
Advances in the Cystic Fibrosis Drug Development Pipeline
 and
and
Abstract
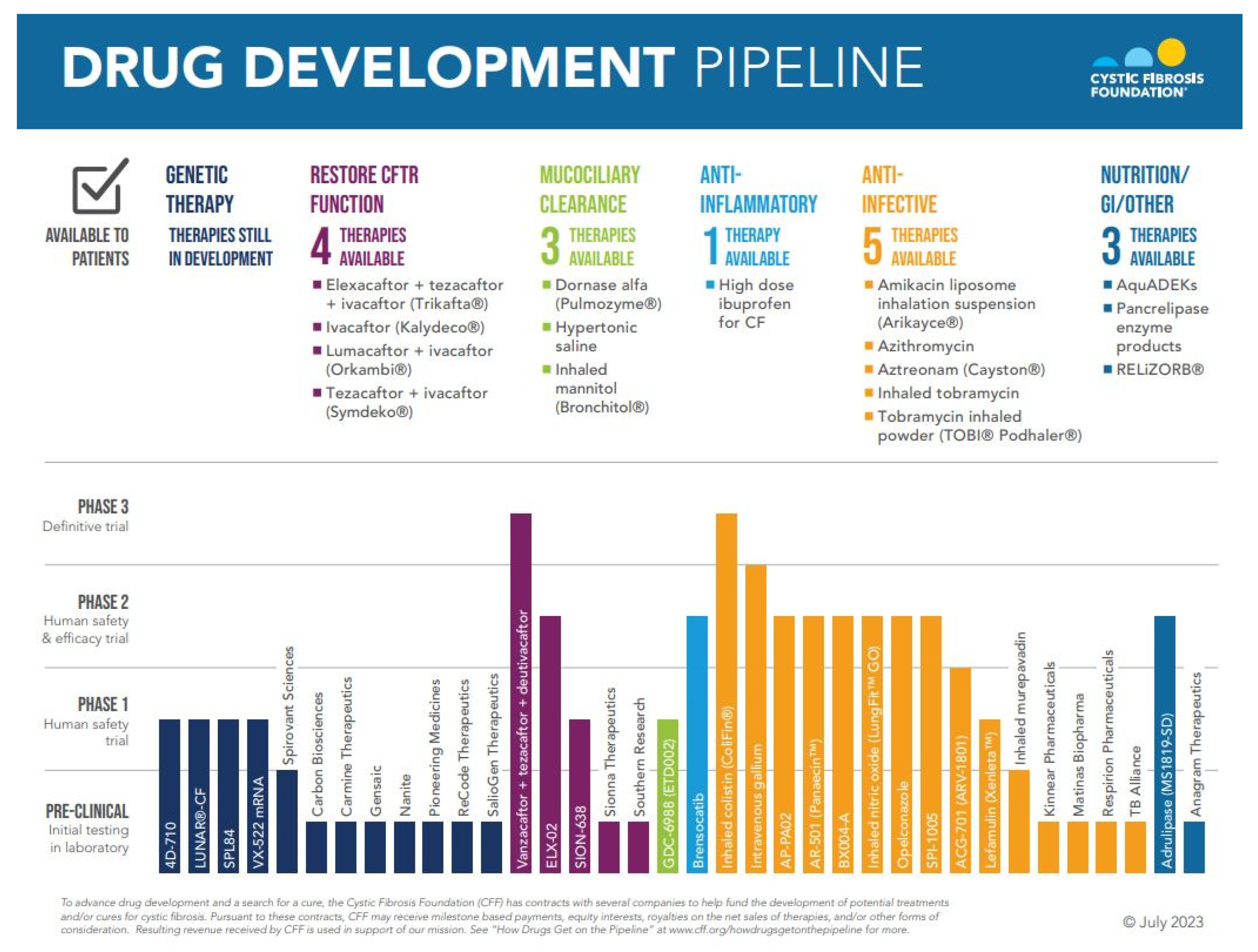
:1. Introduction
2. Restoration of CFTR Function with CFTR Modulator Therapy
3. Genetic Therapies
3.1. RNA Based Therapies
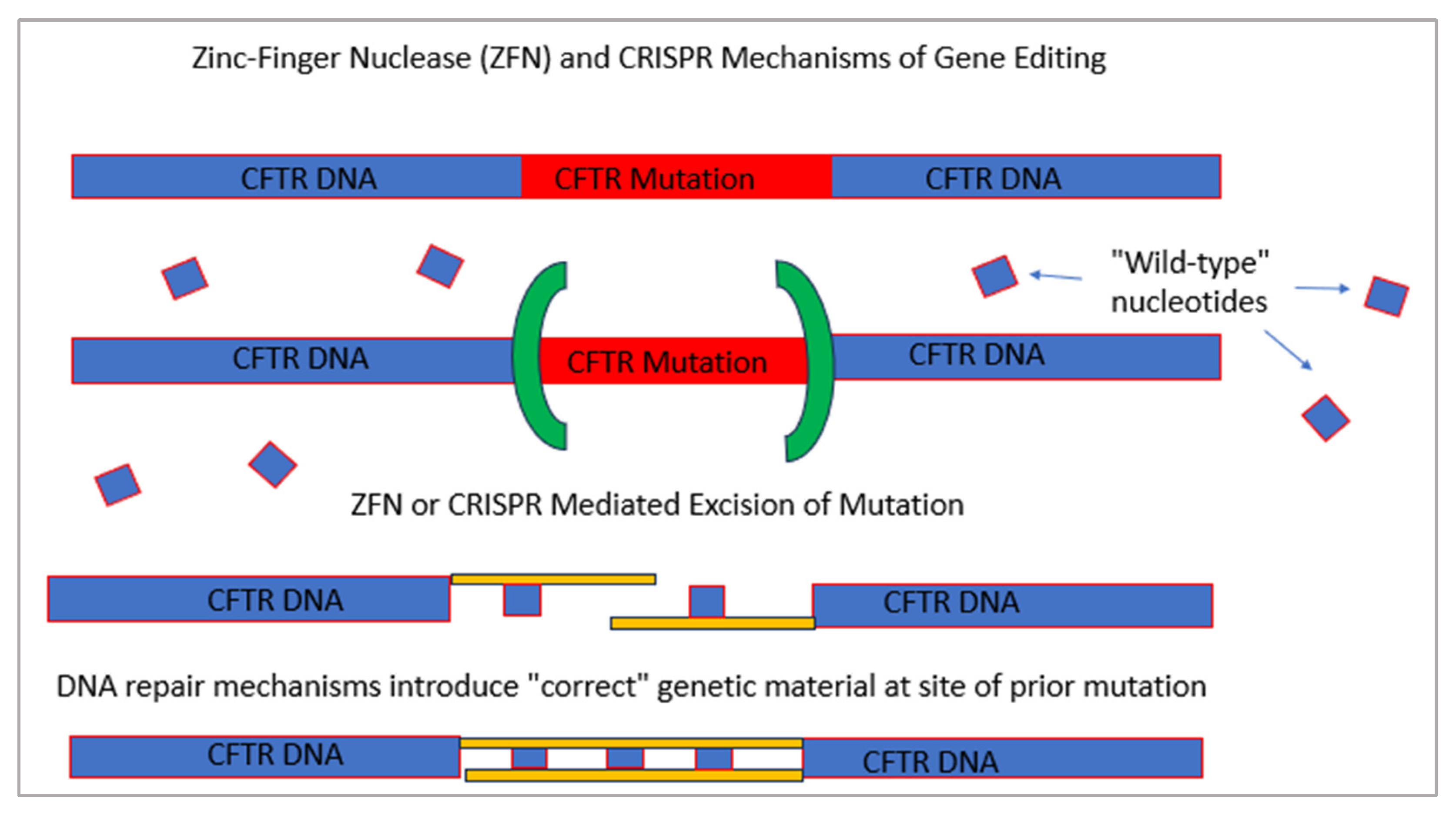
3.2. Gene Therapy
4. Antimicrobial Therapies
4.1. Oral and Intravenous Antibiotics
4.2. Inhaled Antibiotics
4.2.1. Tobramycin inhalation solution
4.2.2. Aztreonam Lysine (Cayston)
4.2.3. Liposomal Amikacin (Arikayce®)
4.2.4. Colistimethate Sodium (Colistin)
4.2.5. Inhaled Vancomycin
4.2.6. Inhaled Fluoroquinolones
4.3. Inhaled Murepavadin
4.4. Phage Therapy
4.5. Other Antimicrobial Therapies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimicrobial Therapies | Mechanism of Action | Targeted Pathogen |
|---|---|---|
| Inhalation Antibiotics | ||
| Tobramycin 1 | Aminoglycoside; inhibits protein synthesis of Gram-negative bacteria | Pseudomonas aeruginosa |
| Tobramycin dry powder 1 | ||
| Aztreonam 1 | Monobactam; inhibits cell wall synthesis against aerobic Gram-negative bacteria | |
| Levofloxacin 1 | Flouroquinolone; directly inhibits bacterial DNA synthesis | |
| Vancomycin 1 | Glycopeptide; inhibits Gram-positive bacterial cell wall synthesis | MRSA |
| Colistimethate 4 | Polymyxin derivative; disrupts the integrity of the bacterial cell membrane of most Gram-negative bacteria | Gram negative |
| Murepavadin 3 | Antimicrobial peptidomimetic; binds to the lipopolysaccharide transport protein in Gram-negative bacteria and causes cell death | Pseudomonas aeruginosa |
| Phage Therapy Studied Targets | ||
| Pseudomonas aeruginosa 2 | Viruses that infect specific bacteria and replicate the viral genome, ultimately lysing the host bacteria and killing it | As noted |
| Mycobacterium abscessus 3 | ||
| Staphylococcal aureus 3 | ||
| Achromobacter xylosoxidans 3 | ||
| Methicillin resistant Staphylcoccus aureus 3 | ||
| Other | ||
| Galium 2 | Disrupts iron-dependent processes necessary for bacterial survival and reproduction | Pseudomonas aeruginosa MAC Mycobacterium abscessus |
| Nitric oxide 2 | Gas compound that breaks down biofilms and kills bacteria | MAC Mycobacterium abscessus |
| Sodium fusidate 2 | Protein synthesis inhibitor that prevents bacterial replication | MRSA Pseudomonas aeruginosa Staphylococcal aureus |
| Lefamulin 2 | A pleuromutilin antibacterial compound that binds to bacterial ribosome, preventing the binding of transfer RNA and inhibiting the production of protein | MRSA Staphylococcal aureus |
| Glycopolymer SNSP113 2 | Disrupts bacterial biofilms; has anti-inflammatory properties | Staphylococcal aureus, MAC Mycobacterium abscessus |
| Opelconazole 2 | Synthetic triazole; prevents the formation of the fungi cell membrane, resulting in lysis | Aspergillus fumigatus |
5. Anti-Inflammatory Therapies
6. Mucolytics and Mucociliary Clearance
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Naehrlich, L. The Changing Face of Cystic Fibrosis and Its Implications for Screening. Int. J. Neonatal Screen. 2020, 6, 54. [Google Scholar] [CrossRef]
- Frantzen, T.; Barsky, S.; LaVecchia, G.; Marowitz, M.; Wang, J. Evolving Nutritional Needs in Cystic Fibrosis. Life 2023, 13, 1431. [Google Scholar] [CrossRef] [PubMed]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Burton, B.; Huang, C.-J.; Worley, J.; Cao, D.; Johnson, J.P.; Urrutia, A.; Joubran, J.; Seepersaud, S.; Sussky, K.; et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J. Cyst. Fibros. 2012, 11, 237–245. [Google Scholar] [CrossRef]
- De Boeck, K.; Munck, A.; Walker, S.; Faro, A.; Hiatt, P.; Gilmartin, G.; Higgins, M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J. Cyst. Fibros. 2014, 13, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.M.; Barry, P.J. Lumacaftor/ivacaftor for patients homozygous for Phe508del-CFTR: Should we curb our enthusiasm? Thorax 2015, 70, 615–616. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; Van Der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Cystic Fibrosis Foundation. Patient Registry 2021 Annual Data Report; Cystic Fibrosis Foundation: Bethesda, MD, USA, 2021. [Google Scholar]
- Uluer, A.Z.; MacGregor, G.; Azevedo, P.; Indihar, V.; Keating, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Rubenstein, R.C.; et al. Safety and efficacy of vanzacaftor-tezacaftor-deutivacaftor in adults with cystic fibrosis: Randomised, double-blind, controlled, phase 2 trials. Lancet Respir. Med. 2023, 11, 550–562. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05033080 (accessed on 1 March 2023).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04853368 (accessed on 2 March 2023).
- Cystic Fibrosis Foundation. Available online: https://www.cff.org/press-releases/2019-10/cystic-fibrosis-foundation-launches-500-million-path-cure (accessed on 20 July 2023).
- Sharma, J.; Du, M.; Wong, E.; Mutyam, V.; Li, Y.; Chen, J.; Wangen, J.; Thrasher, K.; Fu, L.; Peng, N.; et al. A small molecule that induces translational readthrough of CFTR nonsense mutations by eRF1 depletion. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.K.; Mullenders, J.; Pott, J.; Boj, S.F.; Landskroner-Eiger, S.; Goddeeris, M.M. Targeting G542X CFTR nonsense alleles with ELX-02 restores CFTR function in human-derived intestinal organoids. J. Cyst. Fibros. 2021, 20, 436–442. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04135495 (accessed on 1 March 2023).
- Rowe, S.; Zuckerman, J.; Dorgan, D.; Lascano, J.; McCoy, K.; Jain, M.; Schechter, M.; Lommatzsch, S.; Indihar, V.; Lechtzin, N.; et al. Inhaled mRNA therapy for treatment of cystic fibrosis: Interim results of a randomized, double-blind, placebo-controlled phase 1/2 clinical study. J. Cyst. Fibros. 2023, 1. [Google Scholar] [CrossRef] [PubMed]
- NACFC 2023 PLENARY 1: Hope for All: Addressing the Needs of Those with Untreated CF Mutations. Available online: https://www.youtube.com/watch?v=k-H_hqkhu24 (accessed on 7 August 2023).
- Available online: https://pipelinereview.com/index.php/2023013182590/DNA-RNA-and-Cells/Arcturus-Therapeutics-Announces-Clinical-Trial-Application-for-ARCT-032-Received-Approval-to-Proceed-into-First-in-Human-Studies-to-Treat-Cystic-Fibrosis.html (accessed on 7 August 2023).
- Ishimaru, D.; Boudko, D.; Meleshkevitch, E.A.; Sidhu, M.S.; Poniatowski, J.R.; Gao, P.; Molla, T.; Comini, S.; Lister, H.; Coquelin, M.; et al. Functional Rescue of CFTR by Aerosolized Delivery of Optimized CFTR mRNA Using ReCode-LPNs in Primary Human Bronchial Epithelial Cells Derived from Patients with Cystic Fibrosis. Abstrct 507; ATS 2022. Available online: https://recodetx.com/wp-content/uploads/2022/10/ATS-Poster-Daniella-15Apr2022_FINAL-Oct2022.pdf (accessed on 7 August 2023).
- Cystic Fibrosis Foundation. Drug Development Pipeline. Available online: https://apps.cff.org/Trials/Pipeline/details/10204/VX-522 (accessed on 7 August 2023).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05668741 (accessed on 2 March 2023).
- Dana, H.; Chalbatani, G.M.; Mahmoodzadeh, H.; Karimloo, R.; Rezaiean, O.; Moradzadeh, A.; Mehmandoost, N.; Moazzen, F.; Mazraeh, A.; Marmari, V.; et al. Molecular Mechanisms and Biological Functions of siRNA. Int. J. Biomed. Sci. 2017, 13, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Tagalakis, A.D.; Munye, M.M.; Ivanova, R.; Chen, H.; Smith, C.M.; Aldossary, A.M.; Rosa, L.Z.; Moulding, D.; Barnes, J.L.; Kafetzis, K.N.; et al. Effective silencing of ENaC by siRNA delivered with epithelial-targeted nanocomplexes in human cystic fibrosis cells and in mouse lung. Thorax 2018, 73, 847–856. [Google Scholar] [CrossRef]
- Conte, G.; Costabile, G.; Baldassi, D.; Rondelli, V.; Bassi, R.; Colombo, D.; Linardos, G.; Fiscarelli, E.V.; Sorrentino, R.; Miro, A.; et al. Hybrid Lipid/Polymer Nanoparticles to Tackle the Cystic Fibrosis Mucus Barrier in siRNA Delivery to the Lungs: Does PEGylation Make the Difference? ACS Appl. Mater. Interfaces 2022, 14, 7565–7578. [Google Scholar] [CrossRef]
- Patricia, I. First Clinical Trial Begins for RNAbased therapy SPL84 for CF. Cystic Fibrosis News Today, 15 December 2022. 15 December.
- Egan, M.E. Non-Modulator Therapies: Developing a Therapy for Every Cystic Fibrosis Patient. Clin. Chest Med. 2022, 43, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Pranke, I.; Golec, A.; Hinzpeter, A.; Edelman, A.; Sermet-Gaudelus, I. Emerging Therapeutic Approaches for Cystic Fibrosis. From Gene Editing to Personalized Medicine. Front. Pharmacol. 2019, 10, 121. [Google Scholar] [CrossRef]
- Vidović, D.; Carlon, M.S.; da Cunha, M.F.; Dekkers, J.F.; Hollenhorst, M.I.; Bijvelds, M.J.C.; Ramalho, A.S.; Haute, C.V.D.; Ferrante, M.; Baekelandt, V.; et al. rAAV-CFTRΔR Rescues the Cystic Fibrosis Phenotype in Human Intestinal Organoids and Cystic Fibrosis Mice. Am. J. Respir. Crit. Care Med. 2016, 193, 288–298. [Google Scholar] [CrossRef]
- Yan, Z.; Feng, Z.; Sun, X.; Zhang, Y.; Zou, W.; Wang, Z.; Jensen-Cody, C.; Liang, B.; Park, S.Y.; Qiu, J.; et al. Human Bocavirus Type-1 Capsid Facilitates the Transduction of Ferret Airways by Adeno-Associated Virus Genomes. 8 May 2017. Available online: https://europepmc.org/backend/ptpmcrender.fcgi?accid=PMC5567599&blobtype=pdf (accessed on 2 March 2023).
- Alton, E.W.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Calcedo, R.; et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fi brosis: A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015, 3, 684–691. [Google Scholar] [CrossRef]
- Crane, A.M.; Kramer, P.; Bui, J.H.; Chung, W.J.; Li, X.S.; Gonzalez-Garay, M.L.; Hawkins, F.; Liao, W.; Mora, D.; Choi, S.; et al. Targeted Correction and Restored Function of the CFTR Gene in Cystic Fibrosis Induced Pluripotent Stem Cells. Stem Cell Rep. 2015, 4, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Bednarski, C.; Tomczak, K.; Hövel, B.V.; Weber, W.-M.; Cathomen, T. Targeted Integration of a Super-Exon into the CFTR Locus Leads to Functional Correction of a Cystic Fibrosis Cell Line Model. PLoS ONE 2016, 11, e0161072. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Koo, B.-K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional Repair of CFTR by CRISPR/Cas9 in Intestinal Stem Cell Organoids of Cystic Fibrosis Patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed]
- McNeer, N.A.; Anandalingam, K.; Fields, R.J.; Caputo, C.; Kopic, S.; Gupta, A.; Quijano, E.; Polikoff, L.; Kong, Y.; Bahal, R.; et al. Nanoparticles that deliver triplex-forming peptide nucleic acid molecules correct F508del CFTR in airway epithelium. Nat. Commun. 2015, 6, 6952. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Stanley, G.; Modak, M.; Koff, J.L.; Turner, P.E. Bacteriophage therapy for infections in CF. Pediatr. Pulmonol. 2021, 56, S4–S9. [Google Scholar] [CrossRef]
- Blanchard, A.C.; Waters, V.J. Microbiology of Cystic Fibrosis Airway Disease. Semin. Respir. Crit. Care Med. 2019, 40, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Chmiel, J.; Rosenfeld, M.; Davis, S.D. Cystic Fibrosis: A Multi-Organ System Approach; Humana Press Incorporated: Cham, Switzerland, 2020. [Google Scholar]
- Akkerman-Nijland, A.M.; Akkerman, O.W.; Grasmeijer, F.; Hagedoorn, P.; Frijlink, H.W.; Rottier, B.L.; Koppelman, G.H.; Touw, D.J. The pharmacokinetics of antibiotics in cystic fibrosis. Expert. Opin. Drug Metab. Toxicol. 2021, 17, 53–68. [Google Scholar] [CrossRef]
- Mogayzel, P.J., Jr.; Naureckas, E.T.; Robinson, K.A.; Brady, C.; Guill, M.; Lahiri, T.; Lubsch, L.; Matsui, J.; Oermann, C.M.; Ratjen, F.; et al. Cystic Fibrosis Foundation Pulmonary Guideline*. Pharmacologic Approaches to Prevention and Eradication of InitialPseudomonas aeruginosaInfection. Ann. Am. Thorac. Soc. 2014, 11, 1640–1650. [Google Scholar] [CrossRef]
- Suh, G.A.; Lodise, T.P.; Tamma, P.D.; Knisely, J.M.; Alexander, J.; Aslam, S.; Barton, K.D.; Bizzell, E.; Totten, K.M.C.; Campbell, J.L.; et al. Considerations for the Use of Phage Therapy in Clinical Practice. Antimicrob. Agents Chemother. 2022, 66, e0207121. [Google Scholar] [CrossRef]
- Saiman, L.; Marshall, B.C.; Mayer-Hamblett, N.; Burns, J.L.; Quittner, A.L.; Cibene, D.A.; Coquillette, S.; Fieberg, A.Y.; Accurso, F.J.; Campbell, P.W., III; et al. Azithromycin in Patients with Cystic Fibrosis Chronically Infected with Pseudomonas aeruginosa: A randomized controlled trial. JAMA 2003, 290, 1749–1756. [Google Scholar] [CrossRef]
- Goss, C.H.; Heltshe, S.L.; West, N.E.; Skalland, M.; Sanders, D.B.; Jain, R.; Barto, T.L.; Fogarty, B.; Marshall, B.C.; VanDevanter, D.R.; et al. A Randomized Clinical Trial of Antimicrobial Duration for Cystic Fibrosis Pulmonary Exacerbation Treatment. Am. J. Respir. Crit. Care Med. 2021, 204, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05548283 (accessed on 2 May 2023).
- Taccetti, G.; Francalanci, M.; Pizzamiglio, G.; Messore, B.; Carnovale, V.; Cimino, G.; Cipolli, M. Cystic Fibrosis: Recent Insights into Inhaled Antibiotic Treatment and Future Perspectives. Antibiotics 2021, 10, 338. [Google Scholar] [CrossRef] [PubMed]
- I Restrepo, M.; Keyt, H.; Reyes, L.F. Aerosolized Antibiotics. Respir. Care 2015, 60, 762–773; discussion 771–773. [Google Scholar] [CrossRef]
- Máiz, L.; Girón, R.M.; Olveira, C.; Quintana, E.; Lamas, A.; Pastor, D.; Cantón, R.; Mensa, J. Inhaled antibiotics for the treatment of chronic bronchopulmonary Pseudomonas aeruginosa infection in cystic fibrosis: Systematic review of randomised controlled trials. Expert. Opin. Pharmacother. 2013, 14, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.P.; Durmowicz, A.G.; Field, A.; Flume, P.A.; VanDevanter, D.R.; Mayer-Hamblett, N. Developing Inhaled Antibiotics in Cystic Fibrosis: Current Challenges and Opportunities. Ann. Am. Thorac. Soc. 2019, 16, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Foundation. Eradication of Initial P. aeruginosa Clinical Care Guidelines. Available online: https://www.cff.org/eradication-initial-p-aeruginosa-clinical-care-guidelines (accessed on 10 January 2023).
- NIH. Clinical Trials.gov. Available online: https://clinicaltrials.gov/ (accessed on 2 May 2023).
- Puvvadi, R.; Mikkelsen, H.; McCahon, L.; Grogan, S.; Ditcham, W.; Reid, D.W.; Lamont, I.; Stick, S.M.; Clements, B. Role of Tris-CaEDTA as an adjuvant with nebulised tobramycin in cystic fibrosis patients with Pseudomonas aeruginosa lung infections: A randomised controlled trial. J. Cyst. Fibros. 2021, 20, 316–323. [Google Scholar] [CrossRef]
- McCoy, K.S.; Quittner, A.L.; Oermann, C.M.; Gibson, R.L.; Retsch-Bogart, G.Z.; Montgomery, A.B. Inhaled Aztreonam Lysine for Chronic Airway Pseudomonas aeruginosa in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.; Chaudary, N. The Use of Amikacin Liposome Inhalation Suspension (Arikayce) in the Treatment of Refractory Nontuberculous Mycobacterial Lung Disease in Adults. Drug Des. Dev. Ther. 2020, 14, 2287–2294. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=Cystic+Fibrosis&term=amikacin+pseudomonas&cntry=&state=&city=&dist= (accessed on 2 May 2023).
- Bilton, D.; Pressler, T.; Fajac, I.; Clancy, J.P.; Sands, D.; Minic, P.; Cipolli, M.; Galeva, I.; Quittner, A.L.; Liu, K.; et al. Amikacin liposome inhalation suspension for chronic Pseudomonas aeruginosa infection in cystic fibrosis. J. Cyst. Fibros. 2020, 19, 284–291. [Google Scholar] [CrossRef]
- Uttley, L.; Harnan, S.; Cantrell, A.; Taylor, C.; Walshaw, M.; Brownlee, K.; Tappenden, P. Systematic review of the dry powder inhalers colistimethate sodium and tobramycin in cystic fibrosis. Eur. Respir. Rev. 2013, 22, 476–486. [Google Scholar] [CrossRef]
- Patel, S.; Preuss, C.V.; Bernice, F. Vancomycin. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459263/ (accessed on 8 August 2023).
- Waterer, G.; Lord, J.; Hofmann, T.; Jouhikainen, T. Phase I, Dose-Escalating Study of the Safety and Pharmacokinetics of Inhaled Dry-Powder Vancomycin (AeroVanc) in Volunteers and Patients with Cystic Fibrosis: A New Approach to Therapy for Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2020, 64, e01776-19. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03181932?term=aerovanc&draw=2&rank=2 (accessed on 2 May 2023).
- Dasenbrook, E.C. Nebulized Vancomycin for Eradication of Persistent MRSA in Patients with Cystic Fibrosis. Consult QD. Available online: https://consultqd.clevelandclinic.org/nebulized-vancomycin-for-eradication-of-persistent-mrsa-in-patients-with-cystic-fibrosis/ (accessed on 15 January 2023).
- Muhlebach, M.S.; Beckett, V.; Popowitch, E.; Miller, M.B.; Baines, A.; Mayer-Hamblett, N.; Zemanick, E.T.; Hoover, W.C.; VanDalfsen, J.M.; Campbell, P.; et al. Microbiological efficacy of early MRSA treatment in cystic fibrosis in a randomised controlled trial. Thorax 2017, 72, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Caldwell, E.; Mayer-Hamblett, N.; Goss, C.H.; Muhlebach, M.S. Eradication of early MRSA infection in cystic fibrosis: A novel study design for the STAR-ter trial. ERJ Open Res. 2022, 8, 00190–02022. [Google Scholar] [CrossRef] [PubMed]
- Clinicaltrials.gov. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03489629 (accessed on 2 May 2023).
- Podder, V.; Sadiq, N.M. Levofloxacin. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK545180/ (accessed on 8 August 2023).
- Flume, P.A.; VanDevanter, D.R.; Morgan, E.E.; Dudley, M.N.; Loutit, J.S.; Bell, S.C.; Kerem, E.; Fischer, R.; Smyth, A.R.; Aaron, S.D.; et al. A phase 3, multi-center, multinational, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of levofloxacin inhalation solution (APT-1026) in stable cystic fibrosis patients. J. Cyst. Fibros. 2016, 15, 495–502. [Google Scholar] [CrossRef]
- Martin-Loeches, I.; Dale, G.E.; Torres, A. Murepavadin: A new antibiotic class in the pipeline. Expert. Rev. Anti-Infect. Ther. 2018, 16, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chang, R.Y.K.; Britton, W.J.; Chan, H.-K. Advances in the development of antimicrobial peptides and proteins for inhaled therapy. Adv. Drug Deliv. Rev. 2022, 180, 114066. [Google Scholar] [CrossRef] [PubMed]
- CysticFibrosisFoundation.org. Available online: https://apps.cff.org/Trials/Pipeline/details/10181/Inhaled-Murepavadin (accessed on 2 May 2023).
- Martínez-Gallardo, M.J.; Villicaña, C.; Yocupicio-Monroy, M.; Alcaraz-Estrada, S.L.; León-Félix, J. Current knowledge in the use of bacteriophages to combat infections caused by Pseudomonas aeruginosa in cystic fibrosis. Folia Microbiol. 2022, 68, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Trend, S.; Fonceca, A.M.; Ditcham, W.G.; Kicic, A.; Cf, A. The potential of phage therapy in cystic fibrosis: Essential human-bacterial-phage interactions and delivery considerations for use in Pseudomonas aeruginosa-infected airways. J. Cyst. Fibros. 2017, 16, 663–670. [Google Scholar] [CrossRef]
- Gibson, S.B.; Green, S.I.; Liu, C.G.; Salazar, K.C.; Clark, J.R.; Terwilliger, A.L.; Kaplan, H.B.; Maresso, A.W.; Trautner, B.W.; Ramig, R.F. Constructing and Characterizing Bacteriophage Libraries for Phage Therapy of Human Infections. Front. Microbiol. 2019, 10, 2537. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04596319 (accessed on 2 May 2023).
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05010577 (accessed on 2 May 2023).
- Kutateladze, M.; Adamia, R. Phage therapy experience at the Eliava Institute. Med. Mal. Infect. 2008, 38, 426–430. [Google Scholar] [CrossRef]
- Kvachadze, L.; Balarjishvili, N.; Meskhi, T.; Tevdoradze, E.; Skhirtladze, N.; Pataridze, T.; Adamia, R.; Topuria, T.; Kutter, E.; Rohde, C.; et al. Evaluation of lytic activity of staphylococcal bacteriophage Sb-1 against freshly isolated clinical pathogens. Microb. Biotechnol. 2011, 4, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Aslam, S.; Courtwright, A.M.; Koval, C.; Lehman, S.M.; Morales, S.; Furr, C.L.; Rosas, F.; Brownstein, M.J.; Fackler, J.R.; Sisson, B.M.; et al. Early clinical experience of bacteriophage therapy in 3 lung transplant recipients. Am. J. Transpl. 2019, 19, 2631–2639. [Google Scholar] [CrossRef] [PubMed]
- Law, N.; Logan, C.; Yung, G.; Furr, C.-L.L.; Lehman, S.M.; Morales, S.; Rosas, F.; Gaidamaka, A.; Bilinsky, I.; Grint, P.; et al. Successful adjunctive use of bacteriophage therapy for treatment of multidrug-resistant Pseudomonas aeruginosa infection in a cystic fibrosis patient. Infection 2019, 47, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Visaggio, D.; Frangipani, E.; Hijazi, S.; Pirolo, M.; Leoni, L.; Rampioni, G.; Imperi, F.; Bernstein, L.; Sorrentino, R.; Ungaro, F.; et al. Variable Susceptibility to Gallium Compounds of Major Cystic Fibrosis Pathogens. ACS Infect. Dis. 2022, 8, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02354859 (accessed on 2 May 2023).
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03669614 (accessed on 2 May 2023).
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04294043 (accessed on 2 May 2023).
- Thomson, R.M.; Morgan, L.C.; Burke, A.; A Colin, A.; Tal, A. Home-based treatment of nontuberculous mycobacteria pulmonary disease via a novel nitric oxide generator and delivery system. Chest 2022, 162, A470–A471. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05101915 (accessed on 2 May 2023).
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04685720 (accessed on 2 May 2023).
- Beyond air, LungfitTM Go. 2023. Available online: https://www.beyondair.net/technology/ (accessed on 2 May 2023).
- Fernandes, P. Fusidic Acid: A Bacterial Elongation Factor Inhibitor for the Oral Treatment of Acute and Chronic Staphylococcal Infections. Cold Spring Harb. Perspect. Med. 2016, 6, a025437. [Google Scholar] [CrossRef] [PubMed]
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05641298 (accessed on 2 May 2023).
- Food and Drug Administration. Xenelta (Lefamulin) Prescribing Data. 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211672s000,211673s000lbl.pdf (accessed on 24 April 2023).
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05225805 (accessed on 2 May 2023).
- Narayanaswamy, V.P.; Townsend, S.M.; Loughran, A.J.; Wiesmann, W.; Baker, S. Polycationic Glycopolymer Demonstrates Activity Against Persisters and Biofilms of Non-tuberculosis Mycobacteria Cystic Fibrosis Clinical Isolates in vitro. Front. Microbiol. 2022, 13, 821820. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 121383526, Opelconazole. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Opelconazole (accessed on 26 April 2023).
- Clinicaltrials.gov. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT05037851 (accessed on 2 May 2023).
- Chmiel, J.F.; Konstan, M.W.; Elborn, J.S. Antibiotic and Anti-Inflammatory Therapies for Cystic Fibrosis. Cold Spring Harb. Perspect. Med. 2013, 3, a009779. [Google Scholar] [CrossRef]
- Cohen, T.S.; Prince, A. Cystic fibrosis: A mucosal immunodeficiency syndrome. Nat. Med. 2012, 18, 509–519. [Google Scholar] [CrossRef]
- Chmiel, J.F.; Konstan, M.W. Inflammation and Anti-Inflammatory Therapies for Cystic Fibrosis. Clin. Chest Med. 2007, 28, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in cystic fibrosis: An update. Pediatr. Pulmonol. 2018, 53, S30–S50. [Google Scholar] [CrossRef] [PubMed]
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflmmation in cystic fibrosis ling disease: Pathogensis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Bruscia, E.M.; Bonfield, T.L. Update on Innate and Adaptive Immunity in Cystic Fibrosis. Clin. Chest Med. 2022, 43, 603–615. [Google Scholar] [CrossRef]
- Perrem, L.; Ratjen, F. Designing Clinical Trials for Anti-Inflammatory Therapies in Cystic Fibrosis. Front. Pharmacol. 2020, 11, 576293. [Google Scholar] [CrossRef]
- Konstan, M.W.; Byard, P.J.; Hoppel, C.L.; Davis, P.B. Effect of High-Dose Ibuprofen in Patients with Cystic Fibrosis. N. Engl. J. Med. 1995, 332, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Gaggar, A.; Chen, J.; Chmiel, J.F.; Dorkin, H.L.; Flume, P.A.; Griffin, R.; Nichols, D.; Donaldson, S.H. Inhaled alpha 1 -proteinase inhibitor therapy in patients with cystic fibrosis. J. Cyst. Fibros. 2016, 15, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Chmiel, J.F.; Flume, P.; Downey, D.G.; Dozor, A.J.; Colombo, C.; Mazurek, H.; Sapiejka, E.; Rachel, M.; Constantine, S.; Conley, B.; et al. Safety and efficacy of lenabasum in a phase 2 randomized, placebo-controlled trial in adults with cystic fibrosis. J. Cyst. Fibros. 2021, 20, 78–85. [Google Scholar] [CrossRef]
- Elborn, J.; Horsley, A.; MacGregor, G.; Bilton, D.; Grosswald, R.; Ahuja, S.; Springman, E. Phase I Studies of Acebilustat: Biomarker Response and Safety in Patients with Cystic Fibrosis. Clin. Transl. Sci. 2017, 10, 28–34. [Google Scholar] [CrossRef]
- Chalmers, J.D.; Haworth, C.S.; Metersky, M.L.; Loebinger, M.R.; Blasi, F.; Sibila, O.; O’donnell, A.E.; Sullivan, E.J.; Mange, K.C.; Fernandez, C.; et al. Phase 2 Trial of the DPP-1 Inhibitor Brensocatib in Bronchiectasis. N. Engl. J. Med. 2020, 383, 2127–2137. [Google Scholar] [CrossRef]
- Barth, P.; Bruijnzeel, P.; Wach, A.; Kessler, O.S.; Hooftman, L.; Zimmermann, J.; Naue, N.; Huber, B.; Heimbeck, I.; Kappeler, D.; et al. Single dose escalation studies with inhaled POL6014, a potent novel selective reversible inhibitor of human neutrophil elastase, in healthy volunteers and subjects with cystic fibrosis. J. Cyst. Fibros. 2020, 19, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Henke, M.O.; Ratjen, F. Mucolytics in cystic fibrosis. Paediatr. Respir. Rev. 2007, 8, 24–29. [Google Scholar] [CrossRef]
- Fuchs, H.J.; Borowitz, D.S.; Christiansen, D.H.; Morris, E.M.; Nash, M.L.; Ramsey, B.W.; Rosenstein, B.J.; Smith, A.L.; Wohl, M.E. Effect of Aerosolized Recombinant Human DNase on Exacerbations of Respiratory Symptoms and on Pulmonary Function in Patients with Cystic Fibrosis. N. Engl. J. Med. 1994, 331, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Witt, D.M.; Anderson, L. Dornase alfa: A new option in the management of cystic fibrosis. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1996, 16, 40–48. [Google Scholar]
- Donaldson, S.; Bennett, W.; Zeman, K.; Knowles, M.; Tarran, R.; Boucher, R. Mucous Clearance and Lung Function in Cystic Fibrosis with Hypertonic Saline. N. Engl. J. Med. 2006, 354, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Flume, P.A.; Amelina, E.; Daines, C.L.; Charlton, B.; Leadbetter, J.; Guasconi, A.; Aitken, M.L. Efficacy and safety of inhaled dry-powder mannitol in adults with cystic fibrosis: An international, randomized controlled study. J. Cyst. Fibros. 2021, 20, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Ermund, A.; Meiss, L.N.; Gustafsson, J.K.; Hansson, G.C. Hyper-osmolarity and calcium chelation: Effects on cystic fibrosis mucus. Eur. J. Pharmacol. 2015, 764, 109–117. [Google Scholar] [CrossRef]
- Danahay, H.; Gosling, M. TMEM16A: An Alternative Approach to Restoring Airway Anion Secretion in Cystic Fibrosis? Int. J. Mol. Sci. 2020, 21, 2386. [Google Scholar] [CrossRef]
- Cystic Fibrosis Foundation, Community Voice. Available online: https://www.cff.org/get-involved/community-voice (accessed on 28 April 2023).
- Mayer-Hamblett, N.; Ratjen, F.; Russell, R.; Donaldson, S.H.; Riekert, K.A.; Sawicki, G.S.; Odem-Davis, K.; Young, J.K.; Rosenbluth, D.; Taylor-Cousar, J.L.; et al. Discontinuation versus continuation of hypertonic saline or dornase alfa in modulator treated people with cystic fibrosis (SIMPLIFY): Results from two parallel, multicentre, open-label, randomised, controlled, non-inferiority trials. Lancet Respir Med 2023, 11, 329–340. [Google Scholar] [CrossRef]
- Saunders, C.; Jensen, R.; Robinson, P.D.; Stanojevic, S.; Klingel, M.; Short, C.; Davies, J.C.; Ratjen, F. Integrating the multiple breath washout test into international multicentre trials. J. Cyst. Fibros. 2020, 19, 602–607. [Google Scholar] [CrossRef]




| CFTR Modulators | mRNA-Based Therapies | Gene-Based Therapies |
|---|---|---|
| Ivacaftor 1 | Aminoglycoside read-through nonsense mutation (ELX-02) 2 | CFTR Gene Transfer Vectors:
|
| Lumacaftor/Ivacaftor 1 | Depletion of termination factor, eRF1 (SRI-37240) 3 | |
| Tezacaftor/Ivacaftor 1 | Inhaled CFTR mRNA (MRT5005 2 VX-522 2 ARCT-032 2 ReCode 3) | |
| Elexacaftor/Tezacaftor/Ivacaftor 1 | Short-interfering RNAs (siRNAs) 3 | Zinc-finger nucleases (ZFN) 3 |
| Deuticaftor (VX561) 2 | Transcription activator-like effector nucleases (TALENS) 3 | |
| Vanzacaftor (VX-121)/tezacaftor/ Deuticaftor 2 | Clustered Regularly Interspersed Palindromic Repeats (CRISPR)/CRISPR-associated nuclease 9 (CAS9) 3 | |
| Navocaftor (ABBV-3067), galicaftor (ABBV-2222) and ABBV-576 2 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esposito, C.; Kamper, M.; Trentacoste, J.; Galvin, S.; Pfister, H.; Wang, J. Advances in the Cystic Fibrosis Drug Development Pipeline. Life 2023, 13, 1835. https://doi.org/10.3390/life13091835
Esposito C, Kamper M, Trentacoste J, Galvin S, Pfister H, Wang J. Advances in the Cystic Fibrosis Drug Development Pipeline. Life. 2023; 13(9):1835. https://doi.org/10.3390/life13091835
Chicago/Turabian StyleEsposito, Christine, Martin Kamper, Jessica Trentacoste, Susan Galvin, Halie Pfister, and Janice Wang. 2023. "Advances in the Cystic Fibrosis Drug Development Pipeline" Life 13, no. 9: 1835. https://doi.org/10.3390/life13091835