Hot Spring Metagenomics




Abstract
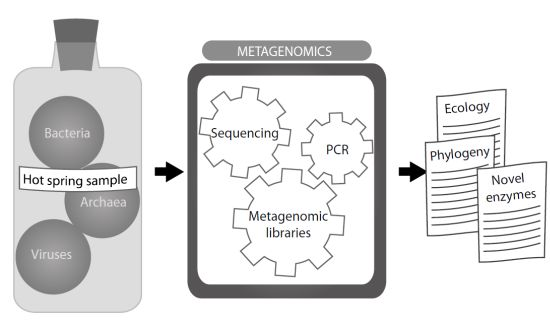
:
{kind=link}
1. Introduction
2. Bacteria and Archaea
2.1. PCR Approach
2.2. Metagenomic Libraries
3. Viruses
4. Novel Thermophilic Enzymes
5. Conclusions
Acknowledgments
References and Notes
- Chien, A.; Edgar, D.B.; Trela, J.M. Deoxyribonucleic acid polymerase from the extreme thermophile Thermus aquaticus. J. Bacteriol. 1976, 127, 1550–1557. [Google Scholar]
- Marsh, C.L.; Larsen, D.H. Characterization of some thermophilic bacteria from the hot springs of Yellowstone National Park. J. Bacteriol. 1953, 65, 193–197. [Google Scholar]
- Amann, R.I.; Ludwig, W.; Schleifer, K.H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 1995, 59, 143–169. [Google Scholar]
- Ghosh, D.; Bal, B.; Kashyap, V.K.; Pal, S. Molecular phylogenetic exploration of bacterial diversity in a Bakreshwar (India) hot spring and culture of Shewanella-related thermophiles. Appl. Environ. Microbiol. 2003, 69, 4332–4336. [Google Scholar] [CrossRef]
- Barns, S.M.; Fundyga, R.E.; Jeffries, M.W.; Pace, N.R. Remarkable archaeal diversity detected in a Yellowstone National Park hot spring environment. Proc. Natl. Acad. Sci. USA 1994, 91, 1609–1613. [Google Scholar]
- Barns, S.M.; Delwiche, C.F.; Palmer, J.D.; Pace, N.R. Perspectives on archaeal diversity, thermophily and monophyly from environmental rRNA sequences. Proc. Natl. Acad. Sci. USA 1996, 93, 9188–9193. [Google Scholar] [CrossRef]
- Meyer-Dombard, D.R.; Shock, E.L.; Amend, J.P. Archaeal and bacterial communities in geochemically diverse hot springs of Yellowstone National Park, USA. Geobiology 2005, 3, 211–227. [Google Scholar] [CrossRef]
- Huber, H.; Hohn, M.J.; Rachel, R.; Fuchs, T.; Wimmer, V.C.; Stetter, K.O. A new phylum of Archaea represented by a nanosized hyperthermophilic symbiont. Nature 2002, 417, 63–67. [Google Scholar]
- Stetter, K.O. A brief history of the discovery of hyperthermophilic life. Biochem. Soc. Trans. 2013, 41, 416–420. [Google Scholar] [CrossRef]
- Hobel, C.F.V.; Marteinsson, V.T.; Hreggvidsson, G.O.; Kristjansson, J.K. Investigation of the microbial ecology of intertidal hot springs by using diversity analysis of 16S rRNA and chitinase genes. Appl. Environ. Microbiol. 2005, 71, 2771–2776. [Google Scholar] [CrossRef]
- Atanassov, I.; Dimitrova, D.; Stefanova, K.; Tomova, A.; Tomova, I.; Lyutskanova, D.; Stoilova-Disheva, M.; Radeva, G.; Danova, I.; Kambourova, M. Molecular characterization of the Archaeal diversity in Vlasa hot spring, Bulgaria, by using 16S rRNA and glycoside hydrolase family 4 genes. Biotechnol. Biotecnol. Equip. 2010, 24, 1979–1985. [Google Scholar] [CrossRef]
- Miller, S.R.; Strong, A. . Bar-Coded pyrosequencing reveals shared bacterial community properties along the temperature gradients of two alkaline hot springs in Yellowstone National Park. App. Environ. Microbiol. 2009, 4565–4572. [Google Scholar] [CrossRef]
- Valverde, A.; Tuffin, M.; Cowan, D.A. Biogeography of bacterial communities in hot springs: A focus on the actinobacteria. Extremophiles 2012, 16, 669–679. [Google Scholar] [CrossRef]
- Tekere, M.; Lötter, A.; Olivier, J.; Jonker, N.; Venter, S. Metagenomic analysis of bacterial diversity of Siloam hot water spring, Limpopo, South Africa. Afr. J. Biotechnol. 2011, 10, 18005–18012. [Google Scholar]
- Mardanov, A.V.; Gumerov, V.M.; Beletsky, A.V.; Perevalova, A.A.; Karpov, G.A.; Bonch-Osmolovskaya, E.A.; Ravin, N.V. Uncultured archaea dominate in the thermal groundwater of Uzon Caldera, Kamchatka. Extremophiles 2011, 15, 365–372. [Google Scholar] [CrossRef]
- Jiménez, D.J.; Andreote, F.D.; Chaves, D.; Montaña, J.S.; Osorio-Forero, C.; Junca, H.; Zambrano, M.M.; Baena, S. Structural and functional insights from the metagenome of an acidic hot spring microbial planktonic community in the Colombian Andes. PLoS One 2012, 7, e52069. [Google Scholar]
- Kanokratana, P.; Chanapan, S.; Pootanakit, K.; Eurwilaichitr, L. Diversity and abundance of Bacteria and Archaea in the Bor Khlueng Hot Spring in Thailand. J. Basic Microbiol. 2004, 44, 430–444. [Google Scholar] [CrossRef]
- Papke, R.T.; Ramsing, N.B.; Bateson, M.M.; Ward, D.M. Geographical isolation in hot spring cyanobacteria. Environ. Microbiol. 2003, 5, 650–659. [Google Scholar] [CrossRef]
- Shah, N.; Tang, H.; Doak, T.G.; Ye, Y. Comparing bacterial communities inferred from 16S rRNA gene sequencing and shotgun metagenomics. Pac. Symp. Biocomput. 2011, 165–176. [Google Scholar]
- Ward, D.M.; Cohan, F.M.; Bhaya, D.; Heidelberg, J.F.; Kühl, M.; Grossman, A. Genomics, environmental genomics and the issue of microbial species. Heredity 2008, 100, 207–219. [Google Scholar] [CrossRef]
- Tindall, B.J.; Rosselló-Móra, R.; Busse, H.-J.; Ludwig, W.; Kampfer, P. Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Syst. Evol. Micr. 2010, 60, 249–266. [Google Scholar] [CrossRef] [Green Version]
- Bhaya, D.; Grossman, A.R.; Steunou, A.S.; Khuri, N.; Cohan, F.M.; Hamamura, N.; Melendrez, M.C.; Bateson, M.M.; Ward, D.M.; Heidelberg, J.F. Population level functional diversity in a microbial community revealed by comparative genomic and metagenomic analyses. ISME J. 2007, 1, 703–713. [Google Scholar] [CrossRef]
- Klatt, C.G.; Wood, J.M.; Rusch, D.B.; Bateson, M.M.; Hamamura, N.; Heidelberg, J.F.; Grossman, A.R.; Bhaya, D.; Cohan, F.M.; Kühl, M.; Bryant, D.A.; Ward, D.M. Community ecology of hot spring cyanobacterial mats: predominant populations and their functional potential. ISME J. 2011, 5, 1262–1278. [Google Scholar] [CrossRef]
- Bryant, D.A.; Costas, A.M.; Maresca, J.A.; Chew, A.G.; Klatt, C.G.; Bateson, M.M.; Tallon, L.J.; Hostetler, J.; Nelson, W.C.; Heidelberg, J.F.; Ward, D.M. Candidatus Chloracidobacterium thermophilum: An Aerobic Phototrophic Acidobacterium. Science 2007, 317, 523–526. [Google Scholar] [CrossRef]
- Liu, Z.; Klatt, C.G.; Ludwig, M.; Rusch, D.B.; Jensen, S.I.; Kühl, M.; Ward, D.M.; Bryant, D.A. Candidatus Thermochlorobacter aerophilum: An aerobic chlorophotoheterotrophic member of the phylum Chlorobi defined by metagenomics and metatranscriptomics. ISME J. 2012, 6, 1869–1882. [Google Scholar] [CrossRef]
- Anantharaman, K.; Breier, J.A.; Sheik, C.S.; Dick, G.J. Evidence for hydrogen oxidation and metabolic plasticity in widespread deep-sea sulfur-oxidizing bacteria. Proc. Natl. Acad. Sci. USA 2013, 110, 330–335. [Google Scholar] [CrossRef]
- Youssef, N.H.; Blainey, P.C.; Quake, S.R.; Elshahed, M.S. Partial genome assembly for a candidate division OP11 single cell from an anoxic spring (Zodletone Spring, Oklahoma). Appl. Environ. Microbiol. 2011, 77, 7804–7814. [Google Scholar] [CrossRef]
- Liu, Z.; Klatt, C.G.; Wood, J.M.; Rusch, D.B.; Ludwig, M.; Wittekindt, N.; Tomsho, L.P.; Schuster, S.C.; Ward, D.M.; Bryant, D.A. Metatranscriptomic analyses of chlorophototrophs of a hot-spring microbial mat. ISME J. 2011, 5, 1279–1290. [Google Scholar] [CrossRef]
- Nelson, W.C.; Wollerman, L.; Bhaya, D.; Heidelberg, J.F. Analysis of insertion sequences in thermophilic cyanobacteria: exploring the mechanisms of establishing, maintaining, and withstanding high insertion sequence abundance. Appl. Environ. Microbiol. 2011, 77, 5458–5466. [Google Scholar] [CrossRef]
- Heidelberg, J.F.; Nelson, W.C.; Schoenfeld, T.; Bhaya, D. Germ warfare in a microbial mat community: CRISPRs provide insights into the co-evolution of host and viral genomes. PLoS One 2009, 4, e4169. [Google Scholar]
- Xie, W.; Wang, F.; Guo, L.; Chen, Z.; Sievert, S.M.; Meng, J.; Huang, G.; Li, Y.; Yan, Q.; Wu, S.; Wang, X.; Chen, S.; He, G.; Xiao, X.; Xu, A. Comparative metagenomics of microbial communities inhabiting deep-sea hydrothermal vent chimneys with contrasting chemistries. ISME J. 2011, 5, 414–426. [Google Scholar] [CrossRef]
- Swingley, W.D.; Meyer-Dombard, D.R.; Shock, E.L.; Alsop, E.B.; Falenski, H.D.; Havig, J.R.; Raymond, J. Coordinating Environmental Genomics and Geochemistry Reveals Metabolic Transitions in a Hot Spring Ecosystem. PLoS ONE 2012, 7, e38108. [Google Scholar]
- Schoenfeld, T.; Patterson, M.; Richardson, P.M.; Wommack, K.E.; Young, M.; Mead, D. Assembly of Viral Metagenomes from Yellowstone Hot Springs. Appl. Environ. Microbiol. 2008, 74, 4164–4174. [Google Scholar] [CrossRef]
- Servín-Garcidueñas, L.E.; Peng, X.; Garrett, R.A.; Martínez-Romero, E. Genome sequence of a novel archaeal rudivirus recovered from a mexican hot spring. Genome Announc. 2013, 1, e00040–12. [Google Scholar]
- Garrett, R.A.; Prangishvili, D.; Shah, S.A.; Reuter, M.; Stetter, K.O.; Peng, X. Metagenomic analyses of novel viruses and plasmids from a cultured environmental sample of hyperthermophilic neutrophiles. Environ. Microbiol. 2010, 12, 2918–2930. [Google Scholar] [CrossRef]
- Snyder, J.C.; Bateson, M.M.; Lavin, M.; Young, M.J. Use of Cellular CRISPR (Clusters of Regularly Interspaced Short Palindromic Repeats) Spacer-Based Microarrays for Detection of Viruses in Environmental Samples. Appl. Environ. Microbiol. 2010, 76, 7251–7258. [Google Scholar] [CrossRef]
- Pride, D.T.; Schoenfeld, T. Genome signature analysis of thermal virus metagenomes reveals Archaea and thermophilic signatures. BMC Genet. 2008, 9, 420. [Google Scholar]
- Bolduc, B.; Shaughnessy, D.P.; Wolf, Y.I.; Koonin, E.; Roberto, F.F.; Young, M. Identification of novel positive-strand RNA viruses by metagenomic analysis of archaea- 2 dominated Yellowstone Hot Springs. J. Virol. 2012, 86, 5562–5573. [Google Scholar] [CrossRef]
- Garcia Costas, A.M.; Liu, Z.; Tomsho, L.P.; Schuster, S.C.; Ward, D.M.; Bryant, D.A. Complete genome of Candidatus Chloracidobacterium thermophilum, a chlorophyll-based photoheterotroph belonging to the phylum Acidobacteria. Environ. Microbiol. 2012, 14, 177–190. [Google Scholar] [CrossRef]
- Diemer, G.S.; Stedman, K.M. A novel virus genome discovered in an extreme environment suggests recombination between unrelated groups of RNA and DNA viruses. Biol. Direct. 2012, 7, 13. [Google Scholar] [CrossRef]
- He, Y.Z.; Fan, K.Q.; Jia, C.J.; Wang, Z.J.; Pan, W.B.; Huang, L.; Yang, K.Q.; Dong, Z.Y. Characterization of a hyperthermostable Fe-superoxide dismutase from hot spring. Appl. Microbiol. Biotechnol. 2007, 75, 367–376. [Google Scholar] [CrossRef]
- Wemheuer, B.; Taube, R.; Akyol, A.; Wemheuer, F.; Daniel, R. Microbial Diversity and Biochemical Potential Encoded by Thermal Spring Metagenomes Derived from the Kamchatka Peninsula. Archaea 2013, 2013, 136714. [Google Scholar]
- Gabor, E.M.; Alkema, W.B.L.; Janssen, D.B. Quantifying the accessibility of the metagenome by random expression cloning techniques. Environ. Microbiol. 2004, 6, 879–886. [Google Scholar] [CrossRef]
- Tirawongsaroj, P.; Sriprang, R.; Harnpichamchai, P.; Thongaram, T.; Champreda, V.; Tanapongpipat, S.; Pootanakit, K.; Eurwilaichitr, L. Novel thermophilic and thermostable lipolytic enzymes from a Thailand hot spring metagenomic library. J. Biotechnol. 2008, 133, 42–49. [Google Scholar]
- Sharma, P.K.; Singh, K.; Singh, R.; Capalash, N.; Ali, A.; Mohammad, O.; Kaur, J. Characterization of a thermostable lipase showing loss of secondary structure at ambient temperature. Mol. Biol. Rep. 2012, 39, 2795–2804. [Google Scholar] [CrossRef]
- Sharma, P.K.; Kumar, R.; Kumar, R.; Mohammad, O.; Singh, R.; Kaur, J. Engineering of a metagenome derived lipase toward thermal tolerance: Effect of asparagine to lysine mutation on the protein surface. Gene 2012, 491, 264–271. [Google Scholar] [CrossRef]
- Moser, M.J.; DiFrancesco, R.A.; Gowda, K.; Klingele, A.J.; Sugar, D.R.; Stocki, S.; Mead, D.A.; Schoenfeld, T.W. Thermostable DNA Polymerase from a Viral Metagenome Is a Potent RT-PCR Enzyme. PLoS ONE 2012, 7, e38371. [Google Scholar] [CrossRef]
- Kemp, P.F.; Aller, J.Y. Bacterial diversity in aquatic and other environments: What 16S rDNA libraries can tell us. FEMS Microbiol. Ecol. 2004, 47, 161–177. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
López-López, O.; Cerdán, M.E.; González-Siso, M.I. Hot Spring Metagenomics. Life 2013, 3, 308-320. https://doi.org/10.3390/life3020308
López-López O, Cerdán ME, González-Siso MI. Hot Spring Metagenomics. Life. 2013; 3(2):308-320. https://doi.org/10.3390/life3020308
Chicago/Turabian StyleLópez-López, Olalla, María Esperanza Cerdán, and María Isabel González-Siso. 2013. "Hot Spring Metagenomics" Life 3, no. 2: 308-320. https://doi.org/10.3390/life3020308