3.2. Effect of Acid Concentration and Type
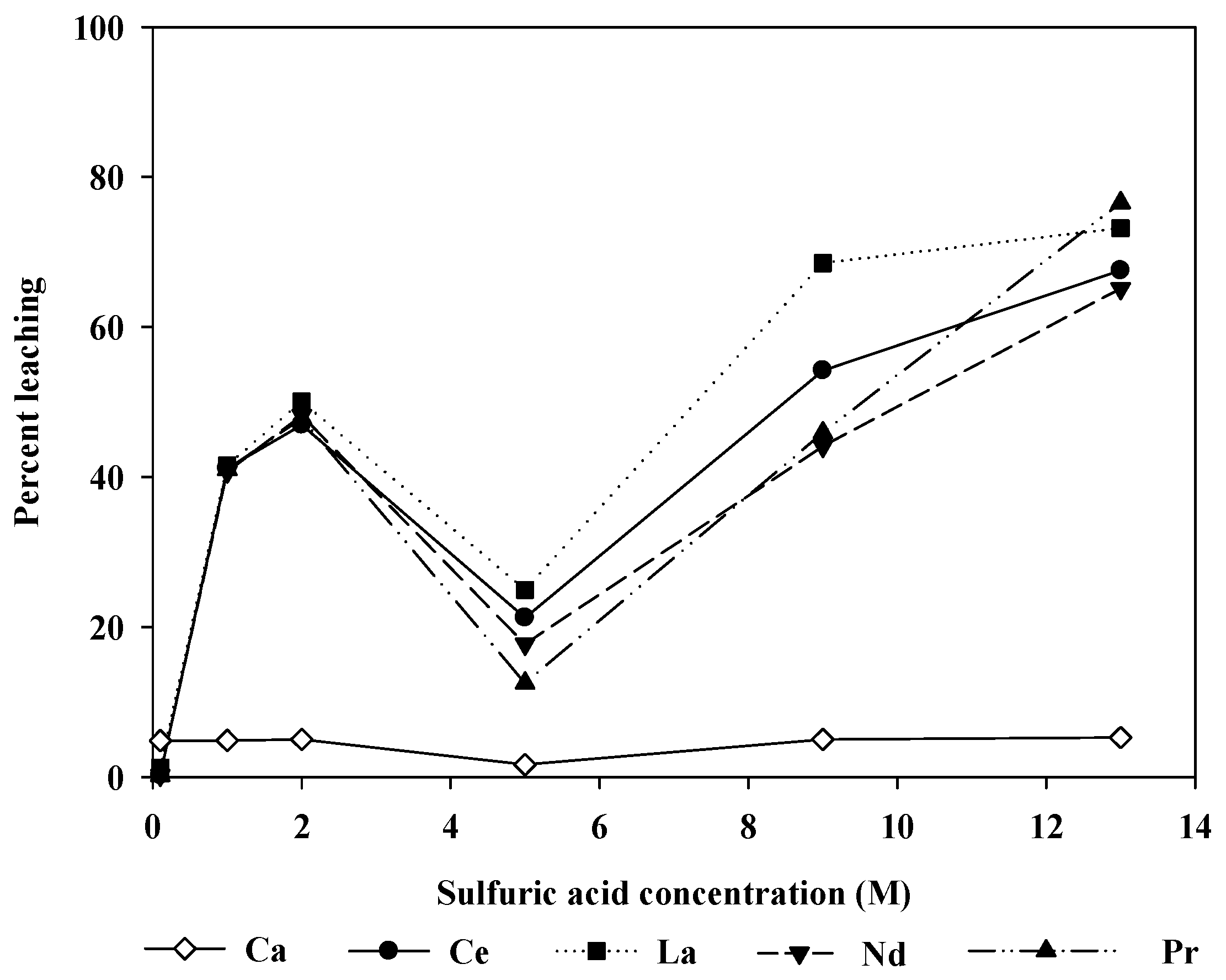
Figure 4 shows the change in the leaching efficiency with the sulfuric acid concentration. The experiments were run for 5 h at 20 °C. At low acid concentrations, the leaching level increased with increasing acid concentration. However, the leaching level decreased at 5.0 M. This may result from reprecipitation of REEs via calcium sulfate formation. It is known that calcium sulfate hydrates (gypsum, hemihydrate, and anhydrite) are readily formed wherever calcium and sulfate are present together in aqueous solutions [
16].
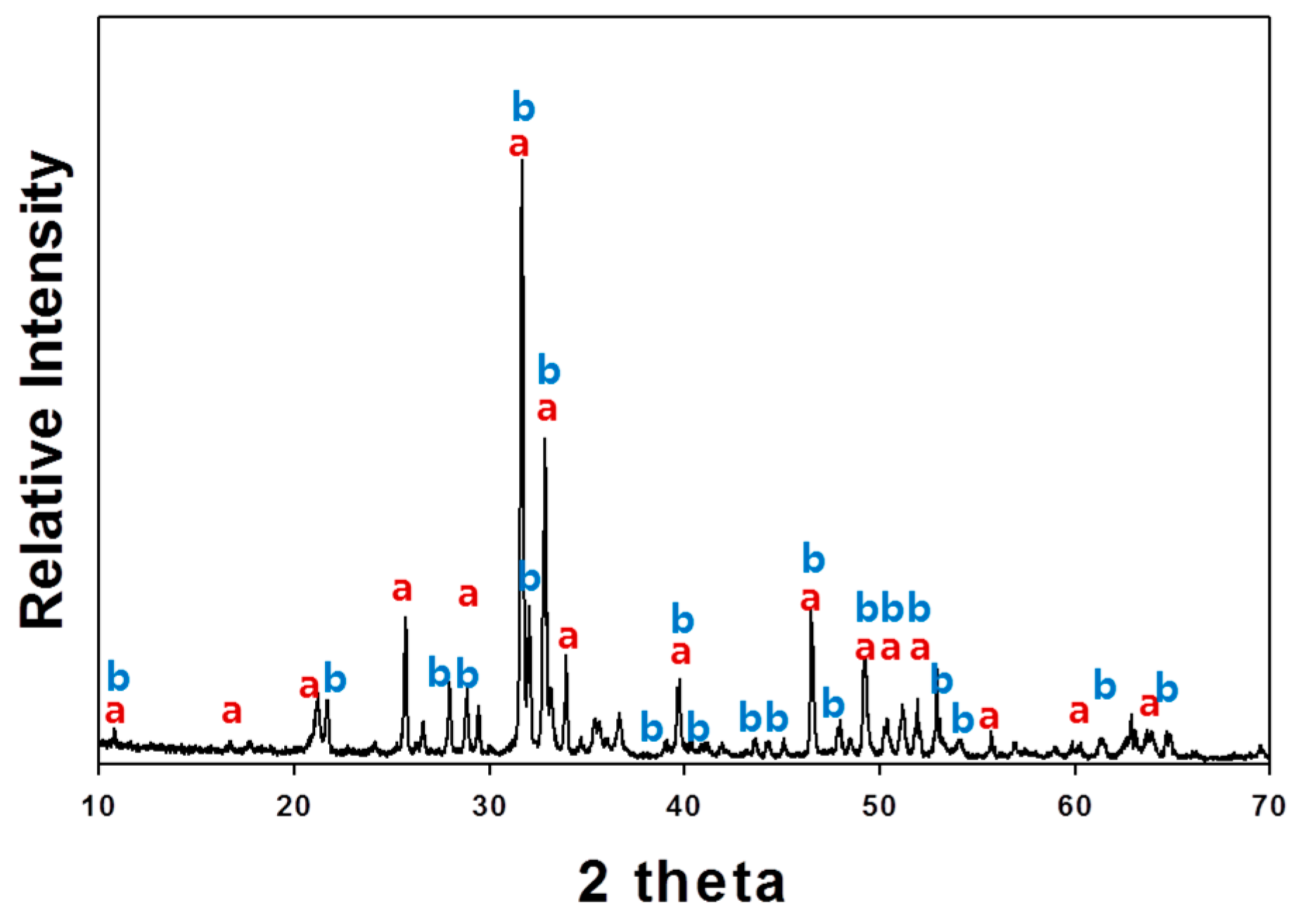
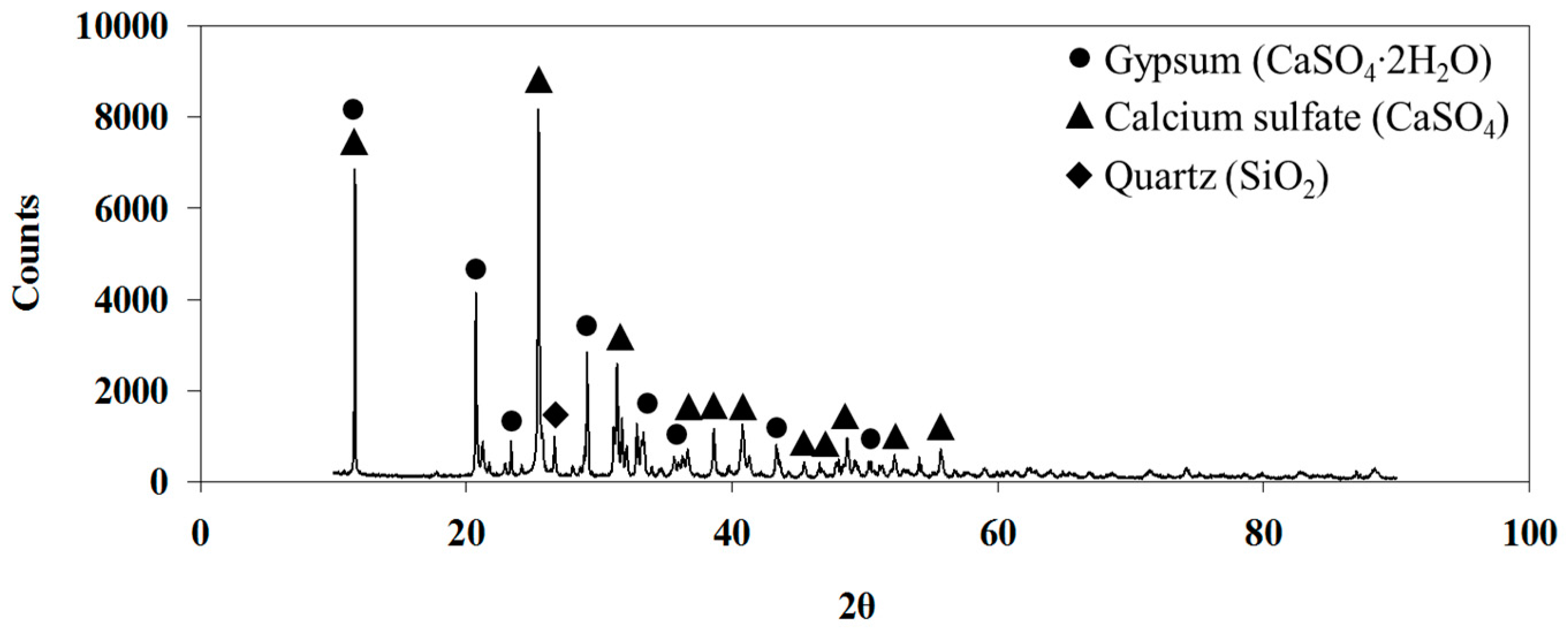
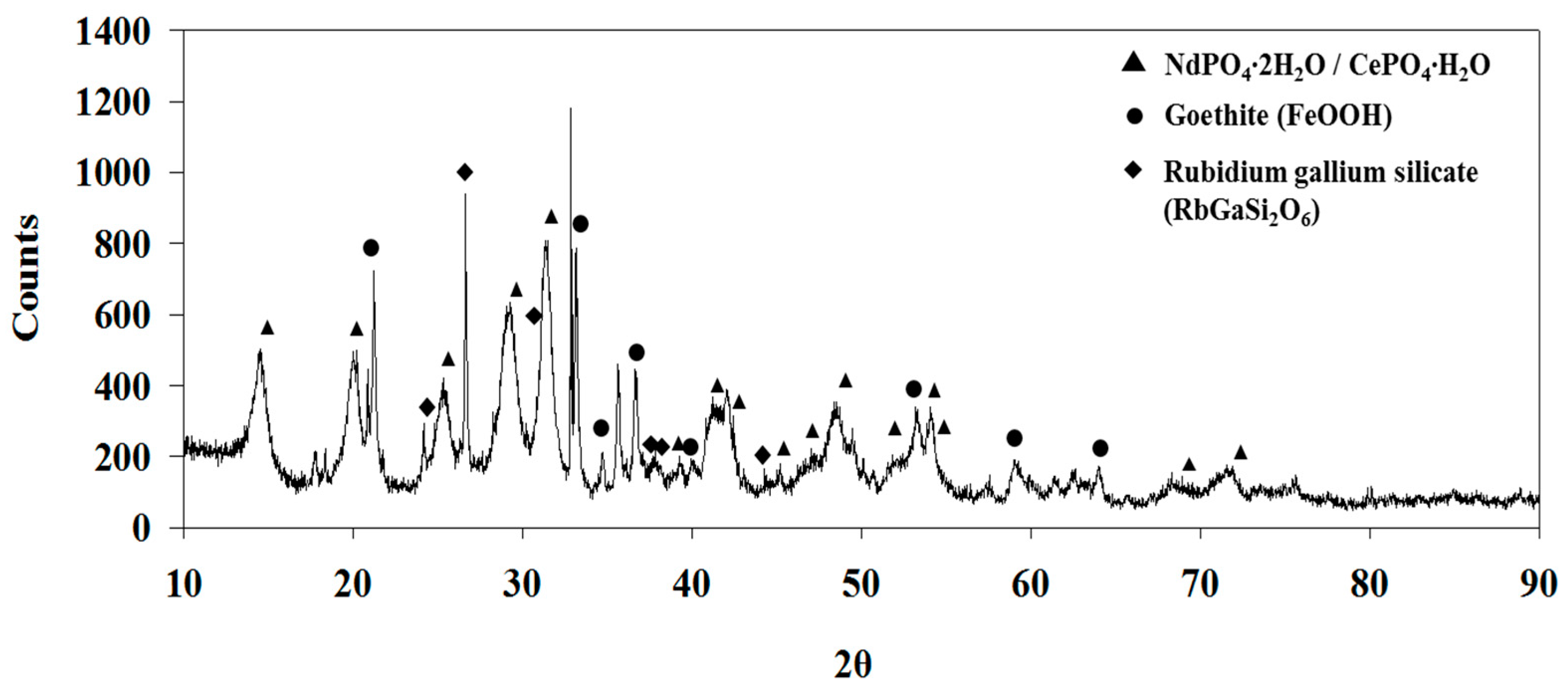
Indeed, the XRD analysis of the residue after leaching shows the presence of gypsum (CaSO
4∙2H
2O) and anhydrite (CaSO
4) (
Figure 5). This residue may contain REEs by isomorphous substitutions for Ca
2+ [
8,
12,
17,
18], because Ca and REEs have similar ionic radii. To confirm the existence of REEs in the precipitated gypsum and CaSO
4, the leaching residue was dried in atmosphere and was washed with water. The solution phase from this washing was found to contain a significant amount of Ca and REEs (1.03 g/L Ca, 0.08 g/L Ce, 0.03 g/L La, 0.03 g/L Nd, 0.02 g/L Pr, and 0.009 g/L Y), which indicates that the precipitated calcium sulfate hydrates contain REEs by ion substitution. The Ca concentration seems to be higher than the solubility of gypsum (2.4 g/L), but the solution may contain free calcium ions and associated calcium sulfate neutral species [CaSO
4(aq)] dissolved from anhydrite [
16,
19].
On the other hand, when the concentration of sulfuric acid further increased above 5.0 M, the leaching level increased again. It can be postulated that the increase in the acidity may change the solution chemistry to limit the precipitation of calcium sulfate hydrates. To examine more closely the relationship between the leaching patterns and H
2SO
4 concentration, the Ca speciation was calculated for the Ca-SO
4-H
2O system using thermodynamic data available in the literature [
20,
21,
22]:
There are seven species in the solution: OH−, H2SO4, HSO4−, SO42−, Ca2+, Ca(OH)+, CaSO4(aq). Therefore, three more equations in addition to Equations (1)–(4) are required to calculate the equilibrium composition. Two additional equations used were mass balance equations for Ca species and sulfate species. One additional equation could be the charge balance equation, but in our calculations, the OH− concentrations were assumed to be known and were calculated from the final pH of the solutions in the actual experiment. The total Ca concentration was assumed to be 0.5 M considering the number of moles of Ca in the ore. The equilibrium composition was then calculated under different levels of total sulfate concentrations (1.0, 2.0, 5.0, 9.8, and 13.0 M). If the calculated values were greater than any of the Ksp values [Equations (5)–(7)], the Ca2+ concentration was recalculated using the Equations (5)–(7) and the calculations were repeated until the calculated values satisfied all conditions. The exact solution was obtained by an iterative method using “Solver” function in MS Excel (Microsoft, Redmond, WA, USA).
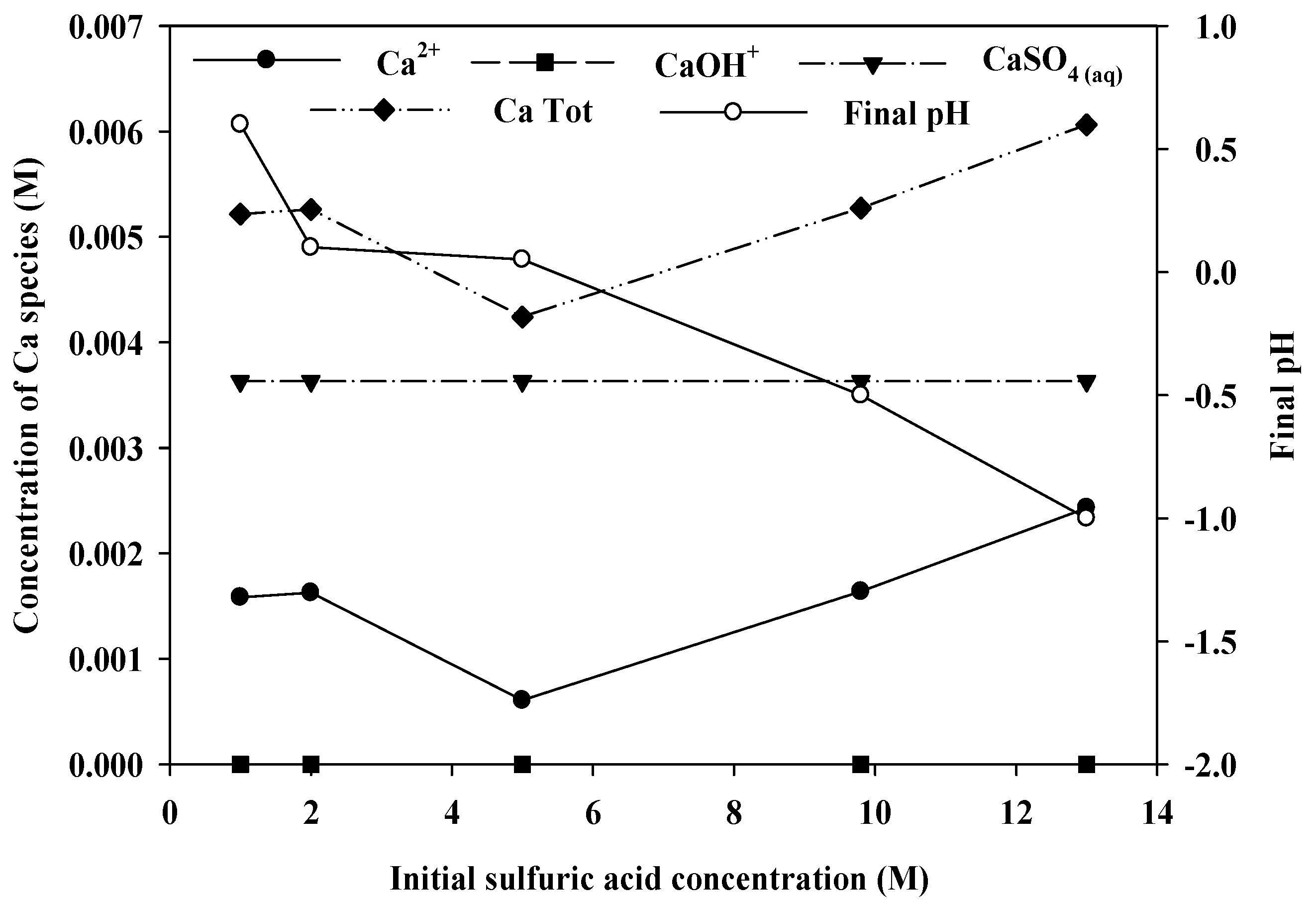
Figure 6 shows the calculation results, showing the distribution of Ca species as a function of the initial sulfuric acid concentration. In this highly acidic condition, Ca was present mainly in the form of Ca
2+ and CaSO
4(aq). The concentration of CaSO
4(aq) does not change with the sulfuric acid concentration, as the solution become saturated owing to the high concentrations of Ca
2+ and SO
42−. On the other hand, the concentration of Ca
2+ changes with the sulfuric acid concentration, showing a minimum at 5.0 M. The final pH of the solution decreased initially with increasing the sulfuric acid concentration, but remained the same from 2.0 to 5.0 M of sulfuric acid. However, with more addition of sulfuric acid, the final pH decreased again, which accompanies the increases in the concentration of Ca
2+. It indicates that the concentration of Ca
2+ species changed in a complicated manner due to the competing balance between the precipitation and dissolution of Ca, which is affected by H
+, SO
42− and Ca
2+ in the solution. Accordingly, the total soluble Ca species varies with the sulfuric acid concentration in a pattern very similar to the leaching pattern. This result suggests that sulfuric acid may not be a good leaching agent for this ore, as the side reaction, Ca(REE) sulfate precipitation, limits the REE leaching level, which did not exceed 80%, even when a large excess of acid (13.0 M) was used.
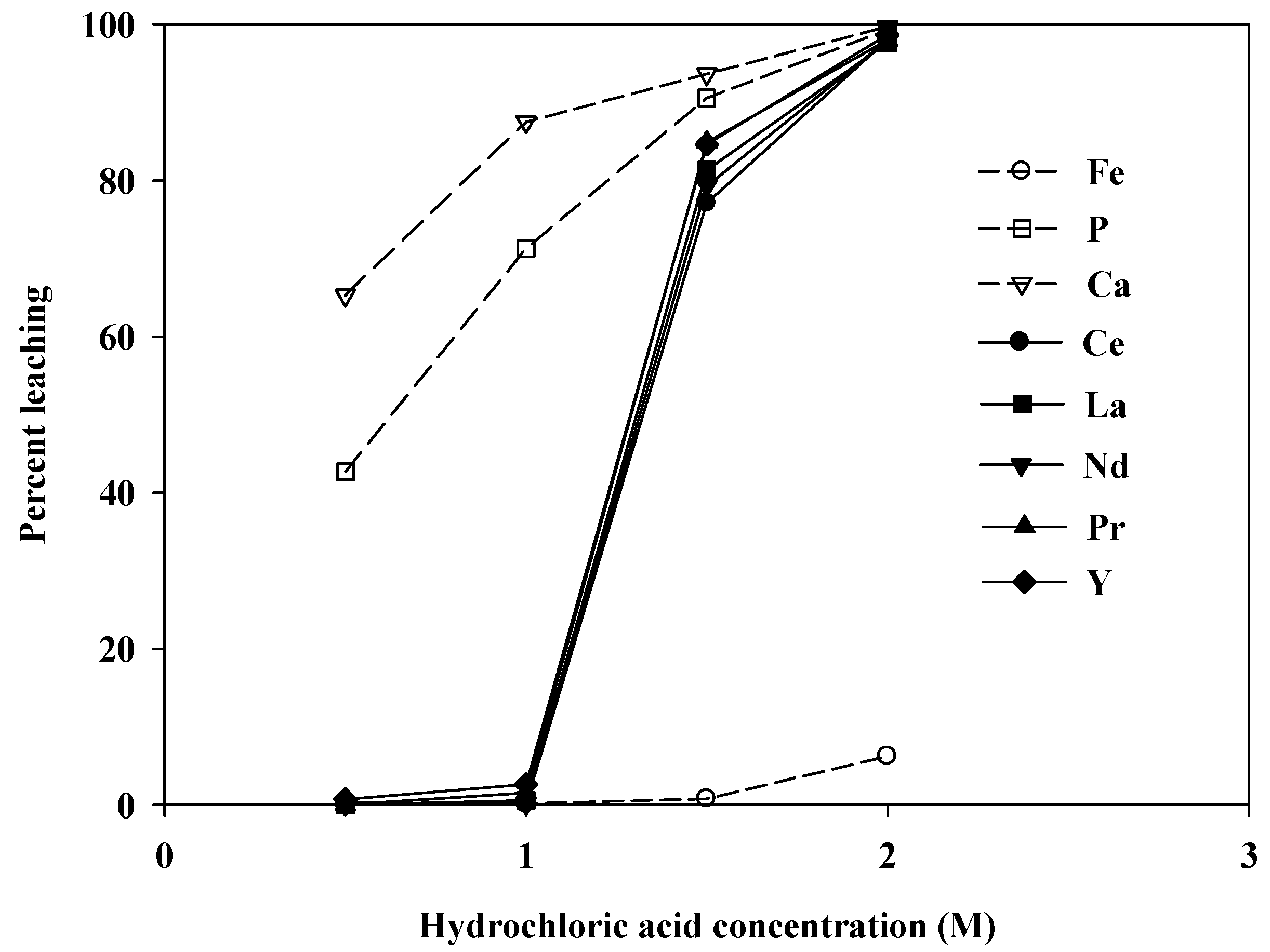
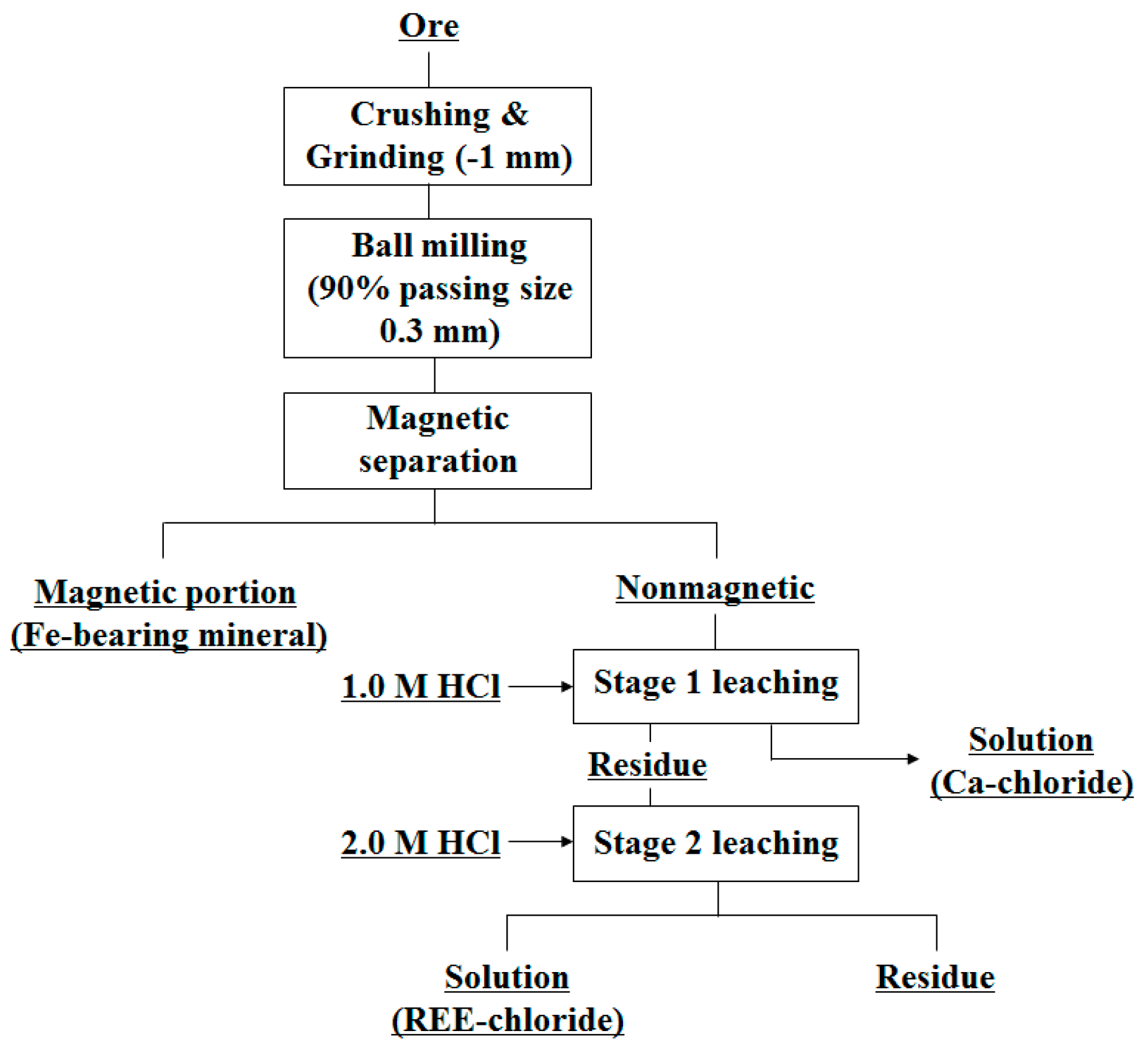
Figure 7 shows the variation in the leaching levels with the initial concentration of hydrochloric acid. Almost no REEs were leached when the concentration was less than 1.0 M. At higher concentrations of hydrochloric acid, the leaching levels increased sharply. At an acid concentration of 2.0 M, nearly 100% of the REEs were leached. However, the amount of Ca in the leachate was also very high (27,480 mg/L), which would undoubtedly cause complications during REE recovery by either precipitation or solvent extraction.
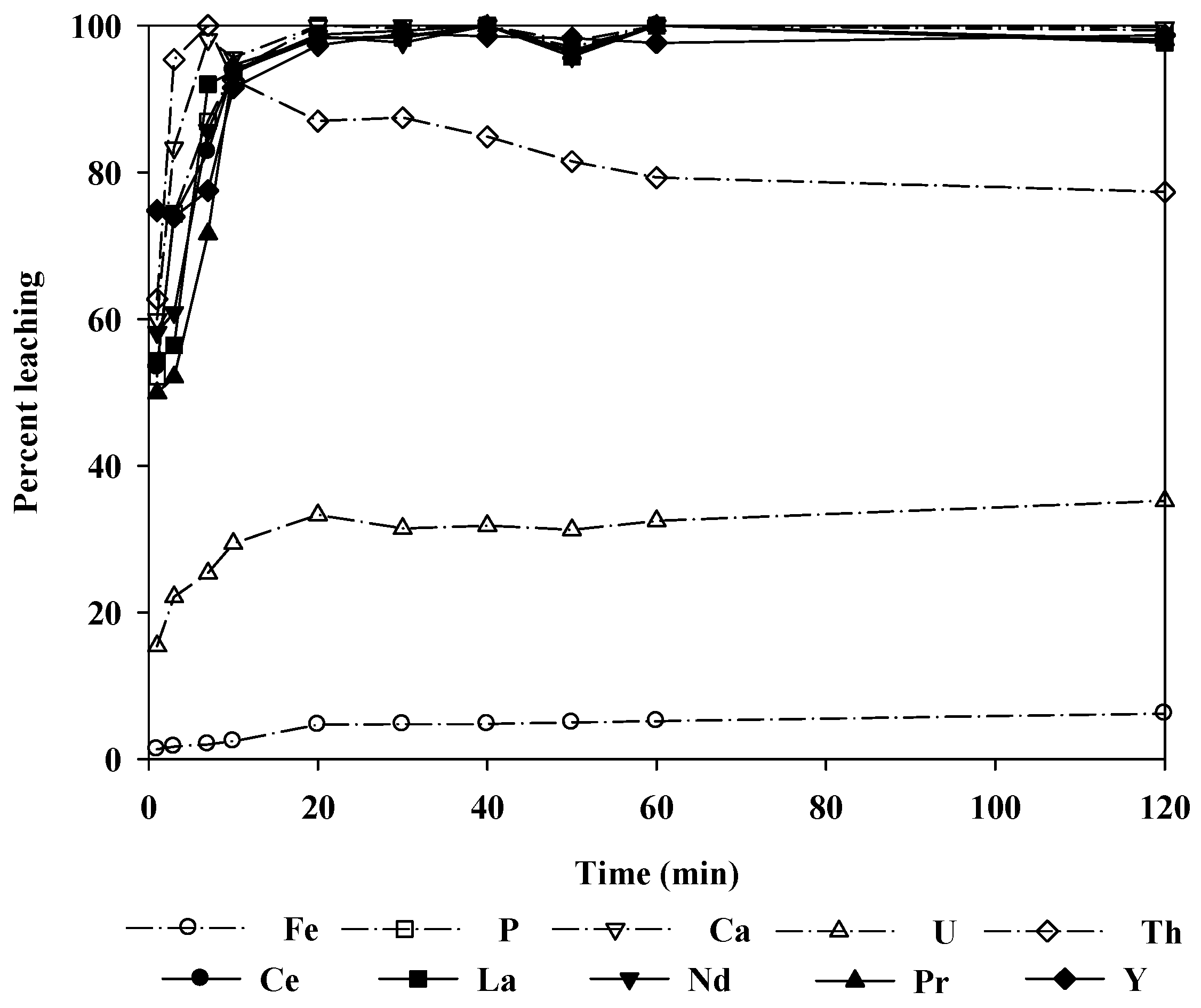
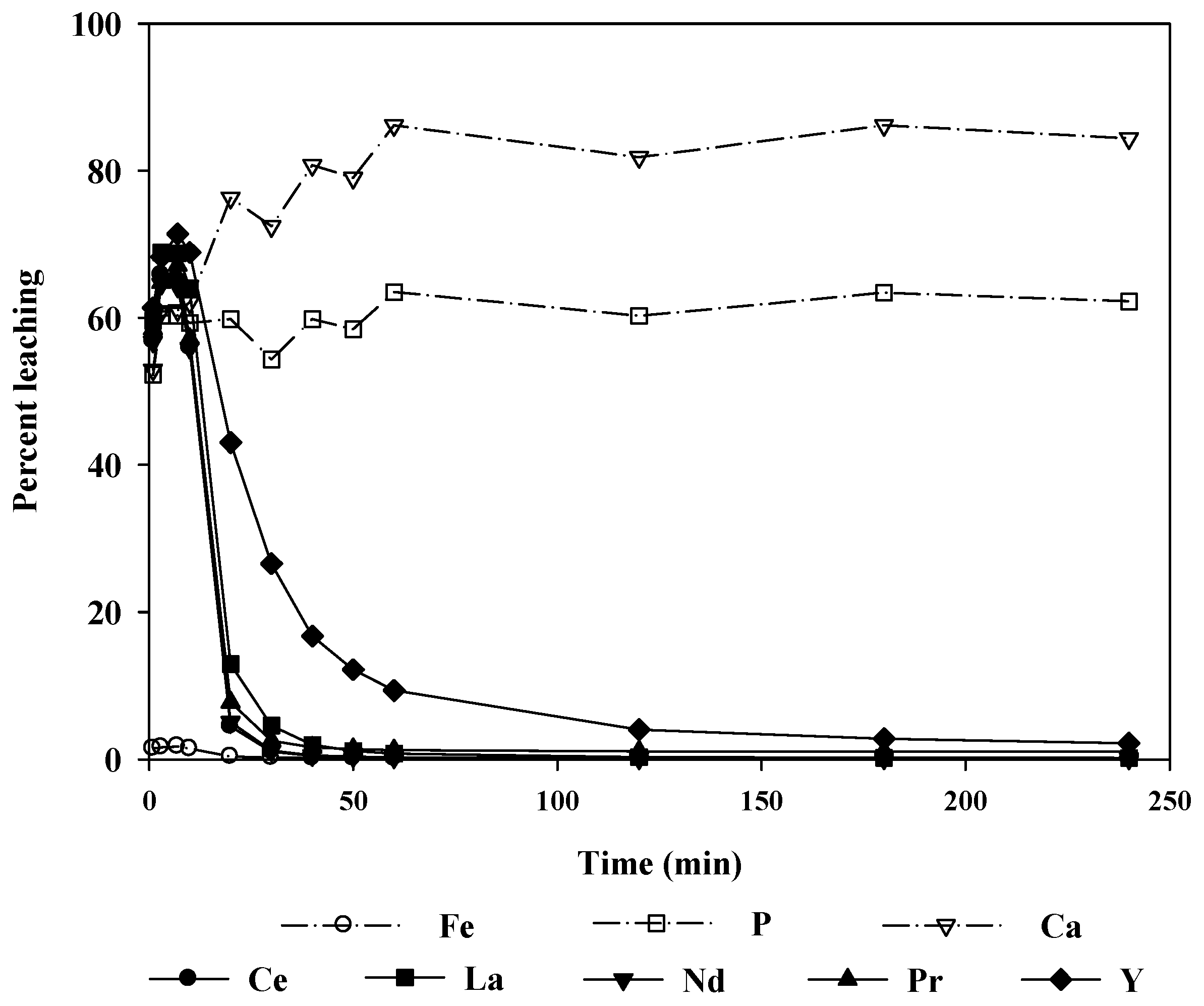
Figure 8 shows the change in the leaching levels with time when 2.0 M hydrochloric acid was used as the leaching agent. More than 90% of the REEs were leached in 10 min. At 1 h, almost 100% of the REEs were leached, and the reaction reached equilibrium in 2 h. It could be noted that the rate of the leaching reaction was quite fast. Thus, it was sufficient to leach the ore at room temperature for a relatively short time.
Unfortunately, the radioactive elements U and Th were also leached with the REEs, although the level of radioactivity was statistically insignificant for the ore treated in this study (U: 45 mg/L, Th: 24 mg/L). Additionally, the Th leaching level decreased with time because Th precipitated as phosphate in the relatively low pH region. Thus, the radioactivity of the leaching solution could be further decreased. Note, however, that if the radioactivity of the ore was significant, the removal of radioactive elements from the leachate would require the use of separation techniques such as selective precipitation or the solvent extraction method.
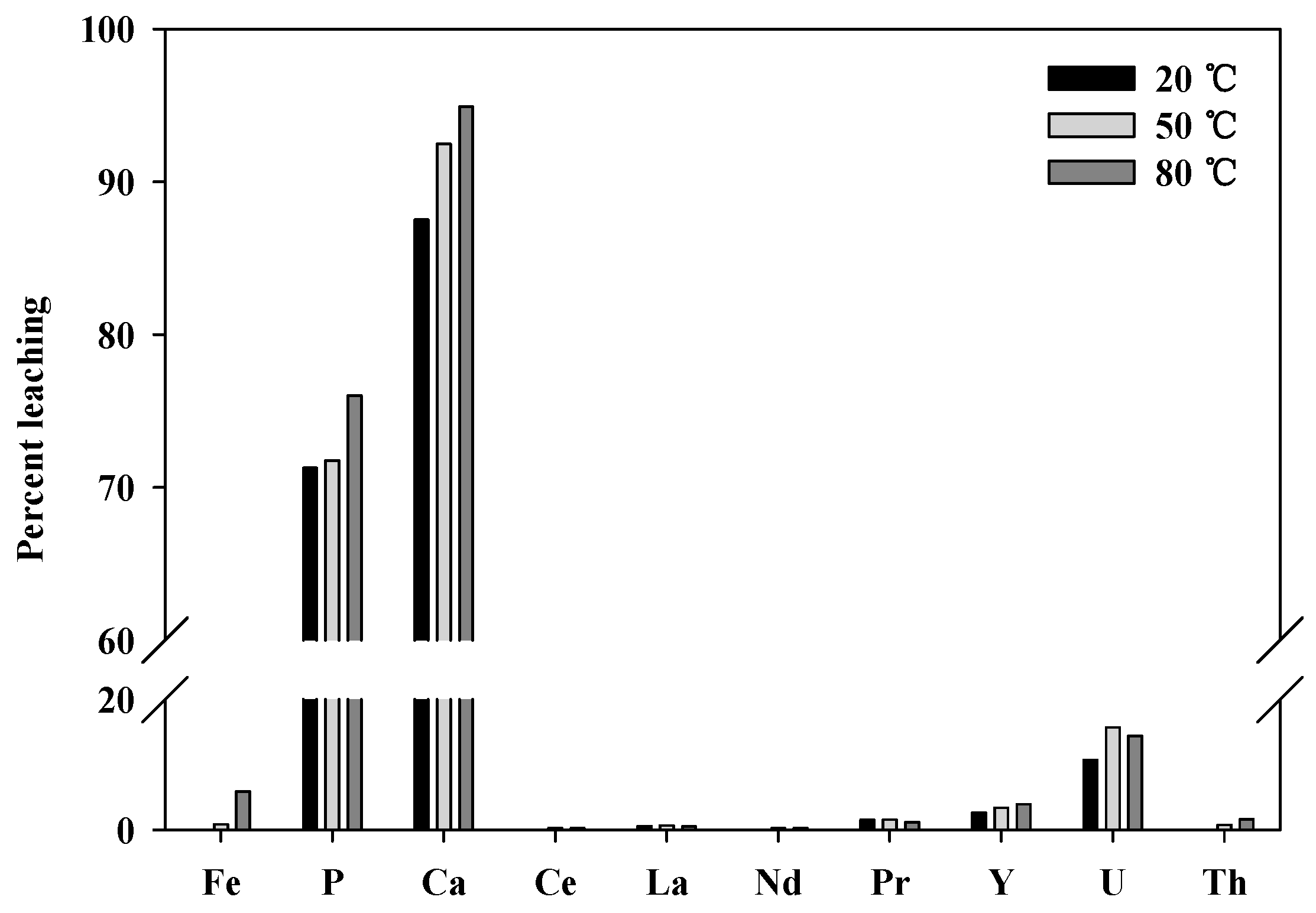
Leaching tests were conducted at elevated temperatures to determine whether the leaching levels and reaction rate could be improved. Because almost 100% of the REEs were leached using 2.0 M hydrochloric acid in an hour, tests of the temperature dependence were conducted using 1.0 M hydrochloric acid at 20, 50, and 80 °C. As shown in
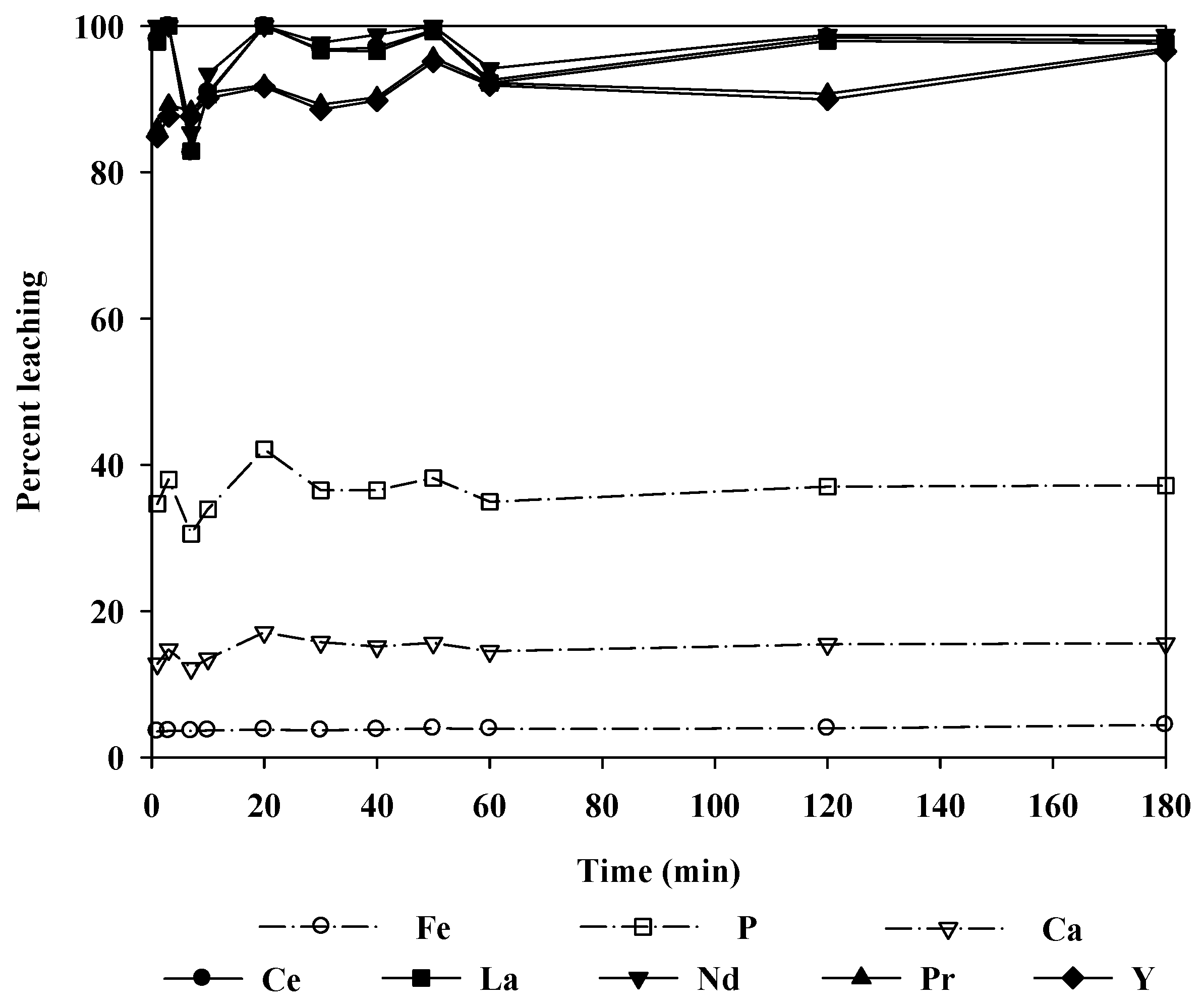
Figure 9, the overall leaching levels for all REEs did not increase significantly; however, other impurity ions (Fe, P, Ca, and U) were leached more as the temperature increased. Therefore, it is preferable to operate with a higher concentration of hydrochloric acid at room temperature than to operate at elevated temperatures.
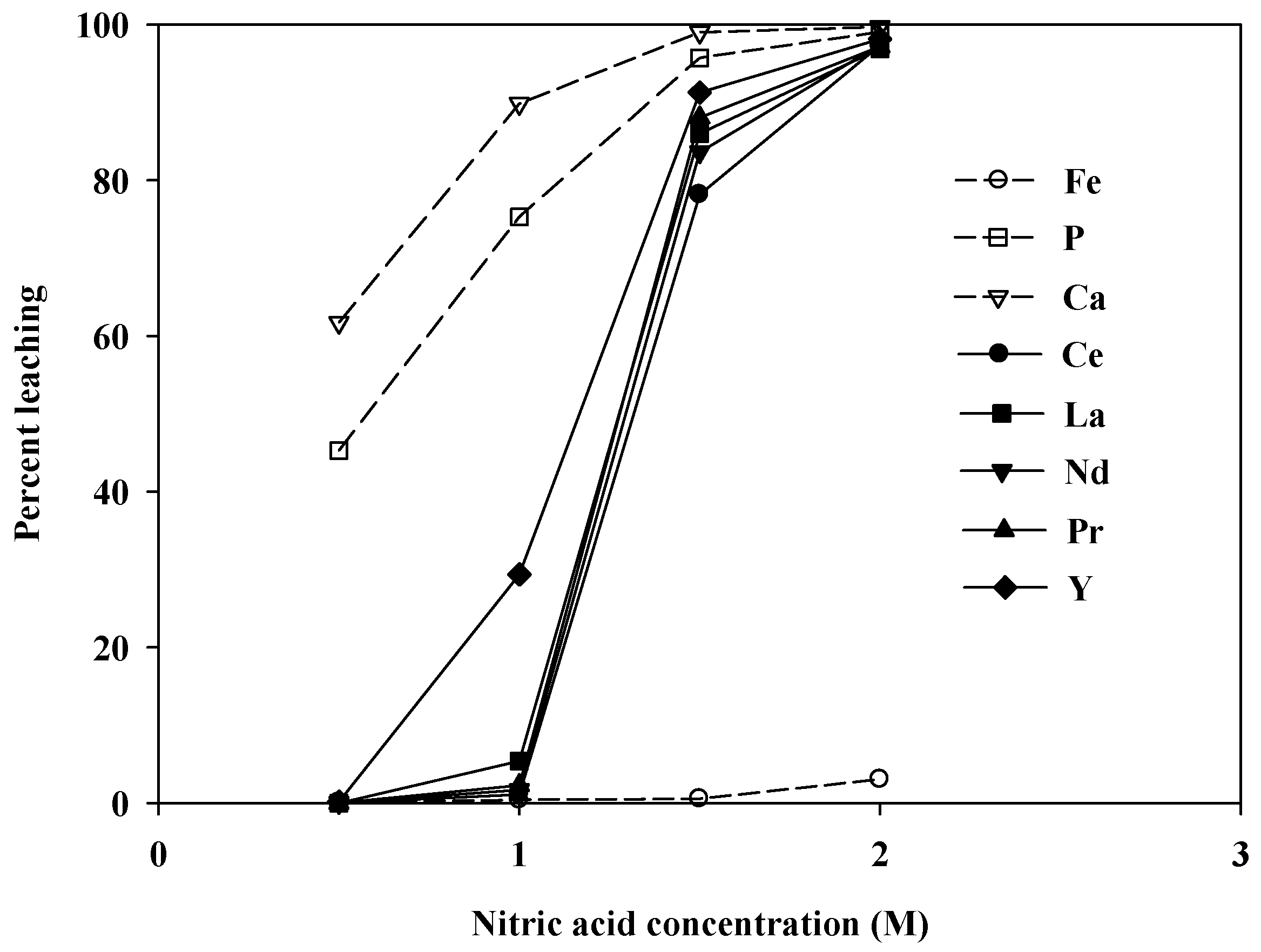
Figure 10 shows the leaching levels at varying concentrations of nitric acid. No REEs, aside from Y, were leached at 1.0 M, whereas impurities such as P and Ca were leached quite well. However, as the acid concentration was increased to 2.0 M, almost 100% of the REEs were leached. These results were similar to the hydrochloric acid case. Because nitric acid is generally more expensive than hydrochloric acid, it was not considered in later sections. Consequently, hydrochloric acid was chosen as the optimum leaching agent for this apatite REE ore, and the effects of other variables on the leaching efficiency were investigated using hydrochloric acid in later experiments.


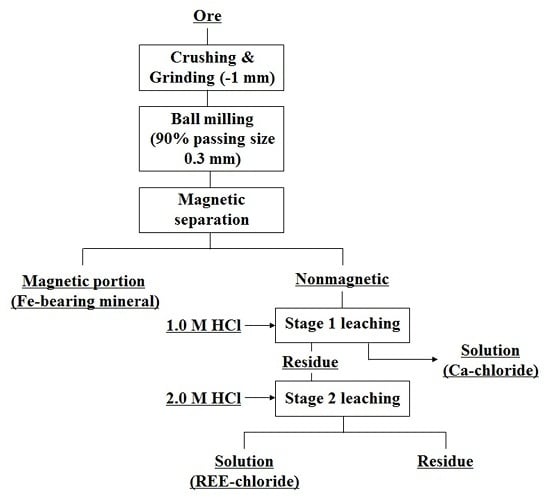















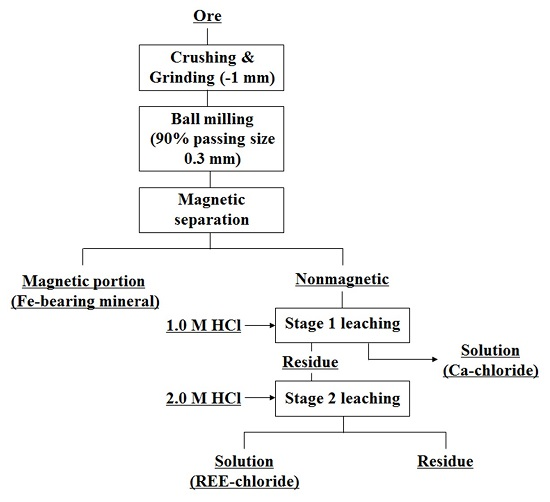{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}