Abstract
Prebiotic organic synthesis catalyzed by Earth-abundant metal sulfides is a key process for understanding the evolution of biochemistry from inorganic molecules, yet the catalytic functions of sulfides have remained poorly explored in the context of the origin of life. Past studies on prebiotic chemistry have mostly focused on a few types of metal sulfide catalysts, such as FeS or NiS, which form limited types of products with inferior activity and selectivity. To explore the potential of metal sulfides on catalyzing prebiotic chemical reactions, here, the chemical diversity (variations in chemical composition and phase structure) of 304 natural metal sulfide minerals in a mineralogy database was surveyed. Approaches to rationally predict the catalytic functions of metal sulfides are discussed based on advanced theories and analytical tools of electrocatalysis such as proton-coupled electron transfer, structural comparisons between enzymes and minerals, and in situ spectroscopy. To this end, we introduce a model of geoelectrochemistry driven prebiotic synthesis for chemical evolution, as it helps us to predict kinetics and selectivity of targeted prebiotic chemistry under “chemically messy conditions”. We expect that combining the data-mining of mineral databases with experimental methods, theories, and machine-learning approaches developed in the field of electrocatalysis will facilitate the prediction and verification of catalytic performance under a wide range of pH and Eh conditions, and will aid in the rational screening of mineral catalysts involved in the origin of life.
1. Introduction
The origin of life on Earth is generally envisioned as having started from abiotic syntheses of basic building blocks requisite for metabolism and replication [1,2,3,4,5]. Metal sulfides have been proposed as key players in these prebiotic processes in several scenarios, such as the “iron–sulfur world” by Wächtershäuser [6,7], the “iron–sulfur membrane model” by Russell and Hall [5,8,9] and more recently by Lane and Martin [10], the “zinc world” hypothesis by Mulkidjanian and Galperin [11,12], and the “geoelectrochemistry driven origin of life” by Nakamura, Yamamoto [13,14,15,16,17,18], and Barge [19,20,21]. Metal sulfides are ubiquitous in reducing environments, including sulfidic ores [22,23], deep-sea hydrothermal vent deposits [8,24,25,26], sulfide-rich euxinic sediment environments (e.g., black sea) [27,28,29], and black shale [30,31,32]. In deep-sea hydrothermal vent environments, where massive sulfide deposits are produced, many dissolved transition metals are concentrated in hydrothermal fluids mainly as chloride complexes [22,23]. The generally low solubility of metal sulfides possibly limited the bio-availability of metal ions in the early ocean and has led to the assumption that the availability of metal ions in the ocean may have constrained metabolic pathways in early life [33,34,35].
Deep-sea hydrothermal systems associated with the serpentinization of ultramafic rocks are among the most plausible geological settings for life to have originated [8,10,13,15,17,18,36,37]. The temperature, pH, and chemical composition differences between hydrothermal fluid and seawater generate a steep redox gradient across the sulfide-rich vent rocks, thereby serving as the driving force for prebiotic chemistry [5,8,9]. In addition, the generation of a chemical potential gradient across electrically conductive metal sulfides provides a continuous and unidirectional supply of high-energy electrons from the inside of the vent to the outside of the rock wall, as has been proposed based on the electrochemical analysis of hydrothermal vent minerals [17,19,21] and the electrochemical potential mapping of deep-sea hydrothermal fields [13,17,18]. In this environment, the high-energy electrons continuously supplied by the natural geoelectrochemical reactor trigger CO2 reduction to CO and CH4 and nitrate and nitrite reduction to NO, N2O, and NH3, as demonstrated in recent laboratory investigations [14,15,16,38]. Under the continuous flow of electrical current from a hot, reductive hydrothermal fluid to cold seawater, the ocean-vent interface is ideal for prebiotic chemistry, as organic molecules crucial for the life’s origin, including α-keto acids, amino acids, and oligonucleotides are typically unstable at high temperatures [5,8]. In addition, the unique nano- and micro-scale structures of metal sulfides in hydrothermal vent environments may have promoted the concentration and organization of the synthesized molecules, preventing them from diffusing into seawater [39,40].
The thermo- and electro-catalytic properties of metal sulfides are critical for understanding how the pH, temperature, and Eh disequilibria between hydrothermal fluids and seawater trigger prebiotic organic synthesis. In mineralogy, metal sulfides have been studied mainly with respect to crystallography, thermodynamic and high-pressure phase transition, air stability, electric and magnetic properties, trace element incorporation, and mineral conversion [41,42]. Their thermo-catalytic activities towards hydrogenation, hydrodesulfurization and hydrodenitrogenation have also been studied under gas-phase, high temperature conditions in petrochemistry since the 1920s [43,44]. However, the thermal and electrochemical catalysis of metal sulfides remain poorly explored in the context of the origin of life, particularly under simulated hydrothermal vent conditions.
To synthesize macromolecules (phospholipids, oligo-nucleotides, and peptides) and their respective building blocks (fatty acids, glycerol, ribose, nucleobases, nucleotides, and amino acids) from simple molecules (CO2, N2, NO3−/NO2−, and H2) to form a complex prebiotic chemical network, efficient mineral catalysts must be identified, which requires extensive screening efforts. Although several studies have used sulfides as catalysts for abiotic carbon and nitrogen fixation and peptide bond formation [14,15,16,38,45,46,47,48,49,50], limited types of organic products were typically formed and the reported activities and selectivity were generally much lower than those found in contemporary biological systems. These problems suggest that new types of earth-abundant mineral catalysts that function efficiently under geologically relevant conditions are needed for prebiotic synthesis.
Numerous minerals are identified in natural environments yearly, contributing to a huge library of mineral catalysts, and the number of unknown minerals awaiting discovery is predicted to be even larger [51,52]. As will be presented in this review, natural metal sulfides in mineralogy databases show marked variation in chemical composition, crystal structure and metal/sulfur valence states, and therefore have the potential for a wide variety of catalytic functions. However, prebiotic experiments conducted to date have rarely taken into account these diversities, and have typically only used a limited number of single-metal sulfides, such as FeS and NiS, with a few exceptions [14,15,53], most likely due to the lack of the general principle to predict the catalytic function of natural mineral samples. Notably, in the field of heterogeneous catalysis, earth-abundant transition metal sulfides have attracted keen attention as cost–effective catalysts for energy conversion applications [54]. In particular, the hydrogen evolution, hydrogen oxidation, CO2 reduction, nitrate/nitrite reduction, and N2 reduction catalyzed by metals, metal oxides, and metal sulfides have been analyzed to identify the structural and electronic features of intermediates involved in the targeted reactions and determine the reaction pathways. Moreover, the application of the advanced theory of proton-coupled electron transfer allows prediction of how the activity and selectivity of prebiotic catalysts are potentially influenced by solution pH and Eh [55,56,57].
In the present review, we highlight the significance of the chemical diversity and complexity of natural metal sulfides for facilitating prebiotic chemical evolution and propose rational approaches for screening mineral catalysts from mineralogy databases. First, prebiotic synthesis experiments conducted to date using sulfides as catalysts will be summarized and the chemical diversity of metal sulfides in the mineralogy database will be surveyed with emphasis on Cu, Co, Fe, Mn, Mo, Ni, V, and W as the metal components, as these elements contain multiple accessible oxidation states and are involved in biological redox reactions with sulfur as a pervasive ligand. Transition metal sulfides have been proposed to have an evolutionary significance as protoenzyme catalysts [4,16,58,59]. Further, the structure-function relationship of metal sulfides is discussed based on material and catalysis engineering research findings. In addition, studies on metal sulfides with well-defined crystal phases, chemical composition, and defect structures are reviewed. Finally, theoretical and experimental approaches for probing structure-function relationships (i.e., how the structure of active sites dictates the overall performance of catalysts) and predicting heterogeneous catalytic properties are introduced. By presenting this integrated information, we hope to promote the screening and performance prediction of sulfide catalysts in prebiotic processes that preceded the origin of life.
2. Catalysis on Metal Sulfides Relevant to Prebiotic Chemistry
2.1. Thermal Catalysis on Metal Sulfides
Experiments on sulfide-promoted prebiotic reactions reported to date can be divided into two categories: non-catalytic (stoichiometric) and catalytic. A representative example of a non-catalytic process is associated with Wächtershäuser’s proposal of an “iron–sulfur world”, where FeS oxidation from mackinawite to pyrite (FeS + H2S → FeS2 + 2H+ + 2e−) provides reducing energy for organic syntheses. Huber and Wächtershäuser demonstrated that the reductive amination of α-keto acids to the corresponding α-amino acids (e.g., pyruvate + NH3 + 2H+ + 2e− → alanine + H2O) occurs on freshly precipitated FeS, which was simultaneously oxidized to Fe3+ [50]. Interestingly, the reaction showed a strong metal specificity, as replacing FeS with Ag2S, CoS, Cr2S3, CuS, MgS, MnS, NiS, or ZnS did not result in amino acid formation. Heinen and Lauwers showed that CO2 was reduced to alkane thiols in a serum bottle containing FeS, H2S, water, and pure CO2 gas or a mixture of N2 and CO2 gases [60], with the formation of pyrite and H2 as byproducts.
In addition to carbon fixation, ammonia serves as an important nitrogen source for the synthesis of amino acids, nucleotides, and other N-containing biomolecules [1]. Therefore, prebiotic ammonia synthesis has been investigated by reducing either nitrate/nitrite anions or dinitrogen. FeS can reduce nitrate and nitrite to ammonia in an acidic solution (pH 4) with the simultaneous oxidation of FeS to Fe2O3 [46]. The reaction was inhibited effectively in the presence of either Cl−, SO42− or H2PO4−, likely owing to the low binding affinity of nitrate on the catalyst surface. Brandes et al. [61] reported that pyrite and pyrrhotite reduce nitrate and nitrite to ammonia at a high yield (21%~89%) at temperatures between 300 and 700 °C and pressures of 0.1–0.4 GPa. These researchers also observed that Fe/H2O/N2 or Fe3O4/HCOOH/N2 systems reduce dinitrogen to ammonia at optimal temperatures of 700 and 500 °C, respectively, and a pressure of 0.1 GPa, albeit with a lower yield than that obtained from nitrate/nitrite reduction [61]. Based on these results, an oceanic or hydrothermal source for reduced nitrogen on early Earth was proposed. Dörr et al. [62] demonstrated the reduction of N2 to ammonia on freshly precipitated FeS using H2S as a reductant under milder conditions (under 1 bar N2 at 70–80 °C) with a yield of 0.1%. The Fe-S cluster-like surface moieties, which resemble the active center of FeMoS nitrogenase, were proposed to play a key role in the N2 activation.
For catalytic conversions of carbon and nitrogen-bearing molecules, Huber and Wächtershäuser reported that FeS, NiS, and (Fe,Ni)S provide favorable surface sites for activated thioester (CH3-CO-SCH3) formation from CO and CH3SH [47] and for amino acid polymerization via CO and H2S condensation to COS [49]. The former reaction resembles that found in the reductive acetyl-coenzyme A pathway, in which Ni and Fe centers are responsible for binding CH3SH and CO, respectively. Cody et al. [63] examined the condensation of alkylthiol (RSH; R = nonyl group) and CO under high temperature (250 °C) and pressure (200 MPa) condition in the presence of FeS and detected a small amount of pyruvate formation. FeS was inferred to activate pyruvate synthesis by forming a carbonylated iron sulfur complex (Fe2(RS)2(CO)6). Further, Cody et al. [53] assayed several nickel, cobalt, iron, copper, and zinc containing sulfides for hydrocarboxylation under the same high temperature and pressure condition and found that carboxylic acid was synthesized via carbonyl insertion at metal-sulfide-bound alkyl groups. With the exception of CuS, all of the metal sulfides promoted hydrocarboxylation. Because many transition metal complexes, including tertiary phosphate complexes of Co2+, Fe2+, Mo, and Ni2+, CH3Mn(CO)5 [64,65,66], are capable of mediating carbonyl insertion, similar chemical reactions can potentially proceed on various types of metal sulfides. In presence of FeS minerals (pyrrhotite, troilite, pyrite, and arseno-pyrite) together with H2, H2S, and CO2, Novikov et al. [67] demonstrated the conversion of pyruvate to lactate and/or reduced aldol compounds at more moderate temperatures (25–110 °C). These reactions were considered to be important because of their participation in extant metabolic pathways. Notably, addition of NH4Cl induced the reductive amination of pyruvate to alanine. Metal sulfides can also facilitate the condensation of formamide to purine and pyrimidine nucleic bases. Saladino et al. [68] conducted a heating experiment of formamide in the presence of FeS and (Fe,Cu)S minerals as catalysts at 160 °C for 48 h. In contrast to purine as the only product in the absence of any catalyst, several types of precursors of primary metabolism, such as purine and adenine, and of nucleobase isomers, such as isocytosine, 2-aminopurine, and 2(1H)-pyrimidinone, were synthesized. The presence of iron was a crucial factor for the synthesis of purine derivatives. Sulfides with both Fe and Cu, including chalcopyrite FeCuS2 and tetrahedrite (Fe,Cu,Sb)S, show enhanced yield towards isocytosine and 2(1H)-pyrimidinone products.
2.2. Electrocatalysis on Metal Sulfides
Recently, a model of geoelectrochemistry driven organic synthesis was proposed based on the detection of electrical current flow in natural deep-sea hydrothermal fields [13,17,18] and demonstration of the relevant organic syntheses in a laboratory setting [14,15,16,19,21,38]. The temperature, pH, and chemical composition differences between hydrothermal fluid and seawater generate a steep redox gradient across sulfide-rich vent rocks (Figure 1). This unique geoelectrochemical environment results in continuous and unidirectional electron flow from the internal hydrothermal fluid to the external chimney rock wall by coupling the oxidation of H2 or H2S in the hydrothermal fluid with the reduction of oxidative species in sea water, such as H2O, CO2, and nitrate/nitrite. The wide range of redox potentials and pHs in this environment control the kinetics and selectivity of target reactions even under chemically messy conditions, circumventing the lack of stereoselectivity seen in modern enzymatic systems [14,15,16]. As the reaction energetics and kinetics are dependent on multiple parameters (Eh, pH, and T), the distribution of products can be tuned based on the wide variation of geochemical environmental parameters, which have the potential to be rationally predicted based on the theory of proton-coupled electron transfer [55,56,57]. In addition, if the electrochemical fuel cell model (two-container model for prebiotic reactions; Figure 1) is applied to prebiotic reactions, even strong endothermic reactions can be initiated, as the pH (ΔpH) and temperature gradients (ΔT) across the electrically conductive, yet thermally insulative vent wall [68] can generate high-energy electrons at cold, ocean-vent interfaces [13,14,15,16,17,69].
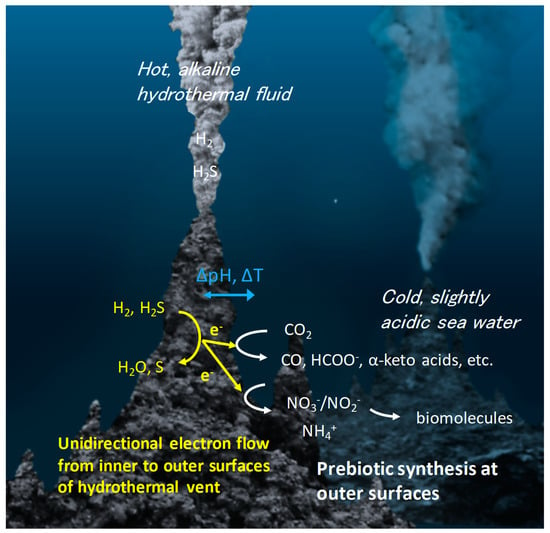
Figure 1.
Model of geoelectrochemical prebiotic synthesis for chemical evolution. In deep-sea hydrothermal vents, electrons generated from the oxidation of H2 and H2S in the hydrothermal fluids flow towards the seawater-rock interface through the porous, conductive metal sulfide chimney minerals. The reduction of molecules, such as CO2, NO3−, NO2−, at the seawater-rock interface is catalyzed by metal sulfides to generate various types of life’s building blocks, which further undergo condensation or polymerization to bio-essential polymers.
Based on the electrochemical fuel cell model, Yamaguchi et al. [15] demonstrated the electrocatalytic reduction of CO2 to CO and CH4 on greigite (Fe3S4) under simulated alkaline hydrothermal vent conditions. As CO2 reduction will not proceed without overcoming the high energetic barrier associated with the first electron uptake step (‒1.9 V vs. the standard hydrogen electrode, SHE at pH 7) and also competes with the thermodynamically and kinetically more favorable reduction of protons to H2, the Faraday efficiency for CO production (CO2 + 2H+ + 2e− → CO + H2O) is low (<0.02% in total at ‒1.3 V vs. SHE) at pH 5.5. However, by doping FeS with Ni to form FeNi2S4 (violarite), the CO2 electroreduction selectivity is enhanced by ~85-fold concurrently with a lowered onset potential (‒0.5 V vs. SHE, pH 5.5). CO2 reduction activity is further enhanced by modifying Ni-doped FeS catalyst with amine compounds [15]. Yamaguchi et al. have also found that the onset potential for CO2 reduction on Ni-doped FeS catalyst is ~180 mV more negative than the redox couple of H+/H2 at pH 5.5, suggesting that a naturally occurring proton motive force (PMF), which is equivalent to a 3–4 pH gradient, is sufficient to initiate CO2 reduction to CO when the hydrothermal fluid pH is higher than 9. Note that violarite resembles the NiFe4S4 active center of carbon monoxide dehydrogenase, and the amine species further enhance the CO2 reduction activity, similar to the peptide amine group surrounding the enzymatic active center (ligand-assisted catalysis). Amine-induced enhancement was associated with the ability of violarite to donate electrons to the anti-bonding π* orbital of CO2, which is essential for the efficient electrochemical activation of CO2 [69].
A study by Kitadai et al. [14] using various single-metal sulfides (Ag2S, CdS, CoS, CuS, FeS, MnS, MoS2, NiS, PbS, WS2, and ZnS) demonstrated that CdS and Ag2S function as excellent CO production catalysts (CO2 + 2H+ + 2e‒ → CO + H2O). The Faraday efficiency was as high as 40% on CdS at ≤−1.0 V (vs. SHE) at pH 5.5–6. The selective production of CO from CO2 by CdS and Ag2S is remarkable, because even though CO is frequently used as a carbon source for organic synthesis through thermocatalysis, CO is considered to have been present in only trace amounts in the Hadean atmosphere [70]. However, direct CO2 fixation into C2‒C3 compounds, such as acetate and pyruvate, has not been demonstrated in the presence of metal sulfides under electrochemical conditions. One exception was reported by Roldan et al. [38], who demonstrated CO2 reduction on highly faceted greigite nanoparticles into a series of C1‒C3 compounds, including formic acid, acetic acid, methanol, and pyruvic acid. In contrast to the reports by Yamaguchi [15] and Kitadai [14], Roldan et al. [38] cycled the electrode potential continuously during electrochemical CO2 reduction between 0.2 and −0.8 V vs. SHE (pH 6.5) at a slow rate (1 mV s−1), which may have promoted the formation of C‒C bonds.
In the geoelectricity driven prebiotic catalysis model, trace metal elements, such as Mo and W, could have played a significant role in prebiotic chemistry, as the continuous supply of electrons to the metal sites maintains the catalytic cycle. For example, Li et al. [16] demonstrated that molybdenum sulfide (MoS2) and Mo-doped greigite serve as electrocatalysts for reducing nitrate and nitrite to ammonia. It was found that in the presence of anions, such as phosphate, sulfate, iron sulfide impairs nitrate reduction ability, similar to the inhibition observed on FeS by Summers et al. [46]. In contrast, molybdenum sulfide can catalyze both nitrate and nitrite reduction over a wide pH range (pH 3–11) in the presence of phosphate and sulfate anions. In a neutral medium using molybdenum sulfide as the catalyst, ammonia was generated through nitrate reduction at an onset potential of 0 V vs. RHE (reversible hydrogen electrode). At more positive potential regions (0.6~0 V vs. RHE), not only ammonia, but also strong oxidants such as nitric oxide (NO) and nitrous oxide (N2O) were generated from nitrite and showed a potential-dependent distribution. This result suggests that the proton motive force is not required for ammonia synthesis. Notably, greigite doped with 5 atom % Mo efficiently activated nitrite reduction, suggesting that even at trace amounts, Mo can function as a catalytic site.
As a short summary, previous prebiotic chemical syntheses conducted using metal sulfides as the catalysts showed that in addition to iron sulfide, other transition metal sulfides can potentially function as C and N fixation catalysts with even higher activity than iron sulfides. The catalytic performance of CO2 reduction varies largely depending on the doping elements, crystal size, and surface structure of greigite, as well as the pH and Eh conditions [14,15,38]. Therefore, due to the chemical diversity of metal sulfides, in addition to the influence of pH and Eh on catalytic activity, suitable catalysts in mineralogy database with optimized activity can be potentially identified by screening reaction conditions. Effective stoichiometric C and N fixation requires an abundant source of electrons. In addition, Fe2+ was likely a dominant dissolved/colloidal metal species in the early ocean. Nevertheless, if geoelectrical currents had supplied energy to trigger prebiotic reactions, even trace amounts of metal elements could have continuously catalyzed the reaction in the geoelectrochemical environments in the Hadean ocean floor [16].
The above rationale and experimental evidence suggest that prebiotic metal sulfide catalysts should be screened not only based on the abundance of the metal, but also the electrocatalytic activity. In comparison to iron sulfides, the electrochemical and electrocatalytic properties of other transition metal sulfides and their mixtures remain poorly understood, with a limited number of studies conducted to date [14,15,53]. Proposed reaction mechanisms of these compounds are based on their resemblance with the relevant enzymatic processes, such as those mediated by carbon monoxide dehydrogenase [15], acetyl-CoA synthase [53,63], and nitrogenase [62], although no sufficient spectroscopic evidence and/or valence state information has been provided. The underlying reaction mechanisms should therefore be investigated in more detail for guiding catalyst screening based on functional, rather than structural relevance to well-characterized enzymes.
3. Chemical Diversity of Metal Sulfides in Mineralogy Libraries
As the coordination and electronic structure of mineral surfaces largely determines catalytic activity, here we evaluate the chemical diversity of naturally occurring metal sulfides to aid in the screening and identification of suitable prebiotic sulfide catalysts. A special emphasis was placed on the sulfides containing Co, Cu, Fe, Mn, Mo, Ni, V and/or W, as they are well-known redox-active elements and present in the active centers of enzymes catalyzing carbon and nitrogen conversions, such as those mediated by carbon monoxide dehydrogenase (Fe, Ni, Cu and Mo), a cobalt cobalamin cofactor for methyl transfer in acetyl-CoA synthase (Co), nitrogenase (Mo, V and Fe), nitrate reductase (Mo), nitrite reductase (Fe, Cu, and Mo), formate dehydrogenase (Mo and W), aldehyde oxidoreductase (Mo and W), and nitrous oxide reductase (Cu). In these enzymatic processes, transition metals switch oxidation states for charge accumulation, coordinate to a variety of substrates and intermediates, and also catalyze enzymatic redox reactions involving simple inorganic molecules, such as H2, AsO2−, Sxn‒, as substrates, which function as coupling reactions for energy conversion and conservation in organisms. Sulfur is a pervasive ligand in the first coordination sphere of the enzymatic cofactors, although cobalt cobalamin is an exception, as tetrapyrrole serves as the ligand. The ubiquitous role of metals and sulfur in metabolic processes has led to the hypothesis that metal sulfides worked as “proto-enzymes” for prebiotic organic syntheses, and sustained the continuous chemical evolution toward functioning in complex biochemical systems [4,16,58,59].
Among the 5327 mineral species discovered to date (as officially listed by the International Mineralogical Association (IMA); http://rruff.info/ima/, as of 18 May 2018), 304 metal sulfides contain Co, Cu, Fe, Mn, Mo, Ni, V and/or W with 135,434 species-locality pairs. We evaluated the diversity and distribution of these metal sulfides based on the following criteria:
- (1)
- Locality frequency. This parameter reflects the availability of minerals in natural environments. Although detection limits and metal-specific biases reflecting the metal’s economic value may exist, an estimate of the probability of natural occurrence is useful for evaluating the feasibility of mineral-promoted prebiotic reactions.
- (2)
- Multiple metal composition. Naturally observed metal sulfides are commonly coupled to more than one transition metal and occasionally exhibit ternary compositions. Because cation substitutions and the presence of dual metal surface sites influence catalytic activities of minerals, as described in Section 2.2, Section 4 and Section 5, metal-specific behaviors and tendency to form multiple compositions are important factors for screening potential mineral catalysts.
- (3)
- Crystal structure and valence state of dominating elements. These two properties, particularly the valence state, control the electronic characteristics of the mineral. Valence state information is available for 89% of the localities of metal sulfides in the database (http://rruff.info/ima/).
3.1. Distribution of Transition Metal Sulfides
The locality count indicates the frequency that a certain mineral will be found within a wide range of geological localities. Although this parameter partially reflects the natural occurrence probability within the ability of human detection, it is useful for evaluating the relative importance of the minerals as geochemical catalysts. Among the 304 metal sulfides considered, Fe-containing sulfides dominate the library (Figure 2). Pyrite (Fe2+S−2) is the most frequently found species, followed by chalcopyrite (Cu+Fe3+S2−2), pyrrhotite (Fe2+5Fe3+2S2−8), arsenopyrite (Fe2+(AsS)2−), molybdenite (Mo4+S2−2), chalcocite (Cu+2S2−), marcasite (Fe2+S−2), bornite (Cu+5Fe3+S2−4), and tetrahedrite (Cu+10Cu2+2Sb3+4S2−13). The locality counts of these minerals exceed 5000 and together, these minerals comprise ~80% of the sulfide minerals discovered to date. The nine most frequently observed minerals may have been present since the middle Archean eon (~3500 Ma) or perhaps even earlier, although directly dated sulfides are rare.
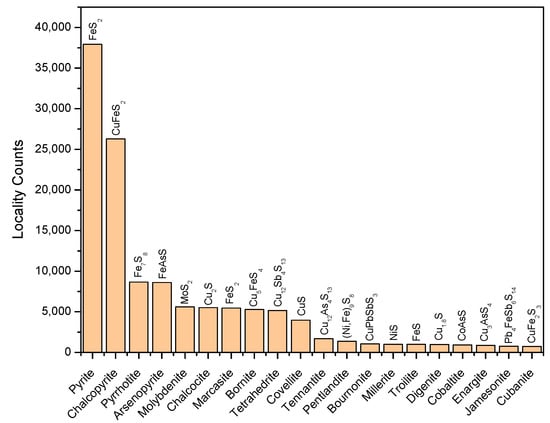
Figure 2.
Metal sulfide distribution in natural environments. The 20 most frequently observed species are ranked in order of locality counts. The chemical composition of each species is shown.
Among transition metal sulfide species in the library, ~39% (119 species) contain iron, and are found in ~74% of localities (Figure S1), suggesting that iron was a pervasively occurring element during sulfide mineral formation. Co, Mn, Mo, W and V are mostly seen in Fe-free sulfides (>93% relative to the respective sulfides’ locality counts), whereas Cu and Ni show a high tendency to coexist with Fe. Cu-Fe- and Ni-Fe-containing sulfides are seen in 59% and 45% of localities of Cu- and Ni-sulfides, respectively, and exhibit structural variations accounting for 32% and 42% of the total number of Cu- and Ni-sulfide species, respectively. Nevertheless, Fe-free Cu and Ni single-sulfides exhibit high locality counts (24,085 and 2848, respectively), among which chalcocite (Cu+2S2−) and millerite (Ni2+S2−) are particularly prevalent (5520 and 1007 localities, respectively). Thus, although Cu and Ni tend to easily incorporate into Fe-bearing sulfides, their Fe-free sulfide forms are also ubiquitous. Please refer to Figures S1–S8 for more information regarding the spatial distribution and compositional diversity of metal sulfides.
3.2. Single-Metal Sulfides
Single-metal sulfide minerals have been mainly investigated as key prebiotic catalysts [14,16,38,46,47,48,49,50]. By studying this family of minerals, their intrinsic metal-specific properties can be derived and compared, as was shown by Kitadai et al. [14] through the intensive screening of catalysts for CO2 fixation. Single-metal sulfides of Fe, Cu, and Ni show marked variations in crystal structure, chemical composition and metal/sulfur valence states. Among 45 Fe single-metal sulfides (Figure 3), two pairs of polymorphs are present with the chemical compositions of FeS2 (pyrite and marcasite) and FeS (troilite and keilite). Pyrite and marcasite have indirect band gaps of 0.7 and 0.4 eV, respectively [71]. Orthorhombic marcasite exhibits an anisotropic property that results in a unique sulfur surface site (sulfur trimers) on a specific surface orientation [72]. In contrast to pyrite on which S2− is firstly oxidized, the sulfur trimer species on marcasite are most easily oxidized.
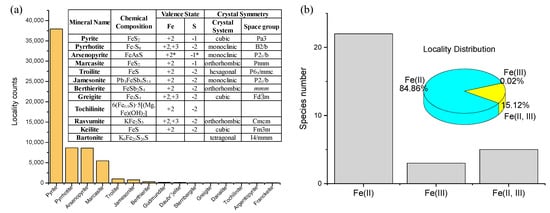
Figure 3.
(a) Distribution and chemical diversity (chemical composition, Fe/S valence states, and crystal symmetry) of Fe single-metal sulfides. (b) Relative abundances and locality distribution of Fe2+-, Fe3+-, and Fe2+ plus Fe3+-sulfides. Note that the valence state of arsenopyrite (Fe3+(AsS)3−) in the RRUFF mineral database is examined as (Fe2+(AsS)2−) in this review based on electric spectroscopic observations [76,77].
Fe single-metal sulfides in 91% of localities have valence state information. Sulfides having Fe2+ alone dominate the mineral family (84.86%), followed by those containing both Fe2+ and Fe3+ (15.12%), and by three sulfides that only contain Fe3+ (0.02%). Sulfides composed of a mixture of Fe2+ and Fe3+, such as pyrrhotite (Fe2+5Fe3+2S2−8) and greigite (Fe2+Fe3+2S2−4), typically exhibit magnetic properties [73,74,75].
Cu-, Co-, and Ni-single sulfides also display diverse chemical and physical properties (Figures S9–11). As many as 119 species were identified in Cu single-metal sulfides, of which 27 species have valence state information (73% relative to the locality counts of Cu single-metal sulfides). In addition, 18 species contain only Cu+ (59%), four (tetrahedrite, tennantite, famatinite, and kuramite) are formed from Cu+ and Cu2+ (40%), and the remaining 5 species possess only Cu2+ (<0.6%). The percentages in parentheses represent the respective mineral distributions relative to the locality counts of Cu single-metal sulfides with valence state information.
Co single-metal sulfides form 10 different structures, including five Co3+-sulfides with two sulfosalt polymorphs, Co3+(AsS)3− (cobaltite and alloclasite) and Co3+(SbS)3− (willyamite, costibite, and paracostibite). Linnaeite (Co2+Co3+2S4) is the only species having a mixed valence state. The environmental distributions of Co3+-, Co3+ plus Co2+-, and Co2+-sulfides are 78.93%, 18.47%, and 2.59%, respectively, relative to the total locality counts of Co single-metal sulfides.
For Ni single-metal sulfides, the valence state of Ni is always +2, with the exception of polydymite, which has both Ni2+ and Ni3+ (Ni2+Ni3+2S2−4).
In Mn, Mo, and W single-metal sulfides, the valence states of metals are conserved, with only Mn2+, Mo4+, and W4+ being observed.
For V single-metal sulfides, three species are known (patrónite (VS4), colimaite (K3VS4), and yushkinite (Mg,Al)(OH)2VS2), in which the valence state of vanadium is +4 or +5. The occurrence of these species in natural environments is rare, and V-bearing sulfides are typically found as Cu–V binary forms (95% in terms of locality).
Generally, metals with low valence states predominate the library of metal sulfides, with the exception of Co, allowing these minerals to act as an electron source or a catalytic center for charge accumulation during redox conversion. In addition, sulfur with a −1 valence state, which typically exists as a disulfur (S2)2− ligand, can act as an electron-accepting site, and thus has the potential to be involved in redox catalysis. The participation of not only metal sites, but also sulfur ligands, during redox catalysis has been extensively reported and will be discussed in Section 4.3.
3.3. Binary- and Ternary-Metal Sulfides
As described in Section 2.2, the doping of a secondary metal in iron sulfide can enhance the activity and selectivity of CO2 reduction [15]. Sulfides with binary metal composition can thus potentially serve as superior catalysts to their single-metal counterparts. In addition, as binary metal sites are present in several extant enzymatic systems, such as Ni-S-Fe hydrogenase, Ni-S-Fe carbon monoxide dehydrogenase (CODH), Mo-S-Cu CODH, Fe-S-Mo nitrogenase, and Fe-S-V nitrogenase, it has been suggested that binary metal centers can synergistically function as Lewis acid and base sites for facilitating catalysis. For example, Fe and Ni bind CO2 in a bidentate mode via O and C atoms, respectively [78].
Cu-bearing binary sulfides exhibit 193 structures among 58,932 localities and include 52 species of Cu-Fe binary sulfides (59% in terms of locality) (Table S1). Calcopyrite (Cu1+Fe3+S2−2) and bornite (Cu1+5Fe3+S2−4) are the most prevalent forms in this category. Sulfides with Fe and Cu as substitutional cations are also known, but are rare in terms of distribution (<1% of localities).
Ni-bearing binary sulfides with 38 structures have been identified among 5185 localities. Of these species, 10 are Ni-Fe binary sulfides (45% in terms of locality) (Table S1), of which 7 contain Ni and Fe as substitutional cations with an Ni/Fe ratio of 0–35 atomic % being reported [79]. These seven sulfides cover 75% of localities of Ni-Fe binary sulfides with pentlandite (Ni,Fe)9S8 being the most prevalent form. The remaining 28 species possess Fe and Ni with fixed stoichiometries (e.g., violarite (FeNi2S4)), in which Fe and Ni occupy independent crystal lattice sites. The capability of Ni-Fe binary sulfides to have both fixed and varied Ni/Fe ratios in their structures is a unique characteristic different from that of Cu-Fe binary sulfides, in which Cu and Fe tend to form specific structures with fixed stoichiometries.
A number of ternary-metal sulfides with combinations of Fe–Cu–Ni (seven species), Fe–Cu–Mo (two species; tarkianite ((Cu,Fe)(Re,Mo)4S8) and maikainite (Cu+10Fe2+3Mo4+Ge4+3S2−16)), and Fe–Cu–W (one species; ovamboite, Cu+10Fe2+3W4+Ge4+3S2−16) have been characterized (Table S2). The occurrence of ternary systems is uncommon (77 localities or 0.05% of the total locality counts of metal sulfides) and it is notable that binary Fe-Mo and Fe-W sulfides are not found in nature, indicating that Cu is important for the incorporation of Mo and W into FeS structures.
3.4. X-ray Amorphous Metal Sulfides
As a clear XRD pattern is required to identify crystal structures, X-ray amorphous species are rarely included in mineralogy databases (Table S3 lists the eight amorphous species recorded in the RRUFF database). Jordisite (MoS2), the second most prevalent Mo single-metal sulfide, is the only amorphous metal sulfide recorded. Using high-resolution transmittance electron microscopy (TEM), jordisite exhibits a low crystallinity, but still shows a layered structure with interlayer spacing of ≈6 Å resembling that in hexagonal molybdenite (MoS2) [32]. Mo sulfide is also seen in anaerobic sulfidic sediments and basins [28,30,80,81,82], and its speciation is controlled by the O2 and HS− concentrations in these environments [28,29]. Mo sulfide associated with Cretaceous deep-sea sediments and with other black shales is characterized by the molecular structure of the Mo center coordinated with sulfur and oxygen, as resolved by extended X-ray absorption fine structure spectroscopy [30].
Despite of the low number of amorphous species in the database, metal sulfide species with a low crystallinity are not rare. Aqueous clusters of FeS, ZnS and CuS constitute a major fraction of the dissolved metal load in anoxic oceanic, sedimentary, freshwater and deep ocean-vent environments, and may be possible building blocks during mineral formation [83]. Luther et al. [83] reported that Fe-S, Zn-S, Ag-S clusters with sizes <3 nm form in the presence of low concentrations (μM order) of metallic ions and sulfides and show remarkable kinetic stability. For example, the half-life of as-formed Ag-S clusters with respect to O2 oxidation is 360 days. In addition, these nanoclusters show a wider band gap in comparison to their bulk counterparts owing to the quantum confinement effect. Cu-S clusters also show wide variation in metal/sulfur stoichiometry (neutral Cu3S3 and anionic Cu4S6 clusters). Nanoclusters with varied metal/sulfur stoichiometry are either neutral or negatively charged and are soluble in aqueous solutions. With the advance of atomic resolution structural characterization techniques, such as X-ray absorption spectroscopy and transmittance electron microscopy, low-crystallinity metal sulfide species are expected to be more frequently discovered in geochemical environments. As such amorphous materials adopt a metastable state and have numerous “dangling bonds”, the unique band structure and surface properties of X-ray amorphous metal sulfides are expected to have unique catalytic properties in comparison to that of their bulk counterparts.
4. Impact of Metal Sulfide Chemical Diversity on Catalysis
The previous section summarized the marked chemical diversities of natural transition metal sulfides, as exhibited by the large variations in chemical composition, crystal structure and valence states of the component metal and sulfur species. The chemical diversity of metal sulfide minerals partially reflects the “messy” feature of geological environments. In the context of prebiotic chemistry, such diversity among the available catalysts could have generated messy chemistry with an enormous number of possible reactions, leading to the formation of complex prebiotic organic synthesis networks and, ultimately, autocatalytic reaction networks [84,85]. This raises the question of what types of sulfides, or their combinations, could have been involved in prebiotic processes toward the origin of life?
To extract valuable information from mineral databases for the screening of prebiotic catalysts, a critical guiding principle is the “structure–function relationship”, including the key reaction intermediate(s) and physicochemical factor(s) dictating catalytic activity. As was introduced in Section 1 and Section 2.2, electrocatalytic processes can occur in hydrothermal vent environments for driving prebiotic synthesis. More importantly, the experimental methods and theories developed in the electrocatalysis field can be generally applied for all redox and a portion of non-redox catalytic processes. For instance, redox processes occurring in enzymes have been extensively studied using electrochemical methods [86,87,88], thereby allowing detailed elucidation of the electron and proton- transfer mechanisms. To provide a better understanding of structure–function relationships toward rational catalyst screening, here, we review the electrocatalytic properties of sulfides with well-defined structures that have been extensively studied in engineering fields.
4.1. CO2 Reduction
CO2 electroreduction has been examined on various types of catalysts, including metals, alloys, metal–organic complexes, homogeneous molecular catalysts, and metal sulfides, in both aqueous solutions and organic solvents [89,90,91,92]. The products of these reactions are typically simple C1 compounds, such as CO and formate, in water or C2 compounds, such as oxalate, in aprotic solvents [89,90], although Cu was reported to efficiently catalyze CO2 reduction to hydrocarbons, including methane and ethylene, in water [93,94].
Metal sulfides generally exhibit lower catalytic activity for CO2 reduction than their metal counterparts [14,15,94], but the presence of favorable active sites on metal sulfides is computationally predicted to facilitate the CO2 reduction by stabilizing the key reaction intermediates [95]. The edge sites of MoS2 and Ni-doped MoS2 preferentially bind COOH* and CHO* to the bridging S atoms and CO* to the metal sites. The binding of intermediates on different sites is expected to decrease the overpotential owing to the intrinsic thermodynamic adsorption energy scaling relations between different intermediates [96,97]. In fact, selective CO2 electroreduction to CO has been reported on MoS2 in water containing 4 mol% of an ionic liquid (1-ethyl-3-methylimidazolium tetrafulorobrate; EMIM-BF4) [98]. CO production occurred at an onset potential as low as 0.054 V (vs. RHE) and the Faraday efficiency increased from <10% to close to 100% by increasing the overpotential from 0.2 to 0.76 V. Notably, no appreciable amount of CO was observed in the absence of ionic liquid. Density functional theory (DFT) calculations suggested that the d-orbital electrons of Mo dominating near the Fermi energy level are freely transferred to reactants adsorbed on edge Mo sites. EMIM ions were inferred to stabilize CO2 via hydrogen bonding to form an [EMIM-CO2]+ complex and inhibit the conversion of CO2 to HCO3− and CO32−. The complexation also lowers the energy barrier for electron transfer from MoS2 to the physisorbed CO2 on MoS2.
Besides the artificial catalytic systems, the mechanistic implications of CO2 reduction can be obtained from analyzing the catalytic process in biological enzymatic systems. The reversible transformation of CO2 to CO is catalyzed by a highly asymmetric [NiFe4S4] cluster in anaerobic carbon monoxide dehydrogenase (CODH), which operates at potentials close to the thermodynamic potential of the CO2/CO couple (−0.52 V vs. SHE, pH 7) and contains bifunctional metal sites. In Ni-Fe-containing CODH, high-resolution crystallographic structure determination [78] revealed that CO2 undergoes two-electron reduction after binding to Ni and Fe sites with a μ2, η2 mode, and the intermediate with bent-bound CO22− is stabilized by substantial π backbonding. The bifunctionality of Ni and Fe to serve as nucleophilic and electrophilic sites, respectively, plays a key role in activating CO2 and promoting electron transfer from the metal to CO2. A short length of Ni-C bonds suggests that the Ni center has high nucleophilicity, which likely originates from the integration of Ni into the Fe/S scaffold. Based on these enzymatic properties and mechanisms, sulfide minerals bearing Ni and Fe, which have an Ni site of square planar symmetry and a single unsaturated site, could potentially act as CO2 reduction catalysts.
4.2. Ammonia Synthesis from Nitrate/Nitrite or Dinitrogen Reduction
Electrochemical ammonia synthesis by nitrate/nitrite reduction was recently studied on synthesized molybdenum and iron sulfides [16,99]. Nitrate and nitrite reduction catalyzed by molybdenum sulfide have an evolutionary relevance with respect to enzymatic catalysis, because all nitrate reductase enzymes and one family of nitrite reductases rely on molybdenum-pterin (Mo-S2) active centers, in which Mo is coordinated by sulfur atoms of a pterin group [100]. He et al. [99] found that product selectivity of nitrite reduction on molybdenum sulfide is pH dependent with respect to the formation of NO, N2O and NH4+. Operando electron paramagnetic resonance (EPR) and Raman spectroscopies during nitrite reduction revealed that an oxo-ligated Mo(V) species serves as the key intermediate controlling the nitrite reduction selectivity [99]. Oxo-ligated Mo(V) species changes its configuration from an isotropic to distorted geometry in response to pH change, suggesting that these species decouple electron transfer (ET) from proton transfer (PT). This unique property results in a volcano-type pH-dependent selectivity of N2O formation, where the optimal pH for this reaction is close to the pKa of MoV-oxo species, at which a kinetic balance is anticipated to exist between ET and PT.
Dinitrogen reduction is another candidate pathway to generate ammonia (N2 + 6e− + 8H+ → 2NH4+), and several electrocatalytic systems using Fe2O3/CNT [101], Fe/CNT [102], and Ru/C [103], among others, as catalysts have been reported to function in aqueous solution at room temperature and ambient pressure. Typically, low Faradaic efficiency (<1%) was exhibited and H2 was generated as the main byproduct. The reaction rate was enhanced by increasing the temperature or optimizing the potential. Higher Faradaic efficiencies (4.02–10.1%) were recently reported by engineering the catalysts [104,105]. For example, through amorphourization of an Au nanocatalyst, structural distortion was observed, which was proposed to increase the concentration of active sites in the catalyst with higher binding affinities towards N2. To our knowledge, no metal sulfide has been reported to perform electrocatalytic N2 reduction. As FeMoS cofactor of nitrogenase catalyzes N2 reduction under ambient temperature and pressure, several Mo-containing molecular [106,107] and heterogeneous (MoS2, MoO3, MoN) [108,109,110] catalysts have been synthesized for N2 reduction. For example, Yandulov et al. [106] reported that a Mo triamidoamine complex catalyzes N2 reduction using decamethyl chromocene and pyridinium salts as electron and proton sources, respectively, at ambient temperature and 1 bar N2 in an aprotic organic medium. Based on the chemical properties of the isolated intermediates, the reaction is proposed to proceed via a distal associative mechanism, wherein the catalytic intermediates are sequentially protonated and reduced. A single molybdenum center is proposed to function as the active site and cycle from Mo(III) to Mo(VI) states. Note that the displacement of ammonia by N2 is the rate-determining step on this molecular catalyst, which differs from heterogeneous Au [104] and MoS2 [109] catalysts, where the reductive protonation of bound N2* is considered to be the rate limiting step based on DFT calculations to weaken the N≡N triple bond.
4.3. Hydrogen Evolution and Oxidation Reactions
The H2 evolution reaction (HER; 2H+ + 2e− → H2 or 2H2O + 2e− → H2 + 2OH−) has been the most intensively studied electrochemical reaction catalyzed by metal sulfides. As mentioned above, H2 evolution tends to compete with the reduction of N2, CO2 and NO3− in regions of large overpotential. For this reason, understanding the HER mechanism is expected to aid in the rational design and selection of catalysts and conditions for the selective N2, CO2 and NO3− reduction through suppression of HER. HER has been examined on various sulfides, including Fe [111], Co [112], Mo [113,114,115,116,117,118], and W [119,120] single-metal sulfides, Ni/Fe [121] and Co/Fe [122] binary-metal sulfides, Co-based binary-metal sulfides (with Cu, Ni, Zn) [123], and pyrite-type minerals [124]. Mo sulfide possesses a laminated lattice structure that is highly amenable to electric and structural modifications, and its catalytic property has therefore been investigated in different crystal phases, including hexagonal MoS2 (2H-MoS2), chemically exfoliated metallic MoS2 (1T MoS2), amorphous MoSx, Mo-S clusters (Mo3S4−), and Mo-Cu binary-metal sulfide (Cu2MoS4) [113,114,115,116,117,118]), and with various degrees of non-stoichiometric defects [115,117], doping [125,126], and hybridization [127,128,129]. From the results of spectroscopic analyses, including in situ and ex situ X-ray absorption [130], X-ray photoelectron spectroscopy (XPS) [131], EPR [132] and Raman spectroscopy [113,132], hexagonal MoS2 nanoparticles appear to have catalytic activities on edge sites, rather than on basal planes [114,115,132,133,134,135]. This possibility has been supported by several experimental studies that have used [Mo-S2]-bearing compounds, such as [Mo3S13]2− and [(PY5Me2)MoS2]2+, which mimic edge sites [118,135,136,137].
Experimental evidence also suggests that terminal di-sulfur (S-S)tr species are critical for the HER on amorphous MoSx [113,130,131]. (S-S)tr species provide thermodynamically favorable adsorption sites for hydrogen (∆rGo ~0) by forming unsaturated –S sites through the reductive cleavage of the S-S bond [131,134]. The formation of a molybdenum hydride moiety (Mo-H) also triggers the HER on amorphous MoSx [132] and on Mo3S4− [138]. Recently, operando Raman spectroscopy revealed a dynamic structural change of a Mo tri-nuclear cluster characterized by Mo-Mo bond weakening and the concurrent emergence of a terminal S-S ligand, indicating that both multinuclear Mo-Mo bonds and terminal S-S ligands play synergistic roles in facilitating the HER [139]. A similar mechanism involving exposed disulfur ligands as active sites was proposed to explain the efficient HER that proceeds on amorphous CoSx [112]. In both WS2 and MoS2, the 2H phase material converts into a metallic 1T phase when chemically exfoliated to monolayer thickness. The metallic 1T phase exhibits superior HER activity because of favorable electron transfer rate as well as the thermoneutral adsorption of H [119]. Based on these mechanistic studies, sulfides with S22− moieties, such as pyrite (FeS2), marcasite (FeS2), covellite (Cu+4Cu2+2(S2)2S2), vaesite (NiS2), and cattierite (CoS2), are promising catalysts for H2 evolution owing to the redox activity and suitable adsorption energy of the of S-S species towards Hads.
The reverse reaction of the HER, namely the hydrogen oxidation reaction (HOR), is an important half reaction that is predicted to have occurred at the inner chimney wall–hydrothermal fluid interface of geoelectrochemical reactors, and to have supplied electrons for driving reductive organic reactions at the mineral–seawater interface (Figure 1). The HOR is catalyzed efficiently by Pt-group metals, such as Pt, Pd, Ir [140,141] in acidic medium. Moderate HOR activities have also been shown using Ni-based catalysts, such as Ni [142,143], Ni binary alloys (NiMo, NiTi) [144], and ternary metallic CoNiMo catalyst [145] in alkaline medium, and by Ni complex molecules [146] in organic medium. Based on DFT calculations, the H adsorption energy on the surface dictates the HOR activity. Pt shows thermoneutral adsorption, whereas Ni displays relatively higher adsorption energy (stronger binding). By alloying Ni with other metals, the H binding energy was tuned owing to the changes of the d-orbitals of Ni, which facilitates the HOR [144,145]. To date, no metal sulfide has been reported to catalyze the HOR, although numerous types of metal sulfides efficiently catalyze the HER, as mentioned above. This contrasts greatly with Fe-S-Ni hydrogenase, which reversibly and efficiently catalyzes both the HER and the HOR [88], and is also predicted to thermoneutrally adsorb H based on DFT calculations [134]. Based on Koper’s prediction [56], the presence of only one intermediate (generated through an electrochemical step) in a 2e−/2H+ reaction will allow the catalyst to effectively perform both the forward and backward reactions, given that thermoneutral adsorption of the intermediate occurs on the catalyst surface. One possible explanation for the inertness of metal sulfide towards the HOR is that another non-electrochemical step, such as the surface binding of H2, non-redox dissociation of H-H bond, or deactivation of the catalyst surface, is rate limiting. Because of the importance of the hydrogen oxidation reaction for geoelectrochemical organic syntheses, the HOR catalyzed by metal sulfides warrants further investigation.
The electrocatalytic studies reviewed above demonstrate that metal sulfides have the potential to function as versatile catalysts for redox reactions that are relevant to prebiotic synthesis. Several key factors of metal sulfide catalysts can be summarized here. First, in situ spectroscopic analyses have suggested that not only single-metal sites, but also heterogeneous surface moieties, such as those with metal-metal, metal-sulfur, metal-hydride, and sulfur-sulfur bonds, play important and occasionally synergistic roles in promoting specific reactions. This is a remarkable feature of metal sulfide catalysts that differs from that observed in metal complex-mediated reaction systems, in which the redox chemistry is mainly dominated by the valence change on single-metal centers. Moreover, a surface dominated by metal sites is likely preferable for CO2 and nitrate electroreduction on metal sulfides, as (di-)sulfur-dominated surface sites will be active for H2 evolution. Furthermore, the doping of a secondary metal was theoretically predicted to tune the binding energies of the reaction intermediates, such as CHO*, COOH*, and H*, thereby enhancing the CO2 reduction selectivity by suppressing competing H2 evolution [95]. Therefore, the binary- and ternary-metal sulfides in the mineralogy database, which includes 193 Cu-bearing binary-metal sulfides species, 38 Ni-bearing binary-metal sulfide species, and 10 types of ternary-metal sulfides, are anticipated to show unique activities in comparison with their single-metal sulfide counterparts. The microscopic mechanism of decoupled electron and proton transfer represents a new mechanistic aspect to mutli-electron, multi-proton reactions [55,56,57] and provides a rational approach to tuning catalytic reactivity and selectivity over wide pH and Eh ranges.
5. Rational Screening of Catalysts
The examples shown in Section 4 highlight the physicochemical factors influencing the catalytic activity of specific types of metal sulfide catalysts. To establish a complex chemical network composed of life’s building blocks and their polymers, significant efforts are required to screen and identify efficient mineral catalysts for activating simple molecules (CO2, N2, NO3−/NO2−, H2, etc.). Although a few prebiotic studies have attempted to screen metal sulfide catalysts [14,15,53], approaches for rational screening have not yet been described. In this section, we briefly review an in silico approach, followed by machine-learning methods (so-called catalytic informatics) for predicting the catalytic activity of electrocatalysts.
Electrocatalysis occurs via the adsorption of substrates on active surface site(s), followed by the sequential conversion of the substrates into bound intermediates, and finally, the desorption of products from the catalytic surface. Efficient catalysts are expected to have adsorption energy towards the reactants and intermediates that are neither too high nor too low, because too weak binding will not drive effective conversion, whereas too strong binding will lead to the poisoning of active sites (the so-called Sabatier principle, d-band theory). Recently, the prediction based on the Sabatier principle has become a more powerful approach owing to the pioneering research by Nørskov et al. [96,125,147]. Instead of experimentally identifying the binding energy of a key intermediate, they developed the DFT methods to calculate the adsorption energy. This approach has been widely applied to rationalize the experimentally observed tendency of CO2 electroreduction reactivity on various metal catalysts and even predict new catalysts with better performance. For example, Figure 4a–c illustrates typical reaction pathways towards various products, particularly CH4, CO, HCOO−, CH3OH, and CH3CH2OH, as was summarized by Kortlever et al. [91]. Peterson and Nørskov [96] calculated the CO2-to-CH4 reduction pathway on a series of fcc transition metal (211) surfaces and showed that Cu locates at the top of the activity plot and is hence the best catalyst (Figure 4d). On Cu (211), the energetic barrier between the CO2 → COOH* conversion and that of the CO*→CHO* step was well balanced. The CO*→CHO* process is typically the potential-determining step and leads to a high overpotential (on the order of 1 V). The CO2 reduction pathways towards the products CO, C2H4, and CH3CH2OH were also calculated on Cu (100) and Cu (211) [148,149]. Generally, negatively charged intermediates, such as the adsorbed CO2− anion intermediate and adsorbed (CO)2− anionic dimer, are critical for C1 and C2 compound formation.
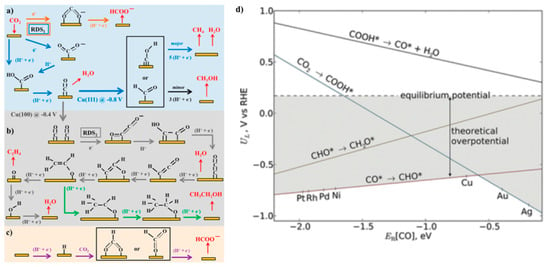
Figure 4.
(a–c) Possible reaction pathways for the electrocatalytic reduction of CO2 to products on transition metals and molecular catalysts (adapted from Reference [91]): (a) reaction pathways from CO2 to CO, CH4 (blue arrows), CH3OH (black arrows), and HCOO− (orange arrows); (b) reaction pathways from CO2 to ethylene (gray arrows) and ethanol (green arrows); (c) reaction pathway of CO2 insertion into a metal−H bond yielding formate (purple arrows). Species in black are adsorbates, whereas those in red are reactants or products in solution. Potentials are reported versus RHE, RDS indicates the rate-determining step, and (H+ + e−) indicates steps in which either concerted or separated proton−electron transfer takes place. (d) Limiting potentials (UL) for elementary proton-transfer steps in the reduction of CO2 to CH4 (adapted from Reference [96]). Each line represents the calculated potential at which the indicated elementary reaction step is neutral with respect to free energy and as a function of the carbon monoxide affinity (EB[CO]) of the electrocatalyst. The theoretical overpotential is defined as the potential difference between the most-negative limiting potential line and the equilibrium potential for the reduction of CO2 to CH4 (+0.17 V versus RHE), as highlighted in gray.
Certain alloys facilitate redox reactions with greater efficiencies than the single-metal counterparts, as has been demonstrated for CO oxidation on Pt-alloy, methanation on Fe-Ni alloy, and ethylene oxidation on Ag-alloy [147]. Particularly, CO2 electroreduction on Cu-Ag alloy shows a higher selectivity for hydrocarbon and oxygenate (ethylene, ethanol, and propanol) formation in comparison with that on Cu [150]. He et al. [151] screened several binary- and ternary-metal alloys (InxM1−x; M = Fe, Co, Ni, Cu, Zn) for CO2 reduction to CO and found that InxCu1−x alloys with x = 0.2–0.5 exhibited the highest activity for the CO production (90% selectivity). The CO evolution on InxM1−x occurred with the trend Fe < Co < Zn < Ni < Cu, whereas that for the H2 evolution followed the trend Zn < Cu < Ni < Co < Fe. The activity trend for the CO formation was consistent with the sequence of CO adsorption energy on the catalyst surfaces calculated by DFT, thus verifying the validity of CO adsorption energy as the key descriptor of catalytic activity. In these alloys, the secondary metal changes the average energy of the d-electrons (εd–εF; the d-band center with respect to the Fermi level) in the primary metal, resulting in either weakening or strengthening of key intermediate adsorptions. [147]. In addition to CO2 reduction, the catalytic activity for H2 oxidation [140,152,153] and N2 reduction [154,155,156,157] have also been examined by DFT calculations with d-band theory.
For reactions involving multiple intermediates, such as CO2 reduction to CH4 or alcohols, and N2 reduction to ammonia, the adsorption energy scaling relationship of chemisorbates commonly leads to intrinsic overpotentials [96,158]. It is possible to lower the onset potential of these reactions through the selective binding of intermediates on different sites or with different binding modes by decoupling the scaling relationship of the binding energies of intermediates [96]. By applying DFT calculations to a mineral with a known crystal structure and chemical composition, the electronic structure of the bulk catalyst and surface sites, including band gap and Fermi levels, can be derived for determining the adsorption energy of key intermediates, which can be used for predicting the activity and identifying superior catalysts (Figure 5). The Materials Project database [159] (https://materialsproject.org/) has been developed for this purpose and is supported by the U.S. Department of Energy (DOE). This database contains the crystal structures of 83,989 inorganic compounds (as of 31 July 2018). Using high-throughput computing, the band structures of 52,179 compounds have been calculated and can be used for calculating adsorption energy.
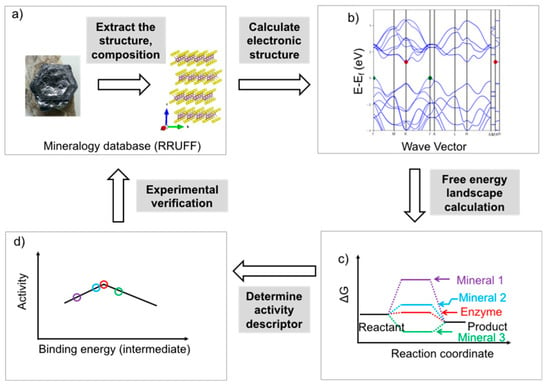
Figure 5.
Framework of the methodology proposed for the rational screening of mineral catalysts using DFT calculations. (a) Select a series of minerals and extract the structure and chemical composition information from the RRUFF mineralogy database (the mineral shown here is molybdenite, MoS2, and was obtained from the mindat website: https://www.mindat.org/search.php?name=Molybdenite; the crystal structure was created using BIOVIA Draw software); (b) Calculate the electronic structure of the bulk catalyst and surface, and obtain information of the band gap, Fermi level, conductivity, and other electronic properties. The band structure shown here was obtained from the Materials Project database for molybdenite: https://materialsproject.org/; (c) Calculate the free energy landscape for the specific reaction (here, the reaction pathway scheme assumes that only one intermediate is involved); (d) Determine the activity descriptor (binding energy of the key intermediate). Using this approach, the relative activity profile for a series of minerals can be obtained. The predicted activity profile can be verified using experimental methods to validate the optimized computational model and obtain more precise structure-activity relationships.
It is to be noted here that the aforementioned in silico approaches have now become a more exhaustive search for new catalysts owing to the integration to machine-learning methods. For example, Ma et al. developed a machine-learning-augmented chemisorption model that enables fast and accurate prediction of CO2 reduction activity of metal alloys [160] (Figure 6). With this model and the d-band theory, they identified promising new catalysts for selective reduction of CO2 to C2 species [160]. Ulissi et al. [161] developed a neural-network-based surrogate to share information between activity estimates with an order of magnitude fewer explicit DFT calculations. This approach allows for an order of magnitude reduction in the number of DFT calculations required and thus rapid prediction of catalysts even for highly complicated bimetallic electrocatalyst for CO2 reduction. Besides the use of in silico data for machine learning, a recent study by Shao-Horn and coworkers [162] showed the potential to utilize experimental data from various literature for catalyst prediction after standardizing with the activity of a material common in all studies. This approach can be considered as a technical breakthrough in this field, because one of the difficulties hindering catalyst informatics was the lack of data in a uniform structure. The implementation of an internal standard allows access to the wealth of experimental data accumulated to date, which may lead to a more widespread application of machine-learning techniques. More recently, Ooka et al. developed the method to utilize genetic sequence information for catalyst development [163]. In contrast to experimentally-determined activity which varies depending on the evaluation method (current vs. overpotential, or pH conditions), the unambiguity of the gene sequence allows for a more robust statistical analysis.

Figure 6.
(a) Rational screening of CO adsorption energy on the second-generation core-shell alloy surfaces using the developed neural network model. (b) The parity plot shows a comparison of the CO adsorption energies on selected Cu monolayer alloys calculated by the neural network model and self-consistent density functional theory (DFT) (adapted from Reference [160]).
Owing to advances in protein crystallography, high-resolution enzyme structures are becoming increasingly available (e.g., 132,902 protein structures are deposited in the Protein Bank Database as of 9 August 2018). The Protein Bank Database provides structural information for numerous metal-sulfur enzymes, from which the metal-sulfur distance, sulfur-metal-sulfur angles, and coordination symmetry around the metal center can then be determined. The LUMO and HOMO energy levels of enzyme active centers can be calculated using quantum chemical methods based on simplified enzyme structural models [164,165]. These parameters can be compared with the corresponding crystal structures and band energies of inorganic minerals [71,166,167]. Such comparisons between the electronic structure of minerals and enzymes would aid the screening and identification of suitable mineral catalysts for specific target reactions.
Structure–function relationships can be reliably predicted by combining molecular-level information with quantum chemical calculations. As reviewed in Section 4, heterogeneous sulfide catalysts typically have surface states that are distinct from those of single-metal sulfides and can only be characterized by spectroscopic measurements. In situ spectroscopic monitoring of the structural evolution of catalysts provides molecular information of key reaction intermediates. The well-defined molecular structure of active species can further be applied in DFT calculations to derive the descriptor (adsorption energy of key intermediate) of the activity. This is particularly important in cases where the catalyst structure changes during charge accumulation and/or new intermediate adsorbates are formed. Amorphous materials are another example to be examined spectroscopically for elucidating reaction mechanisms [113,130,131].
Despite the recent advancement of catalytic screening mentioned above, several challenges remain to be made in order to effectively apply these approaches. Specifically, for DFT calculations, determining the descriptor in multi-step reactions is more difficult compared to single-step reactions, as the descriptor changes depending on the reaction conditions, such as the atmosphere and solvent composition, pressure, pH, and temperature. The amount of surface coverage by adsorbates and adsorbate–adsorbate interactions also adds to the complexity of such computations. In addition, during the reaction process, the original surface structure may be modified. Spectroscopic investigation of the intermediate processes is needed to trace these potential structural changes and provide an accurate molecular basis for DFT calculations.
6. Perspectives
Mineral-mediated catalysis is at the center of chemical evolution involved in the emergence of life. Interdisciplinary knowledge from the fields of catalysis engineering and mineralogy is needed to rationally explore catalytic processes that occur in plausible geochemical settings. The vast chemical diversity of metal sulfides is associated with marked variations in catalytic properties, which are influenced by metal composition, crystal structure, valence state, and defect structure. However, this diversity provides a possible solution to the current problem in prebiotic chemistry, where usually an inferior activity, poor selectivity of catalytic processes and low chemical diversity among the generated products were shown. As most studies have examined limited types of catalysts, considerable efforts are needed to examine the properties of various types of metal sulfides and their combinations, and to determine concrete structure–function relationships that can be used as a guide to search mineralogy databases for target catalysts. Modern computational chemistry, operando spectroscopy, and machine-learning approaches are critical for catalysis engineering by establishing the key “descriptor” of activity. Further advances in the understanding of electron and proton transfer and d-band theory, which is generally considered to be the most suitable approach for predicting electrocatalytic performance, will aid in the screening of mineral catalysts for prebiotic reactions crucial for the origin of life.
Supplementary Materials
The following material is available online at http://www.mdpi.com/2075-1729/8/4/46/s1, Figure S1: Species-locality distribution of Fe-containing sulfides (top 20). Figure S2: Species-locality distribution of Ni-containing sulfides (top 12). Figure S3: Species-locality distribution of Cu-containing sulfides (top 10). Figure S4: Species-locality distribution of Co-containing sulfides. Figure S5: Species-locality distribution of Mo-containing sulfides. Figure S6: Species-locality distribution of Mn-containing sulfides. Figure S7: Species-locality distribution of W-containing sulfides. Figure S8: Species-locality distribution of V-containing sulfides. Figure S9: (a) Locality-mineral distribution of Cu single-metal sulfide species and chemical diversity with regards to chemical composition, Cu/S valence states and crystal symmetry; (b) plots of the distribution of species type and locality counts for species containing Cu(I), Cu(II) and Cu(I,II) valence states. Figure S10: (a) Locality-mineral distribution of Ni single-metal sulfide species and chemical diversity with regards to chemical composition, Ni/S valence states and crystal symmetry; (b) plots of the distribution of species type and locality counts for species containing Ni(II), Ni(III) and Ni(II,III) valence states. Figure S11: (a) Locality-mineral distribution of Co single-metal sulfide species and chemical diversity with regards to chemical composition, Co/S valence states and crystal symmetry; (b) plots of the distribution of species type and locality counts for species containing Co(II), Co(III) and Co(II,III) valence states. Table S1: Chemical properties of Fe-Cu and Fe-Ni binary-sulfide minerals. Table S2: Sulfide minerals with ternary compositions. Table S3: X-ray amorphous mineral species in the RRUFF database.
Author Contributions
Y.L. and R.N. conceived and designed this study. Y.L. surveyed the chemical diversity of sulfide minerals from the RRUFF database. Y.L., N.K., and R.N. searched and discussed the chemical aspects relating to sulfide-catalyzed prebiotic synthesis. All authors contributed to the writing and editing of the paper.
Funding
This work was supported by the Astrobiology Center Program of the National Institutes of Natural Sciences (NINS) (Grant Number AB292004).
Acknowledgments
The authors thank Shawn E. McGlynn of Earth-Life Science Institute, Michael J. Russell of NASA Jet Propulsion Laboratory, and Ken Takai and Masahiro Yamamoto of the Japan Agency for Marine-Earth Science and Technology (JAMSTEC) for discussions about prebiotic chemistry and origin of life theory at deep-sea hydrothermal vents. The authors thank Shaunna M. Morrison and Robert M. Hazen of Carnegie Institution for introducing RRUFF database and discussing about mineral-mediated catalysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kitadai, N.; Maruyama, S. Origins of building blocks of life: A review. Geosci. Front. 2018, 9, 1117–1153. [Google Scholar] [CrossRef]
- James Cleaves, H., II; Michalkova Scott, A.; Hill, F.C.; Leszczynski, J.; Sahai, N.; Hazen, R. Mineral–organic interfacial processes: Potential roles in the origins of life. Chem. Soc. Rev. 2012, 41, 5502. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Russell, M.J. On the origin of biochemistry at an alkaline hydrothermal vent. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2007, 362, 1887–1925. [Google Scholar] [CrossRef] [PubMed]
- Cody, G.D. Geochemical Connections to Primitive Metabolism. Elements 2005, 1, 139–143. [Google Scholar] [CrossRef]
- Russell, M.J.; Hall, A.J.; Mellersh, A.R. On the Dissipation of Thermal and Chemical Energies on the Early Earth. In Natural and Laboratory-Simulated Thermal Geochemical Processes; Springer: Dordrecht, The Netherlands, 2003; pp. 325–388. [Google Scholar]
- Wächtershäuser, G. On the Chemistry and Evolution of the Pioneer Organism. Chem. Biodivers. 2007, 4, 584–602. [Google Scholar] [CrossRef] [PubMed]
- Wächtershäuser, G. Before Enzymes and Templates: Theory of Surface Metabolism. Microbiol. Rev. 1988, 52, 452–484. [Google Scholar] [PubMed]
- Russell, M.J.; Hall, A.J. The emergence of life from iron monosulphide bubbles at a submarine hydrothermal redox and pH front. J. Geol. Soc. Lond. 1997, 154, 377–402. [Google Scholar] [CrossRef]
- Russell, M.J.; Hall, A.J. The onset and early evolution of life. In Memoir 198: Evolution of Early Earth’s Atmosphere, Hydrosphere, and Biosphere—Constraints from Ore Deposits; Geological Society of America: Boulder, CO, USA, 2006; pp. 1–32. [Google Scholar]
- Lane, N.; Martin, W.F. The origin of membrane bioenergetics. Cell 2012, 151, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Mulkidjanian, A.Y.; Galperin, M.Y. On the origin of life in the Zinc world. 2. Validation of the hypothesis on the photosynthesizing zinc sulfide edifices as cradles of life on Earth. Biol. Direct 2009, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Mulkidjanian, A.Y. On the origin of life in the Zinc world: 1. Photosynthesizing, porous edifices built of hydrothermally precipitated zinc sulfide as cradles of life on Earth. Biol. Direct 2009, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Nakamura, R.; Takai, K. Deep-Sea Hydrothermal Fields as Natural Power Plants. ChemElectroChem 2018, 5, 2162–2166. [Google Scholar] [CrossRef]
- Kitadai, N.; Nakamura, R.; Yamamoto, M.; Takai, K.; Li, Y.; Yamaguchi, A.; Gilbert, A.; Ueno, Y.; Yoshida, N.; Oono, Y. Geoelectrochemical CO production: Implications for the autotrophic origin of life. Sci. Adv. 2018, 4, eaao7265. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Yamamoto, M.; Takai, K.; Ishii, T.; Hashimoto, K.; Nakamura, R. Electrochemical CO2 Reduction by Ni-containing Iron Sulfides: How Is CO2 Electrochemically Reduced at Bisulfide-Bearing Deep-sea Hydrothermal Precipitates? Electrochim. Acta 2014, 141, 311–318. [Google Scholar] [CrossRef]
- Li, Y.; Yamaguchi, A.; Yamamoto, M.; Takai, K.; Nakamura, R. Molybdenum Sulfide: A Bioinspired Electrocatalyst for Dissimilatory Ammonia Synthesis with Geoelectrical Current. J. Phys. Chem. C 2017, 121, 2154–2164. [Google Scholar] [CrossRef]
- Nakamura, R.; Takashima, T.; Kato, S.; Takai, K.; Yamamoto, M.; Hashimoto, K. Electrical Current Generation across a Black Smoker Chimney. Angew. Chem. 2010, 122, 7858–7860. [Google Scholar] [CrossRef]
- Yamamoto, M.; Nakamura, R.; Oguri, K.; Kawagucci, S.; Suzuki, K.; Hashimoto, K.; Takai, K. Generation of Electricity and Illumination by an Environmental Fuel Cell in Deep-Sea Hydrothermal Vents. Angew. Chem. 2013, 125, 10958–10961. [Google Scholar] [CrossRef]
- Barge, L.M.; Kee, T.P.; Doloboff, I.J.; Hampton, J.M.P.; Ismail, M.; Pourkashanian, M.; Zeytounian, J.; Baum, M.M.; Moss, J.A.; Lin, C.-K.; et al. The fuel cell model of abiogenesis: a new approach to origin-of-life simulations. Astrobiology 2014, 14, 254–270. [Google Scholar] [CrossRef] [PubMed]
- Barge, L.M.; Branscomb, E.; Brucato, J.R.; Cardoso, S.S.S.; Cartwright, J.H.E.; Danielache, S.O.; Galante, D.; Kee, T.P.; Miguel, Y.; Mojzsis, S.; et al. Thermodynamics, Disequilibrium, Evolution: Far-From-Equilibrium Geological and Chemical Considerations for Origin-Of-Life Research. Orig. Life Evol. Biospheres 2017, 47, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Barge, L.M.; Abedian, Y.; Russell, M.J.; Doloboff, I.J.; Cartwright, J.H.E.; Kidd, R.D.; Kanik, I. From Chemical Gardens to Fuel Cells: Generation of Electrical Potential and Current Across Self-Assembling Iron Mineral Membranes. Angew. Chem. 2015, 127, 8302–8305. [Google Scholar] [CrossRef]
- Sangster, D.F. Precambrian volcanogenic massive sulfide deposits in Canada: A review. Geol. Surv. Can. Pap. 1972, 72–22, 1–43. [Google Scholar]
- Hazen, R.M.; Papineau, D.; Bleeker, W.; Downs, R.T.; Ferry, J.M.; McCoy, T.J.; Sverjensky, D.A.; Yang, H. Mineral evolution. Am. Mineral. 2008, 93, 1693–1720. [Google Scholar] [CrossRef]
- Von Damm, K. Seafloor Hydrothermal Activity: Black Smoker Chemistry And Chimneys. Annu. Rev. Earth Planet. Sci. 1990, 18, 173–204. [Google Scholar] [CrossRef]
- Reed, M.H. Sulfide Mineral Precipitation from Hydrothermal Fluids. Rev. Mineral. Geochem. 2006, 61, 609–631. [Google Scholar] [CrossRef]
- Macleod, G.; McKeown, C.; Hall, A.J.; Russell, M.J. Hydrothermal and oceanic pH conditions of possible relevance to the origin of life. Orig. Life Evol. Biospheres 1994, 24, 19–41. [Google Scholar] [CrossRef]
- Dahl, T.W.; Wirth, S.B. Molybdenum isotope fractionation and speciation in a euxinic lake—Testing ways to discern isotope fractionation processes in a sulfidic setting. Chem. Geol. 2017, 460, 84–92. [Google Scholar] [CrossRef]
- Helz, G.R.; Bura-Nakić, E.; Mikac, N.; Ciglenečki, I. New model for molybdenum behavior in euxinic waters. Chem. Geol. 2011, 284, 323–332. [Google Scholar] [CrossRef]
- Erickson, B.E.; Helz, G.R. Molybdenum(VI) speciation in sulfidic waters: Stability and lability of thiomolybdates. Geochim. Cosmochim. Acta 2000, 64, 1149–1158. [Google Scholar] [CrossRef]
- Helz, G.R.R.; Miller, C.V.V.; Charnock, J.M.M.; Mosselmans, J.F.W.F.W.; Pattrick, R.A.D.; Garner, C.D.D.; Vaughan, D.J.J. Mechanism of molybdenum removal from the sea and its concentration in black shales: EXAFS evidence. Geochim. Cosmochim. Acta 1996, 60, 3631–3642. [Google Scholar] [CrossRef]
- Wilde, P.; Lyons, T.W.; Quinby-Hunt, M.S. Organic carbon proxies in black shales: molybdenum. Chem. Geol. 2004, 206, 167–176. [Google Scholar] [CrossRef]
- Kao, L.S.; Peacor, D.R.; Coveney, J.; Zhao, G.; Dungey, K.E.; Curtis, M.D.; Penner-Hahn, J.E. A C/MoS2mixed-layer phase (MoSC) occurring in metalliferous black shales from Southern China, and new data on jordisite. Am. Mineral. 2001, 86, 852–861. [Google Scholar] [CrossRef]
- Anbar, A.D. Oceans. Elements and evolution. Science 2008, 322, 1481–1483. [Google Scholar] [CrossRef] [PubMed]
- Glass, J.B.; Wolfe-Simon, F.; Anbar, A.D. Coevolution of metal availability and nitrogen assimilation in cyanobacteria and algae. Geobiology 2009, 7, 100–123. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.K.; Jelen, B.I.; Giovannelli, D.; Raanan, H.; Falkowski, P.G. Metal availability and the expanding network of microbial metabolisms in the Archaean eon. Nat. Geosci. 2017, 10, 629–636. [Google Scholar] [CrossRef]
- Russell, M.J.; Daniel, R.M.; Hall, A.J. On the emergence of life via catalytic iron-sulphide membranes. Terra Nov. 1993, 5, 343–347. [Google Scholar] [CrossRef]
- Russell, M.J.; Daia, D.E.; Hall, A.J. The emergence of life from FeS bubbles at alkaline hot springs in an acid ocean. In Molecular Evolution and the Origin of Life; Wiegel, J., Michael, A.W.W., Eds.; Taylor & Francis: London, UK, 1998; pp. 77–126. [Google Scholar]
- Roldan, A.; Hollingsworth, N.; Roffey, A.; Islam, H.-U.; Goodall, J.B.M.; Catlow, C.R.A.; Darr, J.A.; Bras, W.; Sankar, G.; Holt, K.B.; et al. Bio-inspired CO2 conversion by iron sulfide catalysts under sustainable conditions. Chem. Commun. 2015, 51, 7501–7504. [Google Scholar] [CrossRef] [PubMed]
- Hazen, R.M. Mineral surfaces and the prebiotic selection and organization of biomolecules. Am. Mineral. 2006, 91, 1715–1729. [Google Scholar] [CrossRef]
- Orgel, L.E. Polymerization on the rocks: Theoretical introduction. Orig. Life Evol. Biospheres 1998, 28, 227–234. [Google Scholar] [CrossRef]
- Shatynski, S.R. The thermochemistry of transition metal sulfides. Oxid. Met. 1977, 11, 307–320. [Google Scholar] [CrossRef]
- Moh, G.H. High-temperature metal sulfide chemistry. In Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 1978; pp. 107–151. ISBN 978-3-540-35680-6. [Google Scholar]
- Chianelli, R.R.; Daage, M.; Ledoux, M.J. Fundamental Studies of Transition-Metal Sulfide Catalytic Materials. Adv. Catal. 1994, 40, 177–232. [Google Scholar] [CrossRef]
- Chianelli, R.R. Periodic trends transition metal sulfide catalysis: Intuition and theory. Oil Gas Sci. Technol. 2006, 61, 503–513. [Google Scholar] [CrossRef]
- Summers, D.P.; Khare, B. Nitrogen Fixation on Early Mars and Other Terrestrial Planets: Experimental Demonstration of Abiotic Fixation Reactions to Nitrite and Nitrate. Astrobiology 2007, 7, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Summers, D.P. Ammonia Formation By The Reduction Of Nitrite/Nitrate By FeS: Ammonia Formation Under Acidic Conditions. Orig. Life Evol. Biospheres 2005, 35, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Wachterhauser, G. Activated Acetic Acid by Carbon Fixation on (Fe, Ni) S under Primordial Conditions. Science 1997, 276, 245–247. [Google Scholar] [CrossRef]
- Huber, C.; Wächtershäuser, G. Α-Hydroxy and Α-Amino Acids Under Possible Hadean, Volcanic Origin-of-Life Conditions. Science 2006, 314, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Wächtershäuser, G. Peptides by Activation of Amino Acids with CO on (Ni,Fe)S Surfaces: Implications for the Origin of Life. Science 1998, 281, 670–672. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Wächtershäuser, G. Primordial reductive amination revisited. Tetrahedron Lett. 2003, 44, 1695–1697. [Google Scholar] [CrossRef]
- Hazen, R.M.; Hystad, G.; Golden, J.J.; Hummer, D.R.; Liu, C.; Downs, R.T.; Morrison, S.M.; Ralph, J.; Grew, E.S. Cobalt mineral ecology. Am. Mineral. 2017, 102, 108–116. [Google Scholar] [CrossRef]
- Liu, C.; Hystad, G.; Golden, J.J.; Hummer, D.R.; Downs, R.T.; Morrison, S.M.; Ralph, J.P.; Hazen, R.M. Chromium mineral ecology. Am. Mineral. 2017, 102, 612–619. [Google Scholar] [CrossRef]
- Cody, G.D.; Boctor, N.Z.; Brandes, J.A.; Filley, T.R.; Hazen, R.M.; Yoder, H.S. Assaying the catalytic potential of transition metal sulfides for abiotic carbon fixation. Geochim. Cosmochim. Acta 2004, 68, 2185–2196. [Google Scholar] [CrossRef]
- Roger, I.; Shipman, M.A.; Symes, M.D. Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat. Rev. Chem. 2017, 1, 0003. [Google Scholar] [CrossRef]
- Koper, M.T.M. Theory of the transition from sequential to concerted electrochemical proton-electron transfer. Phys. Chem. Chem. Phys. 2013, 15, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Koper, M.T.M. Theory of multiple proton–electron transfer reactions and its implications for electrocatalysis. Chem. Sci. 2013, 4, 2710–2723. [Google Scholar] [CrossRef]
- Koper, M.T.M. Volcano Activity Relationships for Proton-Coupled Electron Transfer Reactions in Electrocatalysis. Top. Catal. 2015, 58, 1153–1158. [Google Scholar] [CrossRef]
- Nitschke, W.; McGlynn, S.E.; Milner-White, E.J.; Russell, M.J. On the antiquity of metalloenzymes and their substrates in bioenergetics. Biochim. Biophys. Acta Bioenerg. 2013, 1827, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.J.; Barge, L.M.; Bhartia, R.; Bocanegra, D.; Bracher, P.J.; Branscomb, E.; Kidd, R.; McGlynn, S.; Meier, D.H.; Nitschke, W.; et al. The drive to life on wet and icy worlds. Astrobiology 2014, 14, 308–343. [Google Scholar] [CrossRef] [PubMed]
- Heinen, W.; Lauwers, A.M. Organic sulfur compounds resulting from the interaction of iron sulfide, hydrogen sulfide and carbon dioxide in an anaerobic aqueous environment. Orig. Life Evol. Biospheres 1996, 26, 131–150. [Google Scholar] [CrossRef]
- Brandes, J.A.; Boctor, N.Z.; Cody, G.D.; Cooper, B.A.; Hazen, R.M.; Yoder, H.S. Abiotic nitrogen reduction on the early Earth. Nature 1998, 395, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Dörr, M.; Käßbohrer, J.; Grunert, R.; Kreisel, G.; Brand, W.A.; Werner, R.A.; Geilmann, H.; Apfel, C.; Robl, C.; Weigand, W. A Possible Prebiotic Formation of Ammonia from Dinitrogen on Iron Sulfide Surfaces. Angew. Chem. Int. Ed. 2003, 42, 1540–1543. [Google Scholar] [CrossRef] [PubMed]
- Cody, G.D. Primordial Carbonylated Iron-Sulfur Compounds and the Synthesis of Pyruvate. Science 2000, 289, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Booth, B.G.; Chatt, J. The reactions of carbon monoxide and nitric oxide with tertiary phosphine complexes of iron (II), cobalt (II) and nickel (II). J. Chem. Soc. 1962, 2099–2106. [Google Scholar] [CrossRef]
- Butler, I.S.; Basolo, F.; Pearson, R.G. Kinetics Studies of Carbon Monoxide Insertion Reactions. Reactions of Cyclopentadienyl(methyl)iron Dicarbonyl and Cyclopentadienyl(methyl) Molybdenum Tricarbonyl with Phosphines and Phosphites. Inorg. Chem. 1967, 6, 2074–2079. [Google Scholar] [CrossRef]
- Calderazzo, F.; Cotton, F.A. Carbon Monoxide Insertion Reactions. I. The Carbonylation of Methyl Manganese Pentacarbonyl and Decarbonylation of Acetyl Manganese Pentacarbonyl. Inorg. Chem. 1962, 1, 30–36. [Google Scholar] [CrossRef]
- Novikov, Y.; Copley, S.D. Reactivity landscape of pyruvate under simulated hydrothermal vent conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 13283–13288. [Google Scholar] [CrossRef] [PubMed]
- Ang, R.; Khan, A.U.; Tsujii, N.; Takai, K.; Nakamura, R.; Mori, T. Thermoelectricity Generation and Electron-Magnon Scattering in a Natural Chalcopyrite Mineral from a Deep-Sea Hydrothermal Vent. Angew. Chem. Int. Ed. Engl. 2015, 54, 12909–12922. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Li, Y.; Takashima, T.; Hashimoto, K.; Nakamura, R. CO2 Reduction Using an Electrochemical Approach from Chemical, Biological, and Geological Aspects in the Ancient and Modern Earth; Springer: Cham, Switzerland, 2016; pp. 213–228. [Google Scholar]
- Trail, D.; Watson, E.B.; Tailby, N.D. The oxidation state of Hadean magmas and implications for early Earth’s atmosphere. Nature 2011, 480, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Bullett, D.W. Electronic structure of 3d pyrite- and marcasite-type sulphides. J. Phys. C Solid State Phys. 1982, 15, 6163–6174. [Google Scholar] [CrossRef]
- Uhlig, I.; Szargan, R.; Nesbitt, H.; Laajalehto, K. Surface states and reactivity of pyrite and marcasite. Appl. Surf. Sci. 2001, 179, 222–229. [Google Scholar] [CrossRef]
- Snowball, I.F. Magnetic hysteresis properties of greigite (Fe3S4) and a new occurrence in Holocene sediments from Swedish Lappland. Phys. Earth Planet. Inter. 1991, 68, 32–40. [Google Scholar] [CrossRef]
- Rochette, P.; Fillion, G.; Mattéi, J.L.; Dekkers, M.J. Magnetic transition at 30–34 Kelvin in pyrrhotite: Insight into a widespread occurrence of this mineral in rocks. Earth Planet. Sci. Lett. 1990, 98, 319–328. [Google Scholar] [CrossRef]
- Boller, H. IUCr On the synthesis, crystal chemistry and magnetic properties of rasvumite and related compounds. Acta Crystallogr. Sect. A Found. Crystallogr. 2004, 60, s47. [Google Scholar] [CrossRef]
- Silva, J.C.M.; De Abreu, H.A.; Duarte, H.A. Electronic and structural properties of bulk arsenopyrite and its cleavage surfaces—A DFT study. RSC Adv. 2015, 5, 2013–2023. [Google Scholar] [CrossRef]
- Jones, R.A.; Nesbitt, H.W. XPS evidence for Fe and As oxidation states and electronic states in loellingite (FeAs2). Am. Mineral. 2002, 87, 1692–1698. [Google Scholar] [CrossRef]
- Fesseler, J.; Jeoung, J.H.; Dobbek, H. How the [NiFe4S4] Cluster of CO Dehydrogenase Activates CO2 and NCO-. Angew. Chem. Int. Ed. 2015, 54, 8560–8564. [Google Scholar] [CrossRef] [PubMed]
- van Velzen, A.J.; Dekkers, M.J.; Zijderveld, J.D.A. Magnetic iron-nickel sulphides in the Pliocene and Pleistocene marine marls from the Vrica section (Calabria, Italy). Earth Planet. Sci. Lett. 1993, 115, 43–55. [Google Scholar] [CrossRef]
- Bostick, B.C.; Fendorf, S.; Helz, G.R. Differential adsorption of molybdate and tetrathiomolybdate on pyrite (FeS2). Environ. Sci. Technol. 2003, 37, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Tribovillard, N.; Riboulleau, A.; Lyons, T.; Baudin, F. Enhanced trapping of molybdenum by sulfurized marine organic matter of marine origin in Mesozoic limestones and shales. Chem. Geol. 2004, 213, 385–401. [Google Scholar] [CrossRef]
- Vorlicek, T.P.; Kahn, M.D.; Kasuya, Y.; Helz, G.R. Capture of molybdenum in pyrite-forming sediments: role of ligand-induced reduction by polysulfides. Geochim. Cosmochim. Acta 2004, 68, 547–556. [Google Scholar] [CrossRef]
- Luther, G.W.; Rickard, D.T. Metal sulfide cluster complexes and their biogeochemical importance in the environment. J. Nanopart. Res. 2005, 7, 389–407. [Google Scholar] [CrossRef]
- Guttenberg, N.; Virgo, N.; Chandru, K.; Scharf, C.; Mamajanov, I. Bulk measurements of messy chemistries are needed for a theory of the origins of life. Philos. Trans. A Math. Phys. Eng. Sci. 2017, 375, 20160347. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, D.S. Messy biology and the origins of evolutionary innovations. Nat. Chem. Biol. 2010, 6, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.J.; Richardson, D.J.; Butt, J.N. Catalytic Protein Film Voltammetry from a Respiratory Nitrate Reductase Provides Evidence for Complex Electrochemical Modulation of Enzyme Activity. Biochemistry 2001, 40, 11294–11307. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J. Elucidating the mechanisms of coupled electron transfer and catalytic reactions by protein film voltammetry. Biochim. Biophys. Acta 2006, 1757, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Hoeben, F.J.M.; Meijer, F.S.; Dekker, C.; Albracht, S.P.J.; Heering, H.A.; Lemay, S.G. Toward Single-Enzyme Molecule Electrochemistry: [NiFe]-Hydrogenase Protein Film Voltammetry at Nanoelectrodes. ACS Nano 2008, 2, 2497–2504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hu, Y.; Ma, L.; Zhu, G.; Wang, Y.; Xue, X.; Chen, R.; Yang, S.; Jin, Z. Progress and Perspective of Electrocatalytic CO2 Reduction for Renewable Carbonaceous Fuels and Chemicals. Adv. Sci. 2018, 5, 1700275. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Robert, M.; Savéant, J.-M. Catalysis of the electrochemical reduction of carbon dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.; Shen, J.; Schouten, K.J.P.; Calle-Vallejo, F.; Koper, M.T.M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, T.; Choi, J.; Yasuda, H. Transformation of Carbon Dioxide Transformation of Carbon Dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Dinh, C.; Burdyny, T.; Kibria, M.G.; Seifitokaldani, A.; Gabardo, C.M.; García de Arquer, F.P.; Kiani, A.; Edwards, J.P.; De Luna, P.; Bushuyev, O.S.; et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Kikuchi, K.; Suzuki, S. Production of CO and CH4 in electrochemical reduction of CO2 at metal electrodes in aqueous hydrogencarbonate solution. Chem. Lett. 1985, 14, 1695–1698. [Google Scholar] [CrossRef]
- Chan, K.; Tsai, C.; Hansen, H.A.; Nørskov, J.K. Molybdenum Sulfides and Selenides as Possible Electrocatalysts for CO2 Reduction. ChemCatChem. 2014, 6, 1899–1905. [Google Scholar] [CrossRef]
- Peterson, A.A.; Nørskov, J.K. Activity Descriptors for CO2 Electroreduction to Methane on Transition-Metal Catalysts. J. Phys. Chem. Lett. 2012, 3, 251–258. [Google Scholar] [CrossRef]
- Shi, C.; Hansen, H.A.; Lausche, A.C.; Nørskov, J.K. Trends in electrochemical CO2 reduction activity for open and close-packed metal surfaces. Phys. Chem. Chem. Phys. 2014, 16, 4720–4727. [Google Scholar] [CrossRef] [PubMed]
- Asadi, M.; Kumar, B.; Behranginia, A.; Rosen, B.A.; Baskin, A.; Repnin, N.; Pisasale, D.; Phillips, P.; Zhu, W.; Haasch, R.; et al. Robust carbon dioxide reduction on molybdenum disulphide edges. Nat. Commun. 2014, 5, 4470. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Li, Y.; Ooka, H.; Go, Y.K.; Jin, F.; Kim, S.H.; Nakamura, R. Selective Electrocatalytic Reduction of Nitrite to Dinitrogen Based on Decoupled Proton-Electron Transfer. J. Am. Chem. Soc. 2018, 140, 2012–2015. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.B.; Moura, J.J.G. How Biology Handles Nitrite. Chem. Rev. 2014, 114, 5273–5357. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Perathoner, S.; Ampelli, C.; Mebrahtu, C.; Su, D.; Centi, G. Room-Temperature Electrocatalytic Synthesis of NH3 from H2O and N2 in a Gas-liquid-Solid Three-Phase Reactor. ACS Sustain. Chem. Eng. 2017, 5, 7393–7400. [Google Scholar] [CrossRef]
- Chen, S.; Perathoner, S.; Ampelli, C.; Mebrahtu, C.; Su, D.; Centi, G. Electrocatalytic Synthesis of Ammonia at Room Temperature and Atmospheric Pressure from Water and Nitrogen on a Carbon-Nanotube-Based Electrocatalyst. Angew. Chem. Int. Ed. 2017, 56, 2699–2703. [Google Scholar] [CrossRef] [PubMed]
- Kordali, V.; Kyriacou, G.; Lambrou, C. Electrochemical synthesis of ammonia at atmospheric pressure and low temperature in a solid polymer electrolyte cell. Chem. Commun. 2000, 1673–1674. [Google Scholar] [CrossRef]
- Bao, D.; Zhang, Q.; Meng, F.-L.; Zhong, H.-X.; Shi, M.-M.; Zhang, Y.; Yan, J.-M.; Jiang, Q.; Zhang, X.-B. Electrochemical Reduction of N2 under Ambient Conditions for Artificial N2 Fixation and Renewable Energy Storage Using N2 /NH3 Cycle. Adv. Mater. 2017, 29, 1604799. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-J.; Bao, D.; Shi, M.-M.; Wulan, B.-R.; Yan, J.-M.; Jiang, Q. Amorphizing of Au Nanoparticles by CeO x -RGO Hybrid Support towards Highly Efficient Electrocatalyst for N2 Reduction under Ambient Conditions. Adv. Mater. 2017, 29, 1700001. [Google Scholar] [CrossRef] [PubMed]
- Yandulov, D.V. Catalytic Reduction of Dinitrogen to Ammonia at a Single Molybdenum Center. Science 2003, 301, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Schrock, R.R. Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Acc. Chem. Res. 2005, 38, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Ji, X.; Ren, X.; Cui, G.; Li, L.; Xie, F.; Wang, H.; Li, B.; Sun, X. MoO3 nanosheets for efficient electrocatalytic N2 fixation to NH3. J. Mater. Chem. A 2018, 6, 12974–12977. [Google Scholar] [CrossRef]
- Zhang, L.; Ji, X.; Ren, X.; Ma, Y.; Shi, X.; Tian, Z.; Asiri, A.M.; Chen, L.; Tang, B.; Sun, X. Electrochemical Ammonia Synthesis via Nitrogen Reduction Reaction on a MoS2 Catalyst: Theoretical and Experimental Studies. Adv. Mater. 2018, 30, 1800191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ji, X.; Ren, X.; Luo, Y.; Shi, X.; Asiri, A.M.; Zheng, B.; Sun, X. Efficient Electrochemical N2 Reduction to NH3 on MoN Nanosheets Array under Ambient Conditions. ACS Sustain. Chem. Eng. 2018, 6, 9550–9554. [Google Scholar] [CrossRef]
- Di Giovanni, C.; Wang, W.A.; Nowak, S.; Grenèche, J.M.; Lecoq, H.; Mouton, L.; Giraud, M.; Tard, C. Bioinspired iron sulfide nanoparticles for cheap and long-lived electrocatalytic molecular hydrogen evolution in neutral water. ACS Catal. 2014, 4, 681–687. [Google Scholar] [CrossRef]
- Kornienko, N.; Resasco, J.; Becknell, N.; Jiang, C.M.; Liu, Y.S.; Nie, K.; Sun, X.; Guo, J.; Leone, S.R.; Yang, P. Operando Spectroscopic Analysis of an Amorphous Cobalt Sulfide Hydrogen Evolution Electrocatalyst. J. Am. Chem. Soc. 2015, 137, 7448–7455. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Ting, L.R.L.; Neo, P.H.L.; Zhang, Y.-J.; Peterson, A.A.; Yeo, B.S. Operando Raman Spectroscopy of Amorphous Molybdenum Sulfide (MoSx) during the Electrochemical Hydrogen Evolution Reaction: Identification of Sulfur Atoms as Catalytically Active Sites for H + Reduction. ACS Catal. 2016, 6, 7790–7798. [Google Scholar] [CrossRef]
- Ting, L.R.L.; Deng, Y.; Ma, L.; Zhang, Y.-J.; Peterson, A.A.; Yeo, B.S. Catalytic Activities of Sulfur Atoms in Amorphous Molybdenum Sulfide for the Electrochemical Hydrogen Evolution Reaction. ACS Catal. 2016, 6, 861–867. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, H.; Li, S.; Wang, R.; Sun, X.; Zhou, M.; Zhou, J.; Lou, X.W.D.; Xie, Y. Defect-rich MoS2 ultrathin nanosheets with additional active edge sites for enhanced electrocatalytic hydrogen evolution. Adv. Mater. 2013, 25, 5807–5820. [Google Scholar] [CrossRef] [PubMed]
- Merki, D.; Fierro, S.; Vrubel, H.; Hu, X. Amorphous molybdenum sulfide films as catalysts for electrochemical hydrogen production in water. Chem. Sci. 2011, 2, 1262–1267. [Google Scholar] [CrossRef]
- Lukowski, M.A.; Daniel, A.S.; Meng, F.; Forticaux, A.; Li, L.; Jin, S. Enhanced Hydrogen Evolution Catalysis from Enhanced Hydrogen Evolution Catalysis from Chemically Exfoliated Metallic MoS2 Nanosheets. J. Am. Chem. Soc. 2013, 135, 10274–10277. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, T.F.; Bonde, J.; Zhang, J.; Ooi, B.L.; Andersson, K.; Ulstrup, J.; Chorkendorff, I. Hydrogen evolution on supported incomplete cubane-type [Mo3S4]4+ electrocatalysts. J. Phys. Chem. C 2008, 112, 17492–17498. [Google Scholar] [CrossRef]
- Voiry, D.; Yamaguchi, H.; Li, J.; Silva, R.; Alves, D.C.B.; Fujita, T.; Chen, M.; Asefa, T.; Shenoy, V.B.; Eda, G.; et al. Enhanced catalytic activity in strained chemically exfoliated WS2 nanosheets for hydrogen evolution. Nat. Mater. 2013, 12, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Voiry, D.; Ahn, S.J.; Kang, D.; Kim, A.Y.; Chhowalla, M.; Shin, H.S. Two-dimensional hybrid nanosheets of tungsten disulfide and reduced graphene oxide as catalysts for enhanced hydrogen evolution. Angew. Chem. Int. Ed. 2013, 52, 13751–13754. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Li, G.; Wang, Z.; Zhu, H.; Zhang, T.; Xiao, S.; Guo, W.; Yang, S. Metallic Iron–Nickel Sulfide Ultrathin Nanosheets As a Highly Active Electrocatalyst for Hydrogen Evolution Reaction in Acidic Media. J. Am. Chem. Soc. 2015, 137, 11900–11903. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Gong, M.; Chou, H.L.; Pan, C.J.; Chen, H.A.; Wu, Y.; Lin, M.C.; Guan, M.; Yang, J.; Chen, C.W.; et al. Highly active and stable hybrid catalyst of cobalt-doped FeS2 nanosheets-carbon nanotubes for hydrogen evolution reaction. J. Am. Chem. Soc. 2015, 137, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.F.; Song, J.; Li, K.; Tahir, M.; Wang, Y.T.; Pan, L.; Wang, L.; Zhang, X.; Zou, J.J. Hollow Cobalt-Based Bimetallic Sulfide Polyhedra for Efficient All-pH-Value Electrochemical and Photocatalytic Hydrogen Evolution. J. Am. Chem. Soc. 2016, 138, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Faber, M.S.; Lukowski, M.A.; Ding, Q.; Kaiser, N.S.; Jin, S. Earth-abundant metal pyrites (FeS2, CoS2, NiS2, and their alloys) for highly efficient hydrogen evolution and polysulfide reduction electrocatalysis. J. Phys. Chem. C 2014, 118, 21347–21356. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.; Chan, K.; Nørskov, J.K.; Abild-Pedersen, F. Rational design of MoS2 catalysts: tuning the structure and activity via transition metal doping. Catal. Sci. Technol. 2015, 5, 246–253. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, J.; Li, S.; Grote, F.; Zhang, X.; Zhang, H.; Wang, R. Controllable Disorder Engineering in Oxygen-Incorporated MoS2 Ultrathin Nanosheets for Efficient Hydrogen Evolution. J. Am. Chem. Soc. 2013, 135, 17881–17888. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhuo, J.; Du, K.; Chen, B.; Zhu, Z.; Shao, Y.; Li, M. Electrochemically fabricated polypyrrole and MoSx copolymer films as a highly active hydrogen evolution electrocatalyst. Adv. Mater. 2014, 26, 3761–3766. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Dou, K.; Kaun, C.; Kuang, Q.; Yang, S. MoSe nanosheets and their graphene hybrids: Synthesis, characterization and hydrogen evolution reaction studies. Mater. Chem. A 2014, 2, 360–364. [Google Scholar] [CrossRef]
- Li, D.J.; Maiti, U.N.; Lim, J.; Choi, D.S.; Lee, W.J.; Oh, Y.; Lee, G.Y.; Kim, S.O. Molybdenum sulfide/N-doped CNT forest hybrid catalysts for high-performance hydrogen evolution reaction. Nano Lett. 2014, 14, 1228–1233. [Google Scholar] [CrossRef] [PubMed]
- Lassalle-Kaiser, B.; Merki, D.; Vrubel, H.; Gul, S.; Yachandra, V.K.; Hu, X.; Yano, J. Evidence from in Situ X-ray Absorption Spectroscopy for the Involvement of Terminal Disulfide in the Reduction of Protons by an Amorphous Molybdenum Sulfide Electrocatalyst. J. Am. Chem. Soc. 2015, 137, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Casalongue, H.G.S.; Benck, J.D.; Tsai, C.; Karlsson, R.K.B.; Kaya, S.; Ng, M.L.; Pettersson, L.G.M.; Abild-Pedersen, F.; Nørskov, J.K.; Ogasawara, H.; et al. Operando Characterization of an Amorphous Molybdenum Sulfide Nanoparticle Catalyst during the Hydrogen Evolution Reaction. J. Phys. Chem. C 2014, 118, 29252–29259. [Google Scholar] [CrossRef]
- Tran, P.D.; Tran, T.V.; Orio, M.; Torelli, S.; Truong, Q.D.; Nayuki, K.; Sasaki, Y.; Chiam, S.Y.; Yi, R.; Honma, I.; et al. Coordination polymer structure and revisited hydrogen evolution catalytic mechanism for amorphous molybdenum sulfide. Nat. Mater. 2016, 15, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, T.F.; Jorgensen, K.P.; Bonde, J.; Nielsen, J.H.; Horch, S.; Chorkendorff, I. Identification of Active Edge Sites for Electrochemical H2 Evolution from MoS2 Nanocatalysts. Science 2007, 317, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Hinnemann, B.; Moses, P.G.; Bonde, J.; Jorgensen, K.P.; Nielsen, J.H.; Horch, S.; Chorkendorff, I.; Nørskov, J.K. Biomimetic Hydrogen Evolution: MoS2 Nanoparticles as Catalyst for Hydrogen Evolution. J. Am. Chem. Soc. 2005, 127, 5308–5309. [Google Scholar] [CrossRef] [PubMed]
- Karunadasa, H.I.; Montalvo, E.; Sun, Y.; Majda, M.; Long, J.R.; Chang, C.J. A Molecular MoS2 Edge Site Mimic for Catalytic Hydrogen Generation. Science 2012, 335, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Hijazi, A.; Kemmegne-Mbouguen, J.C.; Floquet, S.; Marrot, J.; Fize, J.; Artero, V.; David, O.; Magnier, E.; Pegot, B.; Cadot, E. Tuning the electrocatalytic hydrogen evolution reaction promoted by [Mo2O2S2]-based molybdenum cycles in aqueous medium. Dalton Trans. 2013, 42, 4848–4858. [Google Scholar] [CrossRef] [PubMed]
- Kibsgaard, J.; Jaramillo, T.F.; Besenbacher, F. Building an appropriate active-site motif into a hydrogen-evolution catalyst with thiomolybdate [Mo3S13]2− clusters. Nat. Chem. 2014, 6, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.A.; Saha, A.; Raghavachari, K. Bond Activation and Hydrogen Evolution from Water through Reactions with M3S4 (M = Mo, W) and W3S3 Anionic Clusters. J. Phys. Chem. A 2017, 121, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Nakamura, R. Structural change of molybdenum sulfide facilitates the electrocatalytic hydrogen evolution reaction at neutral pH as revealed by in situ Raman spectroscopy. Chin. J. Catal. 2018, 39, 401–406. [Google Scholar] [CrossRef]
- Durst, J.; Siebel, A.; Simon, C.; Hasché, F.; Herranz, J.; Gasteiger, H.A. New insights into the electrochemical hydrogen oxidation and evolution reaction mechanism. Energy Environ. Sci. 2014, 7, 2255–2260. [Google Scholar] [CrossRef]
- Vasić, D.D.; Pašti, I.A.; Mentus, S.V. DFT study of platinum and palladium overlayers on tungsten carbide: Structure and electrocatalytic activity toward hydrogen oxidation/evolution reaction. Int. J. Hydrogen Energy 2013, 38, 5009–5018. [Google Scholar] [CrossRef]
- Kiros, Y.; Schwartz, S. Long-term hydrogen oxidation catalysts in alkaline fuel cells. J. Power Sources 2000, 87, 101–105. [Google Scholar] [CrossRef]
- Zhuang, Z.; Giles, S.A.; Zheng, J.; Jenness, G.R.; Caratzoulas, S.; Vlachos, D.G.; Yan, Y. Nickel supported on nitrogen-doped carbon nanotubes as hydrogen oxidation reaction catalyst in alkaline electrolyte. Nat. Commun. 2016, 7, 10141. [Google Scholar] [CrossRef] [PubMed]
- Mund, K.; Richter, G.; von Sturm, F. Titanium-Containing Raney Nickel Catalyst for Hydrogen Electrodes in Alkaline Fuel Cell Systems. J. Electrochem. Soc. 1977, 124, 1–6. [Google Scholar] [CrossRef]
- Sheng, W.; Bivens, A.P.; Myint, M.; Zhuang, Z.; Forest, R.V.; Fang, Q.; Chen, J.G.; Yan, Y. Non-precious metal electrocatalysts with high activity for hydrogen oxidation reaction in alkaline electrolytes. Energy Environ. Sci. 2014, 7, 1719–1724. [Google Scholar] [CrossRef]
- Wilson, A.D.; Newell, R.H.; McNevin, M.J.; Muckerman, J.T.; Rakowski DuBois, M.; DuBois, D.L. Hydrogen Oxidation and Production Using Nickel-Based Molecular Catalysts with Positioned Proton Relays. J. Am. Chem. Soc. 2006, 128, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Nørskov, J.K.; Bligaard, T.; Rossmeisl, J.; Christensen, C.H. Towards the computational design of solid catalysts. Nat. Chem. 2009, 1, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Montoya, J.H.; Peterson, A.A.; Nørskov, J.K. Insights into C-C Coupling in CO2 Electroreduction on Copper Electrodes. ChemCatChem 2013, 5, 737–742. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Koper, M.T.M. Theoretical Considerations on the Electroreduction of CO to C2 Species on Cu(100) Electrodes. Angew. Chem. Int. Ed. 2013, 52, 7282–7285. [Google Scholar] [CrossRef] [PubMed]
- Gurudayal, G.; Bullock, J.; Srankó, D.F.; Towle, C.M.; Lum, Y.; Hettick, M.; Scott, M.C.; Javey, A.; Ager, J. Efficient solar-driven electrochemical CO2 reduction to hydrocarbons and oxygenates. Energy Environ. Sci. 2017, 10, 2222–2230. [Google Scholar] [CrossRef]
- He, J.; Dettelbach, K.E.; Salvatore, D.A.; Li, T.; Berlinguette, C.P. High-Throughput Synthesis of Mixed-Metal Electrocatalysts for C2 Reduction. Angew. Chem. Int. Ed. 2017, 56, 6068–6072. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Zhuang, Z.; Gao, M.; Zheng, J.; Chen, J.G.; Yan, Y. Correlating hydrogen oxidation and evolution activity on platinum at different pH with measured hydrogen binding energy. Nat. Commun. 2015, 6, 5848. [Google Scholar] [CrossRef] [PubMed]
- Skúlason, E.; Tripkovic, V.; Björketun, M.E.; Gudmundsdóttir, S.; Karlberg, G.; Rossmeisl, J.; Bligaard, T.; Jónsson, H.; Nørskov, J.K. Modeling the Electrochemical Hydrogen Oxidation and Evolution Reactions on the Basis of Density Functional Theory Calculations. J. Phys. Chem. C 2010, 114, 18182–18197. [Google Scholar] [CrossRef]
- Howalt, J.G.; Bligaard, T.; Rossmeisl, J.; Vegge, T. DFT based study of transition metal nano-clusters for electrochemical NH3 production. Phys. Chem. Chem. Phys. 2013, 15, 7785–7795. [Google Scholar] [CrossRef] [PubMed]
- Skúlason, E.; Bligaard, T.; Gudmundsdóttir, S.; Studt, F.; Rossmeisl, J.; Abild-Pedersen, F.; Vegge, T.; Jónsson, H.; Nørskov, J.K. A theoretical evaluation of possible transition metal electro-catalysts for N2 reduction. Phys. Chem. Chem. Phys. 2012, 14, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Abghoui, Y.; Garden, A.L.; Hlynsson, V.F.; Björgvinsdóttir, S.; Ólafsdóttir, H.; Skúlason, E. Enabling electrochemical reduction of nitrogen to ammonia at ambient conditions through rational catalyst design. Phys. Chem. Chem. Phys. 2015, 17, 4909–4918. [Google Scholar] [CrossRef] [PubMed]
- van der Ham, C.J.M.; Koper, M.T.M.; Hetterscheid, D.G.H. Challenges in reduction of dinitrogen by proton and electron transfer. Chem. Soc. Rev. 2014, 43, 5183–5191. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, Y.; Koper, M.T.M.; Calle-Vallejo, F. Bond-making and breaking between carbon, nitrogen, and oxygen in electrocatalysis. J. Am. Chem. Soc. 2014, 136, 15694–15701. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Ma, X.; Li, Z.; Achenie, L.E.K.; Xin, H. Machine-learning-augmented chemisorption model for CO2 electroreduction catalyst screening. J. Phys. Chem. Lett. 2016, 6, 3528–3533. [Google Scholar] [CrossRef] [PubMed]
- Ulissi, Z.W.; Tang, M.T.; Xiao, J.; Liu, X.; Torelli, D.A.; Karamd, M.; Cummins, K.; Hahn, C.; Lewis, N.S.; Jaramillo, T.F.; et al. Machine-learning methods enable exhaustive searches for active bimetallic facets and reveal active site motifs for CO2 reduction. ACS Catal. 2017, 7, 6600–6608. [Google Scholar] [CrossRef]
- Hong, W.T.; Welsch, R.E.; Shao-Horn, Y. Descriptors of oxygen-evolution activity for oxides: A statistical evaluation. J. Phys. Chem. C 2016, 120, 78–86. [Google Scholar] [CrossRef]
- Ooka, H.; Hashimoto, K.; Nakamura, R. Design strategy of multi-electron transfer catalysts based on a bioinformatic analysis of oxygen evolution and reduction enzymes. Mol. Inf. 2018, 37, 1700139. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, I.K.; Enemark, J.H. EPR Studies of Oxo–Molybdenum(V) Complexes with Sulfur Donor Ligands: Implications for the Molybdenum Center of Sulfite Oxidase. Inorg. Chem. 1996, 35, 4873–4882. [Google Scholar] [CrossRef] [PubMed]
- Inscore, F.E.; McNaughton, R.; Westcott, B.L.; Helton, M.E.; Jones, R.; Dhawan, I.K.; Enemark, J.H.; Kirk, M.L. Spectroscopic Evidence for a Unique Bonding Interaction in Oxo-Molybdenum Dithiolate Complexes: Implications for σ Electron Transfer Pathways in the Pyranopterin Dithiolate Centers of Enzymes. Inorg. Chem. 1999, 38, 1401–1410. [Google Scholar] [CrossRef]
- Roldan, A.; Santos-Carballal, D.; de Leeuw, N.H. A comparative DFT study of the mechanical and electronic properties of greigite Fe3S4 and magnetite Fe3O4. J. Chem. Phys. 2013, 138, 204712. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, D.J.; Tossell, J.A. Electronic structures of sulfide minerals-Theory and experiment. Phys. Chem. Miner. 1983, 9, 253–262. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).