
Evolutionary Limitation and Opportunities for Developing tRNA Synthetase Inhibitors with 5-Binding-Mode Classification


Abstract
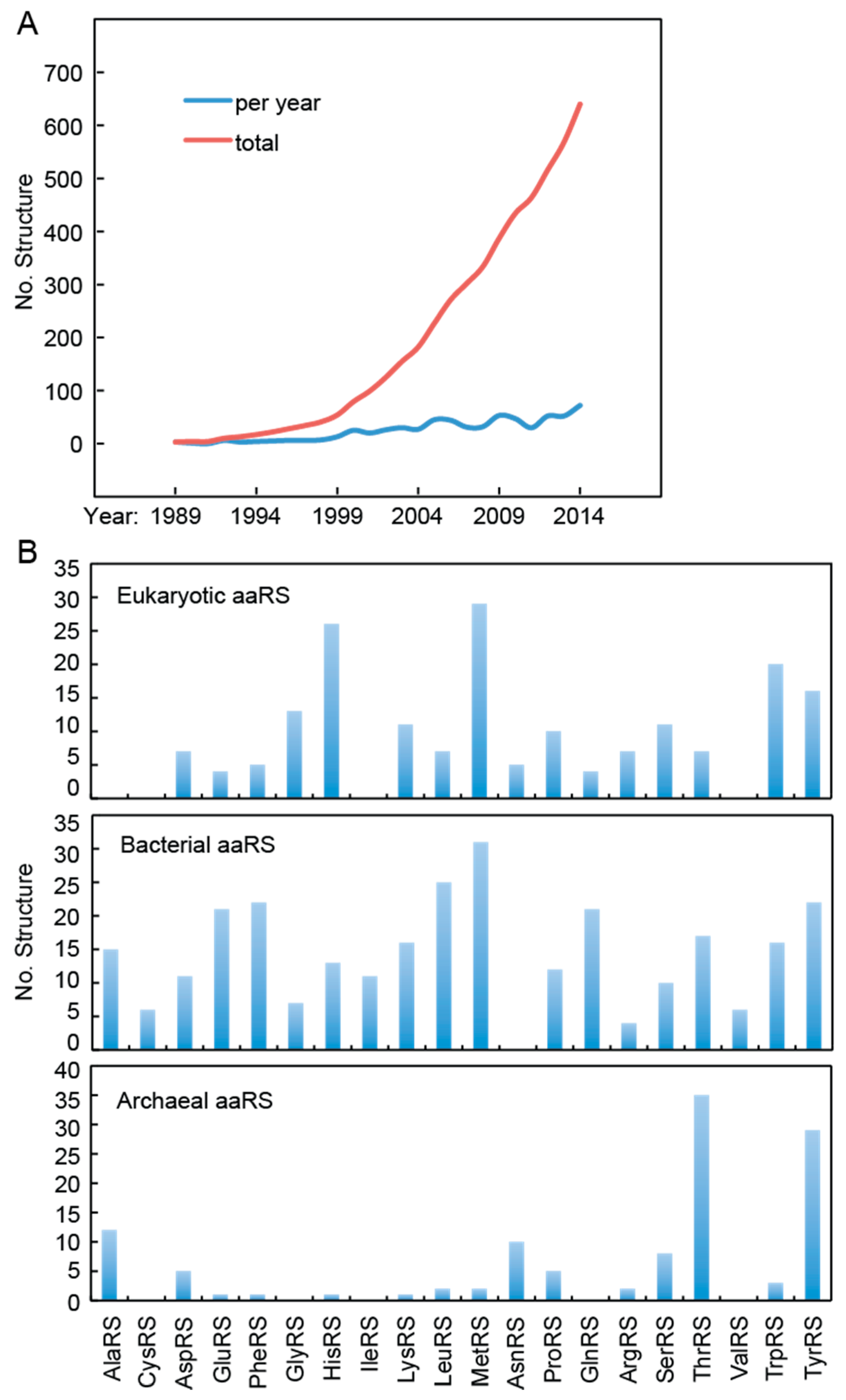
:1. Introduction
2. aaRSs as Target for Disease Therapy

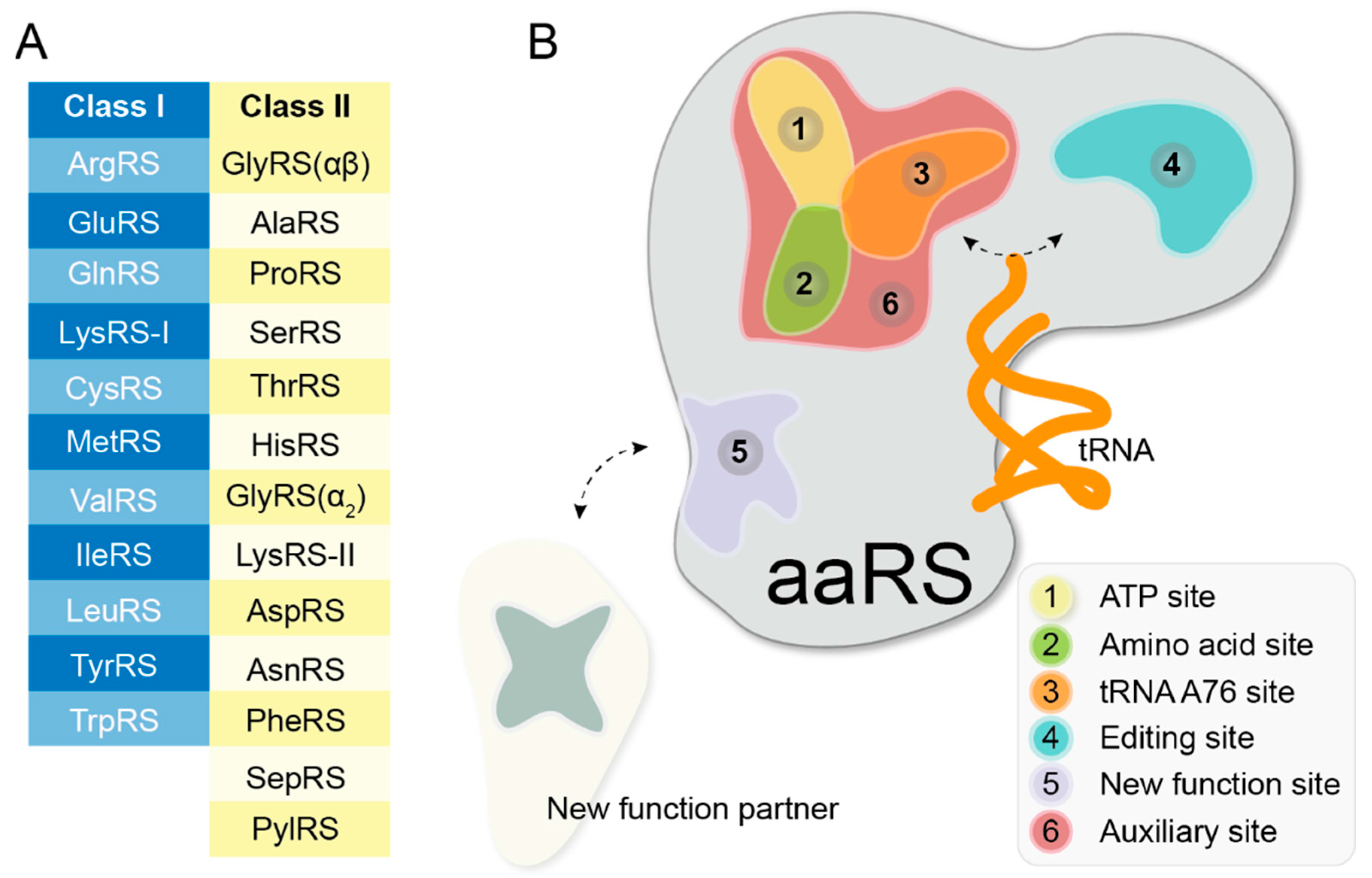
3. Classification of aaRS Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
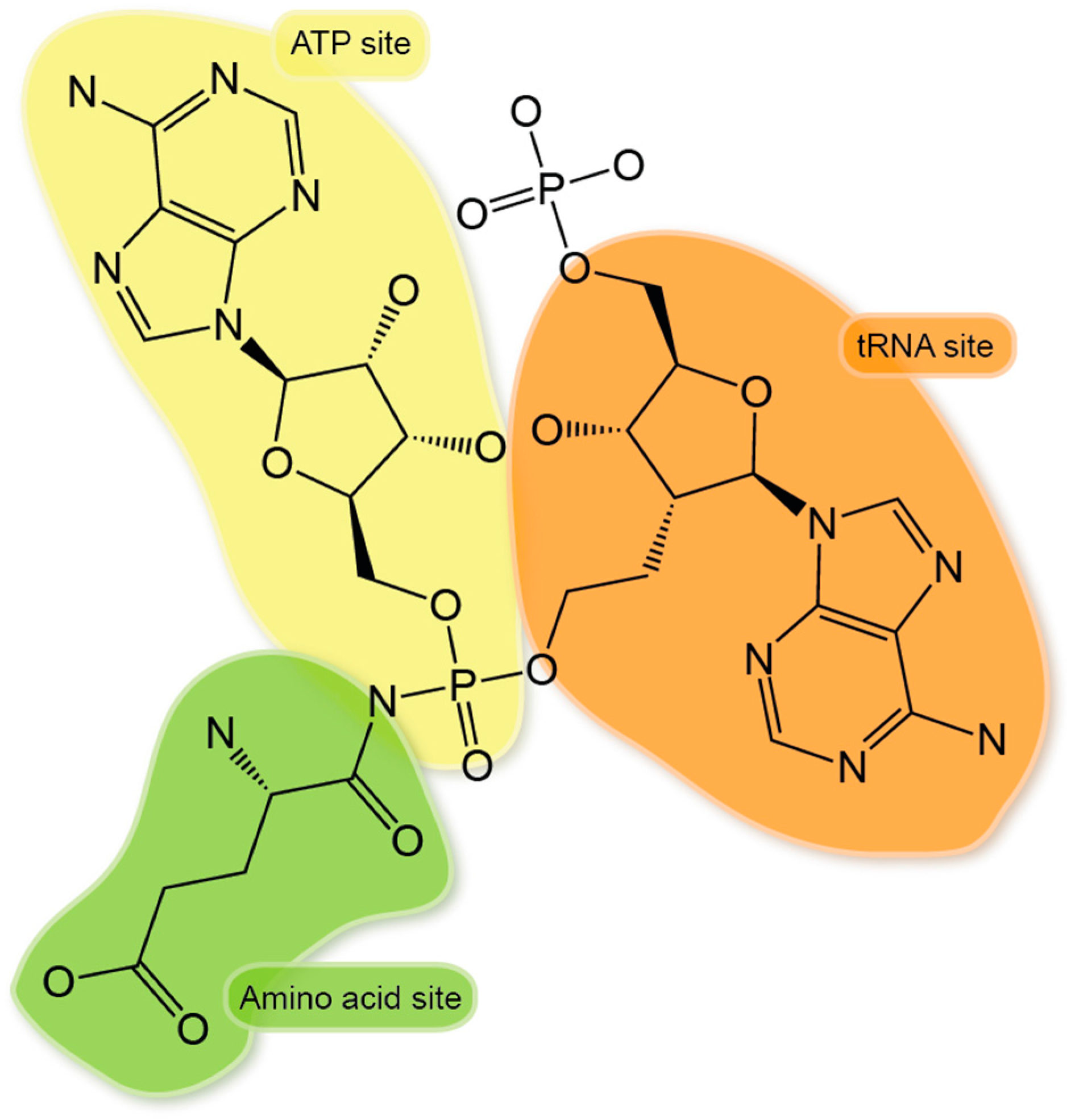{kind=link}
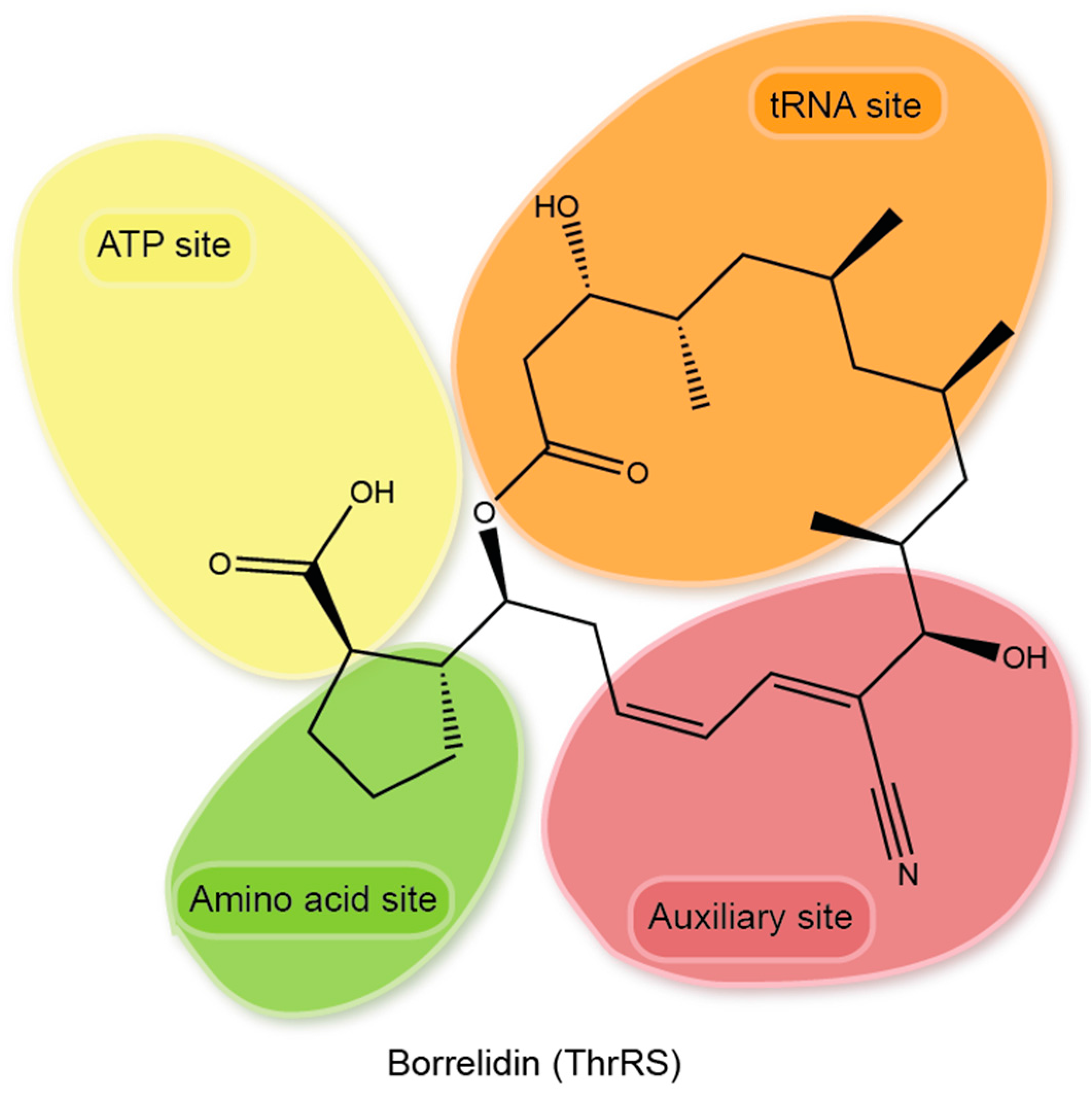{kind=link}
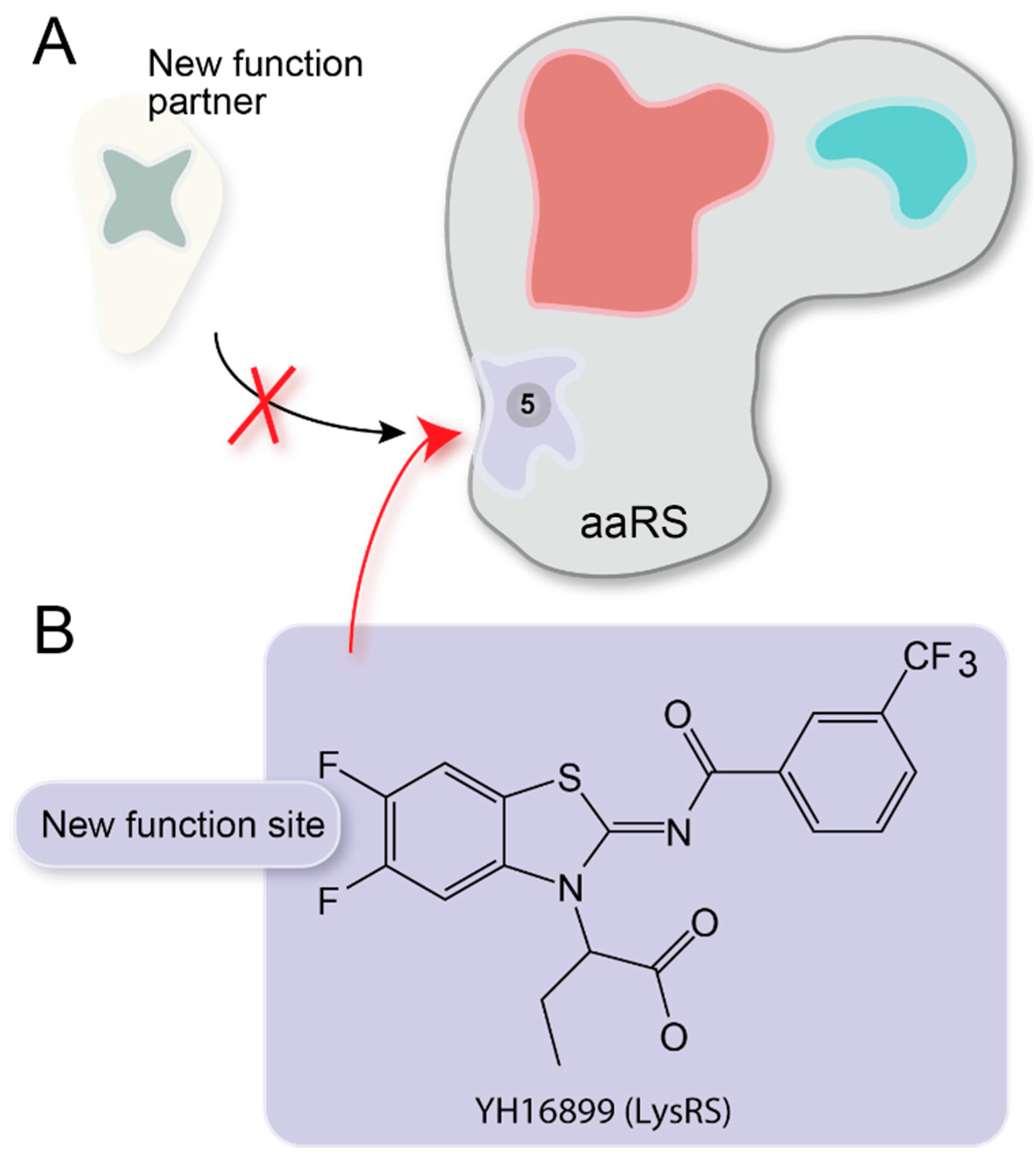{kind=link}
| Chemicals | Acylation | Editing | Other | Notes | |||
|---|---|---|---|---|---|---|---|
| ATP | aa | tRNA | auxiliary | ||||
| Class I. Single Active Site Inhibitor | |||||||
| AMPCPP and AMPPNP | √ | AMPCPP and AMPPNP are non/slow hydrolyzable ATP analogues. | |||||
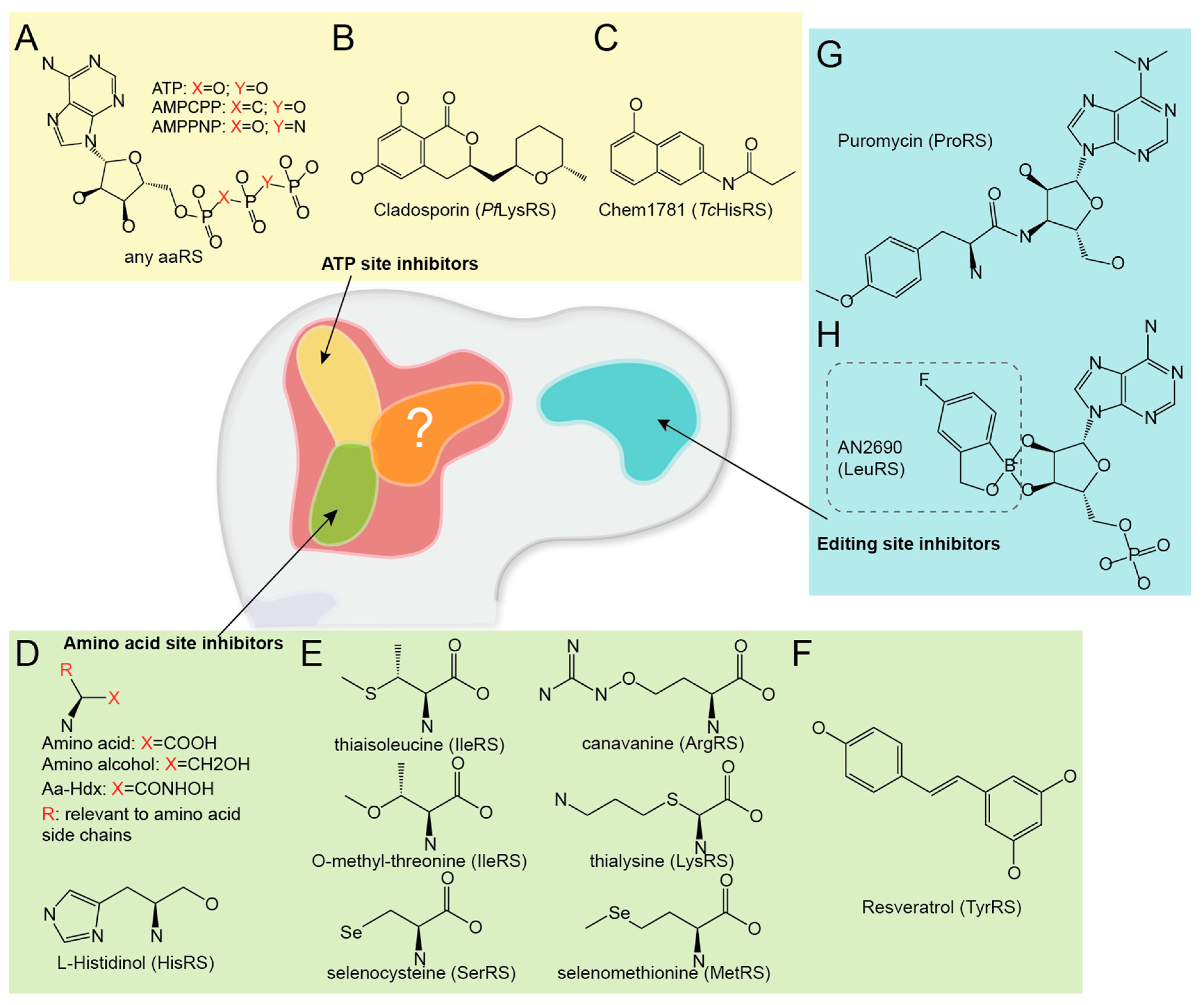
| Cladosporin (CP) | √ | CP partially mimics ATP and specifically inhibits Plasmodium falciparum LysRS [82]. | |||||
| Chem1781 | √ | Chem1781 is one of the 15 chemical fragments, which bind Trypanosoma cruzi HisRS [83]. | |||||
| Amino alcohols (aa-ol) and Amino acid hydroxamates (aa-Hdx) | √ | aa-ols and aa-Hdxs are non-reactive amino acid analogues. | |||||
| Non proteinogenic amino acids | √ | Some non-proteinogenic amino acids can compete with proteinogenic amino acid for the binding and reaction. | |||||
| Resveratrol | √ | Resveratrol is a widely used nutrition supplement, inhibits human TyrRS aminoacylation, but activates TyrRS’ non-translational function that stimulates PARPI [84]. | |||||
| 2-Aminoquinolin-8-ol | √ | This is a potential allosteric aaRS inhibitor, which traps Clostridium difficile MetRS in a “non-aminoacylation conformation” [85]. | |||||
| AN2690 | √ | AN2690 is a broad-spectrum antifungal compound recently approved for onychomycosis treatment. It adducts tRNA at the editing site of fungal LeuRS, inhibits aminoacylation activity by disrupting tRNA turn over [74]. | |||||
| Puromycin | √ | Puromycin is a well-known ribosome-targeting antibiotic. It can also bind to PheRS editing site [86]. | |||||
| Class II. Dual Active Site Inhibitor | |||||||
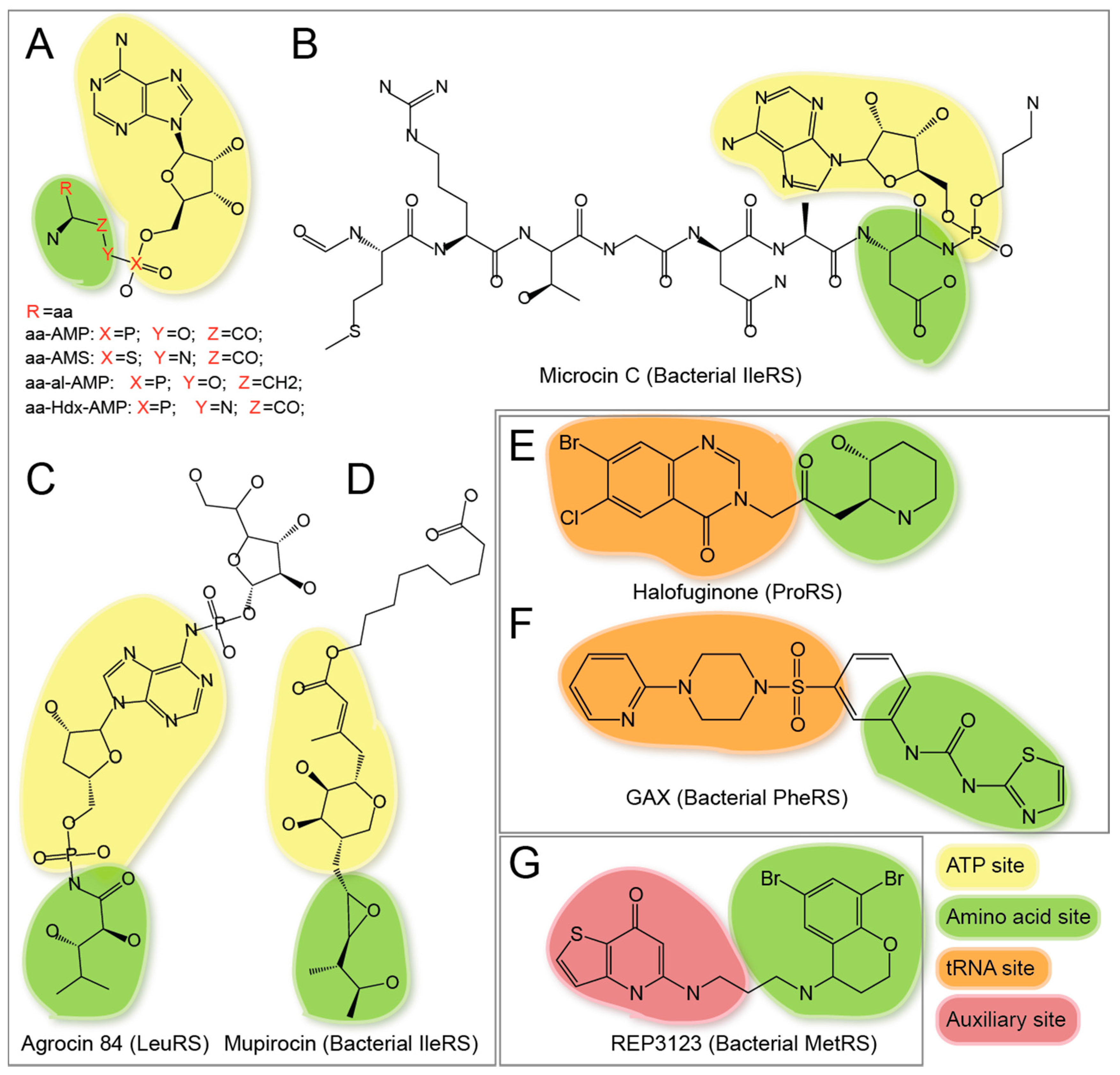
| aa-AMS(s), aa-ol-AMP(s), and aa-Hdx-AMP(s) | √ | √ | These are reaction intermediate aa-AMP analogues with high binding affinity to aaRSs. | ||||
| Quinazoline derivatives of Thr-AMS | √ | √ | These inhibitors were developed from Thr-AMS with improved selectivity for bacterial ThrRS [87]. | ||||
| Thiazole sulfametes | √ | √ | These compounds contain a thiazole moiety instead of adenine in aa-AMS [88]. | ||||
| Microcin C | √ | √ | The in vivo processed product of Microcin C is a non-hydrolyzable aspartyl-adenylate analogue that inhibits AspRS in bacteria [89]. | ||||
| Agrocin 84 | √ | √ | The in vivo processed product of Agrocin 84 is a non-hydrolyzable leucyl-adenylate analogue that inhibits LeuRS in Agrobacterium tumefaciens [90]. | ||||
| Mupirocin | √ | √ | Mupirocin structurally mimics Ile-AMP, inhibits Gram-positive bacteria growth, and is the first approved aaRS inhibitor drug for human [75]. | ||||
| Febrifugine | √ | √ | Febrifugine is a bioactive natural product extracted from root of the hydrangea Dichroa febrifuga Lour used in traditional Chinese medicine, inhibiting ProRS and possessing antimalarial activity [91]. | ||||
| Halofuginone (HF) | √ | √ | HF is a halogenated derivative of febrifugine, inhibits ProRS in mammalian system, induces antifibrotic activities in fibroblasts through inhibition of T helper 17 cell differentiation [68],[92],[93]. HF has obtained FDA’s orphan drug designation. | ||||
| Phenyl-thiazolylurea-sulfonamides | √ | √ | This is a novel class of specific bacterial PheRS inhibitors [94]. | ||||
| REP3123 and its analogues | √ | √ | This is a novel class of bacterial specific MetRS inhibitors [95]. | ||||
| Class III. Triple Active Site Inhibitor | |||||||
| Triple active site inhibitors | √ | √ | √ | This is a conceptual class of aaRS inhibitors proposed in this paper. This kind of inhibitors may be useful for ArgRS/GluRS/GlnRS, whose 3 substrates bind to aaRSs synergistically. | |||
| Class IV. Multi-Site Inhibitor | |||||||
| Borrelidin (BN) and its analogues. | √ | √ | √ | √ | BN has a unique 18-member macrolide ring structure, inhibits bacterial and eukaryotic ThrRS through an induced-fit mechanism, and occupies all three substrate-binding sites and an extra area in the active site cavity, inducing a significant conformational change to ThrRS [96]. BN derivative BC220 showed significantly improved druggability as an antimalarial [97]. | ||
| Class V. Non-Translational Function Inhibitor | |||||||
| YH16899 | √ | YH16899 inhibits non-translational function of LysRS for cancer cell migration [72]. | |||||
| Ungrouped Inhibitors | |||||||
| 4-(2-Nitro-l-propenyl)-1,2-benzenediol | This is a potential AlaRS inhibitor obtained from structural-based virtual screening [98]. | ||||||
| Spirocyclic furan and pyrrolidine inhibitors | These inhibitors were obtained from high throughput screening (HTS) to inhibit Enterococcus faecalis and Staphylococcus aureus PheRS [99]. | ||||||
| Pyrazoles | HTS identified a series of pyrazoles, which selectively inhibit bacterial MetRS [100]. | ||||||
| Oxazolone-dipeptides | Two HTS identified oxazolone-dipeptides and their analogues showed selective inhibition on bacterial MetRS [101]. | ||||||
3.1. Single Site Inhibitors

3.1.1. ATP Site Inhibitors

3.1.2. Amino Acid Site Inhibitors
3.1.3. Editing Site Inhibitors
3.2. Dual Site Inhibitors
3.2.1. ATP-Amino Acid Dual Site Inhibitors

3.2.2. Amino Acid-tRNA Dual Site Inhibitors
3.2.3. Amino Acid—“Auxiliary” Pocket Dual Site Inhibitors
3.3. Triple Site Inhibitor

3.4. Multi Site Inhibitor

3.5. Non-Translational Function Inhibitor

3.6. Other Ungrouped Inhibitors
4. Perspective/Conclusions
Acknowledgments
Conflicts of Interest
References
- Ibba, M.; Soll, D. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000, 69, 617–650. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.W., Jr. Cognition, mechanism, and evolutionary relationships in aminoacyl-tRNA synthetases. Annu. Rev. Biochem. 1993, 62, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Lacey, J.C., Jr.; Wickramasinghe, N.S.; Cook, G.W. Experimental studies on the origin of the genetic code and the process of protein synthesis: A review update. Orig. Life Evol. Biosph. 1992, 22, 243–275. [Google Scholar] [CrossRef] [PubMed]
- Saks, M.E.; Sampson, J.R. Evolution of tRNA recognition systems and tRNA gene sequences. J. Mol. Evol. 1995, 40, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, R. Evolution of the aminoacyl-tRNA synthetases and the origin of the genetic code. J. Mol. Evol. 1995, 40, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Giege, R. The early history of tRNA recognition by aminoacyl-tRNA synthetases. J. Biosci. 2006, 31, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Sheppard, K.; Soll, D. Amino acid modifications on tRNA. Acta Biochim. Biophys. Sin. (Shanghai) 2008, 40, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Moghal, A.; Mohler, K.; Ibba, M. Mistranslation of the genetic code. FEBS Lett. 2014, 588, 4305–4310. [Google Scholar] [CrossRef] [PubMed]
- Beuning, P.J.; Musier-Forsyth, K. Transfer RNA recognition by aminoacyl-tRNA synthetases. Biopolymers 1999, 52, 1–28. [Google Scholar] [CrossRef]
- Seligmann, H. Pocketknife tRNA hypothesis: Anticodons in mammal mitochondrial tRNA side-arm loops translate proteins? BioSystems 2013, 113, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H. Putative anticodons in mitochondrial tRNA sidearm loops: Pocketknife tRNAs? J. Theor. Biol. 2014, 340, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Ikeda, K.T.; Noro, E.; Hiraoka, K.; Tomita, M.; Kanai, A. Precise mapping and dynamics of tRNA-derived fragments (tRFs) in the development of Triops cancriformis (tadpole shrimp). BMC Genet. 2015, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Hopfield, J.J.; Yamane, T.; Yue, V.; Coutts, S.M. Direct experimental evidence for kinetic proofreading in amino acylation of tRNAIle. Proc. Natl. Acad. Sci. USA 1976, 73, 1164–1168. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.X.; Baltzinger, M.; Remy, P. Fast kinetic study of yeast phenylalanyl-tRNA synthetase: Role of tRNAPhe in the discrimination between tyrosine and phenylalanine. Biochemistry 1984, 23, 4109–4116. [Google Scholar] [CrossRef] [PubMed]
- Yamane, T.; Hopfield, J.J. Experimental evidence for kinetic proofreading in the aminoacylation of tRNA by synthetase. Proc. Natl. Acad. Sci. USA 1977, 74, 2246–2250. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.; Moras, D. The fidelity of the translation of the genetic code. Acta Biochim. Pol. 2001, 48, 323–335. [Google Scholar] [PubMed]
- Bullwinkle, T.; Lazazzera, B.; Ibba, M. Quality control and infiltration of translation by amino acids outside of the genetic code. Annu. Rev. Genet. 2014, 48, 149–166. [Google Scholar] [PubMed]
- Guo, M.; Schimmel, P. Structural analyses clarify the complex control of mistranslation by tRNA synthetases. Curr. Opin. Struct. Biol. 2012, 22, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H. Positive and negative cognate amino acid bias affects compositions of aminoacyl-tRNA synthetases and reflects functional constraints on protein structure. BIO 2012, 2, 16. [Google Scholar] [CrossRef]
- Seligmann, H. Do anticodons of misacylated tRNAs preferentially mismatch codons coding for the misloaded amino acid? BMC Mol. Biol. 2010, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, T.K.; Kapil, C.; Khan, S.; Jairajpuri, M.A.; Sharma, V.; Santoni, D.; Silvestrini, F.; Pizzi, E.; Sharma, A. A genomic glimpse of aminoacyl-tRNA synthetases in malaria parasite Plasmodium falciparum. BMC Genomics 2009, 10, 644. [Google Scholar] [CrossRef] [PubMed]
- Vondenhoff, G.H.; Van Aerschot, A. Aminoacyl-tRNA synthetase inhibitors as potential antibiotics. Eur. J. Med. Chem. 2011, 46, 5227–5236. [Google Scholar] [CrossRef] [PubMed]
- Pham, J.S.; Dawson, K.L.; Jackson, K.E.; Lim, E.E.; Pasaje, C.F.; Turner, K.E.; Ralph, S.A. Aminoacyl-tRNA synthetases as drug targets in eukaryotic parasites. Int. J. Parasitol Drugs Drug Resist. 2014, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mudge, S.J.; Williams, J.H.; Eyre, H.J.; Sutherland, G.R.; Cowan, P.J.; Power, D.A. Complex organisation of the 5'-end of the human glycine tRNA synthetase gene. Gene 1998, 209, 45–50. [Google Scholar] [CrossRef]
- Dias, J.; Octobre, G.; Kobbi, L.; Comisso, M.; Flisiak, S.; Mirande, M. Activation of human mitochondrial lysyl-tRNA synthetase upon maturation of its premitochondrial precursor. Biochemistry 2012, 51, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Echevarria, L.; Clemente, P.; Hernandez-Sierra, R.; Gallardo, M.E.; Fernandez-Moreno, M.A.; Garesse, R. Glutamyl-tRNAGln amidotransferase is essential for mammalian mitochondrial translation in vivo. Biochem. J. 2014, 460, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.; Fox, P.L. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol. Med. 2013, 5, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Datt, M.; Sharma, A. Evolutionary and structural annotation of disease-associated mutations in human aminoacyl-tRNA synthetases. BMC Genomics 2014, 15, 1063. [Google Scholar] [CrossRef] [PubMed]
- Park, S.G.; Schimmel, P.; Kim, S. Aminoacyl tRNA synthetases and their connections to disease. Proc. Natl. Acad. Sci. USA 2008, 105, 11043–11049. [Google Scholar] [CrossRef] [PubMed]
- Simons, C.; Griffin, L.B.; Helman, G.; Golas, G.; Pizzino, A.; Bloom, M.; Murphy, J.L.; Crawford, J.; Evans, S.H.; Topper, S.; et al. Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am. J. Hum. Genet. 2015, 96, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Taft, R.J.; Vanderver, A.; Leventer, R.J.; Damiani, S.A.; Simons, C.; Grimmond, S.M.; Miller, D.; Schmidt, J.; Lockhart, P.J.; Pope, K.; et al. Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am. J. Hum. Genet. 2013, 92, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Puffenberger, E.G.; Jinks, R.N.; Sougnez, C.; Cibulskis, K.; Willert, R.A.; Achilly, N.P.; Cassidy, R.P.; Fiorentini, C.J.; Heiken, K.F.; Lawrence, J.J.; et al. Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS One 2012, 7, e28936. [Google Scholar] [CrossRef] [PubMed]
- Santos-Cortez, R.L.; Lee, K.; Azeem, Z.; Antonellis, P.J.; Pollock, L.M.; Khan, S.; Irfanullah; Andrade-Elizondo, P.B.; Chiu, I.; Adams, M.D.; et al. Mutations in KARS, encoding lysyl-tRNA synthetase, cause autosomal-recessive nonsyndromic hearing impairment DFNB89. Am. J. Hum. Genet. 2013, 93, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.P.; McGettigan, P.; Lynam-Lennon, N.; McDermott, M.; Regan, R.; Conroy, J.; Bourke, B.; O'Sullivan, J.; Crushell, E.; Lynch, S.; et al. Identification of a mutation in LARS as a novel cause of infantile hepatopathy. Mol. Genet. Metab. 2012, 106, 351–358. [Google Scholar] [CrossRef] [PubMed]
- van Meel, E.; Wegner, D.J.; Cliften, P.; Willing, M.C.; White, F.V.; Kornfeld, S.; Cole, F.S. Rare recessive loss-of-function methionyl-tRNA synthetase mutations presenting as a multi-organ phenotype. BMC Med. Genet. 2013, 14, 106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ling, J.; Barcia, G.; Jing, L.; Wu, J.; Barry, B.J.; Mochida, G.H.; Hill, R.S.; Weimer, J.M.; Stein, Q.; et al. Mutations in QARS, encoding glutaminyl-tRNA synthetase, cause progressive microcephaly, cerebral-cerebellar atrophy, and intractable seizures. Am. J. Hum. Genet. 2014, 94, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.I.; Salomons, G.S.; Rodenburg, R.J.; Pouwels, P.J.; Schieving, J.H.; Derks, T.G.; Fock, J.M.; Rump, P.; van Beek, D.M.; van der Knaap, M.S.; et al. Mutations in RARS cause hypomyelination. Ann. Neurol. 2014, 76, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Schimmel, P. Essential nontranslational functions of tRNA synthetases. Nat. Chem. Biol. 2013, 9, 145–153. [Google Scholar] [PubMed]
- Gonzalez, M.; McLaughlin, H.; Houlden, H.; Guo, M.; Yo-Tsen, L.; Hadjivassilious, M.; Speziani, F.; Yang, X.L.; Antonellis, A.; Reilly, M.M.; et al. Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1247–1249. [Google Scholar] [CrossRef] [PubMed]
- Hyun, Y.S.; Park, H.J.; Heo, S.H.; Yoon, B.R.; Nam, S.H.; Kim, S.B.; Park, C.I.; Choi, B.O.; Chung, K.W. Rare variants in methionyl- and tyrosyl-tRNA synthetase genes in late-onset autosomal dominant Charcot-Marie-Tooth neuropathy. Clin. Genet. 2014, 86, 592–594. [Google Scholar] [CrossRef] [PubMed]
- Niehues, S.; Bussmann, J.; Steffes, G.; Erdmann, I.; Kohrer, C.; Sun, L.; Wagner, M.; Schafer, K.; Wang, G.; Koerdt, S.N.; et al. Impaired protein translation in Drosophila models for Charcot-Marie-Tooth neuropathy caused by mutant tRNA synthetases. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Grice, S.J.; Sleigh, J.N.; Motley, W.W.; Liu, J.L.; Burgess, R.W.; Talbot, K.; Cader, M.Z. Dominant, toxic gain-of-function mutations in gars lead to non-cell autonomous neuropathology. Hum. Mol. Genet. 2015, 24, 4397–4406. [Google Scholar] [CrossRef] [PubMed]
- Seburn, K.L.; Nangle, L.A.; Cox, G.A.; Schimmel, P.; Burgess, R.W. An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-Tooth 2D mouse model. Neuron 2006, 51, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Dallabona, C.; Diodato, D.; Kevelam, S.H.; Haack, T.B.; Wong, L.J.; Salomons, G.S.; Baruffini, E.; Melchionda, L.; Mariotti, C.; Strom, T.M.; et al. Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology 2014, 82, 2063–2071. [Google Scholar] [CrossRef] [PubMed]
- Gotz, A.; Tyynismaa, H.; Euro, L.; Ellonen, P.; Hyotylainen, T.; Ojala, T.; Hamalainen, R.H.; Tommiska, J.; Raivio, T.; Oresic, M.; et al. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Scheper, G.C.; van der Klok, T.; van Andel, R.J.; van Berkel, C.G.; Sissler, M.; Smet, J.; Muravina, T.I.; Serkov, S.V.; Uziel, G.; Bugiani, M.; et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat. Genet. 2007, 39, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Elo, J.M.; Yadavalli, S.S.; Euro, L.; Isohanni, P.; Gotz, A.; Carroll, C.J.; Valanne, L.; Alkuraya, F.S.; Uusimaa, J.; Paetau, A.; et al. Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum. Mol. Genet. 2012, 21, 4521–4529. [Google Scholar] [CrossRef] [PubMed]
- Pierce, S.B.; Chisholm, K.M.; Lynch, E.D.; Lee, M.K.; Walsh, T.; Opitz, J.M.; Li, W.; Klevit, R.E.; King, M.C. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 6543–6548. [Google Scholar] [CrossRef] [PubMed]
- Pierce, S.B.; Gersak, K.; Michaelson-Cohen, R.; Walsh, T.; Lee, M.K.; Malach, D.; Klevit, R.E.; King, M.C.; Levy-Lahad, E. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am. J. Hum. Genet. 2013, 92, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Bayat, V.; Thiffault, I.; Jaiswal, M.; Tetreault, M.; Donti, T.; Sasarman, F.; Bernard, G.; Demers-Lamarche, J.; Dicaire, M.J.; Mathieu, J.; et al. Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol. 2012, 10, e1001288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belostotsky, R.; Ben-Shalom, E.; Rinat, C.; Becker-Cohen, R.; Feinstein, S.; Zeligson, S.; Segel, R.; Elpeleg, O.; Nassar, S.; Frishberg, Y. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am. J. Hum. Genet. 2011, 88, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Sasarman, F.; Nishimura, T.; Thiffault, I.; Shoubridge, E.A. A novel mutation in YARS2 causes myopathy with lactic acidosis and sideroblastic anemia. Hum. Mutat. 2012, 33, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Riley, L.G.; Cooper, S.; Hickey, P.; Rudinger-Thirion, J.; McKenzie, M.; Compton, A.; Lim, S.C.; Thorburn, D.; Ryan, M.T.; Giege, R.; et al. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia—MLASA syndrome. Am. J. Hum. Genet. 2010, 87, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; You, S.; Hwang, D. Aminoacyl-tRNA synthetases and tumorigenesis: More than housekeeping. Nat. Rev. Cancer 2011, 11, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Rodova, M.; Ankilova, V.; Safro, M.G. Human phenylalanyl-tRNA synthetase: Cloning, characterization of the deduced amino acid sequences in terms of the structural domains and coordinately regulated expression of the alpha and beta subunits in chronic myeloid leukemia cells. Biochem. Biophys. Res. Commun. 1999, 255, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Kushner, J.P.; Boll, D.; Quagliana, J.; Dickman, S. Elevated methionine-tRNA synthetase activity in human colon cancer. Proc. Soc. Exp. Biol. Med. 1976, 153, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Scandurro, A.B.; Weldon, C.W.; Figueroa, Y.G.; Alam, J.; Beckman, B.S. Gene microarray analysis reveals a novel hypoxia signal transduction pathway in human hepatocellular carcinoma cells. Int. J. Oncol. 2001, 19, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Park, S.G.; Kim, H.J.; Min, Y.H.; Choi, E.C.; Shin, Y.K.; Park, B.J.; Lee, S.W.; Kim, S. Human lysyl-tRNA synthetase is secreted to trigger proinflammatory response. Proc. Natl. Acad. Sci. USA 2005, 102, 6356–6361. [Google Scholar] [CrossRef] [PubMed]
- Tsun, Z.Y.; Possemato, R. Amino acid management in cancer. Semin. Cell Dev. Biol. 2015, 43. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Liu, P.; Wei, W. mTOR signaling in tumorigenesis. Biochim. Biophys. Acta 2014, 1846, 638–654. [Google Scholar] [CrossRef] [PubMed]
- Koromilas, A.E. Roles of the translation initiation factor eIF2alpha serine 51 phosphorylation in cancer formation and treatment. Biochim. Biophys. Acta 2015, 1849, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Feun, L.G.; Kuo, M.T.; Savaraj, N. Arginine deprivation in cancer therapy. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.F.; Ohnuma, T. Lessons from the study of induced alterations in amino acids in patients with cancer. Cancer Treat. Rep. 1979, 63, 1013–1018. [Google Scholar] [PubMed]
- Appel, I.M.; van Kessel-Bakvis, C.; Stigter, R.; Pieters, R. Influence of two different regimens of concomitant treatment with asparaginase and dexamethasone on hemostasis in childhood acute lymphoblastic leukemia. Leukemia 2007, 21, 2377–2380. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Huang, J.; Sui, M. Targeting arginine metabolism pathway to treat arginine-dependent cancers. Cancer Lett. 2015, 364, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Prayson, R.; Farver, C. Antisynthetase syndrome: Not just an inflammatory myopathy. Cleve. Clin. J. Med. 2013, 80, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Carlson, T.J.; Pellerin, A.; Djuretic, I.M.; Trivigno, C.; Koralov, S.B.; Rao, A.; Sundrud, M.S. Halofuginone-induced amino acid starvation regulates Stat3-dependent Th17 effector function and reduces established autoimmune inflammation. J. Immunol. 2014, 192, 2167–2176. [Google Scholar] [CrossRef] [PubMed]
- Sundrud, M.S.; Koralov, S.B.; Feuerer, M.; Calado, D.P.; Kozhaya, A.E.; Rhule-Smith, A.; Lefebvre, R.E.; Unutmaz, D.; Mazitschek, R.; Waldner, H.; et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science 2009, 324, 1334–1338. [Google Scholar] [CrossRef] [PubMed]
- Sajish, M.; Zhou, Q.; Kishi, S.; Valdez, D.M., Jr.; Kapoor, M.; Guo, M.; Lee, S.; Kim, S.; Yang, X.L.; Schimmel, P. Trp-tRNA synthetase bridges DNA-PKcs to PARP-1 to link IFN-gamma and p53 signaling. Nat. Chem. Biol. 2012, 8, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, R.; Jia, J.; Arif, A.; Ray, P.S.; Fox, P.L. The GAIT system: A gatekeeper of inflammatory gene expression. Trends Biochem. Sci. 2009, 34, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Ofir-Birin, Y.; Fang, P.; Bennett, S.P.; Zhang, H.M.; Wang, J.; Rachmin, I.; Shapiro, R.; Song, J.; Dagan, A.; Pozo, J.; et al. Structural switch of lysyl-tRNA synthetase between translation and transcription. Mol. Cell 2013, 49, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.G.; Lee, J.Y.; Kwon, N.H.; Fang, P.; Zhang, Q.; Wang, J.; Young, N.L.; Guo, M.; Cho, H.Y.; Mushtaq, A.U.; et al. Chemical inhibition of prometastatic lysyl-tRNA synthetase-laminin receptor interaction. Nat. Chem. Biol. 2014, 10, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.J.; Nyborg, J.; Reid, B.R.; Blow, D.M. The crystal structure of tyrosyl-transfer RNA synthetase at 2-7 A resolution. J. Mol. Biol. 1976, 105, 577–586. [Google Scholar] [CrossRef]
- Rock, F.L.; Mao, W.; Yaremchuk, A.; Tukalo, M.; Crepin, T.; Zhou, H.; Zhang, Y.K.; Hernandez, V.; Akama, T.; Baker, S.J.; et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science 2007, 316, 1759–1761. [Google Scholar] [CrossRef] [PubMed]
- Silvian, L.F.; Wang, J.; Steitz, T.A. Insights into editing from an ile-tRNA synthetase structure with tRNAile and mupirocin. Science 1999, 285, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Sun, L.; Yang, X.L.; Schimmel, P. ATP-directed capture of bioactive herbal-based medicine on human tRNA synthetase. Nature 2013, 494, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Gadakh, B.; Van Aerschot, A. Aminoacyl-tRNA synthetase inhibitors as antimicrobial agents: A patent review from 2006 till present. Expert Opin. Ther. Pat. 2012, 22, 1453–1465. [Google Scholar] [CrossRef] [PubMed]
- WHO Technical Report Series 985. The Selection and Use of Essential Medicines. Available online: http://apps.who.int/iris/bitstream/10665/112729/1/WHO_TRS_985_eng.pdf (accessed on 7 December 2015).
- Jarvie, B.D.; Leslie, K.E.; Peregrine, A.S.; Duffield, T.F.; Weese, J.S. Preliminary evaluation of the efficacy of halofuginone lactate (Halocur (R)) as an aid in the prevention of cryptosporidiosis in Ontario dairy calves. J. Anim. Sci. 2004, 82, 405–406. [Google Scholar]
- Pines, M.; Spector, I. Halofuginone—the multifaceted molecule. Molecules 2015, 20, 573–594. [Google Scholar] [CrossRef] [PubMed]
- Anonymous Tavaborole Topical Solution (Kerydin) for Onychomycosis. Med. Lett. Drugs Ther. 2015, 57, 35–36.
- Fang, P.; Han, H.; Wang, J.; Chen, K.; Chen, X.; Guo, M. Structural Basis for Specific Inhibition of tRNA Synthetase by an ATP Competitive Inhibitor. Chem. Biol. 2015, 22, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.Y.; Kallur Siddaramaiah, L.; Ranade, R.M.; Nguyen, J.; Jian, T.; Zhang, Z.; Gillespie, J.R.; Buckner, F.S.; Verlinde, C.L.; Fan, E.; et al. A binding hotspot in Trypanosoma cruzi histidyl-tRNA synthetase revealed by fragment-based crystallographic cocktail screens. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 1684–1698. [Google Scholar] [CrossRef] [PubMed]
- Sajish, M.; Schimmel, P. A human tRNA synthetase is a potent PARP1-activating effector target for resveratrol. Nature 2015, 519, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.Y.; Kim, J.E.; Shibata, S.; Ranade, R.M.; Yu, M.; Liu, J.; Gillespie, J.R.; Buckner, F.S.; Verlinde, C.L.; Fan, E.; et al. Distinct states of methionyl-tRNA synthetase indicate inhibitor binding by conformational selection. Structure 2012, 20, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Tworowski, D.; Klipcan, L.; Peretz, M.; Moor, N.; Safro, M.G. Universal pathway for posttransfer editing reactions: Insights from the crystal structure of TtPheRS with puromycin. Proc. Natl. Acad. Sci. USA 2015, 112, 3967–3972. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.; Hilgers, M.T.; Cunningham, M.L.; Borchardt, A.; Locke, J.B.; Abraham, S.; Haley, G.; Kwan, B.P.; Hall, C.; Hough, G.W.; et al. Identification of bacteria-selective threonyl-tRNA synthetase substrate inhibitors by structure-based design. J. Med. Chem. 2013, 56, 1748–1760. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Y.; Hill, J.M.; Yu, G.; Wang, W.; Kluge, A.F.; Wendler, P.; Gallant, P. Synthesis and structure-activity relationships of a series of novel thiazoles as inhibitors of aminoacyl-tRNA synthetases. Bioorg. Med. Chem. Lett. 1999, 9, 375–380. [Google Scholar] [CrossRef]
- Vondenhoff, G.H.; Dubiley, S.; Severinov, K.; Lescrinier, E.; Rozenski, J.; Van Aerschot, A. Extended targeting potential and improved synthesis of Microcin C analogs as antibacterials. Bioorg. Med. Chem. 2011, 19, 5462–5467. [Google Scholar] [CrossRef] [PubMed]
- Reader, J.S.; Ordoukhanian, P.T.; Kim, J.G.; de Crecy-Lagard, V.; Hwang, I.; Farrand, S.; Schimmel, P. Major biocontrol of plant tumors targets tRNA synthetase. Science 2005, 309, 1533. [Google Scholar] [CrossRef] [PubMed]
- Takaya, Y.; Tasaka, H.; Chiba, T.; Uwai, K.; Tanitsu, M.; Kim, H.S.; Wataya, Y.; Miura, M.; Takeshita, M.; Oshima, Y. New type of febrifugine analogues, bearing a quinolizidine moiety, show potent antimalarial activity against Plasmodium malaria parasite. J. Med. Chem. 1999, 42, 3163–3166. [Google Scholar] [CrossRef] [PubMed]
- Samant, B.S.; Sukhthankar, M.G. Synthesis and comparison of antimalarial activity of febrifugine derivatives including halofuginone. Med. Chem. 2009, 5, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Keller, T.L.; Zocco, D.; Sundrud, M.S.; Hendrick, M.; Edenius, M.; Yum, J.; Kim, Y.J.; Lee, H.K.; Cortese, J.F.; Wirth, D.F.; et al. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat. Chem. Biol. 2012, 8, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Beyer, D.; Kroll, H.P.; Endermann, R.; Schiffer, G.; Siegel, S.; Bauser, M.; Pohlmann, J.; Brands, M.; Ziegelbauer, K.; Haebich, D.; et al. New class of bacterial phenylalanyl-tRNA synthetase inhibitors with high potency and broad-spectrum activity. Antimicrob. Agents Chemother. 2004, 48, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Critchley, I.A.; Green, L.S.; Young, C.L.; Bullard, J.M.; Evans, R.J.; Price, M.; Jarvis, T.C.; Guiles, J.W.; Janjic, N.; Ochsner, U.A. Spectrum of activity and mode of action of REP3123, a new antibiotic to treat Clostridium difficile infections. J. Antimicrob. Chemother. 2009, 63, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Yu, X.; Jeong, S.J.; Mirando, A.; Chen, K.; Chen, X.; Kim, S.; Francklyn, C.S.; Guo, M. Structural basis for full-spectrum inhibition of translational functions on a tRNA synthetase. Nat. Commun. 2015, 6, 6402. [Google Scholar] [CrossRef] [PubMed]
- Novoa, E.M.; Camacho, N.; Tor, A.; Wilkinson, B.; Moss, S.; Marin-Garcia, P.; Azcarate, I.G.; Bautista, J.M.; Mirando, A.C.; Francklyn, C.S.; et al. Analogs of natural aminoacyl-tRNA synthetase inhibitors clear malaria in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E5508–E5517. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Sharma, A.; Jamwal, A.; Sharma, V.; Pole, A.K.; Thakur, K.K.; Sharma, A. Uneven spread of cis- and trans-editing aminoacyl-tRNA synthetase domains within translational compartments of, P. falciparum. Sci. Rep. 2011, 1, 188. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Y.; Finn, J.; Hill, J.M.; Wang, Z.G.; Keith, D.; Silverman, J.; Oliver, N. A series of spirocyclic analogues as potent inhibitors of bacterial phenylalanyl-tRNA synthetases. Bioorg. Med. Chem. Lett. 2004, 14, 1339–1342. [Google Scholar] [CrossRef] [PubMed]
- Finn, J.; Mattia, K.; Morytko, M.; Ram, S.; Yang, Y.; Wu, X.; Mak, E.; Gallant, P.; Keith, D. Discovery of a potent and selective series of pyrazole bacterial methionyl-tRNA synthetase inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 2231–2234. [Google Scholar] [CrossRef]
- Tandon, M.; Coffen, D.L.; Gallant, P.; Keith, D.; Ashwell, M.A. Potent and selective inhibitors of bacterial methionyl tRNA synthetase derived from an oxazolone-dipeptide scaffold. Bioorg. Med. Chem. Lett. 2004, 14, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Ibba, M.; Soll, D. Aminoacyl-tRNAs: Setting the limits of the genetic code. Genes Dev. 2004, 18, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.T.; Chong, Y.E.; Guo, M.; Yang, X.L. Crystal structures and biochemical analyses suggest a unique mechanism and role for human glycyl-tRNA synthetase in Ap4A homeostasis. J. Biol. Chem. 2009, 284, 28968–28976. [Google Scholar] [CrossRef] [PubMed]
- Hoepfner, D.; McNamara, C.W.; Lim, C.S.; Studer, C.; Riedl, R.; Aust, T.; McCormack, S.L.; Plouffe, D.M.; Meister, S.; Schuierer, S.; et al. Selective and specific inhibition of the plasmodium falciparum lysyl-tRNA synthetase by the fungal secondary metabolite cladosporin. Cell Host Microbe 2012, 11, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, M.H.; Hansen, B.S. Control of initiation of protein synthesis in human cells. Evidence for a role of uncharged transfer ribonucleic acid. J. Biol. Chem. 1973, 248, 7087–7096. [Google Scholar] [PubMed]
- Litt, M.; Weiser, K. Histidine transfer RNA levels in Friend leukemia cells: Stimulation by histidine deprivation. Science 1978, 201, 527–529. [Google Scholar] [CrossRef] [PubMed]
- Warrington, R.C.; Fang, W.D.; Zhang, L.U. L-histidinol reverses resistance to cisplatinum and other antineoplastics in a tumorigenic epithelial cell line. Anticancer Res. 1996, 16, 3641–3646. [Google Scholar] [PubMed]
- Busiello, V.; Di Girolamo, M.; De Marco, C. Thiaisoleucine and protein synthesis. Biochim. Biophys. Acta 1979, 561, 206–214. [Google Scholar] [CrossRef]
- Walden, H. Selenium incorporation using recombinant techniques. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigo, R.; Gladyshev, V.N. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Gambini, J.; Ingles, M.; Olaso, G.; Lopez-Grueso, R.; Bonet-Costa, V.; Gimeno-Mallench, L.; Mas-Bargues, C.; Abdelaziz, K.M.; Gomez-Cabrera, M.C.; Vina, J.; et al. Properties of Resveratrol: In Vitro and In Vivo Studies about Metabolism, Bioavailability, and Biological Effects in Animal Models and Humans. Oxid. Med. Cell. Longev. 2015, 2015, 837042. [Google Scholar] [CrossRef] [PubMed]
- Buryanovskyy, L.; Fu, Y.; Boyd, M.; Ma, Y.; Hsieh, T.C.; Wu, J.M.; Zhang, Z. Crystal structure of quinone reductase 2 in complex with resveratrol. Biochemistry 2004, 43, 11417–11426. [Google Scholar] [CrossRef] [PubMed]
- Klabunde, T.; Petrassi, H.M.; Oza, V.B.; Raman, P.; Kelly, J.W.; Sacchettini, J.C. Rational design of potent human transthyretin amyloid disease inhibitors. Nat. Struct. Biol. 2000, 7, 312–321. [Google Scholar] [PubMed]
- Davies, D.R.; Mamat, B.; Magnusson, O.T.; Christensen, J.; Haraldsson, M.H.; Mishra, R.; Pease, B.; Hansen, E.; Singh, J.; Zembower, D.; et al. Discovery of leukotriene A4 hydrolase inhibitors using metabolomics biased fragment crystallography. J. Med. Chem. 2009, 52, 4694–4715. [Google Scholar] [CrossRef] [PubMed]
- Pineda-Sanabria, S.E.; Robertson, I.M.; Sykes, B.D. Structure of trans-resveratrol in complex with the cardiac regulatory protein troponin, C. Biochemistry 2011, 50, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.T.; Gertz, M.; Steegborn, C. Crystal structures of Sirt3 complexes with 4'-bromo-resveratrol reveal binding sites and inhibition mechanism. Chem. Biol. 2013, 20, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Nguyen, G.T.; Fischer, F.; Suenkel, B.; Schlicker, C.; Franzel, B.; Tomaschewski, J.; Aladini, F.; Becker, C.; Wolters, D.; et al. A molecular mechanism for direct sirtuin activation by resveratrol. PLoS One 2012, 7, e49761. [Google Scholar] [CrossRef] [PubMed]
- Calleri, E.; Pochetti, G.; Dossou, K.S.; Laghezza, A.; Montanari, R.; Capelli, D.; Prada, E.; Loiodice, F.; Massolini, G.; Bernier, M.; et al. Resveratrol and its metabolites bind to PPARs. ChemBioChem 2014, 15, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Shafqat, N.; Muniz, J.R.; Pilka, E.S.; Papagrigoriou, E.; von Delft, F.; Oppermann, U.; Yue, W.W. Insight into S-adenosylmethionine biosynthesis from the crystal structures of the human methionine adenosyltransferase catalytic and regulatory subunits. Biochem. J. 2013, 452, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Nwachukwu, J.C.; Srinivasan, S.; Bruno, N.E.; Parent, A.A.; Hughes, T.S.; Pollock, J.A.; Gjyshi, O.; Cavett, V.; Nowak, J.; Garcia-Ordonez, R.D.; et al. Resveratrol modulates the inflammatory response via an estrogen receptor-signal integration network. Elife 2014, 3, e02057. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, R.; Yokoyama, S. Structural basis for substrate recognition by the editing domain of isoleucyl-tRNA synthetase. J. Mol. Biol. 2006, 359, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Dock-Bregeon, A.C.; Rees, B.; Torres-Larios, A.; Bey, G.; Caillet, J.; Moras, D. Achieving error-free translation; the mechanism of proofreading of threonyl-tRNA synthetase at atomic resolution. Mol. Cell 2004, 16, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.; Kruparani, S.P.; Pal, B.; Dock-Bregeon, A.C.; Dwivedi, S.; Shekar, M.R.; Sureshbabu, K.; Sankaranarayanan, R. Post-transfer editing mechanism of a D-aminoacyl-tRNA deacylase-like domain in threonyl-tRNA synthetase from archaea. EMBO J. 2006, 25, 4152–4162. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.; Kamarthapu, V.; Kruparani, S.P.; Deshmukh, M.V.; Sankaranarayanan, R. Mechanistic insights into cognate substrate discrimination during proofreading in translation. Proc. Natl. Acad. Sci. USA 2010, 107, 22117–22121. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Muthukumar, S.; Kuncha, S.K.; Routh, S.B.; Yerabham, A.S.; Hussain, T.; Kamarthapu, V.; Kruparani, S.P.; Sankaranarayanan, R. Specificity and catalysis hardwired at the RNA-protein interface in a translational proofreading enzyme. Nat. Commun. 2015, 6, 7552. [Google Scholar] [CrossRef] [PubMed]
- Lipovsek, D.; Pluckthun, A. In-vitro protein evolution by ribosome display and mRNA display. J. Immunol. Methods 2004, 290, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Nissen, P.; Hansen, J.; Ban, N.; Moore, P.B.; Steitz, T.A. The structural basis of ribosome activity in peptide bond synthesis. Science 2000, 289, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Cusack, S.; Yaremchuk, A.; Tukalo, M. The crystal structure of the ternary complex of, T.thermophilus seryl-tRNA synthetase with tRNA(Ser) and a seryl-adenylate analogue reveals a conformational switch in the active site. EMBO J. 1996, 15, 2834–2842. [Google Scholar] [PubMed]
- Cassio, D.; Lemoine, F.; Waller, J.P.; Sandrin, E.; Boissonnas, R.A. Selective inhibition of aminoacyl ribonucleic acid synthetases by aminoalkyl adenylates. Biochemistry 1967, 6, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Sakurama, H.; Takita, T.; Mikami, B.; Itoh, T.; Yasukawa, K.; Inouye, K. Two crystal structures of lysyl-tRNA synthetase from Bacillus stearothermophilus in complex with lysyladenylate-like compounds: Insights into the irreversible formation of the enzyme-bound adenylate of L-lysine hydroxamate. J. Biochem. 2009, 145, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Belrhali, H.; Yaremchuk, A.; Tukalo, M.; Larsen, K.; Berthet-Colominas, C.; Leberman, R.; Beijer, B.; Sproat, B.; Als-Nielsen, J.; Grubel, G.; et al. Crystal structures at 2.5 angstrom resolution of seryl-tRNA synthetase complexed with two analogs of seryl adenylate. Science 1994, 263, 1432–1436. [Google Scholar] [CrossRef] [PubMed]
- Pope, A.J.; Lapointe, J.; Mensah, L.; Benson, N.; Brown, M.J.; Moore, K.J. Characterization of isoleucyl-tRNA synthetase from Staphylococcus aureus. I: Kinetic mechanism of the substrate activation reaction studied by transient and steady-state techniques. J. Biol. Chem. 1998, 273, 31680–31690. [Google Scholar] [CrossRef] [PubMed]
- Hurdle, J.G.; O'Neill, A.J.; Chopra, I. Prospects for aminoacyl-tRNA synthetase inhibitors as new antimicrobial agents. Antimicrob. Agents Chemother. 2005, 49, 4821–4833. [Google Scholar] [CrossRef] [PubMed]
- Chopra, S.; Palencia, A.; Virus, C.; Tripathy, A.; Temple, B.R.; Velazquez-Campoy, A.; Cusack, S.; Reader, J.S. Plant tumour biocontrol agent employs a tRNA-dependent mechanism to inhibit leucyl-tRNA synthetase. Nat. Commun. 2013, 4, 1417. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.; Mellows, G. Interaction of pseudomonic acid A with Escherichia coli B isoleucyl-tRNA synthetase. Biochem. J. 1980, 191, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, H.; Kagasaki, T.; Torikata, A.; Tanaka, N.; Fujimoto, K.; Hata, T.; Furukawa, Y.; Takahashi, S. Thiomarinols B and C, new antimicrobial antibiotics produced by a marine bacterium. J. Antibiot. (Tokyo) 1995, 48, 907–909. [Google Scholar] [CrossRef]
- Shiozawa, H.; Shimada, A.; Takahashi, S. Thiomarinols D, E, F and G, new hybrid antimicrobial antibiotics produced by a marine bacterium; isolation, structure, and antimicrobial activity. J. Antibiot. (Tokyo) 1997, 50, 449–452. [Google Scholar] [CrossRef]
- Brown, M.J.; Mensah, L.M.; Doyle, M.L.; Broom, N.J.; Osbourne, N.; Forrest, A.K.; Richardson, C.M.; O'Hanlon, P.J.; Pope, A.J. Rational design of femtomolar inhibitors of isoleucyl tRNA synthetase from a binding model for pseudomonic acid-A. Biochemistry 2000, 39, 6003–6011. [Google Scholar] [CrossRef] [PubMed]
- Abibi, A.; Ferguson, A.D.; Fleming, P.R.; Gao, N.; Hajec, L.I.; Hu, J.; Laganas, V.A.; McKinney, D.C.; McLeod, S.M.; Prince, D.B.; et al. The role of a novel auxiliary pocket in bacterial phenylalanyl-tRNA synthetase druggability. J. Biol. Chem. 2014, 289, 21651–21662. [Google Scholar] [CrossRef] [PubMed]
- Critchley, I.A.; Ochsner, U.A. Recent advances in the preclinical evaluation of the topical antibacterial agent REP8839. Curr. Opin. Chem. Biol. 2008, 12, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.Y.; Kim, J.E.; Wetzel, A.B.; de van der Schueren, W.J.; Shibata, S.; Ranade, R.M.; Liu, J.; Zhang, Z.; Gillespie, J.R.; Buckner, F.S.; et al. Structures of Trypanosoma brucei methionyl-tRNA synthetase with urea-based inhibitors provide guidance for drug design against sleeping sickness. PLoS Negl. Trop. Dis. 2014, 8, e2775. [Google Scholar] [CrossRef] [PubMed]
- Carmo, J.; Marques, S.; Chapim, I.; Túlio, M. A.; Rodrigues, J.P.; Bispo, M.; Chagas, C. Leaping Forward in the Treatment of Clostridium Difficile Infection: Update in 2015. GE Portuguese, J. Gastroenterolo. 2015, 22, 259–267. [Google Scholar] [CrossRef]
- Freist, W.; Gauss, D.H.; Ibba, M.; Soll, D. Glutaminyl-tRNA synthetase. Biol. Chem. 1997, 378, 1103–1117. [Google Scholar] [PubMed]
- Freist, W.; Gauss, D.H.; Soll, D.; Lapointe, J. Glutamyl-tRNA sythetase. Biol. Chem. 1997, 378, 1313–1329. [Google Scholar] [PubMed]
- Blais, S.P.; Kornblatt, J.A.; Barbeau, X.; Bonnaure, G.; Lague, P.; Chenevert, R.; Lapointe, J. tRNAGlu increases the affinity of glutamyl-tRNA synthetase for its inhibitor glutamyl-sulfamoyl-adenosine, an analogue of the aminoacylation reaction intermediate glutamyl-AMP: Mechanistic and evolutionary implications. PLoS One 2015, 10, e0121043. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Jampolsky, L.M.; Goldberg, M.W. Borrelidin, a new antibiotic with antiborrelia activity and penicillin enhancement properties. Arch. Biochem. 1949, 22, 476–478. [Google Scholar] [PubMed]
- Dewan, V.; Wei, M.; Kleiman, L.; Musier-Forsyth, K. Dual role for motif 1 residues of human lysyl-tRNA synthetase in dimerization and packaging into HIV-1. J. Biol. Chem. 2012, 287, 41955–41962. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.F.; Mirando, A.C.; Wilkinson, B.; Francklyn, C.S.; Lounsbury, K.M. Secreted Threonyl-tRNA synthetase stimulates endothelial cell migration and angiogenesis. Sci. Rep. 2013, 3, 1317. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, K.; Slike, B.M.; Hood, J.; Otani, A.; Ewalt, K.L.; Friedlander, M.; Cheresh, D.A.; Schimmel, P. A human aminoacyl-tRNA synthetase as a regulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Delarue, M.; Moras, D. The aminoacyl-tRNA synthetase family: Modules at work. Bioessays 1993, 15, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Ribas de Pouplana, L.; Schimmel, P. Two classes of tRNA synthetases suggested by sterically compatible dockings on tRNA acceptor stem. Cell 2001, 104, 191–193. [Google Scholar] [CrossRef]
- Francklyn, C.; Musier-Forsyth, K.; Martinis, S.A. Aminoacyl-tRNA synthetases in biology and disease: New evidence for structural and functional diversity in an ancient family of enzymes. RNA 1997, 3, 954–960. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, P.; Guo, M. Evolutionary Limitation and Opportunities for Developing tRNA Synthetase Inhibitors with 5-Binding-Mode Classification. Life 2015, 5, 1703-1725. https://doi.org/10.3390/life5041703
Fang P, Guo M. Evolutionary Limitation and Opportunities for Developing tRNA Synthetase Inhibitors with 5-Binding-Mode Classification. Life. 2015; 5(4):1703-1725. https://doi.org/10.3390/life5041703
Chicago/Turabian StyleFang, Pengfei, and Min Guo. 2015. "Evolutionary Limitation and Opportunities for Developing tRNA Synthetase Inhibitors with 5-Binding-Mode Classification" Life 5, no. 4: 1703-1725. https://doi.org/10.3390/life5041703