
On Analogies in Proton-Transfers for Pyrimidine Bases in the Gas Phase (Apolar Environment)—Cytosine Versus Isocytosine
Department of Chemistry, Warsaw University of Life Sciences (SGGW), ul. Nowoursynowska 159c, 02-776 Warszawa, Poland
Symmetry 2023, 15(2), 342; https://doi.org/10.3390/sym15020342
Submission received: 3 January 2023
/
Revised: 20 January 2023
/
Accepted: 22 January 2023
/
Published: 26 January 2023
(This article belongs to the Special Issue Symmetry in Acid-Base Chemistry II)
Abstract
:Inter- and intra-molecular proton-transfers between functional groups in nucleobases play a principal role in their interactions (pairing) in nucleic acids. Although prototropic rearrangements (intramolecular proton-transfers) for neutral pyrimidine bases are well documented, they have not always been considered for their protonated and deprotonated forms. The complete isomeric mixtures in acid-base equilibria and in acidity–basicity parameters have not yet been examined. Taking into account the lack of literature and data, research into the question of prototropy for the ionic (protonated and deprotonated) forms has been undertaken in this work. For the purposes of this investigation, two isomeric pyrimidine bases (C—cytosine and iC—isocytosine) were chosen. They exhibit analogous (symmetrical) general acid-base equilibria (intermolecular proton-transfers). Being similar polyfunctional tautomeric systems, C and iC possess two labile protons and five conjugated tautomeric sites. However, positions of exo groups are different. Consequently, structural conversions such as prototropy, rotational, and geometrical isomerism of exo groups (=O/−OH and =NH/−NH2) and their intramolecular interactions with endo groups (=N−/>NH) possible in neutral C and iC and in their ionic forms lead to some differences in compositions of isomeric mixtures. By application of quantum–chemical methods to the isolated (in vacuo) species, stability of all possible neutral and ionic isomers has been examined and the candidate isomers selected. The complete isomeric mixtures have been considered for the first time for di-deprotonated, mono-deprotonated, mono-protonated, and di-protonated forms. Protonation–deprotonation reactions have been analyzed in the gas phase that models non-polar environment. The gas-phase microscopic (kinetic) and macroscopic (thermodynamic) acidity–basicity parameters have been estimated for each step of acid-base equilibria. When proceeding from di-anion to di-cation in four steps of protonation–deprotonation reaction, the macroscopic proton affinities for C and iC differ by less than 10 kcal mol−1. Their DFT-calculated values are as follows: 451 and 457, 340 and 339, 228 and 224, and 100 and 104 kcal mol−1, respectively. Differences between the microscopic proton affinities for analogous isomers of C and iC seem to be larger for the exo than endo groups. Owing to variations of relative stabilities for neutral and ionic isomers, in some cases they are even larger than 10 kcal mol−1.
1. Introduction
The polyfunctional pyrimidine base cytosine (C), a structural building part of nucleic acids (RNA and DNA), and isocytosine (iC), structurally identical with the pyrimidine fragment of the nucleobase guanidine [1], are constitutional isomers that display some structural analogies as well as some structural differences. First of all, they differ by positions of the exo groups and by positions of the labile protons in the canonical forms, favored in aqueous environment (Figure 1). These differences affect acidity and basicity of functional groups that influence their interactions (pairing) in nucleic acids.
On the other hand, the pyrimidine bases C and iC contain the same number of exo and endo functional groups, labile protons, and conjugated tautomeric sites. Consequently, they exhibit prototropic conversions such as amide-iminol and amine-imine, and additionally keto-enol in iC and imine-enamine in C [2,3,4,5]. Two exo groups at 2- and 4-positions (=O/−OH and =NH/−NH2) possess different heteroatoms; however, they demonstrate similar electronic properties (electron-accepting/electron-donating). Two endo groups at 1- and 3-positions (=N−/>NH) are of the same type and show analogous electronic effects (electron-accepting/electron-donating). The four functional groups in C and iC are conjugated with one endo =CH− group at 5-position. These structural similarities dictate acid-base equilibria for pyrimidine bases.
The prototropic conversions, always accompanied by π-electron migrations [4], lead to nine possible tautomeric forms for neutral C and iC (Table 1) that possess analogous positions of labile protons [6,7]. Taking into account conformational isomerism of the exo −OH group and configurational isomerism of the exo =NH− group, twenty-one isomers are possible for C and iC. Quantum-chemical calculations carried out for all isomers of neutral C and iC [6,7] confirmed very weak stability of the neutral CH tautomers that possess one labile proton at the endo C5 atom (isomers 35, 57, and 58 in Table 1). Hence, only six tautomers with two labile protons at N and/or O atoms can be treated as candidate structures for the neutral bases and can be considered in the tautomeric mixtures of C and iC. In the literature, one can also find some reports on zwitterionic forms of pyrimidine bases; however, zwitterions are exceptionally rare isomers in vacuo.
Although prototropy has been extensively studied to explain the structural rearrangements for neutral pyrimidine bases, cytosine and isocytosine [6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27], this phenomenon has frequently been omitted for protonated and deprotonated forms. Investigating gas-phase acidity–basicity, and the favored sites of acid-base reactions, very often the microscopic acid-base parameters have been theoretically estimated for potential sites in the favored isomers [28,29,30,31,32]. To our knowledge, there is only a few reports for protonated pyrimidine bases, published by Tureček, Salapin, Maître, and their co-workers [33,34]. They investigated experimentally and theoretically products of protonation reaction, and suggested that the pyrimidine base cytosine can change isomeric preferences upon protonation.
Taking into account these interesting documents and the lack of literature data for the complete acid base-equilibria of the chosen pyrimidine bases (Figure 2), tautomeric conversions have been re-examined not only for the neutral molecules (AH2B), but also for the deprotonated {(ABH)− and (AB)2−} and protonated forms {(AH2BH)+ and (AH2BH2)2+} in vacuo (non-polar environment). In this way, the gas-phase acidity–basicity parameters that correspond to particular sites in individual isomers as well as to tautomeric mixtures could be estimated. Some analogies and differences in proton-transfer reactions could also be discussed.
All possible ionic isomers of C and iC have been analyzed in this work. For investigation, prototropic transformations as well as isomeric conversions of the exo groups, i.e., conformational isomerism about C−OH and configurational isomerism about C=NH, have been considered. The ionic tautomers–rotamers have been derived as products of the four steps protonation–deprotonation reactions (Figure 2) of the isomeric mixtures consisting of twenty one isomers of neutral C and iC. Using quantum–chemical methods described below in Methodology section, the structures of the complete isomeric ionic forms have been determined, their relative stabilities analyzed, and their contributions in the isomeric mixtures of (ABH)−, (AB)2−, (AH2BH)+, and (AH2BH2)2+ calculated. Acidity–basicity of functional groups has been analyzed for selected candidate isomers, and acidity–basicity parameters estimated for the potential protonation–deprotonation sites. For isomeric mixtures, acidity–basicity parameters of the four steps of protonation–deprotonation reactions given in Figure 2 have also been examined. Details of their prediction are included below in Methodology section.
The differences in the acidity–basicity parameters of the functional groups affect the intramolecular proton-transfers (prototropic conversions) in neutral and ionic bases. They also dictate the intramolecular interactions of the exo and endo groups, and consequently, the isomeric preferences. In the other words, they influence the tautomeric equilibria and the composition of the tautomeric (isomeric) mixtures. They also play an important role in the intermolecular interactions (pairing) of isomeric bases in nucleic acids as well as in their intermolecular proton-transfers leading to possible nucleic-acid mutations. The acidity–basicity parameters for the selected isomers of neutral and ionic C and iC can also be useful in explanations of their structural trends in molecular biology and in analyses of mechanisms of their biotransformations and various biochemical processes in biochemistry, whereas those for their isomeric mixtures can help to examine various structure-activity relations in physical organic, acid-base, and medicinal chemistry.
2. Methodology
Starting from the complete tautomeric mixtures of neutral pyrimidine bases, all possible isomeric mono- and di-deprotonated forms as well as all possible isomeric mono- and di-protonated species have been considered and quantum chemical calculations carried out for them in vacuo. First, to save computation time and to select the candidate isomers, their stabilities in the ground state have been analyzed at the semi-empirical level of theory using the Austin Model 1 method (AM1 [35]). The AM1 method has already been used in the literature for tautomeric biomolecules, including pyrimidine and purine bases [36]. For ionic tautomers–rotamers of C and iC with all positive frequencies, the energy differences (ΔE) have been calculated between the considered and preferred structures, separately for mono-anions (AHB)−, di-anions (AB)2−, mono-cations (AH2BH)+ and di-cations (AH2BH2)2+. To select the potential isomers for ionic species, the Mezey et al. rule [37,38] has been employed. According to this rule, the lowest limit of significant ΔE difference between favored and rare isomers should not be higher than 10 kcal mol−1. Taking this rule into account, all isomers deprotonated at the endo =CH− and exo =NH sites, as well as all isomers protonated at the C atoms in endo =CH−, >C=O, and >C-OH, and those protonated at the N and O atoms in endo >NH, exo −NH2, and exo −OH have been neglected. Their AM1-calculated ΔEs are considerably higher than 10 kcal mol−1. For the ionic tautomeric mixtures, we selected only the candidate isomers, i.e., those deprotonated at endo >NH, exo −NH2, and exo −OH, and also those protonated at exo and endo N-imino, and also at exo O-carbonyl.
Next, to detect the major, minor, and rare isomers, calculations have been carried out without any geometry restrictions for selected structures at the higher level of theory. We employed the density functional theory (DFT) method [39] with the Becke three-parameter hybrid exchange functional with the non-local correlation functional of Lee, Yang, and Parr (B3LYP) [40,41], and the 6-311+G(d,p) basis set with the diffuse and polarization functions [42]. For calculations, the Gaussian 03 programs have been applied [43]. The DFT level of theory has been chosen by Rodgers and his co-workers for an analysis of protonated forms of nucleic acid fragments [44], as well as by Koppel, Leito, Maksić, and their co-workers for estimation of acidity–basicity parameters for nitrogen bases in the gas phase [45,46]. It has also been selected by us for investigations of other tautomeric nucleobases, including adenine, xanthine, caffeine, and purine itself [47,48,49,50]. Because this level of theory has been employed previously for all tautomers–rotamers of neutral C and iC (Table 1) [6,7], and here for ionic forms, the complete acid-base equilibria could be analyzed. The use of the same level of theory for different neutral and ionic forms of tautomeric systems, help to understand not only structural isomeric phenomena, but also thermochemistry of various proton-transfer processes in which isomerism plays a pivotal role.
Vibrational frequencies and thermochemical parameters such as the electronic energy (E), enthalpy (H = E + pV), entropy (S), and Gibbs free energy called here Gibbs energy (G = H − TS for T = 298.15 K) have been calculated for all selected ionic isomers at the same DFT level as that used for their geometry optimization. Perusal of the DFT results indicates that the optimized structures possess all real frequencies and correspond to the energy minima. The relative quantities (ΔE, ΔH, TΔS, and ΔG) have been calculated separately for ionic isomers of mono-anions (ABH)−, di-anions (AB)2−, mono-cations (AH2BH)+, and di-cations (AH2BH2)2+. The composition of the neutral and ionic isomeric mixtures of C and iC have be estimated on the basis of ΔGi calculated for individual neutral and ionic isomers using equation (1), where xi is the isomer mole-fraction [51,52,53,54]. Note that ΔGi can be different for neutral and ionic isomers, and thus the calculated xi cannot be the same for ionic and neutral species. Equation (1) is, therefore, a general relation between xi and ΔGi for neutral or ionic isomers. However, it should be understood as five different relations: xin vs. ΔGin, xida vs. ΔGida, xima vs. ΔGima, ximc vs. ΔGimc, xidc vs. ΔGidc, where the abbreviations n, da, ma, mc, and dc being in superscript correspond to different pyrimidine base forms: neutral, di-anionic, mono-anionic, mono-cationic, and di-cationic, respectively.
xi≈ {exp(−ΔGi/RT)}/{Σ1n [exp(−ΔGi/RT)]}
Gas-phase acidity–basicity has been calculated for potential sites of protonation–deprotonation in individual isomers as well as for the tautomeric mixtures. To distinguish these quantities, two different terms have been employed, microscopic (kinetic) and macroscopic (thermodynamic) acidity–basicity, respectively. For their estimations, the following procedures have been applied. For each step of the reversible deprotonation-protonation reactions (Figure 2), microscopic gas-phase basicity parameters such as proton affinity (PAmicro) or gas-phase basicity (GBmicro) can be calculated using Equations (2) or (3), respectively [55,56].
PAmicro = Hi(base form) + H(H+) − Hi(acid form)
GBmicro = Gi(base form) + G(H+) − Gi(acid form)
The macroscopic basicity parameters (PAmacro or GBmacro) can be found on the basis of Equations (4) or (5), respectively. They correspond to the step of acid-base reactions given in Figure 2, in which the isomeric mixtures of the corresponding acid and base forms, and also their isomer mole-fractions xi have been taken into account. In Equations (2)−(5), H(H+) = 1.48 kcal mol−1 and G(H+) = −6.28 kcal mol−1 [57,58]. Hi and Gi refer to the enthalpy and Gibbs energy for 298 K, respectively, estimated for the corresponding acid and base forms of the isomer i.
PAmacro = Σ1nxi(base form)Hi(base form) + H(H+) − Σ1nxi(acid form)Hi(acid form)
GBmacro = Σ1nxi(base form)Gi(base form) + G(H+) − Σ1nxi(base form)Gi(base form)
According to the Brönsted and Lowry theory applied to the gas phase, basicity parameters (PA = ΔHbase or GB = ΔGbase) for the neutral or ionic base form are equal to the corresponding acidity parameters for the conjugate-acid form in the enthalpy (ΔHacid = ΔHbase) or Gibbs energy (ΔGacid = ΔGbase) scales, respectively [55]. Hence, the calculated microscopic PAs or GBs refer to both basicity and acidity of the conjugate acid-base pair of individual isomers, whereas the calculated macroscopic PAs or GBs correspond to both basicity and acidity of the isomeric-mixture of the base and acid forms.
3. Results and Discussion
3.1. Tautomers–rotamers Selected for Neutral Bases
Among twenty-one isomers possible for nine tautomers of neutral C and iC [6,7], fourteen structures (Figure 3) possessing the labile protons at N and O atoms have been selected in this work. They form the isomeric mixture corresponding to AH2B in acid-base equilibria (Figure 2). For tautomers possessing exo −OH and/or exo =NH, two extreme conformational and/or configurational isomers (a and b) have been considered, respectively. The CH-isomers containing one labile proton at the endo C5 atom (see in Table 1) have been omitted, owing to their high relative Gibbs energies (ΔG > 10 kcal mol−1) estimated in the gas phase at the DFT(B3LYP)/6-311+G(d,p) level [6,7]. Taking into account the well-recognized push–pull effects in the tautomeric amide-iminol {O=C(R)−NH− ⇄ HO−C(R)=N−) and amidine (−N=C(R)−NH2 ⇄ −HN−C(R)=NH} parts, the candidate site of protonation and deprotonation can be easily indicated without calculations. In the protonation reaction, the sp2 hybridized =O and/or =N− can gain a proton, whereas >NH, −NH2, and/or −OH containing the sp3 hybridized heteroatoms can lose the labile proton. In Figure 3, the selected sites of protonation and deprotonation in the fourteen selected isomer of C and iC have been indicated by red and blue arrows, respectively.
The values of ΔG included in Figure 3 show variations in isomeric stability resulting not only from prototropy, conformational, and configurational isomerism, but also from possible intramolecular interactions (favorable or unfavorable) between the exo and endo groups. Larger variations are caused by prototropy (from 0 to 33 kcal mol−1) than by conformational and configurational isomerism (from 1 to 9 kcal mol−1). The smallest effects take place for these a and b pairs, for which the same type of interactions are possible (favorable or unfavorable in both isomers). The highest effects occur when a favorable interaction in one isomer changes into an unfavorable repulsion in the other one and vice versa.
Additional perusal of ΔGs estimated for all selected isomers shows some analogy in the isomeric preferences for cytosine and isocytosine. Only six tautomers–rotamers of neutral cytosine (C-13a, C-13b, C-18, C-38, C-78a, and C-78b) and only six tautomers–rotamers of neutral isocytosine (iC-13a, iC-13b, iC-17, iC-37, iC-78a, and iC-78b) possess ΔG ≤ 10 kcal mol−1. This indicates that the isomeric mixture of neutral C and iC can consist mainly of these six isomers (two imino-oxo, two amino-oxo, and two amino-hydroxy forms). Taking the Mezey et al. rule [37,38] into account, the other isomers of neutral C and iC can be neglected in the isomeric mixtures in the gas phase (or non-polar environment). The canonical C-18 isomer is the major form for cytosine, and the other ones can be considered as minor or rare forms. In the case of isocytosine, the DFT-calculated canonical iC-37 isomer has slightly lower Gibbs energy than iC-78a. However, both isomers can be treated as major forms. The other isomers are minor or rare forms.
It should be mentioned here that experimental methods do not give the complete picture on relative stabilities of all possible isomers in tautomeric mixtures. The reasons are as follows: (i) prototropic conversions are frequently very fast and reversible processes, (ii) tautomerism is often very sensitive to the environment, (iii) experimental methods have various limits of identification, (iv) only signals of major isomers (1–100%) of significant intensities can be well detected, (v) signals of minor isomers (0.01–1%) can be too small and difficult to distinguish from the background, and (vi) rare isomers (<0.01 %) are usually undetectable. Hence, the number of identified isomers can be different using different methods such as ultraviolet (UV), infrared (IR), matrix isolation IR, IR laser in helium nanodroplets, Raman, microwave (MV), nuclear magnetic resonance (NMR), mass spectrometry (MS), core-level X-ray photoemission, resonant multi-photon ionization (REMPI), near-edge X-ray absorption, etc.
For example, depending on the method employed for gaseous cytosine, one (C-18), two (C-18 and C-78) or three (C-18, C-78, and C-13) tautomers have been identified [8,10,16,17,18]. Five isomers of gaseous cytosine (C-18, two conformational isomers a and b of C-78, and two configurational isomers a and b of C-13) have only been found by Alonso et al. [9], who applied a laser ablation molecular beam FT-MW spectroscopy. For crystal cytosine, only the canonical isomer C-18 has been detected [23,24], whereas for hydrated cytosine, the isomer C-38 has been additionally observed [15,19,20,21,22].
In the case of neutral isocytosinie, using various spectroscopic techniques different tautomers have also been identified when proceeding from the gas phase to aqueous solution, and next to the solid state [7,11,12,13,14,15,25,26,27]. The tautomer iC-78 predominates in vacuo or non-polar environment (e.g., lipids), whereas iC-37 is favored in aqueous solution. Interestingly, two tautomeric amino-oxo forms (iC-17 and iC-37) have been found in the crystal state [25,26].
3.2. Isomerism in Deprotonated Bases
Some interesting structures for mono- and di-anionic pyrimidine bases that play an important role in chemistry of organic calixarenes have been reported in the literature by Lippert, Sanz Miguel, Freisinger, and their co-workers [59,60]. Acting as N-ligands, like aliphatic di-amines (e.g., ethylenediamine) and aromatic di-imines (e.g., 2,2′-bipyridine), they can form various multinuclear complexes, even structures with mixed-metal (Ptx, Pdy), mixed-nucleobase (uracil, cytosine), and mixed-amine (ethylenediamine, 2,2′-bipyridine).
Note that the di-deprotonated forms (AB)2− of the pyrimidine bases C and iC have no labile proton. Thus, no prototropic conversion takes place in them. Only configurational isomerism is possible for the exo =NH group. The geometrical isomers a and b of di-deprotonated cytosine (da-C) and isocytosine (da-iC), being a consequence of configurational isomerism about the C=N bond, are given in Figure 4. Their ΔG values calculated at the DFT level are also included in this figure. We can see that the ΔG values of the configurational isomers are not very large. They are considerably lower than 10 kcal mol−1. This means that the two di-anionic isomers a and b can be considered in the isomeric mixtures of da-C-0 and da-iC-0 with different preference of the isomer a and b, respectively. To our knowledge, there is no documented example in the literature for the pyrimidine bases on their di-anionic forms in the gas-phase, and no comparison can be made.
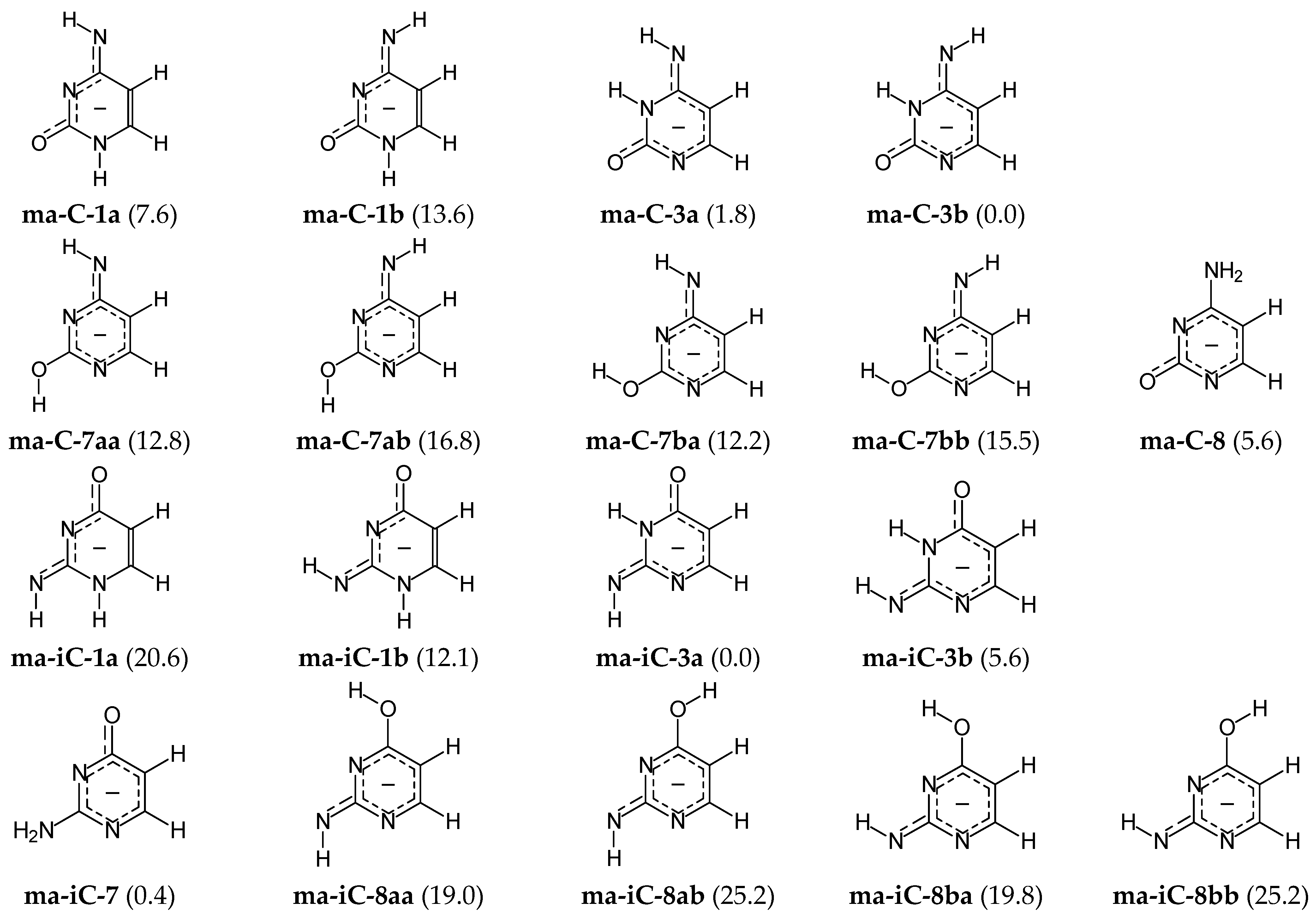
A different situation occurs for the mono-deprotonated pyrimidine bases, mono-anions (AHB)− of C and iC. They contain only one labile proton that can move between five conjugated sites. The number of tautomers for the C and iC mono-anions (ma-C and ma-iC, respectively) is smaller than that for neutrals because of lower number of the labile protons, and also, because two or more neutral isomers can be deprotonated to the same common mono-anion. Consequently, tautomeric conversions in mono-anions of C and iC lead to five possible prototropic tautomers. Like for neutral C and iC, the CH tautomers can be omitted in the isomeric mixtures of mono-anions. Their ΔE values are between 15 and 20 kcal mol−1 at the AM1 level. Hence, only four tautomers with the labile proton at N or O, i.e., at 1-, 3-, 7-, or 8-position, have been examined at the DFT level. Their conformational and configurational isomers, and also their ΔGs are included in Figure 5.
For mono-anionic isomers of cytosine (ma-C), the variations of ΔGs are not very large (0 < ΔG < 17 kcal mol−1), whereas those for isocytosine (ma-iC) are considerably larger (0 < ΔG < 26 kcal mol−1). The difference in stability of the anionic forms involves different number of potential isomers that significantly affect properties of the isomeric mixtures ma-C and ma-iC. Four isomers of mono-anionic cytosine (ma-C-1a, ma-C-3a, ma-C-3b, and ma-C-8) possess ΔGs lower than 10 kcal mol−1, whereas only three isomers (ma-iC-3a, ma-iC-3b, and ma-iC-7) significantly participate in the isomeric mixture of mono-deprotonated isocytosine. The ΔG value (12.1 kcal mol−1) of ma-iC-1b (exhibiting some analogous favorable intramolecular interactions like ma-C-1a) seems to be too high and cannot influence significantly acid-base properties of isocytosine. However, taking into account the analogy with cytosine mono-anion, ma-iC-1b can be considered for the isomeric mixture ma-iC in acid-base equilibria.
DFT calculations show additionally that the favored mono-anionic isomer ma-C-3b cannot be directly formed from the preferred neutral canonical form C-18. Direct deprotonation reaction of C-18 at N1H or N8H leads to three possible isomers ma-C-8, ma-C-1a, and ma-C-1b. All of them possess considerably higher Gibbs energies than the favored one (by more than 5 kcal mol−1) and can be considered rather as rare forms. The change of isomeric preferences shows that prototropy occurs in mono-anionic C. After deprotonation of the canonical isomer C-18 at N1H to the isomer ma-C-8, the labile proton at N8H can move to N3 and the isomer ma-C-3b of the lowest Gibbs energy can be formed. Note that Gibbs energy of its geometrical isomer ma-C-3a is close to that of the favored one (ΔG 1.8 kcal mol−1), indicating that the two geometrical isomers are the major forms in the isomeric mixture of mono-anionic cytosine. The major isomers (ma-C-3a and ma-C-3b) can also be formed from the rare neutral isomer C-38 in deprotonation reaction at N8H, as well as from the minor isomers C-13a or C-13b by deprotonation at N1H.
In the literature, usually, the N1 atom has been proposed as the favored site of cytosine deprotonation in the canonical neutral tautomer C-18, and the isomer ma-C-8 considered as the favored form in the gas phase [61]. Prototropy in the deprotonated form has not been taken into account. Hence, this is not in agreement with our DFT investigations on the complete isomeric mixture of mono-deprotonated cytosine for which ma-C-3 tautomer is favored in the gas phase. On the other hand, some mono-anionic isomers of cytosine (ma-C-8, ma-C-1b, ma-C-3a, and ma-C-3b) have been selected for interaction with the metal-cation Ba2+ and investigated by both theory (DFT) and experiment (IRMPD) [62]. The lowest Gibbs energy in the gas phase has been found for the ma-C-8−Ba2+ adduct with metal-cation chelated by N1 and O7 atoms. However, comparison of the calculated and experimental spectra in the 1550-1630 cm−1 spectral region indicated that the ma-C-8−Ba2+ adduct of higher Gibbs energy with metal-cation chelated by N3 and O7 atoms cannot be neglected in the isomeric mixture in the gas phase. This result is very interesting for metal-cation adducts, but not for proton-transfer chemistry. It is well recognized that metal-cation can change the isomeric preference in tautomeric system and the favored isomer in adduct can be different than that in isolated tautomeric system. Lack of data in the literature for mono-anionic tautomers–rotamers of cytosine in vacuo makes any comparison of our DFT results impossible.
A different situation takes place in the case of isocytosine. From our DFT calculations result, the two major mono-anionic isomers ma-iC-3a and ma-iC-7 (ΔG 0.4 kcal mol−1) can be formed from the two major neutral isomers iC-37 and iC-78a (or partially from iC-78b) in deprotonation at N7H and O8H, respectively. On the other hand, deprotonation of the major neutral isomer iC-37 at N7 leads to the rare mono-anionic isomer ma-iC-3b. This isomer can also be formed from the rare neutral isomers iC-13b and iC-37 in deprotonation at N1 and N7, respectively.
In our previous work on isocytosine-charged radicals [63], we tested deprotonation reaction of three neutral isocytosine isomers (iC-17, iC-37, and iC-78a). Considering seven among nine mono-anionic tautomers–rotamers studied here, we found analogous stability-order for them at the DFT level. However, G4 calculations performed for the two major mono-anionic isomers showed their reverse stabilities. The isomer ma-iC-7 has slightly lower Gibbs energy than ma-iC-3a at the G4 level. This indicates that the two isomers ma-iC-3a and ma-iC-7 participate in the isomeric mixture in similar amount. In an aqueous solution, only ma-iC-7 seems to be favored, and ma-iC-3a can be treated as a rare isomer. The solvation effect has been investigated by 1H, 13C, and 15N NMR experiments, and also by PCM calculations [27,63].
3.3. Isomerism in Protonated Bases
Owing to n-π conjugation in the tautomeric moieties (push-pull effect), the sp2-hybridized heteroatoms (=N− and =O) are candidate protonation-sites in the neutral pyrimidine bases C and iC (Figure 3). Mono-protonation of the potential sites in fourteen neutral isomers lead to the formation of nine mono-cationic isomers (Figure 6). They contain three labile protons and the same number of conjugated tautomeric sites—five, such as their neutral forms. Isomers with one labile proton at C5 have been neglected in calculations. Additionally, isomers protonated at the sp3-hybridized endo and exo amino N atoms, exo hydroxy O atoms, and also sp2-hybridized endo C atoms, even in the favored neutral forms have also been omitted. Tureček and co-workers [33] investigating mono-protonation at N8H2, C5H, and C6H in C-18 and C-78 proved by quantum–chemical calculations performed at different levels of theory (B3LYP, MP2, B3-MP2, CCSD(T) with less or more extended basis sets) that the N-sp3 and C-sp2 atoms are the last ones for the proton gain. They exhibit very weak basicities. Their protonated structures are insignificant in the isomeric mixture of protonated cytosine. Analogous conclusion has been derived by Lee and co-workers [32] on the basis of DFT calculations of basicity parameters for N-sp3 and O-sp3 in C-78a, and C-78b and N-sp3 in C-18, C-13, C-78a, and C-78b.
The ΔG values calculated for selected nine mono-cationic isomers (mc-C and mc-iC) vary from 0 to 33 kcal mol−1 for cytosine and from 0 to 24 kcal mol−1 for isocytosine. This suggests that the position-change of exo groups in the isomeric C and iC influences the relative stability of mono-cationic isomers. However, the same number of mono-cationic isomers with ΔG lower than 10 kcal mol−1 can dictate acid-base properties of pyrimidine bases, four of mc-C (mc-C-138, mc-C-178a, mc-C-178b, and mc-C-378a) and four of mc-iC (mc-iC-137, mc-iC-178a, mc-iC-178b, and mc-iC-378b). The three isomers selected for mono-cationic cytosine, major mc-C-138, major mc-C-178b, and rare mc-C-178a can be formed from the favored canonical isomer C-18 in protonation reaction at N3, O7b, and O7a, respectively, whereas the fourth rare isomer mc-C-378a is possible when the major isomer C-78a or rare isomer C-38 are protonated at N3 or O7a, respectively. Analogously for isocytosine, the major isomer mc-iC-178a can be directly formed from the major neutral isomer iC-78a in the protonation reaction at N1. Mono-protonation of the canonical form iC-37 at N1 or O8b leads to the rare isomers mc-iC-137 and mc-iC-378b, respectively.
Note that Tureček and co-workers [33], investigating theoretically the protonated forms of four candidate isomers of neutral cytosine (C-13b, C-18, C-78a, and C-78b), considered three potential mono-cationic isomers (mc-C-138, mc-C-178b, and mc-C-378a) and examined their relative stabilities using various levels of theory (B3LYP, MP2, B3-MP2, and CCSD(T) with less or more extended basis sets). This choice of isomers was sufficient to select the two major isomers mc-C-138 and mc-C-178b for protonated cytosine with not very different absolute relative energies (ΔE 0–1.6 kcal mol−1), and to subsequently study their dissociations in the gas phase by both theory and experiment using different theoretical methods and various experimental techniques of mass spectrometry. Cytosine mono-cations were generated by chemical ionization and fast atom bombardment, and dissociations of mono-cations examined by tandem mass spectrometry. In a completely theoretical work by employing different levels of the MP2 and MP4 methods, Leszczynski and co-workers [30] also examined stability of some mono-cations. They considered three neutral cytosine isomers: the canonical form (C-18) and two other minor isomers (C-13b and C-78a), and products of their protonation at heteroatoms. For the isomeric mixture of protonated cytosine, they also proposed a coexistence of the two major isomers mc-C-138 and mc-C-178b (ΔG 0.1–1.2 kcal mol−1) with slight preference of mc-C-178b. The close stability of the two major mono-cationic isomers mc-C-138 and mc-C-178b in the gas phase have been confirmed by Salapin and co-workers [34], who used both theoretical (DFT) and experimental (IRMPD) methods. The isomers mc-C-138 and mc-C-178a have been identified in the 1000–2000 cm−1 spectral region.
In the case of isocytosine, the relative stabilities of the three potential mono-cationic isomers mc-iC-137, mc-iC-178a, and mc-iC-378b considered as protonated forms of selected three neutral isomers iC-17, iC-37, and iC-78a have been previously studied in our laboratory at the DFT level [7] and analogous stability-order has been reported as that found here. G4-calculations [63], performed for the two isomers mc-iC-137 and mc-iC-178a, confirmed the DFT-calculated stability-order and showed without any doubt that mc-iC-178a has the lowest Gibbs energy for protonated isocytosine in the gas phase. However, the mono-cationic isomer mc-iC-137 predominates in aqueous solution as confirmed by 1H, 13C, and 15N NMR experiments and by PCM calculations [27,63].
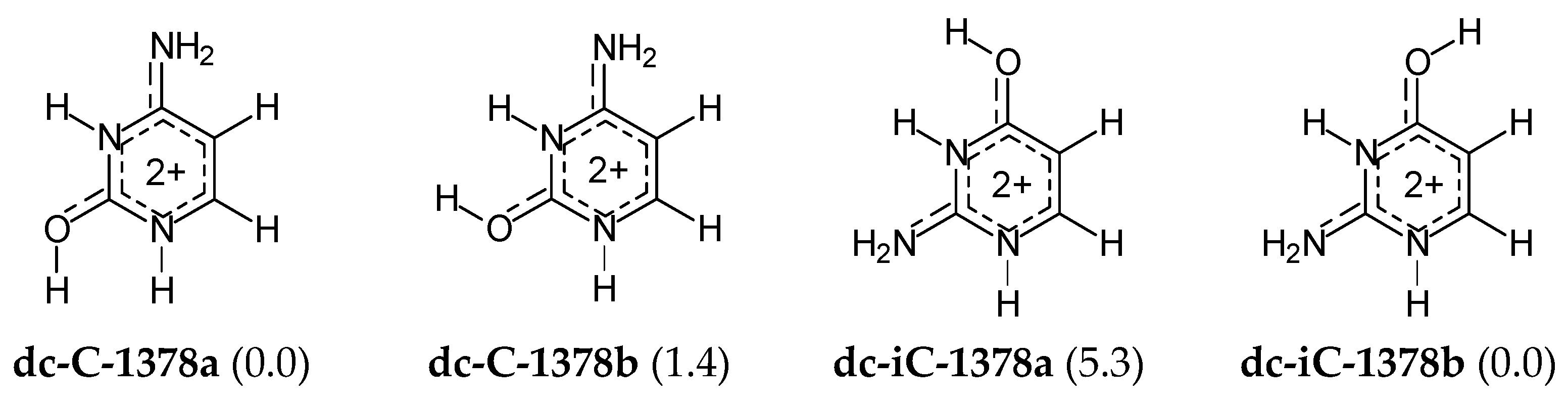
For di-protonated forms of cytosine and isocytosine, the isomeric mixtures are very simple, like for their di-deprotonated species. Neglecting the CH tautomers with ΔG > 10 kcal mol−1, only two geometrical isomers for each base (Figure 7) have been considered in DFT-calculations for the isomeric mixtures. Their Gibbs energies do not differ very much, and thus both isomers contribute significantly to acid-base properties of the isomeric mixture of di-cationic cytosine (dc-C) and di-cationic isocytosine (dc-iC). There is no literature report for di-cationic forms of cytosine and isocytosine in the gas phase so no comparison can be made.
3.4. Microscopic Acidity–Basicity Parameters
Gas-phase acidity–basicity parameters, PA and/or GB defined as the enthalpy and/or Gibbs energy changes (Equations (2) and (3), respectively) for reversible protonation–deprotonation reactions (given in Figure 2) can be analyzed from two view-points, i.e., on the microscopic (kinetic) and/or macroscopic (thermodynamic) scales. The microscopic acidity–basicity refers to equilibrium between the acid and base forms of selected isomer and helps to discuss the favored protonation and deprotonation sites in individual isomer. When tautomer–rotamer exists at sufficient long time to be isolated and analyzed in the gas phase, the microscopic acidity–basicity parameters can be even measured for the favored site(s) [32]. For this reason, the microscopic PA and GB are called kinetic parameters.
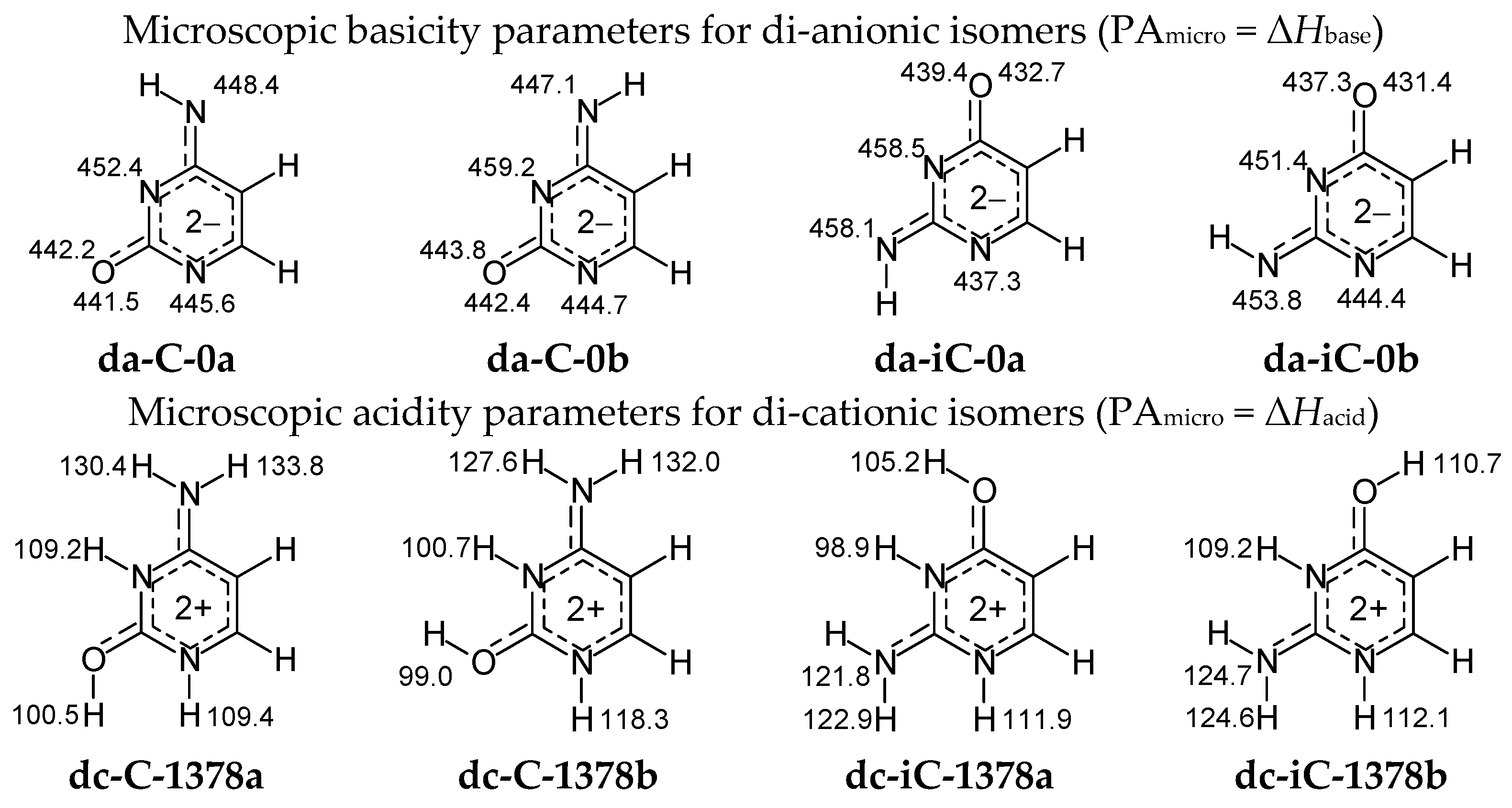
Figure 8 summarizes the microscopic acidity–basicity parameters in the enthalpy scale (PA = ΔHbase = ΔHacid) for protonation–deprotonation reactions estimated here according to Equation (2) at the DFT level for di-anionic and di-cationic isomeric forms of cytosine and isocytosine. Calculations have been carried out for each potential sites of protonation and deprotonation reaction, respectively, in the selected isomers of da-C, da-iC, dc-C, and dc-iC.
The highest basicity-parameter value for the di-anionic forms indicates the strongest basicity and the favored site of protonation: N3 in both isomers of da-C, whereas N3 in a and N7 in b of da-iC. Note that PAs of N3 and N7 in da-iC do not differ very much. The order of basicity parameters for heteroatoms in di-anionic cytosine and isocytosine isomers corresponds to the stability-order of the corresponding mono-anionic forms. On the other hand, the lowest acidity-parameter value for the di-cationic forms shows the strongest acidity as well as the preferred site of deprotonation: O7H in both isomers of dc-C, and N3H in both isomers of dc-iC. Note that acidities of N3H in dc-C-1378b and O8H in dc-iC-1378b are very close to those of O7H and N3H, respectively. The order of acidity parameters for NH and OH in di-cationic cytosine and isocytosine isomers refers to the stability-order of their mono-cationic isomers. In the literature, the is no document on gas-phase acidity–basicity parameters of di-anionic and di-cationic forms of cytosine and isocytosine and no comparison can be made.
For neutral C and iC isomers, microscopic basicity-parameters have been estimated for the sp2-hybridized heteroatoms =N− and =O, and acidity-parameters for–NH− and –OH with the sp3-hybridized heteroatoms. The DFT-calculated parameters in the enthalpy scale (PA = ΔHbase = ΔHacid) are included in Figure 9. Variations of their values clearly show a strong relation between acidity–basicity and intramolecular proton-transfer (prototropy). Generally, =N− seems to be more basic (higher PA) than =O for cytosine isomers. Some exception is the favored canonical form C-18, for which the O7b-carbonyl possesses basicity close to that of the imino N3. Analogous trend takes place for isocytosine with one exception of the rare isomer iC-17, for which =O8b seems to be more basic than =N3−. In deprotonation reaction, –OH displays higher acidity (lower PA) than −NH− for both C and iC. When –OH is absent in isomer, −NH− can be deprotonated, but the favored site depend on the structure of isomer.
The canonical forms play a very significant role in the isomeric mixtures of C and iC. In the cytosine isomer C-18, =N3– (or =O7b) seems to be the favored site of protonation, and –N1H– is preferentially deprotonated, whereas =N1– favorably gains a proton, and –N7bH– (or –N3H–) loses its labile proton in the isocytosine isomer iC-37. In the favored isomer iC-78a, =N1– is also more basic than =N3–, whereas –O8aH is more acidic then –N7H2. The more basic and more acidic sites participate preferentially in protonation and deprotonation reactions, respectively, for isolated isomers.
In the literature, one can find theoretical microscopic basicity parameters for the canonical forms and for some other isomers of pyrimidine bases [7,28,29,30,31,32,33,63]. For example, Tureček and co-workers [33] performing calculations at different levels of theory (B3LYP, MP2, B3-MP2, CCSD(T) with various basis sets) for mono-protonated cytosine showed that the microscopic PA values of the two potential protonation sites (N3 and O7b) in the canonical isomer C-18 strongly depend on the level of calculations. They are in the following ranges: 225–235 kcal mol−1 for N3 and 226–235 kcal mol−1 for O7b. However, at each level of theory, the difference between PAs of N3 and O7b in C-18 is very small (ca. 0–1 kcal mol−1) so that both sites can chelate the proton. Analogous PAs at the B3LYP/6-31+G(d) level (226.5 and 225.5 kcal mol−1 for N3 and O7b in C-18, respectively) and absolute PA difference (1 kca mol−1) have been reported by Lee and co-workers [32], and also earlier by Leszczynski and co-workers [30], who applied various MP2 and MP4 levels of theory to calculate PAs for N3 and O7b in C-18 (227.2–230.8 and 228.3–230.6 kcal mol−1, respectively). Investigating protonation of C5H, C6H and N7H2 groups in C-18, Tureček and co-workers [33] confirmed additionally by calculations of their PAs that these sites display exceptionally weak basicity at each level of applied theory. Similarly, Lee and co-workers [32], calculating PAs for selected sp2- and sp3-hybridized N and O in four cytosine isomers (C-18, C-13, C-78a, and C-78b) at the DFT level, confirmed higher PA values (stronger basicity) for =N– and =O than for >N– and –O–. In our previous work on prototropy in neutral isocytosine [7], we reported the DFT-calculated microscopic PAs for N1, N3, and O8 sites in three selected isomers of neutral cytosine (ic-17, iC- 37, and ic-78a). The PAs order is the same as that in Figure 9.
In the case of mono-deprotonation reaction of neutral cytosine, Lee and co-workers [32] calculated the microscopic acidity-parameters for the candidate sites in four major and minor neutral cytosine isomers (C-18, C-13, C-78a, and C-78b) at the B3LYP/6-31+G(d) level. For the canonical isomer C-18, the order of acidity-parameters (in enthalpy scale) is analogous to that studied here, and their values are as follows: 343.3 (N1H), 346.6 (N8bH) and 352.6 (N8aH) kcal mol−1, indicating also the highest acidity of the endo N1H group. For other isomers, analogous microscopic acidity–basicity parameters have been reported as those found here. The level of theory applied by Lee and co-workers give the values only slightly lower that that employed in this work.
For isocytosine, our earlier calculations performed at the G4 level [63] for favored isomer predict almost the same microscopic acidity: 340.6 kcal mol−1 for O8H in iC-78a as that at the DFT level (339.9 kcal mol−1). Difference in the G4- and DFT-calculated PA values is not larger than 1 kcal mol−1. An exception is the major isomer iC-37, for which N3H seems to lose the proton first than N7bH at the G4 level. However, differences in the microscopic acidities (338.4 and 339.6 kcal mol−1 at the G4 level, whereas 339.1 and 338.7 kcal mol−1 at the DFT level, respectively) are not very large (ca. 1 kcal mol−1). This suggests that the two sites in iC-37 can lose the proton with similar probability. It would be interesting to confirm this theoretical prediction in the future by appropriate experiment(s) performed for mono-anionic isocytosine in the gas phase.
3.5. Acid-Base Equilibria for Pyrimidine Bases
For simple bifunctional tautomeric compounds of the following general formula: HA−X=B ⇄ A=X−BH, containing only one labile proton that move between only two conjugated sites (A and B), like in acetaldehyde, formamide, formamidine, etc., acid-base equilibria are very simple. Protonation and deprotonation reactions, analogous to those for amphiprotic compounds, lead to one cation and one anion, respectively, common for the two prototropic tautomers. Only π-electrons and charge in ions are better delocalized than in neutral tautomers. According to Brønsted and Lowry theory, acid-base equilibria for simple bifunctional tautomeric compounds can be written as follows [2,4,64]: (AXB)− + 2H+ ⇄ (HA−X=B ⇄ A=X−BH) + H+ ⇄ (HAXBH)+. These equilibria have frequently been considered in explanation of the intramolecular proton-transfer (prototropy) mechanism [4].
However, for polyfunctional tautomeric systems, like pyrimidine bases, the situation is more complex. Containing two labile protons and five conjugated sites, neutral bases exist in the mixture of twenty-one tautomers–rotamers that can be protonated or deprotonated. Possibility of formation of different cations and anions in proton-transfer reactions explains the existence of various ionic isomers for the tautomeric molecules. Consequently, acid-base equilibria, generally written for pyrimidine bases in Figure 2, are more complex than for bifunctional derivatives. The abbreviations AH2B, (AB)2−, (AHB)−, (AH2BH)+, and (AH2BH2)2+ in Figure 2 refer to the corresponding tautomeric mixtures for neutral C or iC and for their ionic forms, respectively.
Figure 10 shows four-steps for acid-base equilibria of tautomeric cytosine and isocytosine: two steps of deprotonation and two steps of protonation. For simplicity, geometrical isomerism of the exo =NH group about the C=N double bond and rotational isomerism of the exo −OH group about the C−O single bond are not displayed. Exceptionally rare isomers are omitted in the isomeric mixtures of both the neutral and ionic forms. Only the structures, selected tautomers for DFT calculations, are included in this scheme, i.e., one structure for (AB)2−, four tautomers for (AHB)−, six tautomers for AH2B, four tautomers for (AH2BH)+, and one structure for (AH2BH2)2+.
3.6. Macroscopic Acidity–Basicity Parameters
The macroscopic acidity–basicity parameters correspond to deprotonation-protonation equilibria between the isomeric mixtures of the acid and base forms. They are determined mainly by the major and minor isomers. The rare forms have usually small effect on their values. When all isomeric conversions in the acid and base forms are in equilibrium with intermolecular proton-transfer during acidity–basicity measurements in the gas phase, the experimental acidity–basicity parameters for tautomeric systems refer to the isomeric mixtures of the acid and base forms, and may be compared with theoretically estimated macroscopic acidity–basicity parameters. For this reason, the macroscopic PA and GB for tautomeric systems are named thermodynamic parameters.
It has been shown above (Figure 3) that at least six tautomers–rotamers for neutral cytosine (C-13a, C-13b, C-18, C-38, C-78a, and C-78b) and at least six tautomers–rotamers for neutral isocytosine (iC-13a, iC-13b, iC-17, iC-37, iC-78a, and iC-78b) should contribute to acid-base properties of C and iC. For anionic bases, the following isomers are the most important: two geometrical isomers of di-deprotonated C (da-C-a and da-C-b) and iC (da-iC-a and da-iC-b) that exist in the di-anionic isomeric mixtures (Figure 4), and at least four isomers of mono-deprotonared C (ma-C-1a, ma-C-3a, ma-C-3b, and ma-C-8) and three isomers of mono-deprotonared iC (ma-iC-3a, ma-iC-3b, and ma-iC-7) that should be considered in the isomeric mixture of mono-anionic bases (Figure 5). The isomeric mixtures of mono-protonated cytosine (Figure 6) consist at least of four isomers (mc-C-138, mc-C-178a, mc-C-178b, and mc-C-378a). The same is true for mono-protonated isocytosine, four isomers can be considered (mc-iC-137, mc-iC-178a, mc-iC-178b, and mc-iC-378b). For di-protonated bases (Figure 7), two geometrical isomers should be taken in the isomeric mixtures of cytosine (da-C-a and da-C-b) and isocytosine (da-iC-a and da-iC-b). Selection of the potential neutral and ionic isomers has been based on the Mezey et al. rule [37,38].
The abbreviations of selected neutral and ionic isomers for cytosine and isocytosine and their DFT-calculated percentage contents (xi) estimated according to Equation (1) are given in Table 2. The exact structures of chosen isomers in this table are the same as those in Figure 4, Figure 5, Figure 6 and Figure 7. Differences in acid-base properties of functional groups and intramolecular interactions in tautomeric species dictate xi of individual isomers. Because functional groups in tautomeric (conjugated) systems upon protonation–deprotonation can change their acidity–basicity, and consequently intramolecular interactions between them can also change, the composition of tautomeric mixtures of neutral and ionic forms and contributions of potential isomers (xi) also vary. These variations can be clearly seen regarding the xi data in Table 2.
For di-anionic bases, the isomers da-C-0a and da-iC-0b dominate in the isomeric mixtures. Although exo groups have different positions, the favored di-anionic isomers seem to be stabilized by an analogous intramolecular interaction between exo NH and endo N3. Note that N3 possesses higher basicity than N1 in di-deprotonated forms (Figure 8).
The mono-anionic bases prefer the structures of the isomers ma-C-3b and ma-iC-3a. In this case, the two isomers seem to be preferentially stabilized by an intramolecular interaction between exo N and endo N3H. Additionally, intramolecular interactions between exo NH2 and both endo N1 and N3 seem to stabilize the isomer ma-iC-7 that significantly participates in the isomeric mixture of ma-iC as an additional major isomer. So favorable interactions for the ma-C-8 isomer are not possible due to repulsion between exo NH2 and endo C5H, which increases the Gibbs energy of this isomer and makes it less significant (rare form) in the mixture of ma-C isomers.
Mono-protonation of cytosine and isocytosine leads to the different tautomeric preferences, mc-C-138 and mc-iC-178a, respectively. The mc-iC-137 isomer—protonated form of the canonical tautomer iC-37—is rather a rare isomer. This difference in the favored tautomers can be a consequence of different internal effects. The protonated canonical isomer of cytosine (mc-C-138) is stabilized by the favorable intramolecular interactions between exo O and two acidic endo N1H and N3H. However, the protonated canonical isomer of isocytosine (mc-iC-137) is stabilized by the interaction between exo O and endo N3H and C5H (less acidic group). On the other hand, mc-C-178b—a second major tautomer in the isomeric mixture of protonated cytosine—is stabilized by an analogous intramolecular interaction between exo OH and endo N3 like the favored mc-iC-178a. The preferred isomers of di-cationic cytosine and isocytosine, dc-C-1378a and dc-iC-1378b are stabilized by analogous intramolecular interactions between exo O and endo N3H.
Taking into account the DFT-estimated percentage contents (xi) and thermochemical parameters (Hi and Gi) for all selected isomers, the macroscopic acidity–basicity parameters (PAmacro and GBmacro) have been estimated for the isomeric mixtures using Equations (4) and (5). Their values, summarized in Table 3, indicate some differences in thermodynamic acid-base properties of the two pyrimidine bases. However, the absolute differences between macroscopic PAs (or GBs) are not larger than 6 kcal mol−1.
In the literature, there is no article on theoretical estimation of the macroscopic (thermodynamic) acidity–basicity parameters for the complete isomeric mixtures of neutral and ionic cytosine. There are only some microscopic parameters estimated for particular acid-base sites in the canonical form of neutral cytosine and in its potential isomers (vide infra) [28,29,30,31,32,33]. Some exception is an estimation of Leszczynski and co-workers [30], who considering only three neutral isomers (C-18, C-13b, and C-78a) and two mono-cationic forms (mc-C-138 and mc-C-178b), proposed the average PA for cytosine (225.3–230.8 kcal mol−1, calculated at various MP2 and MP4 levels). Concerning the experimental literature-data, one can find acidity–basicity parameters only for neutral cytosine, measured in different laboratories by various kinetic and equilibrium methods. For mono-deprotonation reaction of neutral base AH2B ⇄ (AXB)− + H+, proton acidity parameters have been measured by Lee and co-workers [32] using two procedures, bracketing and extended Cooks kinetic methods, and the following PA values have been reported 342 ± 3 and 343 ± 3 kcal mol−1, respectively. The GB value of 335 ± 3 kcal mol−1 has been found by the bracketing method. Analogous PA (340 kcal mol−1) has been earlier derived for cytosine by Chen et al. [66] employing electron impact spectra and acid dissociation constants determined in DMSO. For mono-protonation reaction of neutral cytosine AH2B + H+ ⇄ (HAXBH)+, Hunter and Lias [65] in 1998 compiled the PAs and GBs data, earlier reported by other chemists, and proposed for cytosine the following values 227.0 and 219.4 kcal mol−1, respectively. Ten year later Lee and co-workers [32] investigated proton basicity of neutral cytosine and found PA 228 ± 3 and GB 220 ± 3 kcal mol−1 by the bracketing method, and PA 227 ± 3 kcal mol−1 by the extended Cooks kinetic method. The calculated here macroscopic acidity–basicity parameters for neutral cytosine are close to those experimentally determined.
To our knowledge, the literature experimental gas-phase acidity–basicity parameters for isocytosine have not yet been documented [55]. Preliminary prediction of proton acidity and proton basicity at the G4 level for neutral isocytosine considering only the favored isomers leads to the following PA values: 339.2 and 223.2 kcal mol−1 in the enthalpy scale for (AHB)− + H+ ⇄ AH2B and AH2B + H+ ⇄ (AH2BH)+ reactions, respectively [63]. They are close to those estimated in this work at the DFT level. It should also be mentioned here that the experimental pKa values for the two steps of acid-base equilibria in aqueous solution (9.6 and 4.0 [27]) correspond also to the macroscopic pKas.
4. Conclusions
Taking the limits of experimental methods into account, quantum–chemical methods are sole tools for complete structural-investigation of the very complex polyfunctional tautomeric system, complete examination of very complex acid-base equilibria, and complete estimation of acidity–basicity parameters in both the micro and macro scales for both individual isomers and tautomeric mixtures, respectively. These methods applied to the two isomeric polyfucntional tautomeric pyrimidine bases, cytosine and isocytosine in vacuo, gave the possibility to (i) indicate the structures of all major, minor, and rare isomers in the tautomeric mixtures of neutral (Figure 3) and ionic forms (Figure 4, Figure 5, Figure 6 and Figure 7) in non-polar environment (e.g., lipids), (ii) estimate their percentage contents (Table 2), (iii) predict kinetic acidity–basicity of all potential sites of protonation–deprotonation in individual neutral and ionic isomers (Figure 8 and Figure 9), as well as (iv) investigate acid-base equilibria (Figure 10), and (v) calculate thermodynamic acidity–basicity parameters for isomeric mixtures (Table 3).
Although cytosine and isocytosine display some structural difference (e.g., different positions of the exo groups), they exhibit particular symmetry for neutral and ionic forms in various isomeric conversions (Table 1, Figure 3, Figure 4, Figure 5, Figure 6 and Figure 7), number of isomers in isomeric mixtures (Figure 3, Figure 4, Figure 5, Figure 6 and Figure 7), and number of steps in general acid-base equilibria (Figure 2). Some analogies exist also in internal stabilization between functional exo and endo groups of the favored isomers and push–pull effects on acidity–basicity order of tautomeric groups. Consequently, these similarities and differences lead to the following major ionic isomers: ma-C-3b and ma-C-3a for mono-anionic cytosine, ma-iC-3a and ma-iC-7 for mono-anionic isocytosine, mc-C-138 and mc-C-178b for mono-cationic cytosine, mc-iC-178a for mono-cationic isocytosine, da-C-0a for di-anionic cytosine, da-iC-0b and da-iC-0a for di-anionic isocytosine, dc-C-1378a and dc-C-1378b for di-cationic cytosine, and da-iC-1378b for di-cationic isocytosine.
The slight differences in the contribution of the major isomers are in relation with internal effects of conjugated tautomeric groups that affect their acidity–basicity parameters. In consequence, these parameters estimated at the DFT level for the four steps of acid-base reaction when proceeding from di-anion to di-cation (Figure 10) have slightly different GB values for cytosine and isocytosine in the Gibbs energy scale: 446.6 and 449.5, 332.7 and 332.1, 220.6 and 216.2, and 92.8 and 96.6 kcal mol−1, respectively (Table 3). It should be signaled here that for polyfunctional tautomeric system being in equilibrium between intramolecular proton-transfers (prototropy) and intermolecular proton-transfers (acid-base equilibria), a conclusion on the protonation–deprotonation sites is difficult to be derived, particularly in the case of pyrimidine bases when the tautomeric mixtures of neutral and ionic forms contain a few isomers and the tautomeric preferences change upon the intermolecular proton-transfers.
On the other hand, the favored sites of protonation and deprotonation could be determined for isolated isomers and the following important conclusions on internal effects derived. For the canonical tautomers of neutral C and iC owing to the push-pull effect in the amidine (H2N−C(R)=N−) and amide (−HN−C(R)=O) groups, the endo N3 and N1 atoms compete in the proton-gain with the exo O7b and O8a atoms, whereas the endo N1H and N3H groups compete in the proton-loss with the exo N8Ha and N7Hb, respectively. In the case of di-anionic isomers of C, N3 is firstly protonated, whereas N3 competes in the proton-gain with N7 in those of iC. For di-cationic isomers of C, O7H is firstly deprotonated in a, and N3H competes in the proton-loss with O7H in b. A slightly different trend occurs for di-cationic isomers of iC, where N3H firstly loses the proton in a, and N3H competes in the proton-loss with O8H in b. Differences in acidity–basicity parameters of particular sites in individual isomers of neutral and ionic C and iC are higher (in some cases even >10 kcal mol−1) than those of their tautomeric mixtures (<6 kcal mol−1).
Funding
This work received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
As a professor emeritus the author thanks the Warsaw University of Life Sciences (SGGW) for some kind of support.
Conflicts of Interest
The author declares no conflict of interest.
Sample Availability
Not applicable.
References
- Saenger, W. Principles of Nucleic Acid Structure; Springer: New York, NY, USA, 1994. [Google Scholar]
- Elguero, J.; Marzin, C.; Katritzky, A.R.; Linda, P. The Tautomerism of Heterocycles; Academic Press: New York, NY, USA, 1976. [Google Scholar]
- Kwiatkowski, J.S.; Person, W.B. Theoretical Biochemistry and Molecular Biology; Beveridge, D.L., Lavery, R., Eds.; Academic Press: New York, NY, USA, 1990; pp. 153–171. [Google Scholar]
- Raczyńska, E.D.; Kosińska, W.; Ośmiałowski, B.; Gawinecki, R. Tautomeric Equilibria in Relation to pi-Electron Delocalization. Chem. Rev. 2005, 105, 3561–3612. [Google Scholar] [CrossRef]
- Shukla, M.K.; Leszczynski, J. Tautomerism in Nucleic Acid Bases and Base Pairs: A Brief Overview. WIREs Comput. Mol. Sci. 2013, 3, 637–649. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Sapuła, M.; Zientara-Rytter, K.; Kolczyńska, K.; Stępniewski, T.M.; Hallmann, M. DFT Studies on the Favored and Rare Tautomers of Neutral and Redox Cytosine. Struct. Chem. 2016, 27, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Raczyńska, E.D. Quantum-Chemical Studies on the Favored and Rare Isomers of Isocytosine. Comput. Theor. Chem. 2017, 1121, 58–67. [Google Scholar] [CrossRef]
- Brown, R.D.; Godfrey, P.D.; McNaughton, D.; Pierlot, A.P. Tautomers of Cytosine by Microwave Spectroscopy. J. Am. Chem. Soc. 1989, 111, 2308–2310. [Google Scholar] [CrossRef]
- Alonso, J.L.; Vaquero, V.; Peña, I.; López, J.C.; Mata, S.; Caminati, W. All Five Forms of Cytosine Revealed in the Gas Phase. Angew. Chem. Int. Ed. 2013, 52, 2331–2334. [Google Scholar] [CrossRef]
- Szczesniak, M.; Szczepaniak, K.; Kwiatkowski, J.S.; KuBulat, K.; Person, W.B. Matrix Isolation Infrared Studies of Nucleic Acid Constituents. 5. Experimental Matrix-Isolation and Theoretical an Initio SCF Molecular Orbital Studies of the Infrared Spectra of Cytosine Monomers. J. Am. Chem. Soc. 1988, 110, 8319–8330. [Google Scholar] [CrossRef]
- Stepanian, S.G.; Sheina, G.G.; Radchenko, E.D.; Blagoi, Y.P. Infrared Spectra and Tautomerism of Isocytosine in Argon Matrix. Zh. Fiz. Khim. 1989, 63, 3008–3014. [Google Scholar]
- Vranken, H.; Smets, J.; Maets, G.; Lapinski, L.; Nowak, M.J.; Adamowicz, L. Infrared Spectra and Tautomerism of Isocytosine; an Ab Initio and Matrix Isolation Study. Spectrochim. Acta A 1994, 50, 875–889. [Google Scholar] [CrossRef]
- Ivanov, A.Y.; Stepanian, S.G.; Adamowicz, L. Tautomeric Transitions of Isocytosine Isolated in Argon and Neon Matrices Induced by UV Irradiation. J. Mol. Struct. 2012, 1025, 92–104. [Google Scholar] [CrossRef]
- Ha, T.K.; Keller, H.J.; Gunde, R.; Gunthard, H.H. Quantum Chemical Study of Structure and Stability of All 14 Isomers of Isocytosine. J. Mol. Struct. 1996, 376, 375–397. [Google Scholar] [CrossRef]
- Gorb, L.; Podolyan, Y.; Leszczynski, J. A Theoretical Investigation of Tautomeric Equilibria and Proton Transfer in Isolated and Monohydrated Cytosine and Isocytosine Molecules. J. Mol. Struct. (Theochem) 1999, 487, 47–55. [Google Scholar] [CrossRef]
- Nir, E.; Müller, M.; Grace, L.I.; de Vries, M.S. REMPI Spectroscopy of Cytosine. Chem. Phys. Lett. 2002, 355, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.Y.; Dong, F.; Miller, R.E. Multiple Tautomers of Cytosine Identified and Characterized by Infrared Laser Spectroscopy in Helium Nanodroplets: Probing Structure Using Vibrational Transition Moment Angles. Phil. Trans. R. Soc. A 2005, 363, 393–413. [Google Scholar] [CrossRef]
- Feyer, V.; Plekan, O.; Richter, R.; Coreno, M.; Vall-llosera, G.; Prince, K.C.; Trofimov, A.B.; Zaytseva, I.L.; Moskovskaya, T.E.; Gromov, E.V.; et al. Tautomerism in Cytosine and Uracil: An Experimental and Theoretical Core Level Spectroscopic Study. J. Phys. Chem. A 2009, 113, 5736–5742. [Google Scholar] [CrossRef]
- Dreyfus, M.; Bensaude, O.; Dodin, G.; Dubois, J.E. Tautomerism in Cytosine and 3-Methylcytosine. A Thermodynamic and Kinetic Study. J. Am. Chem. Soc. 1976, 98, 6338–6349. [Google Scholar] [CrossRef]
- Trygubenko, S.A.; Bogdan, T.V.; Rueda, M.; Orozco, M.; Luque, F.J.; Šponer, J.; Slavíček, P.; Hobza, P. Correlated Ab Initio Study of Nucleic Acid Bases and Their Tautomers in the Gas Phase, in a Microhydrated Environment and in Aqueous Solution. Part 1. Cytosine. Phys. Chem. Chem. Phys. 2002, 4, 4192–4203. [Google Scholar] [CrossRef]
- Freedman, H.; Nguyen, H.N.; Truong, T.N. A Study of the Tautomeric Equilibria of 2-Hydroxypyridine/2-Oxopyridine and of Cytosine in Water Using the Coupled Reference Interaction Site Model(RISM)/Molecular Dynamics (MD) Approach. J. Phys. Chem. B 2004, 108, 19043–19048. [Google Scholar] [CrossRef]
- Mazzuca, D.; Marino, T.; Russo, N.; Toscano, M. A Theoretical Study on Tautomerization Processes of Dehydrated and Monohydrated Cytosine. J. Mol. Struct. (Theochem) 2007, 811, 161–167. [Google Scholar] [CrossRef]
- Baker, D.; Marsh, R.E. The Crystal Structure of Cytosine. Acta Cryst. 1964, 17, 1581–1587. [Google Scholar] [CrossRef]
- McClure, R.J.; Craven, B.M. New Investigations of Cytosine and Its Monohydrate. Acta Cryst. B 1973, 29, 1234–1238. [Google Scholar] [CrossRef]
- McConnell, J.F.; Sharma, B.D.; Marsch, R.E. Co-crystallization of Two Tautomers: Crystal Structure of Isocytosine. Nature 1964, 203, 399–400. [Google Scholar] [CrossRef]
- Partalone, G.; Colapietro, M. Redetermination of Isocytosine. Acta Crystallogr. Sect. E 2007, 63, 01869–01871. [Google Scholar] [CrossRef]
- Dračínský, M.; Jansa, P.; Ahonen, K.; Buděsínský, M. Tautomerism and the Protonation/Deprotonation of Isocytosine in Liquid- and Solid-States Studied by NMR Spectroscopy and Theoretical Calculations. Eur. J. Org. Chem. 2011, Issue 8, 1544–1551. [Google Scholar] [CrossRef]
- Colominas, C.; Luque, F.J.; Orozco, M. Tautomerism and Protonation of Guanine and Cytosine. Implications in the Formation of Hydrogen-Bonded Complexes. J. Am. Chem. Soc. 1996, 118, 6811–6821. [Google Scholar] [CrossRef]
- Russo, N.; Toscano, M.; Grand, A.; Jolibois, F. Protonation of Thymine, Cytosine, Adenine, and Guanine DNA Nucleic Acid Bases: Theoretical Investigation into the Framework of Density Functional Theory. J. Comput. Chem. 1998, 19, 989–1000. [Google Scholar] [CrossRef]
- Podolyan, Y.; Grob, L.; Leszczynski, J. Protonation of Nucleic Acid Bases. A Comprehensive Post-Hartree-Fock Study of the Energetic and Proton Affinities. J. Phys. Chem. A 2000, 104, 7346–7352. [Google Scholar] [CrossRef]
- Chandra, A.K.; Michalska, D.; Wysokiñsky, R.; Zeegers-Huyskens, T. Theoretical Study of the Acidity and Basicity of the Cytosine Tautomers and Their 1:1 Complexes with Water. J. Phys. Chem. A 2004, 108, 9593–9600. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Li, T.; Amegayibor, F.S.; Cardoso, D.S.; Fu, Y.; Lee, J.K. Gas-Phase Thermochemical Properties of Pyrimidine Nucleobases. J. Org. Chem. 2008, 73, 9283–9291. [Google Scholar] [CrossRef]
- Yao, C.; Tureček, F.; Polce, M.J.; Wesdemiotis, C. Proton and Hydrogen Atom Adducts to Cytosine. An Experimental and Computational Study. Int. J. Mass Spectrom. 2007, 265, 106–123. [Google Scholar] [CrossRef]
- Salapin, J.-Y.; Guillaumont, S.; Tortajada, J.; MacAleese, L.; Lemaire, J.; Maitre, P. Infrared Spectra of Protonated Uracil, Thymine and Cytosine. Chem. Phys. Chem. 2007, 8, 2235–2244. [Google Scholar] [CrossRef] [Green Version]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. Development and Use of Quantum Mechanical Molecular Models. 76. AM1: A New General Purpose Quantum Mechanical Molecular Model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Karelson, M. AM1 Calculations of Reaction Field Effects on the Tautomeric Equilibria of Nucleic Acid Pyrimidine and Purine Bases and Their 1-Methyl Analogs. J. Am. Chem. Soc. 1991, 113, 1561–1566. [Google Scholar] [CrossRef]
- Mezey, P.G.; Ladik, J.J. A Non-Empirical Molecular Orbital Study on the Relative Stabilities of Adenine and Guanine Tautomers. Theor. Chim. Acta 1979, 52, 129–145. [Google Scholar] [CrossRef]
- Mezey, P.G.; Ladik, J.J.; Barry, M. Non-Empirical SCF MO Studies on the Protonation of Biopolymer Constituents II. Protonation of Adenine, Guanine and their Tautomeric Forms. Theor. Chim. Acta 1980, 54, 251–258. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecular Orbital Theory; Oxford University Press: New York, NY, USA, 1998. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; and Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1993, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Hehre, W.J.; Radom, L.; Schleyer, P.V.R.; Pople, J.A. Ab Initio Molecular Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian-03, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Wu, R.R.; Yang, B.; Frieler, C.E.; Berden, G.; Oomens, J.; Rodgers, M.T. Diverse Mixtures of 2,4-Dihydroxy Tautomers and O4 Protonated Conformers of Uridine and 2’-Deoxyuridine Coexist in the Gas Phase. Phys. Chem. Chem. Phys. 2015, 17, 25978–25988. [Google Scholar] [CrossRef] [Green Version]
- Leito, I.; Koppel, I.A.; Koppel, I.; Kaupmees, K.; Tshepelevitsh, S.; Saame, J. Basicity Limits of Neutral Organic Superbases. Angew. Chem. Int. Ed. 2015, 54, 9262–9265. [Google Scholar] [CrossRef]
- Maksić, Z.B.; Kovačević, B.; Vianello, R. Advances in Determining the Absolute Proton Affinities of Neutral Organic Molecules in the Gas Phase and Their Interpretation: A Theoretical Account. Chem. Rev. 2012, 112, 5240–5270. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Makowski, M.; Zientara-Rytter, K.; Kolczyńska, K.; Stępniewski, T.M.; Hallmann, M. Quantum-Chemical Studies on the Favored and Rare Tautomers of Neutral and Redox Adenine. J. Phys. Chem. A 2013, 117, 1548–1559. [Google Scholar] [CrossRef] [PubMed]
- Raczyńska, E.D.; Kurpiewski, J.; Igielska, M.; Kamińska, B. Quantitative Description of Bond Lengths Alternation for Caffeine—Effects of Ionization, Proton-Transfer, and Noncovalent Interaction. Comput. Theor. Chem. 2020, 1180, 112811. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Kamińska, B. Structural and Thermochemical Consequences of Prototropy and Ionization for the Biomolecule Xanthine in Vacuo. J. Chem. Thermodyn. 2022, 171, 106788. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Gal, J.F.; Maria, P.C.; Kamińska, B.; Igielska, M.; Kurpiewski, J.; Juras, W. Purine Tautomeric Preferences and Bond-length Alternation in Relation with Protonation-Deprotonation and Alkali Metal Cationization. J. Mol. Model. 2020, 26, 93. [Google Scholar] [CrossRef] [Green Version]
- Raczyńska, E.D.; Gal, J.-F.; Maria, P.-C.; Michalec, P.; Zalewski, M. Exceptionally High Proton and Lithium Cation Gas-Phase Basicity of the Anti-Diabetic Drug Metformin. J. Phys. Chem. A 2017, 121, 8706–8718. [Google Scholar] [CrossRef]
- Raczyńska, E.D. Quantum-Chemical Search for Keto Tautomers of Azulenols in Vacuo and Aqueous Solution. Symmetry 2021, 13, 497. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Gal, J.-F.; Maria, P.-C.; Saeidian, H. Push-Pull Effect on the Gas-Phase Basicity of Nitriles. Transmission of the Resonance Effect by Methylenecyclopropene and Cyclopropenimine π-Systems Substituted by Two Identical Strong Electron-Donors. Symmetry 2021, 13, 1554. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Gal, J.-F.; Maria, P.-C.; Sakhawat, G.S.; Fahim, M.Q.; Saeidian, H. Nitriles with High Gas-Phase Basicity—Part II Transmission of the Push-Pull Effect through Methylenecyclopropene and Cyclopropenimine Scaffolds Intercalated between Different Electron Donnor(s) and the Cyano N-Protonation Site. Molecules 2022, 27, 4370. [Google Scholar] [CrossRef]
- Linstrom, P.J.; Mallard, W.G. (Eds) NIST Chemistry WebBook, NIST Standard Reference Database No. 69; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2014. Available online: http://webbook.nist.gov/chemistry (accessed on 17 January 2023).
- Raczyńska, E.D.; Gal, J.-F.; Maria, P.-C. Enhanced Basicity of Push-Pull Nitrogen Bases in the Gas Phase. Chem. Rev. 2016, 116, 13454–13511. [Google Scholar] [CrossRef]
- Bartmess, J.E. Thermodynamics of the Electron and the Proton. J. Phys. Chem. 1994, 98, 6420–6424, Erratum in J. Phys. Chem. 1995, 98, 6755. [Google Scholar] [CrossRef]
- Fifen, J.J.; Dhaouadi, Z.; Nsangou, M. Revision of the Thermodynamics of the Proton in the Gas Phase. J. Phys. Chem. A 2014, 118, 11090–11097. [Google Scholar] [CrossRef]
- Gil Bardají, E.; Freisinger, E.; Costisella, B.; Schalley, C.A.; Brüning, W.; Sabat, M.; Lippert, B. Mixed-Metal (Platinium, Palladium), Mixed-Pyrimidine (Uracil, Cytosine) Self-Assembling Metallacalix[n]arenes: Dynamic Combinatorial Chemistry with Nucleobases and Metal Species. Chem. Eur. J. 2007, 13, 6019–6039. [Google Scholar] [CrossRef] [PubMed]
- Vellé, A.; Cebollada, A.; Lippert, B.; Sanz Miguel, P.J. Topology of Metallacalix[4]arenes with Uracil and Cytosine Ligands: Favorable and Unfavorable Assemblies. New J. Chem. 2016, 40, 5914–5919. [Google Scholar] [CrossRef]
- Nichols, C.M.; Wang, Z.-C.; Lineberger, W.C.; Bierbaum, V.M. Gas-Phase Reactions of Deprotonated Nucleobases with H, N, and O Atoms. J. Phys. Chem. Lett. 2019, 10, 4863–4867. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Oritz, A.; Taccone, M.; Maitre, P.; Rossa, M.; Pino, G. On Interaction between Deprotonated Cytosine [C(-H)]− and Ba2+: Infrered Multiphoton Spectroscopy and Dynamics. ChemPhysChem 2020, 21, 2571–2582. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Makowski, M. Effects of Positive and Nagative Ionization on Prototropy in Pyrimidine Bases: An Unusual Case of Isocytosine. J. Phys. Chem. A 2018, 122, 7863–7879. [Google Scholar] [CrossRef]
- King, E.J. Acid-Base Equilibria; Pergamon Press: Oxford, UK, 1965. [Google Scholar]
- Hunter, E.P.L.; Lias, S.G. Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update. J. Phys. Chem. Ref. Data 1998, 27, 413–656. [Google Scholar] [CrossRef]
- Chen, E.C.M.; Herder, C.; Chen, E.S. The Experimental and Theoretical Gas Phase Acidities of Adenine, Guanine, Cytosine, Uracil, Thymine and Halouracils. J. Mol. Struct. 2006, 798, 126–133. [Google Scholar] [CrossRef]
Figure 1.
Canonical forms of neutral cytosine (C) and isocytosine (iC) containing two labile protons and five tautomeric conjugated sites (indicated in blue and red color, respectively).
Figure 1.
Canonical forms of neutral cytosine (C) and isocytosine (iC) containing two labile protons and five tautomeric conjugated sites (indicated in blue and red color, respectively).

Figure 2.
Acid-base equilibria for pyrimidine bases.

Figure 3.
Selected isomers for neutral cytosine (C) and neutral isocytosine (iC), their potential sites of deprotonation and protonation (indicated by blue and red arrows, respectively), and their ΔGs (given in parentheses in kcal mol−1 for 298.15 K, calculated at the DFT level [6,7]).

Figure 4.
Geometrical isomers for di-deprotonated cytosine (da-C) and isocytosine (da-iC), and their ΔGs (given in parentheses in kcal mol−1 for 298.15 K, and calculated at the DFT level).
Figure 4.
Geometrical isomers for di-deprotonated cytosine (da-C) and isocytosine (da-iC), and their ΔGs (given in parentheses in kcal mol−1 for 298.15 K, and calculated at the DFT level).

Figure 5.
Selected isomers for mono-deprotonated cytosine (ma-C) and isocytosine (ma-iC), and their ΔGs (given in parentheses in kcal mol−1 for 298.15 K, and calculated at the DFT level).
Figure 5.
Selected isomers for mono-deprotonated cytosine (ma-C) and isocytosine (ma-iC), and their ΔGs (given in parentheses in kcal mol−1 for 298.15 K, and calculated at the DFT level).

Figure 6.
Selected isomers for mono-protonated cytosine (mc-C) and isocytosine (mc-iC) and their ΔGs (given in parentheses in kcal mol−1 for 298.15 K, and calculated at the DFT level).
Figure 6.
Selected isomers for mono-protonated cytosine (mc-C) and isocytosine (mc-iC) and their ΔGs (given in parentheses in kcal mol−1 for 298.15 K, and calculated at the DFT level).

Figure 7.
Geometrical isomers for di-protonated cytosine (da-C) and isocytosine (da-iC), and their ΔGs (given in parentheses in kcal mol−1 for 298.15 K, and calculated at the DFT level).
Figure 7.
Geometrical isomers for di-protonated cytosine (da-C) and isocytosine (da-iC), and their ΔGs (given in parentheses in kcal mol−1 for 298.15 K, and calculated at the DFT level).

Figure 8.
DFT-estimated microscopic basicity parameters for heteroatoms N and O in di-anionic isomers (da-C and da-iC) and acidity parameters for NH and OH in di-cationic species (dc-C and dc-iC) of pyrimidine bases (in enthalpy scale for 298 K in kcal mol−1).
Figure 8.
DFT-estimated microscopic basicity parameters for heteroatoms N and O in di-anionic isomers (da-C and da-iC) and acidity parameters for NH and OH in di-cationic species (dc-C and dc-iC) of pyrimidine bases (in enthalpy scale for 298 K in kcal mol−1).

Figure 9.
DFT-estimated microscopic basicity and acidity parameters for selected neutral C and iC isomers (in enthalpy scale: PAmicro = ΔHbase in red color and PAmicro = ΔHacid in blue color, respectively, for 298 K in kcal mol−1).
Figure 9.
DFT-estimated microscopic basicity and acidity parameters for selected neutral C and iC isomers (in enthalpy scale: PAmicro = ΔHbase in red color and PAmicro = ΔHacid in blue color, respectively, for 298 K in kcal mol−1).

Figure 10.
Acid-base equilibria for pyrimidine bases (C: X = O and Y = NH, iC: X = NH and Y = O).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Nine prototropic tautomers of cytosine (C) and isocytosine (iC).
| Abbreviations for Tautomers | Positions of Labile Protons in Neutral Pyrimidine Bases | General Names of Tautomers | ||
|---|---|---|---|---|
| C | iC | C | iC | |
| 13 | N1N3 | N1N3 | imino-oxo | imino-oxo |
| 17 | N1O7 | N1N7 | imino-hydroxy | amino-oxo |
| 18 | N1N8 | N1O8 | amino-oxo | imino-hydroxy |
| 37 | N3O7 | N3N7 | imino-hydroxy | amino-oxo |
| 38 | N3N8 | N3O8 | amino-oxo | imino-hydroxy |
| 78 | O7N8 | N7O8 | amino-hydroxy | amino-hydroxy |
| 35 | N3C5 | N3C5 | CH-imino-oxo | CH-imino-oxo |
| 57 | O7C5 | N7C5 | CH-imino-hydroxy | CH-amino-oxo |
| 58 | N8C5 | O8C5 | CH-amino-oxo | CH-imino-hydroxy |
Table 2.
DFT-calculated amounts (xi in %) for selected neutral and ionic isomers of cytosine and isocytosine.
Table 2.
DFT-calculated amounts (xi in %) for selected neutral and ionic isomers of cytosine and isocytosine.
| Form | Isomer | xi | Isomer | xi |
|---|---|---|---|---|
| (AB)2− | da-C-0a | 99.97 | da-iC-0a | 10.14 |
| da-C-0b | 0.03 | da-iC-0b | 89.86 | |
| (AHB)− | ma-C-1a | 2.7∙10−4 | ma-iC-1b | 8.7∙10−8 |
| ma-C-3a | 4.52 | ma-iC-3a | 67.40 | |
| ma-C-3b | 95.48 | ma-iC-3b | 5.4∙10−3 | |
| ma-C-8 | 8.0∙10−3 | ma-iC-7 | 32.59 | |
| AH2B | C-13a | 0.12 | iC-13a | 1.4∙10−4 |
| C-13b | 2.04 | iC-13b | 1.3∙10−3 | |
| C-18 | 86.66 | iC-17 | 1.7∙10−6 | |
| C-38 | 6.6∙10−4 | iC-37 | 20.60 | |
| C-78a | 8.87 | iC-78a | 79.35 | |
| C-78b | 2.30 | iC-78b | 0.05 | |
| (AH2BH)+ | mc-C-138 | 71.21 | mc-iC-137 | 5.6∙10−3 |
| mc-C-178a | 3.4∙10−5 | mc-iC-178a | 99.96 | |
| mc-C-178b | 28.79 | mc-iC-178b | 0.04 | |
| mc-C-378a | 2.1∙10−5 | mc-iC-378b | 2.8∙10−4 | |
| (AH2BH2)2+ | dc-C-1378a | 91.73 | dc-iC-1378a | 0.01 |
| dc-C-1378b | 8.27 | dc-iC-1378b | 99.99 |
Table 3.
DFT-calculated macroscopic acidity–basicity parameters for tautomeric cytosine and isocytosine (PA and GB in kcal mol−1 for 298 K).
Table 3.
DFT-calculated macroscopic acidity–basicity parameters for tautomeric cytosine and isocytosine (PA and GB in kcal mol−1 for 298 K).
| Acid-Base Equilibrium | PAmacro (C) | GBmacro (C) | PAmacro (iC) | GBmacro (iC) |
|---|---|---|---|---|
| (AB)2− + H+ ⇄ (AHB)− | 451.1 | 446.6 | 457.1 | 449.5 |
| (AHB)− + H+ ⇄ AH2B | 339.8 a | 332.7 b | 339.5 c | 332.1 |
| AH2B + H+ ⇄ (AH2BH)+ | 228.6 d | 220.6 e | 223.8 f | 216.2 |
| (AH2BH)+ + H+ ⇄ (AH2BH2)2+ | 100.5 | 92.8 | 104.4 | 96.6 |
a Experimental value 342 ± 3 (bracketing method) and 343 ± 3 kcal mol−1 (extended Cooks kinetic method) [32]. b Experimental value 335 ± 3 kcal mol−1 (bracketing method) [32]. c Theoretically derived value for mixtures of major isomers 339.2 kcal mol−1 (G4 method) [63]. d Experimental value 228 ± 3 (bracketing method) [32], 227 ± 3 (extended Cooks kinetic method) [32], and 227.0 kcal mol−1 (evaluated by Hunter and Lias on the basis of earlier literature kinetic and equilibrium experiments) [65]; theoretically derived average value 225.3–230.8 kcal mol−1 (various MPn methods) [30]. e Experimental value 220 ± 3 (bracketing method) [32] and 219.0 kcal mol−1 (evaluated by Hunter and Lias on the basis of earlier literature kinetic or equilibrium experiments) [65]. f Theoretically derived value for mixtures of major isomers 223.2 kcal mol−1 (G4 method) [63].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Raczyńska, E.D. On Analogies in Proton-Transfers for Pyrimidine Bases in the Gas Phase (Apolar Environment)—Cytosine Versus Isocytosine. Symmetry 2023, 15, 342. https://doi.org/10.3390/sym15020342
AMA Style
Raczyńska ED. On Analogies in Proton-Transfers for Pyrimidine Bases in the Gas Phase (Apolar Environment)—Cytosine Versus Isocytosine. Symmetry. 2023; 15(2):342. https://doi.org/10.3390/sym15020342
Chicago/Turabian StyleRaczyńska, Ewa D. 2023. "On Analogies in Proton-Transfers for Pyrimidine Bases in the Gas Phase (Apolar Environment)—Cytosine Versus Isocytosine" Symmetry 15, no. 2: 342. https://doi.org/10.3390/sym15020342
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.