
Gene-Family Extension Measures and Correlations



Abstract
:
1. Introduction
2. Methods
2.1. COGs Database and Input for Ranking
2.2. Kemeny Rank Aggregation Approach
2.3. Ranking Methods
2.3.1. Average Ranking Method
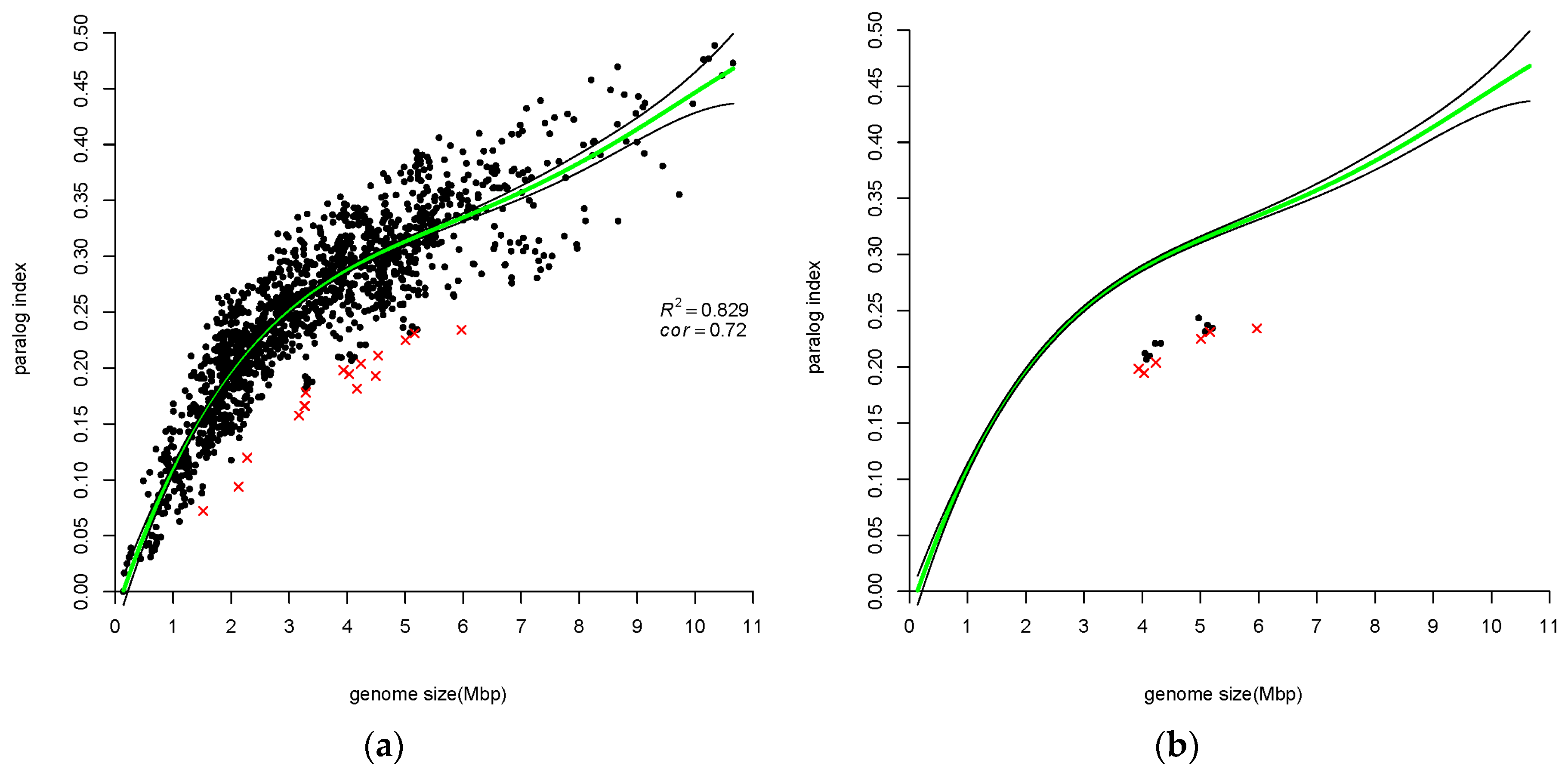
2.3.2. Paralog Index
2.3.3. Index of Multi-Paralogous Families
2.3.4. Sort Ranking
2.4. Regression Analysis and Outlier Detection
2.5. Correlation between GFE Measures
3. Results
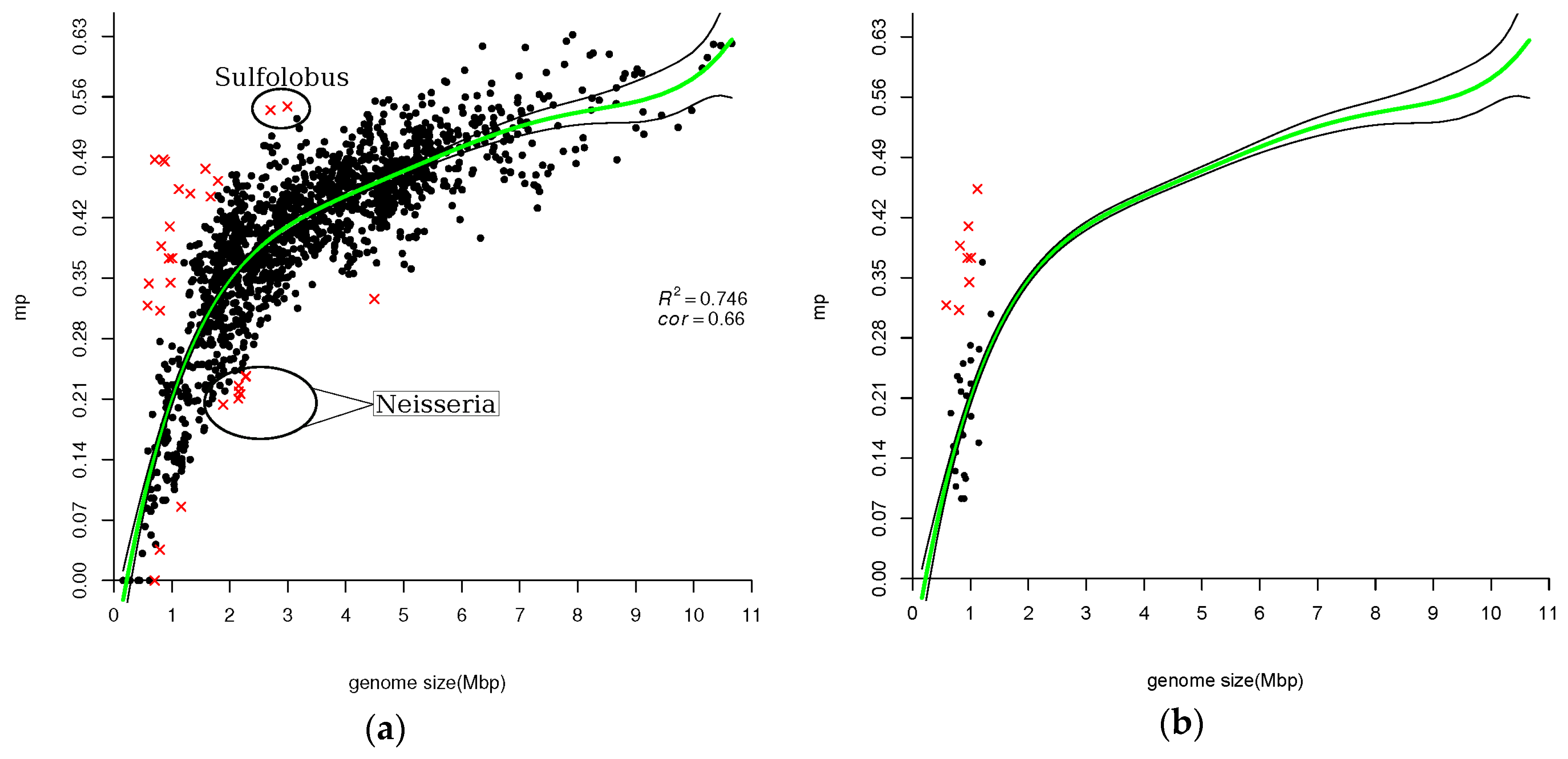
3.1. Number of Paralogs is Correlated with Genome Size
3.1.1. Percentage of Paralogous Gene Families is Correlated with Genome Size
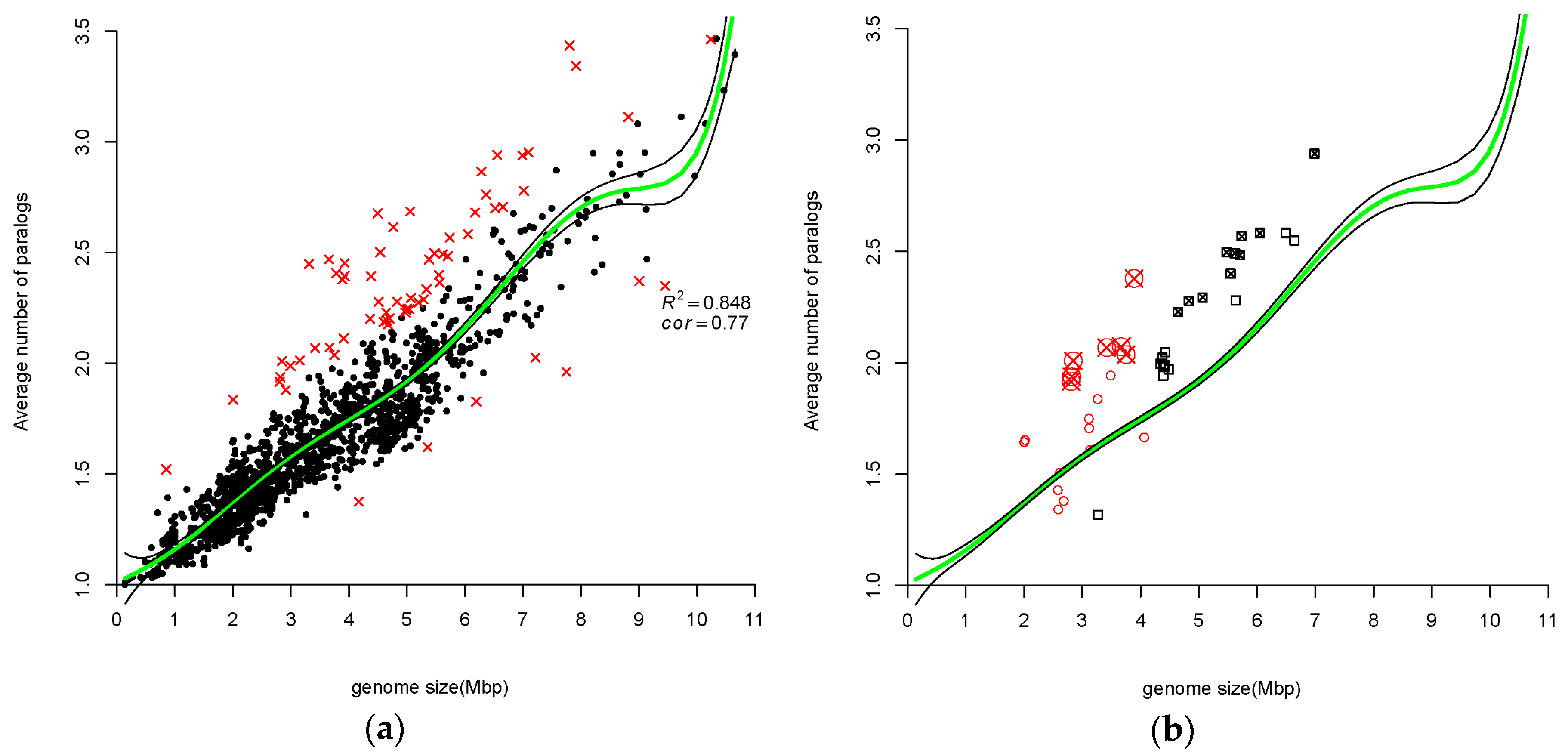
3.1.2. Average Number of Paralogs Correlate with Genome Size
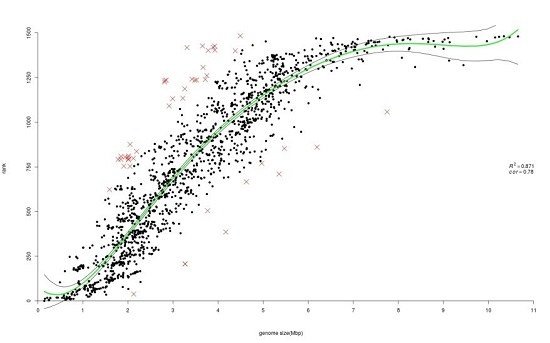
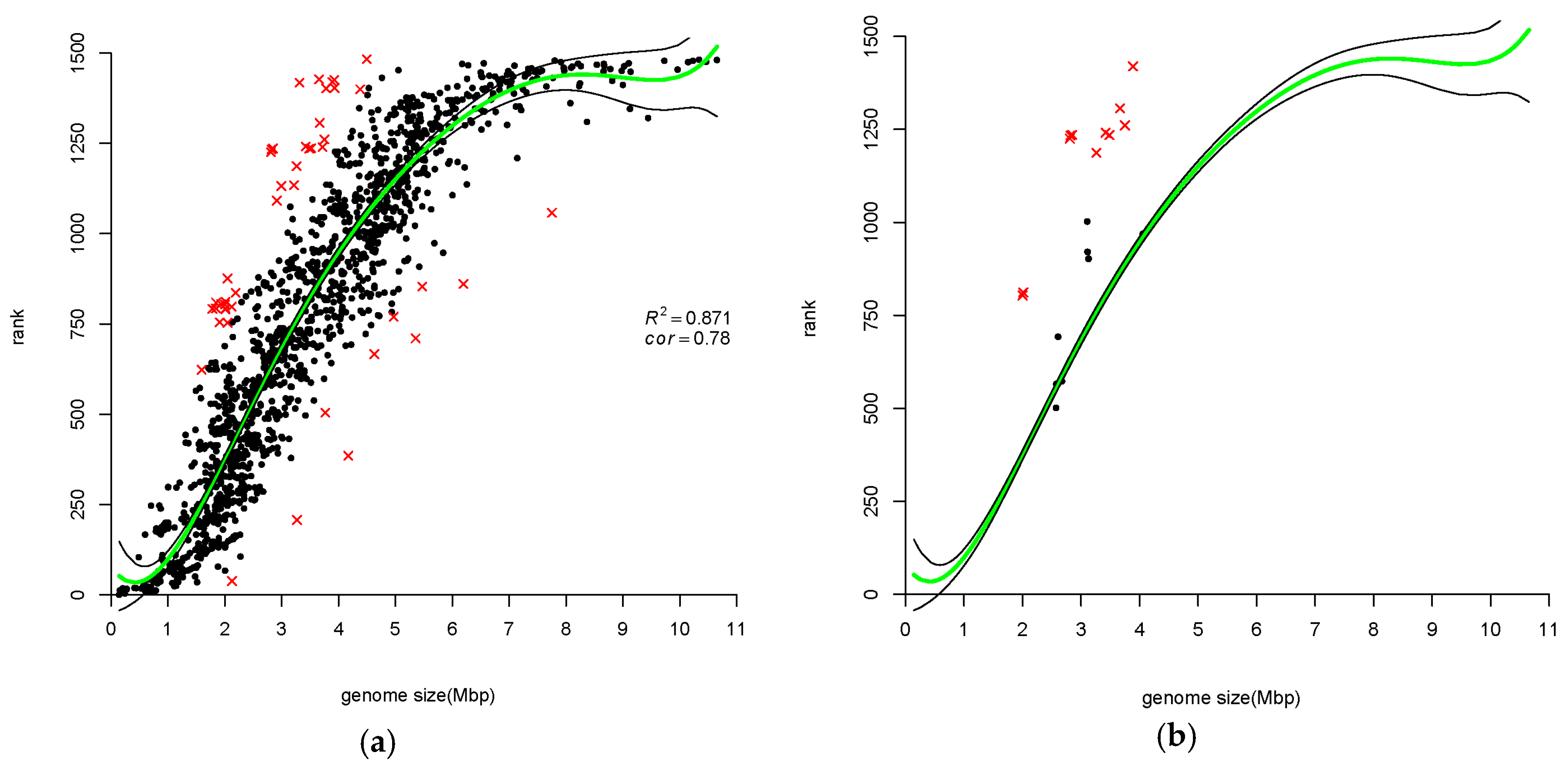
3.1.3. Ranking of Prokaryotic Genomes Based on Gene-Family Size Confirms Correlation with Genome Size
3.2. Fraction of Larger Gene-Families
4. Discussion
4.1. Number of Gene Copies Is Correlated with Genome Size
4.2. Atypical Genomes
4.2.1. Mycoplasmas
4.2.2. Mycobacterium
4.2.3. Halophiles
4.2.4. Pirellula
4.2.5. Orientia tsutsugamushi
4.3. Ranking Methods
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| COG | Cluster of Orthologous Groups of proteins |
| HGT | Horizontal Gene Transfer |
| GFE | gene-family extension |
| Mbp | millions of base pairs |
| OT | Orientia tsutsugamushi |
| p.i. | the number of protein-coding gene families having more than one copy divided by the total number of COG-annotated protein-coding gene families |
| ave | average size of protein-coding gene families, including singletons |
| mp | the number of protein-coding gene families having more than two copies divided by the number of protein-coding gene families having more than one copy |
References
- Brenner, S.E.; Hubbard, T.; Murzin, A.; Chothia, C. Gene duplications in H. Influenzae. Nature 1995, 378, 140. [Google Scholar] [CrossRef] [PubMed]
- Huynen, M.A.; van Nimwegen, E. The frequency distribution of gene family sizes in complete genomes. Mol. Biol. Evol. 1998, 15, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Jordan, I.K.; Makarova, K.S.; Spouge, J.L.; Wolf, Y.I.; Koonin, E.V. Lineage-specific gene expansions in bacterial and archaeal genomes. Genome Res. 2001, 11, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Pushker, R.; Mira, A.; Rodriguez-Valera, F. Comparative genomics of gene-family size in closely related bacteria. Genome Biol. 2004, 5, R27. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.A. Orthologs and paralogs—We need to get it right. Genome Biol. 2001, 2, interactions1002.1001–interactions1002.1003. [Google Scholar] [CrossRef] [PubMed]
- Fitch, W.M. Distinguishing homologous from analogous proteins. Syst. Zool. 1970, 19, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A genomic perspective on protein families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The cog database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Natale, D.A.; Garkavtsev, I.V.; Tatusova, T.A.; Shankavaram, U.T.; Rao, B.S.; Kiryutin, B.; Galperin, M.Y.; Fedorova, N.D.; Koonin, E.V. The cog database: New developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001, 29, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The cog database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Hooper, S.D.; Berg, O.G. Duplication is more common among laterally transferred genes than among indigenous genes. Genome Biol. 2003, 4, R48. [Google Scholar] [CrossRef] [PubMed]
- Snel, B.; Bork, P.; Huynen, M. Genome evolution. Gene fusion versus gene fission. TIG 2000, 16, 9–11. [Google Scholar] [CrossRef]
- Snel, B.; Bork, P.; Huynen, M.A. The identification of functional modules from the genomic association of genes. Proc. Natl. Acad. Sci. USA 2002, 99, 5890–5895. [Google Scholar] [CrossRef] [PubMed]
- Kunin, V.; Ouzounis, C.A. The balance of driving forces during genome evolution in prokaryotes. Genome Res. 2003, 13, 1589–1594. [Google Scholar] [CrossRef] [PubMed]
- Yanai, I.; Camacho, C.J.; DeLisi, C. Predictions of gene family distributions in microbial genomes: Evolution by gene duplication and modification. Phys. Rev. Lett. 2000, 85. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; Kunin, V.; Ouzounis, C.A. Protein families and tribes in genome sequence space. Nucleic Acids Res. 2003, 31, 4632–4638. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Vandepoele, K.; Simillon, C.; Van de Peer, Y. Gene duplication and biased functional retention of paralogs in bacterial genomes. Trends Microbiol. 2004, 12, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Hooper, S.D.; Berg, O.G. On the nature of gene innovation: Duplication patterns in microbial genomes. Mol. Biol. Evol. 2003, 20, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Bolshoy, A.; Tatarinova, T. Methods of combinatorial optimization to reveal factors affecting gene length. Bioinform. Biol. Insights 2012, 6, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Kemeny, J.G. Mathematics without numbers. Daedalus 1959, 88, 571. [Google Scholar]
- Kemeny, J.G.; Snell, J.L. Mathematical Models in the Social Sciences; The MIT Press: Cambridge, UK, 1972. [Google Scholar]
- Young, H.P.; Levenglick, A. A consistent extension of condorcet’s election principle. SIAM J. Appl. Math. 1978, 35, 285–300. [Google Scholar] [CrossRef]
- Bartholdi, I.; Tovey, C.A.; Trick, M.A. Voting schemes for which it can be difficult to tell who won the election. Soc. Choice Welf. 1989, 6, 157–165. [Google Scholar] [CrossRef]
- Dwork, C.; Kumar, R.; Naor, M.; Sivakumar, D. Rank aggregation methods for the web. In Proceedings of the 10th International Conference on World Wide Web (WWW01), Hong Kong, China, 1–5 May 2001; p. 613.
- Tatarinova, T.; Salih, B.; Dien Bard, J.; Cohen, I.; Bolshoy, A. Lengths of orthologous prokaryotic proteins are affected by evolutionary factors. BioMed Res. Int. 2015, 2015, 786861. [Google Scholar] [CrossRef] [PubMed]
- Zucchini, W. An introduction to model selection. J. Math. Psychol. 2000, 44, 41–61. [Google Scholar] [CrossRef] [PubMed]
- Tukey, J.W. Exploratory Data Analysis; Addison-Wesley: Boston, MA, USA, 1977. [Google Scholar]
- DeCoursey, W.J. Statistics and Probability for Engineering Applications with Microsoft Excel; Newnes: Burlington, ON, Canada, 2003. [Google Scholar]
- R Core Team. R: A language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Kim, S. Ppcor: An R package for a fast calculation to semi-partial correlation coefficients. Commun. Stat. Appl. Methods 2015, 22, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Bolshoy, A.; Salih, B.; Cohen, I.; Tatarinova, T. Ranking of prokaryotic genomes based on maximization of sortedness of gene lengths. J. Data Min. Genom. Proteom. 2014, 5, 151. [Google Scholar] [CrossRef]
- Singh, P.; Cole, S.T. Mycobacterium leprae: Genes, pseudogenes and genetic diversity. Future Microbiol. 2011, 6, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Benjak, A.; Schuenemann, V.J.; Herbig, A.; Avanzi, C.; Busso, P.; Nieselt, K.; Krause, J.; Vera-Cabrera, L.; Cole, S.T. Insight into the evolution and origin of leprosy bacilli from the genome sequence of Mycobacterium lepromatosis. Proc. Natl. Acad. Sci. USA 2015, 112, 4459–4464. [Google Scholar] [CrossRef] [PubMed]
- Akinola, R.O.; Mazandu, G.K.; Mulder, N.J. A quantitative approach to analyzing genome reductive evolution using protein-protein interaction networks: A case study of Mycobacterium leprae. Front. Genet. 2016, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- McGuire, A.M.; Weiner, B.; Park, S.T.; Wapinski, I.; Raman, S.; Dolganov, G.; Peterson, M.; Riley, R.; Zucker, J.; Abeel, T.; et al. Comparative analysis of mycobacterium and related actinomycetes yields insight into the evolution of Mycobacterium tuberculosis pathogenesis. BMC Genom. 2012, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Perez, G.; Mira, A.; Nyiro, G.; Pasic, L.; Rodriguez-Valera, F. Adapting to environmental changes using specialized paralogs. TIG 2008, 24, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Gloeckner, F.O.; Kube, M.; Bauer, M.; Teeling, H.; Lombardot, T.; Ludwig, W.; Gade, D.; Beck, A.; Borzym, K.; Heitmann, K.; et al. Complete genome sequence of the marine Planctomycete pirellula sp. Strain 1. Proc. Natl. Acad. Sci. USA 2003, 100, 8298–8303. [Google Scholar] [CrossRef] [PubMed]
- Bratlie, M.S.; Johansen, J.; Sherman, B.T.; Huang da, W.; Lempicki, R.A.; Drablos, F. Gene duplications in prokaryotes can be associated with environmental adaptation. BMC Genom. 2010, 11, 588. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.H.; Kim, H.R.; Lee, J.H.; Kim, S.Y.; Kim, J.; Cha, S.; Kim, S.Y.; Darby, A.C.; Fuxelius, H.H.; Yin, J.; et al. The Orientia tsutsugamushi genome reveals massive proliferation of conjugative type IV secretion system and host-cell interaction genes. Proc. Natl. Acad. Sci. USA 2007, 104, 7981–7986. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Yamashita, A.; Kurokawa, K.; Morimoto, T.; Ogawa, M.; FUKuhara, M.; Urakami, H.; Ohnishi, M.; Uchiyama, I.; Ogura, Y.; et al. The whole-genome sequencing of the obligate intracellular bacterium Orientia tsutsugamushi revealed massive gene amplification during reductive genome evolution. DNA Res. 2008, 15, 185–199. [Google Scholar] [PubMed]
- Nakayama, K.; Kurokawa, K.; Fukuhara, M.; Urakami, H.; Yamamoto, S.; Yamazaki, K.; Ogura, Y.; Ooka, T.; Hayashi, T. Genome comparison and phylogenetic analysis of Orientia tsutsugamushi strains. DNA Res. 2010, 17, 281–291. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | p.i. | Size (Mb) | Atypical Genomes |
|---|---|---|---|
| 34.8 | 0.072 | 1.516 | Ehrlichia ruminantium Welgevonden |
| 38.3 | 0.094 | 2.127 | Orientia tsutsugamushi Boryong |
| 106 | 0.120 | 2.279 | Treponema pallidum SS14 |
| 379 | 0.158 | 3.168 | Prevotella melaninogenica ATCC25845 |
| 207 | 0.166 | 3.268 | Mycobacterium leprae Br4923 |
| 208 | 0.166 | 3.268 | Mycobacterium leprae TN |
| 611 | 0.178 | 3.286 | Brucella abortus bv 19941 |
| 769 | 0.198 | 3.939 | Vibrio cholera M662 |
| 763 | 0.194 | 4.033 | Vibrio cholera O1 biovar ElTor N16961 |
| 385 | 0.182 | 4.171 | Sodalis glossinidius morsitans |
| 820 | 0.204 | 4.236 | Vibrio cholera MJ1236 |
| 1483 | 0.193 | 4.494 | Candidatus Cloacamonas acidaminovorans Evry |
| 787 | 0.211 | 4.532 | Aliivibrio salmonicida LFI1238 |
| 1072 | 0.225 | 5.008 | Vibrio vulnificus MO624O |
| 1281 | 0.231 | 5.166 | Vibrio parahaemolyticus RIMD2210633 |
| 1293 | 0.234 | 5.969 | Vibrio harveyi ATCCBAA1116 |
| Rank | Ave | Size (Mb) | Atypical Genomes |
|---|---|---|---|
| 246.8 | 1.521 | 0.853 | Onion yellows phytoplasma OYM uid58015 |
| … | |||
| 1225.1 | 1.915 | 2.809 | Halalkalicoccus jeotgali B3 uid50305 |
| 1233.4 | 1.936 | 2.821 | Halogeometricum borinquense DSM |
| 1235.3 | 2.008 | 2.848 | Haloferax volcanii DS2 uid46845 |
| 1091.1 | 1.878 | 2.914 | Halophilic archaeon DL31 uid72619 |
| 1240.8 | 2.067 | 3.420 | Haloarcula marismortui ATCC 43049 uid57719 |
| 1306.5 | 2.071 | 3.668 | Halopiger xanaduensis SH6 uid68105 |
| 1260.9 | 2.036 | 3.752 | Natrialba magadii ATCC 43099 uid46245 |
| 1419.5 | 2.378 | 3.889 | Haloterrigena turkmenica DSM 5511 |
| … | |||
| 948.4 | 2.228 | 4.644 | Mycobacterium JDM601 uid67369 |
| 1074.7 | 2.277 | 4.830 | Mycobacterium aviumparatuberculosis K10 |
| 1211.8 | 2.293 | 5.067 | Mycobacterium abscessus uid61613 |
| 1074.9 | 2.495 | 5.475 | Mycobacterium avium 104 uid57693 |
| 1275.8 | 2.399 | 5.548 | Mycobacterium gilvum Spyr1 uid61403 |
| 1303.6 | 2.491 | 5.620 | Mycobacterium gilvum PYRGCK uid59421 |
| 1306.9 | 2.483 | 5.705 | Mycobacterium MCS uid58465 |
| 1320.9 | 2.567 | 5.737 | Mycobacterium KMS uid58491 |
| 1319.4 | 2.582 | 6.048 | Mycobacterium JLS uid58489 |
| 1449.2 | 2.938 | 6.988 | Mycobacterium smegmatis MC2155 uid57701 |
| … | |||
| 1477.8 | 3.463 | 10.237 | Amycolatopsis mediterranei U32 uid50565 |
| Rank | Size (Mb) | Atypical Genomes |
|---|---|---|
| 622.8 | 1.591 | Candidatus Korarchaeum cryptofilum OPF8 |
| … | ||
| 803.4 | 2.001 | Halobacterium salinarum R1 |
| 811.5 | 2.014 | Halobacterium NRC1 |
| 1225.1 | 2.809 | Halalkalicoccus jeotgali B3 |
| 1233.4 | 2.821 | Halogeometricum borinquense DSM11551 |
| 1235.3 | 2.848 | Haloferax volcanii DS2 |
| 1091.1 | 2.914 | Halophilic archaeon DL31 |
| 1186.8 | 3.261 | Halorubrumlacus profundi ATCC 49239 |
| 1240.8 | 3.420 | Haloarcula marismortui ATCC 43049 |
| 1235.0 | 3.484 | Haloarcula hispanica ATCC 33960 |
| 1306.5 | 3.668 | Halopiger xanaduensis SH6 |
| 1260.9 | 3.752 | Natrialba magadii ATCC 43099 |
| 1419.5 | 3.889 | Haloterrigena turkmenica DSM 5511 |
| … | ||
| 1057.6 | 7.750 | Trichodesmium erythraeum IMS101 |
| Rank | mp | Size (Mb) | Atypical Genomes |
|---|---|---|---|
| 31.2 | 0.32 | 0.580 | Mycoplasma genitalium G37 |
| 166.6 | 0.34 | 0.602 | Candidatus Phytoplasma Mali |
| 21.9 | 0.00 | 0.706 | Candidatus Blochmannia floridanus |
| 246.7 | 0.49 | 0.707 | Aster yellows witches broom phytoplasma AYWB |
| 11.5 | 0.04 | 0.792 | Candidatus Blochmannia pennsylvanicus BPEN |
| 183.6 | 0.31 | 0.799 | Mycoplasma synoviae 53 |
| 31.8 | 0.39 | 0.816 | Mycoplasma pneumoniae M129 |
| 246.8 | 0.49 | 0.853 | Onion yellows phytoplasma OY M |
| 167.2 | 0.48 | 0.880 | Candidatus Phytoplasma australiense |
| 192.8 | 0.37 | 0.948 | Mycoplasma bovis Hubei 1 |
| 199.5 | 0.41 | 0.964 | Mycoplasma pulmonis UAB CTIP |
| 191.8 | 0.34 | 0.978 | Mycoplasma fermentans JER |
| 297.6 | 0.37 | 1.007 | Mycoplasma agalactiae |
| 186.2 | 0.45 | 1.119 | Mycoplasma fermentans M64 |
| 77.1 | 0.09 | 1.161 | Candidatus Ruthia magnifica Cm Calyptogena magnifica |
| 420.9 | 0.45 | 1.317 | Thermosphaera aggregans DSM 11486 |
| 411.0 | 0.48 | 1.580 | Staphylothermus hellenicus DSM 12710 |
| 358.7 | 0.44 | 1.667 | Gardnerella vaginalis ATCC 14019 |
| 481.4 | 0.46 | 1.796 | Streptococcus thermophilus CNRZ1066 |
| 196.2 | 0.20 | 1.887 | Haemophilus influenzae PittGG |
| 156.9 | 0.21 | 2.145 | Neisseria meningitidis alpha14 |
| 158.0 | 0.22 | 2.153 | Neisseria meningitidis 053442 |
| 154.3 | 0.23 | 2.154 | Neisseria gonorrhoeae FA 1090 |
| 160.0 | 0.22 | 2.184 | Neisseria meningitidis Z2491 |
| 166.2 | 0.24 | 2.272 | Neisseria meningitidis MC58 |
| 105.6 | 0.24 | 2.279 | Treponema pallidum SS14 |
| 859.3 | 0.54 | 2.702 | Sulfolobus islandicus Y G 57 14 |
| 1131.5 | 0.55 | 2.992 | Sulfolobus solfataricus P2 |
| 1483.0 | 0.33 | 4.494 | Candidatus Cloacamonas acidaminovorans Evry |
| Genome Name | Np | NO | NC | 1 | 2 | 3 | >3 | mp |
|---|---|---|---|---|---|---|---|---|
| M. agalactiae PG2 | 742 | 267 | 475 | 335 | 42 | 10 | 4 | 14/56 |
| M. agalactiae uid46679 | 813 | 291 | 522 | 332 | 42 | 15 | 10 | 25/67 |
| M. arthritidis 158L3 1 | 631 | 214 | 417 | 347 | 20 | 3 | 3 | 6/26 |
| M. bovis Hubei 1 | 801 | 279 | 522 | 346 | 37 | 11 | 11 | 22/59 |
| M. bovis PG45 | 765 | 239 | 526 | 354 | 43 | 9 | 7 | 16/59 |
| M. capricolum ATCC 27343 | 812 | 236 | 576 | 390 | 58 | 10 | 7 | 17/65 |
| M. conjunctivae | 692 | 272 | 420 | 323 | 39 | 0 | 4 | 4/43 |
| M. crocodyli MP145 | 689 | 199 | 490 | 380 | 37 | 6 | 4 | 10/47 |
| M. fermentans JER | 797 | 247 | 550 | 388 | 38 | 8 | 12 | 20/58 |
| M. fermentans M64 | 1049 | 459 | 590 | 383 | 35 | 11 | 18 | 29/64 |
| M. gallisepticum R low | 763 | 274 | 489 | 357 | 43 | 4 | 6 | 10/53 |
| M. genitalium G37 | 475 | 91 | 384 | 330 | 15 | 4 | 3 | 7/22 |
| M. haemofelis Langford 1 | 1545 | 1258 | 287 | 230 | 16 | 2 | 1 | 3/19 |
| M. hominis ATCC 23114 | 523 | 145 | 378 | 315 | 21 | 1 | 4 | 5/26 |
| M. hyopneumoniae 232 | 691 | 254 | 437 | 331 | 39 | 1 | 3 | 4/43 |
| M. hyopneumoniae 7448 | 657 | 214 | 443 | 333 | 38 | 1 | 4 | 5/43 |
| M. hyopneumoniae J | 657 | 186 | 471 | 344 | 44 | 2 | 4 | 6/50 |
| M. hyorhinis HUB 1 | 658 | 194 | 464 | 339 | 36 | 7 | 2 | 9/45 |
| M. leachii PG50 | 882 | 316 | 566 | 398 | 50 | 9 | 8 | 17/67 |
| M. mobile 163K | 633 | 183 | 450 | 370 | 26 | 6 | 2 | 8/34 |
| M. mycoides capri LC 95010 | 922 | 303 | 619 | 400 | 55 | 6 | 14 | 20/75 |
| M. mycoides SC PG1 | 1017 | 325 | 692 | 397 | 55 | 15 | 16 | 31/86 |
| M. penetrans HF 2 | 1037 | 379 | 658 | 447 | 54 | 10 | 14 | 30/84 |
| M. pneumoniae M129 | 648 | 203 | 445 | 359 | 19 | 6 | 6 | 12/31 |
| M. pulmonis UAB CTIP | 782 | 222 | 560 | 387 | 36 | 8 | 17 | 25/61 |
| M. putrefaciens KS1 | 650 | 176 | 474 | 379 | 34 | 4 | 3 | 7/41 |
| M. suis Illinois | 845 | 592 | 253 | 209 | 14 | 0 | 2 | 2/16 |
| M. suis KI3806 | 794 | 553 | 241 | 212 | 11 | 1 | 1 | 2/13 |
| M. synoviae 53 | 659 | 180 | 479 | 357 | 33 | 10 | 5 | 15/48 |
| U. parvum serovar 3 ATCC 27815 | 609 | 196 | 413 | 346 | 25 | 1 | 2 | 3/28 |
| U. parvum serovar 3 ATCC 700970 | 614 | 173 | 441 | 360 | 29 | 3 | 2 | 5/34 |
| U. urealyticum serovar 10 ATCC 33699 | 646 | 230 | 416 | 342 | 25 | 3 | 2 | 5/30 |
| Rank | Ave | Size (Mb) | Atypical Genomes |
|---|---|---|---|
| 861 | 1.827 | 6.196 | Pirellula staleyi DSM_6068_uid43209 |
| 1341 | 2.024 | 7.215 | Hahella chejuensis KCTC_2396_uid58483 |
| 1058 | 1.961 | 7.750 | Trichodesmium erythraeum IMS101_uid57925 |
| 1411 | 2.370 | 9.004 | Myxococcus fulvus HW_1_uid68443 |
| 1319 | 2.349 | 9.446 | Haliangium ochraceum DSM_14365_uid41425 |
| p.i. | Ave | Rank | mp | Genome Size | |
|---|---|---|---|---|---|
| p.i. | 0.57 | 0.57 | 0.46 | 0.72 | |
| ave | 0.57 | 0.61 | 0.52 | 0.77 | |
| rank | 0.57 | 0.61 | 0.38 | 0.78 | |
| mp | 0.46 | 0.52 | 0.38 | 0.66 |
| Genome | ORFans | COGs | p.i. | Ave | Rank | Mp | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||||||
| A | 10 | 1 | 1 | 1 | 1 | 16 | 0.2 | 4 | 1 | 1.0 |
| B | 8 | 1 | 1 | 2 | 2 | 4 | 0.6 | 2 | 2 | 0.3 |
| C | 20 | 1 | 1 | 1 | 6 | 6 | 0.4 | 3 | 3 | 0.7 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carmi, G.; Bolshoy, A. Gene-Family Extension Measures and Correlations. Life 2016, 6, 30. https://doi.org/10.3390/life6030030
Carmi G, Bolshoy A. Gene-Family Extension Measures and Correlations. Life. 2016; 6(3):30. https://doi.org/10.3390/life6030030
Chicago/Turabian StyleCarmi, Gon, and Alexander Bolshoy. 2016. "Gene-Family Extension Measures and Correlations" Life 6, no. 3: 30. https://doi.org/10.3390/life6030030
APA StyleCarmi, G., & Bolshoy, A. (2016). Gene-Family Extension Measures and Correlations. Life, 6(3), 30. https://doi.org/10.3390/life6030030