Microfluidic Reactors for Carbon Fixation under Ambient-Pressure Alkaline-Hydrothermal-Vent Conditions
by
 ,
,
Victor Sojo
1,2,3,* ,
,
Aya Ohno
1,
Shawn E. McGlynn
1,4,5,
Yoichi M.A. Yamada
1 and
Ryuhei Nakamura
1,4 1
RIKEN Center for Sustainable Resource Science, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
2
Systems Biophysics, Ludwig-Maximilian University of Munich, Munich 80799, Germany
3
Institute for Advanced Study, Berlin. Wallotstr. 19, Berlin 14193, Germany
4
Earth-Life Science Institute, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan
5
Blue Marble Space Institute of Science, Seattle, WA 98154, USA
*
Author to whom correspondence should be addressed.
Life 2019, 9(1), 16; https://doi.org/10.3390/life9010016
Submission received: 20 December 2018
/
Revised: 22 January 2019
/
Accepted: 25 January 2019
/
Published: 1 February 2019
(This article belongs to the Special Issue The Origin and Early Evolution of Life: Prebiotic Chemistry of Biomolecules)
Abstract
:The alkaline-hydrothermal-vent theory for the origin of life predicts the spontaneous reduction of CO2, dissolved in acidic ocean waters, with H2 from the alkaline vent effluent. This reaction would be catalyzed by Fe(Ni)S clusters precipitated at the interface, which effectively separate the two fluids into an electrochemical cell. Using microfluidic reactors, we set out to test this concept. We produced thin, long Fe(Ni)S precipitates of less than 10 µm thickness. Mixing simplified analogs of the acidic-ocean and alkaline-vent fluids, we then tested for the reduction of CO2. We were unable to detect reduced carbon products under a number of conditions. As all of our reactions were performed at atmospheric pressure, the lack of reduced carbon products may simply be attributable to the low concentration of hydrogen in our system, suggesting that high-pressure reactors may be a necessity.
1. Introduction
From its very start, life required reduced organic molecules. In a minimalistic scenario for abiogenesis (i.e. the emergence of life), one source of such molecules was the reduction of CO2, in a process overall similar (and potentially homologous) to the modern enzyme-facilitated pathways of extant autotrophic cells [1,2,3,4,5,6,7,8]. A number of reducing agents (i.e. sources of electrons) for reducing CO2 were possible on the early Earth, but multiple reasons make hydrogen (H2) a good candidate. This is discussed at length elsewhere (see [5,8,9,10] and references therein), but two points are worth mentioning here, namely: (1) hydrogen is formed spontaneously in the Earth’s crust via the “serpentinization” of ultramafic-rock minerals such as olivine [9,11,12,13]; and (2) it is used to reduce CO2 by members of both archaea and bacteria, in their respective versions of the Wood–Ljungdahl (WL) or acetyl Co-A pathway [5,14,15].
This process results in CO2 fixation and the production of ATP within a single, linear, metabolic pathway [5,16,17], so it has been suggested as a potential candidate for the metabolism of the last universal common ancestor (LUCA). However, one problem with extrapolating this scenario towards the origin of life is that, while the overall pathway of carbon fixation via the WL pathway is exergonic, the initial reaction between H2 and CO2 is not spontaneous under standard abiotic conditions [18]. Confirming these thermodynamic predictions, numerous experimental electrochemical results show that CO2 reduction is indeed disfavored under most observational conditions, requiring overpotentials of at least 180 mV in order to overcome the initial endergonic steps [19].
However, under putative ancient alkaline-vent conditions, CO2 would have been dissolved in slightly acidic ocean waters (pH 5~7), whereas H2 would have been a product of serpentinization, emanating as part of the efflux of the alkaline vent, itself rich in OH— (pH 9~12).
The geologically sustained pH difference across the vent minerals provided an additional electrochemical driving force, potentially circumventing the lack of reducing power of H2 for CO2 reduction [19]. The reduction of CO2 to formic acid (HCOOH, its first 2-electron reduction product) involves protonation, so it would have been favored in acidic ocean waters. In turn, the oxidation of H2 releases protons (H+), which would have been favored in the alkaline waters that contained the dissolved H2 [20]. The two fluids would have been separated by the mineral precipitates of the vent, which included iron (and nickel) sulfides (Fe(Ni)S) as well as silicates (as reviewed in [21]). Reduced on the inside and oxidized on the outside, a situation analogous to an electrochemical cell would have existed between the two sides. The electrons released from H2 would then hypothetically travel through the electrically conductive Fe(Ni)S network [19], and drive the reduction of CO2 on the other side (Figure 1). This contrast between the pH of the two solutions matches the polarity of modern cells, and it has been suggested as a potential driver of the origins of both membrane bioenergetics and carbon fixation [6,7,8,20,22,23,24].
The pH gradients have also recently been shown to hold in the microscale, at up to six pH‑unit differences [25], suggesting the potential of microfluidic devices to study the reduction of CO2 with H2 under these conditions.
The immobilization of a catalytic boundary by the meeting of two fluids has been demonstrated using a microfluidic reactor [26], so we envisioned that this methodology could be applied to the formation of catalytic Fe(Ni)S clusters at the interface, elaborating on previous results [25]. This effectively mimics the ancient alkaline-vent conditions by the in-situ creation of an electrochemical cell between the oxidized CO2 in the acidic-ocean side and the reduced H2 in the alkaline-vent side (Figure 1).
Here, we present preliminary results in our study of the potential reduction of CO2 with H2. We used microfluidics to simulate the mixing of oceanic and serpentinizing fluids under the putative conditions of ancient alkaline hydrothermal vents—although notably at atmospheric pressure.
We simulated the two sides of the vent system by mixing fluids containing combinations of Fe2+/Ni2+ and CO2 for the acidic-ocean side; whereas the alkaline-vent simulant contained HS— and bubbled H2 (Figure 2).
2. Methods
The microfluidic reactor systems were assembled using custom chips with a Y-shape design, etched from glass by the Institute of Microchemical Technology Co., Ltd. The channels in the system were half-pipes with a width of 100 ± 2.5 µm and a maximum depth of 40 ± 1 µm.
At one tip of the “Y”, we input the analog of the acidic-ocean fluid, with the alkaline-vent analog being input at the other tip (as summarized in the diagram of Figure 2). The compositions of the fluids, presented in Table 1 and detailed in the Results, are similar to those reported elsewhere [10].
Because of the sensitivity of Fe2+ to oxygen in air, and in order to mimic the anoxic Hadean conditions at the origin of life more closely, the water in all of the experiments was de-gassed by boiling for 5 min and then cooling under constant argon bubbling for 30 min. The salts were weighed and then kept as solids under positive pressure of argon. The necessary amounts of de-aerated water were added, and the solutions bubbled with argon for another 15 minutes, with the exception of the H2-containing solutions, which were bubbled with H2. The final pH of the H2-containing solution was ~11, whereas that of the carbonic solution was ~6. Gas-tight syringes were then filled (all with a maximum volume of 1 mL, from Hamilton USA).
The full system setup is presented in Figure 3. The simulants of the acidic-ocean and alkaline-vent fluids were driven into the system using syringe pumps at adjustable flow rates, generally between 0.2 and 20 µL/min. The pumps were modular BabyBee Syringe Drive units from Bioanalytical Systems Inc. (BASi, West Lafayette, IN, USA), regulated by a BeeHive Syringe Drive Controller, also from BASi. The temperature was measured using an infrared thermometer, and regulated using a standard heating plate. A stainless-steel block was laid directly onto the heating plate, with the glass reactor laid on top of the block (Figure 3c, middle). The reactor chip was held in a custom-made stainless-steel casing (Figure 3a–c). The formation of the precipitates was followed using an inline USB microscope (Figure 3b, middle) connected to a standard laptop computer.
Reaction blanks were taken by running water through the chip on both inlets, after the precipitation reaction had taken place, but before adding any CO2/NaHCO3.
NMR spectra (1H and 13C) were determined using a JEOL spectrometer with a 600 MHz magnet.
3. Results
To facilitate microfluidic mixing and simulate the mixing of alkaline-vent and oceanic fluids, we replicated previous concentrations [10] and separated each of the fluids into independently controlled gas-tight syringes. For the acidic side, three inflows were used (see Table 1 for further details), as follows:
- De-aerated water (to achieve parallel flow and take sample blanks).
- FeCl2 (50 mM) and NiCl2 (5 mM).
- De-aerated water bubbled with CO2 (at atmospheric pressure), or alternatively dissolved NaHCO3 (100 mM), acidified with HCl (1 M) to pH 6.
Conversely, the two alkaline-side syringes contained the following fluids:
- De-aerated water.
- De-aerated water with Na2S (10 mM), K2HPO4 (10 mM), and Na2Si3O7 (10 mM), bubbled with H2 (at atmospheric pressure), and at a final pH of ~11.
After attaining parallel flow by letting water run from both inlets for 20 min, the mixing of the metal and sulfide fluids produced a thin dark precipitate at the interface. By changing either or both of the two fluids back to the water syringes, the thickness of the precipitate could be closely controlled (Figure 4).
Once the precipitates were formed at the interface, the inflows were swapped to the respective syringes containing CO2 and H2 (which, in the latter case, was the same as that for the sulfide in most of our reactions). The reaction conditions were controlled to last between 30 min and 24 h, and temperatures between laboratory conditions (~25 °C) and 70 °C (Table 1 and Methods).
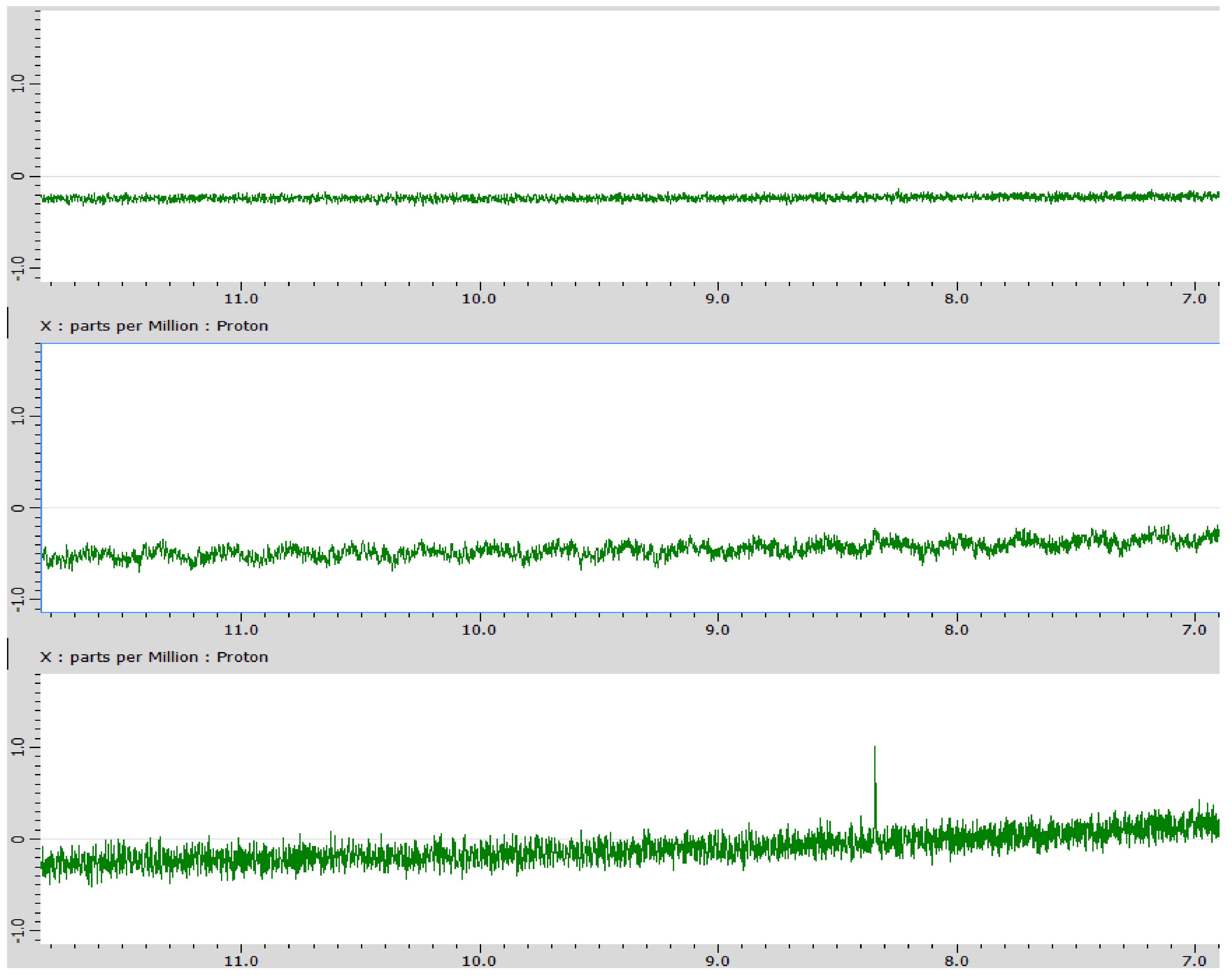
Analysis using 1H-NMR showed a peak in the formic acid region (~8.3 ppm, Figure A1). To assess whether the peak in this result was indeed due to formic acid, 13C‑labelled sodium bicarbonate (NaH13CO3), acidified to pH 6, was used. A peak in the formic-acid region (~164 ppm) of the 13C-NMR spectrum (Figure A2) was however shown to correspond to unreacted NaH13CO3 itself. This was confirmed by spiking with 13C‑labelled formic acid (H13COOH, Figure A3). The 1H spectrum for 13C‑labelled formic acid should show a splitting of the original singlet into a doublet. However, the doublet is not observed in the reaction efflux with NaH13CO3, appearing only when 13C‑labelled formic acid was added externally (Figure A4). The original peak at ~8.3 ppm in the 1H spectrum remains unidentified.
Overall, we did not detect reduced carbon products under the conditions that we tested. Varying conditions (Table 1), including concentrations, thickness of the precipitates, reaction temperature, reaction times, or doping the Fe(Ni)S precipitates with heteroatoms such as Mo(VI), produced no detectable difference.
4. Discussion
We aimed to probe the reduction of CO2 with H2 under putative ancient alkaline-hydrothermal-vent conditions.
In contrast to previous work [10], in which formate and formaldehyde were reported, we do not detect any soluble reduced carbon products under our experimental conditions. A separate set of experiments conducted with a larger-scale system modeled after previous work [27] also failed to yield detectable CO2 reduction (Chang and McGlynn, unpublished).
In view of this, it is important to stress that our experimental conditions were not exhaustive, and they failed to replicate alkaline vents in at least one crucial aspect—pressure.
The solubility of hydrogen is extremely low at ambient pressures, and decreases sharply as temperature rises towards the boiling point of water [28]. We bubbled H2 at atmospheric pressure, prior to the reaction, and did not continue bubbling during the reaction once the desired volume was stored in a gas-tight syringe. Since the H2-containing fluid in our experiments is at pH 11 and the CO2-containing fluid at pH 6, 1 bar of H2 at room temperature is predicted to be sufficient to reduce CO2, a reaction whose favorability would be enhanced with greater pressure as a result of the gain in concentrations [29,30]. The magnitude of the overpotential needed to overcome any kinetic barriers however remains unknown, and it is possible that high-pressure reactors are a necessity to overcome these barriers and evaluate the possibility of H2-powered reduction of CO2 under alkaline-vent conditions. Similarly, continuous bubbling of H2 (as in previous work [10]) may be necessary, given the high volatility of H2 gas.
Notably, recent results show that reduced carbon products are undetectable under similar reaction conditions using pure metals as catalysts [31,32], instead staying bound to the catalysts until concentrated KOH is used to remove them. It is therefore plausible (although it would need to be shown) that any reduced products that we may have formed remained bound to the Fe(Ni)S precipitates. We also note that we did not investigate potential gas-phase products in our study, leaving these as possibilities to be explored.
The electrical potential from the pH gradient under alkaline-vent conditions has been measured at the microscale [25], but it remains to be shown that it can indeed drive otherwise unfavorable redox reactions. Thus, to test the validity of the electrochemical-cell concept—irrespective of its relevance to the origin of life—further (potentially less geologically relevant) experiments could include using stronger reducing and oxidizing agents, at varying concentrations. The utilization of high-pressure reactors to achieve higher concentrations of dissolved gas may be especially important to test for H2-driven reductions.
Broadly, our scheme for overcoming the exergonic steps of carbon reduction relies on the separation of solutions at different conditions coupled to the ability to transfer electrons, which is not unique to pH gradients at alkaline vents—thermal gradients and reducing agents other than H2 are also possible drivers of reduction [29,30].
The alkaline-hydrothermal-vent theory has come under criticism in recent years [33,34,35]. These issues have been addressed elsewhere [36], but it is important to note here that we do not see our results as either disproving the alkaline-vent theory, or providing support for alternative theories for the origin of life. Most simply, they suggest that the catalysts that appropriately lower the kinetic barriers have not been implemented as of yet, or that more realistic conditions—crucially higher pressures, particularly for H2—need to be evaluated to more closely in order ascertain the viability of the hypotheses tested here. These are lines of enquiry that we are pursuing, as are other researchers in the field.
Author Contributions
V.S. planned and performed the research with experimental support from A.O. All authors designed the experiments and analyzed the results. V.S., S.E.M., Y.M.A.Y., and R.N. wrote the manuscript.
Funding
V.S. acknowledges financial support from the Japan Society for the Promotion of Science (JSPS) (FY2016-PE-16721), the European Commission Marie Skłodowska—Curie Actions/European Molecular Biology Organization (EMBO) (ALTF-1455-2015), and the Institute for Advanced Study in Berlin. S.E.M. is supported by NSF Award 1724300.
Acknowledgments
The authors thank Reuben Hudson, Nick Lane, Yamei Li, Hideshi Ooka, and Alexandra Whicher for their suggestions.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
Figure A1.
1H spectrum of dissolved NaHCO3 (top), reaction blank (middle), and reaction efflux (bottom). The reaction efflux shows a peak in the formic acid region, slightly over 8.3 ppm.
Figure A1.
1H spectrum of dissolved NaHCO3 (top), reaction blank (middle), and reaction efflux (bottom). The reaction efflux shows a peak in the formic acid region, slightly over 8.3 ppm.

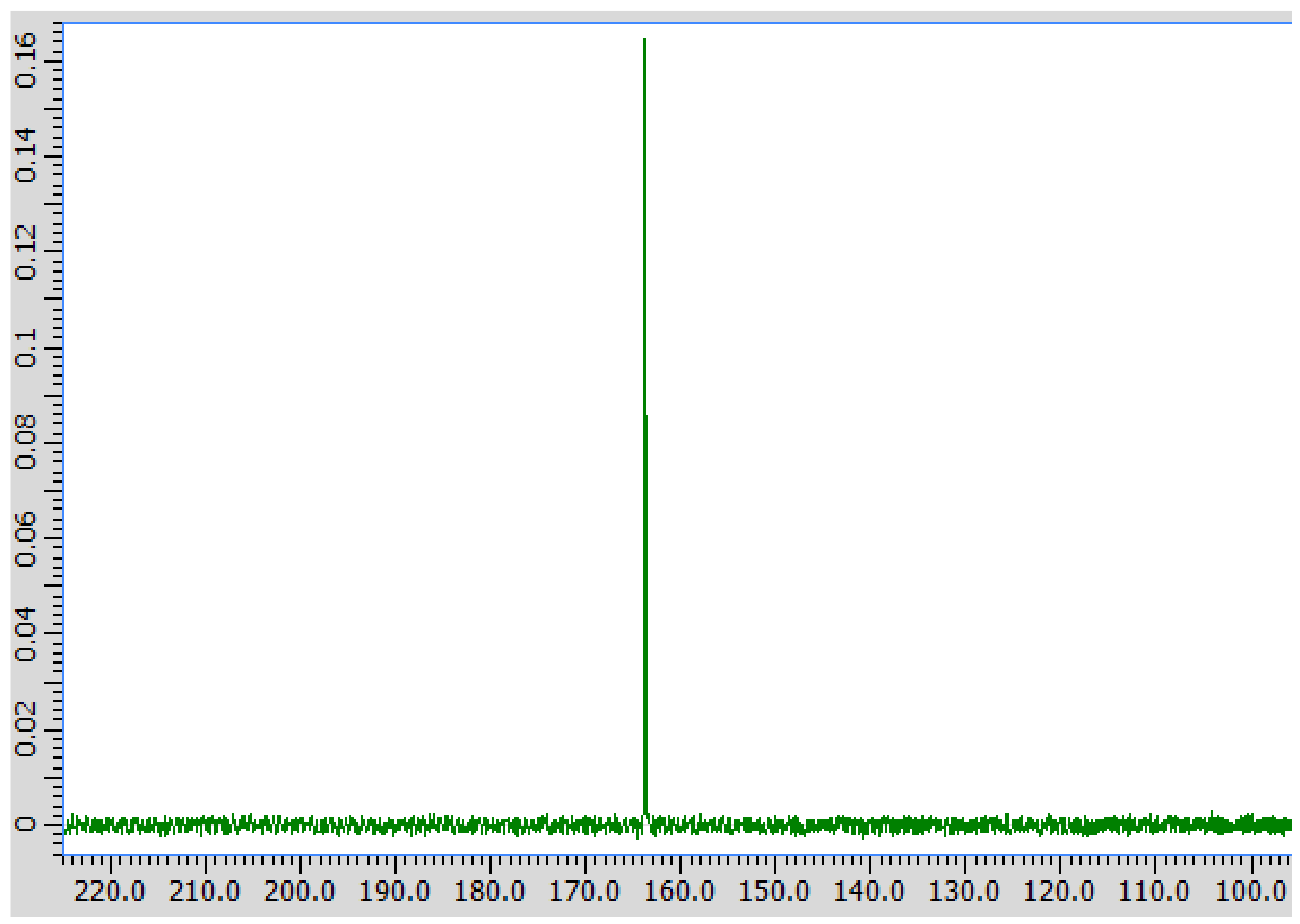
Figure A2.
13C spectrum of reaction efflux, showing a peak in the formic acid region (~164 ppm).

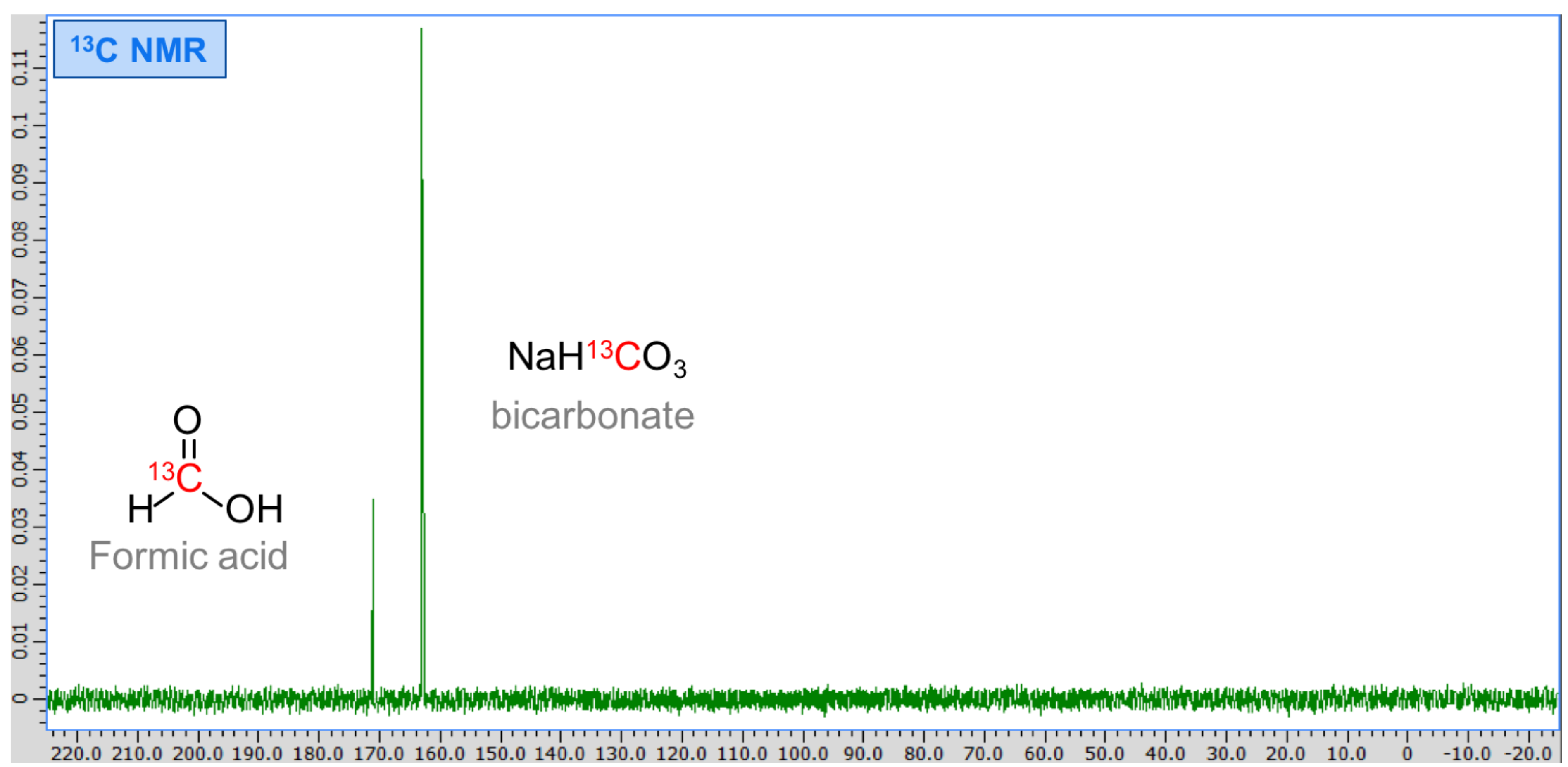
Figure A3.
13C spectrum of reaction efflux with added 13C-labelled formic acid. Instead of a single increased peak, two peaks are visible, thus revealing that formic acid was not produced and the peak to the right corresponds to unreacted bicarbonate in the efflux.
Figure A3.
13C spectrum of reaction efflux with added 13C-labelled formic acid. Instead of a single increased peak, two peaks are visible, thus revealing that formic acid was not produced and the peak to the right corresponds to unreacted bicarbonate in the efflux.

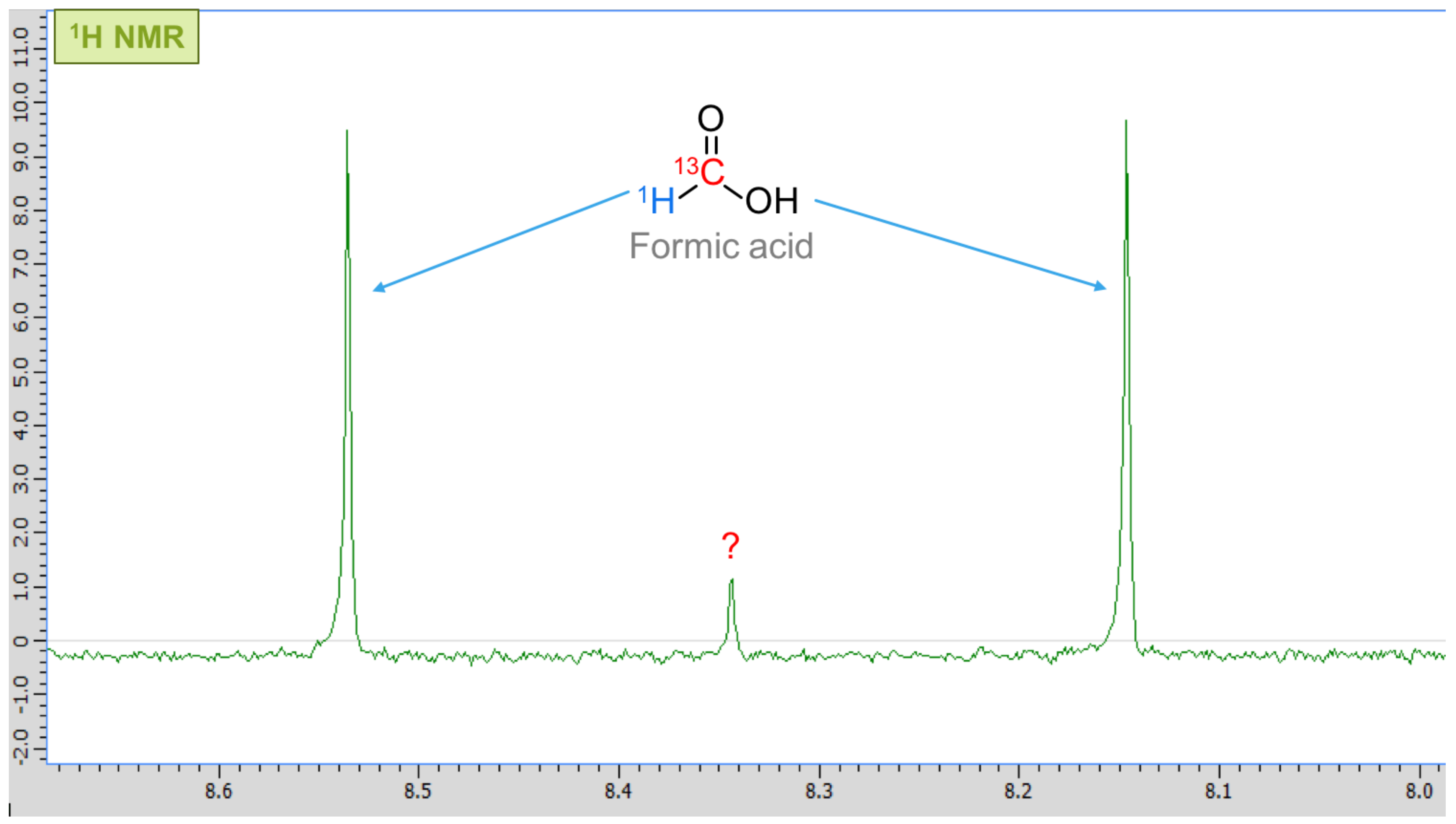
Figure A4.
Adding 13C-labelled formic acid gives a doublet in the 1H spectrum, indicated by the arrows. The peak in the middle (~8.34 ppm) remains unidentified.
Figure A4.
Adding 13C-labelled formic acid gives a doublet in the 1H spectrum, indicated by the arrows. The peak in the middle (~8.34 ppm) remains unidentified.

References
- Wächtershäuser, G. Pyrite formation, the first energy source for life: A hypothesis. Syst. Appl. Microbiol. 1988, 10, 207–210. [Google Scholar] [CrossRef]
- Russell, M.J.; Hall, A.J.; Turner, D. In vitro growth of iron sulphide chimneys: Possible culture chambers for origin-of-life experiments. Terra Nova 1989, 1, 238–241. [Google Scholar] [CrossRef]
- Wächtershäuser, G. Groundworks for an evolutionary biochemistry: The iron-sulphur world. Prog. Biophys. Mol. Biol. 1992, 58, 85–201. [Google Scholar] [CrossRef]
- Maden, B. No soup for starters? Autotrophy and the origins of metabolism. Trends Biochem. Sci. 1995, 20, 337–341. [Google Scholar] [CrossRef]
- Russell, M.J.; Martin, W. The rocky roots of the acetyl-CoA pathway. Trends Biochem. Sci. 2004, 29, 358–63. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Russell, M.J. On the origin of biochemistry at an alkaline hydrothermal vent. Phil. Trans. R. Soc. London B 2007, 362, 1887–1925. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.; Martin, W.F. The origin of membrane bioenergetics. Cell 2012, 151, 1406–1416. [Google Scholar] [CrossRef]
- Sojo, V.; Herschy, B.; Whicher, A.; Camprubí, E.; Lane, N. The origin of life in alkaline hydrothermal vents. Astrobiology 2016, 16, 181–197. [Google Scholar] [CrossRef]
- Sleep, N.H.; Bird, D.K.; Pope, E.C. Serpentinite and the dawn of life. Phil. Trans. R. Soc. London B. Biol. Sci. 2011, 366, 2857–2869. [Google Scholar] [CrossRef] [Green Version]
- Herschy, B.; Whicher, A.; Camprubi, E.; Watson, C.; Dartnell, L.; Ward, J.; Evans, J.R.G.; Lane, N. An origin-of-life reactor to simulate alkaline hydrothermal vents. J. Mol. Evol. 2014, 79, 213–227. [Google Scholar] [CrossRef]
- Sleep, N.H.; Meibom, A.; Fridriksson, T.; Coleman, R.G.; Bird, D.K. H2-rich fluids from serpentinization: Geochemical and biotic implications. Proc. Natl. Acad. Sci. 2004, 101, 12818–12823. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.S.; Baross, J.A.; Delaney, J.R. Volcanoes, fluids, and life at mid-ocean ridge spreading centers. Annu. Rev. Earth Planet. Sci. 2002, 30, 385–491. [Google Scholar] [CrossRef]
- Kelley, D.S.; Karson, J.A.; Früh-Green, G.L.; Yoerger, D.R.; Shank, T.M.; Butterfield, D.A.; Hayes, J.M.; Schrenk, M.O.; Olson, E.J.; Proskurowski, G.; et al. A serpentinite-hosted ecosystem: The Lost City hydrothermal field. Science 2005, 307, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Wood, H.G. Life with CO or CO2 and H2 as a source of carbon and energy. FASEB J. 1991, 5, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Nitschke, W.; Russell, M. Beating the acetyl coenzyme A-pathway to the origin of life. Phil. Trans. R. Soc. B. 2013, 368, 20120258. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.; Allen, J.F.; Martin, W. How did LUCA make a living? Chemiosmosis in the origin of life. BioEssays 2010, 32, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Poehlein, A.; Schmidt, S.; Kaster, A.-K.; Goenrich, M.; Vollmers, J.; Thürmer, A.; Bertsch, J.; Schuchmann, K.; Voigt, B.; Hecker, M.; et al. An ancient pathway combining carbon dioxide fixation with the generation and utilization of a sodium ion gradient for ATP synthesis. PLoS ONE 2012, 7, e33439. [Google Scholar] [CrossRef] [PubMed]
- Maden, B.E. Tetrahydrofolate and tetrahydromethanopterin compared: Functionally distinct carriers in C1 metabolism. Biochem. J. 2000, 350, 609–629. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Yamamoto, M.; Takai, K.; Ishii, T.; Hashimoto, K.; Nakamura, R. Electrochemical CO2 reduction by Ni-containing iron sulfides: How is CO2 electrochemically reduced at bisulfide-bearing deep-sea hydrothermal precipitates? Electrochim. Acta 2014, 141, 311–318. [Google Scholar] [CrossRef]
- Lane, N. The Vital Question: Energy, Evolution, and the Origins of Complex Life; WW Norton & Company: New York, NY, USA, 2015. [Google Scholar]
- Li, Y.; Kitadai, N.; Nakamura, R. Chemical diversity of metal sulfide minerals and its implications for the origin of life. Life 2018, 8, 1–26. [Google Scholar] [CrossRef]
- Martin, W.; Baross, J.; Kelley, D.; Russell, M.J. Hydrothermal vents and the origin of life. Nat. Rev. Microbiol. 2008, 6, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Sojo, V.; Pomiankowski, A.; Lane, N. A bioenergetic basis for membrane divergence in archaea and bacteria. PLoS Biol. 2014, 12, e1001926. [Google Scholar] [CrossRef] [PubMed]
- Lane, N. Bioenergetic constraints on the evolution of complex life. Cold Spring Harb. Perspect. Biol. 2014, 6, a015982. [Google Scholar] [CrossRef]
- Möller, F.M.F.M.; Kriegel, F.; Kieß, M.; Sojo, V.; Braun, D. Steep pH Gradients and Directed Colloid Transport in a Microfluidic Alkaline Hydrothermal Pore. Angew. Chemie Int. Ed. 2017, 56, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.M.A.; Ohno, A.; Sato, T.; Uozumi, Y. Instantaneous click chemistry by a copper-containing polymeric-membrane-installed microflow catalytic reactor. Chem. A Eur. J. 2015, 21, 17269–17273. [Google Scholar] [CrossRef]
- Batista, B.C.; Steinbock, O. Growing inorganic membranes in microfluidic devices: Chemical gardens reduced to linear walls. J. Phys. Chem. C 2015, 119, 27045–27052. [Google Scholar] [CrossRef]
- Pray, H.A.; Schweickert, C.E.; Minnich, B.H. Solubility of hydrogen, oxygen, nitrogen, and helium in water at elevated temperatures. Ind. Eng. Chem. 1952, 44, 1146–1151. [Google Scholar] [CrossRef]
- Kitadai, N.; Nakamura, R.; Yamamoto, M.; Takai, K.; Li, Y.; Yamaguchi, A.; Gilbert, A.; Ueno, Y.; Yoshida, N.; Oono, Y. Geoelectrochemical CO production: Implications for the autotrophic origin of life. Sci. Adv. 2018, 4, eaao7265. [Google Scholar] [CrossRef]
- Ooka, H.; Mcglynn, S.E.; Nakamura, R. Electrochemistry at deep-sea hydrothermal vents: Utilization of the thermodynamic driving force towards the autotrophic origin of life. ChemElectroChem 2018. [Google Scholar] [CrossRef]
- Muchowska, K.B.; Varma, S.J.; Chevallot-Beroux, E.; Lethuillier-Karl, L.; Li, G.; Moran, J. Metals promote sequences of the reverse Krebs cycle. Nat. Ecol. Evol. 2017, 1, 1716–1721. [Google Scholar] [CrossRef] [Green Version]
- Varma, S.J.; Muchowska, K.B.; Chatelain, P.; Moran, J. Native iron reduces CO2 to intermediates and end-products of the acetyl-CoA pathway. Nat. Ecol. Evol. 2018, 2, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.B. Natural pH gradients in hydrothermal alkali vents were unlikely to have played a role in the origin of life. J. Mol. Evol. 2016, 83, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wächtershäuser, G. In praise of error. J. Mol. Evol. 2016, 82, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, J.D. Studies on the origin of life—the end of the beginning. Nat. Rev. Chem. 2017, 1, 0012. [Google Scholar] [CrossRef]
- Lane, N. Proton gradients at the origin of life. BioEssays 2017, 39, 1600217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
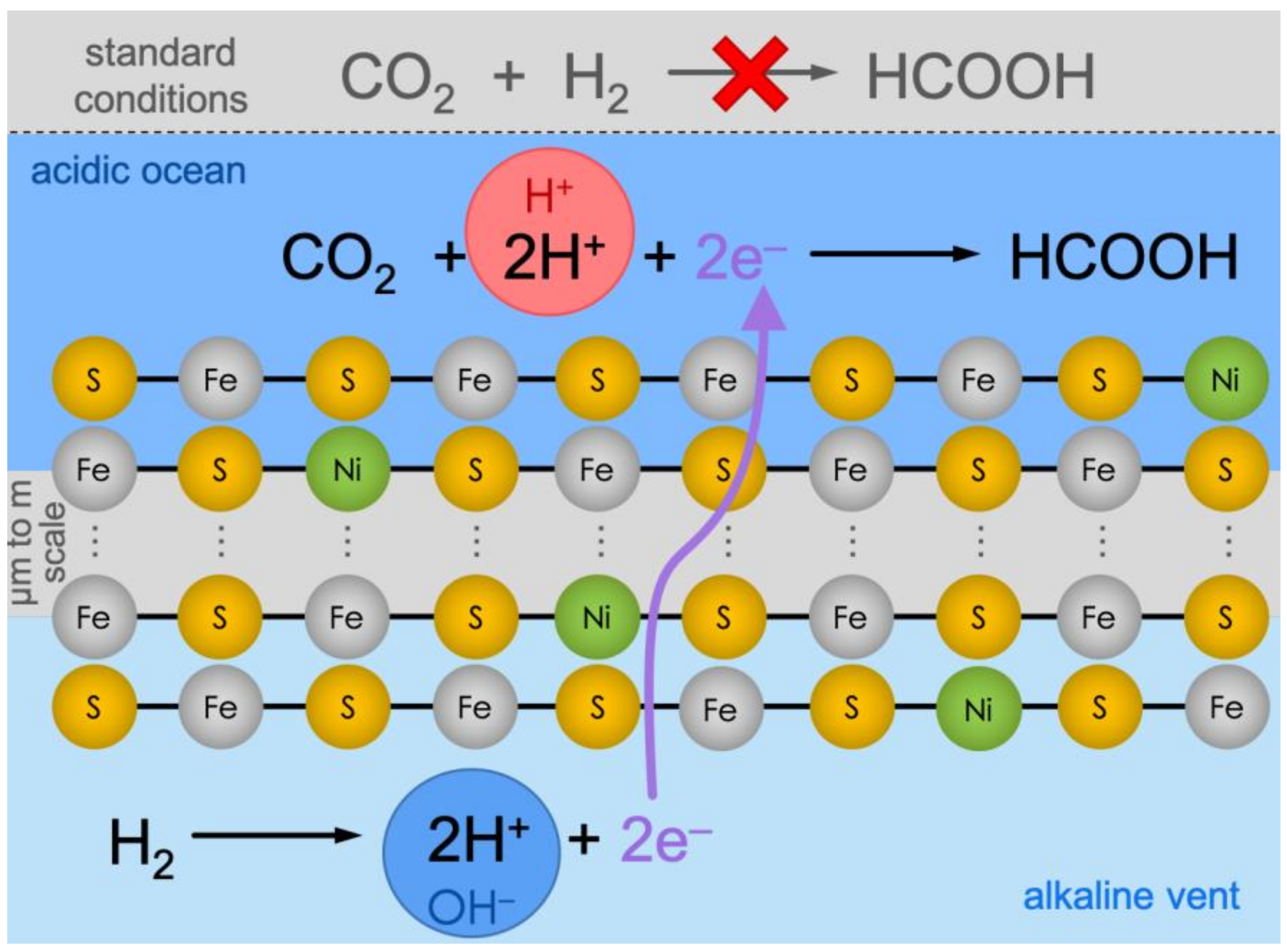
Figure 1.
Under standard conditions (top, grey background), CO2 reduction with H2 is not viable thermodynamically. Conversely, in alkaline vents, the reaction is hypothetically split into two halves, effectively producing an electrochemical cell. On the alkaline-vent side (bottom, light-blue background), the oxidation of H2 to H+ is favored because of the alkaline pH in the vent fluid (symbolized by the blue circle with “OH—”). The electrons would travel through the micrometer-to-meter-scale catalytic Fe(Ni)S precipitate network and meet CO2 at the ocean side (dark-blue background), where the relatively acidic pH (red circle with “H+”) would favor the reduction and protonation towards formic acid (HCOOH).
Figure 1.
Under standard conditions (top, grey background), CO2 reduction with H2 is not viable thermodynamically. Conversely, in alkaline vents, the reaction is hypothetically split into two halves, effectively producing an electrochemical cell. On the alkaline-vent side (bottom, light-blue background), the oxidation of H2 to H+ is favored because of the alkaline pH in the vent fluid (symbolized by the blue circle with “OH—”). The electrons would travel through the micrometer-to-meter-scale catalytic Fe(Ni)S precipitate network and meet CO2 at the ocean side (dark-blue background), where the relatively acidic pH (red circle with “H+”) would favor the reduction and protonation towards formic acid (HCOOH).
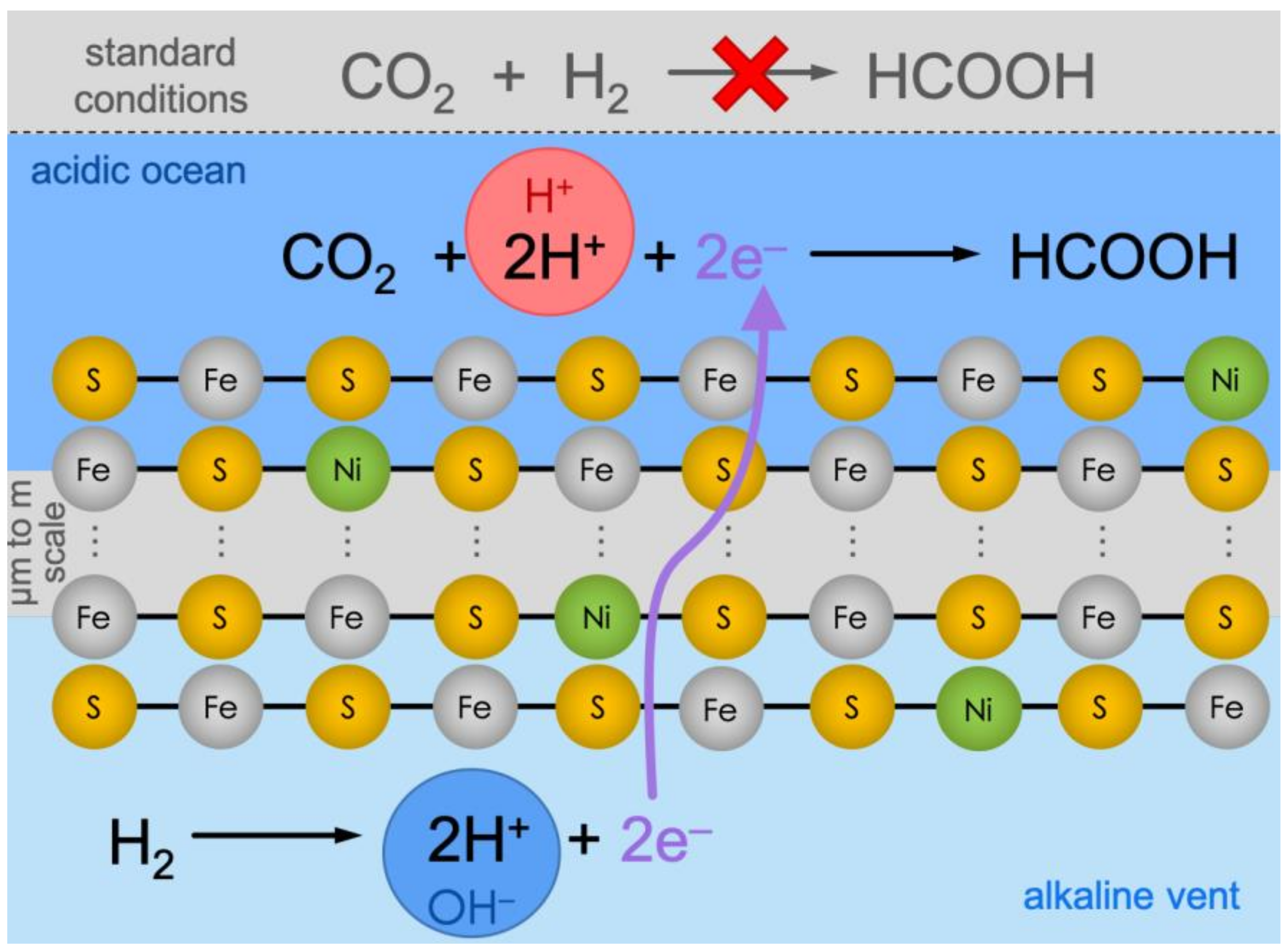
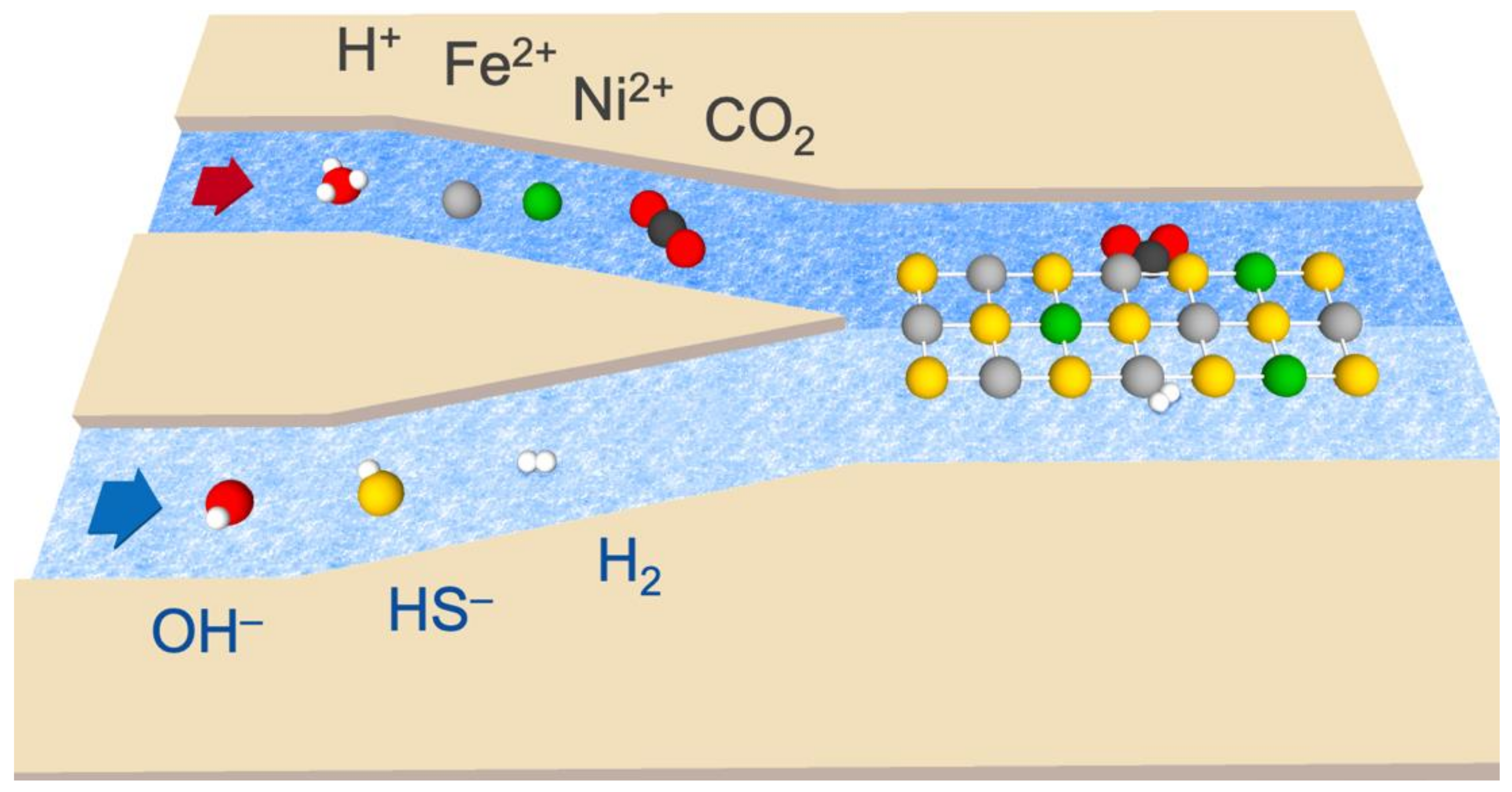
Figure 2.
Diagram of the reaction system, depicting the input of acidic fluid (top, left half) containing H+, Fe2+, Ni2+, and dissolved CO2. The alkaline fluid (bottom, left half) contained OH—, HS—, and dissolved H2. Upon meeting, the fluids form Fe(Ni)S precipitates (represented by the reticulation in the right half), which may serve as catalysts for the indirect redox reaction between H2 and CO2.
Figure 2.
Diagram of the reaction system, depicting the input of acidic fluid (top, left half) containing H+, Fe2+, Ni2+, and dissolved CO2. The alkaline fluid (bottom, left half) contained OH—, HS—, and dissolved H2. Upon meeting, the fluids form Fe(Ni)S precipitates (represented by the reticulation in the right half), which may serve as catalysts for the indirect redox reaction between H2 and CO2.

Figure 3.
Reactor setup. (a) Microfluidic Y-shaped reactor chip with ~100 µm‑width channels, fitted into a stainless-steal holder with screw-in inlets. (b) The two inlets adjusted into position (bottom). A microscope (middle and top) was used to follow the precipitation reaction. (c) Left and far right: BASi drive controller and syringe pumps with Hamilton syringes. Center: reaction chip on the heating plate, with a USB microscope adjusted on top to follow the precipitation.
Figure 3.
Reactor setup. (a) Microfluidic Y-shaped reactor chip with ~100 µm‑width channels, fitted into a stainless-steal holder with screw-in inlets. (b) The two inlets adjusted into position (bottom). A microscope (middle and top) was used to follow the precipitation reaction. (c) Left and far right: BASi drive controller and syringe pumps with Hamilton syringes. Center: reaction chip on the heating plate, with a USB microscope adjusted on top to follow the precipitation.

Figure 4.
Precipitation of Fe(Ni)S at the interface between the acidic and alkaline fluids. By replacing the flow of either (or both) the sulfide or the metals with water, the precipitate could be kept arbitrarily thin.
Figure 4.
Precipitation of Fe(Ni)S at the interface between the acidic and alkaline fluids. By replacing the flow of either (or both) the sulfide or the metals with water, the precipitate could be kept arbitrarily thin.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Conditions tested. The concentrations of NaHCO3 were inevitably lower than shown after acidification to pH 6 with 1 M HCl. In alternative experiments we used CO2 bubbled at atmospheric pressure instead of NaHCO3.
Table 1.
Conditions tested. The concentrations of NaHCO3 were inevitably lower than shown after acidification to pH 6 with 1 M HCl. In alternative experiments we used CO2 bubbled at atmospheric pressure instead of NaHCO3.
| ACIDIC-SIDE CONCENTRATIONS | ALKALINE-SIDE CONCENTRATIONS | ||
|---|---|---|---|
| [FeCl2] | 50 mM | [Na2S] | 10 and 100 mM |
| [NiCl2] | 0 and 10 mM | [K2HPO4] | 10 mM |
| [NaHCO3] | 10, 50, and 100 mM (acidified to pH ~6) | [Na2Si3O7] | 0 and 10 mM |
| [Na2MoO4] | 0 and 1 mM | ||
| CO2 | Bubbled at atmospheric pressure (final pH ~6) | [H2] | Bubbled at atmospheric pressure (final pH ~11) |
| OTHER CONDITIONS | |||
| Reaction durations | ½, 1, 2, 5, 12, and 24 h | Temperature | ~25 (room), 40, 50, 60, and 70 °C |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sojo, V.; Ohno, A.; McGlynn, S.E.; Yamada, Y.M.A.; Nakamura, R. Microfluidic Reactors for Carbon Fixation under Ambient-Pressure Alkaline-Hydrothermal-Vent Conditions. Life 2019, 9, 16. https://doi.org/10.3390/life9010016
AMA Style
Sojo V, Ohno A, McGlynn SE, Yamada YMA, Nakamura R. Microfluidic Reactors for Carbon Fixation under Ambient-Pressure Alkaline-Hydrothermal-Vent Conditions. Life. 2019; 9(1):16. https://doi.org/10.3390/life9010016
Chicago/Turabian StyleSojo, Victor, Aya Ohno, Shawn E. McGlynn, Yoichi M.A. Yamada, and Ryuhei Nakamura. 2019. "Microfluidic Reactors for Carbon Fixation under Ambient-Pressure Alkaline-Hydrothermal-Vent Conditions" Life 9, no. 1: 16. https://doi.org/10.3390/life9010016
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.