
Silent Polymorphisms: Can the tRNA Population Explain Changes in Protein Properties?

Abstract
:1. Introduction
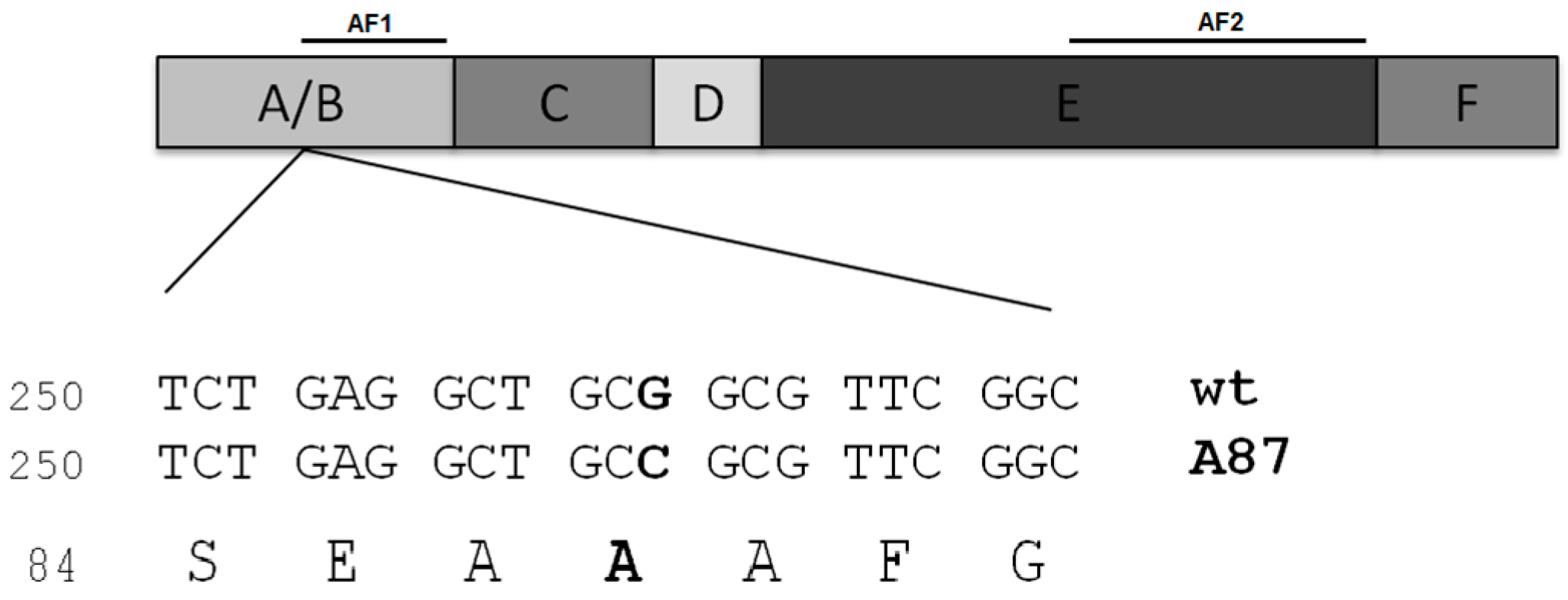
2. Case of Study: ERαAla87 Synonymous Polymorphism
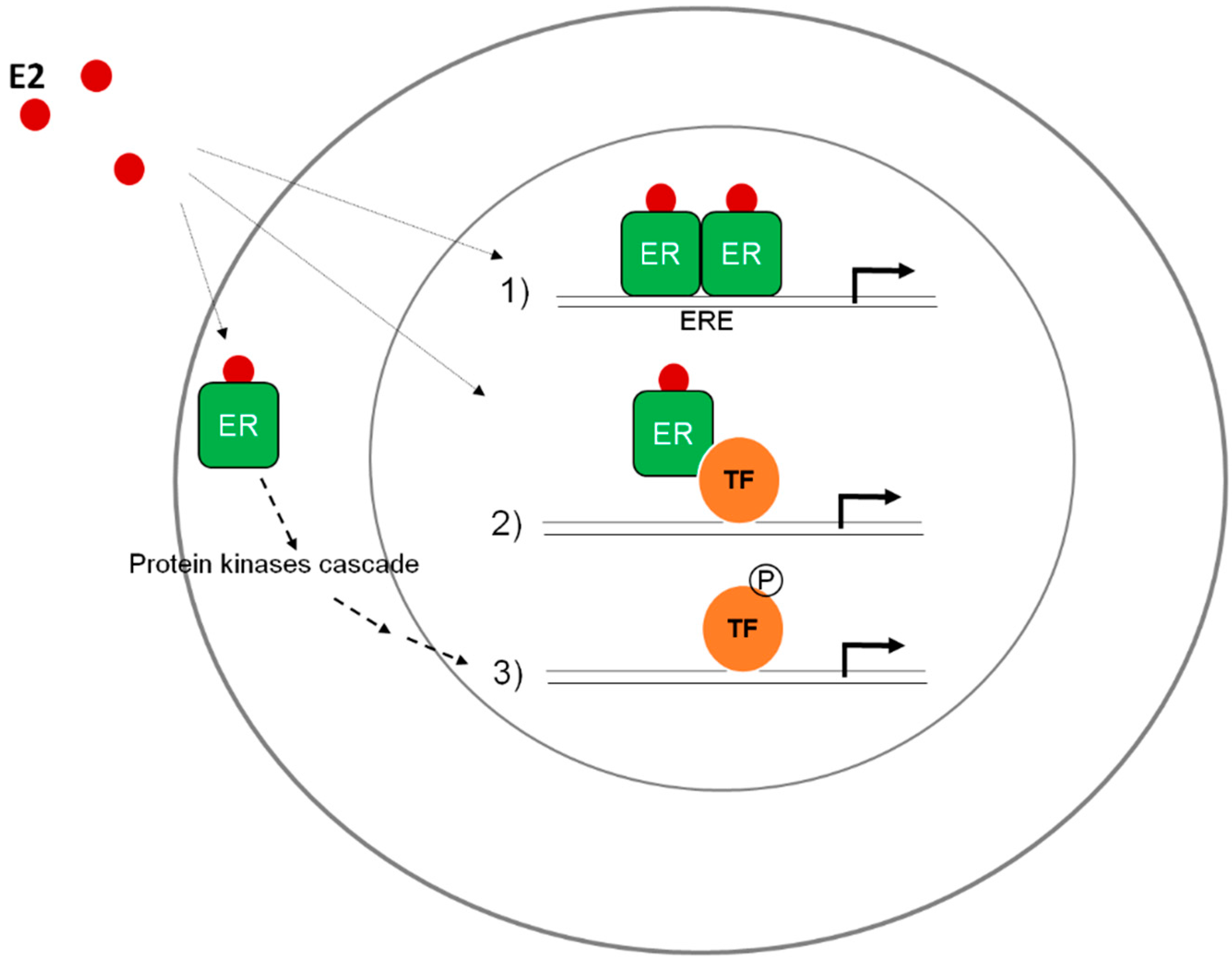
2.1. Differential Functional Properties of ERAla87 in HeLa and HepG2 Cells
2.2. Codons and tRNAs Involved in Decoding Alanine 87
3. tRNA Abundance Can Affect Translation Kinetics and Protein Conformation
3.1. Role of tRNAs in the Conformation of Proteins in Prokaryotes
3.2. tRNAs, Codon Usage and Protein Conformation in Eukaryotes
4. Dynamic Populations of tRNAs
4.1. tRNA Genes Are Differentially Expressed in Different Cell States
4.2. tRNA Post-Transcriptional Modifications: Expanding the Complexity of the tRNA Population
4.3. The tRNA Pool Is Partitioned in Different Cell Compartments
5. General Conclusions
The Sound of Silent Substitutions: The Tale of the Princess and the Pea, and the Case of Synonymous Polymorphism Ala87Ala GCG->GCC in Human ERα
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chaney, J.L.; Clark, P.L. Roles for synonymous codon usage in protein biogenesis. Annu. Rev. Biophys. 2015, 44, 143–166. [Google Scholar] [CrossRef] [PubMed]
- Chamary, J.V.; Parmley, J.L.; Hurst, L.D. Hearing silence: Non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 2006, 7, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Sauna, Z.E.; Kimchi-Sarfaty, C.; Ambudkar, S.V.; Gottesman, M.M. Silent polymorphisms speak: How they affect pharmacogenomics and the treatment of cancer. Cancer Res. 2007, 67, 9609–9612. [Google Scholar] [CrossRef] [PubMed]
- Sauna, Z.E.; Kimchi-Sarfaty, C.; Ambudkar, S.V.; Gottesman, M.M. The sounds of silence: Synonymous mutations affect function. Pharmacogenomics 2007, 8, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Sauna, Z.E.; Kimchi-Sarfaty, C. Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 2011, 12, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Fahraeus, R.; Marin, M.; Olivares-Illana, V. Whisper mutations: Cryptic messages within the genetic code. Oncogene 2015. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Amode, M.R.; Barrell, D.; Beal, K.; Billis, K.; Brent, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; Fitzgerald, S.; et al. Ensembl 2015. Nucleic Acids Res. 2015, 43, D662–D669. [Google Scholar] [CrossRef] [PubMed]
- Gennari, L.; Merlotti, D.; De Paola, V.; Calabro, A.; Becherini, L.; Martini, G.; Nuti, R. Estrogen receptor gene polymorphisms and the genetics of osteoporosis: A huge review. Am. J. Epidemiol. 2005, 161, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Araújo, K.L.; Rezende, L.C.D.d.; Souza, L.S.; Daltoé, R.D.; Madeira, K.P.; Paes, M.F.; Herkenhoff, F.L.; Rangel, L.B.A.; Silva, I.V. Prevalence of estrogen receptor alpha pvuii (c454–397t > c) and xbai (c454a > g) polymorphisms in a population of brazilian women. Braz. Arch. Biol. Technol. 2011, 54, 1151–1158. [Google Scholar]
- Mill, J.; Kiss, E.; Baji, I.; Kapornai, K.; Daroczy, G.; Vetro, A.; Kennedy, J.; Kovacs, M.; Barr, C. Association study of the estrogen receptor alpha gene (esr1) and childhood-onset mood disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008, 147, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Calero, T.; Astrada, S.; Alberti, A.; Horjales, S.; Arnal, J.F.; Rovira, C.; Bollati-Fogolin, M.; Flouriot, G.; Marin, M. The transcriptional activities and cellular localization of the human estrogen receptor alpha are affected by the synonymous ala87 mutation. J. Steroid Biochem. Mol. Biol. 2014, 143, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.P.; Bhaskar, L.; Thampan, R.V. Structural characterization of the goat uterine estrogen receptor activation factor using an endogenous calcium activated neutral protease. Mol. Cell. Endocrinol. 1999, 152, 57–64. [Google Scholar] [CrossRef]
- Mader, S.; Chambon, P.; White, J.H. Defining a minimal estrogen receptor DNA binding domain. Nucleic Acids Res. 1993, 21, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.; Makela, S.; Treuter, E.; Tujague, M.; Thomsen, J.; Andersson, G.; Enmark, E.; Pettersson, K.; Warner, M.; Gustafsson, J.A. Mechanisms of estrogen action. Physiol. Rev. 2001, 81, 1535–1565. [Google Scholar] [PubMed]
- Saville, B.; Wormke, M.; Wang, F.; Nguyen, T.; Enmark, E.; Kuiper, G.; Gustafsson, J.A.; Safe, S. Ligand-, cell-, and estrogen receptor subtype (α/β)-dependent activation at gc-rich (sp1) promoter elements. J. Biol. Chem. 2000, 275, 5379–5387. [Google Scholar] [CrossRef] [PubMed]
- Jakacka, M.; Ito, M.; Weiss, J.; Chien, P.Y.; Gehm, B.D.; Jameson, J.L. Estrogen receptor binding to DNA is not required for its activity through the nonclassical ap1 pathway. J. Biol. Chem. 2001, 276, 13615–13621. [Google Scholar] [PubMed]
- Kushner, P.J.; Agard, D.A.; Greene, G.L.; Scanlan, T.S.; Shiau, A.K.; Uht, R.M.; Webb, P. Estrogen receptor pathways to ap-1. J. Steroid Biochem. Mol. Biol. 2000, 74, 311–317. [Google Scholar] [CrossRef]
- Safe, S.; Kim, K. Non-classical genomic estrogen receptor (er)/specificity protein and er/activating protein-1 signaling pathways. J. Mol. Endocrinol. 2008, 41, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Bjornstrom, L.; Sjoberg, M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol. Endocrinol. 2005, 19, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R. Cell localization, physiology, and nongenomic actions of estrogen receptors. J. Appl. Physiol. 2001, 91, 1860–1867. [Google Scholar] [PubMed]
- Merot, Y.; Metivier, R.; Penot, G.; Manu, D.; Saligaut, C.; Gannon, F.; Pakdel, F.; Kah, O.; Flouriot, G. The relative contribution exerted by af-1 and af-2 transactivation functions in estrogen receptor alpha transcriptional activity depends upon the differentiation stage of the cell. J. Biol. Chem. 2004, 279, 26184–26191. [Google Scholar] [CrossRef] [PubMed]
- Webb, P.; Lopez, G.N.; Uht, R.M.; Kushner, P.J. Tamoxifen activation of the estrogen receptor/ap-1 pathway: Potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol. Endocrinol. 1995, 9, 443–456. [Google Scholar] [PubMed]
- Lewis, J.S.; Jordan, V.C. Selective estrogen receptor modulators (serms): Mechanisms of anticarcinogenesis and drug resistance. Mutat. Res. 2005, 591, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.; Metzger, D.; Chambon, P. Role of the two activating domains of the oestrogen receptor in the cell-type and promoter-context dependent agonistic activity of the anti-oestrogen 4-hydroxytamoxifen. EMBO J. 1990, 9, 2811–2818. [Google Scholar] [PubMed]
- Fan, J.D.; Wagner, B.L.; McDonnell, D.P. Identification of the sequences within the human complement 3 promoter required for estrogen responsiveness provides insight into the mechanism of tamoxifen mixed agonist activity. Mol. Endocrinol. 1996, 10, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Sabarinathan, R.; Tafer, H.; Seemann, S.E.; Hofacker, I.L.; Stadler, P.F.; Gorodkin, J. The rnasnp web server: Predicting snp effects on local RNA secondary structure. Nucleic Acids Res. 2013, 41, W475–W479. [Google Scholar] [CrossRef] [PubMed]
- Sabarinathan, R.; Tafer, H.; Seemann, S.E.; Hofacker, I.L.; Stadler, P.F.; Gorodkin, J. Rnasnp: Efficient detection of local RNA secondary structure changes induced by snps. Hum. Mutat. 2013, 34, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.H.; Huang, H.Y.; Hsu, J.B.; Weng, S.L.; Horng, J.T.; Huang, H.D. An enhanced computational platform for investigating the roles of regulatory RNA and for identifying functional RNA motifs. BMC Bioinform. 2013, 14. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. Gtrnadb: A database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009, 37, D93–D97. [Google Scholar] [CrossRef] [PubMed]
- Murphy, F.V.T.; Ramakrishnan, V. Structure of a purine-purine wobble base pair in the decoding center of the ribosome. Nat. Struct. Mol. Biol. 2004, 11, 1251–1252. [Google Scholar] [CrossRef] [PubMed]
- Czerwoniec, A.; Dunin-Horkawicz, S.; Purta, E.; Kaminska, K.H.; Kasprzak, J.M.; Bujnicki, J.M.; Grosjean, H.; Rother, K. Modomics: A database of RNA modification pathways. 2008 update. Nucleic Acids Res. 2009, 37, D118–D121. [Google Scholar] [CrossRef] [PubMed]
- Dunin-Horkawicz, S.; Czerwoniec, A.; Gajda, M.J.; Feder, M.; Grosjean, H.; Bujnicki, J.M. Modomics: A database of RNA modification pathways. Nucleic Acids Res. 2006, 34, D145–D149. [Google Scholar] [CrossRef] [PubMed]
- Machnicka, M.A.; Milanowska, K.; Osman Oglou, O.; Purta, E.; Kurkowska, M.; Olchowik, A.; Januszewski, W.; Kalinowski, S.; Dunin-Horkawicz, S.; Rother, K.M.; et al. Modomics: A database of RNA modification pathways—2013 update. Nucleic Acids Res. 2013, 41, D262–D267. [Google Scholar] [CrossRef] [PubMed]
- Cochella, L.; Green, R. Wobble during decoding: More than third-position promiscuity. Nat. Struct. Mol. Biol. 2004, 11, 1160–1162. [Google Scholar] [CrossRef] [PubMed]
- Agris, P.F. Bringing order to translation: The contributions of transfer RNA anticodon-domain modifications. EMBO Rep. 2008, 9, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Ciryam, P.; Morimoto, R.I.; Vendruscolo, M.; Dobson, C.M.; O’Brien, E.P. In vivo translation rates can substantially delay the cotranslational folding of the Escherichia coli cytosolic proteome. Proc. Natl. Acad. Sci. USA 2013, 110, E132–E140. [Google Scholar] [CrossRef] [PubMed]
- Komar, A.A. A pause for thought along the co-translational folding pathway. Trends Biochem. Sci. 2009, 34, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Novoa, E.M.; Ribas de Pouplana, L. Speeding with control: Codon usage, tRNAs, and ribosomes. Trends Genet. 2012, 28, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Endres, L.; Dedon, P.C.; Begley, T.J. Codon-biased translation can be regulated by wobble-base tRNA modification systems during cellular stress responses. RNA Biol. 2015, 12, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Ikemura, T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985, 2, 13–34. [Google Scholar] [PubMed]
- Dong, H.; Nilsson, L.; Kurland, C.G. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol. 1996, 260, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Elena, C.; Ravasi, P.; Castelli, M.E.; Peiru, S.; Menzella, H.G. Expression of codon optimized genes in microbial systems: Current industrial applications and perspectives. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.L.; Ceccarelli, E.A. Rare codon content affects the solubility of recombinant proteins in a codon bias-adjusted Escherichia coli strain. Microb. Cell. Fact. 2009, 8. [Google Scholar] [CrossRef] [PubMed]
- Cortazzo, P.; Cervenansky, C.; Marin, M.; Reiss, C.; Ehrlich, R.; Deana, A. Silent mutations affect in vivo protein folding in Escherichia coli. Biochem. Biophys. Res. Commun. 2002, 293, 537–541. [Google Scholar] [CrossRef]
- Hess, A.K.; Saffert, P.; Liebeton, K.; Ignatova, Z. Optimization of translation profiles enhances protein expression and solubility. PLoS ONE 2015, 10, e0127039. [Google Scholar] [CrossRef] [PubMed]
- Thanaraj, T.A.; Argos, P. Protein secondary structural types are differentially coded on messenger RNA. Protein Sci. 1996, 5, 1973–1983. [Google Scholar] [CrossRef] [PubMed]
- Thanaraj, T.A.; Argos, P. Ribosome-mediated translational pause and protein domain organization. Protein Sci. 1996, 5, 1594–1612. [Google Scholar] [CrossRef] [PubMed]
- Zalucki, Y.M.; Beacham, I.R.; Jennings, M.P. Coupling between codon usage, translation and protein export in Escherichia coli. Biotechnol. J. 2011, 6, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Hubalewska, M.; Ignatova, Z. Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat. Struct. Mol. Biol. 2009, 16, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Fedyunin, I.; Lehnhardt, L.; Bohmer, N.; Kaufmann, P.; Zhang, G.; Ignatova, Z. TRNA concentration fine tunes protein solubility. FEBS Lett. 2012, 586, 3336–3340. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.P.; Ciryam, P.; Vendruscolo, M.; Dobson, C.M. Understanding the influence of codon translation rates on cotranslational protein folding. Acc. Chem. Res. 2014, 47, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.P.; Vendruscolo, M.; Dobson, C.M. Kinetic modelling indicates that fast-translating codons can coordinate cotranslational protein folding by avoiding misfolded intermediates. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Wang, T.; Fu, J.; Xiao, G.; Liu, Y. Nonoptimal codon usage influences protein structure in intrinsically disordered regions. Mol. Microbiol. 2015, 97, 974–987. [Google Scholar] [CrossRef] [PubMed]
- Kimchi-Sarfaty, C.; Oh, J.M.; Kim, I.W.; Sauna, Z.E.; Calcagno, A.M.; Ambudkar, S.V.; Gottesman, M.M. A “Silent” Polymorphism in the mdr1 gene changes substrate specificity. Science 2007, 315, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Komar, A.A. Silent snps: Impact on gene function and phenotype. Pharmacogenomics 2007, 8, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T. Ribosome profiling: New views of translation, from single codons to genome scale. Nat. Rev. Genet. 2014, 15, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Gartner, J.J.; Parker, S.C.; Prickett, T.D.; Dutton-Regester, K.; Stitzel, M.L.; Lin, J.C.; Davis, S.; Simhadri, V.L.; Jha, S.; Katagiri, N.; et al. Whole-genome sequencing identifies a recurrent functional synonymous mutation in melanoma. Proc. Natl. Acad. Sci. USA 2013, 110, 13481–13486. [Google Scholar] [CrossRef] [PubMed]
- Dana, A.; Tuller, T. The effect of tRNA levels on decoding times of mRNA codons. Nucleic Acids Res. 2014, 42, 9171–9181. [Google Scholar] [CrossRef] [PubMed]
- Gardin, J.; Yeasmin, R.; Yurovsky, A.; Cai, Y.; Skiena, S.; Futcher, B. Measurement of average decoding rates of the 61 sense codons in vivo. Elife 2014, 3, e03735. [Google Scholar] [CrossRef] [PubMed]
- Charneski, C.A.; Hurst, L.D. Positively charged residues are the major determinants of ribosomal velocity. PLoS Biol. 2013, 11, e1001508. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Yang, J.R.; Pearson, N.M.; Maclean, C.; Zhang, J. Balanced codon usage optimizes eukaryotic translational efficiency. PLoS Genet. 2012, 8, e1002603. [Google Scholar] [CrossRef] [PubMed]
- Pop, C.; Rouskin, S.; Ingolia, N.T.; Han, L.; Phizicky, E.M.; Weissman, J.S.; Koller, D. Causal signals between codon bias, mRNA structure, and the efficiency of translation and elongation. Mol. Syst. Biol. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Gorochowski, T.E.; Ignatova, Z.; Bovenberg, R.A.; Roubos, J.A. Trade-offs between tRNA abundance and mRNA secondary structure support smoothing of translation elongation rate. Nucleic Acids Res. 2015, 43, 3022–3032. [Google Scholar] [CrossRef] [PubMed]
- Quax, T.E.; Claassens, N.J.; Soll, D.; van der Oost, J. Codon bias as a means to fine-tune gene expression. Mol. Cell. 2015, 59, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Minana, B.; Valcarcel, J.; Gabaldon, T.; Lehner, B. Synonymous mutations frequently act as driver mutations in human cancers. Cell 2014, 156, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Garel, J.P.; Hentzen, D.; Schlegel, M.; Dirheimer, G. Structural studies on RNA from Bombyx mori L. I. Nucleoside composition of enriched tRNA species from the posterior silkgland purified by coutercurrent distribution. Biochimie 1976, 58, 1089–1100. [Google Scholar] [CrossRef]
- Hentzen, D.; Chevallier, A.; Garel, J.P. Differential usage of iso-accepting trnaser species in silk glands of bombyx mori. Nature 1981, 290, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Sprague, K.U.; Hagenbuchle, O.; Zuniga, M.C. The nucleotide sequence of two silk gland alanine tRNAs: Implications for fibroin synthesis and for initiator tRNA structure. Cell 1977, 11, 561–570. [Google Scholar] [CrossRef]
- Kurland, C.G. Codon bias and gene expression. FEBS Lett. 1991, 285, 165–169. [Google Scholar] [CrossRef]
- Pang, Y.L.; Abo, R.; Levine, S.S.; Dedon, P.C. Diverse cell stresses induce unique patterns of tRNA up- and down-regulation: tRNA-seq for quantifying changes in tRNA copy number. Nucleic Acids Res. 2014, 42, e170. [Google Scholar] [CrossRef] [PubMed]
- Gingold, H.; Tehler, D.; Christoffersen, N.R.; Nielsen, M.M.; Asmar, F.; Kooistra, S.M.; Christophersen, N.S.; Christensen, L.L.; Borre, M.; Sorensen, K.D.; et al. A dual program for translation regulation in cellular proliferation and differentiation. Cell 2014, 158, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, K.A.; Goodenbour, J.M.; Pan, T. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006, 2, e221. [Google Scholar] [CrossRef] [PubMed]
- Pavon-Eternod, M.; Gomes, S.; Geslain, R.; Dai, Q.; Rosner, M.R.; Pan, T. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 2009, 37, 7268–7280. [Google Scholar] [CrossRef] [PubMed]
- Topisirovic, I.; Sonenberg, N. Distinctive tRNA repertoires in proliferating versus differentiating cells. Cell 2014, 158, 1238–1239. [Google Scholar] [CrossRef] [PubMed]
- Grosjean, H.; de Crecy-Lagard, V.; Marck, C. Deciphering synonymous codons in the three domains of life: Co-evolution with specific tRNA modification enzymes. FEBS Lett. 2010, 584, 252–264. [Google Scholar] [CrossRef] [PubMed]
- El Yacoubi, B.; Bailly, M.; de Crecy-Lagard, V. Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 2012, 46, 69–95. [Google Scholar] [CrossRef] [PubMed]
- Machnicka, M.A.; Olchowik, A.; Grosjean, H.; Bujnicki, J.M. Distribution and frequencies of post-transcriptional modifications in tRNAs. RNA Biol. 2014, 11, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Helm, M.; Alfonzo, J.D. Posttranscriptional RNA modifications: Playing metabolic games in a cell’s chemical legoland. Chem. Biol. 2014, 21, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Jackman, J.E.; Alfonzo, J.D. Transfer RNA modifications: Nature’s combinatorial chemistry playground. Wiley Interdiscip. Rev. RNA 2013, 4, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Gustilo, E.M.; Vendeix, F.A.; Agris, P.F. tRNA’s modifications bring order to gene expression. Curr. Opin. Microbiol. 2008, 11, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Phizicky, E.M.; Alfonzo, J.D. Do all modifications benefit all tRNAs? FEBS Lett. 2010, 584, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Durdevic, Z.; Schaefer, M. tRNA modifications: Necessary for correct tRNA-derived fragments during the recovery from stress? Bioessays 2013, 35, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.G.; Batlle, E.; Ribas de Pouplana, L. Role of tRNA modifications in human diseases. Trends Mol. Med. 2014, 20, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Sarin, L.P.; Leidel, S.A. Modify or die?—RNA modification defects in metazoans. RNA Biol. 2014, 11, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Gehrig, S.; Eberle, M.E.; Botschen, F.; Rimbach, K.; Eberle, F.; Eigenbrod, T.; Kaiser, S.; Holmes, W.M.; Erdmann, V.A.; Sprinzl, M.; et al. Identification of modifications in microbial, native tRNA that suppress immunostimulatory activity. J. Exp. Med. 2012, 209, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Jockel, S.; Nees, G.; Sommer, R.; Zhao, Y.; Cherkasov, D.; Hori, H.; Ehm, G.; Schnare, M.; Nain, M.; Kaufmann, A.; et al. The 2'-o-methylation status of a single guanosine controls transfer RNA-mediated toll-like receptor 7 activation or inhibition. J. Exp. Med. 2012, 209, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Nallagatla, S.R.; Jones, C.N.; Ghosh, S.K.; Sharma, S.D.; Cameron, C.E.; Spremulli, L.L.; Bevilacqua, P.C. Native tertiary structure and nucleoside modifications suppress tRNA’s intrinsic ability to activate the innate immune sensor pkr. PLoS ONE 2013, 8, e57905. [Google Scholar]
- Chan, C.T.; Dyavaiah, M.; DeMott, M.S.; Taghizadeh, K.; Dedon, P.C.; Begley, T.J. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 2010, 6, e1001247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, C.; Begley, T.J.; Dedon, P.C. tRNA modifications regulate translation during cellular stress. FEBS Lett. 2014, 588, 4287–4296. [Google Scholar] [CrossRef] [PubMed]
- Hopper, A.K.; Pai, D.A.; Engelke, D.R. Cellular dynamics of tRNAs and their genes. FEBS Lett. 2010, 584, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, S.; Ignatova, Z. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat. Rev. Genet. 2015, 16, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Laporte, D.; Huot, J.L.; Bader, G.; Enkler, L.; Senger, B.; Becker, H.D. Exploring the evolutionary diversity and assembly modes of multi-aminoacyl-tRNA synthetase complexes: Lessons from unicellular organisms. FEBS Lett. 2014, 588, 4268–4278. [Google Scholar] [CrossRef] [PubMed]
- Park, S.G.; Ewalt, K.L.; Kim, S. Functional expansion of aminoacyl-tRNA synthetases and their interacting factors: New perspectives on housekeepers. Trends Biochem. Sci. 2005, 30, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Raina, M.; Elgamal, S.; Santangelo, T.J.; Ibba, M. Association of a multi-synthetase complex with translating ribosomes in the archaeon thermococcus kodakarensis. FEBS Lett. 2012, 586, 2232–2238. [Google Scholar] [CrossRef] [PubMed]
- Havrylenko, S.; Mirande, M. Aminoacyl-tRNA synthetase complexes in evolution. Int. J. Mol. Sci. 2015, 16, 6571–6594. [Google Scholar] [CrossRef] [PubMed]
- Cannarozzi, G.; Schraudolph, N.N.; Faty, M.; von Rohr, P.; Friberg, M.T.; Roth, A.C.; Gonnet, P.; Gonnet, G.; Barral, Y. A role for codon order in translation dynamics. Cell 2010, 141, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Hussmann, J.A.; Press, W.H. Local correlations in codon preferences do not support a model of tRNA recycling. Cell. Rep. 2014, 8, 1624–1629. [Google Scholar] [CrossRef] [PubMed]
- Hussmann, J.A.; Patchett, S.; Johnson, A.; Sawyer, S.; Press, W.H. Understanding biases in ribosome profiling experiments reveals signatures of translation dynamics in yeast. PLoS Genet. 2015, 11, e1005732. [Google Scholar] [CrossRef] [PubMed]
- Nedialkova, D.D.; Leidel, S.A. Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 2015, 161, 1606–1618. [Google Scholar] [CrossRef] [PubMed]
- Yona, A.H.; Bloom-Ackermann, Z.; Frumkin, I.; Hanson-Smith, V.; Charpak-Amikam, Y.; Feng, Q.; Boeke, J.D.; Dahan, O.; Pilpel, Y. tRNA genes rapidly change in evolution to meet novel translational demands. Elife 2013, 2, e01339. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, D.P.; Connor, C.E.; Wijayaratne, A.; Chang, C.Y.; Norris, J.D. Definition of the molecular and cellular mechanisms underlying the tissue-selective agonist/antagonist activities of selective estrogen receptor modulators. Recent Prog. Horm. Res. 2002, 57, 295–316. [Google Scholar] [CrossRef] [PubMed]
- Horjales, S.; Cota, G.; Senorale-Pose, M.; Rovira, C.; Roman, E.; Artagaveytia, N.; Ehrlich, R.; Marin, M. Translational machinery and protein folding: Evidence of conformational variants of the estrogen receptor alpha. Arch. Biochem. Biophys. 2007, 467, 139–143. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| a. Alanine tRNA Genes in Human Genome According to the Genomic tRNA Database [29] | ||||
| Anticodon (5′ → 3′) | Corresponding Codon (5’ → 3’) | nº of Genes | Genome Codon Usage | |
| AGC | GCT | 30 | 1.84 | |
| GGC | GCC | 1 | 2.77 | |
| CGC | GCG | 5 | 0.74 | |
| UGC | GCA | 10 | 1.58 | |
| b. Wobble Pair Rules Reviewed in [30] | ||||
| tRNA 5’ Anticodon Base | mRNA 3’ Codon Base | ER Ala 87 Polymorphism Recognized by tRNA | ||
| G | U,C | Ala87 | ||
| C | G | wt | ||
| k2C | A | --- | ||
| A | U,C,G>A | wt, Ala87 | ||
| U | U,A,G>C | wt, Ala87 ↓ | ||
| xm5s2U,xm5Um,Um,xm5U | A>G | wt ↓ | ||
| xo5U | U,A,G | wt | ||
| I | A,C,U | Ala87 | ||
| c. Analysis of ERWT and ERAla87 Codon-Anticodon Recognition by Alanine tRNAs Restricted to tRNA Ala Sequences Described in the Literature According to Modomics [31,32,33]. | ||||
| tRNA-AGC: | -without modifications recognizes: wt *, Ala87 * | |||
| -modified A → I recognizes: Ala87 | ||||
| tRNA-CGC: | -without modifications recognizes: wt * | |||
| tRNA-UGC: | -without modifications recognizes: wt, Ala87 ↓ | |||
| -with modifications recognizes: wt | ||||
| tRNA-UGC: | -without modifications recognizes: Ala87 | |||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Calero, T.; Cabrera-Cabrera, F.; Ehrlich, R.; Marín, M. Silent Polymorphisms: Can the tRNA Population Explain Changes in Protein Properties? Life 2016, 6, 9. https://doi.org/10.3390/life6010009
Fernández-Calero T, Cabrera-Cabrera F, Ehrlich R, Marín M. Silent Polymorphisms: Can the tRNA Population Explain Changes in Protein Properties? Life. 2016; 6(1):9. https://doi.org/10.3390/life6010009
Chicago/Turabian StyleFernández-Calero, Tamara, Florencia Cabrera-Cabrera, Ricardo Ehrlich, and Mónica Marín. 2016. "Silent Polymorphisms: Can the tRNA Population Explain Changes in Protein Properties?" Life 6, no. 1: 9. https://doi.org/10.3390/life6010009
APA StyleFernández-Calero, T., Cabrera-Cabrera, F., Ehrlich, R., & Marín, M. (2016). Silent Polymorphisms: Can the tRNA Population Explain Changes in Protein Properties? Life, 6(1), 9. https://doi.org/10.3390/life6010009