Coprecipitation of Co2+, Ni2+ and Zn2+ with Mn(III/IV) Oxides Formed in Metal-Rich Mine Waters


1
Area of Geochemistry and Sustainable Mining, Department of Geological Resources Research, Spanish Geological Survey (IGME), Calera 1, Tres Cantos, 28760 Madrid, Spain
2
Department of Mineralogy and Petrology, Faculty of Science and Technology, University of the Basque Country (UPV/EHU), Apdo. 644, 48080 Bilbao, Spain
*
Author to whom correspondence should be addressed.
Minerals 2019, 9(4), 226; https://doi.org/10.3390/min9040226
Submission received: 19 February 2019
/
Revised: 3 April 2019
/
Accepted: 6 April 2019
/
Published: 10 April 2019
(This article belongs to the Special Issue Acid Mine Drainage Recovery)
Abstract
:Manganese oxides are widespread in soils and natural waters, and their capacity to adsorb different trace metals such as Co, Ni, or Zn is well known. In this study, we aimed to compare the extent of trace metal coprecipitation in different Mn oxides formed during Mn(II) oxidation in highly concentrated, metal-rich mine waters. For this purpose, mine water samples collected from the deepest part of several acidic pit lakes in Spain (pH 2.7–4.2), with very high concentration of manganese (358–892 mg/L Mn) and trace metals (e.g., 795–10,394 µg/L Ni, 678–11,081 µg/L Co, 259–624 mg/L Zn), were neutralized to pH 8.0 in the laboratory and later used for Mn(II) oxidation experiments. These waters were subsequently allowed to oxidize at room temperature and pH = 8.5–9.0 over several weeks until Mn(II) was totally oxidized and a dense layer of manganese precipitates had been formed. These solids were characterized by different techniques for investigating the mineral phases formed and the amount of coprecipitated trace metals. All Mn oxides were fine-grained and poorly crystalline. Evidence from X-Ray Diffraction (XRD) and Scanning Electron Microscopy coupled to Energy Dispersive X-Ray Spectroscopy (SEM–EDX) suggests the formation of different manganese oxides with varying oxidation state ranging from Mn(III) (e.g., manganite) and Mn(III/IV) (e.g., birnessite, todorokite) to Mn(IV) (e.g., asbolane). Whole-precipitate analyses by Inductively Coupled Plasma-Mass Spectrometry (ICP-MS), Inductively Coupled Plasma-Atomic Emission Spectrometry (ICP-AES), and/or Atomic Absorption Spectrometry (AAS), provided important concentrations of trace metals in birnessite (e.g., up to 1424 ppm Co, 814 ppm Ni, and 2713 ppm Zn), while Co and Ni concentrations at weight percent units were detected in asbolane by SEM-EDX. This trace metal retention capacity is lower than that observed in natural Mn oxides (e.g., birnessite) formed in the water column in a circum-neutral pit lake (pH 7.0–8.0), or in desautelsite obtained in previous neutralization experiments (pH 9.0–10.0). However, given the very high amount of Mn sorbent material formed in the solutions (2.8–4.6 g/L Mn oxide), the formation of these Mn(III/IV) oxides invariably led to the virtually total removal of Co, Ni, and Zn from the aqueous phase. We evaluate these data in the context of mine water pollution treatment and recovery of critical metals.
1. Introduction
The potential of manganese oxides to retain many different trace metals from natural and engineered environments is well known and has been recognized in very different settings [1,2,3,4]. Among over 30 different manganese oxide minerals with a MnOx-like formula, birnessite and todorokite are the most profusely studied by their high adsorbing capacity for trace metals like Ni, Co, or Zn [5,6,7,8,9,10,11,12,13,14,15,16,17,18]. In small quantities, these trace metals are micronutrients and thus necessary to sustain life [19]. However, at very high concentrations they become poisonous and can be harmful for aquatic life, as well as for animals and humans [20,21,22]. Therefore, there exists growing interest in finding efficient remediation technologies for removing these metals from polluted environments where they can be present at very high concentrations, such as in mine waters or industrial effluents. On the other hand, Ni and Co are critical raw materials and their use in corrosion-resistant metallic alloys, batteries, catalysis, and metal coatings, make them highly valuable in the modern industry [23]. Their current prices (as for January 2019) are several times higher than those of other base metals (e.g., 17.24 USD/lb for Co or 5.3 USD/lb for Ni, as compared to 2.7 USD/lb for Cu, 1.17 USD/lb for Zn, or 0.86 USD/lb for Al [23]). The price of cobalt has been even higher in the past, reaching peak values of 41 USD/lb in February 2018 [24].
Under this dual perspective (environmental vs. industrial/economic), it may be reasonable and even advisable, to consider possible options for the recovery of these metals from mine waters based on the high adsorption capacity of manganese oxides. However, despite the enormous amounts of studies on trace metal coprecipitation with manganese oxides in the last decades [4,5,6,7,8,9,10,11,12,13], studies focused on trace metal removal via Mn oxides in mine waters are relatively scarce and have been mostly centered in the US [14,15,16,17]. Fuller et al. (2000) and Kay et al. (2001) [14,15] documented the uptake of Ni and Co by Mn oxides formed by Mn(II) oxidation in an acid mine drainage (AMD)-impacted creek in Arizona. Lee et al. (2002) [16] noted a significant removal of Zn, Co, and Ni by sorption onto undefined manganese oxides in waters contaminated with AMD from the Duchtown Mining District in Tennessee. Tani et al. (2004) [17] documented the sorption of Co(II), Ni(II), and Zn(II) on biogenic manganese oxides produced by a Mn-oxidizing fungus. In the Appalachian coal mine drainage treatment systems (Pennsylvania), where Mn concentration is between 27 and 150 mg/L, Tan et al. (2010) [18] found an effective removal of trace metals (Co, Ni and Zn) by MnOx oxides consisting in poorly crystalline birnessite (both hexagonal and triclinic) and todorokite. Trace element portioning in these mine waters took place at pH as low as 5.0 and temperatures as low as 2 °C, but was enhanced at neutral pH and warmer temperatures. The oxidation of Mn(II) was reported to be biologically mediated, and at the same time, strongly catalyzed by mineral surfaces, including previously formed MnOx particles [18]. These authors considered this passive removal of Mn to be considerably cheaper than the active option (consisting in neutralizing the mine waters to pH 9–10 by NaOH addition in treatment plants), which usually requires important costs of chemical reagents [18]. Biological removal of Mn at near neutral pH levels has been a common practice in other Pennsylvanian mining districts since the early 1990’s [19], and Mn concentrations were satisfactorily reduced from a few tens of mg/L to below 2 mg/L Mn (e.g., Shade Mining site in Somerset County [25]). In a review of passive systems for manganese removal in Pennsylvanian mine sites, Rose et al. (2003) observed that open limestone channels showed good Mn removal rates in mine sites with 25–30 mg/l Mn, and thus represented a satisfactory alternative to pyrolusite systems or limestone beds [26]. Outside the US, Bamforth et al. (2006) [27] studied the removal of manganese in coal mine drainage in the Durham County (UK), where Mn(II) was present at low concentrations around 1–3 mg/L. The authors found that Mn precipitation in the form of diverse mineral forms including carbonates (mostly kutnahorite) and amorphous Mn oxy-hydroxides likely consisting of manganite and pirolusite. More recently, Teixeira et al. (2017) [28] conducted lab experiments to compare the efficiency of different oxidants in reducing the concentration of dissolved Mn to less than 1 mg/L. These authors found that the use of NaClO or a combination of the former with H2O2 was effective in removing virtually all dissolved Mn (10 mg/L) in only 60 min, and that using only O2 or H2O2 as oxidant was ineffective [28].
Studies of Mn(II) oxidation and Mn oxide formation in very acidic and metal-concentrated mine drainage from metal mines, where manganese can be present at extremely high concentrations, are virtually non-existent. This is mainly because the extremely acidic conditions prevailing in these sites makes Mn(II) totally conservative (i.e., oxidation does not proceed at measurable rates), and it can only be removed from the solutions in treatment plants by adding lime or sodium hydroxide. Besides, the supposed absence of Mn(II)-oxidizing microbes (most of them known to be neutrophilic [28,29,30,31,32]), a priori that only allows chemical oxidation, and the sorptive capacity of the abiotic mineral products is usually much lower than the highly reactive Mn bio-oxides [8,9,10]. Thus, the first goal of this study was to investigate which Mn oxides are formed during the oxidation of Mn(II) in very concentrated mine waters. A second and important objective was to compare the extent of trace metal coprecipitation with Mn oxides, and the effects of this coprecipitation on metal attenuation in the solution. For this purpose, we used mine water samples from the deep part of several acidic pit lakes in Spain with very high concentration of manganese and trace metals (Figure 1a–c). For comparison purposes, we also used previous data obtained in a circumneutral pit lake in Northern Spain (Reocín mine, Cantabria; Figure 1d), which represents a singular and unique case in Spain of natural Mn oxide formation with associated trapping of Zn, Co, and Ni [33]. The results of our study could provide the basis for future remediation attempts in heavily contaminated mining districts aimed at removing the most soluble metal cations such as Mn(II), Co(II), Ni(II), or Zn(II) which, at present, can only be totally removed by hydroxide formation at very high pH (>9.5–10) in chemical treatment plants.
2. Materials and Methods
2.1. Field Sampling and Preliminary Neutralization Stage
Water samples (2 L each) were taken from the deepest, anoxic part of several acidic pit lakes (Figure 1). Sampled pit lakes included Guadiana (Herrerías) and La Zarza in the Iberian Pyrite Belt mining district (Huelva, SW Spain), and Brunita mine in La Unión mining district in Cartagena (SE Spain). These abandoned mine sites were chosen, among many others existing in Spain, because they contain some of the highest Mn concentrations known in Spanish mine waters (358–892 mg/L Mn; Table 1), as well as very high concentrations of trace metals like Zn (259–624 mg/L), Co (678–11,081 µg/L), and Ni (795–10,394 µg/L) (Table 1). The pH of these lakes is usually 1.9–2.2 near the surface and 2.7–4.2 closer to the bottom. Previous data on water chemistry and precipitate mineralogy from the Reocín mine pit lake have been also included for comparison (Table 1, Figure 1d).
These samples were stored in polyethylene bottles, cool-preserved, and transported to the laboratory for Mn(II) oxidation experiments. All samples were taken from the deep part of the pit lakes under anoxic and reducing conditions, so that dissolved manganese in these waters was considered to exist entirely as Mn(II). In addition, the low pH of these environments theoretically prevents Mn(II) oxidation [34,35,36,37]. Before the Mn(II) oxidation experiments, the acidic mine waters were neutralized to pH 8.0 to remove other, less soluble major cations such as Fe (originally present as Fe[II] and then oxidized to Fe[III]), Al, and Cu in the form of diverse oxy-hydroxysulfates (e.g., schwertmannite, basaluminite, brochantite) without affecting Mn(II) concentration (abiotic oxidation of Mn(II) below pH 8.0 is known to be very slow to negligible [34,35,36,37]). An important part of zinc was also removed at this pH in the form of poorly crystalline silicates (e.g., willemite-like), though the total removal of this element requires pH > 9.0–9.5 [38]. In this neutralization stage, the pH was adjusted by the dropwise addition of 1 M NaOH in glass beakers at room temperature and constant stirring. Once the pH had been stabilized (checked by continuous pH measurement with previously calibrated pH meters from Orion), the precipitated solids were removed by vacuum-filtration using 0.45 and 0.2 µm nitrocellulose membrane filters (Millipore). The removal of trace elements during this neutralization stage (e.g., As, Cr, or Pb by schwertmannite, or Cu and U by basaluminite) has been studied extensively (e.g., [16,38]) and will not be discussed here.
2.2. Batch Experiments of Mn(II) Oxidation
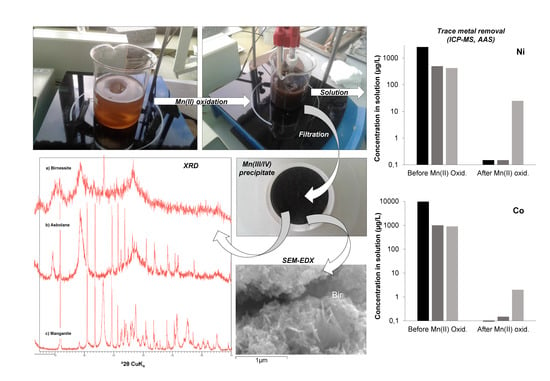
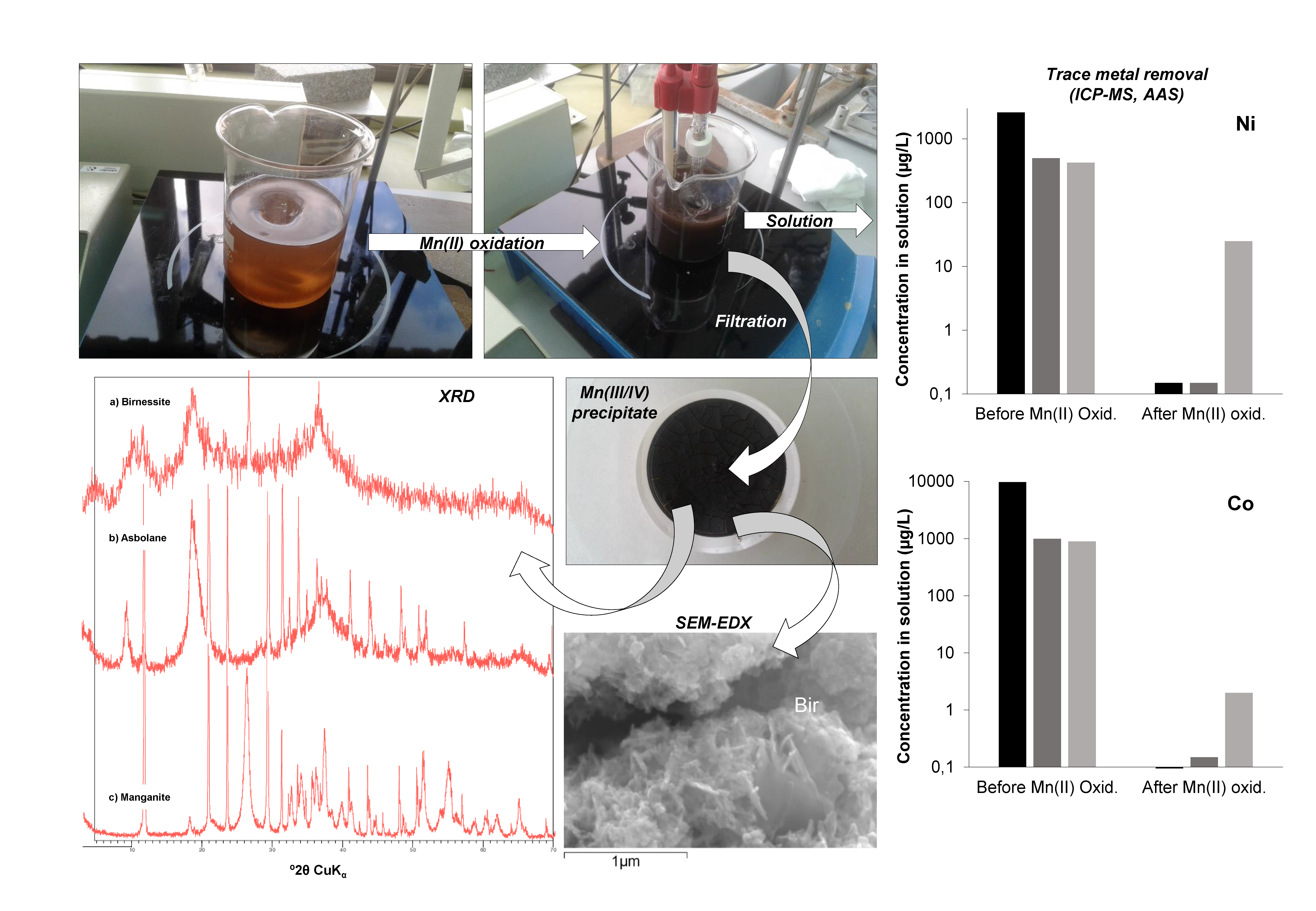
The supernatants obtained in the previous stage and still containing Mn(II) and some other highly soluble cations in solution (including Co, Ni, and part of Zn not precipitated in the previous stage), were subjected to oxidation by continuous stirring in automatic stirring units at room temperature. The beakers were partly closed to avoid excessive evaporation, but oxygen-containing air was allowed to enter the waters through different holes previously made in the plastic caps. Aqueous parameters like pH and redox potential (Eh) were periodically measured to follow the geochemical evolution of the solutions during Mn(II) oxidation. The experiments were also visually controlled, and changes in color and turbidity were annotated to record the appearance of Mn oxides in the different samples (see Figure S1 in Supplementary Materials).
The target pH for all solutions was 9.0 for accelerating the Mn(II) oxidation rate, which increases exponentially in the pH range 8.0–9.5 [35]. However, the oxidation of aqueous manganese and the subsequent precipitation of Mn(III) or Mn(IV), which is generally defined by Reactions (1) or (2) depending on the reaction product formed [3,35,36,37], is a proton-generating process and thus tends to decrease the pH of the parent solutions:
4Mn2+ (aq) + O2 + 6H2O → 4Mn3+OOH(s) + 8H+
Mn2+ (aq) + ½O2 + H2O → Mn4+O2(s) + 2H+
As a consequence, pH was lowered as Mn(II) oxidation proceeded, and this in turn provoked a decrease in the rate of Mn(II) oxidation, which was observed to be negligible below pH 8.0, which is in agreement with previous studies [34,35,36,37]. For this reason, pH required frequent adjustment by adding of a few drops of additional NaOH to maintain the pH between 8.5 and 9.0, and thus sustain the oxidation of Mn(II) at a practical (measurable) rate. At the beginning of the oxidation process, a pale brown colloidal suspension initially formed which, after a few days, turned into a dense, black suspension formed by globular, very fine-grained particles (Figure S1 in Supplementary Materials). The pH increase did not lead to immediate precipitation, and the formation of a black solid took hours or days, indicating that its formation proceeded by oxidative precipitation defined by Reactions (1) and (2).
The oxidation of manganese was considered to be nearly complete in all solutions when Eh and pH were stabilized and an abundant, black precipitate was formed in the bottom of the beakers (Figure S1 in Supplementary Materials). Manganese removal was checked by taking a small sub-sample from the beakers, analyzing it for total Mn concentration by the methods described below, and comparing the results with those measured in the original solutions (which are given in Table 1). The complete oxidation/precipitation of Mn(II) took time intervals between four and five weeks. Considering these time intervals, the resulting average rates of Mn oxide precipitation (which should be nearly equivalent to Mn(II) oxidation rates) were comprised between 2.4 µM L−1 h−1 (solution from Guadiana mine) and 12.3 µM L−1 h−1 (solution from Brunita mine), which are comparable to, though slightly higher than, those reported by Morgan (2005) (e.g., 3.6 µM L−1 h−1 at pH 9.0) [35] or Learman et al. (2011) (e.g., 2.5–3.7 µM L−1 h−1 in the presence of Mn colloids) [31]. It is hypothesized that, because Mn(II) oxidation in the batch reactors proceeded by continuous stirring in dense suspensions with abundant colloids of manganese oxide particles, that oxidation in the final stages of experiments was very likely strongly surface-catalyzed [37]. In addition, considering that the waters had been previously filtered through 0.2 µm pore size and originally consisted in acidic mine water (i.e., supposedly lacking neutrophilic Mn(II)-oxidizing microbes), Mn(II) oxidation was considered to proceed exclusively by an abiotic chemical pathway, either homogeneous or heterogeneous.
The resulting Mn-rich precipitates were recovered by vacuum-filtration using 0.45 µm membrane filters, then washed three times by filtering 50 mL of ultra-pure MilliQ water, air-dried at ambient temperature for at least 48 h, and a fraction was homogenized by gentle milling in an agate mortar mill for their subsequent chemical and mineralogical analyses.
2.3. Mineralogical Identification and Analytical Methods
Synthetic precipitates were analyzed by different techniques at the IGME laboratories at Tres Cantos. A small portion of the samples (10–20 mg) was immediately analyzed by XRD on a PANalytical X’Pert Pro diffractometer (Malvern Panalytical Ltd., Almelo, The Netherlands). XRD conditions included CuKα radiation (40 kV, 40 mA), graphite monochromator and automatic slit, and 2–80 °2θ with 0.02 step size and 1 s per step. The goniometer was calibrated with a silicon standard. The precipitates were also studied on carbon-coated samples by SEM-EDX in a JEOL JSM-7000F scanning electron microscope (JEOL Ltd., Tokyo, Japan) at the SGIker Electron Microscopy Services of the Basque Country University (UPV/EHU). Another fraction of the precipitates was chemically analyzed by ICP-AES, AAS, and ICP-MS in Agilent 7500ce and Varian Vista-MPX instruments (Agilent Technologies Inc., San Clara, CA, USA) after previous acid digestion with HF, HClO4, HNO3, and HCl to determine major cations (e.g., Mn, Na, K, Ca) and trace metals (including Co, Ni, and Zn, but also Ba, Cd, Mo, Se or Pb). Detection limits were usually 1 mg/L for major metals (except Cu and Zn, 0.1 mg/L) and between 0.01 and 0.1 mg/L for most trace elements.
Metal concentrations in the aqueous solutions before and after the experiments were also conducted to quantify Mn removal during Mn(II) oxidation and the influence of Mn(III/IV) precipitation on the concentration of other metal cations (e.g., Co2+, Ni2+, Zn2+). These analyses were also performed using ICP-AES, ICP-MS, and AAS.
2.4. Geochemical Modeling
The geochemical software package PHREEQCI (Version 3.0.5-7748, US Geological Survey, Reston, VA, USA [39]) was used for (1) modeling of ionic species in solution at varying pH, and (2) calculation of saturation indices (SI) of selected Mn-bearing minerals. All the calculations were conducted using the MINTEQA2 thermodynamic database (Version 4.0, USEPA, Washington, DC, USA) [40]. We selected this database because it includes aqueous species of both Co and Ni, two elements that are not integrated in other databases (e.g., PHREEQC.dat, WATEQ4F.dat). Based on the chemical analysis of the waters used for the experiments (Table 1), the ionic activities of all dissolved metals were calculated. The saturation indices of selected manganese oxides were calculated using the corresponding solubility products (log Ksp values) including the MINTQA2.V4 database. The assumptions for the computations (chosen to resemble the experimental conditions) were: T = 25 °C, pe = 13.5, and equilibrium with respect to atmospheric O2 and CO2.
3. Results
3.1. Mineralogy and Chemistry of the Manganese Oxide Precipitates
3.1.1. Mineral Composition Revealed by X-Ray Diffraction and Electron Microscopy
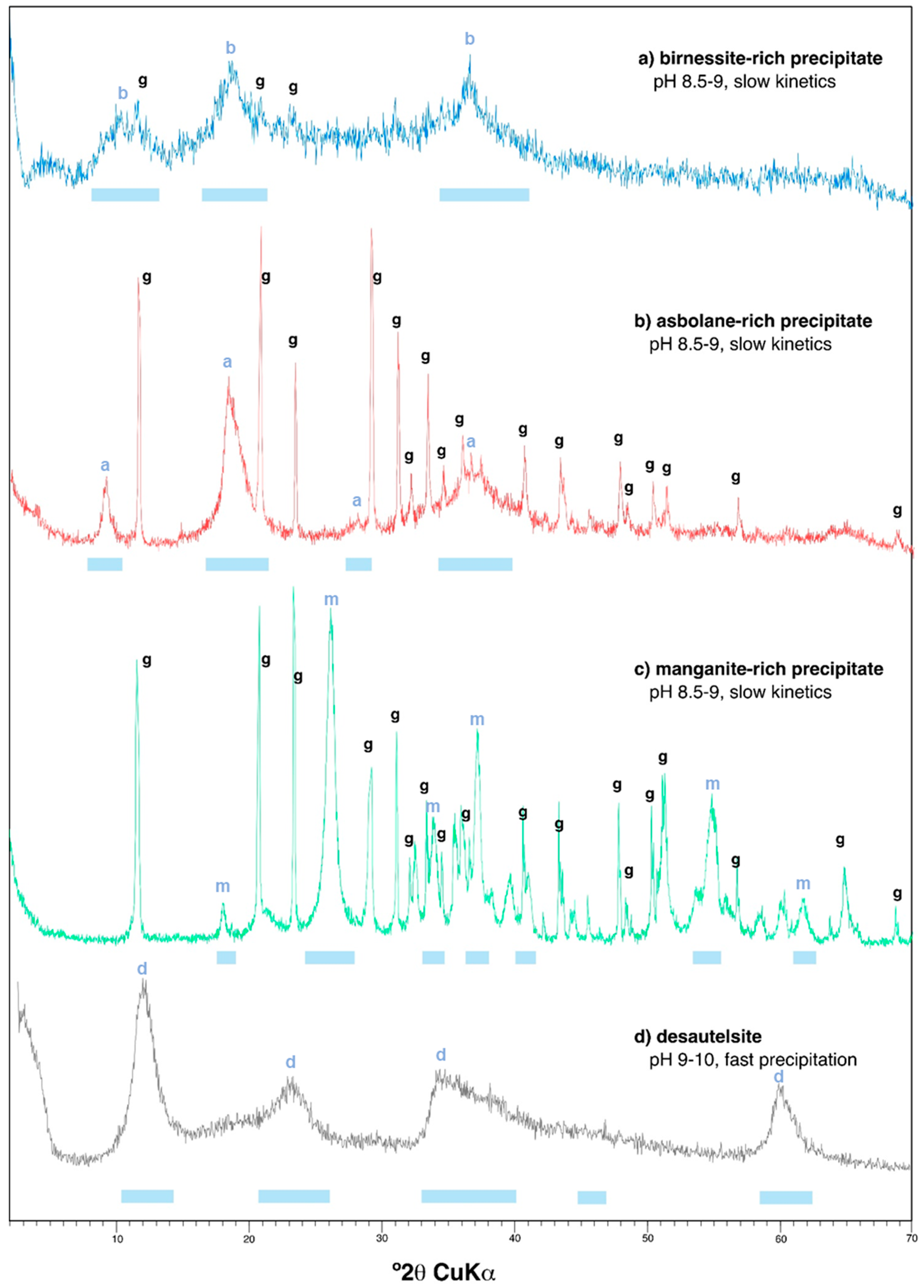
The major mineral phases identified in the Mn precipitates obtained during the batch reaction experiments of Mn(II) oxidation, as identified by XRD and SEM, are illustrated in Figure 2 and Figure 3, and summarized in Table 2 and Table 3. Selected XRD patterns of the precipitates are shown in Figure 2, and characteristic SEM images of some Mn oxides are given in Figure 3. Table 2 provides the ideal composition of the mineral phases identified, as well as those found in natural Mn oxides formed in the Reocín pit lake. Table 3 lists the dominant Mn oxides found in the different solutions, along with their respective conditions of precipitation (pH, laboratory vs. on site, precipitation kinetics), and the trace metal concentrations measured in these manganese oxides.
An important analytical difficulty arose during the mineralogical identification of these fresh Mn precipitates because of the coexistence of highly crystalline phases like gypsum (which was accidentally obtained in two of these Ca- and SO4-rich waters due to solubility saturation above pH 5.5) and the Mn oxides, which tend to be very poorly crystalline and fine-grained. However, it was possible to deduce the presence of certain Mn oxides by both XRD (broad reflections diagnostic of certain Mn phases; Figure 2) and SEM–EDX (observation of crystals or aggregates with well-defined habit and composition; Figure 3).
Even though the experiments were conducted under the same environmental conditions (similar temperature, stirring rate, water volume, etc.) and with waters having analogous chemistries, we obtained evidence of a varied mineralogy of Mn oxides in the different solutions which revealed a distinct Mn(III/IV) ratio (Table 2 and Table 3; Figure 2 and Figure 3). The major mineral phase found in the experiments with water from Guadiana mine, as identified by XRD, was an incipient, poorly-crystalline birnessite which did not provide clear peaks but wide reflections at diagnostic positions (Figure 2a). SEM–EDX investigations of these oxides confirmed XRD results and showed pseudo-tabular habits (Figure 3a,b) and presence of sodium, potassium, and calcium in proportions varying from 0.2 to 3.0 wt. % (not shown). The SEM-–EDX results suggest that these minerals consist in micro- to nano-crystalline birnessite, and probably also todorokite, which are mixed Mn(III/IV) manganates with either layer (birnessite) or tunnel (todorokite) structure incorporating different exchangeable charge-balancing cations such as Na+, K+, or Ca2+ [3]. The oxidation experiments with water from La Zarza mine provided XRD evidence for the formation of asbolane (Figure 2b, Figure 3c,d; see also Figure S1 in Supplementary Materials), a more oxidized, Mn(IV) oxy-hydroxide which has not a constant stoichiometric formula and contains up to several percent units of Co and Ni [11,41]. Finally, the dominant manganese oxide formed upon Mn(II) oxidation in the mine water from Brunita mine likely corresponded to manganite, a Mn(III) oxy-hydroxide (Figure 2c, Figure 3e,f). This variable mineralogy reveals a different oxidation state of dissolved manganese during precipitation, and therefore, a variable kinetics of Mn(II) oxidation (Reactions (1) and (2)), likely depending on factors such as initial Mn concentration in the different solutions or the pH at which Mn oxides precipitated from the parent solutions [34,35,36,37].
The Mn oxides found in this work contrasted with those previously observed in an earlier study in which waters from other acidic pit lakes (in this case, Cueva de la Mora and San Telmo) were subjected to rapid neutralization at higher pH (9–10), and where the Mn minerals formed mostly consisted in desautelsite (a hydrotalcite group mineral with Mn(III) and Mg(II); Figure 2d; see also Figure S2 in Supplementary Materials) and minor quantities of hausmannite and pyrochroite (Table 2 and Table 3) [38]. Birnessite and todorokite are among the most common Mn oxides formed in different environments [1,2,3], and have been also found in the Reocín pit lake (pH 7–8) in Northern Spain, in conjunction with other Mn oxides such as pyrochroite, lithiophorite, or ranciecite (Table 2 and Table 3; Figure S3 in Supplementary Materials) [33]. As discussed below, this varied mineralogy of manganese in the different mine waters (considering both the natural products and the synthetic Mn oxides), likely reflects slightly different geochemical conditions (e.g., pH, Eh) during the oxidative precipitation of dissolved Mn(II) in the mine waters.
From a microscopic and textural perspective, the different Mn(III/IV) oxides formed during Mn(II) oxidation exhibited rather variable morphologies, crystal habit, and grain size (Figure 3). Birnessite was present in the form of tabular plates with diameters ranging from to 200 to 500 nm, and thickness lower than 50 nm (Figure 3a,b). Asbolane formed botryoidal aggregates generated by nanometer-sized globules with diameters in the order of 100–200 nm (Figure 3c,d). Manganite appeared as randomly oriented, tiny laths of nanometric size (100–200 nm in length and 10–20 nm in width), which usually formed larger-size aggregates that could also include sparse large gypsum crystals formed in the solutions during the experiments (Figure 3e,f).
3.1.2. Trace Metal Content of the Mn Oxides
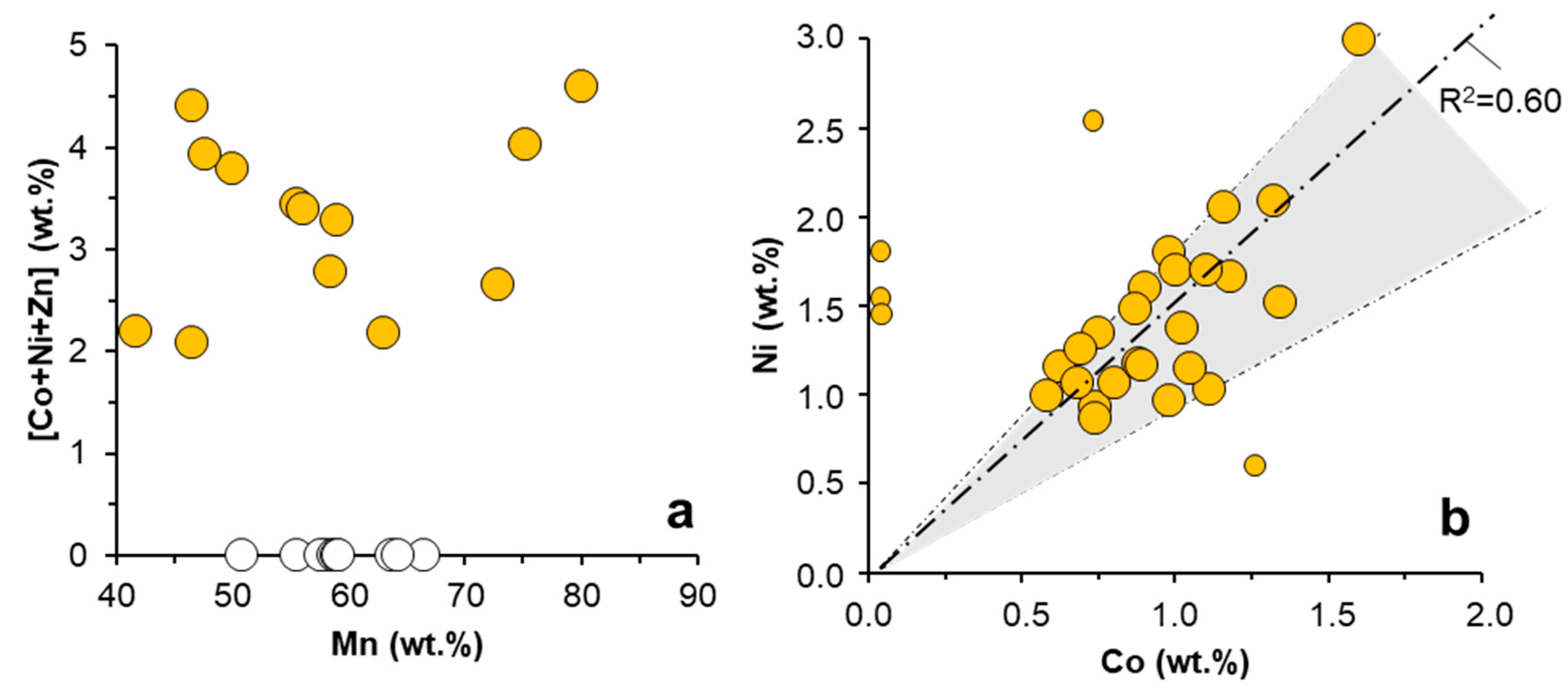
The trace metal content measured by ICP-MS in the Mn oxides formed during the oxidation experiments (Table 3) included important amounts of Ba (up to 2394 ppm), Co (up to 2266 ppm), Ni (up to 3220 ppm), and Zn (up to 2713 ppm), in addition to minor, though still significant, Cd (153–183 ppm) in some samples. It is worth noting, however, that these are bulk-sample concentrations, which due to dilution by some minor but persistent phases like gypsum (Figure 2), may mask higher metal concentrations on a smaller, microscopic scale. This has been confirmed by SEM–EDX analyses on individual grains and crystals of different Mn oxides (Figure 3 and Figure 4). Weight percent values up to 3% Ni and 1.5 wt. % Co were measured by SEM–EDX in globular aggregates of asbolane in the sample from La Zarza, which showed in general (and if outliers are excluded) a fairly good correlation (R2 = 0.60) between these two elements (Figure 4b). On the other hand, neither Co nor Ni was detected by this technique on individual manganite or birnessite crystals formed in the solutions from Brunita or Guadiana mine pit lakes (Figure 4a), likely meaning that these elements were below concentrations about 0.1–0.3 wt. % which usually mark the lower detection limit by EDX in most conventional electron microscopes [42]. Although EDX determinations are semi-quantitative and must be taken with caution, they reveal an important heterogeneity regarding trace metal content in the different crystals analyzed, which may result from slightly varying physico-chemical conditions at the time of Mn(III/IV) precipitation (e.g., pH, reaction time, etc.). Besides, the absence of correlation between Mn and the sum of trace metals (∑[Co + Ni + Zn]; Figure 4a) suggests that the dominant mechanism of trace metal incorporation into Mn oxides would have been the adsorption of these metal ions onto mineral surfaces, which strongly depends on kinetic factors like the reaction time, rather than by structural incorporation into vacancies, ion exchange, or cationic substitution for Mn in the mineral lattice.
Although important, the trace metal content of synthetic Mn oxides is lower than those measured in natural birnessites, and related Mn oxides (todorokite, pyrochroite, lithiophorite, ranciécite) formed at pH 7.0–8.0 in the water column of the Reocín mine pit lake (Table 3). These natural Mn oxides usually show Co and Ni concentrations comparable to those found in the present study (e.g., 420–3099 ppm Co, and 116–675 ppm Ni), but much higher concentrations of Zn (76,029–130,000 ppm Zn), and significant concentrations of many other trace metals (e.g., 24–74 ppm Cd, 51–256 ppm Mo, 53–228 ppm Se, and 10–480 ppm Pb) (Table 3). The concentration of Co, Ni, and Zn in the Mn oxides produced in this study were also slightly lower than those found in desautelsite and hausmannite produced at pH 9.0–10.0 in earlier work based on fast neutralization experiments with Mn(II)-containing mine waters ([38]; Figure S2 in Supplementary Materials), which were in the range of 2981–6566 ppm Co, 1601–3095 ppm Ni, and 3142–6982 ppm Zn (Table 3). In the first case, formation of natural Mn oxides was considered to be catalyzed by Mn(II)-oxidizing microorganisms, and these Mn bio-oxides are known to have adsorption capacities which can be several orders of magnitude higher than those shown by abiotic, chemically produced manganese oxides [3]. In the latter case, higher pH conditions would have favored a higher adsorption of trace metals, since the ion-surface reactions are strongly pH-dependent [4,13].
Overall, the trace metal retention capacity varies significantly among the different Mn oxides, with asbolane having the highest metal-scavenging potential, followed by birnessite and todorokite, and with manganite showing the lowest capacity of metal sorption, in agreement with previous studies (e.g., [1,2,3]).
3.2. Trace Metal (Co, Ni, Zn) Partition between Parent Solutions and Mn Oxides
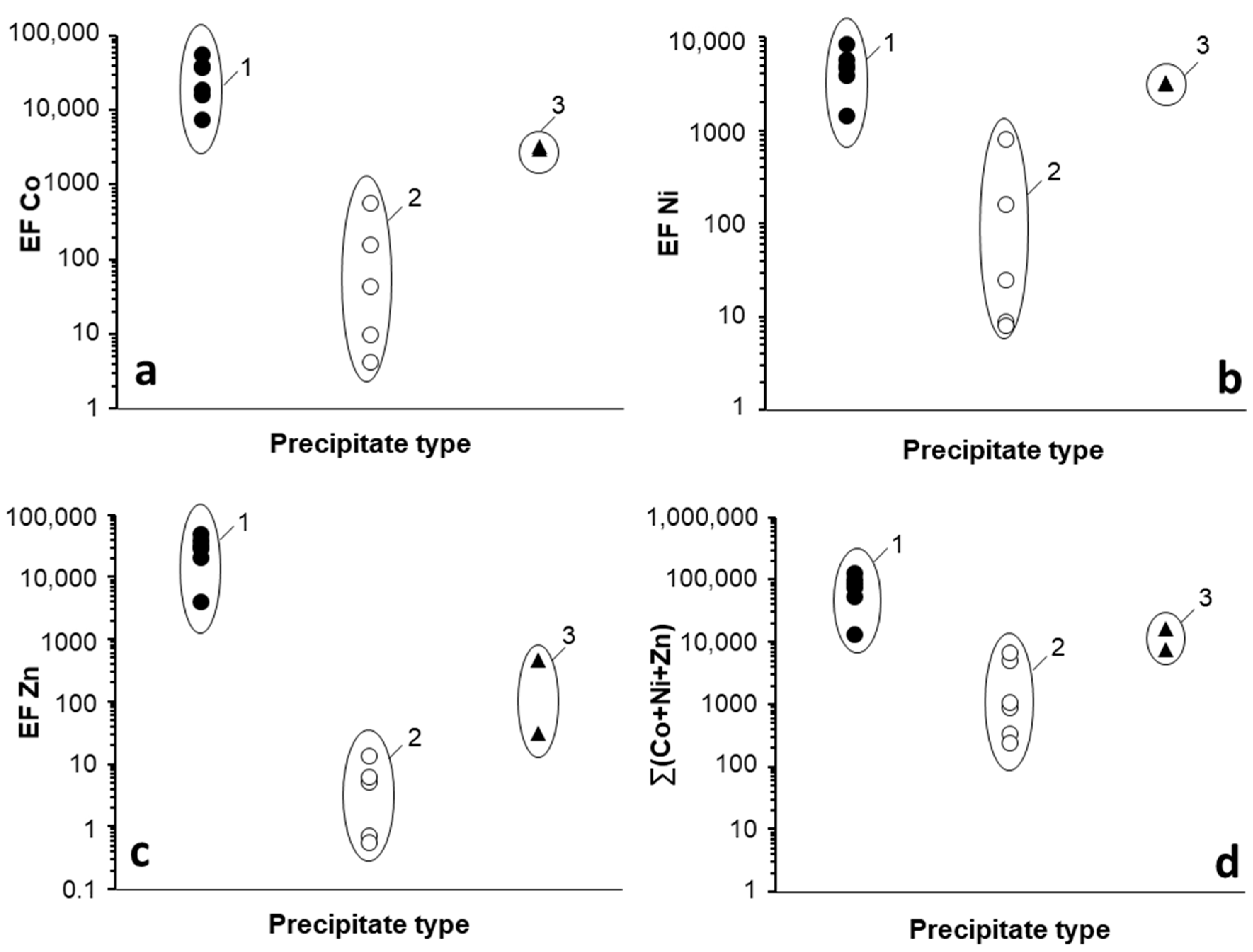
By comparing trace metal concentrations in the Mn oxides included in this study (Table 3) to those of their corresponding parent solutions (either in the lab or in situ; Table 1), enrichment factors (EF) were calculated for Co, Ni, and Zn (Figure 5). Natural Mn oxides (mostly birnessites, in addition to todorokite, pyrochroite, lithiophorite, and ranciécite) showed the highest EF values with respect to their original solutions. These Mn oxides exhibited Co, Ni, and Zn concentrations between 10,000 and 100,000 times those found in the water. The corresponding EF for the experimental Mn(III/IV) oxides obtained in this study ranged from 10 to 1000 for Co and Ni, and between 1 and 10 for Zn (Figure 5), i.e., between 2 and 3 orders of magnitude lower than those of natural Mn oxides. The EF observed in Mn oxides formed in fast neutralization experiments [38] fell between those of experimental and natural oxides.
The extremely high EF calculated for natural Mn oxides formed in Reocín mine derived from the combination of (1) abnormally high Co, Ni, and (most specially) Zn concentrations in the solids, and (2) very low trace metal concentrations in this circumneutral pit lake compared to those found in acidic mine pit lakes (e.g., 50–56 µg/L vs. 678–11,081 µg/L for Co, 76–82 µg/L vs. 795–10,394 mg/L for Ni, and around 3 mg/L vs. 231–624 mg/L for Zn; Table 1). In other words, natural Mn oxides formed in Reocín incorporated higher amounts of trace metals (the case of Zn is outstanding) despite being precipitated from solutions with much lower metallic content. This represents more evidence of the extraordinary capacity of natural Mn oxides (likely, bio-oxides) to scavenge trace metals in the environment, which is in accordance with previous work [3,8,9,10,43,44].
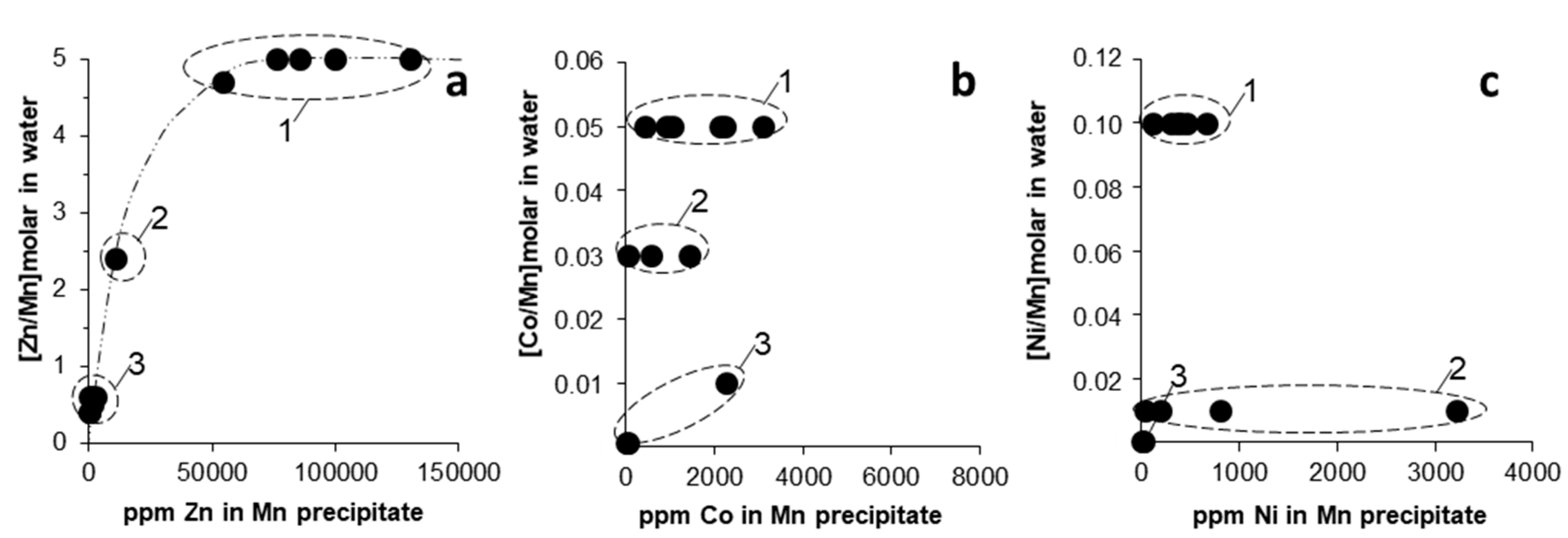
In Figure 6, we plotted the trace metal (Zn, Co, Ni) concentrations in Mn oxides against the corresponding molar ratio of trace metal to Mn in the water (e.g., [Zn/Mn]molar, [Co/Mn]molar, [Ni/Mn]molar). The case of Zn seemed reasonable since it showed an increasing concentration in the solid with an increasing [Zn/Mn]molar ratio in the parent solution (Figure 6a). However, this was not the case for Co and Ni, which showed no obvious relation between trace metal content in Mn oxides and the original metal-to-Mn ratio in the parent solutions (Figure 6b,c). As discussed earlier, this might suggest that factors other than the original water chemistry and metal concentration would have controlled the extent of trace metal incorporation into the solids, the most important being (i) pH, (ii) reactivity of adsorbing solid surface, and (iii) reaction time.
3.3. Effects of Mn(III/IV) Precipitation on Solution Chemistry
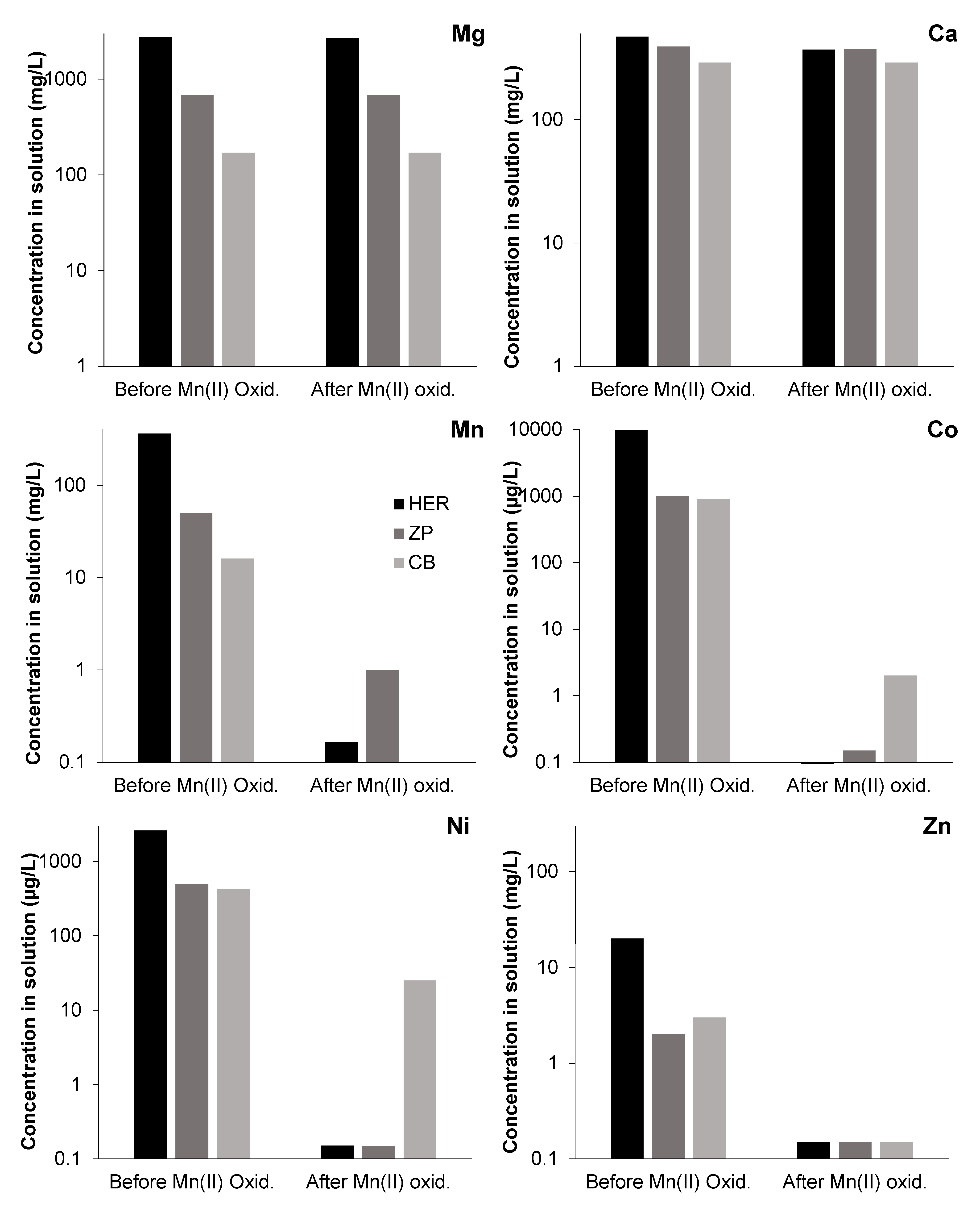
The oxidation of Mn(II) and subsequent formation of mixed Mn(III/IV) oxides exerts a clear control on the concentration of many trace elements originally present in mine waters, especially regarding Co, Ni, and Zn (Figure 7). The plots of Figure 7 show that, in the three mine waters used for the experiments, many major cations such as Mg2+ or Ca2+ behaved conservatively, meaning that they were not significantly incorporated into Mn oxides, and thus their concentrations remained unaffected during the oxidation process.
Conversely, the oxidation of manganese resulted in virtually total precipitation of dissolved Mn in all solutions, and important scavenging of Co, Ni, and Zn (Figure 7). Even though some of Mn oxide precipitates did not show extremely high trace metal concentrations (e.g., manganite in waters from Brunita or some birnessites from Guadiana pit lake, Herrerías; Table 3), the amount of adsorbent Mn solid in the colloidal suspensions was so high (e.g., 2.8–4.6 g/L Mn oxide, depending on the starting Mn concentration and the particular mineral phase formed in the experiments; Table 1) as to imply a high (in some cases, nearly total) partitioning of these trace metals into the Mn oxides.
The evolution of Zn deserves a special mention because in the pre-oxidation stage, its concentration was already significantly diminished with regard to the initial concentration in the original acidic solutions (Table 1 and Figure 7). This is basically due to the fact that Zn precipitated during the previous neutralization stage (at pH > 7.0–7.5) in the form of either willemite or a least stable precursor [38]. In any case, the plot of Figure 7 shows that Zn content that was not removed by direct Zn precipitation, was scavenged by incorporation into Mn oxides.
3.4. Geochemical Modeling
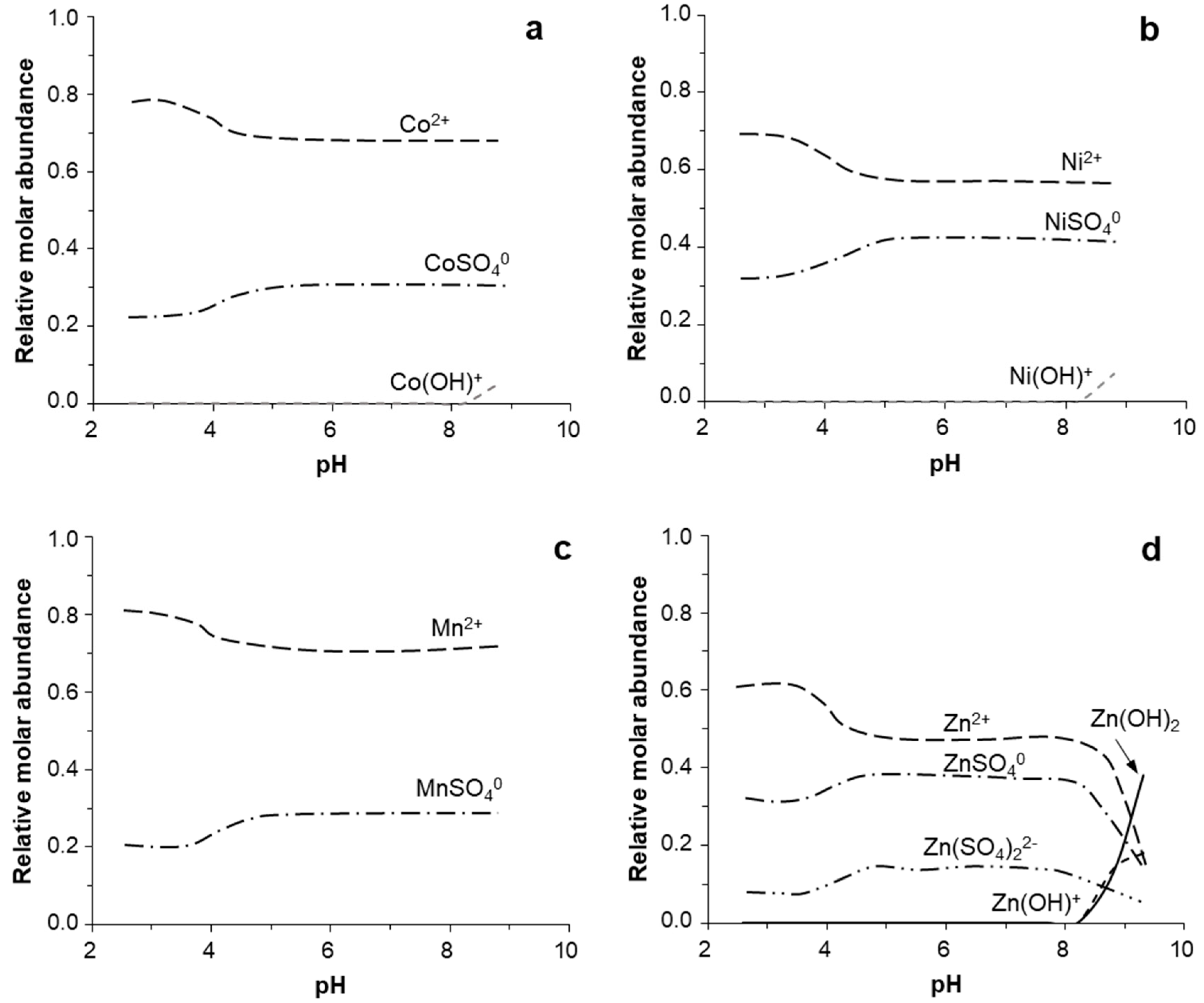
The modeled distribution of ionic species in solution showed that the studied divalent metals (Co[II], Ni[II], Zn[II] in addition to Mn[II]) were mostly present as free cations (e.g., Co2+, Ni2+, Mn2+, Zn2+) over the entire pH range of 2–9 (Figure 8). These free ions represented 60–80% of the total metal concentration, while the remaining was mostly dominated by neutrally-charged sulfate complexes of the type M2+SO40 (e.g., CoSO40, NiSO40). Hydroxyl species of the type Co(OH)+ or Ni(OH)+ became only significant at pH > 9, when they reached up to 10% of the total metal concentration (Figure 8a,b). In the case of Zn, the hydroxide form Zn(OH)2 became the dominant species at pH > 9.0 (Figure 8d). Overall, the presence of Co, Ni, and Zn as free metal ions favored their incorporation into Mn(II) oxides, either by ion substitution for other cations or by adsorption onto negatively charged surfaces [1,2,3,4].
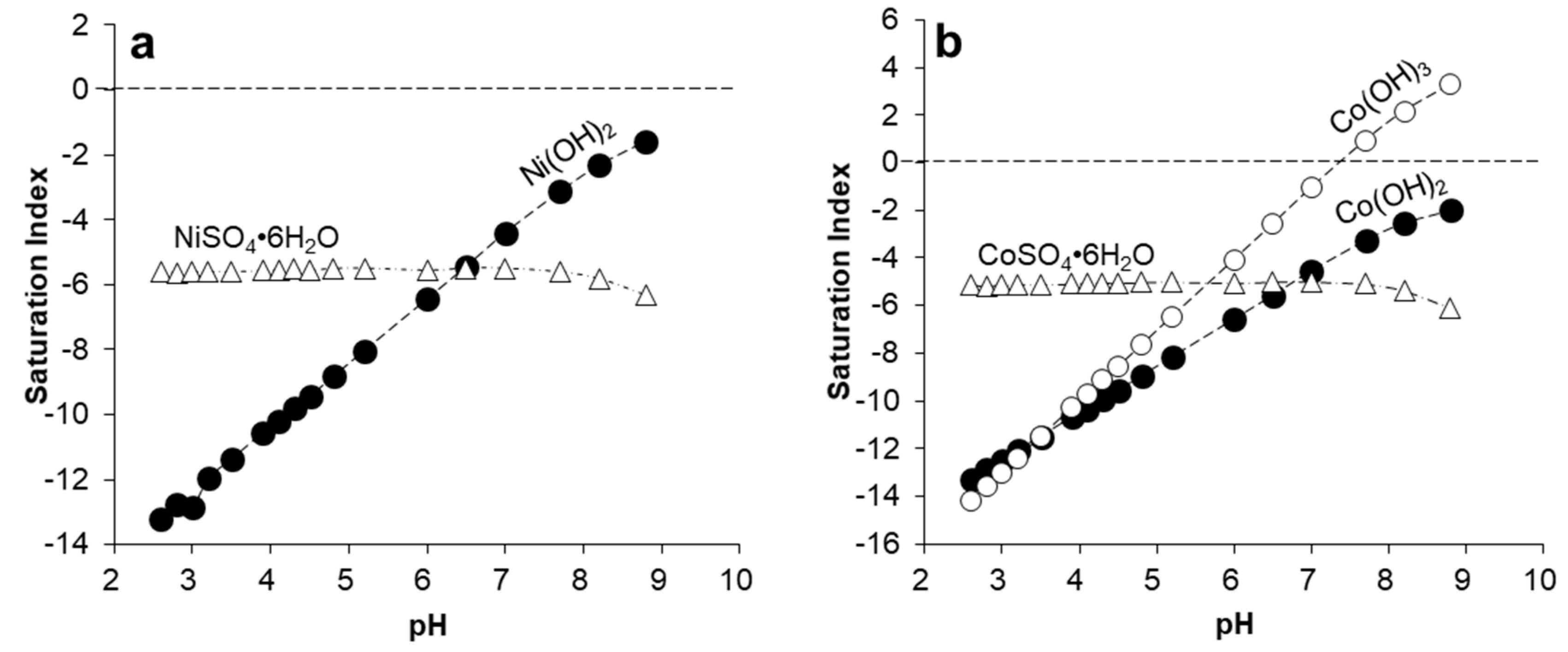
Calculating saturation indices (SI) for some common Ni and Co minerals, including sulfates (e.g., NiSO4·6H2O, CoSO4·6H2O) and hydroxides (e.g., Ni(OH)2, Co(OH)2) revealed that divalent hydroxides would not attain solubility saturation in pH ranging from 2.5 to 9.0, which include the conditions observed for Mn(II) oxidation and Mn(III/IV) oxide precipitation (Figure 9). Precipitation of the Co(III) hydroxide, (Co(OH)3), would be theoretically possible, since the model predicts saturation of this compound at pH > 7.5–8.0 (Figure 9b). However, the formation of this hydroxide would imply the existence of the more oxidized form of cobalt (Co(III)) in the studied mine waters (for which there is no evidence). However, SEM observations did not provide any proof for the existence of a discrete Co solid of this kind. It has been proposed that the oxidation of Co(II) to Co(III) might explain the uptake of cobalt into manganese oxides, since this favors the substitution for Mn(III) or Mn(IV) atoms in the crystal lattice [11]. Figure 9b suggests that the incorporation of Co into manganese oxides is favored at pH > 8.0 due to the metal’s oxidized form. In any case, the removal of Co and Ni cations from mine water solutions is considered to be almost exclusively controlled by coprecipitation with Mn oxides.
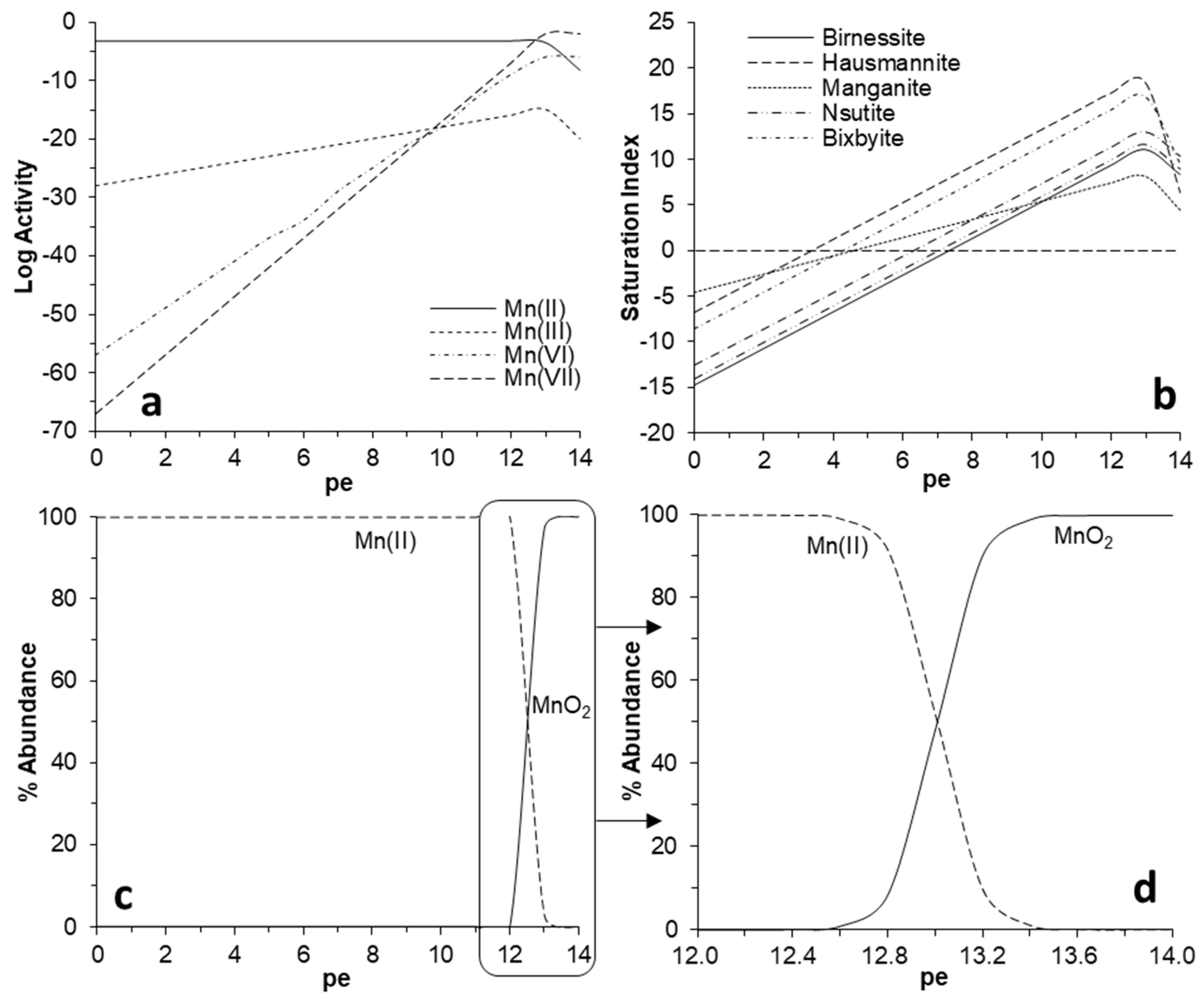
Regarding the behavior of manganese, the geochemical model predicts oxidation of Mn(II) starting at electronic potentials (pe) of at least 12.6 (Eh ≈ 745 mV) for the conditions prevailing in the studied mine waters during the oxidation experiments (Figure 10). The database used in the model (minteqa2.v4) foresees different Mn oxidation states (including Mn[III], Mn[VI], and Mn[VII]; Figure 10a) which have no practical meaning, since the oxidative precipitation of Mn(II) (e.g., through Reaction (1) or (2)) implies the immediate conversion of Mn(II) to solid-phase Mn(III) (e.g., manganite), Mn(IV) (e.g., asbolane), and/or mixed Mn(III/IV) (e.g., birnessite, todorokite) (Figure 10b). Both birnessite and manganite (in addition to some others which have not been identified in the Mn precipitates, such as hausmannite, nsutite or bixbyite) show apparent saturation at significantly lower pe (<8), meaning that their precipitation would have been thermodynamically favored as far as Mn(III) or Mn(IV) (or both) were present in the aqueous phase as a result of Mn(II) oxidation.
In practical terms, and considering only MnO2 as the precipitating manganese phase for simplification, a narrow pe window of 12.6–13.5 (745–800 mV) would mark the conservative to non-conservative behavior of manganese in mine waters (Figure 10c,d). All dissolved Mn(II) would have been oxidized and precipitated in this pe range (as diverse mineral phases depending on the local geochemical conditions) by the continuous aeration and replenishment of O2 to the solutions.
4. Discussion
4.1. Factors Controlling Mn(II) Oxidation and Mn Oxide Mineralogy in the Studied Mine Sites
The singular biogeochemistry of the IPB mine waters represent, at the same time, advantages and disadvantages for the oxidative removal of Mn(II). On one hand, the very low pH of these mine effluents (usually comprised between 1.5 and 3.5; [38]) supposedly prevents the possibility of microbial Mn(II) oxidation, since most known Mn(II)-oxidizing microbes are neutrophilic [29,30,31,32]. Several studies have reported biological Mn(II) oxidation in coal mine sites at pH ~5.0 [18] and Mn(II)-oxidizing microbes have been isolated in mildly acidic (pH 5.0) groundwater in an uranium mine site [45]. However, pH conditions of the IPB mine effluents a priori appear incompatible with the presence of Mn(II)-oxidizing microorganisms. This is an important limitation since Mn bio-oxides show adsorptive capacities, which are orders of magnitude higher than those of their abiotic counterparts [2,3,8,9,10,45]. In the case of the circumneutral Reocín mine pit lake, geochemical conditions would a priori allow the presence of Mn(II)-oxidizing microorganisms that could be greatly catalyzing manganese oxidation. The chemistry of IPB mine waters also includes large amounts of dissolved sulfate (Table 1), which complexes with Mn(II) to form MnSO4 species (Figure 8) that are known to retard Mn(II) oxidation [36]. On the other hand, the very high concentration of dissolved Mn in these waters provides large quantities of Mn oxide surfaces upon oxidation, and this in turn ensures an autocatalytic effect which enhances the heterogeneous oxidation of Mn(II) [37]. The fact that the faster oxidation kinetics were observed in water with the highest Mn(II) concentration supports this idea and suggests that heterogeneous oxidation in Mn oxide surfaces was a major mechanism catalyzing Mn(II) oxidation in the mine water solutions.
It has been argued that the formation, stability, and predominance of certain Mn minerals over others is the result of many different factors, including solution chemistry (e.g., Mn concentration, presence of other cations, pH, Eh), rate of oxidation kinetics, O2 and light availability, or biological mediation, among many others [1,2,3,44]. In addition, manganese oxides are rather unstable and may rapidly transform to other, more stable ones, so that the variable Mn mineralogy observed here could simply reflect a distinct stage of the Mn(II) oxidative precipitation process in different mine waters. Our results show that the oxidation of dissolved Mn(II) in mine waters with relatively similar composition and under identical environmental conditions, can lead to different Mn oxides with variable Mn(III/IV) ratios. Manganite is the more stable and abundant among the MnOOH polymorphs [1], and has been also found as a direct product of biogenic Mn(II) oxidation [9]. It represents an early product of Mn(II) oxidation (with only Mn(III) present in the structure), so its detection in mine water from Corta Brunita represents an indirect, mineralogical evidence of the presence of Mn(III) as an intermediate stage in the Mn(II) oxidation process. There exists very little information on the trace metal adsorption capacity of this mineral, though it is generally considered to be much lower than those of, e.g., birnessite or todorokite [1]. Despite the very fine-grained and poorly crystalline nature of the obtained solids, we also found XRD and SEM–EDX evidence for the presence of micro- to nano-crystalline birnessite, in addition to asbolane. Birnessite and todorokite have been widely reported and studied in the literature as end products of Mn(II) oxidation [1,2,3,5,6,7,12,18]. These two minerals have been also commonly found in the Reocín mine pit lake, along with some others such as ranciecite, lithioforite or pyrochroite in minor proportions (Table 3) [33]. The interest of these two Mn oxides resides in their very high sorptive capacity and well-known affinity for Co and Ni [1,2,3,5,6,7,12,18]. Their mixed valence (Mn(III/IV) indicates an intermediate state of Mn oxidation at the time of Mn oxide nucleation and subsequent mineral growth. Asbolane has been found in cave systems [46,47], but it has been rarely reported in mine-drainage settings. This manganese mineral, however, is the one having the highest Co and Ni concentrations, which can be up to several weight percent units [11,48,49], so its formation implies the highest rate of Co(II) and Ni(II) coprecipitation (per mass unit) among Mn oxides. The presence of asbolane in some of the samples denotes an advanced state of Mn(II) oxidation since this mineral is entirely formed by Mn(IV) [11,48,49].
4.2. Possible Implications for Remediation Technology and Metal Recovery
In the Iberian Pyrite Belt mining district, manganese concentrations in mine drainage are variable—between a few mg/L in the more dilute effluents to as high as 440 mg/L in the more concentrated ones—with an average value around 60 mg/L based on data from 65 different mine effluents [50]. Cobalt and nickel concentrations in these waters can also reach extreme values of 47 mg/L and 17 mg/L, respectively, though the average concentrations of these metals are 3 mg/L for Co and 1 mg/L for Ni [50]. Unlike many other toxic elements which are naturally attenuated downstream of mine sites by coprecipitation with iron and/or aluminum oxy-hydroxides (e.g., As, Cr, Pb, Cu, U) [51,52,53,54,55], manganese remains virtually unaffected by precipitation or sorption because the pH of these waters never exceeds values of 3.5 until they reach the Tinto and Odiel estuary [50]. In a study conducted in 2003–2004, it was calculated that between 1000 and 2600 kg/day (365–950 tons/year) of manganese had been transported to the Atlantic Ocean as dissolved load by tens of mine effluents flowing from the IPB district to the Huelva estuary [50]. At the same time, trace metal loadings transferred every year to the Atlantic Ocean from mine sites were in the order of 145–475 tons for Zn, and ~1.5–5 and 0.5–2 tons for Co and Ni, respectively [50]. Likewise, metal concentrations in surface waters of mine pit lakes of this mine district are comprised in the ranges of 3–254 mg/L for Mn, 5–6670 mg/L for Zn, 1–19 mg/L for Co, and 0.3–7 mg/L for Ni [56], but these metal concentrations can increase exponentially with depth [57,58,59,60,61]. Given the size of many of these pit lakes (e.g., some exceeding 8 × 108 m3) and the volume of acidic and metal-rich water stored in there, these artificial lakes represent an important reserve of base metals [56]. In contrast with fresh water lakes, where Mn is continuously cycled and subjected to oxidation [62,63,64], dissolved Mn in acidic pit lakes and mine effluents (e.g., Iberian Pyrite Belt or La Unión mining districts) is totally conservative by the reasons explained above, having therefore no role in trace metal dynamics. However, the case of Reocín shows that the adsorption/coprecipitation trends observed in Mn(II) oxidation experiments occur in mine sites under geochemically favorable conditions and may represent either (i) a highly efficient mechanism of self-mitigation of metal pollution, and/or (ii) a promising system for metal recovery.
The adsorption and coprecipitation of less soluble trace metals and toxic elements (e.g., As, Cr, Pb, Cu, U) during iron and aluminum oxy-hydroxide is well-known [16,38,51,52,53,54,55] and are being conventionally applied in many neutralization plants installed in mine settings worldwide [65,66]. But new treatment facilities could be designed to remove the most soluble trace metals (including Mn, Co, Ni, and Zn) from these mine waters by favoring oxygenation in aeration ponds at near-neutral pH. This treatment option would help achieve water quality standards with the additional advantage of providing certain economic benefit to the treatment scheme if an adequate technology is developed for the later extraction of metals from the manganese oxides. An efficient removal of dissolved metals (including Mn, Co, Ni, Zn, or Cd) by hydrotalcite precipitation at pH 10 (after MgCl2.6H2O and NaOH addition) has been satisfactorily applied to acidic mine drainage in Australian uranium and metal mines [65,66]. Other experimental studies of AMD treatment by neutralization have shown that the use of seeds (e.g., MgO slurry, ferrite) and higher temperatures (e.g., 60–70 °C) can significantly increase the rate of nickel and cobalt hydroxide precipitation at near-neutral pH values [67,68]. Mubarok and Liberto (2013) reported removal rates of 77% to 85% for dissolved Ni at temperatures of 25 °C at pH 7.0 by using MgO as a neutralizing agent [67], whereas Kefeni et al. (2015) found removal rates of 28% to 48% for Co at pH 7–8.5 and 60 °C using ferrite seeds [68]. The results presented in this study show that the removal of Ni, Co, and Zn can be almost complete with Mn(III/IV) precipitation at pH ~8.5 as the sole treatment mechanism, which obviously represents a great advantage with respect to classical treatment options based on lime, MgO, or NaOH addition in neutralization plants. Even though the extent of metal sorption by these inorganic products is significantly lower than that observed in natural Mn bio-oxides, the extremely high concentrations of dissolved Mn in these mine waters provide an unusually high density of Mn oxide mineral surfaces, which ensures the virtually total scavenging of trace metals if a sufficient reaction time is permitted. Future research could be oriented towards the design of cost-effective mechanisms of metal recovery from these Mn oxides that could help maintain passive treatment facilities. Manganese itself is currently an essential element in modern industry, being currently irreplaceable in strategic sectors like steelmaking [69,70]. Manganese deposits with elevated concentrations of trace metals with technological interest, such as Co or Ni, are currently an important prospective target in seabed exploration [69,71,72,73]. Efficient leaching technologies with acids, bases, or complexing agents have been already applied to recover Co and Ni from Mn oxides forming deep-ocean ferromanganese nodules with satisfactory results [71,72]. Under this perspective, Co-, Ni-, and Zn-rich manganese oxides produced in AMD treatment facilities in metal mining districts such as the IPB could be perceived as a valuable resource within a scheme of circular economy, rather than a simple mine waste product.
Supplementary Materials
The following are available online at https://www.mdpi.com/2075-163X/9/4/226/s1, Figure S1: Pictures on experimental details and XRD pattern of asbolane; Figure S2: Transmission Electron Microscopy (TEM) pictures and XRD patterns of desautelsite obtained in a previous study for comparison purposes; Figure S3: Field aspect and XRD patterns of birnessite-rich precipitate found in the Reocín pit lake.
Author Contributions
Conceptualization, J.S.-E.; methodology, J.S.-E. and I.Y.; validation, J.S.-E.; and I.Y.; formal analysis, J.S.-E. and I.Y.; investigation, J.S.-E.; resources, J.S.-E. and I.Y.; data curation, J.S.-E.; writing—original draft preparation, J.S.-E. and I.Y.; writing—review and editing, J.S.-E. and I.Y.; project administration, J.S.-E.; funding acquisition, J.S.-E.
Funding
This research was funded by the Spanish Ministry of Science, Universities and Innovation through project number CGL2016-74984-R and by the Spanish Geological Survey (project 2639). Previous data on water chemistry and solid phase mineralogy from the Reocín pit lake were obtained during an agreement between XTRATA ZINC and the Spanish Geological Survey.
Acknowledgments
We sincerely thank Jesús Reyes (Geochemistry Lab at IGME) for the chemical analyses of the solutions and solids obtained in the experiments.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Post, J.E. Manganese oxide minerals: Crystal structures and economic and environmental significance. Proc. Natl. Acad. Sci. USA 1999, 96, 3447–3454. [Google Scholar] [CrossRef]
- Tebo, B.M.; Johnson, H.A.; McCarthy, J.K.; Templeton, A.S. Geomicrobiology of Manganese(II) oxidation. Trends Microbiol. 2005, 13, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Tebo, B.M.; Bargar, J.R.; Clement, B.G.; Dick, G.J.; Murray, K.J.; Parker, D.; Verity, R.; Webb, S.M. Biogenic manganese oxides: Properties and Mechanisms of Formation. Annu. Rev. Earth Planet. Sci. 2004, 32, 287–328. [Google Scholar] [CrossRef]
- Tonkin, J.W.; Balistrieri, L.S.; Murray, J.W. Modeling sorption of divalent metal cations on hydrous manganese oxide using the diffuse double layer model. Appl. Geochem. 2004, 19, 29–53. [Google Scholar] [CrossRef]
- Yin, H.; Tan, W.; Zheng, L.; Cui, H.; Qiu, G.; Liu, F.; Feng, X. Characterization of Ni-rich hexagonal birnessite and its geochemical effects on aqueous Pb2+/Zn2+ and As(III). Geochim. Cosmochim. Acta 2012, 93, 47–62. [Google Scholar] [CrossRef]
- Yin, H.; Feng, X.; Qiu, G.; Tan, W.; Liu, F. Characterization of Co-doped birnessites and application for removal of lead and arsenite. J. Hazard. Mater. 2011, 188, 341–349. [Google Scholar] [CrossRef]
- Yin, H.; Liu, F.; Feng, X.; Liu, M.; Tan, W.; Qiu, G. Co2+-exchange mechanism of birnessite and its application for the removal of Pb2+ and As(III). J. Hazard. Mater. 2011, 196, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Tani, Y.; Naitou, H.; Miyata, N.; Seyama, H. Sequestration of Cd(II) and Ni(II) ions on fungal manganese oxides associated with Mn(II) oxidase activity. Appl. Geochem. 2014, 47, 198–208. [Google Scholar] [CrossRef]
- Wang, W.; Shao, Z.; Liu, Y.; Wang, G. Removal of multi-heavy metals using biogenic manganese oxides generated by a deep-sea sedimentary bacterium—Brachybacterium sp. Strain Mn32. Microbiology 2009, 155, 1989–1996. [Google Scholar] [CrossRef]
- Zhou, D.; Kim, D.-G.; Ko, S.O. Heavy metal adsorption with biogenic manganese oxides generated by Pseudomonas putida strain MnB1. J. Ind. Eng. Chem. 2015, 24, 132–139. [Google Scholar] [CrossRef]
- Manceau, A.; Llorca, S.; Calas, G. Crystal chemistry of cobalt and nickel in lithiophorite and asbolane from New Caledonia. Geochim. Cosmochim. Acta 1987, 51, 105–113. [Google Scholar] [CrossRef]
- Manceau, A.; Lanson, B.; Drits, V.A. Structure of heavy metal sorbed birnessite. Part III: Results from powder and polarized extended X-Ray absorption fine structure spectroscopy. Geochim. Cosmochim. Acta 2002, 66, 2639–2663. [Google Scholar] [CrossRef]
- Nicholson, K.; Eley, M. Geochemistry of manganese oxides: Metal adsorption in freshwater and marine environments. In Manganese Mineralization: Geochemistry and Mineralogy of Terrestrial and Marine Deposits; Nicholson, K., Hein, K., Bühn, J.R., Dasgupta, S., Eds.; Geological Society Special Publication No 119; Geological Society of London: London, UK, 1997; pp. 309–326. [Google Scholar] [CrossRef]
- Fuller, C.; Harvey, J. Reactive uptake of trace metals in the hyporheic zone of mining-contaminated stream, Pinal Creek, Arizona. Environ. Sci. Technol. 2000, 34, 1150–1155. [Google Scholar] [CrossRef]
- Kay, J.; Conklin, M.H.; Fuller, C.; O’Day, P.A. Processes of nickel and cobalt uptake by a manganese oxide forming sediment in Pinal Creek, Globe Mining District, Arizona. Environ. Sci. Technol. 2001, 35, 4719–4725. [Google Scholar] [CrossRef]
- Lee, G.; Bigham, J.M.; Faure, G. Removal of trace metals by coprecipitation with Fe, Al and Mn from natural waters contaminated with acid mine drainage in the Ducktown Mining District, Tennessee. Appl. Geochem. 2002, 17, 569–581. [Google Scholar] [CrossRef]
- Tani, Y.; Ohashi, M.; Miyata, N.; Seyama, H.; Iwahori, K.; Soma, M. Sorption of Co(II), Ni(II), and Zn (II) on biogenic manganese oxides produced by a Mn-oxidizing fungus, strain KR21-2. J. Environ. Sci. Health Part A 2004, 39, 2641–2660. [Google Scholar] [CrossRef]
- Tan, H.; Zhang, G.; Heaney, P.; Webb, S.; Burgos, W.D. Characterization of manganese oxide precipitates from Appalachian coal mine drainage treatment systems. Appl. Geochem. 2010, 25, 389–399. [Google Scholar] [CrossRef]
- Wetzel, R.G. Limnology: Lake and River Ecosystems, 3rd ed.; Elsevier-Academic Press: San Diego, CA, USA, 2001; p. 1001. [Google Scholar]
- Fosmire, G.J. Zinc toxicity. Am. J. Clin. Nutr. 1990, 51, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Leyssens, L.; Vinck, B.; Van Der Straeten, C.; Wuyts, F.; Maes, L. Cobalt toxicity in humans—A review of the potential sources and systemic health effects. Toxicology 2017, 387, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Das, K.K.; Das, S.N.; Dhundasi, S.A. Nickel, its adverse health effects & oxidative stress. Indian J. Med. Res. 2008, 128, 412–425. [Google Scholar] [PubMed]
- InfoMine. Available online: http://www.infomine.com (accessed on 21 January 2019).
- London Market Exchange. Available online: https://www.lme.com (accessed on 21 January 2019).
- Brant, D.L.; Ziemkiewicz, P.F. Passive removal of manganese from acid mine drainage. Proc. Am. Soc. Min. Reclam. 1997, 741–744. [Google Scholar] [CrossRef]
- Rose, A.W.; Shah, P.J.; Means, B. Case studies of limestone-bed passive systems for manganese removal from acid mine drainage. Proc. Am. Soc. Min. Reclam. 2003, 1059–1078. [Google Scholar] [CrossRef]
- Bamforth, S.M.; Manning, D.A.C.; Singleton, I.; Younger, P.L.; Johnson, K.L. Manganese removal from mine waters—Investigating the occurrende and importance of manganese carbonates. Appl. Geochem. 2006, 21, 1274–1287. [Google Scholar] [CrossRef]
- Teixeira, L.A.C.; Queiroz, J.P.; Marquez-Sarmiento, C. Oxidative precipitation of manganese from dilute waters. Mine Water Environ. 2017, 36, 452–456. [Google Scholar] [CrossRef]
- Gounot, A.M. Microbial oxidation and reduction of manganese: Consequences in groundwater and applications. FEMS Microbiol. Rev. 1994, 14, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Greene, A.C.; Madgwick, J.C. Microbial formation of manganese oxides. App. Environ. Microbiol. 1991, 57, 1114–1120. [Google Scholar]
- Learman, D.R.; Wankel, S.D.; Webb, S.M.; Martínez, N.; Madden, A.S.; Hansel, C.M. Coupled biotic-abiotic Mn(II) oxidation pathway mediates the formation and structural evolution of biogenic oxides. Geochim. Cosmochim. Acta 2011, 75, 6048–6063. [Google Scholar] [CrossRef]
- Hallberg, K.B.; Johnson, D.B. Biological manganese removal from acid mine drainage in constructed wetlands and prototype bioreactors. Sci Total Environ. 2005, 338, 115–124. [Google Scholar] [CrossRef]
- López-Pamo, E.; Sánchez-España, J.; Santofimia, E.; Reyes, J.; Martín-Rubí, J.A. Limnología físico-química del lago formado durante la inundación de la Corta de Reocín, Cantabria (Marzo 2008–Febrero 2009); Unpublished Report; Instituto Geológico y Minero de España (IGME); Ministerio de Ciencia e Innovación: Madrid, Spain, 2009; p. 116.
- Diem, M.; Stumm, W. Is dissolved Mn2+ being oxidized by O2 in the absence of Mn-bacteria or surface catalysts? Geochim. Cosmochim. Acta 1984, 48, 1571–1573. [Google Scholar] [CrossRef]
- Morgan, A.J. Kinetics of reaction between O2 and Mn(II) species in aqueous solutions. Geochim. Csomochim. Acta 2005, 69, 35–48. [Google Scholar] [CrossRef]
- Hem, J.D. Chemical equilibria and rates of manganese oxidation. In Chemistry of Manganese in Natural Water; Water-Supply Paper 1667-A; U.S. Geological Survey: Washington, DC, USA, 1963. [Google Scholar]
- Martin, S.T. Precipitation and dissolution of iron and manganese oxides. In Environmental Catalysis; Grassian, V.H., Ed.; CRC Press: Boca Raton, FL, USA, 2005; Chapter 4; p. 701. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Yusta, I. Low-crystallinity products of trace-metal precipitation in neutralized pit-lake waters without ferric and aluminous adsorbent: Geochemical modelling and mineralogical analyses. Mineral. Mag. 2015, 79, 781–798. [Google Scholar] [CrossRef]
- Parkurst, D.L.; Appelo, C.A.J. User’s Guide to PHREEQC (Version 2)—A Computer Program for Speciation, Batch-Reaction, One-Domensional Transport, and Inverse Geochemical Calculations; Water-Resources Investigations Report 99-4259; U.S. Geological Survey: Denver, CO, USA, 1999; p. 312.
- Allison, J.D.; Brown, D.S.; Novo-Gradac, J. MINTEQA2/PRODEAFA2, a Geochemical Assessment Model for Environmental Systems: User Manual Supplement for Version 4.0; USEPA: Washington, DC, USA; NERL: Athens, GA, USA, 1999.
- Chuchrov, F.V.; Gorshov, A.I.; Vitovskaya, I.V.; Drits, V.A.; Sivtsov, A.I.; Rudnitskaya, Y.S. Crystallochemical nature of Co-Ni asbolan. Int. Geol. Rev. 1982, 24, 598–604. [Google Scholar] [CrossRef]
- Garratt-Reed, A.; Bell, D.C. Energy-Dispersive X-Ray Analysis in the Electron Microscope; BIOS Scientific Publishers Limited: Oxford, UK, 2003; p. 160. [Google Scholar] [CrossRef]
- Santelli, C.M.; Webb, S.M.; Dohnalkova, A.C.; Hansel, C.M. Diversity of Mn oxides produced by Mn(II)-oxidizing fungi. Geochim. Cosmochim. Acta 2011, 74, 2762–2776. [Google Scholar] [CrossRef]
- Webb, S.M.; Tebo, B.M.; Bargar, J.R. Structural influences of sodium and calcium ions on the biogenic manganese oxides produced by the marine Bacillus Sp.; strain SG-1. Geomicrobiol. J. 2005, 22, 181–193. [Google Scholar] [CrossRef]
- Bohu, T.; Akob, D.M.; Abratis, M.; Lazar, C.S.; Küsel, K. Biological low-pH Mn(II) oxidation in a manganese deposit influenced by metal-rich groundwater. Appl. Environ. Microbiol. 2016, 82, 3009–3021. [Google Scholar] [CrossRef]
- White, W.B.; Scheetz, B.E.; Atkinson, S.D.; Ibberson, D.; Chess, C.A. Mineralogy of Rohrer’s Cave, Pennsylvania. NSS Bull. 1985, 47, 17–27. [Google Scholar]
- Yusta, I.; Castellano, A.; Aranburu, A.; Velasco, F. Adsorción de metales en espeleotemas de Mn-Al-Fe de la Cueva de Lazalday (Zarate-Alava). Macla 2009, 11, 203–204. [Google Scholar]
- Mindat.org. Available online: https://www.mindat.org (accessed on 30 January 2018).
- Mineralogy Database. Available online: https://www.webmineral.com (accessed on 30 November 2018).
- Sánchez-España, F.J.; López Pamo, E.; Santofimia, E.; Aduvire, O.; Reyes, J.; Barettino, D. Acid Mine Drainage in the Iberian Pyrite Belt (Odiel river watershed, Huelva, SW Spain): Geochemistry, Mineralogy and Environmental Implications. Appl. Geochem. 2005, 20–27, 1320–1356. [Google Scholar] [CrossRef]
- Sánchez-España, J.; López-Pamo, E.; Santofimia, E.; Reyes, J.; Martín Rubí, J.A. The natural attenuation of two acidic effluents in Tharsis and La Zarza-Perrunal mines (Iberian Pyrite Belt, Huelva, Spain). Environ. Geol. 2005, 49, 253–266. [Google Scholar] [CrossRef]
- Sánchez-España, J. The behavior of iron and aluminum in acid mine drainage: Speciation, mineralogy, and environmental significance. In Thermodynamics, Solubility and Environmental Issues; Letcher, T., Ed.; Elsevier: Amsterdam, The Netherlands; pp. 137–150.
- Sánchez-Espańa, J.; López-Pamo, E.; Santofimia, E.; Reyes, J.; Martín-Rubí, J.A. The impact of acid mine drainage on the water quality of the Odiel river (Huelva, Spain): Evolution of precipitate mineralogy and aqueous geochemistry along the Concepción-Tintillo segment. Water Air Soil Pollut. 2006, 173, 121–149. [Google Scholar] [CrossRef]
- Sánchez-España, J.; López-Pamo, E.; Santofomia, E.; Reyes, J.; Martín-Rubí, J.A. The removal of dissolved metals by hydroxysulphate precipitates during oxidation and neutralization of acid mine waters, Iberian Pyrite Belt. Aquat. Geochem. 2006, 12–13, 269–298. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Reyes, J. Comparing Schwertmannite and Hydrobasaluminite Dissolution in Ammonium Oxalate (pH 3.0): Implications for Metal Speciation Studies by Sequential Extraction. Minerals 2019, 9, 57. [Google Scholar] [CrossRef]
- Sánchez-España, J.; López-Pamo, E.; Santofimia, E.; Diez-Ercilla, M. The acidic mine pit lakes of the Iberian Pyrite Belt: An approach to their physical limnology and hydrogeochemistry. Appl. Geochem. 2008, 23, 1260–1287. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Yusta, I.; Diez-Ercilla, M. Schwertmannite and hydrobasaluminite: A re-evaluation of therir solubility and control on the iron and aluminum concentration in acidic pit lakes. Appl. Geochem. 2011, 26, 1752–1774. [Google Scholar] [CrossRef]
- Sánchez-España, J.; López-Pamo, E.; Diez, M.; Santofimia, E. Physico-chemical gradients and meromictic stratification in Cueva de la Mora and other acidic pit lakes of the Iberian Pyrite Belt. Mine Water Environ. 2009, 28, 15–19. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Yusta, I.; López, G.A. Schwertmannite to jarosite conversion in the water column of an acidic mine pit lake. Mineral. Mag. 2012, 76, 2659–2682. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Diez, M.; Santofimia, E. Mine pit lakes of the Iberian Pyrite Belt: Some basic limnological, hydrogeochemical and microbiological con-siderations. In Acidic Pit Lakes: The Legacy of Coal and Metal Surface Mines; Geller, W., Schultze, M., Kleinmann, B., Wolkersdorfen, C., Eds.; Springer: Heidelberg, Germany, 2013; pp. 315–342. [Google Scholar]
- Sánchez-España, J.; Boehrer, B.; Yusta, I. Extreme carbon dioxide concentrations in acidic pit lakes provoked by water/rock interaction. Environ. Sci. Technol. 2014, 48, 4273–4281. [Google Scholar] [CrossRef]
- Balistrieri, L.S.; Murray, J.W.; Paul, B. The cycling of iron and manganese in the water column of Lake Sammamish, Washington. Limnol. Oceanogr. 1992, 37, 510–528. [Google Scholar] [CrossRef]
- Kawashima, M.; Takamatsu, T.; Koyama, M. Mechanisms of precipitation of manganese(II) in Lake Biwa, a fresh water lake. Water Resources 1988, 22, 613–618. [Google Scholar] [CrossRef]
- Davison, W.; Woof, C.; Rigg, E. The dynamics of iron and manganese in a seasonally anoxic lake: Direct measurements of fluxes using sediment traps. Limnol. Oceanogr. 1982, 27, 987–1003. [Google Scholar] [CrossRef]
- Douglas, G.B. Contaminant removal from acidic mine pit water via in situ hydrotalcite formation. Appl. Geochem. 2014, 51, 15–22. [Google Scholar] [CrossRef]
- Douglas, G.; Shackleton, M.; Woods, P. Hydrotalcite formation facilitates effective contaminant and radionuclide removal from acidic uranium mine barren lixiviant. Appl. Geochem. 2014, 42, 27–37. [Google Scholar] [CrossRef]
- Mubarok, M.Z.; Lieberto, J. Precipitation of nickel hydroxide from simulated and atmospheric-leach solution of nickel leterite ore. Proc. Earth Planet. Sci. 2013, 6, 457–464. [Google Scholar] [CrossRef]
- Kefeni, K.K.; Msagati, T.M.; Maree, J.P.; Mamba, B.B. Metals and sulphate removal from acid mine drainage in two steps via ferrite sludge and barium sulphate formation. Miner. Eng. 2015, 81, 79–87. [Google Scholar] [CrossRef]
- Cannon, W.F.; Kimball, B.E.; Corathers, L.A. Manganese. In Critical Mineral Resources of the United States-Economic and Environmental Geology and Prospects for Future Supply; Schulz, K.J., DeYoung, J.H., Seal, R.R., Bradley, D.C., Eds.; US Geological Survey Professional Paper 1802; U.S. Geological Survey: Reston, VA, USA, 2017; pp. L1–L28. [Google Scholar] [CrossRef]
- Singerling, S.A.; Tuck, C.A. Ferroalloys [Advanced Release]. In US Geological Survey Minerals Yearbook-2015, Volume I: Metals and Minerals; US Department of the Interior, US Geological Survey: Reston, VA, USA, May 2018; pp. 25.1–25.14. [Google Scholar]
- Mittal, N.K.; Sen, P.K. India’s first medium scale demonstration plant for treating polymetallic nodules. Miner. Eng. 2003, 16, 865–868. [Google Scholar] [CrossRef]
- Zubkov, M.V.; Plucinski, P.K.; Dartiguelongue, A.C.Y.; Lusty, P.A. Metal extraction from deep-ocean mineral deposits. Elements 2018, 14, 319–324. [Google Scholar] [CrossRef]
- Lusty, P.A.J.; Hein, J.R.; Josso, P. Formation and occurrence of ferromanganese crusts: Earth’s storehouse for critical metals. Elements 2018, 14, 313–318. [Google Scholar] [CrossRef]
Figure 1.
Panoramic views of four different pit lakes in Spanish mine districts, which have been selected for this study: (a) La Zarza; (b) Guadiana; (c) Brunita; and (d) Reocín. The water chemistry of these four pit lakes (major cations, sulfate, and trace metals) is given in Table 1.
Figure 1.
Panoramic views of four different pit lakes in Spanish mine districts, which have been selected for this study: (a) La Zarza; (b) Guadiana; (c) Brunita; and (d) Reocín. The water chemistry of these four pit lakes (major cations, sulfate, and trace metals) is given in Table 1.

Figure 2.
X-Ray diffraction (XRD) patterns obtained for precipitates formed during the Mn(II) oxidation experiments at pH 8.5–9.0 under slow precipitation kinetics: (a) brinessite-rich precipitate, (b) asbolane-rich precipitate, and (c) manganite-rich precipitate. The diffractogram of desautelsite (d), obtained by fast precipitation kinetics during neutralization at pH 9–10 in a previous study ([38]) was included for comparison. The blue bars at the bottom of each XRD pattern indicate the positions of diagnostic reflections for the dominant Mn oxide phase in each precipitate (b, birnessite; a, asbolane; m, manganite; d, desautelsite). The label “g” denotes peaks of gypsum, which were also accidentally formed in some solutions during the experiments.
Figure 2.
X-Ray diffraction (XRD) patterns obtained for precipitates formed during the Mn(II) oxidation experiments at pH 8.5–9.0 under slow precipitation kinetics: (a) brinessite-rich precipitate, (b) asbolane-rich precipitate, and (c) manganite-rich precipitate. The diffractogram of desautelsite (d), obtained by fast precipitation kinetics during neutralization at pH 9–10 in a previous study ([38]) was included for comparison. The blue bars at the bottom of each XRD pattern indicate the positions of diagnostic reflections for the dominant Mn oxide phase in each precipitate (b, birnessite; a, asbolane; m, manganite; d, desautelsite). The label “g” denotes peaks of gypsum, which were also accidentally formed in some solutions during the experiments.

Figure 3.
SEM images of Mn oxides formed during oxidation of Mn(II) in controlled experiments with mine waters from Guadiana mine (a,b), La Zarza mine (c,d), and Brunita mine (e,f). Mineral abbreviations: Bir, Birnessite; Asb, Asbolane; Man, Manganite; Gp, Gypsum.
Figure 3.
SEM images of Mn oxides formed during oxidation of Mn(II) in controlled experiments with mine waters from Guadiana mine (a,b), La Zarza mine (c,d), and Brunita mine (e,f). Mineral abbreviations: Bir, Birnessite; Asb, Asbolane; Man, Manganite; Gp, Gypsum.

Figure 4.
[Co + Ni + Zn] vs. Mn content (a) and Ni vs. Co content (b) measured by SEM–EDX in manganese oxides formed during Mn(II) oxidation in different mine waters. Orange circles are analyses conducted in asbolane globular aggregates from La Zarza mine water (see Figure 3c,d) whereas white circles correspond to measurements on manganite nanocrystals formed in AMD from Brunita mine (Figure 3e,f), and on birnessite crystals from Guadiana mine water (Figure 3a,b). The regression line plotted in (b) only considers the points in the shaded area (i.e., outliers excluded).
Figure 4.
[Co + Ni + Zn] vs. Mn content (a) and Ni vs. Co content (b) measured by SEM–EDX in manganese oxides formed during Mn(II) oxidation in different mine waters. Orange circles are analyses conducted in asbolane globular aggregates from La Zarza mine water (see Figure 3c,d) whereas white circles correspond to measurements on manganite nanocrystals formed in AMD from Brunita mine (Figure 3e,f), and on birnessite crystals from Guadiana mine water (Figure 3a,b). The regression line plotted in (b) only considers the points in the shaded area (i.e., outliers excluded).

Figure 5.
Enrichment factors (EF) of Co (a), Ni (b), and Zn (c), and cumulative trace metal content (∑(Co + Ni + Zn)) (d) for Mn oxides formed at different conditions: (1) in situ precipitates formed during slow Mn(II) oxidation in the water column of Reocín pit lake (pH 7.0–8.0) (black circles); (2) precipitates obtained in the lab during slow oxidation of dissolved Mn(II) in mine waters from Guadiana, La Zarza and Brunita (pH 8.5–9.0) (white circles); and (3) precipitates obtained in the lab during fast oxidation kinetics in neutralization experiments (pH 9.0–10.0) (black triangles). The EF values were calculated on the basis of metal (Co, Ni, Zn) concentrations in Mn oxides (Table 3), and the corresponding metal content in parent solutions (Table 1).
Figure 5.
Enrichment factors (EF) of Co (a), Ni (b), and Zn (c), and cumulative trace metal content (∑(Co + Ni + Zn)) (d) for Mn oxides formed at different conditions: (1) in situ precipitates formed during slow Mn(II) oxidation in the water column of Reocín pit lake (pH 7.0–8.0) (black circles); (2) precipitates obtained in the lab during slow oxidation of dissolved Mn(II) in mine waters from Guadiana, La Zarza and Brunita (pH 8.5–9.0) (white circles); and (3) precipitates obtained in the lab during fast oxidation kinetics in neutralization experiments (pH 9.0–10.0) (black triangles). The EF values were calculated on the basis of metal (Co, Ni, Zn) concentrations in Mn oxides (Table 3), and the corresponding metal content in parent solutions (Table 1).

Figure 6.
Binary plots showing the relation between trace metal content ((a) Zn, (b) Co, (c) Ni) in Mn oxides, and the corresponding molar ratio of trace metal-to-Mn in the parent solutions (Table 1) ((a) [Zn/Mn]molar, (b) [Co/Mn]molar, (c) [Ni/Mn]molar). The encircled areas represent the different Mn oxide types (1–3; legend as in Figure 5).
Figure 6.
Binary plots showing the relation between trace metal content ((a) Zn, (b) Co, (c) Ni) in Mn oxides, and the corresponding molar ratio of trace metal-to-Mn in the parent solutions (Table 1) ((a) [Zn/Mn]molar, (b) [Co/Mn]molar, (c) [Ni/Mn]molar). The encircled areas represent the different Mn oxide types (1–3; legend as in Figure 5).
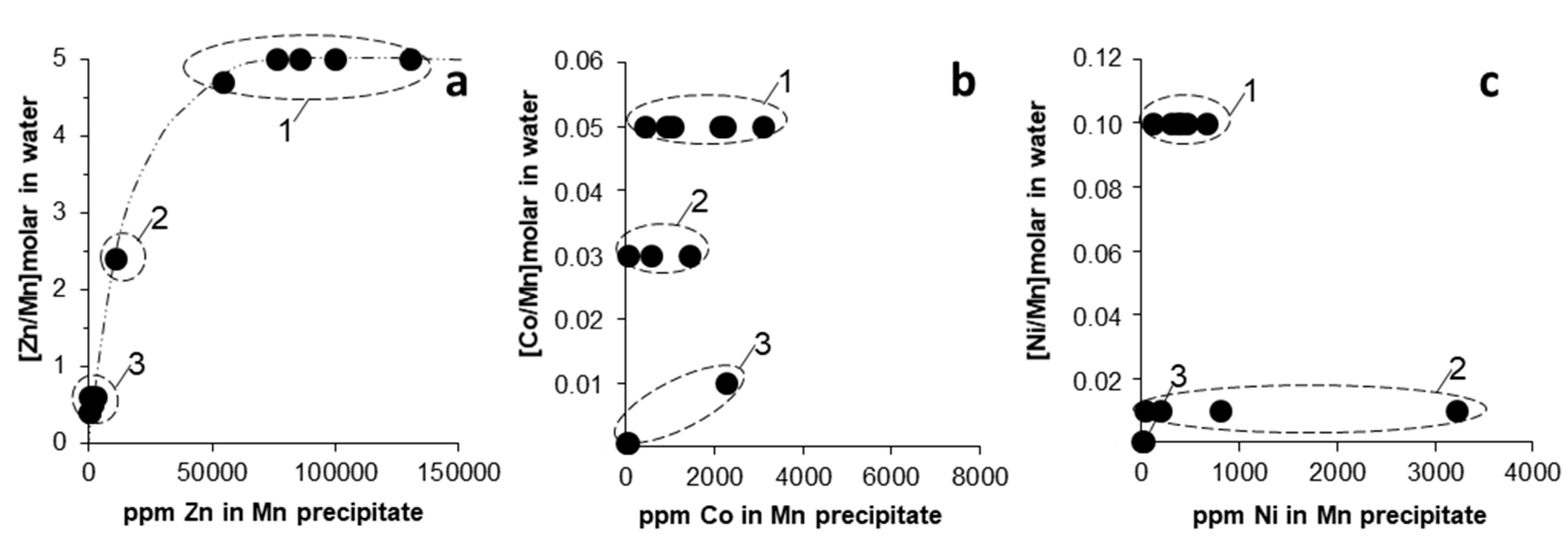
Figure 7.
Metal concentration (Mg, Ca, Mn, Co, Ni, and Zn) measured in mine waters from different pit lakes (Guadiana-Herrerías, HER; La Zarza, ZP; and Brunita, CB), before and after Mn(II) oxidation under continuous stirring.
Figure 7.
Metal concentration (Mg, Ca, Mn, Co, Ni, and Zn) measured in mine waters from different pit lakes (Guadiana-Herrerías, HER; La Zarza, ZP; and Brunita, CB), before and after Mn(II) oxidation under continuous stirring.

Figure 8.
Relative molar abundance of major ionic species of different metals in the pH range 2.0–10.0: (a) Co, (b) Ni, (c) Mn, and (d) Zn. Calculations conducted with PHREEQC using the MinteqA2.v4 thermodynamic database for conditions of [Co(II)] = [Ni(II)] = [Mn(II)] = [Zn(II)] = 10−5 M, [SO42−] = 0.1 M, pe = 12.
Figure 8.
Relative molar abundance of major ionic species of different metals in the pH range 2.0–10.0: (a) Co, (b) Ni, (c) Mn, and (d) Zn. Calculations conducted with PHREEQC using the MinteqA2.v4 thermodynamic database for conditions of [Co(II)] = [Ni(II)] = [Mn(II)] = [Zn(II)] = 10−5 M, [SO42−] = 0.1 M, pe = 12.

Figure 9.
Evolution of the saturation index (SI) for selected Ni (a) and Co (b) mineral phases with pH during neutralization of acidic mine water. Calculations conducted with PHREEQCI using the MinteqA2.v4 thermodynamic database (same conditions as in Figure 8).
Figure 9.
Evolution of the saturation index (SI) for selected Ni (a) and Co (b) mineral phases with pH during neutralization of acidic mine water. Calculations conducted with PHREEQCI using the MinteqA2.v4 thermodynamic database (same conditions as in Figure 8).

Figure 10.
(a) Variation of ionic activities for different Mn species (Mn(II), Mn(III), Mn(VI), Mn(VII)) in the pe range 0–14. (b) Solubility of different manganese oxi-hydroxides (as defined by their corresponding saturation index) in the pe range of 0–14. (c) Relative distribution (in % molar abundance) of Mn(II) vs. MnO2. (d) Enlargement of (c) showing the evolution of Mn species in the critical pe range of 12–14. Calculations conducted with PHREEQCI using the minteqa2.v4 database. For all plots, the conditions were chosen to roughly resemble typical water composition of Guadiana and La Zarza mine pit lakes (T = 25 °C, pH = 8.5, [Mn] = [Na] = 10−3 M, [Ca] = 10−2 M, [K] = 10−4 M).
Figure 10.
(a) Variation of ionic activities for different Mn species (Mn(II), Mn(III), Mn(VI), Mn(VII)) in the pe range 0–14. (b) Solubility of different manganese oxi-hydroxides (as defined by their corresponding saturation index) in the pe range of 0–14. (c) Relative distribution (in % molar abundance) of Mn(II) vs. MnO2. (d) Enlargement of (c) showing the evolution of Mn species in the critical pe range of 12–14. Calculations conducted with PHREEQCI using the minteqa2.v4 database. For all plots, the conditions were chosen to roughly resemble typical water composition of Guadiana and La Zarza mine pit lakes (T = 25 °C, pH = 8.5, [Mn] = [Na] = 10−3 M, [Ca] = 10−2 M, [K] = 10−4 M).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Water chemistry (major cations, sulfate, silica and trace metals) at different depths in several pit lakes in Spain: La Zarza and Guadiana (massive sulfide mines, Huelva), Brunita (Zn-Pb-Fe-Ag mine, Murcia), and Reocín (Zn-Pb mine, Cantabria; taken from [33]).
Table 1.
Water chemistry (major cations, sulfate, silica and trace metals) at different depths in several pit lakes in Spain: La Zarza and Guadiana (massive sulfide mines, Huelva), Brunita (Zn-Pb-Fe-Ag mine, Murcia), and Reocín (Zn-Pb mine, Cantabria; taken from [33]).
| Pit Lake | Depth | Na | K | Mg | Ca | SO4 | SiO2 | Fe | Al | Mn | Zn | Co | Ni |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| m | mg/L | mg/L | mg/L | mg/L | mg/L | mg/L | mg/L | mg/L | mg/L | mg/L | µg/L | µg/L | |
| La Zarza | 0 | 37 | 1 | 756 | 552 | 14,712 | 150 | 3901 | 876 | 317 | 182 | 4126 | 3994 |
| La Zarza | 30 | 39 | 9 | 1190 | 453 | 29,108 | 133 | 10,072 | 1870 | 489 | 523 | 7295 | 6877 |
| La Zarza | 70 | 50 | 16 | 1536 | 510 | 37,044 | 122 | 14,334 | 2178 | 647 | 624 | 11,081 | 10,394 |
| Guadiana | 0 | 48 | 1 | 666 | 530 | 5894 | 62 | 342 | 76 | 158 | 130 | 3379 | 2494 |
| Guadiana | 40 | 91 | 11 | 1394 | 472 | 14,459 | 43 | 3082 | 144 | 270 | 171 | 7117 | 4217 |
| Guadiana | 50 | 106 | 15 | 1834 | 477 | 18,664 | 13 | 4417 | 205 | 314 | 231 | 9456 | 5564 |
| Guadiana | 60 | 83 | 17 | 2351 | 472 | 20,971 | 10 | 4867 | 51 | 358 | 259 | 9724 | 5398 |
| Brunita | 0 | 590 | 2 | 5273 | 580 | 26,092 | 74 | 438 | 72 | 466 | 230 | 656 | 975 |
| Brunita | 12 | 565 | 12 | 5241 | 542 | 27,288 | 67 | 955 | 53 | 536 | 237 | 630 | 936 |
| Brunita | 15 | 344 | 16 | 5237 | 481 | 31,647 | 24 | 3661 | 23 | 814 | 436 | 704 | 863 |
| Brunita | 17 | 330 | 16 | 5184 | 470 | 30,542 | 21 | 4129 | 29 | 892 | 425 | 678 | 795 |
| Reocín | 0 | 42 | 17 | 210 | 556 | 1920 | 4.4 | 0.0 | 0.2 | 0.4 | 2.6 | 50 | 76 |
| Reocín | 8 | 44 | 18 | 211 | 561 | 1940 | 4.5 | 0.1 | 0.3 | 0.5 | 2.6 | 51 | 77 |
| Reocín | 13 | 45 | 18 | 204 | 559 | 1930 | 4.6 | 0.8 | 0.3 | 1.1 | 3.3 | 56 | 82 |
| Reocín | 50 | 41 | 18 | 198 | 571 | 1920 | 4.5 | 1.0 | 0.3 | 1.1 | 3.3 | 55 | 82 |
| Reocín | 90 | 41 | 18 | 199 | 557 | 1940 | 4.7 | 1.1 | 0.2 | 1.2 | 3.3 | 56 | 79 |
Table 2.
Stoichiometric formulae of manganese oxides and oxy-hydroxides identified in different pit lakes in Spain.
Table 2.
Stoichiometric formulae of manganese oxides and oxy-hydroxides identified in different pit lakes in Spain.
| Mineral | Formula |
|---|---|
| Manganite 1 | Mn3+O(OH) |
| Asbolane 1 | Ni0.3Co0.1Ca0.1Mn4+1.5O1.5(OH)2∙0.6(H2O) |
| Hausmannite 1,2 | Mn3+3O4 |
| Desautelsite 2 | Mg6Mn3+2(CO3)(OH)16∙4H2O |
| Birnessite 1,3 | (Na, Ca, K)x(Mn4+Mn3+)2-xO4∙1.5H2O |
| Todorokite 1,3 | (Na, Ca, K)2(Mn4+Mn3+)6O12∙3–4.5H2O |
| Pyrochroite 2,3 | Mn2+(OH)2 |
| Ranciecite 3 | (Ca, Mn2+)Mn4+4O9∙3H2O |
| Lithiophorite 3 | (Al, Li)Mn4+O2(OH)2 |
1 Oxides obtained experimentally in the lab (pH 8.5–9; this study); 2 oxides formed by neutralization (pH 9–10) in a previous study; and 3 natural oxides formed in situ in the Reocín mine pit lake (pH 7–8).
Table 3.
Mineralogy (as deduced by XRD and SEM–EDX), pH range of precipitation, and trace metal content (as measured by ICP-AES, AAS, and/or ICP-MS) of Mn oxides formed by Mn(II) oxidation in Mn-rich waters from different Spanish mine pit lakes (on site or in the lab). Data for natural precipitates found in Reocín pit lake were taken from [33].
Table 3.
Mineralogy (as deduced by XRD and SEM–EDX), pH range of precipitation, and trace metal content (as measured by ICP-AES, AAS, and/or ICP-MS) of Mn oxides formed by Mn(II) oxidation in Mn-rich waters from different Spanish mine pit lakes (on site or in the lab). Data for natural precipitates found in Reocín pit lake were taken from [33].
| Lake | Mineralogy | Conditions | Precip. Kinetics | pH | Ba | Cd | Co | Mo | Ni | Se | Pb | Zn |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ppm | ppm | ppm | ppm | ppm | ppm | ppm | ppm | |||||
| CB | Man | Lab oxidation | Slow (lab) | 8.5–9.0 | 333 | n.d. | 26 | n.d. | 25 | n.d. | n.d. | 279 |
| CB | Man | Lab oxidation | Slow (lab) | 8.5–9.0 | 445 | n.d. | 6 | n.d. | 8 | n.d. | n.d. | 225 |
| ZP | Asb | Lab oxidation | Slow (lab) | 8.5–9.0 | 532 | 153 | 2266 | n.d. | 3220 | n.d. | n.d. | 1101 |
| GUA | Bir | Lab oxidation | Slow (lab) | 8.5–9.0 | 2394 | 183 | 1424 | n.d. | 814 | n.d. | n.d. | 2713 |
| GUA | Bir | Lab oxidation | Slow (lab) | 8.5–9.0 | 2244 | n.d. | 38 | n.d. | 45 | n.d. | n.d. | 1010 |
| GUA | Tod | Lab oxidation | Slow (lab) | 8.5–9.0 | n.m. | n.d. | 560 | n.d. | 200 | n.d. | n.d. | 132 |
| CM | Des, Haus | Lab neutralization | Fast (lab) | 9.0–10.0 | n.m. | n.d. | 6566 | n.d. | 3095 | n.d. | n.d. | 6982 |
| ST | Des, Haus | Lab neutralization | Fast (lab) | 9.0–10.0 | n.m. | n.d. | 2981 | n.d. | 1601 | n.d. | n.d. | 3142 |
| Reo | Bir | Natural precipitate | Slow (on site) | 7.0–8.0 | n.m. | 53 | 2125 | 123 | 675 | 228 | 177 | 10,533 |
| Reo | Bir | Natural precipitate | Slow (on site) | 7.0–8.0 | n.m. | 65 | 2215 | 180 | 390 | 222 | 218 | 99,813 |
| Reo | Tod, Ran | Natural precipitate | Slow (on site) | 7.0–8.0 | n.m. | 74 | 3099 | 256 | 465 | 195 | 287 | 85,200 |
| Reo | Bir | Natural precipitate | Slow (on site) | 7.0–8.0 | n.m. | 24 | 900 | 100 | 116 | 140 | 239 | 54,060 |
| Reo | Bir | Natural precipitate | Slow (on site) | 7.0–8.0 | n.m. | 66 | 1042 | n.d. | 380 | 53 | 10 | 130,000 |
| Reo | Bir, Lit, Pyr | Natural precipitate | Slow (on site) | 7.0–8.0 | n.m. | 62 | 420 | 51 | 311 | 64 | 480 | 76,029 |
Abbreviations: n.m., not measured; n.d., not detected; Mine locations: CB, Brunita; ZP, La Zarza; GUA, Guadiana; CM; Cueva de la Mora; ST, San Telmo; Reo, Reocín. Mineral names: Bir, birnessite; Tod, todorokite; Ran, ranciecite; Lit, lithioforite; Pyr, pyrochroite; Man, manganite; Asb, asbolane; Des, desautelsite; and Haus, hausmannite.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sánchez-España, J.; Yusta, I. Coprecipitation of Co2+, Ni2+ and Zn2+ with Mn(III/IV) Oxides Formed in Metal-Rich Mine Waters. Minerals 2019, 9, 226. https://doi.org/10.3390/min9040226
AMA Style
Sánchez-España J, Yusta I. Coprecipitation of Co2+, Ni2+ and Zn2+ with Mn(III/IV) Oxides Formed in Metal-Rich Mine Waters. Minerals. 2019; 9(4):226. https://doi.org/10.3390/min9040226
Chicago/Turabian StyleSánchez-España, Javier, and Iñaki Yusta. 2019. "Coprecipitation of Co2+, Ni2+ and Zn2+ with Mn(III/IV) Oxides Formed in Metal-Rich Mine Waters" Minerals 9, no. 4: 226. https://doi.org/10.3390/min9040226
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.