
De Novo Transcriptomic Characterization Enables Novel Microsatellite Identification and Marker Development in Betta splendens

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Material and Sample Collection
2.2. RNA Extraction and Construction and Sequencing of the cDNA Library
2.3. De Novo Assembly
2.4. Annotation and Classification
2.5. SSR Loci Search and Primer Design
2.6. SSR Polymorphism Examination
3. Results
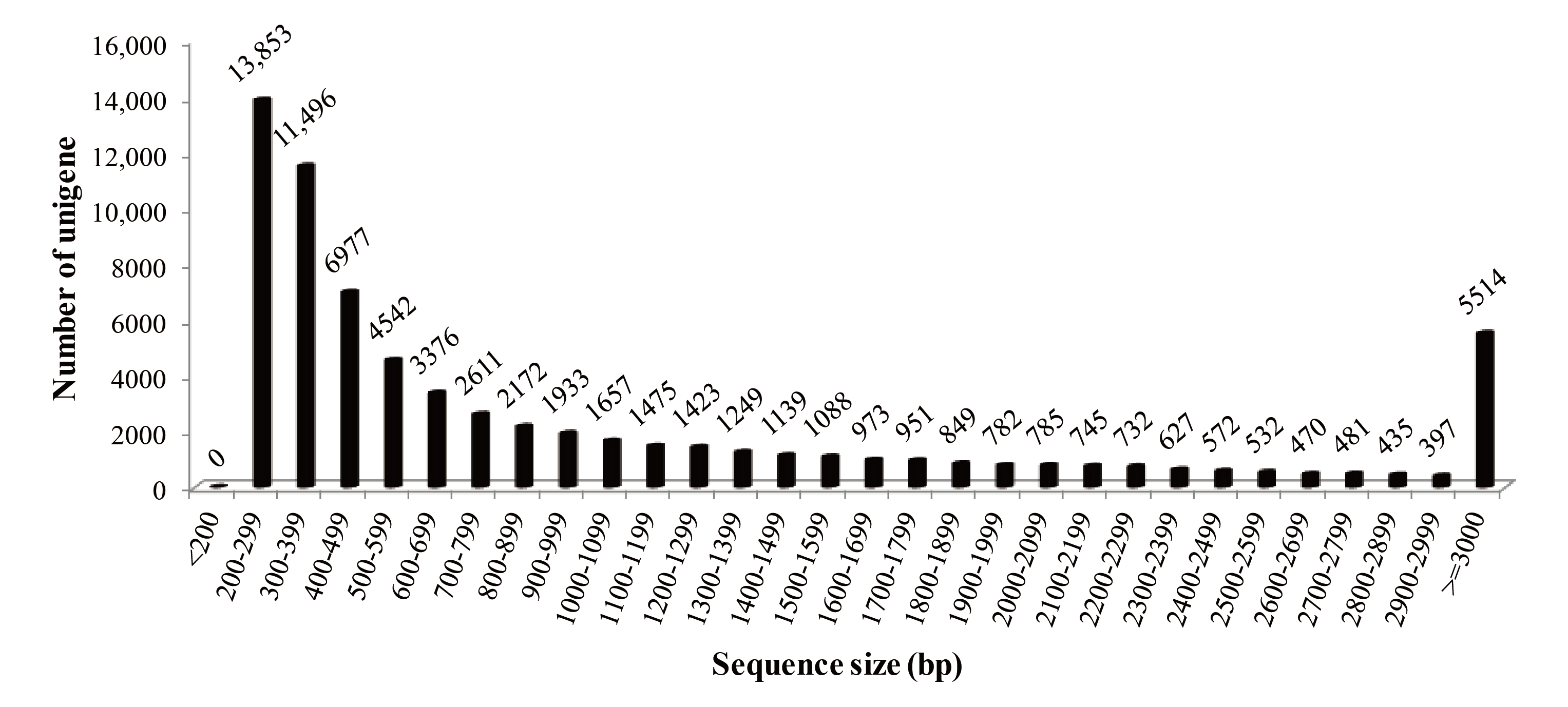
3.1. Sequencing and Assembly
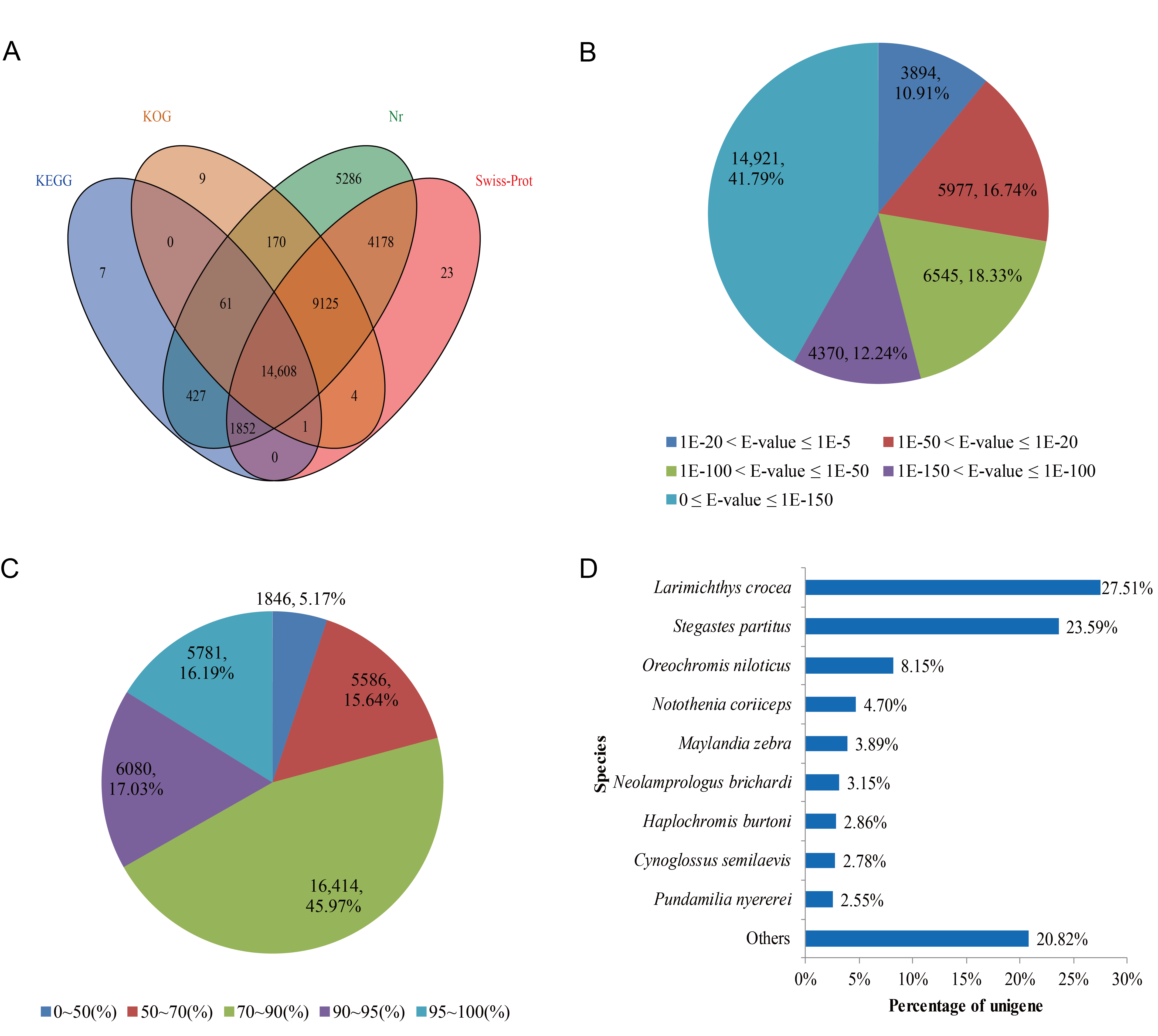
3.2. Functional Annotation
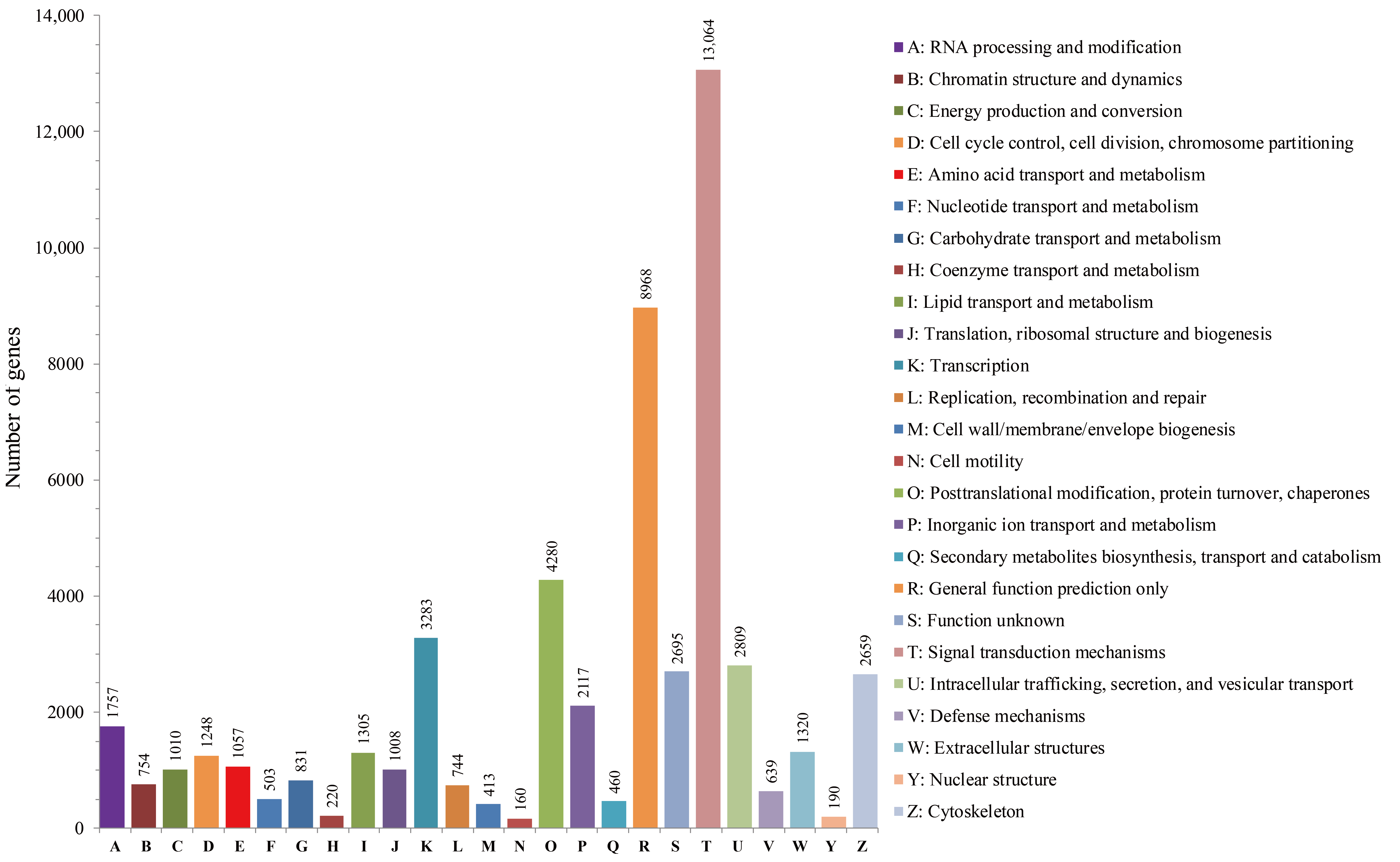
3.3. GO, KEGG Pathway and KOG Classification
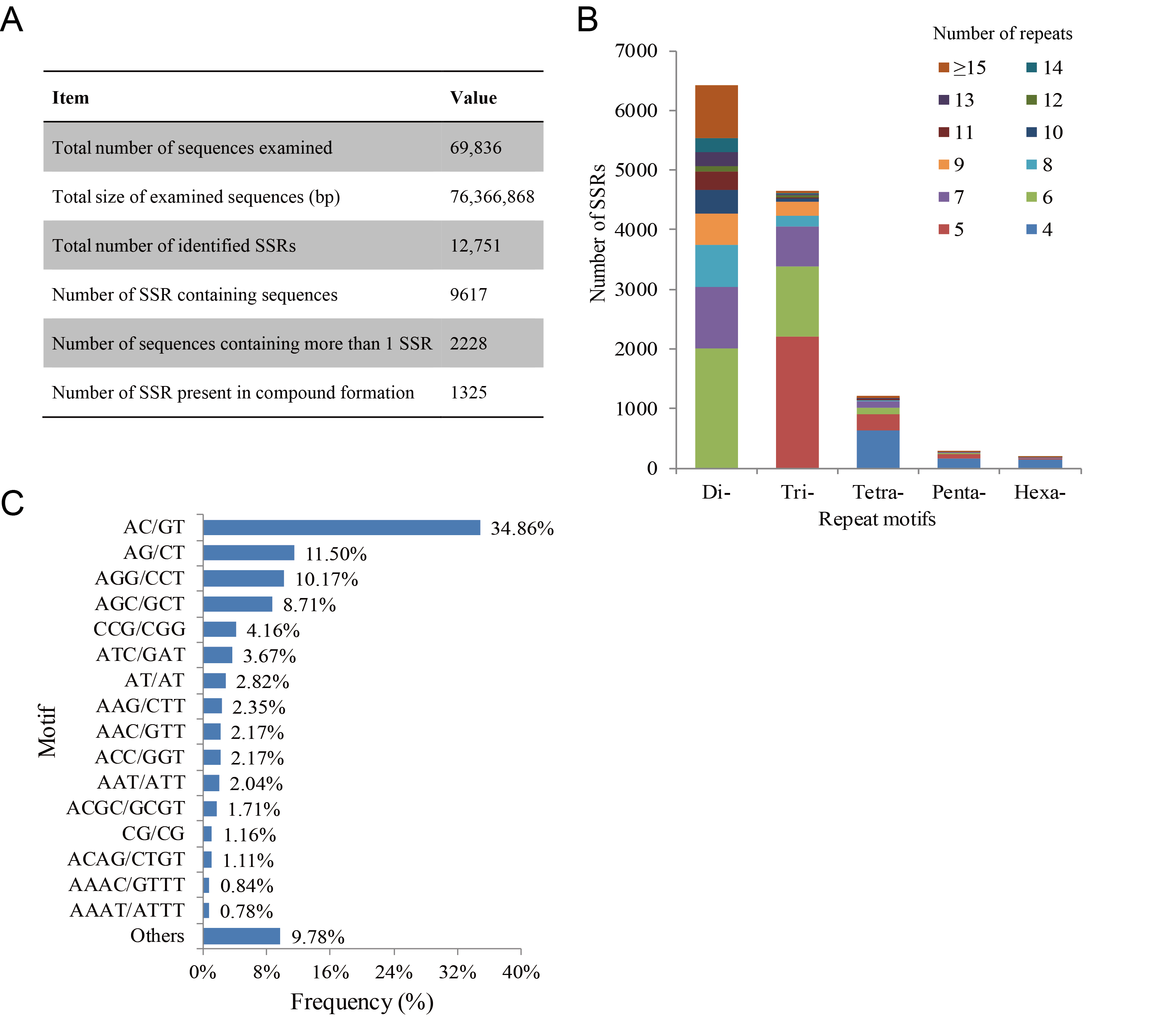
3.4. SSR Loci Identification and Polymorphism Verification
4. Discussion
4.1. Characterization of B. splendens Transcriptome
4.2. SSR Characterization and Marker Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Forsatkar, M.N.; Dadda, M.; Nematollahi, M.A. Lateralization of Aggression during Reproduction in Male Siamese Fighting Fish. Ethology 2015, 121, 1039–1047. [Google Scholar] [CrossRef]
- Jaroensutasinee, M.; Jaroensutasinee, K. Type of intruder and reproductive phase influence male territorial defence in wild-caught Siamese fighting fish. Behav. Process. 2003, 64, 23–29. [Google Scholar] [CrossRef]
- Meejui, O.; Sukmanomon, S.; Na-Nakorn, U. Allozyme revealed substantial genetic diversity between hatchery stocks of Siamese fighting fish, Betta splendens, in the province of Nakornpathom, Thailand. Aquaculture 2005, 250, 110–119. [Google Scholar] [CrossRef]
- Chailertrit, V.; Swatdipong, A.; Peyachoknagul, S.; Salaenoi, J.; Srikulnath, K. Isolation and characterization of novel microsatellite markers from Siamese fighting fish (Betta splendens, Osphronemidae, Anabantoidei) and their transferability to related species, B. smaragdina and B. imbellis. Genet. Mol. Res. 2014, 13, 7157–7162. [Google Scholar] [CrossRef]
- Qin, Y.; Sun, D.; Xu, T.; Liu, X.; Sun, Y. Genetic diversity and population genetic structure of the miiuy croaker, Miichthys miiuy, in the East China Sea by microsatellite markers. Genet. Mol. Res. 2014, 13, 10600–10606. [Google Scholar] [CrossRef]
- Li, C.; Teng, T.; Shen, F.; Guo, J.; Chen, Y.; Zhu, C.; Ling, Q. Transcriptome characterization and SSR discovery in Squaliobarbus curriculus. J. Oceanol. Limnol. 2018, 37, 235–244. [Google Scholar] [CrossRef]
- Dzieweczynski, T.L.; Hebert, O.L. Fluoxetine alters behavioral consistency of aggression and courtship in male Siamese fighting fish, Betta splendens. Physiol. Behav. 2012, 107, 92–97. [Google Scholar] [CrossRef]
- Eisenreich, B.R.; Szalda-Petree, A. Behavioral effects of fluoxetine on aggression and associative learning in Siamese fighting fish (Betta splendens). Behav. Process. 2015, 121, 37–42. [Google Scholar] [CrossRef]
- Forsatkar, M.N.; Nematollahi, M.A.; Brown, C. The toxicological effect of Ruta graveolens extract in Siamese fighting fish: A behavioral and histopathological approach. Ecotoxicology 2016, 25, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Dzieweczynski, T.L.; Hentz, K.B.; Logan, B.; Hebert, O.L. Chronic exposure to 17α-ethinylestradiol reduces behavioral consistency in male Siamese fighting fish. Behaviour 2014, 151, 633–651. [Google Scholar] [CrossRef]
- Regan, M.D.; Dhillon, R.S.; Toews, D.P.L.; Speers-Roesch, B.; Sackville, M.A.; Pinto, S.; Bystriansky, J.S.; Scott, G.R. Biochemical correlates of aggressive behavior in the Siamese fighting fish. J. Zool. 2015, 297, 99–107. [Google Scholar] [CrossRef]
- Fan, G.; Chan, J.; Ma, K.; Yang, B.; Zhang, H.; Yang, X.; Shi, C.; Law, H.C.H.; Ren, Z.; Xu, Q.; et al. Chromosome-level reference genome of the Siamese fighting fish Betta splendens, a model species for the study of aggression. GigaScience 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2009, 11, 31–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garber, M.; Grabherr, M.G.; Guttman, M.; Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 2011, 8, 469–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Chen, H.; Cui, X.; Zhang, K.; Jiang, D.; Deng, S.; Zhu, C.; Li, G. Sequencing, de novo assembly and characterization of the spotted scat Scatophagus argus (Linnaeus 1766) transcriptome for discovery of reproduction related genes and SSRs. J. Oceanol. Limnol. 2018, 36, 1329–1341. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; Garcia-Gomez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Lalitha, S. Primer premier 5. Biotech. Softw. Internet. Rep. 2000, 1, 270–272. [Google Scholar] [CrossRef]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finseth, F.R.; Harrison, R.G. A Comparison of Next Generation Sequencing Technologies for Transcriptome Assembly and Utility for RNA-Seq in a Non-Model Bird. PLoS ONE 2014, 9, e108550. [Google Scholar] [CrossRef] [Green Version]
- Zhenzhen, X.; Li, S.; Dengdong, W.; Chao, F.; Qiongyu, L.; Zihao, L.; XiaoChun, L.; Yong, Z.; Shuisheng, L.; Haoran, L. Transcriptome Analysis of the Trachinotus ovatus: Identification of Reproduction, Growth and Immune-Related Genes and Microsatellite Markers. PLoS ONE 2014, 9, e109419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, J.; Liu, P.; Gao, B.; Wang, Y.; Wang, Z.; Chen, P.; Li, J. Transcriptome Analysis of the Portunus trituberculatus: De Novo Assembly, Growth-Related Gene Identification and Marker Discovery. PLoS ONE 2014, 9, e94055. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Ma, A.; Huang, Z.; Wang, G.; Wang, T.; Xia, D.; Ma, B. Transcriptome Analysis for Identification of Genes Related to Gonad Differentiation, Growth, Immune Response and Marker Discovery in The Turbot (Scophthalmus maximus). PLoS ONE 2016, 11, e0149414. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Hou, R.; Bao, Z.; Du, H.; He, Y.; Su, H.; Zhang, Y.; Fu, X.; Jiao, W.; Li, Y.; et al. Transcriptome Sequencing of Zhikong Scallop (Chlamys farreri) and Comparative Transcriptomic Analysis with Yesso Scallop (Patinopecten yessoensis). PLoS ONE 2013, 8, e63927. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zeng, D.; Chen, X.; Xie, D.; Zhao, Y.; Yang, C.; Li, Y.; Ma, N.; Li, M.; Yang, Q.; et al. Transcriptome Analysis of Litopenaeus vannamei in Response to White Spot Syndrome Virus Infection. PLoS ONE 2013, 8, e73218. [Google Scholar] [CrossRef] [Green Version]
- Malachowicz, M.; Wenne, R.; Burzyński, A. De novo assembly of the sea trout (Salmo trutta m. trutta) skin transcriptome to identify putative genes involved in the immune response and epidermal mucus secretion. PLoS ONE 2017, 12, e0172282. [Google Scholar] [CrossRef]
- Jiang, Y.; Fan, W.; Xu, J. De novo transcriptome analysis and antimicrobial peptides screening in skin of Paa boulengeri. Genes Genom. 2017, 39, 653–665. [Google Scholar] [CrossRef]
- Lu, J.; Luan, P.; Zhang, X.; Xue, S.; Peng, L.; Mahbooband, S.; Sun, X. Gonadal transcriptomic analysis of yellow catfish (Pelteobagrus fulvidraco): Identification of sex-related genes and genetic markers. Physiol. Genom. 2014, 46, 798–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.-Y.; Li, Y.-H.; Yang, S.; Li, Q.-W. De novo Assembly and Characterization of the Global Transcriptome for Rhyacionia leptotubula Using Illumina Paired-End Sequencing. PLoS ONE 2013, 8, e81096. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.-Y.; Yang, H.; Wei, C.-L.; Yu, O.; Zhang, Z.-Z.; Jiang, C.-J.; Sun, J.; Li, Y.-Y.; Chen, Q.; Xia, T.; et al. Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genom. 2011, 12, 131. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.; Lyons, R.E.; Dinh, H.; Hurwood, D.A.; McWilliam, S.; Mather, P.B. Transcriptomics of a Giant Freshwater Prawn (Macrobrachium rosenbergii): De Novo Assembly, Annotation and Marker Discovery. PLoS ONE 2011, 6, e27938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, M.; Li, N.; Niu, B.; Liu, Y.; You, D.; Jiang, D.; Ruan, C.; Qin, Z.; Song, T.; Wang, W. De novo transcriptome analysis of Bagarius yarrelli (Siluriformes: Sisoridae) and the search for potential SSR markers using RNA-Seq. PLoS ONE 2018, 13, e0190343. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.T.; Gao, Z.-X.; Zhao, H.-H.; Yi, S.-K.; Chen, B.-X.; Zhao, Y.-H.; Lin, L.; Liu, X.-Q.; Wang, W.-M. Transcriptome analysis and microsatellite discovery in the blunt snout bream (Megalobrama amblycephala) after challenge with Aeromonas hydrophila. Fish Shellfish. Immunol. 2015, 45, 72–82. [Google Scholar] [CrossRef]
- Gao, Z.; Luo, W.; Liu, H.; Zeng, C.; Liu, X.; Yi, S.; Wang, W. Transcriptome Analysis and SSR/SNP Markers Information of the Blunt Snout Bream (Megalobrama amblycephala). PLoS ONE 2012, 7, e42637. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Li, G.; Mo, Z.; Xiao, P.; Li, J.; Huang, J. De Novo Assembly of the Japanese Flounder (Paralichthys olivaceus) Spleen Transcriptome to Identify Putative Genes Involved in Immunity. PLoS ONE 2015, 10, e0117642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Ling, Q.; Li, C.; Wang, G.; Xu, Z.; Lu, G. Characterization of pikeperch (Sander lucioperca) transcriptome and development of SSR markers. Biochem. Syst. Ecol. 2016, 66, 188–195. [Google Scholar] [CrossRef]
- Chen, X.; Mei, J.; Wu, J.; Jing, J.; Ma, W.; Zhang, J.; Dan, C.; Wang, W.; Gui, J.-F. A Comprehensive Transcriptome Provides Candidate Genes for Sex Determination/Differentiation and SSR/SNP Markers in Yellow Catfish. Mar. Biotechnol. 2014, 17, 190–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, H.; Li, C.; Du, H.; Zhang, S.; Wei, Q. Sequencing and De Novo Assembly of the Gonadal Transcriptome of the Endangered Chinese Sturgeon (Acipenser sinensis). PLoS ONE 2015, 10, e0127332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Kuang, Y.; Lü, W.; Cao, D.; Sun, X. Transcriptome-derived EST–SSR markers and their correlations with growth traits in crucian carp Carassius auratus. Fish. Sci. 2014, 80, 977–984. [Google Scholar] [CrossRef]
- Lu, X.; Luan, S.; Kong, J.; Hu, L.; Mao, Y.; Zhong, S. Genome-wide mining, characterization, and development of microsatellite markers in Marsupenaeus japonicus by genome survey sequencing. Chin. J. Oceanol. Limnol. 2015, 35, 203–214. [Google Scholar] [CrossRef]
- Sha, Z.-X.; Luo, X.-H.; Liao, X.-L.; Wang, S.-L.; Wang, Q.-L.; Chen, S.-L. Development and characterization of 60 novel EST-SSR markers in half-smooth tongue sole Cynoglossus semilaevis. J. Fish Biol. 2010, 78, 322–331. [Google Scholar] [CrossRef]
- Wang, D.; Liao, X.; Cheng, L.; Yu, X.; Tong, J. Development of novel EST-SSR markers in common carp by data mining from public EST sequences. Aquac. 2007, 271, 558–574. [Google Scholar] [CrossRef]
- Luo, W.; Deng, W.; Yi, S.; Wang, W.; Gao, Z. Characterization of 20 polymorphic microsatellites for Blunt snout bream (Megalobrama amblycephala) from EST sequences. Conserv. Genet. Resour. 2012, 5, 499–501. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar] [PubMed]
- Ge, J.; Chen, S.; Liu, C.; Bian, L.; Sun, H.; Tan, J. Characterization of the global transcriptome and microsatellite marker information for spotted halibut Verasper variegatus. Genes Genom. 2016, 39, 307–316. [Google Scholar] [CrossRef]
- Zhang, M.; Nie, J.; Shen, Y.; Xu, X.; Dang, Y.; Wang, R.; Li, J. Isolation and characterization of 25 novel EST-SNP markers in grass carp (Ctenopharyngodon idella). Conserv. Genet. Resour. 2015, 7, 819–822. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, W.; Song, X.; Lin, Q.; Gui, J.-F.; Mei, J. Characterization and Development of EST-SSR Markers Derived from Transcriptome of Yellow Catfish. Mol. 2014, 19, 16402–16415. [Google Scholar] [CrossRef]
- Han, Z.; Ma, X.; Wei, M.; Zhao, T.; Zhan, R.; Chen, W. SSR marker development and intraspecific genetic divergence exploration of Chrysanthemum indicum based on transcriptome analysis. BMC Genom. 2018, 19, 291. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequencing | Assembly | ||||
|---|---|---|---|---|---|
| Item | Female Library | Male Library | Total | Item | Value |
| Raw reads | 54,600,768 | 53,815,332 | 108,416,100 | Unigenes | 69,836 |
| Raw bases (bp) | 6,825,096,000 | 6,726,916,500 | 13,552,012,500 | GC content (%) | 50.66 |
| Clean reads | 53,062,092 | 52,443,394 | 105,505,486 | Total length (bp) | 76,366,868 |
| Clean bases (bp) | 6,632,761,500 | 6,555,424,250 | 13,188,185,750 | N50 length (bp) | 2040 |
| – | – | – | – | Mean length (bp) | 1093.52 |
| SSR ID | Locus | Primer Sequences (5′-3′) | Repeat Motif | Allele Size (bp) | Ta (°C) | Accession No. | Na | NE | Ho | He | PIC | P-HWE | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BsSSR008 | Unigene0061796 | F | GATTCGGACTACGAGCTGGA | (CA)14 | 187 | 60 | MZ615708 | 4 | 2.224 | 0.867 | 0.642 | 0.569 | 0.2670 |
| R | CACATGAAGCTTAGTGGGGG | ||||||||||||
| BsSSR010 | Unigene0009720 | F | CTGGAGAACACGCAGTACGA | (GGACCA)4 | 276 | 55 | MZ615709 | 3 | 2.033 | 0.367 | 0.633 | 0.544 | 0.1735 |
| R | TTCGGATCAGTGTCATGAGG | ||||||||||||
| BsSSR014 | Unigene0028390 | F | GTGACGATGGAGAAAACGGT | (GCC)6 | 149 | 58 | MZ615710 | 3 | 2.400 | 0.833 | 0.657 | 0.570 | 1.0000 |
| R | GCTTTTCTAGGTTTGCACGG | ||||||||||||
| BsSSR019 | Unigene0050924 | F | TCAGAAGAAAGAGGGACGGA | (AC)8 | 280 | 56 | MZ615711 | 3 | 2.020 | 0.700 | 0.595 | 0.513 | 0.0861 |
| R | GAGAGAGTCCAACCTGCACA | ||||||||||||
| BsSSR020 | Unigene0015325 | F | AAGCAGCAACACACGAACAG | (TG)10 | 128 | 56 | MZ615712 | 3 | 1.212 | 0.233 | 0.430 | 0.357 | 0.0364 |
| R | TTGATTCTCTGCACGCTGTC | ||||||||||||
| BsSSR023 | Unigene0021148 | F | ACACAACAGTCTCCCTTCGC | (CGCCA)5 | 203 | 58 | MZ615713 | 5 | 2.727 | 0.724 | 0.682 | 0.617 | 0.1483 |
| R | GGGGGACAATGGGAGAAATA | ||||||||||||
| BsSSR031 | Unigene0041043 | F | AGGAGAGAGTGAGACAGCGG | (AGC)6 | 239 | 56 | MZ615714 | 3 | 1.830 | 0.800 | 0.615 | 0.527 | 0.2250 |
| R | CTGGAAAACAAGGCAGAAGC | ||||||||||||
| BsSSR032 | Unigene0066200 | F | GGCAGCTAAACAACCTCCAG | (TA)6 | 246 | 56 | MZ615715 | 2 | 1.593 | 0.367 | 0.508 | 0.375 | 1.0000 |
| R | CAGGAGCAGCAGACTTTTCC | ||||||||||||
| BsSSR039 | Unigene0010115 | F | GGTCCAAACACAAACCCATC | (GT)10 | 236 | 54 | MZ615716 | 4 | 2.053 | 0.733 | 0.632 | 0.556 | 0.1031 |
| R | GTGCTTCATGCTTGTGCATT | ||||||||||||
| BsSSR042 | Unigene0013687 | F | GGATACAATGAAGGAGCGGA | (CA)9 | 239 | 58 | MZ615717 | 2 | 1.112 | 0.367 | 0.499 | 0.371 | 0.0011* |
| R | GCCTGTATTTGCATGTGGTG | ||||||||||||
| BsSSR043 | Unigene0049069 | F | TTCCGTTTCCTGGACTTGAC | (TTC)5 | 243 | 55 | MZ615718 | 2 | 1.365 | 0.400 | 0.506 | 0.374 | 0.1162 |
| R | GGATGACAGTCCCTGAGAGC | ||||||||||||
| BsSSR044 | Unigene0012215 | F | GGGTTTGCTCCAGTGATTGT | (GT)9 | 262 | 55 | MZ615719 | 4 | 3.200 | 0.400 | 0.617 | 0.541 | 0.0422 |
| R | GTCCACAAGCTTCCCGAATA | ||||||||||||
| BsSSR051 | Unigene0059934 | F | GTGATATCCTGTCACCGCCT | (ATT)8 | 131 | 55 | MZ615720 | 4 | 2.693 | 0.700 | 0.671 | 0.591 | 0.1483 |
| R | GACCAAATTAGCAGGGACGA | ||||||||||||
| BsSSR053 | Unigene0003140 | F | GTTCGGTGGCAGGGTATAAA | (TG)16 | 194 | 58 | MZ615721 | 4 | 1.626 | 0.367 | 0.607 | 0.541 | 0.0001* |
| R | ATGCTTCCTACTGCCCTGTG | ||||||||||||
| BsSSR068 | Unigene0022064 | F | ATGAGAGGAGGAGCAGCAAA | (TGCAGC)4 | 234 | 60 | MZ615722 | 4 | 2.173 | 0.400 | 0.724 | 0.658 | 0.1039 |
| R | GCCTTTGGAAGTGAGACAGG | ||||||||||||
| BsSSR069 | Unigene0008534 | F | TAAGAGCCCAGGTTTTCACG | (CAG)7 | 265 | 55 | MZ615723 | 4 | 2.441 | 0.733 | 0.718 | 0.652 | 0.4257 |
| R | GGCATGTCCTTGATGAGGTT | ||||||||||||
| BsSSR077 | Unigene0033389 | F | GGCTGATTCACCCCAAATAC | (TG)7 | 256 | 53 | MZ615724 | 3 | 2.390 | 0.933 | 0.651 | 0.568 | 0.0076* |
| R | TTCCTTTGCATTGCTCACAG | ||||||||||||
| BsSSR079 | Unigene0039100 | F | CGGAAGCAGCAGTCCTACAT | (GCG)7 | 234 | 57 | MZ615725 | 3 | 1.540 | 0.833 | 0.594 | 0.495 | 0.5415 |
| R | CTGCTGCAGCTCTTTCCTCT | ||||||||||||
| BsSSR081 | Unigene0006030 | F | GTGCGTAAAGCCGAAAGAAG | (GGC)5 | 220 | 55 | MZ615726 | 5 | 3.102 | 0.167 | 0.772 | 0.720 | 0.0692 |
| R | GTGAGGAGACACCGACTGCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Li, X.; Wang, Y.; Zhu, C.; Huang, H.; Yang, W.; Li, G. De Novo Transcriptomic Characterization Enables Novel Microsatellite Identification and Marker Development in Betta splendens. Life 2021, 11, 803. https://doi.org/10.3390/life11080803
Chen H, Li X, Wang Y, Zhu C, Huang H, Yang W, Li G. De Novo Transcriptomic Characterization Enables Novel Microsatellite Identification and Marker Development in Betta splendens. Life. 2021; 11(8):803. https://doi.org/10.3390/life11080803
Chicago/Turabian StyleChen, Huapu, Xiaomeng Li, Yaorong Wang, Chunhua Zhu, Hai Huang, Wei Yang, and Guangli Li. 2021. "De Novo Transcriptomic Characterization Enables Novel Microsatellite Identification and Marker Development in Betta splendens" Life 11, no. 8: 803. https://doi.org/10.3390/life11080803