
Self-Referential Encoding on Modules of Anticodon Pairs—Roots of the Biological Flow System


Abstract
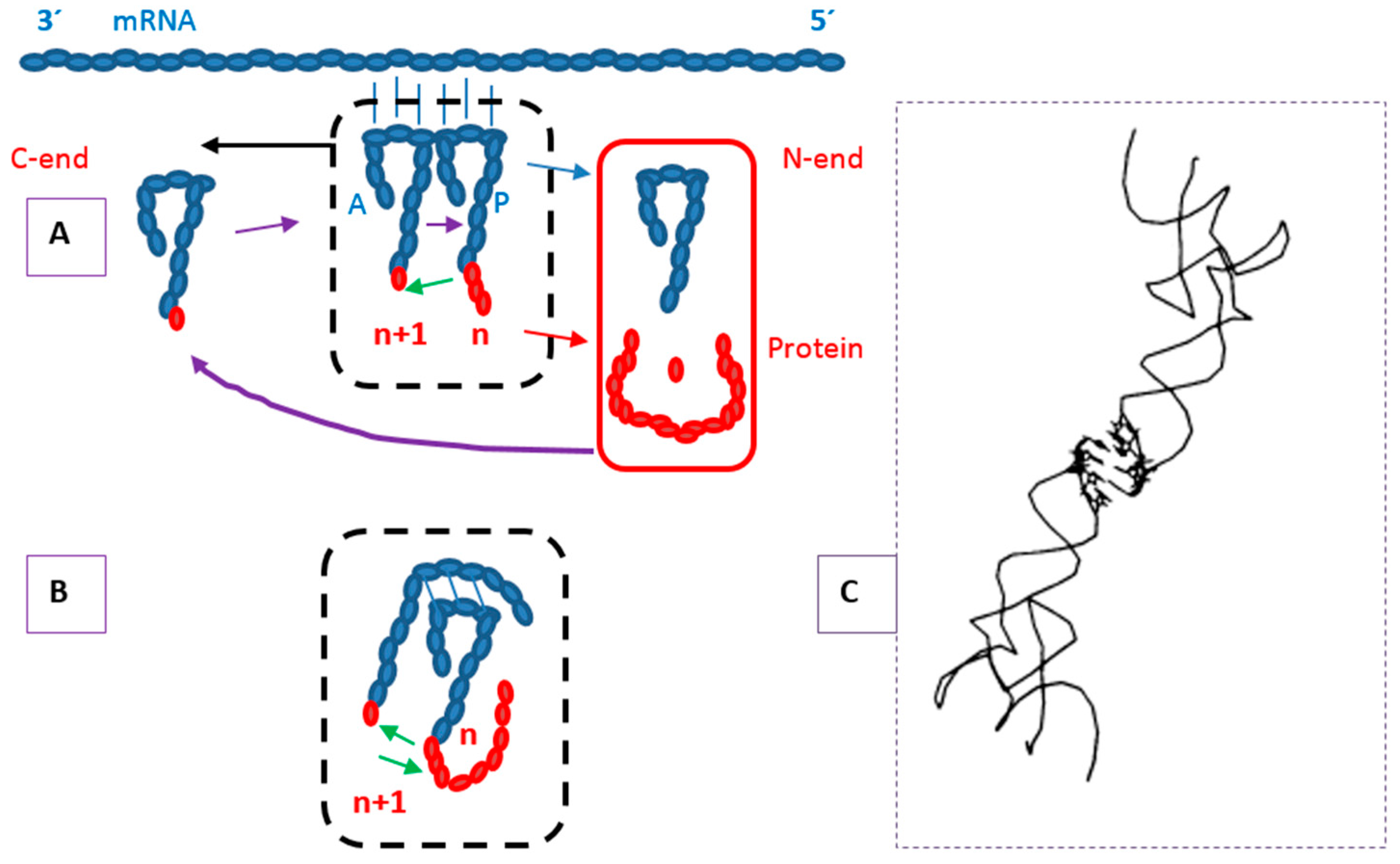
: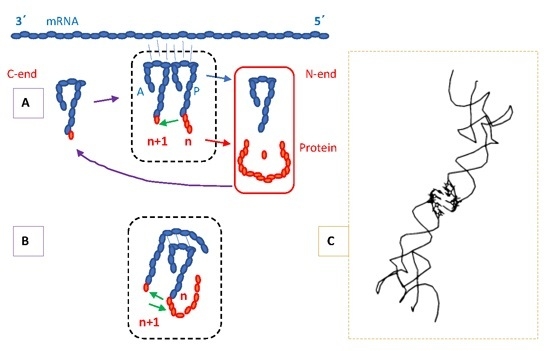
Living beings are metabolic flow systems that self-construct on the basis of memories and adapt/evolve on the basis of constitutive plasticity.Life is the ontogenetic and evolutionary process instantiated by living beings.
1. Context
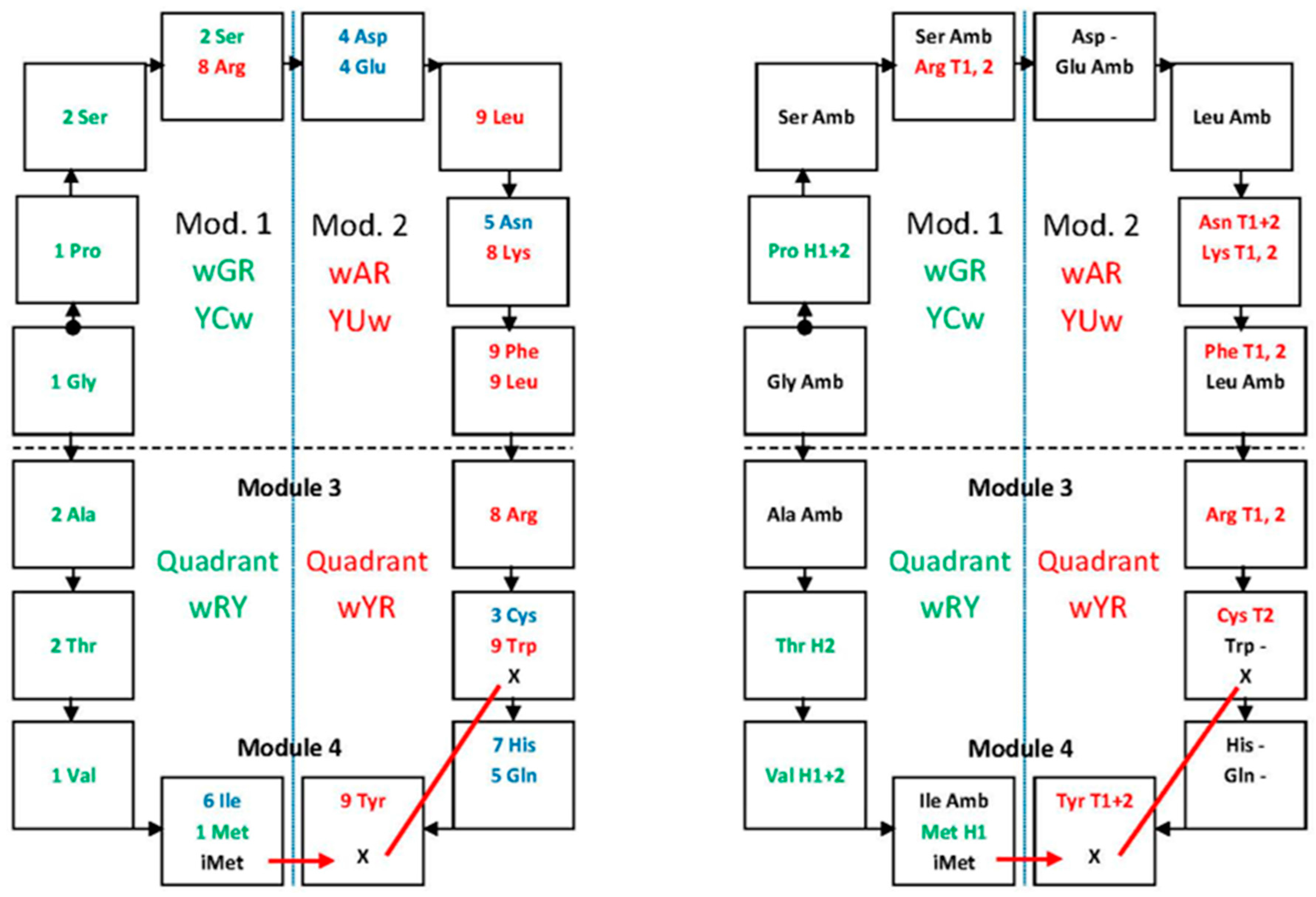
2. Modular Structure in the Genetic Code and Layout of the Text
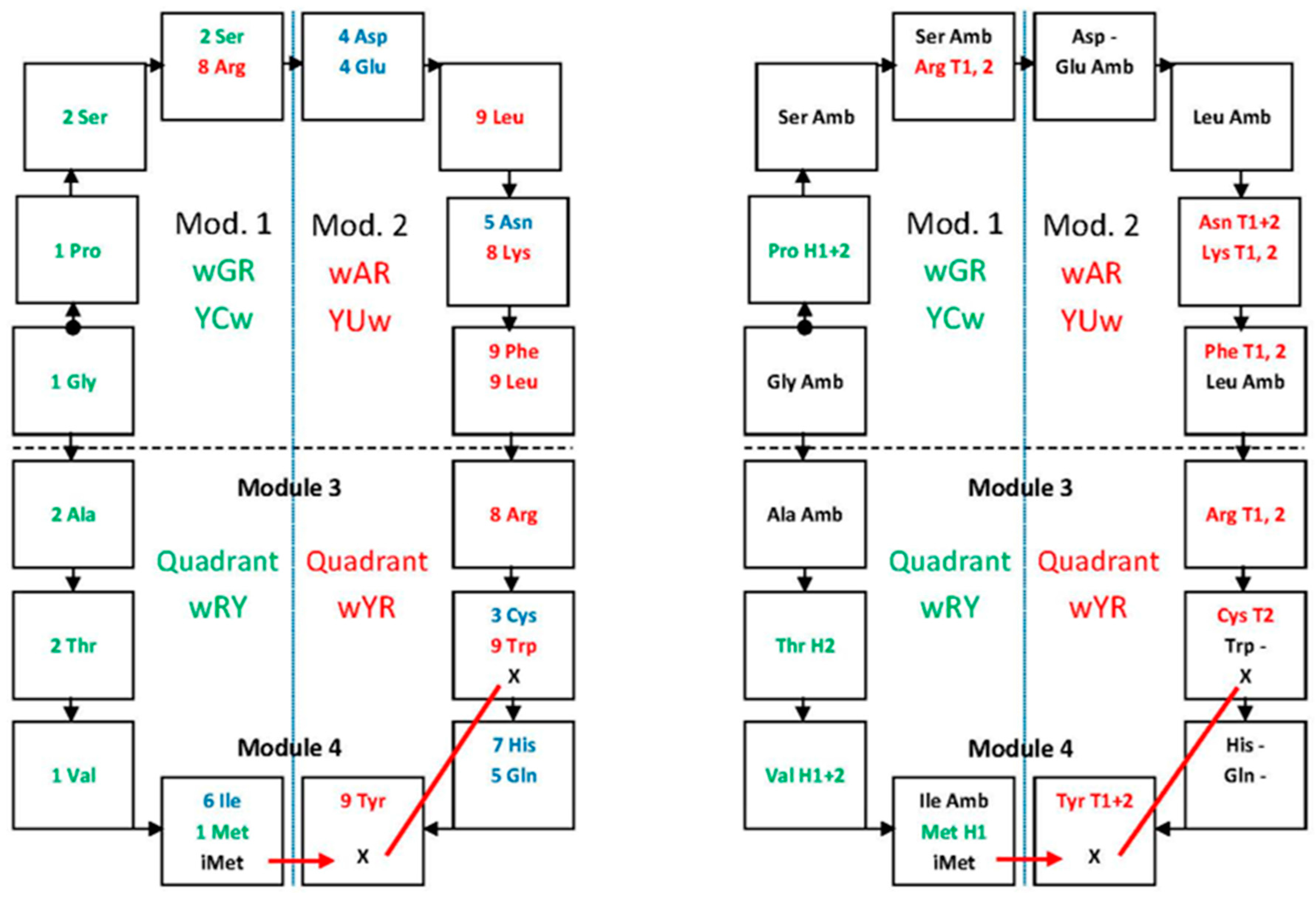
3. Functionality in the Modular Organization
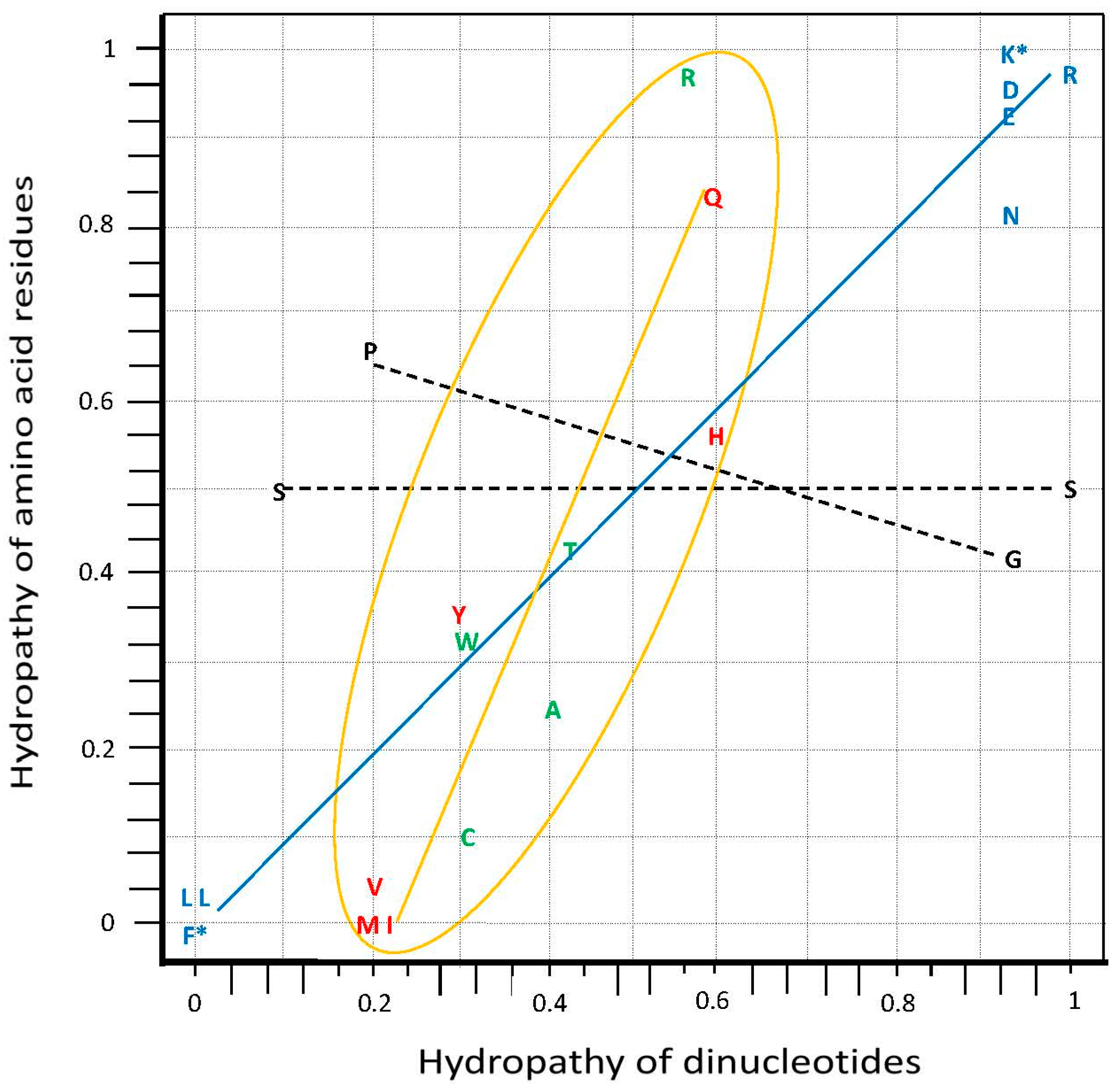
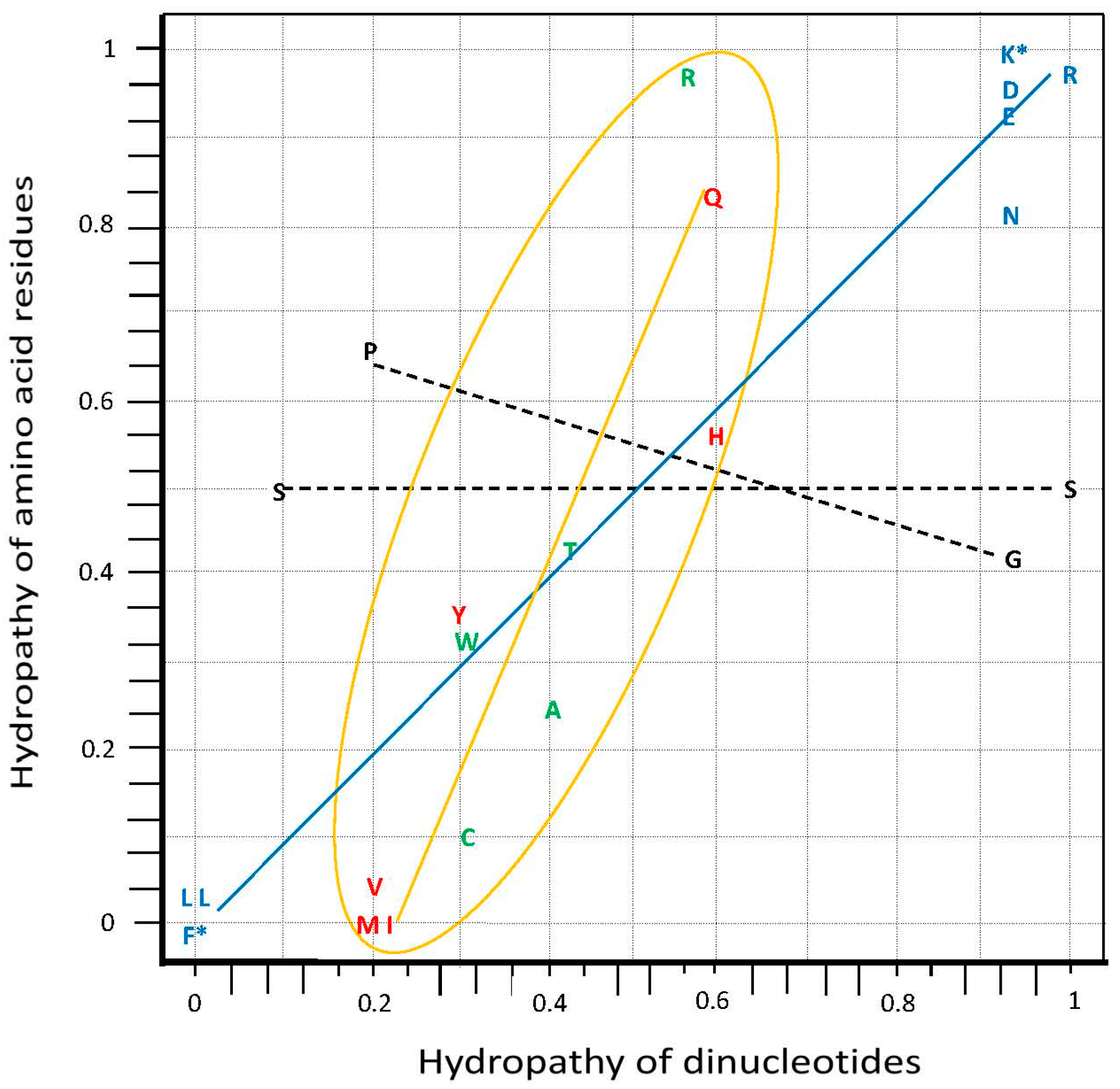
3.1. Hydropathy Correlation
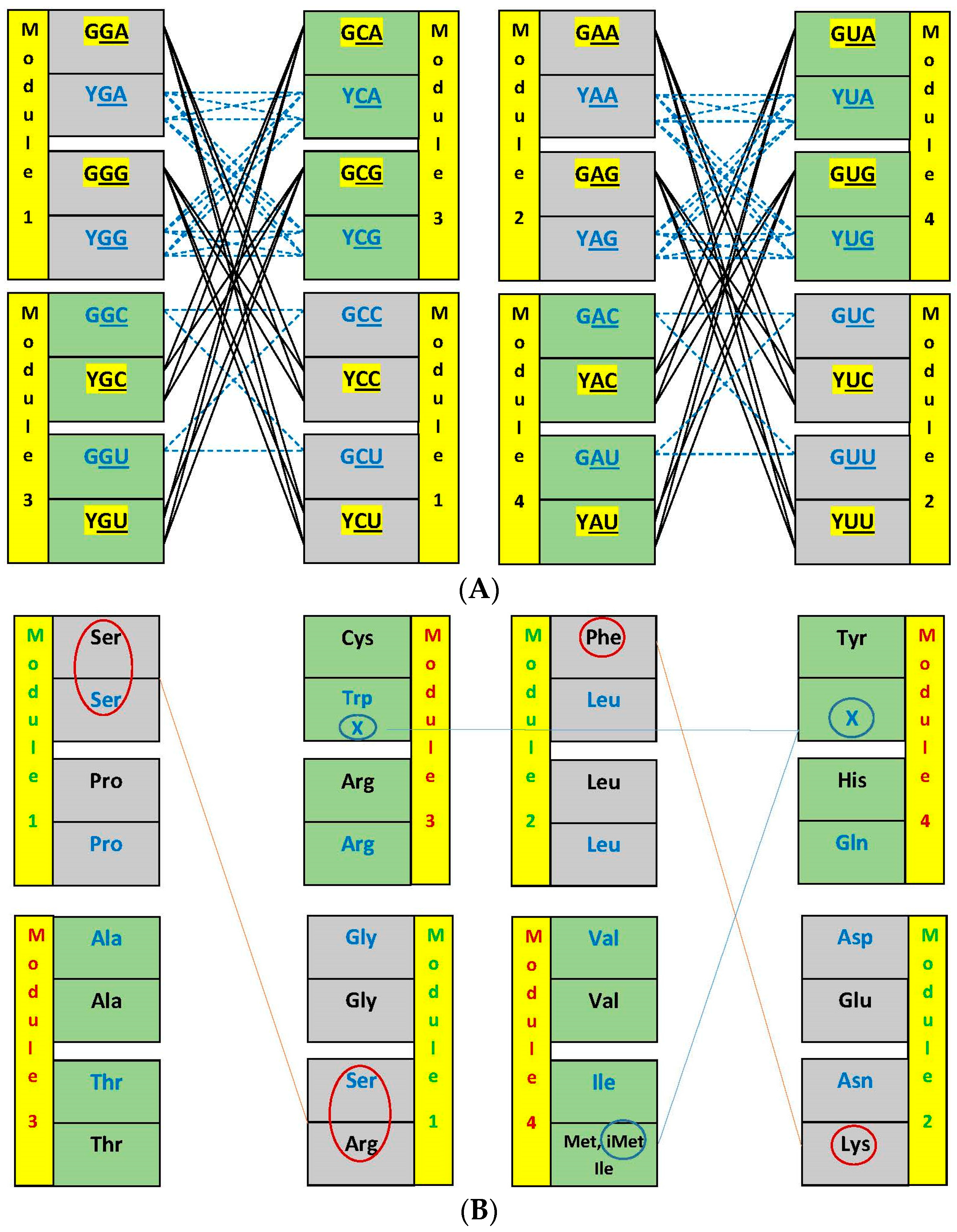
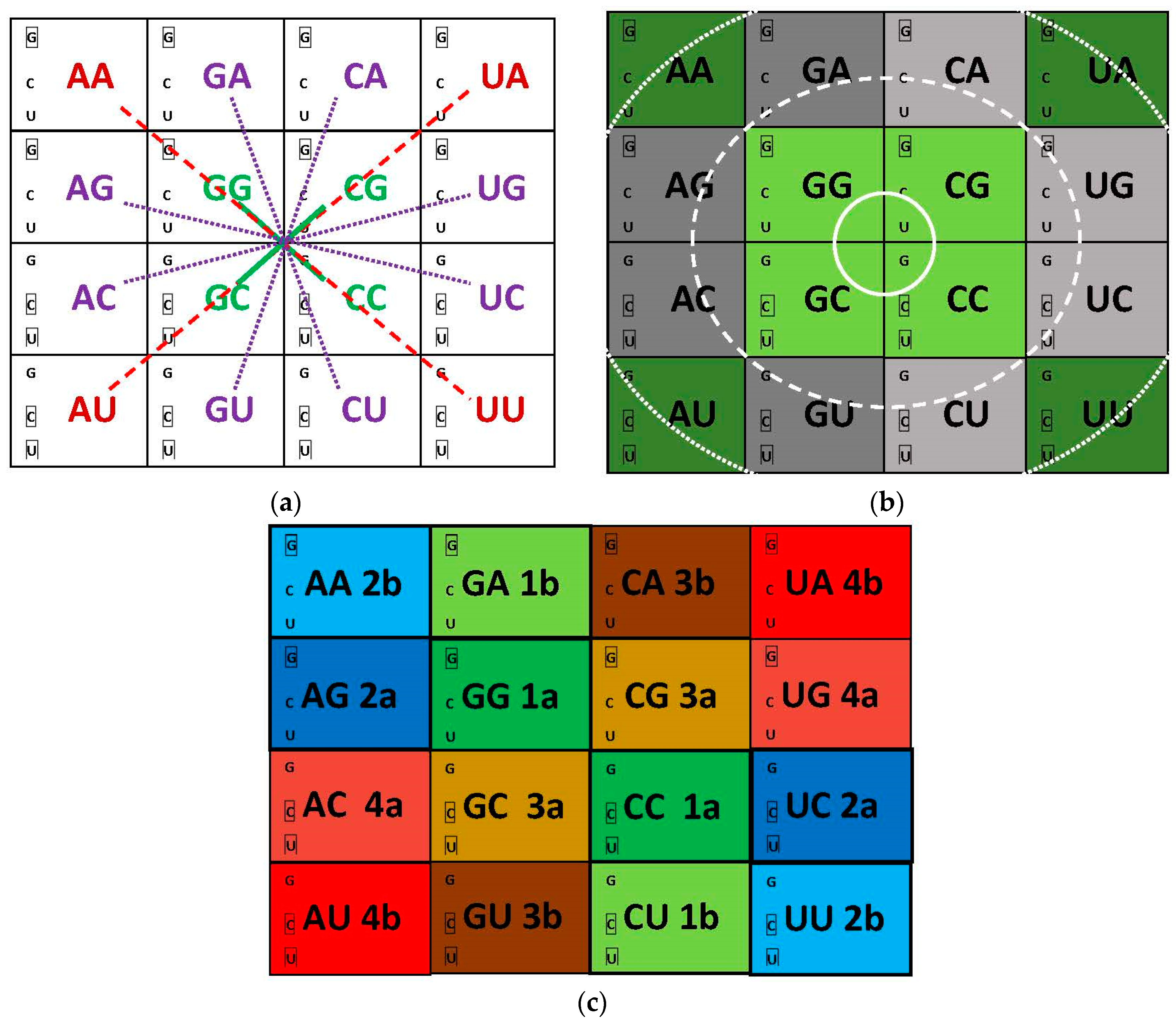
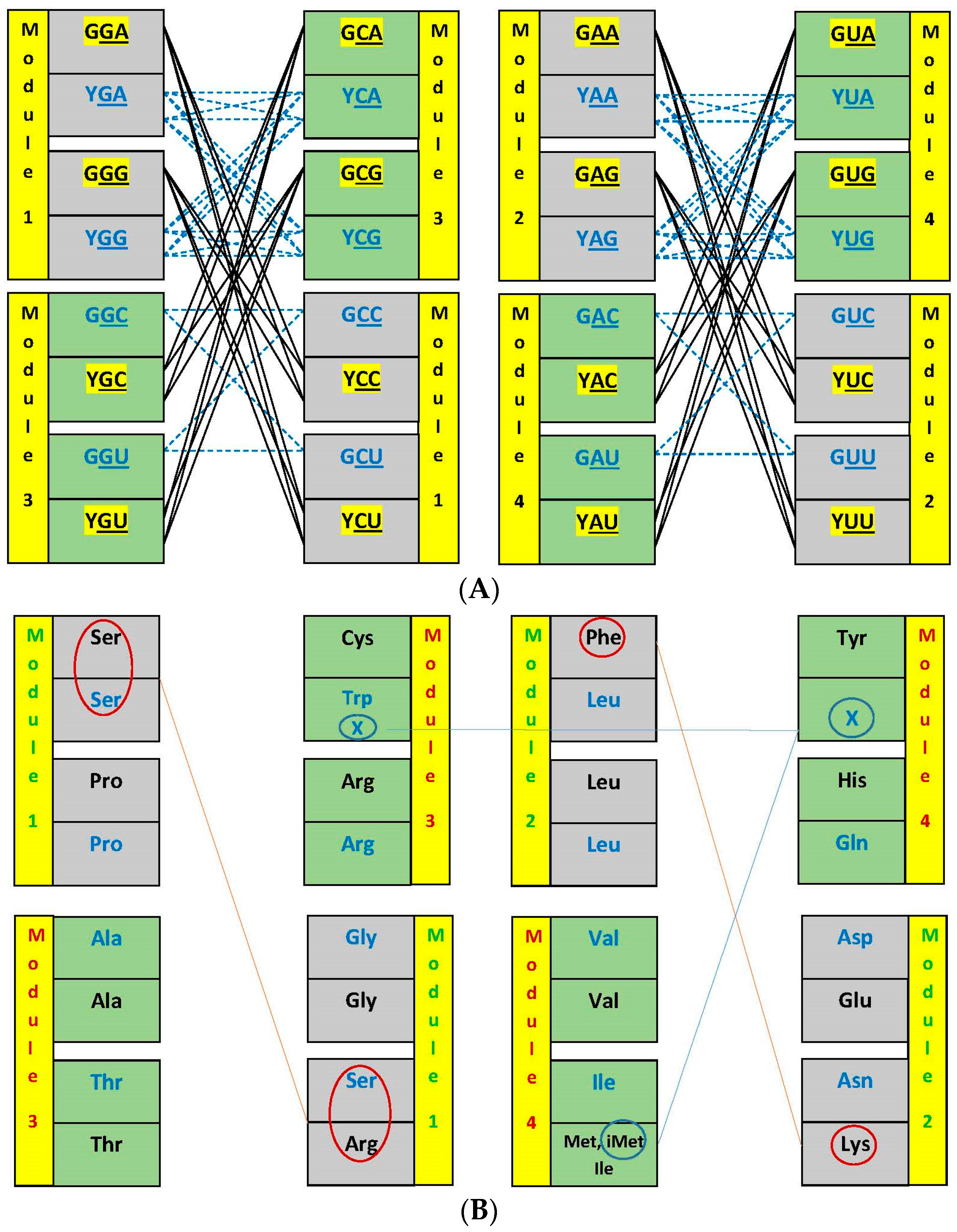
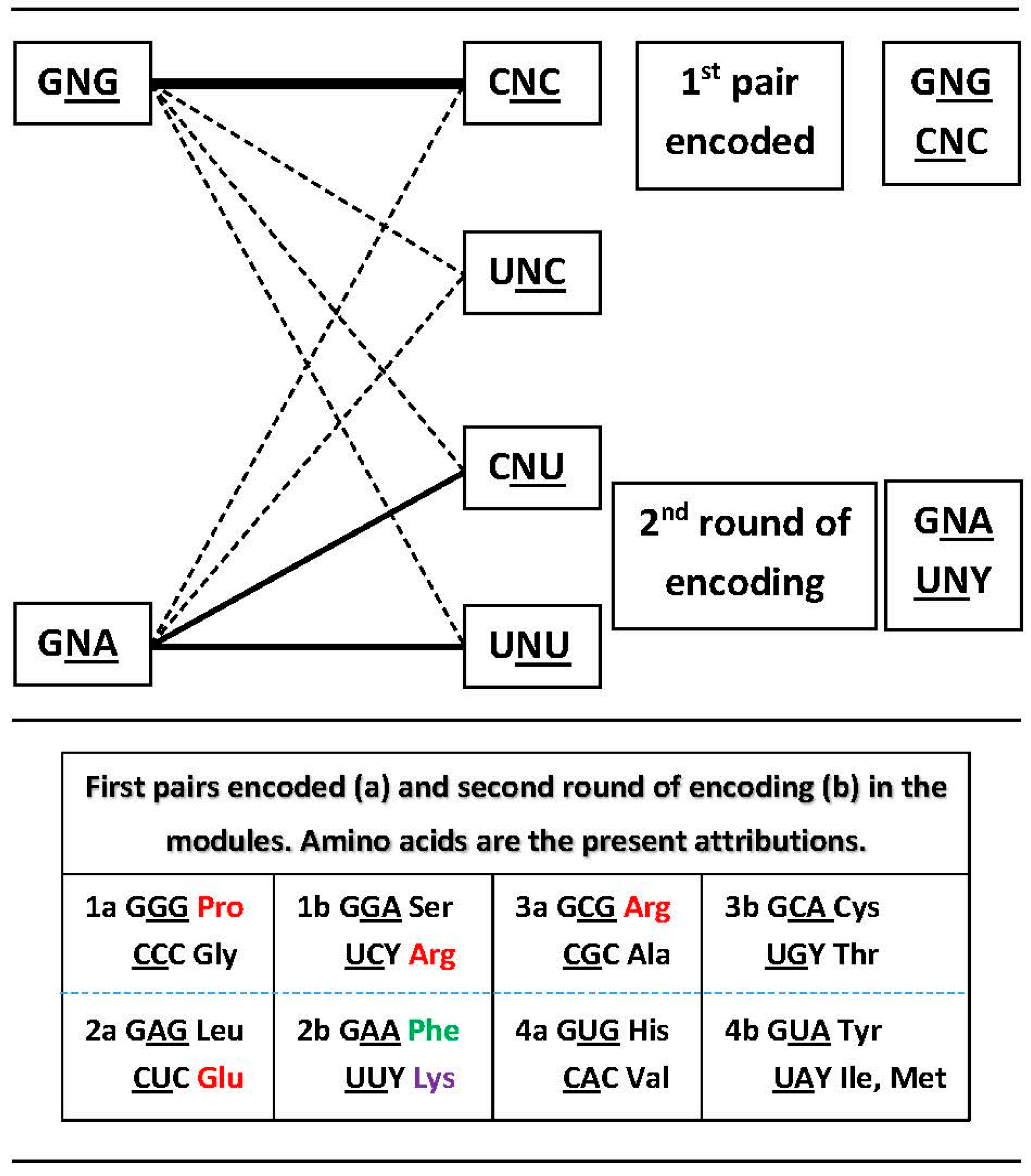
3.2. Networks of Dimers in the Modules and the Encoding Process
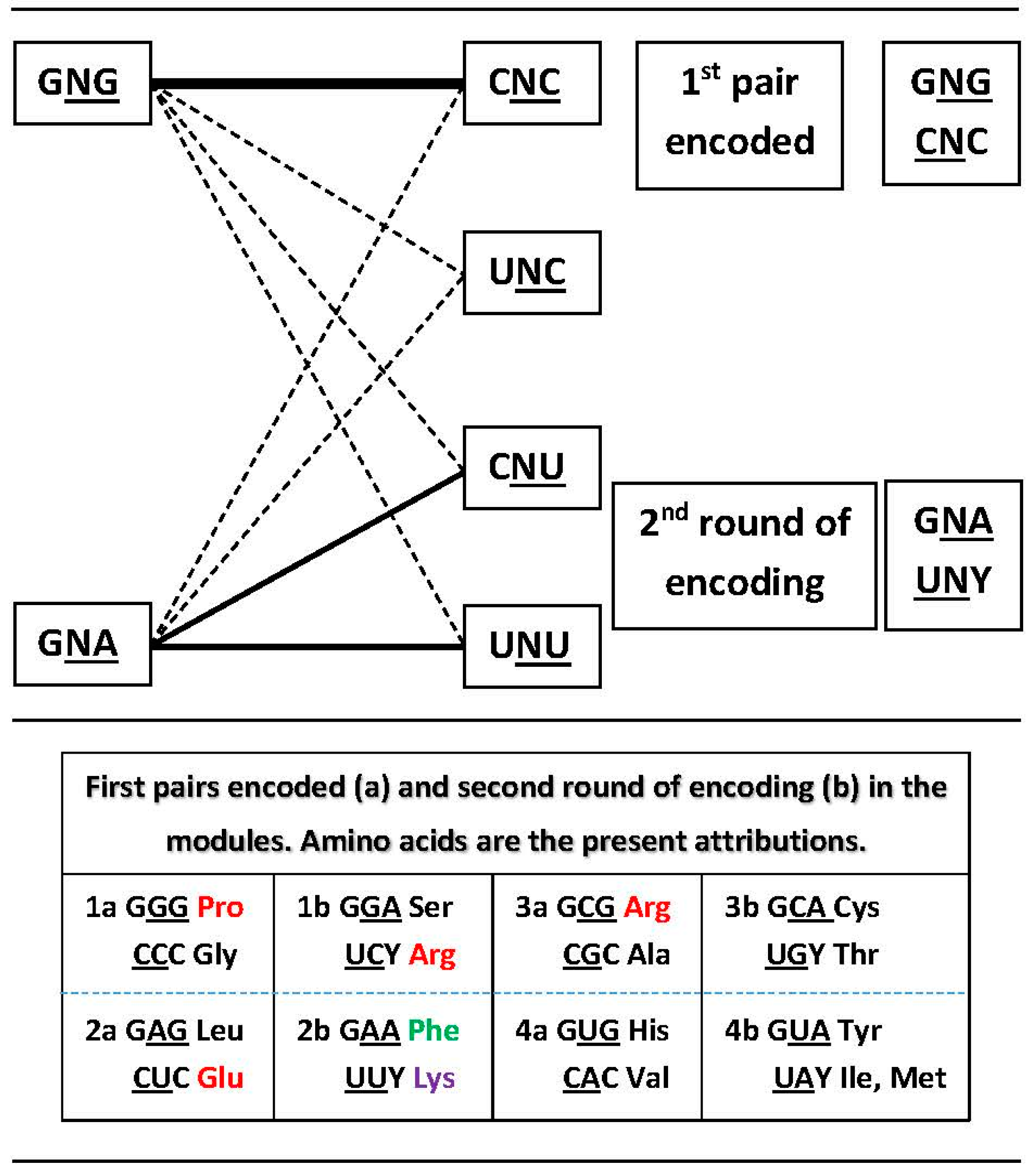
3.3. Network Symmetry-Breaking
- (1)
- Keep the (a) high stability of the G:C pair plus the (b) mono-specificity of C and bi-specificity of G. The choice becomes between A and U, where A is tri-specific and U tetra [21].
- (2)
- Avoid the very weak A:C pair at the laterals of the triplets, in favor of the G:U, which is frequent in RNA. Among the eight dimers in a module, the structure is exactly repetitive. Counting the numbers of lateral base pairs, excluding 5′A makes G:C 6, G:U 6, A:C 2, A:U 2 while excluding 5′U would make G:C 6, G:U 2, A:C 6, A:U 2.
- (3)
- (4)
- Keep U to profit from its tetra-fold ambiguity at decoding, as used in full in today’s vertebrate mitochondria [35].
- (5)
- Presence of A at the 5′ position in the P-site anticodon would, in a not well explained manner, destabilize the codon-anticodon pair in the A-site [36].
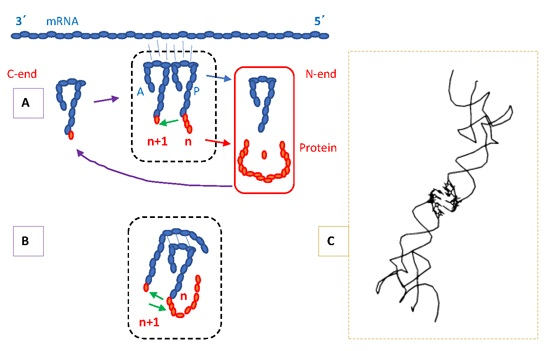
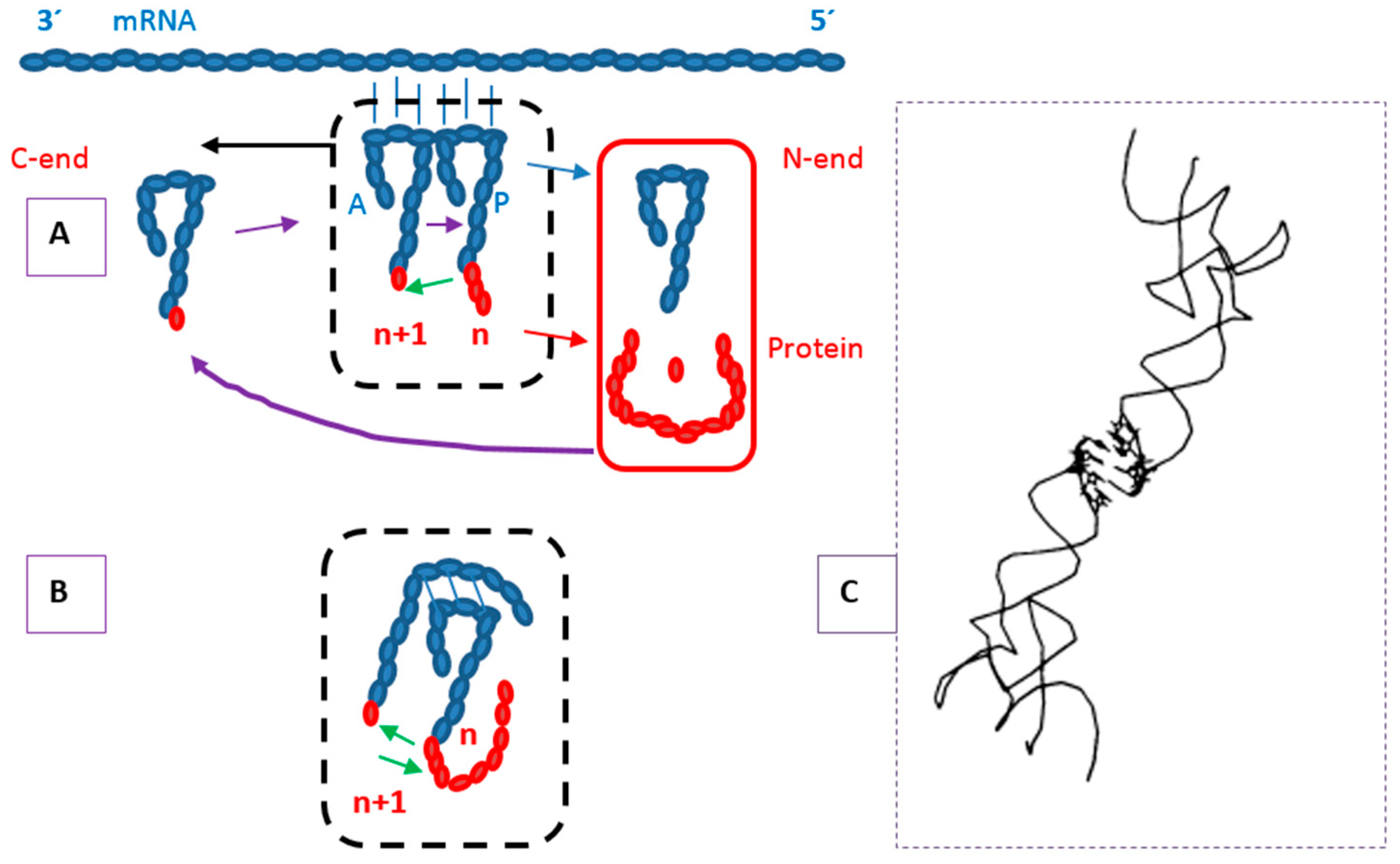
4. Protein Secondary Structure, Nucleic Acid Binding and Composition of Termini
4.1. Secondary Structure
4.2. RNA and DNA Binding
4.3. Protein Termini and the N-End Rule
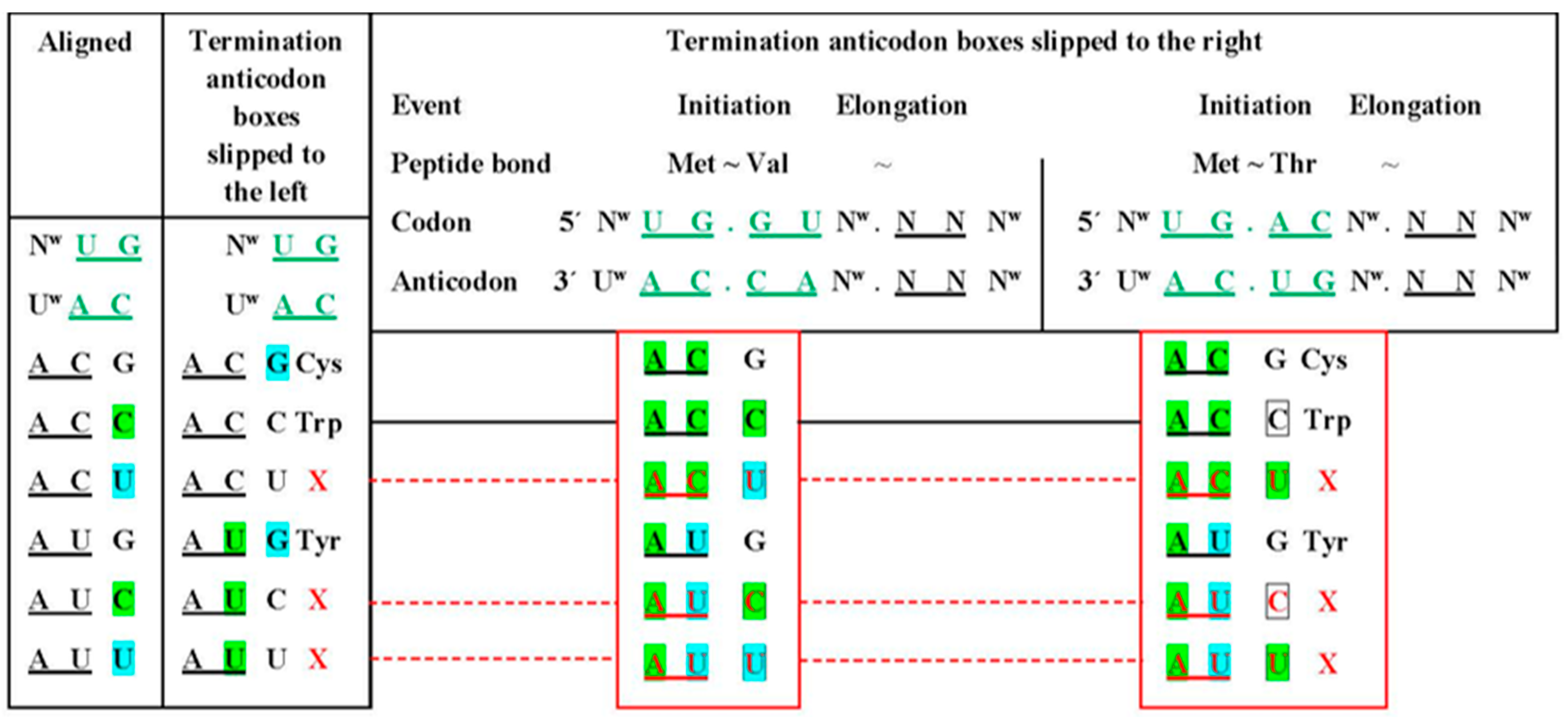
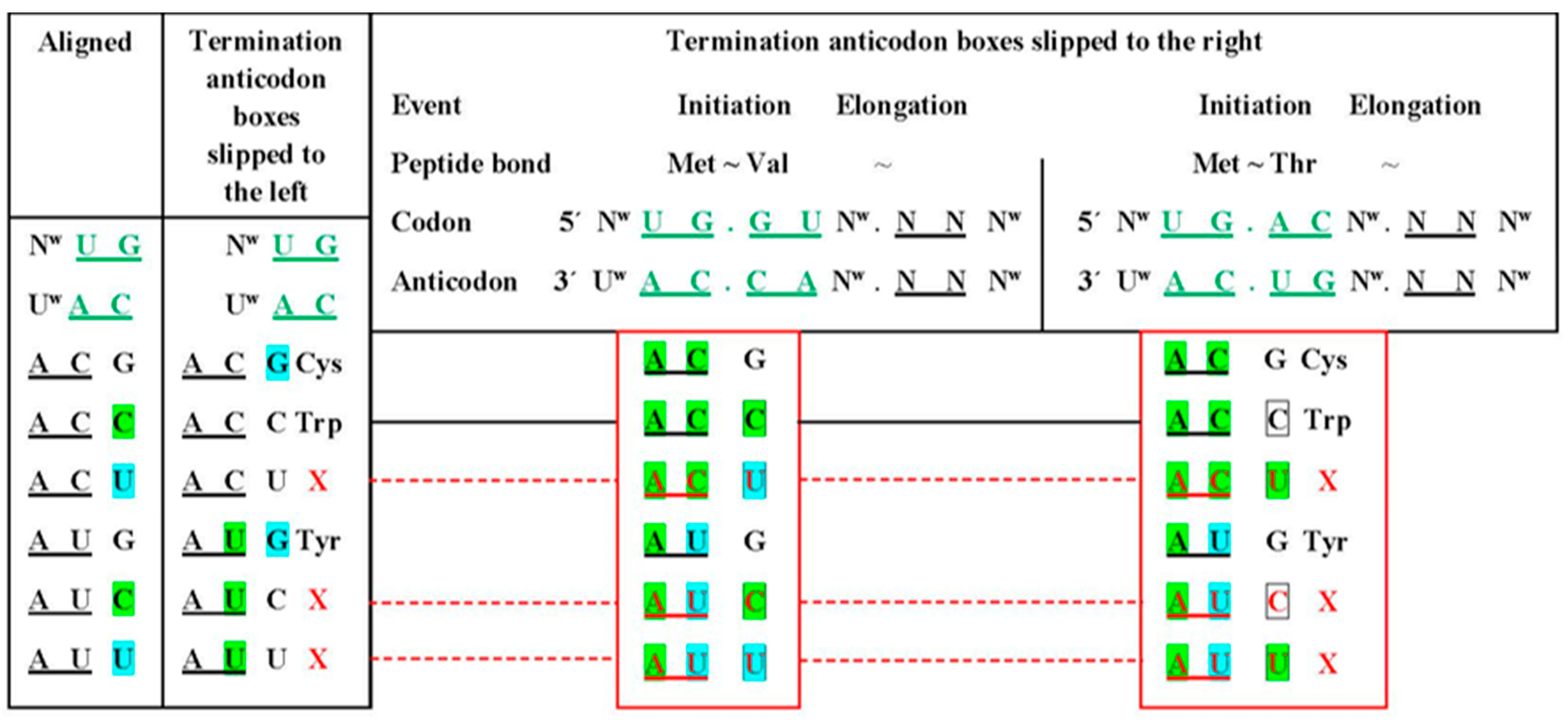
5. Location of the Punctuation Codes
RNP World Instead of RNA World
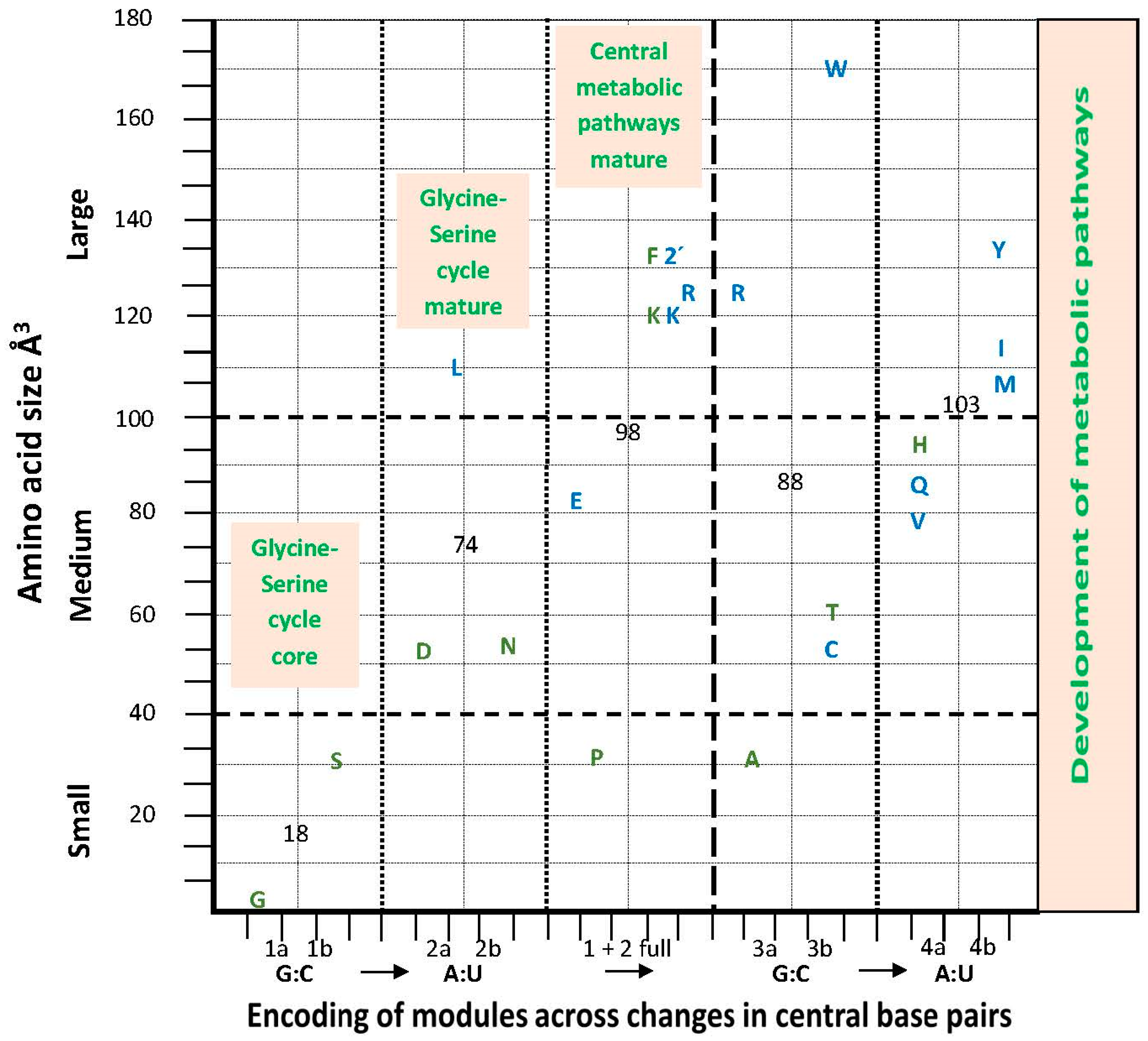
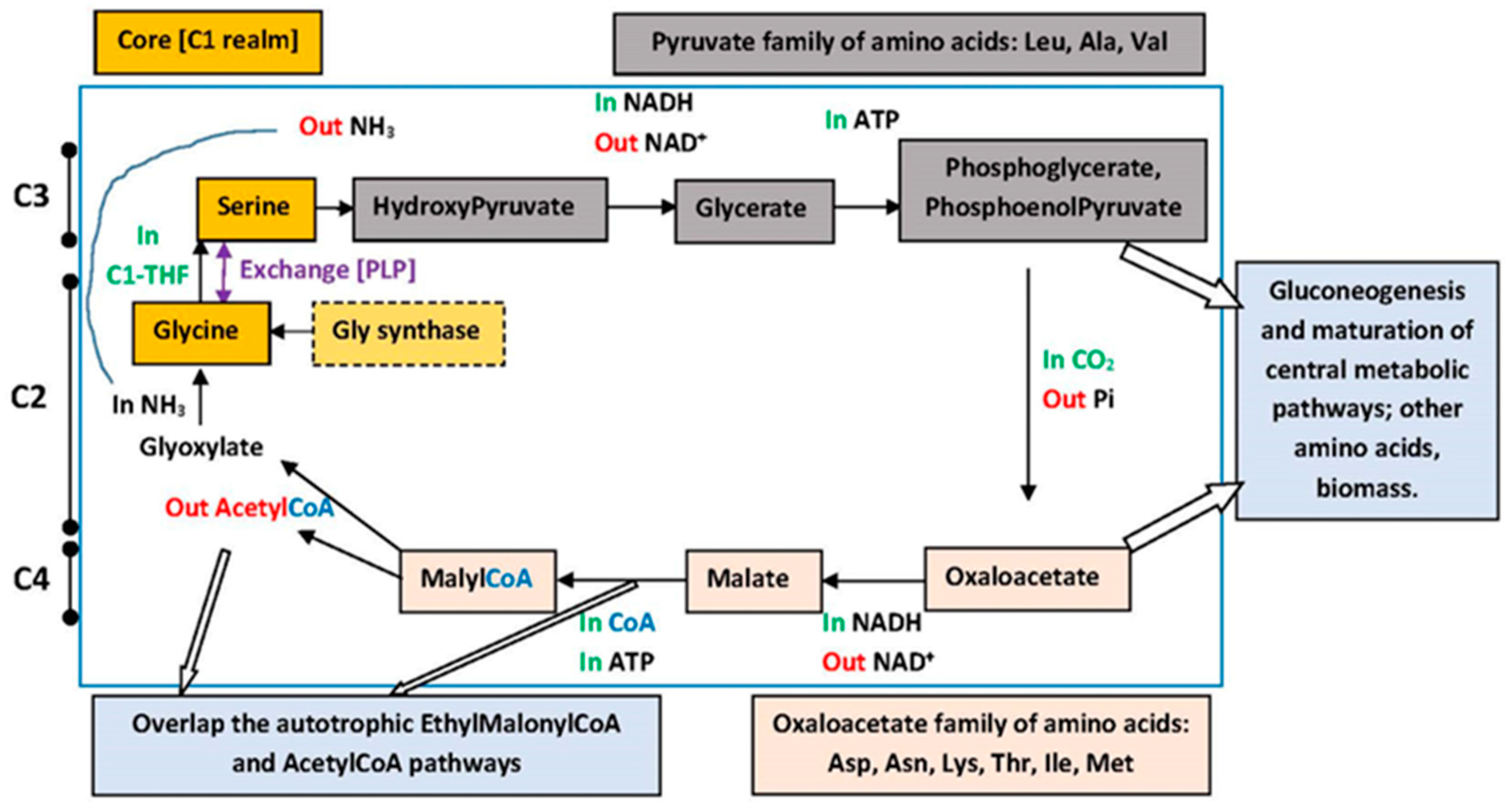
6. Metabolic Pathways
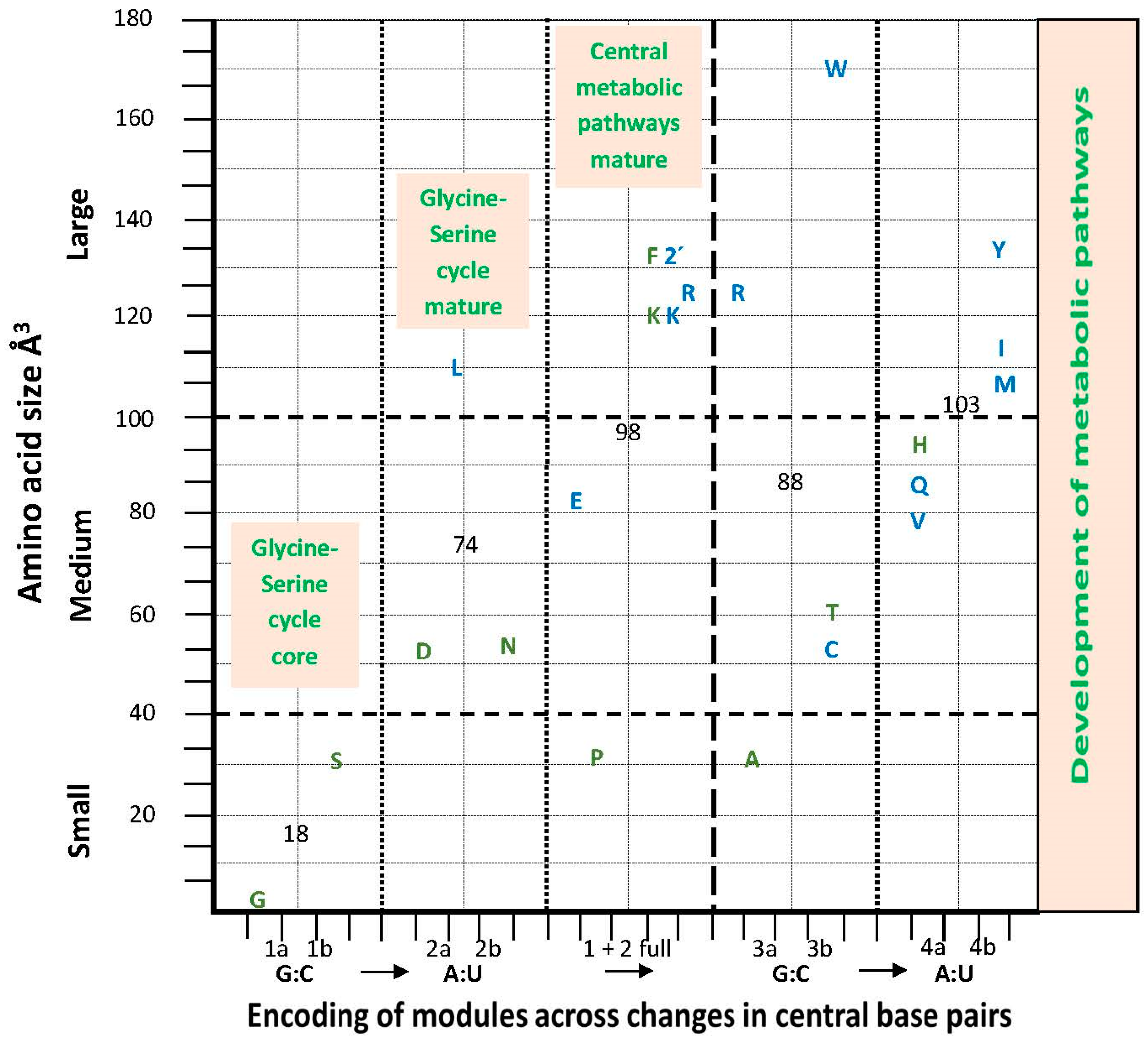
Amino Acid Hydrodynamic Size and the aRS Classes
7. Symmetries and Error-Reduction in Modularity
7.1. Central Bases Compose Standard Base Pairs, Columns Are Divided into Hemi-Columns
7.2. Symmetry in the Distribution of Mono- and Multi-Meaning Boxes
7.3. Symmetry in Modules of Dimers and the Error-Reduction Property
7.4. Metabolic Perspectives
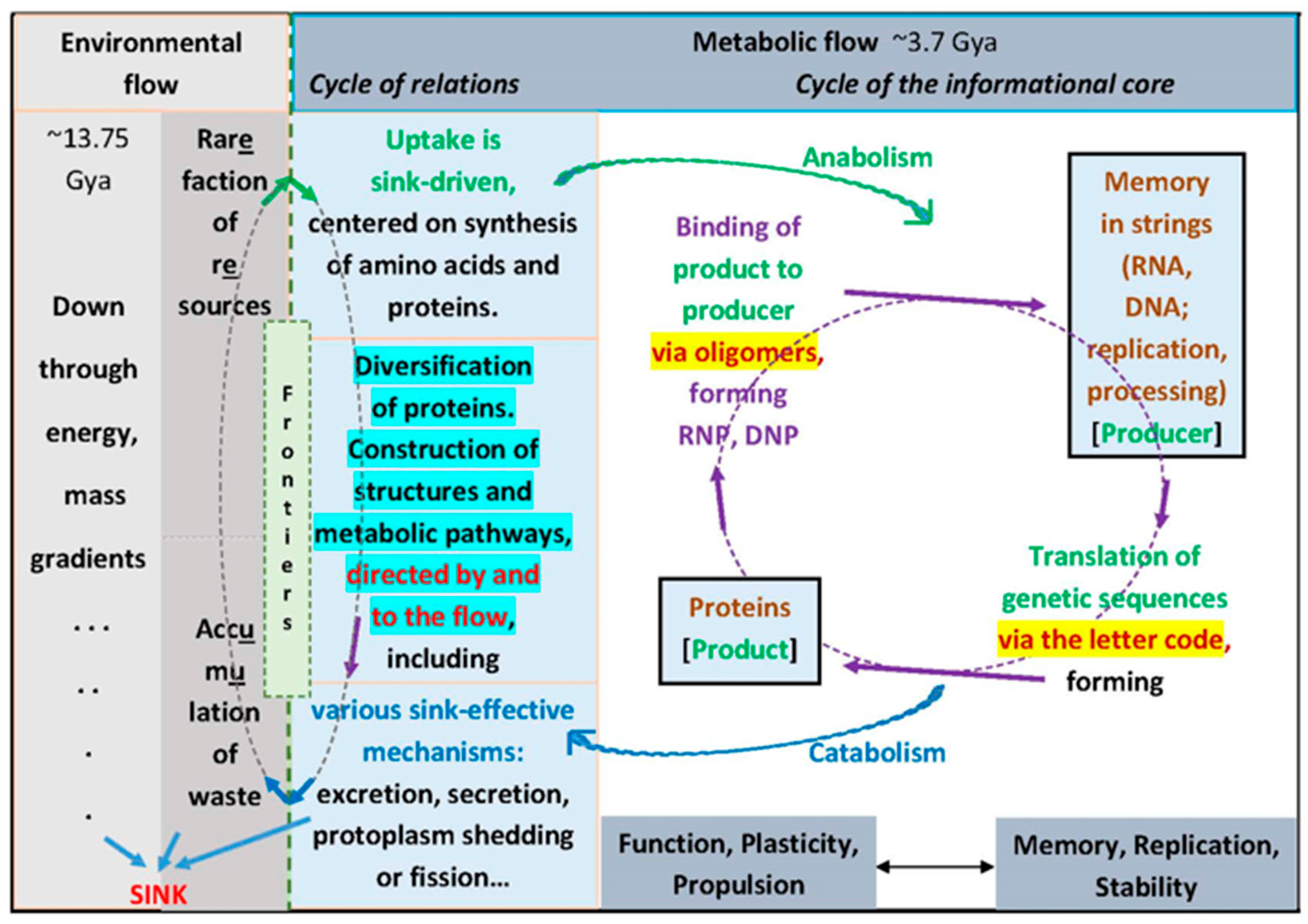
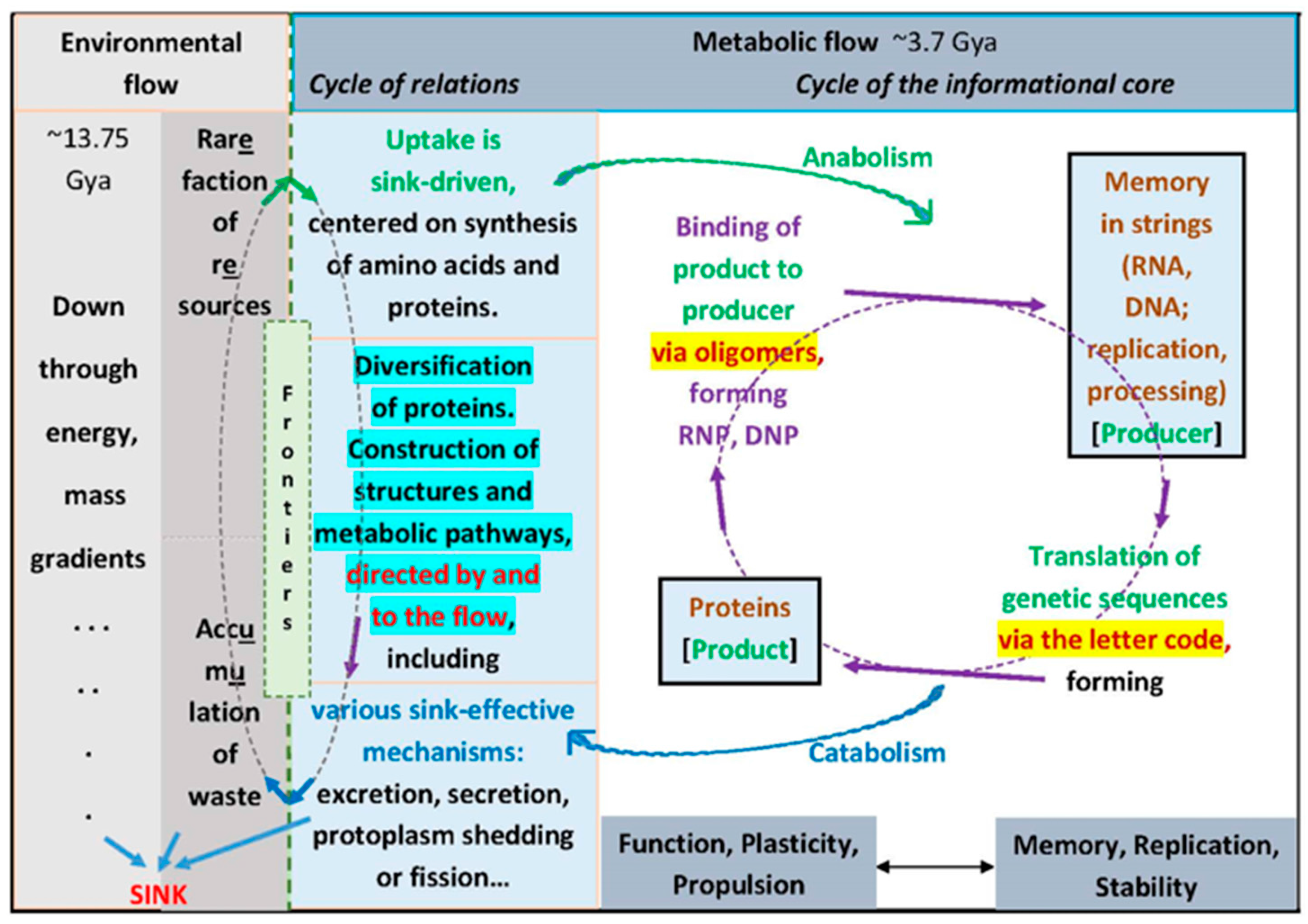
8. The Flow Is the Logic
9. Coda and Direct Tests
Transition to Biological Specificity
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Guimarães, R.C. Essentials in the life process indicated by the self-referential genetic code. Orig. Life Evol. Biosph. 2014, 44, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Farias, S.T.; Rêgo, T.; José, M.V. Origin and evolution of the Peptidyl Transferase Center from 160 proto-tRNAs. FEBS Open Bio. 2014, 4, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K. Origins and early evolution of the tRNA molecule. Life 2015, 5, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Hordijk, W.; Steel, M. Chasing the tail: The emergence of autocatalytic networks. Biosystems 2017, 152, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, R.C. Mutuality in discrete and compositional information: Perspectives for synthetic genetic codes. Cogn. Comput. 2012, 4, 115–139. [Google Scholar] [CrossRef]
- Vitas, M.; Dobovisek, A. On a quest for reverse translation. Found. Chem. 2016, 1–17. [Google Scholar] [CrossRef]
- Moras, D.; Dock, A.C.; Dumas, P.; Westhof, E.; Romby, P.; Ebel, J.-P.; Giegé, R. Anticodon-anticodon interaction induces conformational changes in tRNA: Yeast tRNAASP, a model for tRNA-mRNA recognition. Proc. Natl. Acad. Sci. USA 1986, 83, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Pascal, R.; Pross, A. The logic of life. Orig. Life Evol. Biosph. 2016, 46, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Ptashne, M. Epigenetics: Core misconcept. Proc. Nat. Acad. Sci. USA 2013, 110, 7101–7103. [Google Scholar] [CrossRef] [PubMed]
- Des Marais, D.L.; Hernandez, K.M.; Juenger, T.E. Genotype-by-environment interaction and plasticity: Exploring genomic responses of plants to the abiotic environment. Annu. Rev. Ecol. Evol. Syst. 2013, 44, 5–29. [Google Scholar] [CrossRef]
- Kenkel, C.D.; Matz, M.V. Gene expression plasticity as a mechanism of coral adaptation to a variable environment. Nat. Ecol. Evol. 2016, 1, 0014. [Google Scholar] [CrossRef]
- Murren, C.J.; Auld, J.R.; Callahan, H.; Ghalambor, C.K.; Handelsman, C.A.; Heskel, M.A.; Kingsolver, J.G.; Maclean, H.J.; Masel, J.; Maughan, H.; et al. Constraints on the evolution of phenotypic plasticity: Limits and costs of phenotype and plasticity. Heredity 2015, 115, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.B. Natural pH gradients in hydrothermal alkali vents were unlikely to have played a role in the origin of life. J. Mol. Evol. 2016, 83, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wächtershäuser, G. In praise of error. J. Mol. Evol. 2016, 82, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Füllsack, M. Circularity and the micro-macro difference. Constructivist Foundations 2016, 12, 1. [Google Scholar]
- Lehman, N. The RNA world: 4,000,000,050 years old. Life 2015, 5, 1583–1586. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, R.C. Formation of the Genetic Code—Review and Update as of November 2012. 2013. Available online: http://www.icb.ufmg.br/labs/lbem/pdf/GMRTgeneticodeNov12.pdf. All original publications are available online: https://www.researchgate.net/profile/Romeu_Guimaraes (both sites accessed on December 2016).
- Caetano-Anollés, G.; Wang, M.; Caetano-Anollés, D. Structural phylogenomics retrodicts the origin of the genetic code and uncovers the evolutionary impact of protein flexibility. PLoS ONE 2013, 8, e72225. [Google Scholar] [CrossRef] [PubMed]
- Gulik, P.T.S.; Hoff, W.D. Anticodon modifications in the tRNA set of LUCA and the fundamental regularity in the standard genetic code. PLoS ONE 2016, 11, e0158342. [Google Scholar]
- Lacey, J.C., Jr.; Mullins, D.W., Jr. Experimental studies related to the origin of the genetic code and the process of protein synthesis—A review. Orig. Life Evol. Biosph. 1983, 13, 3–42. [Google Scholar] [CrossRef]
- Osawa, S. Evolution of the Genetic Code; Oxford University Press: Oxford UK, 1995. [Google Scholar]
- Xia, T.; SantaLucia, J., Jr.; Burkard, M.E.; Kierzek, R.; Schroeder, S.J.; Jiao, X.; Cox, C.; Turner, D.H. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry 1998, 37, 14719–14735. [Google Scholar] [CrossRef] [PubMed]
- Farias, S.T.; Moreira, C.H.C.; Guimarães, R.C. Structure of the genetic code suggested by the hydropathy correlation between anticodons and amino acid residues. Orig. Life Evol. Biosph. 2007, 37, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, R.C. Anti-complementary order in the genetic coding system. Int. Conf. Orig. Life 1996, 26, 435–436. [Google Scholar]
- Egholm, M.; Buchardt, O.; Nielsen, P.E.; Berg, R.H. Peptide nucleic acids (PNA). Oligonucleotide analogs with an achiral backbone. J. Am. Chem. Soc. 1992, 114, 1895–1897. [Google Scholar] [CrossRef]
- Francis, B.R. The Hypothesis that the Genetic Code Originated in Coupled Synthesis of Proteins and the Evolutionary Predecessors of Nucleic Acids in Primitive Cells. Life 2015, 5, 467–505. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, H.S. Clues to tRNA evolution from the distribution of class II tRNAs and serine codons in the genetic code. Life 2016, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Rogozin, I.B.; Belinsky, F.; Pavlenko, V.; Shabalina, S.A.; Kristensen, D.M.; Koonin, E.V. Evolutionary switches between two serine codon sets are driven by selection. Proc. Natl. Acad. Sci. USA 2016, 113, 13109–13113. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Zhang, J.; Ehrenberg, M. Genetic code translation displays a linear trade-off between efficiency and accuracy of tRNA selection. Proc. Natl. Acad. Sci. USA 2012, 109, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, R.C.; Erdmann, V.A. Evolution of adenine clustering in 5S ribosomal RNA. Endocyt. Cell Res. 1992, 9, 13–45. [Google Scholar]
- Lee, E.Y.; Lee, H.C.; Kim, H.K.; Jang, S.Y.; Park, S.J.; Kim, Y.H.; Kim, J.H.; Hwang, J.; Kim, J.H.; Kim, T.H.; et al. Infection-specific phosphorylation of glutamyl-prolyl tRNA synthetase induces antiviral immunity. Nat. Immunol. 2016, 17, 1252–1262. [Google Scholar] [CrossRef] [PubMed]
- Beuning, P.J.; Musier-Forsyth, K. Transfer RNA recognition by aminoacyl-tRNA synthetases. Biopolymers 1999, 52, 1–28. [Google Scholar] [CrossRef]
- Jackman, J.E.; Alfonzo, J.D. Transfer RNA modifications: Nature’s combinatorial chemistry playground. WIREs RNA 2013, 4, 35–48. [Google Scholar] [CrossRef] [PubMed]
- A Database of RNA Modifications. Available online: modomics.genesilico.pl/modifications (accessed on 10 November 2016).
- Seligmann, H. Natural mitochondrial proteolysis confirms transcription systematically exchanging/eleting nucleotides, peptides coded by expanded codons. J. Theor. Biol. 2017, 414, 76. [Google Scholar] [CrossRef] [PubMed]
- Lim, V.I. Analysis of action of wobble nucleoside modification on codon-anticodon pairing within the ribosome. J. Mol. Biol. 1994, 240, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Tzul, F.O.; Vasilchuk, D.; Makhatadze, G.I. Evidence for the principle of minimal frustration in the evolution of protein folding landscapes. Proc. Natl. Acad. Sci. USA 2017, E1627–E1632. [Google Scholar] [CrossRef] [PubMed]
- Marck, C.; Grosjean, H. tRNomics: Analysis of tRNA genes from 50 genomes of Eukarya, Archaea, and Bacteria reveals anticodon-sparing strategies and domain-specific features. RNA 2002, 8, 1189–1232. [Google Scholar] [CrossRef] [PubMed]
- Targanski, I.; Cherkasova, V. Analysis of genomic tRNA sets from Bacteria, Archaea and Eukarya points to anticodon-codon hydrogen bonds as a major determinant of tRNA compositional variation. RNA 2008, 14, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Agris, P.F. Decoding the genome: A modified view. Nucleic Acids Res. 2004, 32, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Creighton, T.E. Proteins: Structures and Molecular Properties; WH Freeman: New York, NY, USA, 1993. [Google Scholar]
- Radivojac, P.; Iakoucheva, L.M.; Oldfield, C.J.; Obradovic, Z.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder and functional proteomics. Biophys. J. 2007, 92, 1439–1456. [Google Scholar] [CrossRef] [PubMed]
- Dagliyan, O.; Tarnawski, M.; Chu, D.H.; Shirvanyants, D.; Schlichting, I.; Dokholyan, N.V.; Hahn, K.M. Engineering extrinsic disorder to control protein activity in living cells. Science 2016, 354, 1441–1444. [Google Scholar] [CrossRef] [PubMed]
- Gruszka, D.T.; Mendonça, C.A.T.F.; Paci, E.; Whelan, F.; Hawkhead, J.; Potts, J.R.; Clarke, J. Disorder drives cooperative folding in a multidomain protein. Proc. Natl. Acad. Sci. USA 2016, 113, 11841–11846. [Google Scholar] [CrossRef] [PubMed]
- Tuite, M.F. Remembering the past: A new form of protein-based inheritance. Cell 2016, 167, 302–303. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, R.C.; Moreira, C.H.C. Genetic code—A self-referential and functional model. In Progress in Biological Chirality; Pályi, G., Zucchi, C., Caglioti, L., Eds.; Elsevier: Oxford, UK, 2004; pp. 83–118. [Google Scholar]
- Varschavsky, A. The N-end rule: Functions, mysteries, uses. Proc. Natl. Acad. Sci. USA 1996, 93, 12142–12149. [Google Scholar] [CrossRef]
- Meinnel, T.; Sereno, A.; Giglione, C. Impact of the N-terminal amino acid on targeted protein degradation. Biol. Chem. 2006, 387, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.H.; Hernandez, G.R.; Grant, R.A.; Sauer, R.T.; Baker, T.A. The molecular basis of N-end rule recognition. Mol. Cell 2008, 32, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Oh, S.J.; Lee, B.G.; Song, H.K. Structural basis for dual specificity of yeast N-terminal amidase in the N-end rule pathway. Proc. Natl. Acad. Sci. USA 2016, 113, 12438–12443. [Google Scholar] [CrossRef] [PubMed]
- Berezovsky, I.N.; Kilosanidze, G.T.; Tumanyan, V.G.; Kisselev, L.L. Amino acid composition of protein termini are biased in different manners. Prot. Eng. 1999, 12, 23–30. [Google Scholar] [CrossRef]
- Guimarães, R.C. Two punctuation systems in the genetic code. In First Steps in the Origin of Life in the Universe; Chela-Flores, J., Owen, T., Raulin, F., Eds.; Kluwer: Dordrecht, The Netherlands, 2001; pp. 121–124. [Google Scholar]
- Chen, L.L. The biogenesis and emerging roles of circular RNAs. Nature Rev. Mol. Cell Biol. 2016, 17, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Beier, H.; Grimm, M. Misreading of termination codons in eukaryotes by natural nonsense suppressor tRNAs. Nucl. Acids Res. 2001, 29, 4767–4782. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Friesen, W.J.; Tomizawa, Y.; Leszyk, J.D.; Zhuo, J.; Johnson, B.; Dakka, J.; Trotta, C.R.; Xue, X.; Mutyam, V.; et al. Ataluren stimulates ribosomal selection of near cognate tRNAs to promote nonsense suppression. Proc. Natl. Acad. Sci. USA 2016, 113, 12508–12513. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ma, J.; Lu, W.; Tian, M.; Thauvin, M.; Yuan, C.; Volovitch, M.; Wang, Q.; Holst, J.; Liu, M.; et al. Heritable expansion of the genetic code in mouse and zebrafish. Cell Res. 2016, 27, 294. [Google Scholar] [CrossRef] [PubMed]
- Eltschinger, S.; Bütikofer, P.; Altmann, M. Translation elongation and termination—Are they conserved processes? In Evolution of the Protein Synthesis Machinery and Its Regulation; Hernández, G., Jagus, R., Eds.; Springer: Cham, Switzerland, 2016; pp. 277–311. [Google Scholar]
- Ma, C.; Kurita, D.; Li, N.; Chen, Y.; Himeno, H.; Gao, N. Mechanistic insights into the alternative termination by ArfA and RF2. Nature 2016, 541, 550. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.S.; Gulen, B.; Norris, A.M.; Kovacs, N.A.; Bernier, C.R.; Lanier, K.A.; Fox, G.E.; Harvey, S.C.; Wartel, R.M.; Hud, N.V.; et al. History of the ribosome and the origins of translation. Proc. Natl. Acad. Sci. USA 2015, 112, 15396–15401. [Google Scholar] [CrossRef] [PubMed]
- Colussi, T.M.; Constantino, D.A.; Hammond, J.A.; Ruehle, G.M.; Nix, J.C.; Kieft, J.S. The structural basis of tRNA mimicry and conformational plasticity by a viral RNA. Nature 2014, 511, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Loc’h, J.; Bloud, M.; Réty, S.; Lebaron, S.; Deschamps, P.; Barreille, J.; Jombart, J.; Paganin, J.R.; Delbos, L.; Chardon, F.; et al. RNA mimicry by the Fap7 adenylate kinase in ribosome biogenesis. PLoS Biol. 2014, 12, e1001860. [Google Scholar] [CrossRef] [PubMed]
- Marzi, S.; Romby, P. RNA mimicry, a decoy for regulatory proteins. Mol. Microbiol. 2012, 83, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Ito, K. tRNA mimicry in translation termination and beyond. WIREs RNA 2011, 2, 647–668. [Google Scholar] [CrossRef] [PubMed]
- Suetsuzu, K.H.; Sekine, S.I.; Sakai, H.; Takemoto, C.H.; Terada, T.; Unzai, S.; Tame, J.R.H.; Kuramitsu, S.; Shirouzu, M.; Yokoyama, S. Crystal structure of elongation factor P from Thermus thermophilus HB8. Proc. Natl. Acad. Sci. USA 2004, 101, 9595–9600. [Google Scholar]
- James, N.R.; Brown, A.; Gordiyenko, Y.; Ramakrishnan, V. Translational termination without a stop codon. Science 2016, 354, 1437–1440. [Google Scholar] [CrossRef] [PubMed]
- Zinshteyn, B.; Green, R. When stop makes sense. Science 2016, 354, 1106. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M.; Kwon, M.; Strobel, S.A. Toward ribosomal RNA catalytic activity in the absence of protein. J. Mol. Evol. 2007, 64, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, R.M.; Weixlbaumer, A.; Loakes, D.; Kelley, A.C.; Ramakrishnan, V. Insights into substrate stabilization from snapshots of the peptidyl transferase center of the intact 70S ribosome. Nat. Struct. Mol. Biol. 2009, 16, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.; Reed, C.; Nguyen, D.; Carter, M.; Wang, Y. Detection and formation scenario of citric acid, pyruvic acid, and other possible metabolism precursors in carbonaceous meteorites. Proc. Natl. Acad. Sci. USA 2011, 108, 14015–14020. [Google Scholar] [CrossRef] [PubMed]
- Di Giulio, M. An extension of the coevolution theory of the origin of the genetic code. Biol. Direct 2008, 3, 37. [Google Scholar] [CrossRef] [PubMed]
- Di Giulio, M. An autotrophic origin of the coded amino acids is concordant with the coevolution theory of the genetic code. J. Mol. Evol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hartman, H.; Smith, T.F. The evolution of the ribosome and the genetic code. Life 2014, 4, 227–249. [Google Scholar] [CrossRef] [PubMed]
- Higgs, P.G. A four-column theory for the origin of the genetic code: Tracing the evolutionary pathways that gave rise to an optimized code. Biol. Direct 2009, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Ikehara, K. Evolutionay steps in the emergence of life deduced from the bottom-up approach and ‘GADV hypothesis (top-down approach)’. Life 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Higgs, P.G. Pathways of genetic code evolution in ancient and modern organisms. J. Mol. Evol. 2015, 80, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Trifonov, E.N. Consensus temporal order of amino acids and evolution of the triplet code. Gene 2000, 261, 139–151. [Google Scholar] [CrossRef]
- Trifonov, E.N. The triplet code from first principles. J. Biomol. Struct. Dyn. 2004, 22, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.T.F.; Ng, S.K.; Mat, W.K.; Hu, T.; Xue, H. Coevolution theory of the genetic code at age forty: Pathway to translation and synthetic life. Life 2016, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, R.C. Metabolic basis for the self-referential genetic code. Orig. Life Evol. Biosph. 2011, 41, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdova, L.; Kalyuzhnaya, M.G.; Lidstrom, M.E. The expanding world of methylotrophic metabolism. Annu. Rev. Microbiol. 2009, 63, 477–499. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdova, L. Modularity of methylotrophy, revisited. Environ. Microbiol. 2011, 13, 2603–2622. [Google Scholar] [CrossRef] [PubMed]
- Florio, R.; Salvo, M.L.; Vivoli, M.; Contestabile, R. Serine hydroxymethyltransferase: A model enzyme for mechanistic, structural, and evolutionary studies. Biochim. Biophys. Acta 2011, 1814, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Braakman, R.; Smith, E. The emergence and early evolution of biological carbon-fixation. PLoS Comput. Biol. 2012, 8, e1002455. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Lin, P.P.; Magnusson, L.; Warner, L.; Liao, J.C.; Maness, P.C.; Chou, K.J. CO2-fixing one-carbon metabolism in a cellulose-degrading bacterium Clostridium thermocellum. Proc. Natl. Acad. Sci. USA 2016, 113, 13180–13185. [Google Scholar] [CrossRef] [PubMed]
- Kottakis, F.; Nicolay, B.N.; Roumane, A.; Karnik, R.; Gu, H.; Nagle, J.M.; Boukhali, M.; Hayward, M.C.; Li, Y.Y.; Chen, T.; et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 2016, 539, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.P.; Aurikko, J.P.; Kelly, D.P. A challenge for the 21st century molecular biology and biochemistry: What are the causes of obligate autotrophy and methanotrophy? FEMS Microbiol. Rev. RNA 2004, 28, 335–352. [Google Scholar] [CrossRef]
- Russell, M.J.; Martin, W. The rocky roots of the acetyl-CoA pathway. Trends Bioch. Sci. 2004, 29, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.C.; Neukirchen, S.; Roettger, M.; Mrujavac, N.; Sathi, S.N.; Martin, W.F.; Sousa, F.L. Reply to ‘Is LUCA a thermophilic progenote?’. Nat. Microbiol. 2016, 1, 16230. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Wächtershäuser, G. Activated acetic acid by carbon fixation on (Fe, Ni)S under primordial conditions. Science 1997, 276, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Holm, N.G.; Oze, C.; Mousis, O.; Waite, J.H.; Lepoutre, A.G. Serpentinization and the formation of H2 and CH4 on celestial bodies (planets, moons, comets). Astrobiology 2015, 15, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Grosjean, H.; Westhof, E. An integrated, structure- and energy-based view of the genetic code. Nucleic Acids Res. 2016, 44, 8020–8040. [Google Scholar] [CrossRef] [PubMed]
- Grantham, R. Amino acid difference formula to help explain protein evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Sobolevsky, Y.; Guimarães, R.C.; Trifonov, E.N. Towards functional repertoire of the earliest proteins. J. Biomol. Struct. Dyn. 2013, 31, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Sprinzl, M. Chemistry of aminoacylation and peptide bond formation on the 3′ terminus of tRNA. J. Biosci. 2006, 311, 489–496. [Google Scholar] [CrossRef]
- Ambrogelly, A.; Söll, D.; Nureki, O.; Yokoyama, S.; Ibba, M. Class I lysyl-tRNA synthetases. NCBI Bookshelf. Landes Bioscience: Madame Curie Bioscence Database: Austin TX, USA, 2013. Available online: http://www.ncbi.nlm.nih.gov/books/NBK6444 (accessed on December 2016).
- Xu, X.M.; Carlson, B.A.; Mix, H.; Zhang, Y.; Saira, K.; Glass, R.S.; Barry, M.J.; Gladyshev, V.N.; Hatfield, D.L. Biosynthesis of selenocysteine on its tRNA in eukaryotes. PLoS Biol. 2007, 5, e4. [Google Scholar] [CrossRef] [PubMed]
- Englert, M.; Moses, S.; Hohn, M.; Ling, J.; O′Donohue, P.; Söll, D. Aminoacylation of tRNA 2′- or 3′-hydroxyl by phosphoseryl- and pyrrolysyl-tRNA synthetases. FEBS Lett. 2013, 587, 3360–3364. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.M.; Erdogan, O.; Rodriguez, M.J.; Rivera, K.G.; Williams, T.; Li, L.; Weinreb, V.; Collier, M.; Chandrasekaran, S.N.; Ambrozzio, X.; et al. Functional class I and class II amino acid activating enzymes can be coded by opposite strands of the same gene. J. Biol. Chem. 2015, 290, 19710. [Google Scholar] [CrossRef] [PubMed]
- Bloch, D.P.; McArthur, B.; Widdowson, R.; Spector, D.; Guimarães, R.C.; Smith, J. tRNA-rRNA sequence homologies: A model for the generation of a common ancestral molecule and prospects for its reconstruction. Orig. Life Evol. Biosph. 1984, 14, 571–578. [Google Scholar] [CrossRef]
- Bloch, D.P.; McArthur, B.; Guimarães, R.C.; Smith, J.; Staves, M.P. tRNA-rRNA sequence matches from inter- and intraspecies comparisons suggest common origins for the two RNAs. Braz. J. Med. Biol. Res. 1989, 22, 931–944. [Google Scholar] [PubMed]
- Kanai, A. Disrupted tRNA genes and tRNA fragments: A perspective on tRNA gene evolution. Life 2015, 5, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.; Root-Bernstein, M. The ribosome as a missing link in prebiotic evolution II: Ribosomes encode ribosomal proteins that bind to common regions of their own mRNAs and rRNAs. J. Theor. Biol. 2016, 397, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Macé, K.; Gillet, R. Origins of tmRNA: The missing link in the birth of protein synthesis? Nucl. Acids Res. 2011, 44, 8041–8051. [Google Scholar] [CrossRef] [PubMed]
- Ogle, J.M.; Brodersen, D.E.; Clemens Jr, W.M.; Tarry, M.J.; Carter, A.P.; Ramakrishnan, C.V. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science 2001, 293, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.; Libchaber, A. Degeneracy of the genetic code and stability of the base pair at the second position of the anticodon. RNA 2008, 14, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Massey, S.E. The neutral emergence of error minimized genetic codes superior to the standard genetic code. J. Theor. Biol. 2016, 408, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, R.C. The Self-Referential Genetic Code is Biologic and Includes the Error Minimization Property. Orig. Life Evol. Biosph. 2015, 45, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, R.C.; Moreira, C.H.C.; Farias, S.T. A self-referential model for the formation of the genetic code. Theory Biosci. 2008, 127, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, R.C.; Moreira, C.H.C.; Farias, S.T. Self-referential formation of the genetic system. In The Codes of Life—The Rules of Macroevolution; Barbieri, M., Ed.; Springer: Dordrecht, The Netherlands, 2008; pp. 68–110. [Google Scholar]
- Caetano-Anollés, D.; Caetano-Anollés, G. Piecemeal buildup of the genetic code, ribosomes, and genomes from primordial tRNA building blocks. Life 2016, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Fung, A.W.S.; Payoe, R.; Fahlman, R.D. Perspectives and insights into the competition for aminoacyl-tRNAs between the translational machinery and for tRNA-dependent non-ribosomal peptide bond formation. Life 2016, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Goudry, M.; Saugnet, L.; Belin, P.; Thai, R.; Amoureux, R.; Tellier, C.; Tuphile, K.; Jacqet, M.; Braud, S.; Courçon, M.; et al. Cyclodipeptide synthases are a family of tRNA-dependent peptide bond-forming enzymes. Nat. Chem. Biol. 2009, 5, 414–420. [Google Scholar]
- Mocibob, M.; Ivic, N.; Bilokapic, S.; Maier, T.; Luic, M.; Ban, N.; Durasevic, I.W. Homologs of aminoacyl-tRNA synthetases acylate carrier proteins and provide a link between ribosomal and nonribosomal peptide synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 14585–14590. [Google Scholar] [CrossRef] [PubMed]
- Moutiez, M.; Schmidt, E.; Seguin, J.; Thai, R.; Favry, E.; Belin, P.; Mechulan, Y.; Goudry, M. Unravelling the mechanism of non-ribosomal peptide synthesis by cyclopeptide synthases. Nat. Commun. 2014, 5, 5141–5146. [Google Scholar] [CrossRef] [PubMed]
- Caetano-Anollés, D.; Caetano-Anollés, G. The phylogenomic roots of translation. In Evolution of the Protein Synthesis Machinery and its Regulation; Hernandez, G., Jagus, R., Eds.; Springer: Cham, Switzerland, 2016; pp. 9–30. [Google Scholar]
- Dill, K.A.; Ghosh, K.; Schmit, J.D. Physical limits of cells and proteomes. Proc. Natl. Acad. Sci. USA 2011, 108, 17876–17882. [Google Scholar] [CrossRef] [PubMed]
- Deatherage, B.L.; Cookson, B.T. Membrane vesicle release in bacteria, eukaryotes, and archaea: A conserved yet underappreciated aspect of microbial life. Infect. Immun. 2012, 80, 1948–1957. [Google Scholar] [CrossRef] [PubMed]
- Errington, J. L-form bacteria, cell walls and the origins of life. Open Biol. 2013, 3, 120–143. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Yutin, N.; Bell, S.D.; Koonin, E.V. Evolution of diverse cell division and vesicle formation systems in archaea. Nat. Rev. Microbiol. 2010, 8, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Mercier, R.; Kawai, Y.; Errington, J. Excess membrane synthesis drives a primitive mode of cell proliferation. Cell 2013, 152, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Nyström, T. Spatial protein quality control and the evolution of lineage-specific ageing. Philos. Trans. R. Soc. Lond. B 2011, 366, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, R.C. Emergence of information patterns: In the quantum and biochemical realms. Quantum Biosyst. 2015, 6, 148–159. [Google Scholar]
- Wills, P.R. The generation of meaningful information in molecular systems. Philos. Trans. R. Soc. A 2016, 374, 20150066. [Google Scholar] [CrossRef] [PubMed]
- Callahan, M.P.; Smith, K.E.; Cleaves, H.J., 2nd; Ruzicka, J.; Stern, J.C.; Glavin, D.P.; House, C.H.; Dworkin, J.P. Carbonaceous meteorites contain a wide range of extraterrestrial nucleobases. Proc. Natl. Acad. Sci. USA 2011, 108, 13995–13998. [Google Scholar] [CrossRef] [PubMed]
- Ertem, G.; O´Brien, A.M.S.; Ertem, M.C.; Rogoff, D.A.; Dworkin, J.P.; Johnston, M.V.; Hazen, R.M. Abiotic formation of RNA-like oligomers by montmorillonite catalysis: Part II. Int. J. Astrobiol. 2008, 7, 1–7. [Google Scholar] [CrossRef]
- Powner, M.W.; Gerland, B.; Sutherland, J.D. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 2009, 459, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Santoso, S.; Hwang, W.; Hartman, H.; Zhang, S.G. Self-assembly of surfactant-like peptides with variable glycine tails to form nanotubes and nanovesicles. Nano Lett. 2002, 2, 687–691. [Google Scholar] [CrossRef]
- Rizzotti, M. Early Evolution; Birkhäuser: Basel, Switzerland, 2000. [Google Scholar]
- Kauffman, S.A. The Origins of Order: Self-Organization and Selection in Evolution; Oxford University Press: Oxford, UK, 1993. [Google Scholar]
- Russell, M. Thinking about life: Adding (thermo)dynamic aspects to definitions of life. In Evolution and Transitions in Complexity—The Science of Hierarchical Organization in Nature; Springer: Cham, Switzerland, 2016; pp. 199–202. [Google Scholar]
- Morowitz, H.; Smith, E. Energy flow and the organization of life. Complexity 2007, 13, 51–59. [Google Scholar] [CrossRef]
- Illangasekare, M.; Yarus, M. A tiny RNA that catalyzes both aminoacyl-RNA and peptidyl-RNA synthesis. RNA 1999, 5, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- Yarus, M. The meaning of a minuscule ribozyme. Philos. Trans. R. Soc. Lond. B 2011, 366, 2902–2909. [Google Scholar] [CrossRef] [PubMed]
- Goldford, J.E.; Hartman, H.; Smith, T.F.; Segrè, D. Remnants of an ancient metabolism without phosphate. Cell 2017, 168, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Ricardo, A.; Carrigan, M.A.; Olcott, A.N.; Benner, S.A. Borate Minerals Stabilize Ribose. Science 2004, 303, 196. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | ||||||||
| Wobble | Principal dinucleotide | → | ||||||
| 5’ N | Central | 3’ | Anticodon | |||||
| 3’ | Central | 5’ N | Anticodon | |||||
| Principal dinucleotide | Wobble | ← | ||||||
| Generic R:Y | Standard G:C, A:U | Generic R:Y | Base pair | |||||
| (B) | ||||||||
| Sector | Homogeneous Principal Dinucleotides | |||||||
| Modules | Central G:C | Central A:U | ||||||
| Pairs Elongation | 1a | 1b | 2a | 2b | ||||
| NGG CCN | NGA UCN | NAG CUN | NAA UUN | |||||
| 1st occupier (5′N) | Gly Gly | Ser Ser | Leu Asp | Leu Asn | ||||
| Present (5′N or 5’G/5’Y) | Pro Gly | Ser Ser/Arg | Leu Asp/Glu | Phe/Leu Asn/Lys | ||||
| Sector | Mixed Principal Dinucleotides | |||||||
| Pairs Elongation | 3a | 3b | 4a | 4b | ||||
| NCG CGN | NCA UGN | NUG CAN | NUAUAN | |||||
| 1st occupier (5’N) | Arg Ala | Cys Thr | His Val | Tyr Ile | ||||
| Present (5’N or 5’G/5’Y) | Arg Ala | Cys/Trp Thr | His/Gln Val | Tyr Ile/Met | ||||
| Punctuation | /Met, iMet | |||||||
| /Trp, X | Tyr/X | |||||||
| Sector of Principal Dinucleotide (pDiN) | Homogeneous pDiN | Mixed pDiN | |||
|---|---|---|---|---|---|
| Base size configuration of the anticodon triplets | NRR | Nyy | NRy | NyR | |
| Self-complementary triplet | Triplet | yRR | Gyy | GRy | yyR |
| SC | Purine (R) | •• | • | •• | • |
| lateral bases one R the other y | Pyrimidine (y) | • | •• | • | •• |
| Nonself-complementary triplet | Triplet | GRR | yyy | yRy | GyR |
| NSC | Purine (R) | ••• | • | • • | |
| lateral bases both R or both y | Pyrimidine (y) | ••• | • • | • | |
| aRS | Stage | Anticodon | Nucleotide 73 | Acceptor Arm Pair | ||||
|---|---|---|---|---|---|---|---|---|
| 34 | 35–36 | 1:72 | 2:71 | 3:70 | 4:69 | |||
| Hexa-, Tetracodonic (ancestral states): no interaction with the anticodon or binding through the principal dinucleotide (35–36) only | ||||||||
| Ser | 1 | - | - | + | + | + | + | |
| Leu | 2 | - | + | - | - | - | - | |
| Arg | 3 | - | + | +Weak | - | - | - | - |
| Ala | 4 | - | + | + | + | + | - | |
| Gly | 1 | - | + | + | + | + | +w | - |
| Thr | 4 | - | + | - | + | + | - | - |
| Pro | 3 | - | + | + | + | - | - | - |
| Val | 5 | - | + | + | - | - | - | +w |
| Mono-, Dicodonic (derived states, multi-meaning boxes): binding through the three anticodon positions | ||||||||
| Gln | 5 | + | + | + | + | + | - | |
| Cys | 4 | + | + | - | + | + | - | |
| Met | 6 | + | + | - | + | + | - | |
| Glu | 3 | + | + | + | + | - | - | |
| Asp | 2 | + | + | +w | +w | - | - | |
| Ile | 6 | + | + | - | - | - | +w | |
| Trp | 4 | + | + | - | - | - | - | |
| Asn | 2 | + | + | - | - | - | - | |
| Lys | 3 | + | + | - | - | - | - | |
| Phe | 3 | + | Weak | - | - | - | - | |
| Ambiguous: dicodonic, binding in the ancestral modes | ||||||||
| Tyr | 6 | - | + | + | + | - | - | - |
| His | 5 | +Weak (w) | + | + | - | - | - | |
| Sectors of Anticode Dimers (Wobble + Principal Dinucleotide) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Homogenous Sector, RNP Realm | Mixed Sector, DNP Realm | |||||||
| Modules of tRNA Pairs | 1 G S | 2 L D N | Sector mature E P F K R | Total wRR YYw | 3 R A T C W | 4 V H Q I M Y | Punctu ation iM, X | Total wRY RYw |
| Non-periodical, [34] Coils, Turns | G S | D N | P | 5 | 0 | |||
| Helices | L | E K R | 3–4 | R A | H Q M | 4–5 | ||
| Strands | F | 1 | T C W | V I Y | 6 | |||
| Disorder [35] | S | E P K R | 4–5 | R | Q M | 2–3 | ||
| Borderline, Neutral | G | DN | 3 | A T | H | 3 | ||
| Order | L | F | 2 | C W | V I Y | 5 | ||
| Sectors of Anticode Dimers (Wobble + Principal Dinucleotide) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Homogenous Sector, RNP Realm | Mixed Sector, DNP Realm | |||||||
| Modules of tRNA pairs | 1 G S | 2 L D N | Sector mature E P F K R | Total wRR YYw | 3 R A T C W | 4 V H Q I M Y | Punctu ation iM, X | Total wRY RYw |
| RNA binding [39] | G S | L | P K F | 6 | V M | 2 | ||
| Both | R | 0–1 | R W | I Y Q | 4–5 | |||
| DNA binding | E | 1 | A C T | H | 4 | |||
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardoso Guimarães, R. Self-Referential Encoding on Modules of Anticodon Pairs—Roots of the Biological Flow System. Life 2017, 7, 16. https://doi.org/10.3390/life7020016
Cardoso Guimarães R. Self-Referential Encoding on Modules of Anticodon Pairs—Roots of the Biological Flow System. Life. 2017; 7(2):16. https://doi.org/10.3390/life7020016
Chicago/Turabian StyleCardoso Guimarães, Romeu. 2017. "Self-Referential Encoding on Modules of Anticodon Pairs—Roots of the Biological Flow System" Life 7, no. 2: 16. https://doi.org/10.3390/life7020016
APA StyleCardoso Guimarães, R. (2017). Self-Referential Encoding on Modules of Anticodon Pairs—Roots of the Biological Flow System. Life, 7(2), 16. https://doi.org/10.3390/life7020016