Determination of Au(III) and Ag(I) in Carbonaceous Shales and Pyrites by Stripping Voltammetry

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Apparatus and Electrolysis Procedure
2.2. Reagents and Solutions
2.3. Sample Preparation
2.3.1. Preparation of Samples for the Determination of Ag(I) Ions
2.3.2. Preparation of Samples for Determination of Au(III) Ions
3. Results and Discussion
3.1. Objects of Analysis
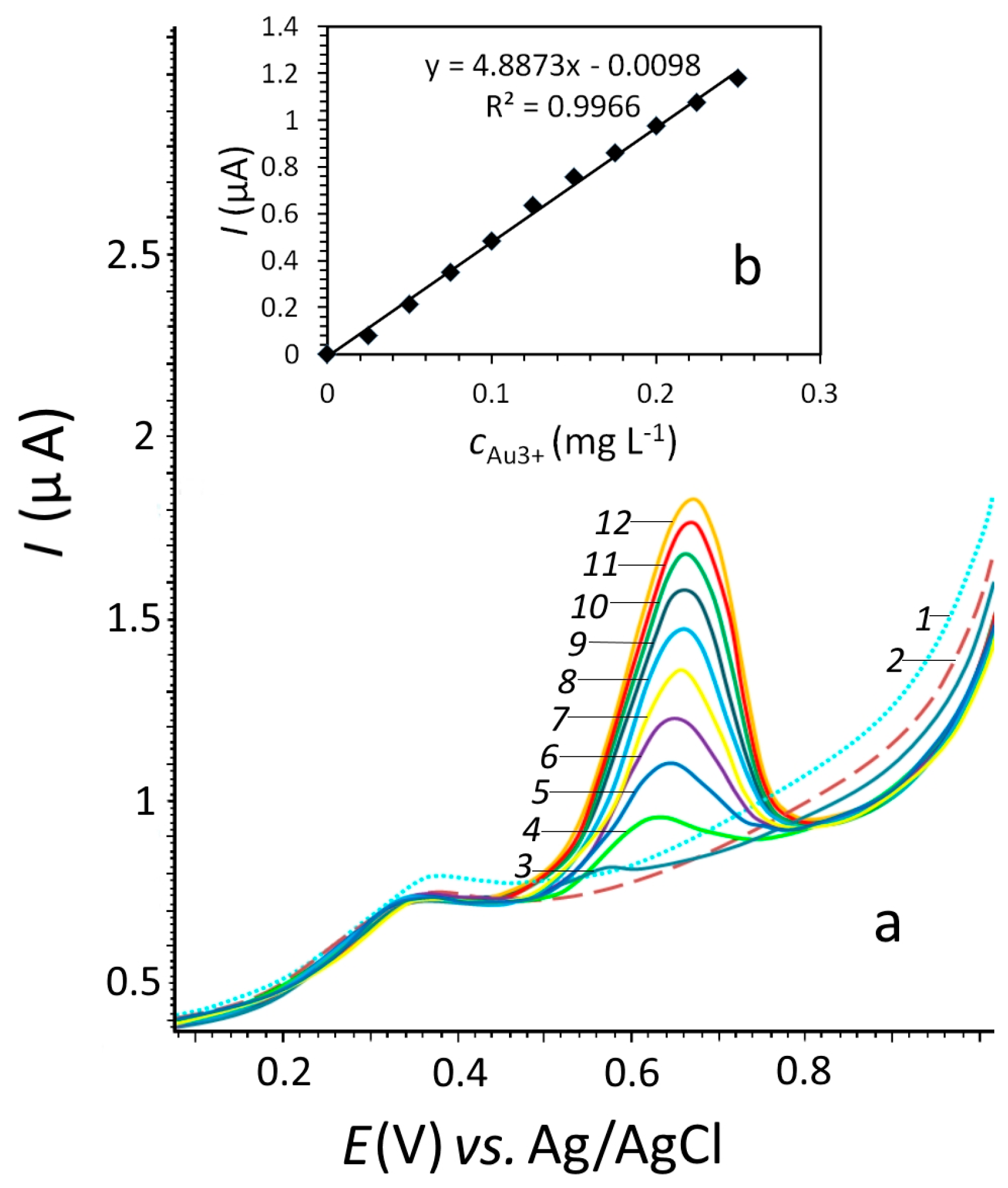
3.2. Determination of Silver(I) ions by Stripping Voltammetry
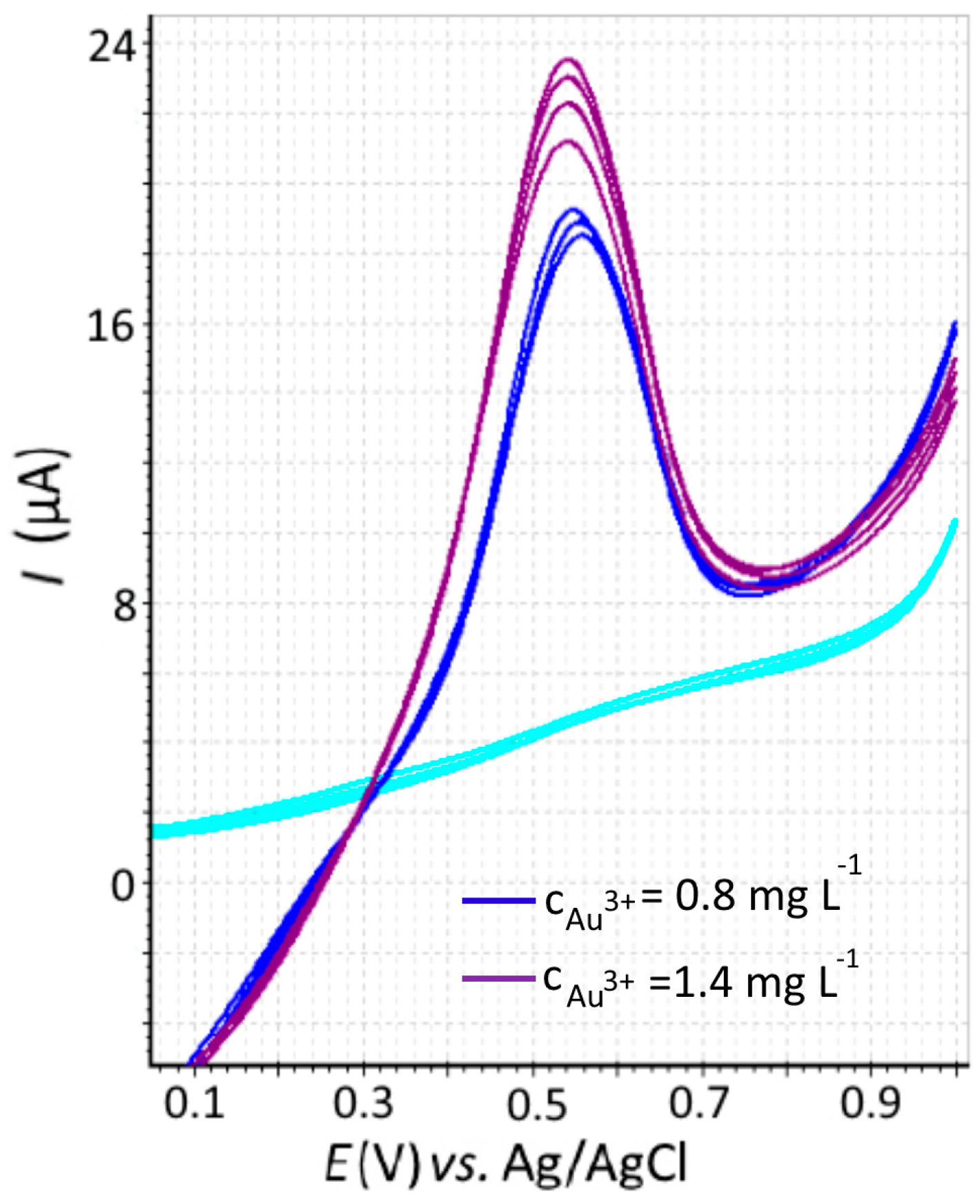
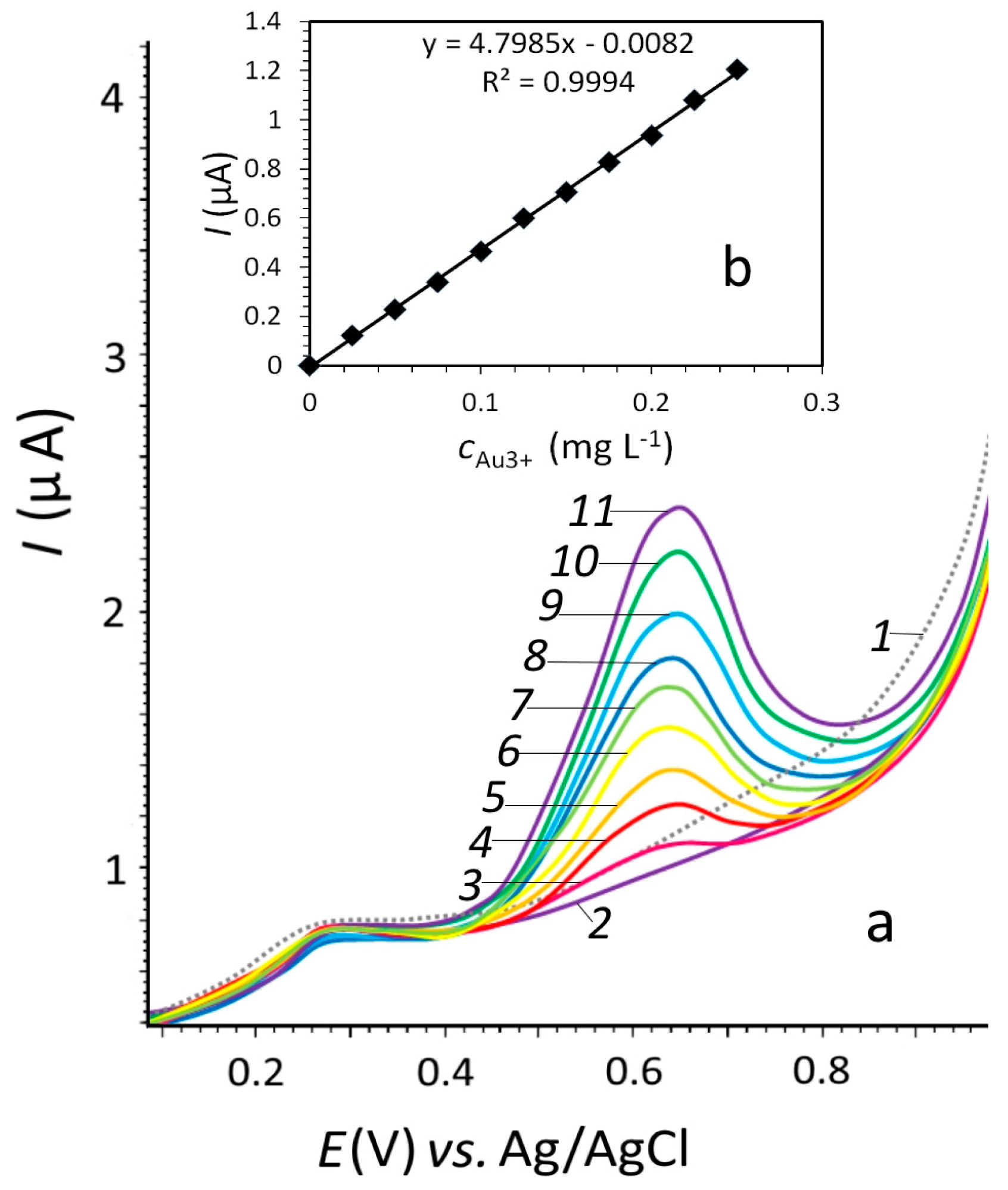
3.3. Determination of Gold(III) Ions by Stripping Voltammetry
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chernyshov, N.M. Noble–metal minerals in ores of the black–shale type in the Voronezh Crystalline Massif, central Russia. Geol. Ore Depos. 2009, 5, 684–697. [Google Scholar] [CrossRef]
- Alexandrova, T. Technological aspects of recovery of precious metals from carbonaceous ores. In Proceedings of the 28th International Mineral Processing Congress (IMPC 2016), Quebec City, QC, Canada, 11–15 September 2016. [Google Scholar]
- Vasilyeva, I.E.; Shabanova, E.V.; Razvozzhaeva, E.A. Noble metals in the insoluble carbonaceous substance of black shales and ores: Direct atomic emission data. Geochem. Int. 2012, 50, 771–776. [Google Scholar] [CrossRef]
- Varshal, G.M. Noble–metal accumulation by carbonaceous material. Geochem. Int. 1995, 32, 47–56. [Google Scholar]
- Zeng, X.; Cai, J.G. Relationship between mineral and organic matter in shales: The case of shahejie formation, Dongying Sag, China. Minerals 2018, 8, 222. [Google Scholar] [CrossRef]
- Dobrowolski, R.; Kuryło, M.; Otto, M.; Mróz, A. Determination of gold in geological materials by carbon slurry sampling graphite furnace atomic absorption spectrometry. Talanta 2012, 99, 750–757. [Google Scholar] [CrossRef]
- Sorokin, A.P.; Eirish, L.V.; Kuz’minykh, V.M. Noble metal mineralization in carbonaceous rocks: A synthesis of data on the Russian Far East. Russ. J. Pac. Geol. 2007, 1, 43–54. [Google Scholar] [CrossRef]
- Mitkin, V.N.; Galizky, A.A.; Korda, T.M. Some observations on the determination of gold and the platinum–group elements in black shales. Geostand. Newsl. 2000, 24, 227–240. Available online: https://onlinelibrary.wiley.com/doi/epdf/10.1111/j.1751-908X.2000.tb00774.x (accessed on 5 November 2018). [CrossRef]
- Berdnikov, N.; Balaram, V.; Cherepanov, A.; Avdeev, D.; Konovalova, N.; Sukharulidze, G. Some observations on the determination of platinum group elements and gold in black shales. Curr. Sci. 2010, 99, 518–521. [Google Scholar]
- Losev, V.N.; Buiko, E.V.; Elsuf’Ev, E.V.; Maznyak, N.V.; Trofimchuk, A.K. Silver(I) sorption by silica gels chemically modified with mercaptopropyl or dipropyl disulfide groups. Russ. J. Inorg. Chem. 2006, 51, 565–568. [Google Scholar] [CrossRef]
- Losev, V.N.; Borodina, E.V.; Buiko, O.V. Sorption-spectrometric determination of palladium and gold using silica chemically modified with dipropyl disulfide groups. J. Anal. Chem. 2014, 69, 413–419. [Google Scholar] [CrossRef]
- Tu, Z.; Lu, S.; Chang, X.; Li, Z.; Hu, Z.; Zhang, L.; Tian, H. Selective solid–phase extraction and separation of trace gold, palladium and platinum using activated carbon modified with ethyl-3-(2-aminoethylamino)-2-chrobut-2-enoate. Microchim. Acta 2011, 173, 231–239. [Google Scholar] [CrossRef]
- Torgov, V.G.; Kostin, G.A.; Us, T.V.; Korda, T.M.; Drapailo, A.B. Palladium and silver extraction with thiacalix[4]arenes and their acyclic analogs from carbonate and ammonia-carbonate solutions. Russ. J. Inorg. Chem. 2015, 60, 372–378. [Google Scholar] [CrossRef]
- Gevorgyan, A.M.; Vanyukov, V.V.; Vakhnenko, S.V. Determination of gold by stripping voltammetry using a modified carbon paste electrode. J. Anal. Chem. 2002, 57, 253–254. [Google Scholar] [CrossRef]
- Davies, T.J. Anodic stripping voltammetry with graphite felt electrodes for the trace analysis of silver. Analyst 2016, 141, 4742–4748. [Google Scholar] [CrossRef] [PubMed]
- Nghi, T.V.; Vydra, F. Stripping voltammetric determination of silver in non-–medium after extraction with dithizone. Collect. Czech. Chem. Commun. 1975, 40, 1485–1489. [Google Scholar] [CrossRef]
- Ustinova, E.M.; Gorchakov, E.V. Determination of platinum and palladium in the presence of other metals in the mineral raw materials. Key En. Mater. 2016, 712, 332–337. [Google Scholar] [CrossRef]
- Doronina, M.S.; Karpov, Y.A.; Baranovskaya, V.B. Combined methods of analysis of metal-containing raw material. J. Inorg. Mater. 2017, 53, 1411–1417. Available online: https://link.springer.com/content/pdf/10.1134%2FS0020168517140047.pdf (accessed on 6 November 2018). [CrossRef]
- Karpov, Y.A.; Baranovskaya, V.B. Problems of analytical control in the production of rare and precious metals. Russ. J. Non-Ferr Met. 2018, 59, 374–384. [Google Scholar] [CrossRef]
- Kosseva, M.R.; Kennedy, J.F. Analytical Voltammetry and Polarography. In Handbook of Analytical Techniques, 2nd ed.; Günzler, H., Williams, A., Eds.; WILEY–VCH Verlag GmbH: Weinheim, Germany, 2001; p. 1182. ISBN 3527301658. Available online: http://web.uni-plovdiv.bg/plamenpenchev/mag/books/anchem/Handbook%20of%20Analytical%20Techniques,%202%20Volume%20Set.pdf (accessed on 7 November 2018).
- Kavanoz, M.; Gulce, H.; Yildiz, A. Anodic stripping voltammetric determination of gold on a polyvinylferrocene coated glassy carbon electrode. Turk. J. Chem. 2004, 28, 287–297. [Google Scholar]
- Meepun, N.; Siriket, S.; Dejmanee, S.; Ratana-Ohpas, R. Stripping chronopotentiometry: An alternative method for the determination of gold in geological samples. Walailak J. Sci. Technol. 2010, 7, 61–67. Available online: https://www.researchgate.net/publication/242349648_53-72-1-PB (accessed on 7 November 2018).
- Monieen, H. Inverse voltammetric determination of small amounts of gold by peak potential measurements. J. Anal. Chem. 1968, 237, 409. [Google Scholar]
- Brainina, K.Z.; Gornostaeva, T.D.; Pronin, V.A. Preconcentration in polarographic analysis discharge–ionization of gold in bromide–chloride solutions. J. Anal. Chem. USSR 1979, 34, 831–836. [Google Scholar]
- Jahandari, S.; Taher, M.A.; Fazelirad, H.; Sheikhshoai, I. Anodic stripping voltammetry of silver(I) using a carbon paste electrode modified with multi-walled carbon nanotubes. Microchim. Acta 2013, 180, 347–354. [Google Scholar] [CrossRef]
- Tashkhourian, J.; Javadi, S.; Ana, F.N. Anodic stripping voltammetric determination of silver ion at a carbon paste electrode modified with carbon nanotubes. Microchim. Acta 2011, 173, 79–84. [Google Scholar] [CrossRef]
- Budnikov, G.K.; Maistrenko, V.N.; Murinov, Y.I. Voltammetry with Modified Electrodes; Science: Moscow, Russia, 1994; p. 236. ISBN 5-02-001842-2. [Google Scholar]
- Barón-Jaimez, J.; Joya, M.R.; Jose Barba Ortega, J. Anodic stripping voltammetry—ASV for determination of heavy metals. J. Phys. Conf. Ser. 2013, 466, 012023. Available online: http://iopscience.iop.org/article/10.1088/1742-6596/466/1/012023/pdf (accessed on 7 November 2018). [CrossRef]
- Dyachenko, E.N.; Kolpakova, N.A.; Oskina, Y.A. Determination of gold by stripping voltammetry in platinum gold ore mineral raw materials for grafite electrode modified by bismuth. Procedia Chem. 2014, 10, 47–50. [Google Scholar] [CrossRef]
- Postnikov, P.S.; Trusova, M.E.; Filimonov, V.D.; Slepchenko, G.B. The new method of covalent grafting of organic functional groups on the graphite/GC electrodes using arenediazonium tosylates. In Proceedings of the 15th European Conference on Composite Materials: Composites at Venice, ECCM 2012, Venice, Italy, 24–28 June 2012. [Google Scholar]
- Sladkov, V.E.; Osipova, E.A. Effect of polyethylenimine on the selectivity of determining silver(I) by stripping voltammetry in the presence of Copper(II). J. Anal. Chem. 2001, 56, 43–46. [Google Scholar] [CrossRef]
- Chooto, P. Modified electrodes for determining trace metal ions. In Applications of the Voltammetry; Stoytcheva, M., Zlatev, R., Eds.; IntechOpen: London, UK, 2017; ISBN 978-953-51-3216-5. Available online: https://www.intechopen.com/books/applications-of-the-voltammetry/modified-electrodes-for-determining-trace-metal-ions (accessed on 5 November 2018). [CrossRef]
- Qi, J.; Zhai, Y.; St-Pierre, J. Effects of Ethylene Glycol and Caprolactam on the ORR and HOR Performances of Pt/C Catalysts. J. Electrochem. Soc. 2016, 163, 618–626. [Google Scholar] [CrossRef]
- Oparin, V.N.; Sekisov, A.G.; Trubachev, A.I. Promising mining technologies for gold placers in transbaikalia. J. Min. Sci. 2017, 53, 489–496. [Google Scholar] [CrossRef]
- Bakanov, V.I.; Larina, N.V. Mathematical model of the surface of electrodeposited metals. In Proceedings of the Electrochemistry and Surface Technology: Abstracts Intern Conference, Moscow, Russia, 4–8 June 2001; p. 147. [Google Scholar]
- Borkhodoev, V.Y. Estimation of limits of detection and determination in X-ray fluorescence analysis by the dependence of the relative standard deviation on analyte concentration. J. Anal. Chem. 2016, 71, 872–877. [Google Scholar] [CrossRef]
- Pyrzyńska, K. Recent developments in the determination of gold by atomic spectrometry techniques. Spectrochim. Acta Part B At. Spectrosc. 2005, 60, 1316–1322. Available online: https://www.sciencedirect.com/science/article/pii/S0584854705002016 (accessed on 8 November 2018). [CrossRef]
- Kaplin, A.A.; Stromberg, A.G.; Pikula, N.P. Use of the method of polarography with accumulation (inversion voltamperometry) in the analysis of macro– and micro–objects. Ind. Lab. (USSR) 1977, 43, 463–480. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SS | Certified Value, g t−1 | Ag Content, g t−1 | Relative Error (%) | ||
|---|---|---|---|---|---|
| AES | ICP–MS | SV | |||
| Copper sludge | 6.38 | 5.8 | 5.5 | 5.6 ± 0.2 | 11.6 |
| Nickel sludge | 0.19 | 0.2 | 0.19 | 0.2 ± 0.02 | 9.9 |
| No. of Experiment | Ag Content, g t−1 | C, g t−1 | ||
|---|---|---|---|---|
| 1 | 13.8 | 11.7 | 11.0 | 12.2 ± 1.6 |
| 2 | 9.5 | 17.9 | 9.09 | 12.2 ± 3.8 |
| 3 | 6.1 | 16.3 | 13.7 | 12.0 ± 3.2 |
| SS | Certified value, g t−1 | Content of Au, g t−1 | Relative Error (%) | ||
|---|---|---|---|---|---|
| AES | ICP–MS | SV (n = 5) | |||
| Copper sludge | 0.21 | 0.25 | 0.26 | 0.25 ± 0.02 | 19.05 |
| Nickel sludge | 0.059 | 0.060 | 0.058 | 0.060 ± 0.02 | 1.69 |
| No. | Value of Gold Contents, g t−1 | Average Content of Au, g t−1 | ||
|---|---|---|---|---|
| 1 | 1.6 | 2.2 | 2.9 | 2.2 ± 0.6 |
| 2 | 1.8 | 1.9 | 2.6 | 2.6 ± 0.7 |
| 3 | 2.0 | 1.8 | 2.7 | 2.2 ± 0.3 |
| No. | Value of Gold Contents, g t−1 | Average Content of Au, g t−1 | ||
|---|---|---|---|---|
| 1 | 1.4 | 0.8 | 0.9 | 1.0 ± 0.2 |
| 2 | 1.2 | 0.7 | 0.8 | 0.9 ± 0.2 |
| 3 | 1.0 | 0.9 | 0.8 | 0.9 ± 0.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolpakova, N.A.; Sabitova, Z.K.; Sachkov, V.I.; Medvedev, R.O.; Nefedov, R.A.; Orlov, V.V. Determination of Au(III) and Ag(I) in Carbonaceous Shales and Pyrites by Stripping Voltammetry. Minerals 2019, 9, 78. https://doi.org/10.3390/min9020078
Kolpakova NA, Sabitova ZK, Sachkov VI, Medvedev RO, Nefedov RA, Orlov VV. Determination of Au(III) and Ag(I) in Carbonaceous Shales and Pyrites by Stripping Voltammetry. Minerals. 2019; 9(2):78. https://doi.org/10.3390/min9020078
Chicago/Turabian StyleKolpakova, Nina A., Zhamilya K. Sabitova, Victor I. Sachkov, Rodion O. Medvedev, Roman A. Nefedov, and Vladislav V. Orlov. 2019. "Determination of Au(III) and Ag(I) in Carbonaceous Shales and Pyrites by Stripping Voltammetry" Minerals 9, no. 2: 78. https://doi.org/10.3390/min9020078
APA StyleKolpakova, N. A., Sabitova, Z. K., Sachkov, V. I., Medvedev, R. O., Nefedov, R. A., & Orlov, V. V. (2019). Determination of Au(III) and Ag(I) in Carbonaceous Shales and Pyrites by Stripping Voltammetry. Minerals, 9(2), 78. https://doi.org/10.3390/min9020078