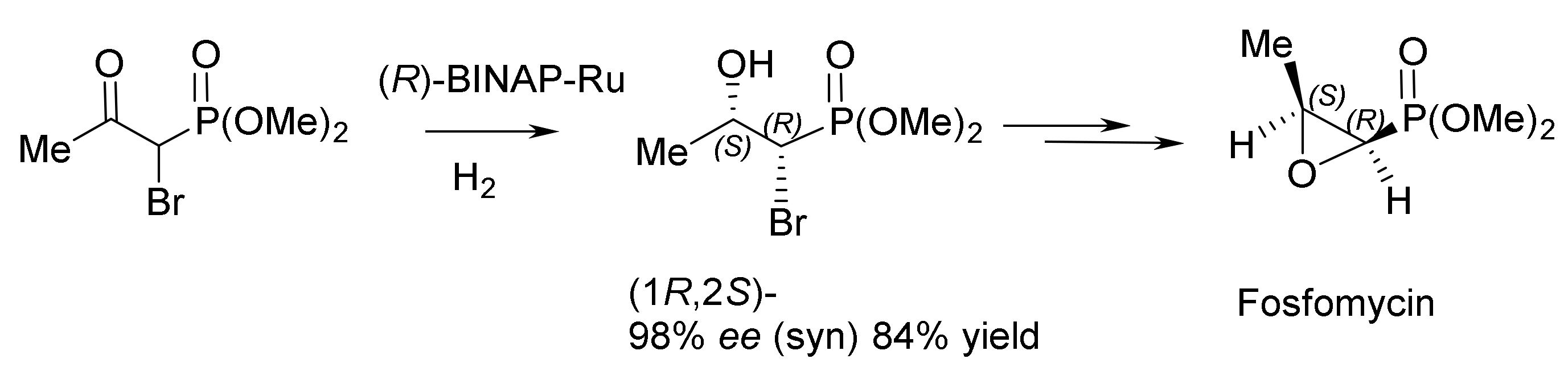
Figure 91.
Asymmetric hydrogenation of β-ketophosphonates catalyzed by BINAP–Ru(II).

Figure 92.
Hydrogenation of ketophosphonates catalyzed by (R)-BINOL/Ru.

Figure 93.
Dynamic kinetic resolution of amido-ketophosphonates.
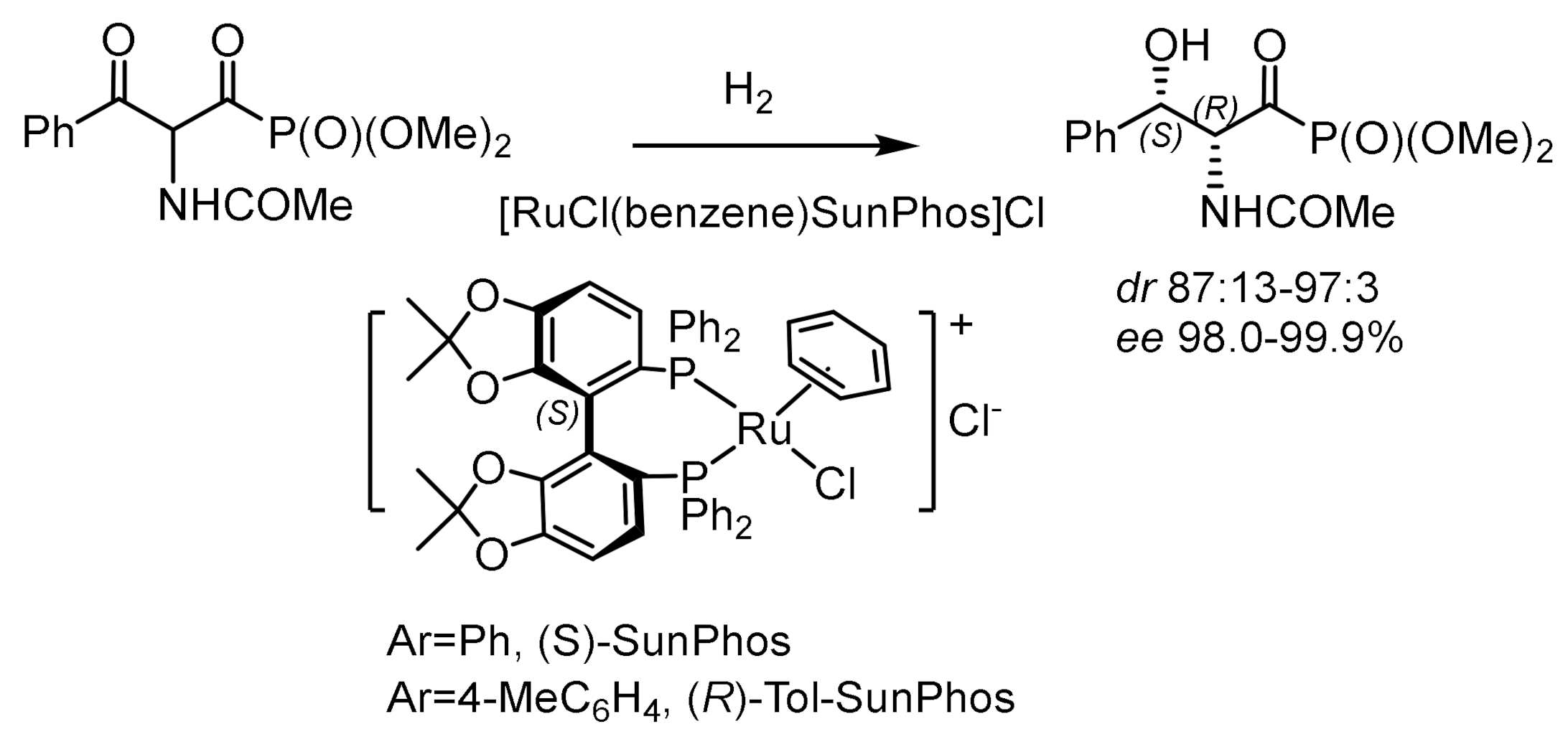
Figure 94.
α–Asymmetric hydrogenation of α-amido-β-ketophosphonates.

Figure 95.
Asymmetric hydrogenation of α-iminophosphonates with a palladium catalyst.
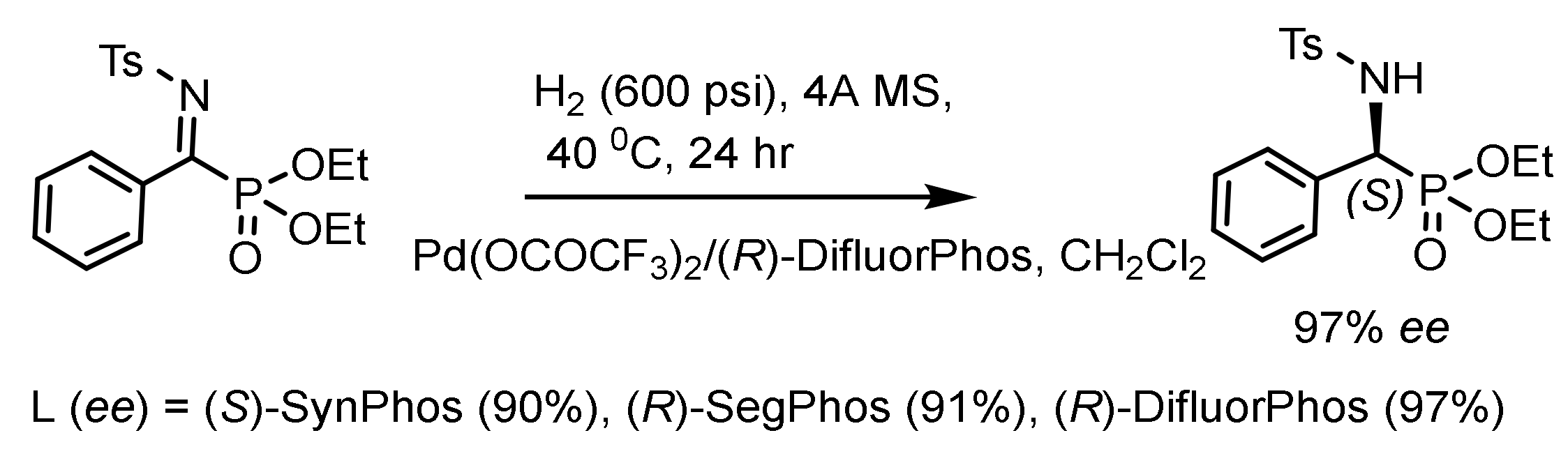
Figure 96.
Enantioselective synthesis of hydroxyphosphonates by catecholborane reduction.
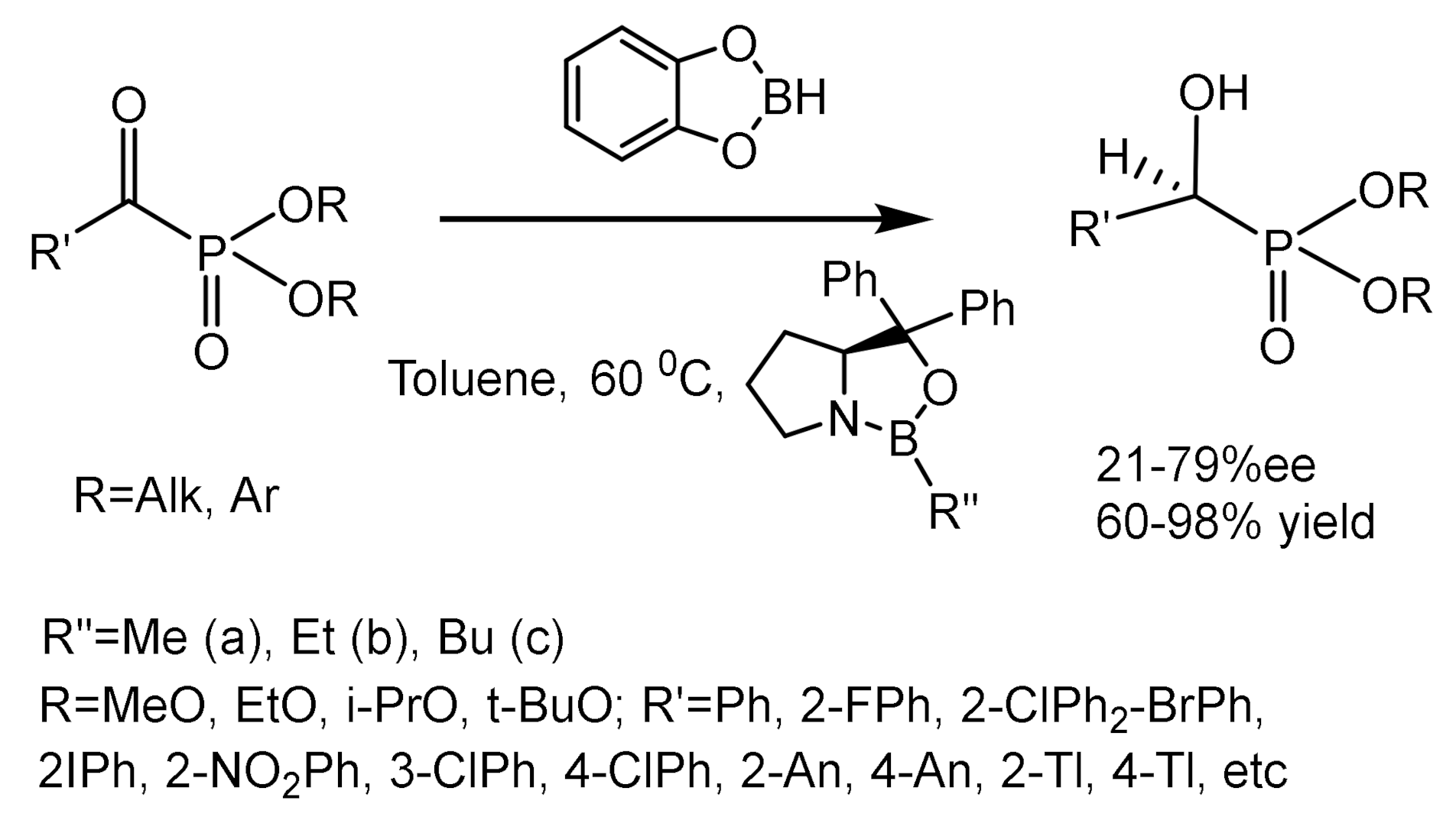
Figure 97.
Enantioselective reduction of 1-imino-2,2,2-trifluoroethylphosphonates.

Figure 98.
Asymmetric synthesis of 2H-azirine-2-phosphonates.

Figure 99.
Recovery of ketophosphonates with the NaBH4–Pro complex.

Figure 100.
Reduction of ketophosphonates with sodium-borohydride–tartaric-acid complex.
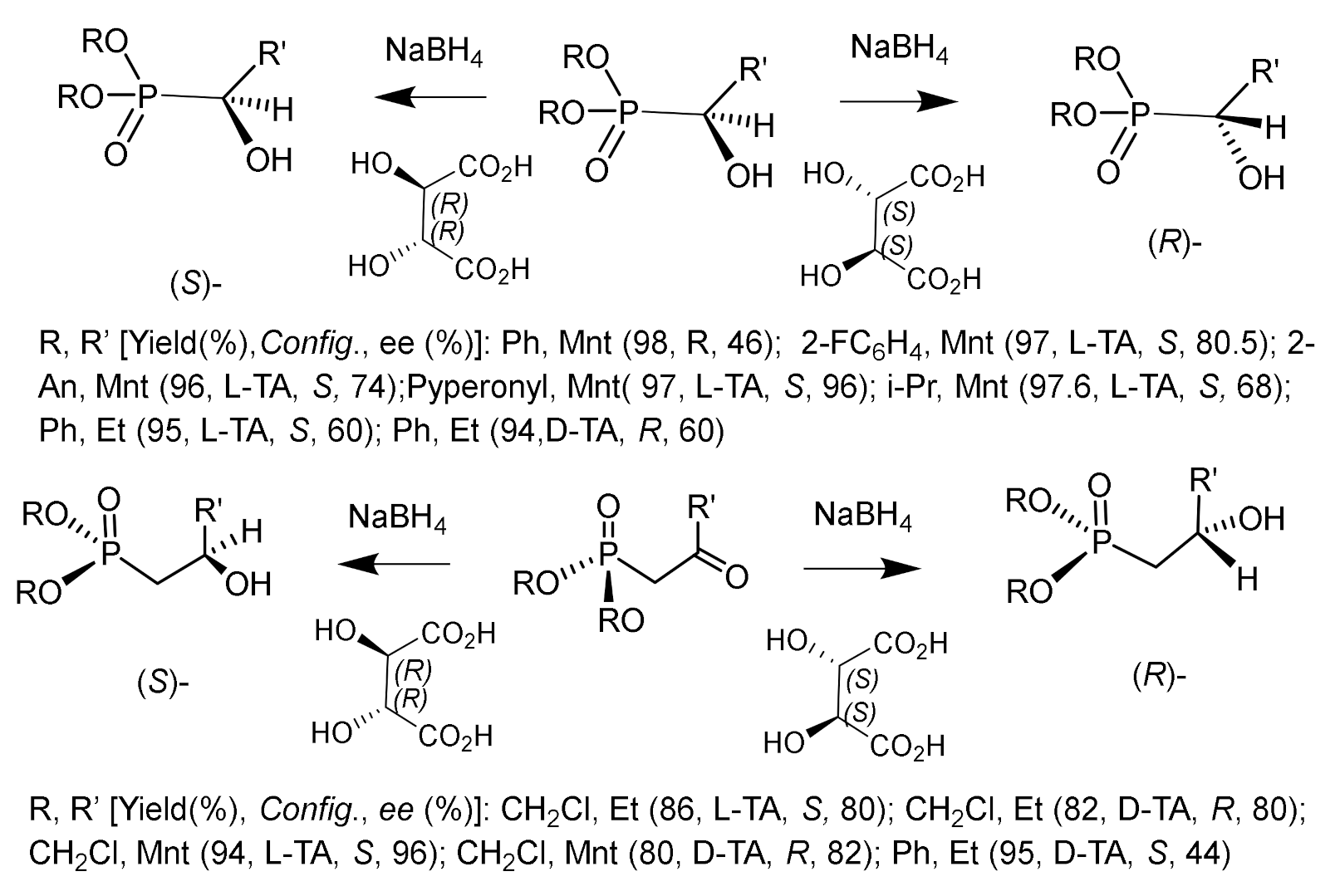
Figure 101.
Synthesis of biologically important β–hydroxyphosphonates. Reaction conditions: (a) NaBH4/L-TA, THF, −30–+20 °C, (b) Me3SiBr/MeOH/Me3N, (c) K2CO3/KI, (d) Bn2NH, H2/Pd-C, (e) NaN3/NH4Cl, (f) Ph3P/Ph3PO.
Figure 101.
Synthesis of biologically important β–hydroxyphosphonates. Reaction conditions: (a) NaBH4/L-TA, THF, −30–+20 °C, (b) Me3SiBr/MeOH/Me3N, (c) K2CO3/KI, (d) Bn2NH, H2/Pd-C, (e) NaN3/NH4Cl, (f) Ph3P/Ph3PO.

Figure 102.
Reduction of acylphosphonates with formic acid and triethylamine catalyzed by a ruthenium complex.
Figure 102.
Reduction of acylphosphonates with formic acid and triethylamine catalyzed by a ruthenium complex.
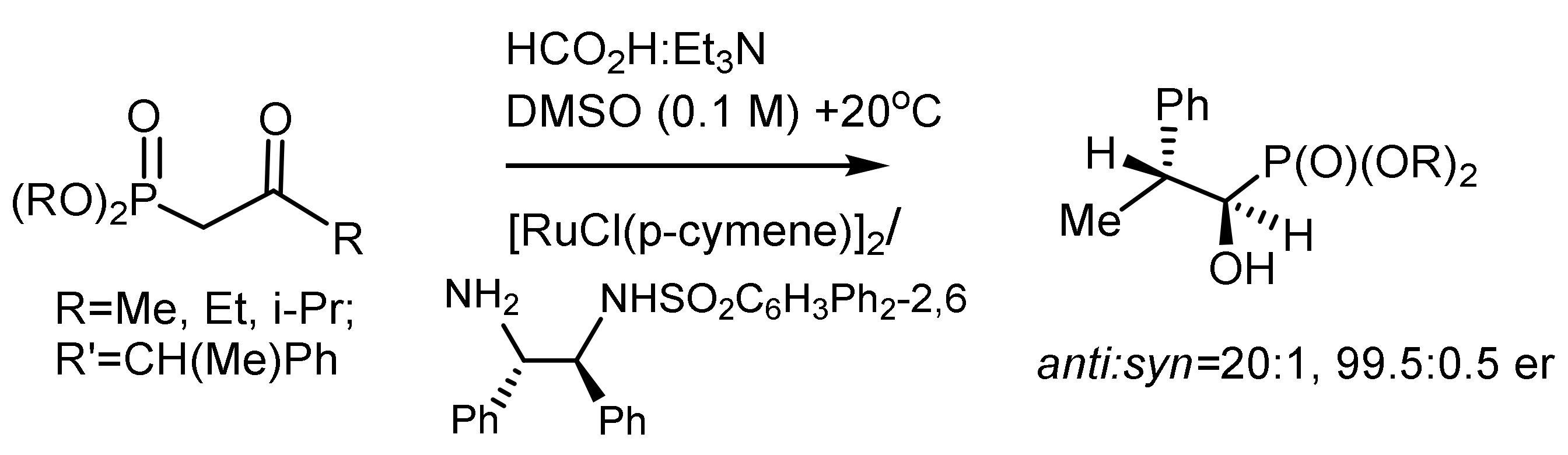
Figure 103.
Reduction of 3-aryl-4-phosphonobutenoates by PMHS in the presence of (S)-Segphos/Cu(OAc) complex.
Figure 103.
Reduction of 3-aryl-4-phosphonobutenoates by PMHS in the presence of (S)-Segphos/Cu(OAc) complex.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kolodiazhna, A.O.; Kolodiazhnyi, O.I. Catalytic Asymmetric Synthesis of C-Chiral Phosphonates. Symmetry 2022, 14, 1758. https://doi.org/10.3390/sym14091758
AMA Style
Kolodiazhna AO, Kolodiazhnyi OI. Catalytic Asymmetric Synthesis of C-Chiral Phosphonates. Symmetry. 2022; 14(9):1758. https://doi.org/10.3390/sym14091758
Chicago/Turabian StyleKolodiazhna, Anastasy O., and Oleg I. Kolodiazhnyi. 2022. "Catalytic Asymmetric Synthesis of C-Chiral Phosphonates" Symmetry 14, no. 9: 1758. https://doi.org/10.3390/sym14091758
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.